
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr. , 16 July 2021
Sec. Pediatric Endocrinology
Volume 9 - 2021 | https://doi.org/10.3389/fped.2021.699129
 Elizabeth Rosenfeld1,2
Elizabeth Rosenfeld1,2 Lauren Mitteer1,2
Lauren Mitteer1,2 Kara Boodhansingh1,2
Kara Boodhansingh1,2 Susan A. Becker1,2Heather McKnight1,2Linda Boyajian1,2Amanda M. Ackermann1,2,3Jennifer M. Kalish2,3,4Tricia R. Bhatti2,5
Susan A. Becker1,2Heather McKnight1,2Linda Boyajian1,2Amanda M. Ackermann1,2,3Jennifer M. Kalish2,3,4Tricia R. Bhatti2,5 Lisa J. States2,6N. Scott Adzick2,7Katherine Lord1,2,3
Lisa J. States2,6N. Scott Adzick2,7Katherine Lord1,2,3 Diva D. De León1,2,3*
Diva D. De León1,2,3*Focal hyperinsulinism (HI) comprises nearly 50% of all surgically treated HI cases and is cured if the focal lesion can be completely resected. Pre-operative localization of the lesion is thus critical. Few cases of hyperinsulinism with multiple focal lesions have been reported, and assessment of the molecular mechanisms driving this rare occurrence has been limited. We present two cases of multifocal HI, each resulting from two independent, pancreatic focal lesions. 18Fluoro-dihydroxyphenylalanine positron emission tomography/computed tomography detected both lesions preoperatively in one patient, whereas identification of the second lesion was an incidental finding during surgical exploration in the other. Complete resection of the focal lesions resulted in cure of the HI in both cases. In each patient, genetic testing of the individual focal lesions revealed different regions of loss of heterozygosity for the maternal 11p15 allele, confirming that each lesion arose from independent somatic events in the setting of a paternally inherited germline ABCC8 mutation. These cases highlight the importance of a multidisciplinary and personalized approach to the management of infants with HI.
Congenital hyperinsulinism (HI) is the most common cause of persistent hypoglycemia in infants and children (1). Prompt diagnosis and initiation of appropriate treatment are crucial to mitigate risk of permanent hypoglycemic brain damage. Inactivating mutations in the genes encoding the β-cell adenosine triphosphate (ATP)-sensitive potassium (KATP) channel, ABCC8 (SUR1) and KCNJ11 (KIR6.2), located on chromosome 11p15.1, are responsible for the most common forms of HI (2). KATP-HI can be classified into two distinct histological forms: a diffuse form, in which all of the pancreatic β-cells are affected, and a focal form of localized islet hyperplasia and dysfunction. While focal HI is more likely to present at an older age and with hypoglycemic seizures than diffuse HI, these two histological forms cannot be distinguished by clinical presentation alone (3). Differentiating between focal and diffuse HI is of paramount importance as surgical excision of the focal lesion is curative. In contrast, the severity of diffuse HI is ameliorated, but not cured, by near-total pancreatectomy, which carries additional risks of diabetes and exocrine pancreatic insufficiency (4).
Focal HI occurs via a “two-hit” mechanism, following the model originally described by Knudson (5), requiring paternal transmission of a recessive loss-of-function mutation in ABCC8 or KCNJ11 and a pancreas-limited somatic loss of the maternal 11p15 region compensated by paternal isodisomy (6). Somatic loss of heterozygosity (LOH) leads to imbalanced expression of imprinted genes that regulate cell growth, including IGF2, H19, and CDKN1C, resulting in the histological findings of focal adenomatous hyperplasia (6). A single paternally inherited recessive KATP mutation has a 94% positive predictive value for focal HI (2). In these cases, preoperative 18fluoro-dihydroxyphenylalanine positron emission tomography/computed tomography (18F-DOPA PET/CT) is utilized to localize the focal lesion and guide surgical excision (7). Owing to the mechanism of focal lesion development, the occurrence of multiple focal lesions is exceedingly rare. Few cases of HI with multiple focal lesions have been reported to date (8–12). Of these reports, only one included molecular genetic information confirming that the two lesions, one pancreatic and one ectopic, arose from independent somatic events (8). We describe two cases of HI due to distinct pancreatic lesions which broaden our understanding of the pathogenic mechanisms underlying multifocal HI.
Clinical information was extracted from the electronic medical records as part of a study approved by our Institutional Review Board. 18F-DOPA PET/CT was performed as previously described under an Investigational New Drug Application with Food and Drug Administration oversight and Institutional Review Board approval (NCT01916148) (13).
Genetic testing was performed in CLIA-approved laboratories. DNA extracted from peripheral blood from patient 1 was subjected to polymerase chain reaction (PCR) amplification and Sanger sequencing of the ABCC8, KCNJ11, and GCK genes, with deletion/duplication analyses for ABCC8 performed using multiplex ligation-dependent probe amplification. Chromosomal single nucleotide polymorphism (SNP) microarrays were performed on DNA extracted from pancreas tissue from patient 1, and from blood and pancreas tissue from patient 2, using the Illumina CytoSNP850Kv1.1 BeadChip. Whole exome sequencing was performed on DNA extracted from peripheral blood from patient 2 using the xGen Whole Exome Panel kit, and findings were confirmed by Sanger sequencing. In addition, deletion and duplication analysis was performed on peripheral blood DNA from patient 2 by next-generation sequencing for a panel of 23 genes, including ABCC8. DNA extracted from pancreas samples from patient 2 underwent PCR amplification and next generation sequencing of the ABCC8, KCNJ11, GCK, GLUD1, HADH, HNF1A, HNF4A, SLC15A1, and UCP2 genes. Methylation analyses of DNA extracted from blood, skin, and pancreas tissue from patient 2 were performed using sodium bisulfite treatment followed by quantitative methylation sensitive PCR (14). The nucleotides of ABCC8 were numbered according to the sequence reported by Nestorowicz et al. that includes the alternatively spliced exon 17 sequence (NCBI accession no. L78224) (15).
Histology studies were performed on pancreatic tissue obtained at the time of surgery from which formalin-fixed, paraffin-embedded sections were mounted on positively charged slides. Sections were stained with hematoxylin and eosin and primary mouse monoclonal antibodies directed against chromogranin (Dako, clone DAK-A3, 1:200) and p57 (Thermo Scientific, p57Kip2 Ab-6, 1:800). Immunohistochemical staining was performed using the Leica Bond Immunohistochemistry Stainer (Leica Biosystems, Inc., Buffalo Grove, IL) following standard protocols and using the Polymer Refine Detection Brown Detection System and DAB Enhancer.
An infant male was born at 38 weeks gestation via spontaneous vaginal delivery complicated by nuchal cord. He had Apgar scores of 4 and 9 at 1 and 5 min of life, respectively, and a birth weight of 3.55 kg (66th percentile, appropriate for gestational age). Shortly after birth, plasma glucose was checked due to jitteriness and was found to be 1.55 mmol/L (28 mg/dL). He was admitted to the neonatal intensive care unit where he had repeated episodes of hypoglycemia. Critical specimens confirmed a diagnosis of HI (Table 1), and treatment was initiated with diazoxide and chlorothiazide, without clinical response even after the diazoxide dose was titrated up to 15 mg/kg/day. Genetic testing of peripheral blood DNA of the patient and his parents revealed a single heterozygous paternally inherited recessive ABCC8 mutation c.3992-9 G>A. 18F-DOPA PET/CT, performed to localize the suspected focal lesion prior to surgery, demonstrated increased uptake in the inferior pancreatic head extending into the uncinate region (Figures 1A,B). He underwent 20% pancreatectomy in which a focal lesion in the inferior aspect of the pancreatic head measuring 1 cm in its largest dimension was excised. Intraoperatively, an additional, non-contiguous lesion in the anterior aspect of the proximal pancreatic body measuring 0.4 cm was identified by palpation and excised. Histopathology showed that both resected regions demonstrated localized islet cell hyperplasia with occasional nucleomegaly surrounded by normal pancreas, consistent with focal HI (Figure 2). Chromosomal SNP array analyses performed on the excised pancreatic tissue revealed a 47.74 Mb mosaic region of LOH of 11p15.5p11.2 (chr11:198,510–47,940,925, hg19) in the head lesion and a 34.1 Mb mosaic region of LOH within chromosome 11p15.5p13 (chr11:198,510–34,265,140, hg19) in the body lesion. Concurrent SNP array analysis performed on unaffected pancreatic tissue resulted as normal. The mosaic LOH was consistent with paternal uniparental disomy in the setting of a paternally inherited heterozygous pathogenic variant in ABCC8 and histologically confirmed focal HI. To assess for resolution of HI, he underwent a fast prior to discharge in which he maintained plasma glucose >3.89 mmol/L (>70 mg/dL) for 9 h with the remainder of results consistent with persistent, mild HI not necessitating additional treatment (plasma glucose 2.44 mmol/L [44 mg/dL] at 21 h of fasting with inappropriately low β-hydroxybutyrate of 1.3 mmol/L and plasma glucose increase of 2.39 mmol/L [43 mg/dL] in response to glucagon administration). Repeat fasting test at 14 months of age demonstrated appropriate rise in β-hydroxybutyrate, undetectable insulin, non-suppressed free fatty acids and insulin growth factor binding protein 1, and lack of glycemic response to glucagon administration consistent with complete HI resolution (Table 1).
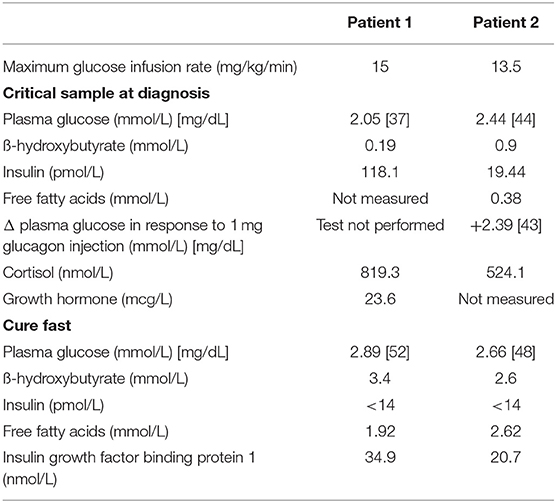
Table 1. Biochemical evaluation at diagnosis and after surgical removal of the lesions.
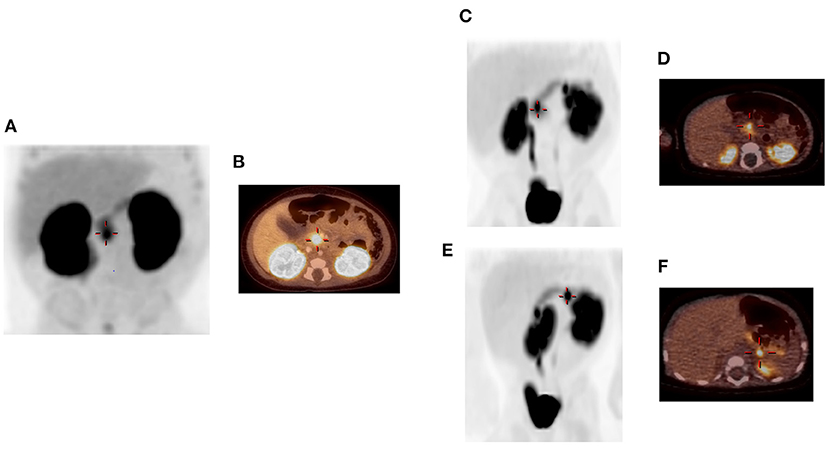
Figure 1. A focal lesion within the pancreatic head/uncinate region (+) is visualized on 3D-MIP image (A) and fused PET/CT image (B) in patient 1. Two focal lesions are identified in patient 2 (C–F). 3D-MIP image at 50 min (C) shows one lesion in the pancreatic head (+) and the other in the proximal pancreatic tail. Fused PET/CT image (D) shows the focal lesion in the pancreatic head (+). Rotated 3D-MIP image (E) permits better visualization of the proximal tail lesion (+), which is shown on fused PET/CT image (F) to be exophytic along posterior margin of pancreas. High uptake in the kidneys and bladder is visualized in all images, as expected, due to renal excretion of the radioisotope. Images were originally published in JNM. States LJ, Davis JC, Hamel SM, Becker SA, Zhuang H. Imaging of congenital hyperinsulinism. J Nucl Med. © SNMMI.
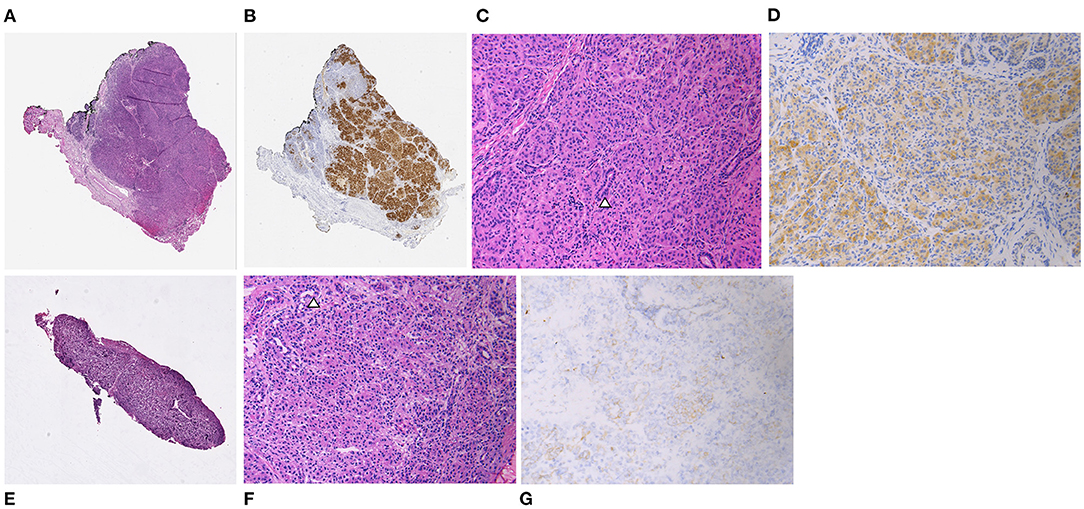
Figure 2. Patient 1. Focal lesions from the pancreatic head [(A) hematoxylin and eosin [H&E], (B) chromogranin immunohistochemical stain [IHC], both at x20 original magnification] and body [(E) H&E, x20 original magnification] composed of localized increases in endocrine tissue with occasional ducts (arrowhead) [(C,F) H&E, x200 original magnification]. Endocrine cells demonstrate loss of nuclear immunoreactivity for p57 in both lesions with non-specific reactivity within the cytoplasm [(D,G) p57 IHC, x200 original magnification].
Patient 2 was born at 39 weeks gestation. She was delivered via uncomplicated, induced vaginal delivery with Apgar scores of 8 and 9 at 1 and 5 min of life, and birth weight of 4.15 kg (97th percentile, large for gestational age). Hypoglycemia was detected on screening performed shortly after birth and persisted. Diagnostic fasting evaluation confirmed HI (Table 1) that was incompletely responsive to both diazoxide 15 mg/kg/day and octreotide 10 mcg/kg/day. A deletion of unknown significance at chromosomal position chr11:17,416,121–17,416,968 (hg19) including exon 36 of ABCC8 was detected on peripheral blood genetic testing and was determined to be paternally inherited. In addition, a single base change in intron 16 (c.2222+15 C>A) of the ABCC8 gene was also identified in peripheral blood DNA; parent of origin was not determined. 18F-DOPA PET/CT revealed two separate foci of increased uptake: an exophytic lesion arising from the posterior aspect of the pancreatic body and a lesion on the medial aspect of the pancreatic head (Figures 1C–F). She subsequently underwent 10% pancreatectomy in which a 0.9 cm lesion in the posterior pancreatic body and an 0.8 cm in the pancreatic head were locally excised with clear surgical margins by frozen section. Final histopathology was consistent with focal islet cell hyperplasia (Figure 3). Terminal mosaic regions of LOH of 11p15.5p11.2 of differing lengths were detected on chromosomal SNP array analyses performed on the resected focal lesions. A 48.83 Mb region (chr11:198,510–49,030,187, hg19) was identified within the pancreatic body lesion and a 47.78 Mb region (chr11:198,510–47,976,882, hg19) within the pancreatic head lesion. SNP analysis of chromosome 11 from the unaffected pancreatic tail tissue was normal. Informed by the prior experience with patient 1, methylation studies were performed on DNA isolated from the focal lesion within the pancreatic body. Results further confirmed paternal uniparental disomy of 11p15.5, with loss of methylation at imprinting control region 2 (IC2) and gain of methylation at IC1. DNA isolated from the patient's blood, skin, and unaffected pancreas showed normal methylation at IC1 and IC2. In addition, next generation sequencing was performed on pancreatic samples from both focal lesions as well as surrounding histologically normal pancreatic tissue. These samples were positive for the exon 36 deletion as well as the single base change in intron 16 (c.2222+15 C>A) of the ABCC8 gene detected in blood. This intronic variant was present at a frequency of 73–86% in affected pancreas suggesting it was on the paternal allele and in cis with the exon 36 deletion. Resolution of HI was demonstrated on fasting evaluation prior to hospital discharge (Table 1).
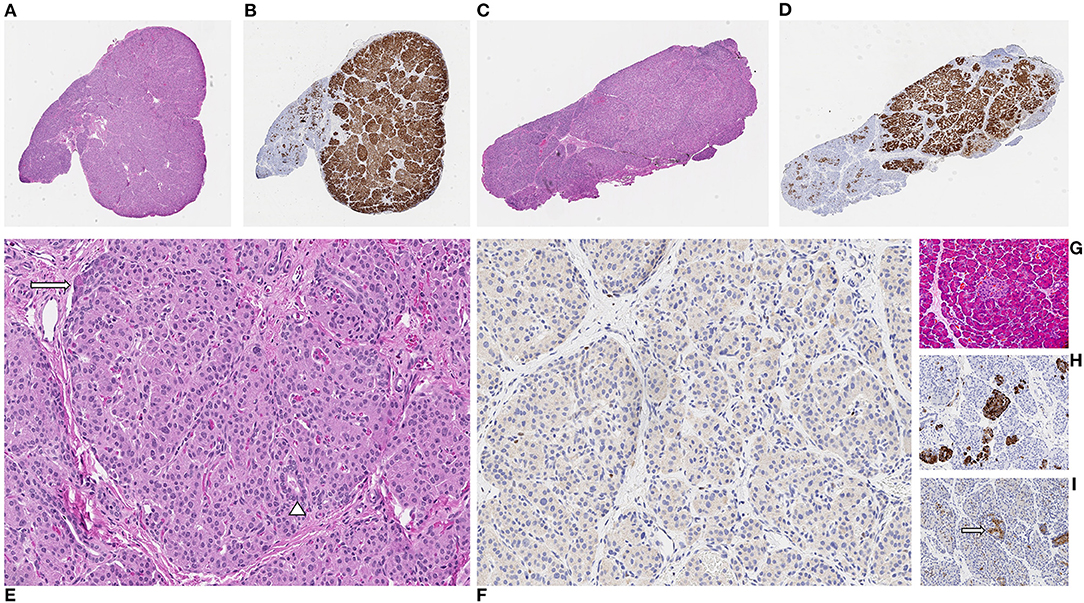
Figure 3. Patient 2. Two distinct focal lesions arising in the pancreatic head (A,B) and body (C,D) each demonstrating localized increases in endocrine tissue which are highlighted on chromogranin immunohistochemical (IHC) stain [(A,C) hematoxylin and eosin [H&E], (B,D) chromogranin IHC, all at x20 original magnification]. Higher-power magnification of head lesion demonstrating increased endocrine cells with rare intermixed acinar cells (arrow) and small ducts (arrowhead) [(E) H&E, x200 original magnification] with loss of nuclear p57 immunoreactivity within lesional cells [(F) p57 IHC, x200 original magnification]. Compared to lesional areas, normal pancreatic tissue shows relatively small islets (arrow) composed of endocrine cells which are positive for chromogranin but with retained nuclear p57 immunoreactivity; non-specific p57 immunoreactivity is observed within the cytoplasm [(G) H&E, (H) chromogranin IHC, (I) p57 IHC, all at x200 original magnification].
Identification of patients with focal HI is of critical importance, as these patients can be cured if the lesion is localized and fully resected. Over the past several decades, improvements in the diagnosis of focal HI have been bolstered both by advances in molecular diagnostic testing and the implementation of 18F-DOPA PET/CT for localization of focal lesions. Focal HI comprises roughly 50% of all surgically treated congenital HI cases (4). In contrast, the presence of multiple focal HI lesions is rare, and few patients with multifocal lesions have been reported to date.
Goossens et al. first reported three cases of “multifocal nesidioblastosis” in 1989 (9). Limited histopathological data is provided, and this report predated our understanding of the genetic and molecular mechanisms responsible for congenital HI. Since then, Ni et al. described a patient with a KCNJ11 mutation found to have one focal lesion in the pancreatic head and another in the pancreatic body (11). Our group previously reported two separate cases, one suspected and one confirmed, of focal HI in which there were both pancreatic and ectopic intestinal focal lesions (10, 12). In each of these cases, the presence of multiple focal lesions was ultimately identified on 18F-DOPA PET/CT imaging, accentuating the utility of this technique in the surgical management of focal HI. However, genetic analysis of the resected tissue was not reported.
To our knowledge, the only report including genetic analyses of tissue from multiple focal lesions is that of Giurgea et al. describing two patients, each with one pancreatic and one ectopic lesion (8). Both patients carried a heterozygous, paternally inherited ABCC8 gene mutation and were cured after complete lesionectomy. Genotyping of affected tissue by fluorescent PCR assay for chromosome 11p markers revealed somatic deletions of the maternal 11p15 region with different deletion break points within the pancreatic and ectopic lesions. No LOH was detected in samples from unaffected pancreatic tissue.
This report provides a detailed description of multiple focal lesions confined to the pancreas, adding to the limited body of literature on multifocal HI. In both patients presented here, analyses of resected pancreatic tissue revealed two distinct focal lesions with different sizes of LOH within 11p15, confirming that the lesions resulted from distinct somatic events in the setting of a paternally inherited, germline recessive ABCC8 mutation. The independence of the somatic events leading to multifocal HI is further supported by the distinct anatomic location of the focal lesions identified in patient 1, which reflects independent spatial origin. In pancreas embryogenesis, the inferior pancreatic head and uncinate process arise from the ventral pancreatic bud, and the remainder of the pancreas is formed from the dorsal pancreatic bud (16). In addition, the relative timing of the somatic event may determine focal lesion size (8, 17). Thus, the somatic event leading to the larger focal lesion in the inferior pancreatic head likely occurred within the ventral pancreatic bud earlier in development, whereas the smaller lesion in the pancreatic body may have resulted from a later somatic event within the dorsal bud. These findings augment those of Giurgea et al. and affirm the “two-hit” pathogenetic mechanism of focal HI.
The c.3992-9 G>A recessive mutation in ABCC8 observed in patient 1 is a well-recognized, founder mutation within the Ashkenazi Jewish population that affects splicing (18). In contrast, the pathogenicity of the ABCC8 changes detected in patient 2 are not well-established. The paternally inherited heterozygous deletion in exon 36 would be predicted to result in a shift in the mRNA reading frame, resulting in an early termination codon. This would affect the nucleotide-binding fold 2 (NBF2) domain of SUR1, mutations in which represent a common molecular mechanism of diazoxide-unresponsive HI (19). The single base change in intron 16 (ABCC8: c.2222+15 C>A) is not predicted to affect splicing [Human Splicing Finder (20)]. However, this variant is rare [allele frequency 0.0007%, gnomAD v3.1 (21)] and has been previously reported (22). Thus, either one of these ABCC8 changes may act as a recessive loss-of-function mutation and represent the first “hit” in this patient.
Based upon the experience at our institution, multiple focal lesions represent ~1% of all focal HI cases (n = 3/264) and 0.3% of all non-syndromic HI cases (n = 3/937) seen. The risk of focal HI in a fetus carrying a paternally inherited recessive KATP mutation (i.e., the risk of somatic paternal uniparental disomy within the pancreas) has been estimated to be 0.37% (1:270) (23). This may suggest that the risk of a second LOH event within the pancreas would also be 0.37%. The incidence of multifocal HI within focal HI cases seen at our institution (1.1%) is nearly three-fold higher than this estimate. The reason for this difference is not clear and may be explained by case ascertainment bias. Alternatively, this may indicate that the risk of pancreas-limited paternal uniparental disomy among carriers of recessive KATP channel mutations is higher than previously estimated.
These cases illuminate the pivotal role of collaboration across the fields of pediatric endocrinology, genetics, radiology, surgery, and pathology in optimizing care for patients with HI. Notably, while 18F-DOPA PET/CT accurately identified both pancreatic focal lesions in patient 2, this was not the case for patient 1 in whom the second focal lesion was identified intraoperatively owing to the surgical expertise and experience at our center. Through this multidisciplinary approach, both patients were cured.
Rarely, patients with the focal form of HI may have more than one focal lesion. This occurs due to separate somatic events resulting in varying lengths of 11p15 LOH unique to each lesion. A multidisciplinary team approach, including specialists with extensive experience caring for patients with HI, plays a critical role in the effective diagnosis and treatment of congenital HI. The importance of this approach to patient care is especially highlighted by rare, atypical, and challenging cases as presented herein.
The relevant original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by Children's Hospital of Philadelphia Institutional Review Board. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
ER wrote the first draft of the manuscript. LM, KB, SB, HM, LB, AA, JK, TB, LS, NA, and KL contributed to the areas relevant to their expertise and edited the manuscript. DDDL conceptualized the work and edited the manuscript. All authors were involved in preparation of the manuscript.
This work was supported by National Institutes of Health Grants R01DK098517 (DDDL) and T32 NS091006-6 (ER).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We acknowledge the patients described in this report, their families, and the dedicated team at the Children's Hospital of Philadelphia Congenital Hyperinsulinism Center.
1. Lord K, De Leon DD. Hyperinsulinism in the neonate. Clin Perinatol. (2018) 45:61–74. doi: 10.1016/j.clp.2017.10.007
2. Snider KE, Becker S, Boyajian L, Shyng SL, MacMullen C, Hughes N, et al. Genotype and phenotype correlations in 417 children with congenital hyperinsulinism. J Clin Endocrinol Metab. (2013) 98:E355–63. doi: 10.1210/jc.2012-2169
3. Lord K, Dzata E, Snider KE, Gallagher PR, De Leon DD. Clinical presentation and management of children with diffuse and focal hyperinsulinism: a review of 223 cases. J Clin Endocrinol Metab. (2013) 98:E1786–9. doi: 10.1210/jc.2013-2094
4. Adzick NS, De Leon DD, States LJ, Lord K, Bhatti TR, Becker SA, et al. Surgical treatment of congenital hyperinsulinism: results from 500 pancreatectomies in neonates and children. J Pediatr Surg. (2018) 54:27–32. doi: 10.1016/j.jpedsurg.2018.10.030
5. Knudson AG Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA. (1971) 68:820–3. doi: 10.1073/pnas.68.4.820
6. Verkarre V, Fournet JC, de Lonlay P, Gross-Morand MS, Devillers M, Rahier J, et al. Paternal mutation of the sulfonylurea receptor (SUR1) gene and maternal loss of 11p15 imprinted genes lead to persistent hyperinsulinism in focal adenomatous hyperplasia. J Clin Invest. (1998) 102:1286–91. doi: 10.1172/JCI4495
7. States LJ, Saade-Lemus S, De Leon DD. 18-F-L 3,4-dihydroxyphenylalanine PET/computed tomography in the management of congenital hyperinsulinism. PET Clin. (2020) 15:349–59. doi: 10.1016/j.cpet.2020.03.004
8. Giurgea I, Sempoux C, Bellanne-Chantelot C, Ribeiro M, Hubert L, Boddaert N, et al. The Knudson's two-hit model and timing of somatic mutation may account for the phenotypic diversity of focal congenital hyperinsulinism. J Clin Endocrinol Metab. (2006) 91:4118–23. doi: 10.1210/jc.2006-0397
9. Goossens A, Gepts W, Saudubray JM, Bonnefont JP, Nihoul F, Heitz PU, et al. Diffuse and focal nesidioblastosis. A clinicopathological study of 24 patients with persistent neonatal hyperinsulinemic hypoglycemia. Am J Surg Pathol. (1989) 13:766–75. doi: 10.1097/00000478-198909000-00006
10. Hussain K, Seppanen M, Nanto-Salonen K, Adzick NS, Stanley CA, Thornton P, et al. The diagnosis of ectopic focal hyperinsulinism of infancy with [18F]-dopa positron emission tomography. J Clin Endocrinol Metab. (2006) 91:2839–42. doi: 10.1210/jc.2006-0455
11. Ni J, Ge J, Zhang M, Hussain K, Guan Y, Cheng R, et al. Genotype and phenotype analysis of a cohort of patients with congenital hyperinsulinism based on DOPA-PET CT scanning. Eur J Pediatr. (2019) 178:1161–9. doi: 10.1007/s00431-019-03408-6
12. Peranteau WH, Bathaii SM, Pawel B, Hardy O, Alavi A, Stanley CA, et al. Multiple ectopic lesions of focal islet adenomatosis identified by positron emission tomography scan in an infant with congenital hyperinsulinism. J Pediatr Surg. (2007) 42:188–92. doi: 10.1016/j.jpedsurg.2006.09.046
13. Laje P, States LJ, Zhuang H, Becker SA, Palladino AA, Stanley CA, et al. Accuracy of PET/CT scan in the diagnosis of the focal form of congenital hyperinsulinism. J Pediatr Surg. (2013) 48:388–93. doi: 10.1016/j.jpedsurg.2012.11.025
14. Baker SW, Duffy KA, Richards-Yutz J, Deardorff MA, Kalish JM, Ganguly A. Improved molecular detection of mosaicism in Beckwith-Wiedemann Syndrome. J Med Genet. (2021) 58:178–84. doi: 10.1136/jmedgenet-2019-106498
15. Nestorowicz A, Wilson BA, Schoor KP, Inoue H, Glaser B, Landau H, et al. Mutations in the sulonylurea receptor gene are associated with familial hyperinsulinism in Ashkenazi Jews. Hum Mol Genet. (1996) 5:1813–22. doi: 10.1093/hmg/5.11.1813
16. Henry BM, Skinningsrud B, Saganiak K, Pekala PA, Walocha JA, Tomaszewski KA. Development of the human pancreas and its vasculature - an integrated review covering anatomical, embryological, histological, and molecular aspects. Ann Anat. (2019) 221:115–24. doi: 10.1016/j.aanat.2018.09.008
17. Fournet JC, Mayaud C, de Lonlay P, Gross-Morand MS, Verkarre V, Castanet M, et al. Unbalanced expression of 11p15 imprinted genes in focal forms of congenital hyperinsulinism: association with a reduction to homozygosity of a mutation in ABCC8 or KCNJ11. Am J Pathol. (2001) 158:2177–84. doi: 10.1016/S0002-9440(10)64689-5
18. Thomas PM, Cote GJ, Wohllk N, Haddad B, Mathew PM, Rabl W, et al. Mutations in the sulfonylurea receptor gene in familial persistent hyperinsulinemic hypoglycemia of infancy. Science. (1995) 268:426–9. doi: 10.1126/science.7716548
19. Shyng SL, Ferrigni T, Shepard JB, Nestorowicz A, Glaser B, Permutt MA, et al. Functional analyses of novel mutations in the sulfonylurea receptor 1 associated with persistent hyperinsulinemic hypoglycemia of infancy. Diabetes. (1998) 47:1145–51. doi: 10.2337/diabetes.47.7.1145
20. Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. (2009) 37:e67. doi: 10.1093/nar/gkp215
21. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. (2020) 581:434–43. doi: 10.1530/ey.17.14.3
22. Li C, Ackermann AM, Boodhansingh KE, Bhatti TR, Liu C, Schug J, et al. Functional and metabolomic consequences of KATP channel inactivation in human islets. Diabetes. (2017) 66:1901–13. doi: 10.2337/db17-0029
Keywords: pancreas, beta cells, islets, KATP channel, hypoglycemia, case report
Citation: Rosenfeld E, Mitteer L, Boodhansingh K, Becker SA, McKnight H, Boyajian L, Ackermann AM, Kalish JM, Bhatti TR, States LJ, Adzick NS, Lord K and De León DD (2021) Case Report: Two Distinct Focal Congenital Hyperinsulinism Lesions Resulting From Separate Genetic Events. Front. Pediatr. 9:699129. doi: 10.3389/fped.2021.699129
Received: 22 April 2021; Accepted: 18 June 2021;
Published: 16 July 2021.
Edited by:
Gianluca Tornese, Institute for Maternal and Child Health Burlo Garofolo (IRCCS), ItalyReviewed by:
Fabio Sirchia, Neurological Institute Foundation Casimiro Mondino (IRCCS), ItalyCopyright © 2021 Rosenfeld, Mitteer, Boodhansingh, Becker, McKnight, Boyajian, Ackermann, Kalish, Bhatti, States, Adzick, Lord and De León. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Diva D. De León, ZGVsZW9uQGNob3AuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.