 Yanfeng Liu1
Yanfeng Liu1 Qingling Zhu
Qingling Zhu- 1Department of Endocrinology, Quanzhou Women and Children's Hospital, Quanzhou, China
- 2Department of Children Health Care, Quanzhou Women and Children's Hospital, Quanzhou, China
The discovery of rare genetic variation through different gene sequencing methods is a very challenging subject in the field of human genetics. A case of a 1-year-old boy with metabolic acidosis and hypokalemia, a small penis, growth retardation, and G-6PD deficiency was reported. Since the clinical symptoms are complex and seem uncorrelated, the authors hypothesized that the child had chromosome or gene problems, and exome sequencing (ES) was applied to samples from him and his parents. Three main locus mutations in three genes were found in the proband, including SLC4A1, FGFR1, and G6PD genes. A missense mutation (c.1766G>T, p.R589 L) was found in exon 14 of SLC4A1 gene, which was a de novo mutation. Another missense mutation (c.1028 A>G, p.H343R) was found in exon 9 of FGFR1 gene, which was also a de novo mutation. These findings further demonstrate the utility of ES in the diagnosis of rare diseases.
Introduction
It is a challenging task to study the pathogenesis of genetic diseases. Exome sequencing (ES), also known as target exome capture, is a genomic analysis method that uses sequence capture technology to capture and enrich whole genome exon region DNA and conduct high-throughput sequencing. Previous studies (1) have shown that ES has high clinical practicability and is cost-effective. It has the highest diagnostic rate for children with genetic heterogeneity or characteristics that overlap multiple diseases (2). Our study reports a male boy who was found to have G-6PD deficiency and a small penis on his 15-day visit and showed growth retardation, metabolic acidosis, and hypokalemia on his 1-year second visit. The proband had no elder brother or sister; his parents were not blood relatives, and both of their phenotypes were normal. To further study the proband's disease, we sequenced the whole exome of the proband and his parents after obtaining consent from the proband's parents.
Case Description
A male boy with normal birth weight and length was found to have G-6PD deficiency and a small penis on his 15-day first visit. Hearing screening passed in both ears. G2P1, the first fetus, was embryo stop pregnancy, with no special maternal pregnancy. At the time of his birth, his mother was 30 years old, and the father was 34 years old. There was no regular physical examination after his birth. The second visit was at 1 year of age with a slow increase in body weight, and the size and length of the penis did not change. His body length was 69.3 cm (−2.5 SD), and his weight was 6.0 kg (−4.1). Clinical evaluation showed a thin and small stature with normal appearance; penile length was 1.5 * 0.5 cm with right testicular volume test approximately 1 ml (the left was not touched), pubic hair Tanner I stage. Blood gas analysis in the outpatient department showed pH 7.226, PCO2 21.60 mmHg, PO2 63.50 mmHg, BE− 16.70 mmol/L, BEECF− 18.8 mmol/L, HCO3− 8.80 mmol/L, K+ 2.72 mmol/L, Na+ 137.5 mmol/L, and Cl− 109.5 mmol/L (hypokalebicarbonate acid correction, which was related to complete examinations). After repeated treatment with acidosis and oral potassium supplementation for 7 days, the blood gas basically returned to normal.
A hormonal basal study revealed low values of follicle-stimulating hormone (FSH), luteinizing hormone (LH), PREG, TESTOST, and estradiol for the proband's sex and chronological age. The gonadotropin-releasing hormone (GnRH) stimulation test elicited a prepubertal LH response with LH and FSH peaks of 0.45 and 7.10 mUI/ml, respectively. The basal value of LH was <0.07 mIU/ml. The human chorionic gonadotropin (hCG) excitation test showed a TESTOST peak of 12.36 ng/dl; the extended hCG excitation test showed that the level of TESTOST was 119.93 ng/dl, both of which suggested no obvious increase in testosterone. The results of inhibin B (INH-B) and anti-Müllerian hormone (AMH) were 21.05 pg/ml and 14.24 ng/ml, respectively, which showed poor function of testicular Sertoli cells. Drostenedione <0.44 nmol/L; dehydroepiandrosterone sulfate <0.1 μg/dl. IGF-1 was 25.0 ng/ml, and IGFBP-3 was 1.7 μg/ml, both of which were lower than in patients of the same race, sex, and age group. Plasma ammonia was elevated at 73.0 μmol/L. Citrulline was elevated at 42.71 μmol/L. A child neuropsychological development assessment suggested a low development quotient. The levels of 17-hydroxyprogesterone and hydrocortisone (COR), adrenocorticotropic hormone (ACTH), alpha-fetoprotein (AFP), carcinoembryonic antigen (CEA), and βHCG were normal. The ACTH cortisol rhythm and thyroid function were normal. The reference ranges of the test results are shown in Table 1.
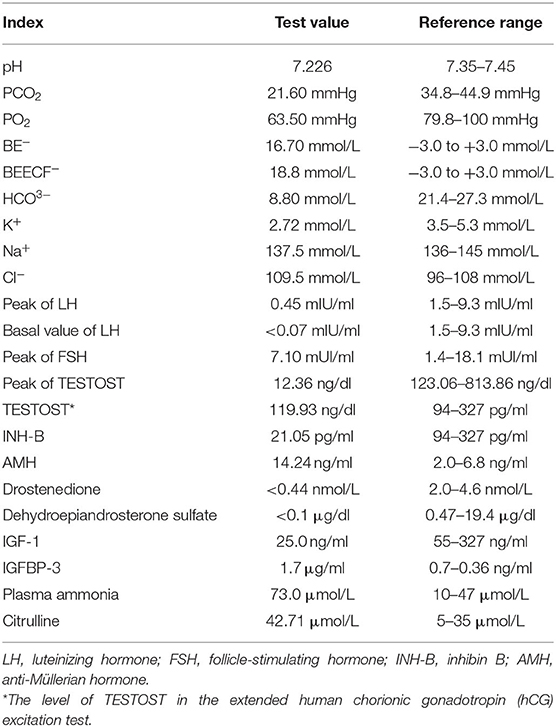
Table 1. Laboratory test results and reference range of the child.
The karyotype was 46, XY. A pituitary MRI scan and enhancement showed that the pituitary gland was small and the pituitary stalk was thin, which implicated pituitary hypoplasia, and the position of the cerebellar tonsil was low. Olfactory bulb MRI showed that both olfactory tracts were asymmetric and that the olfactory sulcus was not clear. Color Doppler ultrasound of the urinary system showed that bilateral renal medulla echoes were significantly enhanced, and calcareous deposition was considered, with spermatic cord effusion on the right region and cryptorchidism on the left region. The child had normal ACTH cortisol rhythm and thyroid function.
The initial diagnoses were as follows: (1) distal renal tubular acidosis (dRTA; type I); (2) growth retardation; (3) G-6PD enzyme deficiency; and (4) congenital small penis.
After the informed consent of family members and the consent of the hospital ethics committee was obtained, the detection of trio-ES in the child and his parents was performed to detect potential variants. Sanger sequencing technology was used to verify the suspected pathogenic mutations.
Methods for Trio-Exome Sequencing
DNA was extracted from peripheral blood, and ES was carried out on the proband and his parents. The coding exons were captured using the xGen® Exome Research Panel v1.0 (IDT). The captured fragments were sequenced using a NovaSeq 6000 sequencer (Illumina, San Diego, CA, USA) to an average depth of 100 reads per target base. The clean data were aligned to the National Center for Biotechnology Information (NCBI) human reference genome (HG19) using the Burrows–Wheeler Aligner (BWA), and variants were determined using GATK. Samtools and Pindel were used to determine single-nucleotide polymorphisms (SNPs) and indels, respectively. The clean data were filtered for further analysis according to the quality of the sequencing. For variant annotation and prediction, non-synonymous substitutions and SNPs with minor allele frequencies (MAFs) lower than 5% were filtered using SIFT. The function of mutated genes and their pathogenicity were analyzed referencing the dbSNP, 1,000 Genomes Project, ExAC, ESP, OMIM, Swiss-var, HGMD, ClinVar, and other disease databases. The variants with unknown pathogenicity of single bases were screened using Provean, SIFT, Polyphen2-HVAR, Polyphen2-HDIV, Mutationtster, and other protein structure prediction software. MaxEntScan was used to screen potential splice sites.
Treatment
After admission, the child was treated with sodium bicarbonate 20 ml qd by intravenous injection for 7 days and 10% KCl 15.5 ml qd for 4 days, later changed to potassium citrate 2 g tid p.o. The medicine after discharge was potassium citrate 2 g tid p.o. The child has regular follow-up visits and taking medications on time without adverse drug events occurring, symptoms are well-controlled, and body length and weight are increasing steadily. The timeline for the treatment and follow-up is shown in Table 2. During the recent follow-up, parents said that the child was more active and his appetite improved significantly.
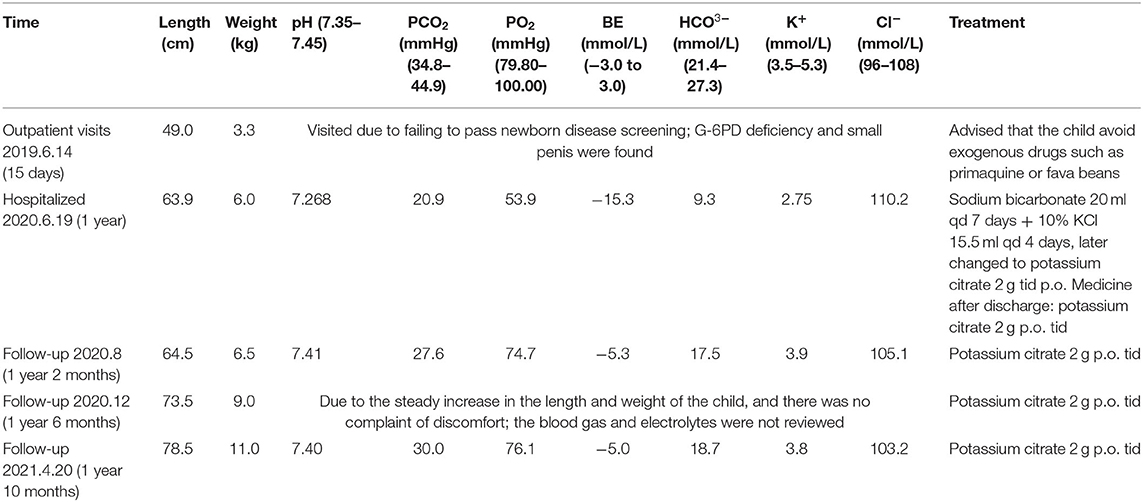
Table 2. Timeline for the treatment and follow-up.
Results
Seven locus mutations in seven genes were found in the proband: SLC4A1, FGFR1, G6PD, GLI3, BCOR, FAT4, and KAT6B.
A missense mutation (c.1766G>T, p.R589 L) was found in exon 14 of SLC4A1 gene of the proband, at base 1,766, in which G was mutated to T, resulting in the mutation of the amino acid residue at position 589 from arginine to leucine. The mutation was not found in either of his parents and was a de novo mutation. After searching the SNP website (https://www.ncbi.nlm.nih.gov/snp/, retrieval date July 2020), we found that the R589 L mutation in the proband had not been reported before, and we named it SLC4A1 Quanzhou according to nomenclature by city (3, 4). In addition, a missense mutation (c.1028 A>G, p.H343R) was found in exon 9 of FGFR1 gene in the proband, which was also a de novo mutation. A missense mutation (c.296G>A, p.R2329H) was found in exon 3 of KAT6B gene, which was observed in the father, so it was a hereditary mutation. Similarly, these two mutations have not been reported. We named them FGFR1 Quanzhou and KAT6B Quanzhou. The other four gene mutation sites were noted in previous reports, as shown in Table 3. The inclusion status and frequency of the three “pathogenic” genes in the database are shown in Table 4.
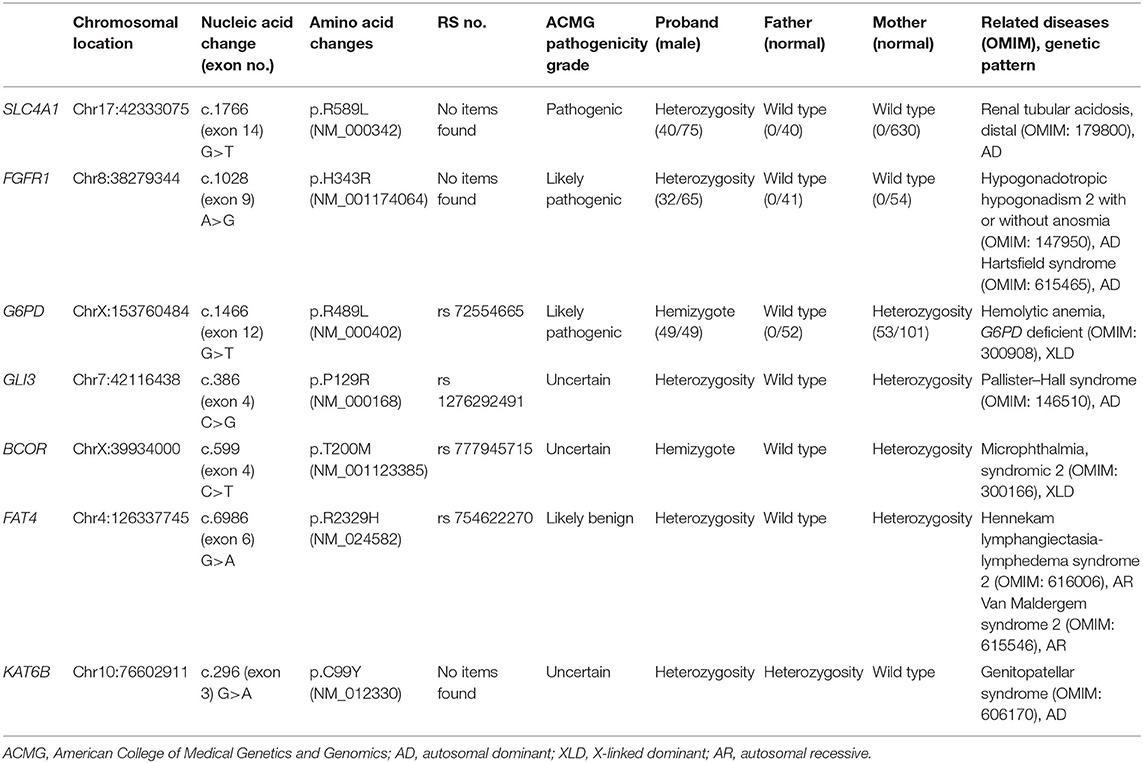
Table 3. Results of exome sequencing.

Table 4. The inclusion status and frequency of the three “pathogenic” genes in the database.
According to the American College of Medical Genetics and Genomics (ACMG) guidelines (2015), SLC4A1 gene mutation was mutated as “pathogenic (PS2 + PM1 + PM2 + PM5 + PP3),” FGFR1 gene mutation was mutated as “likely pathogenic (PS2 + PM1 + PM2 + PP3),” and G6PD gene mutation was mutated as “likely pathogenic (PS1 + PM1 + PP3).” The evidence of the remaining four genes becoming pathogenic was insufficient, but possible pathogenic variation was not ruled out (Table 3). The verification results of gene mutations with high pathogenicity are shown in Figures 1–3.
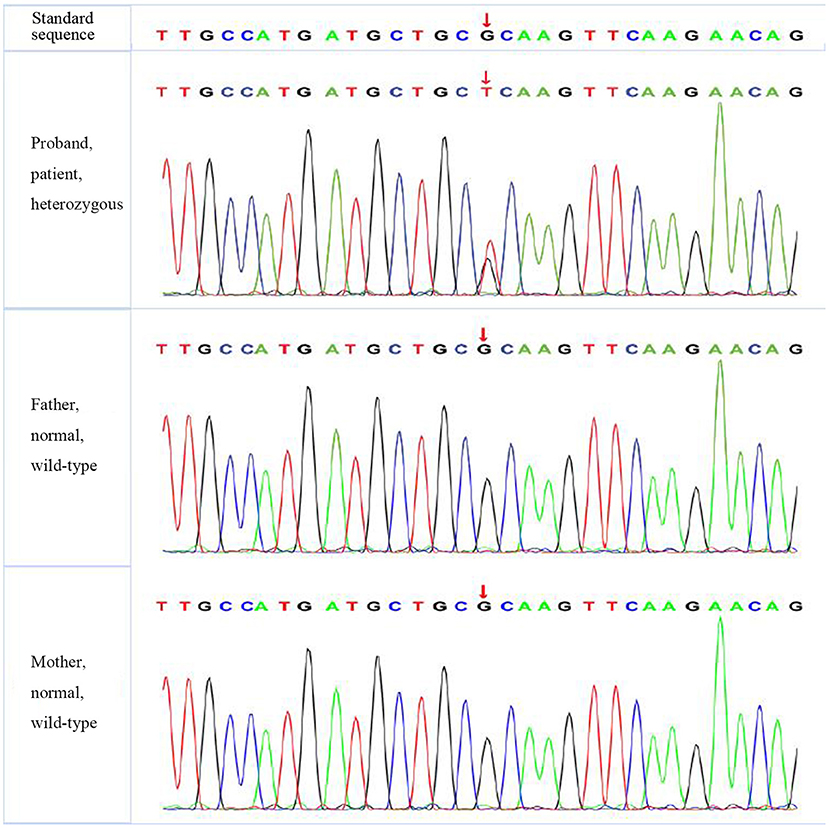
Figure 1. The verification results of SLC4A1. SLC4A1:c.1766 (exon 14) G>T.
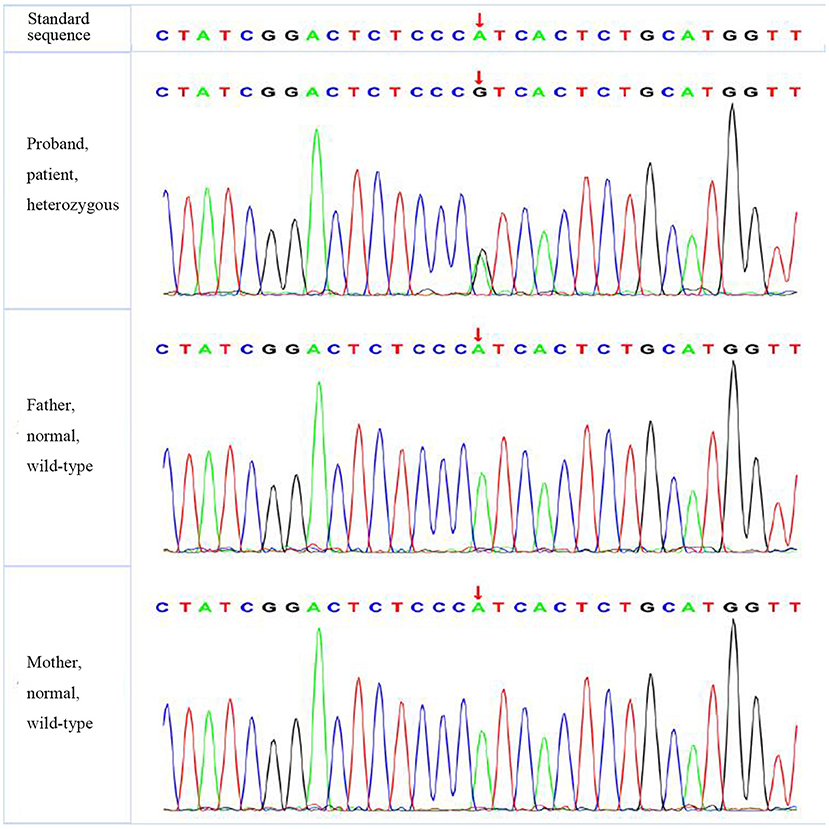
Figure 2. The verification results of FGFR1. FGFR1:c.1028 (exon 9) A>G.

Figure 3. The verification results of G6PD. G6PD:c.1466 (exon 12) G>T.
Discussion
SLC4A1 Gene Mutation: Pathogenic
The 17-kb SLC4A1 gene is located on chromosome 17q21 and has 20 exons. It encodes ion exchange protein anion exchanger 1 (AE1; also called band 3 protein), first reported in 1972 (5). More than 10 mutations have been reported (6) that comprise multiple mutations of 589 loci, including R589 L, R589H, R589C, and R589S, and are considered hot spot mutations. In previous reports, SLC4A1 gene mutation is observed with hereditary dRTA, hereditary spherocytosis, and Southeast Asian ovalocytosis.
SLC4A1, which encodes AE1, has two subtypes. The eAE1 subtype is expressed in red blood cells and plays a role in maintaining the normal morphology of red blood cells. The KAE1 subtype is expressed in the kidney and plays a role in transporting Cl−/HCO. After gene mutation, the activity of Cl−/HCO transport was affected, and the acidification function of distal renal tubules was impaired, which led to acidosis and increased urinary pH-value. Distal renal tubular acidosis caused by SLC4A1 gene mutation is usually caused by autosomal dominant (AD) inheritance. However, in equatorial countries such as Thailand, Malaysia, the Philippines, and Papua New Guinea, autosomal recessive inheritance is observed (7). In addition, it was reported that patients with the A858D homozygous mutation of SLC4A1 had manifestations of both hereditary spherocytosis and dRTA (8).
The main clinical feature of dRTA is that the disorder of H+ secretion in the distal renal tubules causes renal acidification dysfunction, leading to high chlorine metabolic acidosis with normal anions, which is often accompanied by hypokalemia, hypercalciuria, nephrocalcinosis, and kidney stones. With the occurrence of acidosis, growth retardation and rickets can occur and have been observed in children with variants in SLC4A1 p.R589C, p.R589H, and p.R589S. The proband presented with typical manifestations of acidosis, high blood chlorine, low blood potassium, renal calcinosis, and slow growth; and after repeated correction of acidosis and potassium supplementation, the blood gas returned to normal. Previous studies have reported that children with dRTA have a good prognosis after reasonable treatment. However, due to the possibility of deterioration of renal function in adulthood, especially after puberty, it is necessary to be vigilant and follow up regularly. In addition to SLC4A1 gene described in this case, the hereditary pathogenic genes that have been identified to cause dRTA can also be found in ATP6V1B1 and ATP6V0A4 genes. Studies have found that patients with dRTA caused by SLC4A1 gene mutations are mostly diagnosed in adolescence, while those caused by ATP6V1B1 and ATP6V0A4 gene mutations are more commonly identified in infancy and early childhood (9). The age of onset in this study (1 year) was significantly younger than that of SLC4A1 gene mutations reported in previous reports.
FGFR1 Gene Mutation: Likely Pathogenic
Fibroblast growth factor receptor 1 (FGFR1), also known as acidic fibroblast growth factor receptor (aFGFR), is located on chromosome 8p12 and has 19 exons and a molecular weight of 78 kDa. Its mutation or enhanced expression can lead to changes in protein tyrosine kinase activity, thereby enhancing tyrosine kinase phosphorylation and its downstream effects. FGFR1 is involved in the growth, differentiation, migration, apoptosis, angiogenesis, and drug resistance of tumor cells (10). Previous reports focused on the role of FGFR1 in the field of cancer. FGFR1 gene amplification seems to be one of the most common genetic changes in tumors and can be seen in gastric cancer, oral squamous cell carcinoma, ovarian cancer, bladder cancer, and breast cancer. FGFR1 also plays an important role in the development of the reproductive and nervous systems. The role of nerve cell development in the embryonic stage may be related to the development of GnRH neurons and olfactory neurons. FGFR1 protein defects can cause GnRH neuromigration and abnormal olfactory bulb development.
FGFR1 gene mutation was reported to be associated with Kallmann syndrome (KS), which is a disease that results in idiopathic hypogonadotropic hypogonadism (IHH) combined with anosmia or hyposmia (11). Some patients with hypogonadotropic hypogonadism 2 with or without anosmia (IHH) can present specific non-reproductive phenotypes, including cryptorchidism, hypogonadotropic hypogonadism, congenital penile, olfactory loss, osteopenia, developmental delay, agenesis of the corpus callosum, bimanual synkinesis, short stature, hearing impairment, unilateral (occasionally bilateral) renal hypoplasia, cleft lip and palate, loss of teeth, and obesity (12). At present, the common causes of IHH are FGFR1 and KAL1 gene mutations; in addition, the FGFR1 gene is associated with Hartsfield syndrome (13).
In this case, the proband had a congenital small penis (micropenis), which means that the stretched penile length (SPL) was lower than the average of the population of the same age or the same developmental status by 2.5 standard deviations or more, accompanied by deformities such as cryptorchidism, small-volume testes, and scrotal hypoplasia. The normal value of the penis length of newborns measured by Feldman in the United States is 28–42 mm (14). Some researchers in the West have diagnosed a small penis with a penis length of <2 cm in full-term newborns. The normal penile length of full-term newborns in Asia is 26–46 mm, and it is considered that penile length <26 mm is a small penis (15).
Most FGFR1 gene mutations were observed in male patients and were usually found in adolescence, rarely in infancy. The proband had an FGFR1 gene heterozygous de novo mutation, which is consistent with the pathogenesis of AD inheritance, with clinical manifestations of congenital penile, left cryptorchidism, and growth retardation and no significant increase in testosterone in the hCG provocation test, suggesting that the genotype is consistent with the phenotype. According to the ACMG guidelines (2015), the mutation is likely to be pathogenic. In addition, FGFR1 gene is inherited in an AD manner, so heterozygous mutations can cause disease. In this case, FGFR1 gene of the proband is a heterozygous mutation. His parents should be wary that there is a possibility of subsequent development of IHH or Hartsfield syndrome.
G6PD Gene Mutation: Likely Pathogenic
The human G6PD gene, located on chromosome Xq28, has 13 exons and spans 18 kb. G6PD gene plays a critical role in the production of ribose 5-phosphate and the generation of NADpH in the hexose monophosphate pathway. To date, there have been more than 140 mutations in G6PD gene found worldwide (16), and its genotype has the characteristics of regional mutations (17). Previous studies have confirmed that G6PD deficiency has a protective effect for malaria (18). With some environmental factors, such as infection and certain drugs and foods, G6PD deficiency may lead to hemolysis. It is the most common group of human enzyme deficiency syndrome and follows an X-linked incomplete dominant inheritance model.
Most G6PD deficiency patients are asymptomatic for life, but it can also be life-threatening, especially in children (19). The most common clinical manifestations of G6PD deficiency are neonatal jaundice and acute hemolytic anemia caused by exogenous drugs such as primaquine or fava beans. No persistent jaundice or clinical manifestations such as obvious hemolytic anemia were found in the neonatal period of the proband. He was discovered due to “Chinese abnormal screening of neonatal diseases” upon his first visit and was confirmed by ES by chance. The mutation was not found in the proband's father, and a heterozygous mutation was found in his mother, so he was considered to be a hemizygote.
No Sufficient Evidence of Pathogenicity, but Variation With Pathogenicity Cannot Be Excluded
GLI3 gene mutation was reported to be associated with Pallister–Hall syndrome (pHS; MIM 146510) (20), which was characterized by polydactyly, micropenis with undescended or hydroxyplastic testes in males, and urinary dysplasia. BCOR gene mutation is related to oculofaciocardiodental (OFCD) syndrome (OMIM 300166) (21), with clinical manifestations of microphthalmia, congenital cataracts, radiculomegaly, and cardiac and digital abnormalities, as well as mild developmental delay, delayed motor development, and short stature. FAT4 gene mutation can lead to Hennekam lymphangiectasia-lymphedema syndrome 2 (OMIM 616006), and Van Maldergem syndrome 2 (OMIM 615546) (22). The former is mainly manifested as lymphedema, claudication dementia, and developmental delay, while the latter is characterized by intellectual disability, and some may suffer from renal hyperplasia. Both of these diseases may have intellectual disability and developmental delay. In addition, small kidney and genital abnormalities, including micropenis, cryptorchidism, and double scrotum, have been observed in Van Maldergem syndrome (23). KAT6B gene mutation is relevant to genitopatellar syndrome (OMIM 606170) (24), with main manifestations of patellar loss, congenital flexion contracture of lower limbs, psychomotor disorders, and abnormalities of external genitalia and kidneys.
In our study, the proband had the clinical manifestations of delayed growth and development, slow height and weight gain, cryptorchidism, and micropenis, which may have a certain degree of crossover with the four gene-related diseases mentioned above. According to the ACMG guidelines, the four gene mutations were identified as having insufficient evidence of pathogenicity, but possible pathogenic variation was not excluded.
Polygenic Mutation
Many human diseases are likely caused by single-gene and/or multiple-gene mutations, which regulate the occurrence and development of diseases through gene interactions, thereby affecting human health (25). It is not uncommon to have polygenic mutations in the same patient. However, the mechanism of these diseases and the principle of gene interaction are not clear. Multiple mutations tend to have different effects on genes compared with single mutations.
Multiple-gene mutations associated with the same disease have been reported, with incidences of digenic and trigenic variants of 8/48 (16.7%) and 1/48 (2.1%), respectively (26). Various gene mutations may work together in the same pathway to affect the phenotype of the disease, while they may have no or only minimal effect on the phenotype when these heterozygous mutations exist alone.
Multiple mutations in the same gene, also known as compound mutations, have been found to be associated with various genetic diseases. Some studies have also reported that the combined effect of multiple mutations in the same gene leads to more serious cardiovascular disease than a single mutation (27). They believe that these multiple-gene mutations have additive effects on cardiovascular pathology and function and highlight the importance of basic molecular, cellular, and animal model studies in elucidating key pathogenic pathways. However, some studies suggest that multigene mutations can either aggravate the development of the disease or lighten it; the latter is known as a compensatory mutation (28).
Our case is different from the above two cases. In this case, seven gene mutations were detected by the whole-exome test, among which one was pathogenic and two were likely to be pathogenic. The other four genes with insufficient evidence need to be further confirmed. These gene mutations are not the cause of the same disease, and there is no correlation among them that has been reported. However, the cause of this patient's polygenic mutation is unclear. The existence of multiple mutations directly affects the strategy of gene diagnosis. It is important to study the combined effect of multiple mutations. Thus, we look forward to more research on the mechanism of multigene mutations.
Inevitably, there were some limitations to this study. First, we did not have the capacity to conduct functional research; thus, missense mutations could not be verified. In addition, this article focuses on the results of exon detection of this rare polygenic mutation; thus, only the details related to the clinical phenotype and treatment of this child were discussed, not all the relevant details of the related diseases.
The discovery of rare genetic variation through different gene sequencing methods is a very challenging subject in the field of human genetics. Our research confirms the role of ES in the diagnosis of dRTA. With the help of ES, case detection, and diagnosis become easier.
Conclusions
The child needs to take bismuth potassium citrate 2 g qd orally for life, and his parents should be wary of the possibility of subsequent development of IHH or Hartsfield syndrome. In addition, the child should avoid exogenous drugs such as primaquine or fava beans. Our case expanded the gene mutation spectrum and enriched the human gene mutation library. With the expansion of newborn screening programs, including for citrullinemia and G6PD deficiency, numerous asymptomatic infants have been identified, aiding in treatment decisions and genetic counseling. In addition, our study extends the mutation spectrum of dRTA and is helpful in early molecular diagnoses of dRTA. In clinical work, attention should be given to the follow-up of multiple-gene mutations, especially asymptomatic infants identified in infancy.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The studies involving human participants were reviewed and approved by Quanzhou Women and Children's Hospital Ethics Committee. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the participants' legal guardian/next of kin for the publication of this case report.
Author Contributions
YL and ZZ were involved in two sessions with the patient. QZ analyzed the sequencing data and performed the literature research. QZ wrote the first draft of the report, which was edited by all authors. All authors reviewed and agreed to the final version of the article. QZ had the final responsibility for the decision to submit for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank Shengping Yang of Quanzhou Orthopedic-traumatological Hospital (Master of Medicine), the husband of the corresponding author (QZ), for providing assistance with comments on revising the manuscript.
References
1. Tan TY, Dillon OJ, Stark Z, Schofield D, Alam K, Shrestha R, et al. Diagnostic impact and cost-effectiveness of whole-exome sequencing for ambulant children with suspected monogenic conditions. JAMA Pediatr. (2017) 171:855–62. doi: 10.1001/jamapediatrics.2017.1755
2. Theunissen TE, Sallevelt SC, Hellebrekers DM, De Koning B, Hendrickx AT, van den Bosch BJ, et al. Rapid resolution of blended or composite multigenic disease in infants by whole-exome sequencing. J. Pediatrics. (2017) 182:371.e2–4.e2. doi: 10.1016/j.jpeds.2016.12.032
3. Jomoui W, Panichchob P, Rujirachaivej P, Panyasai S, Tepakhan W. Coinheritance of Hb A 2-Melbourne (HBD: c.130G>A) and Hb E (HBB: c.79G>A) in Laos and simultaneous high resolution melt detection of Hb A 2-Melbourne and Hb A 2-Lampang (HBD: c.142G>A) in a single tube. Hemoglobin. (2019) 43: 214–7. doi: 10.1080/03630269.2019.1651332
4. Shao L, Yan X, Dong Q, Lang Y, Yue S, Miao Z, et al. novel SLC4A1 variant in an autosomal dominant distal renal tubular acidosis family with a severe phenotype. Endocrine. (2010) 37:473–8. doi: 10.1007/s12020-010-9340-6
5. Richards P, Wrong OM. Dominant inheritance in a family with familial renal tubular acidosis. Lancet. (1972) 2:998–9. doi: 10.1016/s0140-6736(72)92406-3
6. Deejai N, Wisanuyotin S, Nettuwakul C, Khositseth S, Sawasdee N, Saetai K, et al. Molecular diagnosis of solute carrier family 4 member 1 (SLC4A1) mutation-related autosomal recessive distal renal tubular acidosis. Lab Med. (2019) 50:78–86. doi: 10.1093/labmed/lmy051
7. Khositseth S, Bruce LJ, Walsh SB, Bawazir WM, Ogle GD, Unwin RJ, et al. Tropical distal renal tubular acidosis: clinical and epidemiological studies in 78 patients. QJM. (2012) 105:861–77. doi: 10.1093/qjmed/hcs139
8. Shmukler BE, Kedar PS, Warang P, Desai M, Madkaikar M, Ghosh K, et al. Hemolytic anemia and distal renal tubular acidosis in two Indian patients homozygous for SLC4A1/AE1 mutation A858D. Am J Hematol. (2010) 85:824–8. doi: 10.1002/ajh.21836
9. Palazzo V, Provenzano A, Becherucci F, Sansavini G, Mazzinghi B, Orlandini V, et al. The genetic and clinical spectrum of a large cohort of patients with distal renal tubular acidosis. Kidney Int. (2017) 91:1243–55. doi: 10.1016/j.kint.2016.12.017
10. Yu T, Yang Y, Liu Y, Zhang Y, Xu H, Li M, et al. A FGFR1 inhibitor patent review: progress since 2010. Expert Opin Ther Pat. (2017) 27:439–54. doi: 10.1080/13543776.2017.1272574
11. Choi JH, Oh A, Lee Y, Kim GH, Yoo HW. Functional characteristics of novel FGFR1 mutations in patients with isolated gonadotropin-releasing hormone deficiency. Exp Clin Endocrinol Diabetes. (2020) 129:457–63. doi: 10.1055/a-1151-4800
12. Hardelin JP, Dodé C. The complex genetics of Kallmann syndrome: KAL1, FGFR1, FGF8, PROKR2, PROK2, et al. Sex Dev. (2008) 2:181–93. doi: 10.1159/000152034
13. Courage C, Jackson CB, Owczarek-Lipska M, Jamsheer A, Sowińska-Seidler A, Piotrowicz M, et al. Novel synonymous and missense variants in FGFR1 causing Hartsfield syndrome. Am J Med Genet A. (2019) 179:2447–53. doi: 10.1002/ajmg.a.61354
14. Feldman KW, Smith DW. Fetal phallic growth and penile standards for newborn male infants. J Pediatr. (1975) 86:395–8. doi: 10.1016/s0022-3476(75)80969-3
15. Lian WB, Lee WR, Ho LY. Penile length of newborns in Singapore. J Pediatr Endocrinol Metab. (2000) 13:55–62. doi: 10.1515/jpem.2000.13.1.55
16. Cappellini MD, Fiorelli G. Glucose-6-phosphate dehydrogenase deficiency. Lancet. (2008) 371:64–74. doi: 10.1016/s0140-6736(08)60073-2
17. Lo E, Zhong D, Raya B, Pestana K, Koepfli C, Lee MC, et al. Prevalence and distribution of G6PD deficiency: implication for the use of primaquine in malaria treatment in Ethiopia. Malar J. (2019) 18:340. doi: 10.1186/s12936-019-2981-x
18. Min-Oo G, Gros P. Erythrocyte variants and the nature of their malaria protective effect. Cell Microbiol. (2005) 7:753–63. doi: 10.1111/j.1462-5822.2005.00524.x
19. Luzzatto L, Arese P. Favism and glucose-6-phosphate dehydrogenase deficiency. N Engl J Med. (2018) 378:60–71. doi: 10.1056/nejmra1708111
20. Kariminejad A, Ghaderi-Sohi S, Keshavarz E, Hashemi SA, Parsimehr E, Szenker-Ravi E, et al. A GLI3 variant leading to polydactyly in heterozygotes and Pallister-Hall-like syndrome in a homozygote. Clin Genet. (2020) 97:915–9. doi: 10.1111/cge.13730
21. Ng D, Thakker N, Corcoran CM, Donnai D, Perveen R, Schneider A, et al. Oculofaciocardiodental and Lenz microphthalmia syndromes result from distinct classes of mutations in BCOR. Nat Genet. (2004) 36:411–6. doi: 10.1038/ng1321
22. Alders M, Al-Gazali L, Cordeiro I, Dallapiccola B, Garavelli L, Tuysuz B, et al. Hennekam syndrome can be caused by FAT4 mutations and be allelic to van Maldergem syndrome. Hum Genet. (2014) 133:1161–7. doi: 10.1007/s00439-014-1456-y
23. Neuhann TM, Müller D, Hackmann K, Holzinger S, Schrock E, Di Donato N, et al. further patient with van Maldergem syndrome. Eur J Med Genet. (2012) 55:423–8. doi: 10.1016/j.ejmg.2012.02.012
24. Okano S, Miyamoto A, Fukuda I, Tanaka H, Hata K, Kaname T, et al. Genitopatellar syndrome: the first reported case in Japan. Hum Genome Var. (2018) 5:8. doi: 10.1038/s41439-018-0010-1
25. Loeb KR, Loeb LA. Significance of multiple mutations in cancer. Carcinogenesis. (2000) 21:379–85. doi: 10.1093/carcin/21.3.379
26. Quaynor SD, Bosley ME2, Duckworth CG, Porter KR, Kim SH, Kim HG, et al. Targeted next generation sequencing approach identifies eighteen new candidate genes in normosmic hypogonadotropic hypogonadism and Kallmann syndrome. Mol Cell Endocrinol. (2016) 437:86–96. doi: 10.1016/j.mce.2016.08.007
27. Kelly M, Semsarian C. Multiple mutations in genetic cardiovascular disease: a marker of disease severity? Circ Cardiovasc Genet. (2009) 2:182–90. doi: 10.1161/CIRCGENETICS.108.836478
Keywords: genetic diseases, gene mutation, distal renal tubular acidosis, SLC4A1, FGFR1, KAT6B
Citation: Liu Y, Zheng Z and Zhu Q (2021) Case Report: Identification of Polygenic Mutations by Exome Sequencing. Front. Pediatr. 9:689901. doi: 10.3389/fped.2021.689901
Received: 01 April 2021; Accepted: 09 September 2021;
Published: 21 October 2021.
Edited by:
Zhichao Liu, National Center for Toxicological Research (FDA), United StatesReviewed by:
Tingting Yu, Shanghai Children's Medical Center, ChinaYifan Zhang, University of Arkansas at Little Rock, United States
Copyright © 2021 Liu, Zheng and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qingling Zhu, emh1X3FpbmdsaW5nMjAxMiYjeDAwMDQwOzE2My5jb20=