 Yue Song1,2†
Yue Song1,2† Hua Wang
Hua Wang- 1Department of Pediatrics, West China Second University Hospital, Sichuan University, Chengdu, China
- 2Key Laboratory of Birth Defects and Related Diseases of Women and Children, Ministry of Education, Sichuan University, Chengdu, China
- 3Department of Cardiology, West China Hospital, Sichuan University, Chengdu, China
Background: Lipoprotein glomerulopathy is a rare kidney disease characterized by lipoprotein thrombi in the glomerulus. Here, we report a case of lipoprotein glomerulopathy in a Chinese pediatric patient. Furthermore, we summarized the clinical features and genetic characteristics of lipoprotein glomerulopathy in China.
Case Presentation: An 11-year-old Chinese girl presented with nephrotic syndrome with anemia (98 g/L). After excluding secondary causes, primary nephrotic syndrome was considered. Treatment with prednisone (60 mg/day) did not improve her condition. Renal biopsy showed marked dilation of the capillary lumen with lipoprotein thrombi and positive oil red O staining. Genetic testing revealed the genetic variant c.127C > T (p.R43C), known as the Kyoto mutation of the APOE gene. These findings are consistent with the diagnosis of lipoprotein glomerulopathy. Prednisone was gradually tapered and captopril was initiated. A 2-year follow-up revealed elevated urine protein and serum creatinine levels. We also reviewed 17 pediatric and 156 adult cases of lipoprotein glomerulopathy reported in China from the year of creation to 2021. The most common clinical features were edema, hematuria, hypertriglyceridemia, and increased serum apoE levels. Extra-renal manifestations included anemia, splenomegaly, and cardiac lipoprotein deposition.
Conclusion: APOE Kyoto is the most common mutation in patients with lipoprotein glomerulopathy. In China, homozygosity for E3 (E3/3) is the most common isoform.
Introduction
Lipoprotein glomerulopathy (LPG) is a rare hereditary renal disorder associated with abnormal lipid metabolism induced by variations in plasma apolipoprotein E (apoE) (1, 2). Human apoE is a 34-kDa protein with 299 amino acids that mediate the uptake of triglyceride-rich lipoproteins through both the low-density lipoprotein cholesterol (LDL) receptor and LDL receptor-related protein pathways (3). LPG always presents with proteinuria or nephrotic syndrome (NS). The most important histopathologic manifestation of LPG is the presence of massive eosinophilic lipoprotein thrombi within dilated glomerular capillary lumen with positive oil red O staining (1). The prognosis of patients with LPG is poor due to the absence of specific treatment. Approximately 50% of patients progress to end-stage renal disease within 1–27 years of onset (4). In patients who underwent kidney transplantation, relapse was observed from 5 months to 2 years of follow-up (5).
In China, pediatric LPG cases have rarely been reported. Here, we report a case of LPG in an 11-year-old female with no known family history of renal disease who was admitted to our hospital for a 1-month history of mild edema of the lower limbs. Furthermore, we summarized the clinical features and genetic characteristics of LPG cases in China.
Case Description
An 11-year-old Chinese girl was admitted to our hospital who presented with nephrotic syndrome. She weighed 49 kg and her blood pressure was 120/80 mmHg. Physical examination at admission revealed mild edema of the lower limbs with no associated corneal arcus or xanthoma. Laboratory investigations showed anemia (98 g/L), elevated serum urea (6.7 mmol/L) and creatinine (58 μmol/L), and decreased albumin (21 g/L). Urinalysis revealed proteinuria (4+) and microscopic hematuria (3+). Urinary protein was 2.06 g/24 h. Lipid profile showed elevated total cholesterol (5.04 mmol/L), elevated triglyceride (2.18 mmol/L), and normal LDL-cholesterol (2.24 mmol/L).
Based on laboratory findings, secondary causes of NS were excluded. Anti-nuclear, anti-cardiolipin, anti-neutrophil cytoplasmic, and anti-glomerular basement membrane antibodies were negative. Rheumatoid factor was also negative. Hepatitis B and C serology were negative. In addition, complement C3 and C4 levels were normal. Renal ultrasonography revealed normal-sized kidneys. These findings were consistent with the diagnosis of primary NS.
Despite treatment with prednisone (60 mg/day) and captopril (60 mg/day) for 2 months, her massive proteinuria and microscopic hematuria persisted, thereby prompting further evaluation via percutaneous renal biopsy. Light microscopy showed segmental glomerulosclerosis in three out of nine samples and mild-to-moderate proliferation of the mesangial cells and matrix (Figures 1A,B). The capillary lumina were dilated due to the presence of lipoprotein thrombi with pale staining on periodic acid-methenamine silver (Figure 1C). Oil red O staining confirmed the presence of intraluminal lipid droplets (Figure 1D). Immunofluorescence microscopy revealed that immunoglobulins (i.e., IgG, IgA, and IgM) and complement factor 1q (C1q) were negative. These findings were highly suggestive of LPG.
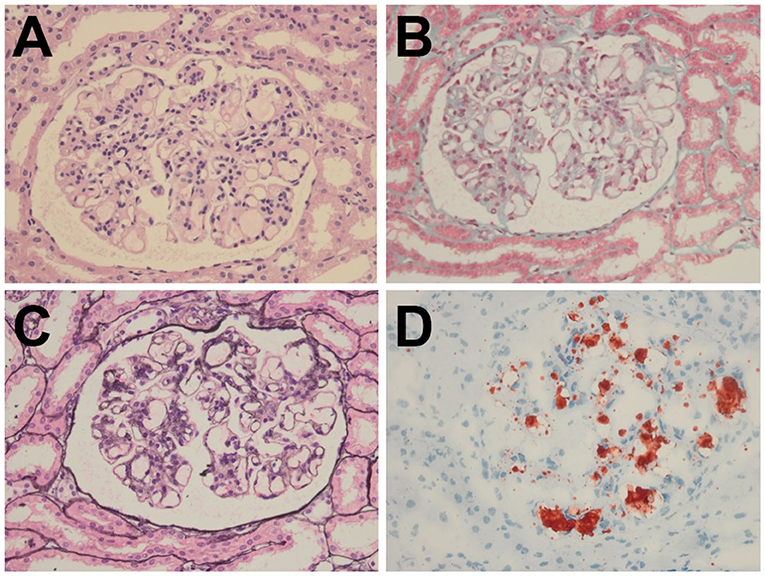
Figure 1. Histopathology of the renal biopsy specimen. (A) H&E (× 400) and (B) Masson (× 400) staining showed segmental glomerulosclerosis with mild-to-moderate proliferation of the mesangial cells and matrix. (C) PASM (× 400) staining revealed marked dilatation of the capillary lumina with lipoprotein thrombi. (D) Oil Red O (× 400) staining confirmed the presence of intraluminal lipid droplets.
Genetic testing confirmed the presence of the c.127C>T (p.R43C) or Kyoto mutation of the APOE gene (Figure 2A). Her parents refused to undergo genetic screening. Therefore, her grandparents were investigated and underwent genetic screening. Her grandfather was asymptomatic and her grandmother was otherwise healthy. The APOE mutation in the patient and her grandfather were heterozygous (Figure 2B). In addition, the patient did not have any siblings.
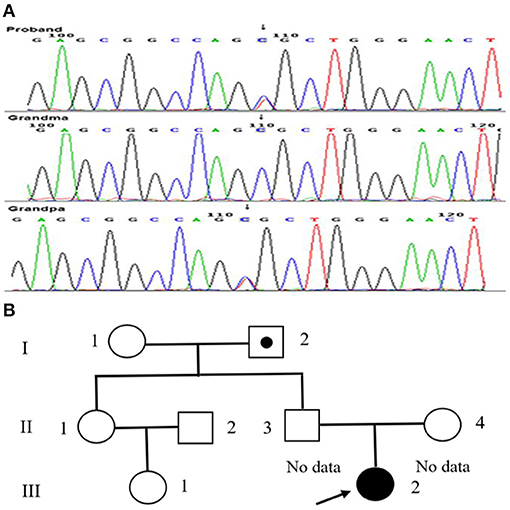
Figure 2. Mutation dictation of APOE and family pedigree of our patient. (A) The mutation in the patient (arrow). (B) The family pedigree of the patient. Proband III-2 is the present patient (arrow). Circles and squares indicate the females and males, respectively. The dotted symbol represents asymptomatic LPG carriers. II-1 and II-2 did not undergo genetic testing for special reasons, both of whom had no history of renal disease.
All of these findings were consistent with the diagnosis of LPG. Prednisone was gradually tapered. Ideally, the patient should have been given captopril and a lipid-lowering agent. However, her grandfather refused the latter.
A 2-year follow-up revealed that her serum creatinine (102 μmol/L) and urine protein levels (3.2 g/24 h) were elevated. Moreover, her lipid profile showed elevated total cholesterol 6.56 mmol/L, triglyceride (2.82 mmol/L), and LDL-cholesterol (3.87 mmol/L).
Discussion
LPG is a rare autosomal dominant renal disease. Although an increasing number of LPG cases have been reported in China, pediatric LPG cases are rare. We described a case of LPG in an 11-year-old girl initially presenting with nephrotic syndrome and anemia. Based on the laboratory examination, there was no evidence suggestive of a secondary cause of NS. However, initial treatment with prednisone did not improve her NS, prompting further evaluation with renal biopsy and genetic testing.
Furthermore, we conducted a literature search of Chinese LPG cases from various databases (i.e., China National Knowledge Infrastructure, Wanfang, WeiPu, PubMed, and Web of Science) from the year of creation to 2021. The language was limited to Chinese and English. Including our case, a total of 18 pediatric cases and 156 adult cases were identified. The retrieved clinical data of these cases are summarized in Tables 1, 2. There were 18 pediatric patients (male = 13; female = 5) with an average age of 12.0 ± 2.90 years (range, 7–17). In contrast, there were 156 adult patients (male = 87; female = 69) with an average age of 35.6 ± 13.8 years (range, 18–72). Furthermore, LPG was mainly diagnosed in teenagers aged 11–17 years and adults aged 31–50 years.
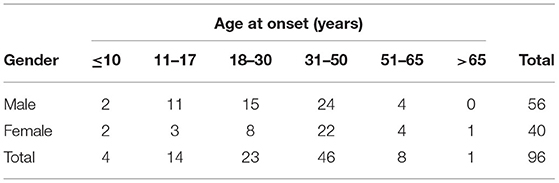
Table 1. Age and sex distribution of 96 patients with LPG in China.
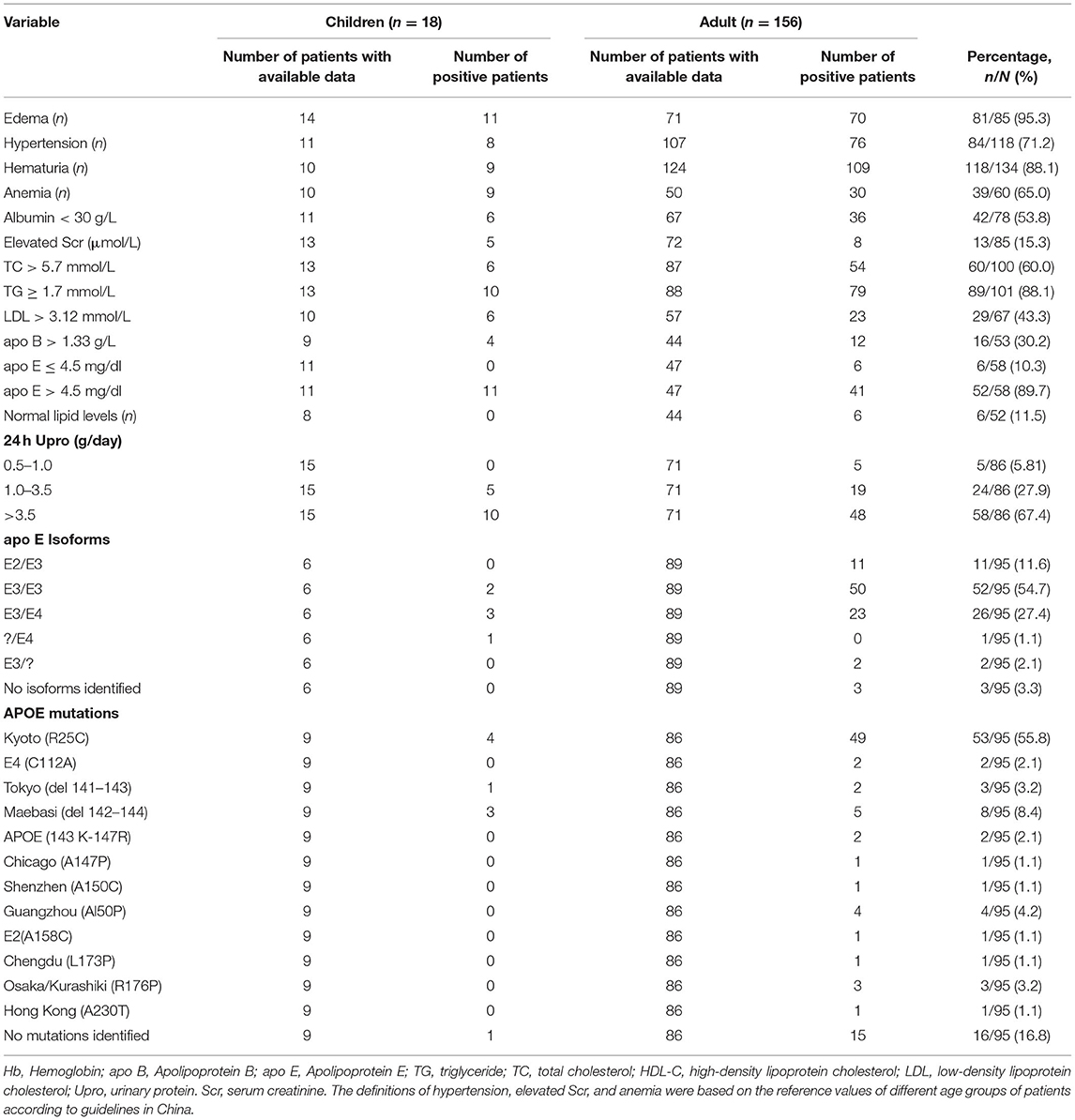
Table 2. Clinical features and genetic characteristics of Chinese LPG patients.
The most common clinical features were edema (95.3%), hematuria (88.1%), hypertriglyceridemia (88.1%), and increased serum apoE levels (89.7%). Other common clinical features were hypertension (71.2%) and anemia (65.0%). However, there were six patients with no clinical signs of systemic hyperlipidemia (e.g., xanthomas and corneal arcus). Furthermore, plasma lipid levels were within normal ranges in these patients (6–8). These findings indicate that the disease is localized primarily in the glomerulus due to the presence of lipoprotein thrombi (9). We also observed that 39 patients presented with unexplained anemia, especially 20 patients with Hb <90 g/L. Among them, one patient presented with severe anemia (Hb 38 g/L) likely due to severe thrombotic microangiopathy (10).
In our case, the patient also had mild anemia, suggesting that this might be an extrarenal manifestation of LPG. In addition, unexplained splenomegaly has been reported in patients with LPG (11, 12). Morris et al. (13) also reported that intravascular cardiac lipoproteinosis may be an extrarenal manifestation of LPG. However, the exact pathophysiologic mechanism of these extrarenal manifestations remains unclear.
The APOE gene is located on chromosome 19q13.2. It is polymorphic with three alleles (i.e., E2, E3, and E4), creating six genotypes (i.e., E2/2, E3/3, E4/4, E2/3, E2/4, and E3/4) (14). In Chinese patients with LPG, homozygosity for E3 (E3/3) is the most common isoform. LPG-associated apoE3 mutations play central roles in the pathogenesis of LPG through protein misfolding, thermodynamic destabilization, and aggregation (15). In the Chinese population, 12 APOE mutation sites have been reported to date, with APOE Kyoto mutation being the most common. Mutations in APOE Chengdu (p.L173P) (16), APOE Guangzhou (p.Rl50P) (17), APOE Shenzhen (p.R150C) (18), and APOE Hong Kong (p.D230Y) (19) have been reported.
In our case, the patient and her grandfather had the same APOE Kyoto mutation. However, her grandfather did not present with the clinical features of LPG. There are two possible explanations for this phenomenon. First, the APOE mutation could have been transmitted as autosomal dominant with incomplete penetrance (17, 20, 21). Second, the Kyoto mutation could have reduced the clearance of apoE from the circulation, requiring a “second hit” before LPG develops (22).
DNA analysis of two LPG patients revealed the co-existence of APOE Kyoto (p.R25C) with E4 (p.C112R) (23) and Hong Kong (p.D230Y) mutations (12). This may have a cumulative effect and ultimately result in phenotypic expression similar to LPG (12). However, 1 pediatric and 15 adult LPG cases did not present with any mutations. In APOE knockout mouse models, Wen et al. (24) demonstrated that APOE Sendai mutation was not necessarily required for the development of LPG. Therefore, APOE mutation is not a necessary factor in the pathogenesis of LPG. However, the underlying mechanisms of LPG remain unclear.
The various treatment strategies done in Chinese LPG cases are presented in Table 3. For patients with LPG, glucocorticoids and immune suppressants were ineffective. The efficacy of lipid-lowering agents is controversial because the types, dosages, courses, and evaluation were variable among different studies (7, 25–27). In addition, few case reports have shown that double-filtration plasmapheresis therapy may be effective (23).
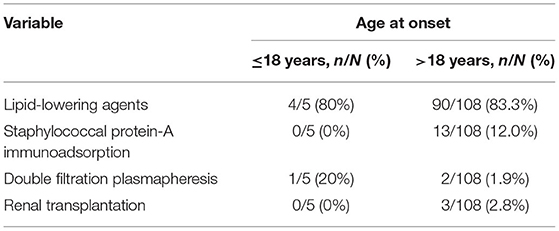
Table 3. Treatment strategy of Chinese LPG patients.
Moreover, Xin et al. (28) administered staphylococcal protein-A immunoadsorption in 13 patients with LPG for 10 sessions of 10 cycles per session. After treatment, urine protein and serum apoE levels decreased. Moreover, the intraglomerular lipoprotein thrombi almost disappeared. Among three patients who underwent kidney transplantation, all relapsed during the follow-up period (17, 29).
In our case, captopril was administered. Although there is no evidence suggesting that angiotensin-converting enzyme inhibitors (ACEi) are effective in the treatment of LPG, it is reasonable to use these drugs to retard the progression of renal disease in patients with persistent proteinuria (12). However, despite treatment, her lipid profile and serum creatinine were still abnormal after 2 years.
We have described a case of a typical LPG with APOE Kyoto mutation in a pediatric patient. Because LPG is difficult to distinguish from primary NS in pediatric patients, it is important to consider LPG as a differential diagnosis, especially in patients who have NS with atypical features (e.g., anemia), significant hypertension, family history of renal disease, or steroid resistance. Renal pathology and genetic testing may be crucial to diagnose the disease early and facilitate timely management.
Author Contributions
YS, CY, and HW designed the study, and drafted and reviewed the manuscript. YS, CY, LL, and HW performed the literature search, made the tables, and reviewed the manuscript. YS and CY contributed equally to this work. All authors read and approved the final manuscript.
Funding
Funding was provided by the National Natural Science Foundation of China (Grant Number: 81401239).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We acknowledge the doctors of the Department of Pathology of West China University Hospital for their help with data collection.
Abbreviations
APOE, apolipoprotein E; LDL, low-density lipoprotein cholesterol; LPG, lipoprotein glomerulopathy; NS, nephrotic syndrome.
References
1. Saito T, Sato H, Kudo K, Oikawa S, Shibata T, Hara Y, et al. Lipoprotein glomerulopathy: glomerular lipoprotein thrombi in a patient with hyperlipoproteinemia. Am J Kidney Dis. (1989) 13:148–53. doi: 10.1016/S0272-6386(89)80134-9
2. Saito T, Oikawa S, Sato H, Sasaki J. Lipoprotein glomerulopathy: renal lipidosis induced by novel apolipoprotein E variants. Nephron. (1999) 83:193–201. doi: 10.1159/000045511
3. Rall SC, Weisgraber KH, Mahley RW. Human apolipoprotein E. The complete amino acid sequence. J Biol Chem. (1982) 257:4171–8. doi: 10.1016/S0021-9258(18)34702-1
4. Saito T, Ishigaki Y, Oikawa S, Yamamoto TT. Role of apolipoprotein E variants in lipoprotein glomerulopathy and other renal lipidoses. Clin Exp Nephrol. (2001) 5:201–8. doi: 10.1007/s10157-001-8014-3
5. Matsunaga A, Furuyama M, Hashimoto T, Toyoda K, Ogino D, Hayasaka K. Improvement of nephrotic syndrome by intensive lipid-lowering therapy in a patient with lipoprotein glomerulopathy. Clin Exp Nephrol. (2009) 13:659–62. doi: 10.1007/s10157-009-0207-1
6. Yang Z, Wu H, Hu Z. Discovery of a Chinese Tibetan patient with lipoprotein glomerulopathy due to APOE Osaka/Kurashiki variant. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. (2020) 37:166–9. doi: 10.3760/cma.j.issn.1003-9406.2020.02.017
7. Yang M, Weng Q, Pan X, Hussain HMJ, Yu S, Xu J, et al. Clinical and genetic analysis of lipoprotein glomerulopathy patients caused by APOE mutations. Mol Genet Genomic Med. (2020) 8:e1281. doi: 10.1002/mgg3.1281
8. Saito T, Sato H, Oikawa S, Kudo K, Kurihara I, Nakayama K, et al. Lipoprotein glomerulopathy. Report of a normolipidemic case and review of the literature. Am J Nephrol. (1993) 13:64–8. doi: 10.1159/000168591
10. Wu Y, Chen X, Yang Y, Wang B, Liu X, Tao Y, et al. A case of lipoprotein glomerulopathy with thrombotic microangiopathy due to malignant hypertension. BMC Nephrol. (2013) 14:53. doi: 10.1186/1471-2369-14-53
11. Hagiwara M, Yamagata K, Matsunaga T, Arakawa Y, Usui J, Shimizu Y, et al. A novel apolipoprotein E mutation, ApoE Tsukuba (Arg 114 Cys), in lipoprotein glomerulopathy. Nephrol Dial Transplant. (2008) 23:381–4. doi: 10.1093/ndt/gfm735
12. Cheung CY, Chan AO, Chan YH, Lee KC, Chan GP, Lau GT, et al. A rare cause of nephrotic syndrome: lipoprotein glomerulopathy. Hong Kong Med J. (2009) 15:57–60.
13. Morris CS, Bois MC, Aust CH, Thomas R, Sethi S, Maleszewski JJ. Intravascular cardiac lipoproteinosis: extrarenal manifestation of lipoprotein glomerulopathy. Cardiovasc Pathol. (2019) 42:6–9. doi: 10.1016/j.carpath.2019.04.006
14. Cambruzzi E, Pêgas KL. Pathogenesis, histopathologic findings and treatment modalities of lipoprotein glomerulopathy: a review. J Bras Nefrol. (2019) 41:393–9. doi: 10.1590/2175-8239-jbn-2018-0148
15. Katsarou M, Stratikos E, Chroni A. Thermodynamic destabilization and aggregation propensity as the mechanism behind the association of apoE3 mutants and lipoprotein glomerulopathy. J Lipid Res. (2018) 59:2339–48. doi: 10.1194/jlr.M088732
16. Wu H, Yang Y, Hu Z. The novel apolipoprotein E mutation ApoE Chengdu (c.518T>C, p.L173P) in a Chinese patient with lipoprotein glomerulopathy. J Atheroscler Thromb. (2018) 25:733–40. doi: 10.5551/jat.41996
17. Luo B, Huang F, Liu Q, Li X, Chen W, Zhou SF, et al. Identification of apolipoprotein E Guangzhou (arginine 150 proline), a new variant associated with lipoprotein glomerulopathy. Am J Nephrol. (2008) 28:347–53. doi: 10.1159/000111828
18. Ku M, Tao C, Zhou AA, Cheng Y, Wan QJ. A novel apolipoprotein E mutation (p.Arg150Cys) in a Chinese patient with lipoprotein glomerulopathy. Chin Med J. (2019) 132:237–9. doi: 10.1097/CM9.0000000000000050
19. Liao MT, Tsai IJ, Cheng HT, Lin WC, Chang YW, Lin YH, et al. A rare cause of childhood-onset nephrotic syndrome: lipoprotein glomerulopathy. Clin Nephrol. (2012) 78:237–40. doi: 10.5414/CN106876
20. Ando M, Sasaki J, Hua H, Matsunaga A, Uchida K, Jou K, et al. A novel 18-amino acid deletion in apolipoprotein E associated with lipoprotein glomerulopathy. Kidney Int. (1999) 56:1317–23. doi: 10.1046/j.1523-1755.1999.00677.x
21. Ogawa T, Maruyama K, Hattori H, Arai H, Kondoh I, Egashira T, et al. A new variant of apolipoprotein E (apo E Maebashi) in lipoprotein glomerulopathy. Pediatr Nephrol. (2000) 14:149–51. doi: 10.1007/s004670050032
22. Rovin BH, Roncone D, McKinley A, Nadasdy T, Korbet SM, Schwartz MM. APOE Kyoto mutation in European Americans with lipoprotein glomerulopathy. N Engl J Med. (2007) 357:2522–4. doi: 10.1056/NEJMc072088
23. Li W, Wang Y, Han Z, Luo C, Zhang C, Xiong J. Apolipoprotein e mutation and double filtration plasmapheresis therapy on a new Chinese patient with lipoprotein glomerulopathy. Kidney Blood Press Res. (2014) 39:330–9. doi: 10.1159/000355810
24. Wen M, Segerer S, Dantas M, Brown PA, Hudkins KL, Goodpaster T, et al. Renal injury in apolipoprotein E-deficient mice. Lab Invest. (2002) 82:999–1006. doi: 10.1097/01.LAB.0000022222.03120.D4
25. Hu Z, Huang S, Wu Y, Liu Y, Liu X, Su D, et al. Hereditary features, treatment, and prognosis of the lipoprotein glomerulopathy in patients with the APOE Kyoto mutation. Kidney Int. (2014) 85:416–24. doi: 10.1038/ki.2013.335
26. Lui DTW, Lee ACH, Yap DYH, Chan GSW, Tan KCB. A young Chinese man with nephrotic syndrome due to lipoprotein glomerulopathy. J Clin Lipidol. (2019) 13:251–3. doi: 10.1016/j.jacl.2018.12.004
27. Zhang P, Matalon R, Kaplan L, Kumar A, Gallo G. Lipoprotein glomerulopathy: first report in a Chinese male. Am J Kidney Dis. (1994) 24:942–50. doi: 10.1016/S0272-6386(12)81066-3
28. Xin Z, Zhihong L, Shijun L, Jinfeng Z, Huiping C, Caihong Z, et al. Successful treatment of patients with lipoprotein glomerulopathy by protein A immunoadsorption: a pilot study. Nephrol Dial Transplant. (2009) 24:864–9. doi: 10.1093/ndt/gfn555
Keywords: lipoprotein glomerulopathy, pediatric, Chinese, nephrotic syndrome, anemia
Citation: Song Y, Yang C, Liu L and Wang H (2021) Case Report: A Pediatric Case of Lipoprotein Glomerulopathy in China and Literature Review. Front. Pediatr. 9:684814. doi: 10.3389/fped.2021.684814
Received: 24 March 2021; Accepted: 26 July 2021;
Published: 27 August 2021.
Edited by:
Joshua Zaritsky, Alfred I. duPont Hospital for Children, United StatesReviewed by:
Sushmita Banerjee, CK Birla Hospitals, IndiaFrancisco Cano, University of Chile, Chile
Copyright © 2021 Song, Yang, Liu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hua Wang, d2FuZ2h1YUBzY3UuZWR1LmNu
†These authors have contributed equally to this work