 Shaili Amatya
Shaili Amatya Meixia Ye
Meixia Ye Lili Yang
Lili Yang Chintan K. Gandhi
Chintan K. Gandhi Rongling Wu
Rongling Wu Beth Nagourney5
Beth Nagourney5 Joanna Floros
Joanna Floros
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pediatr., 04 October 2021
Sec. Genetics of Common and Rare Diseases
Volume 9 - 2021 | https://doi.org/10.3389/fped.2021.682160
This article is part of the Research TopicGenetic and Environmental Factors in the Occurrence of Neonatal Disorders - Volume IView all 10 articles
Background: Neonatal respiratory distress syndrome (RDS), due to surfactant deficiency in preterm infants, is the most common cause of respiratory morbidity. The surfactant proteins (SFTP) genetic variants have been well-studied in association with RDS; however, the impact of SNP-SNP (single nucleotide polymorphism) interactions on RDS has not been addressed. Therefore, this study utilizes a newer statistical model to determine the association of SFTP single SNP model and SNP-SNP interactions in a two and a three SNP interaction model with RDS susceptibility.
Methods: This study used available genotype and clinical data in the Floros biobank at Penn State University. The patients consisted of 848 preterm infants, born <36 weeks of gestation, with 477 infants with RDS and 458 infants without RDS. Seventeen well-studied SFTPA1, SFTPA2, SFTPB, SFTPC, and SFTPD SNPs were investigated. Wang's statistical model was employed to test and identify significant associations in a case-control study.
Results: Only the rs17886395 (C allele) of the SFTPA2 was associated with protection for RDS in a single-SNP model (Odd's Ratio 0.16, 95% CI 0.06–0.43, adjusted p = 0.03). The highest number of interactions (n = 27) in the three SNP interactions were among SFTPA1 and SFTPA2. The three SNP models showed intergenic and intragenic interactions among all SFTP SNPs except SFTPC.
Conclusion: The single SNP model and SNP interactions using the two and three SNP interactions models identified SFTP-SNP associations with RDS. However, the large number of significant associations containing SFTPA1 and/or SFTPA2 SNPs point to the importance of SFTPA1 and SFTPA2 in RDS susceptibility.
Neonatal respiratory distress syndrome (RDS) is the most common cause of respiratory failure in premature infants due to surfactant deficiency (1). However, the infant mortality rate due to RDS was 11.4 per 100,000 live births and accounted for 2% of all infant deaths in 2017 in the United States (2) despite the judicious use of postnatal surfactant along with antenatal steroids (3).
Major risk factors, such as prematurity and low birth weight (BW) along with sex and race (4–7) have been implicated in RDS. Genetic factors have also been associated with RDS by various twins' studies (8, 9). Thus, the susceptibility to RDS is considered multifactorial and/or polygenic (10), with ample evidence in the literature that gene–host-environment interactions may play a large role in the morbidity and mortality associated with this syndrome. The understanding of gene interactions in RDS may help identify novel therapeutic targets for susceptible infants.
Furthermore, it has been noted that infants dying with RDS have low levels of surfactant proteins (SP) (11, 12). SP-A and SP-D are hydrophilic proteins and play an important role in innate immunity and the regulation of inflammatory processes and host defense (13–17). SP-B and SP-C are hydrophobic proteins that enhance the adsorption and spreading of surfactant phospholipid (18). In addition, SP-B is essential for lung function by reducing surface tension and preventing alveolar collapse (19–21). SP B and SP-C are present in the exogenous surfactant used to treat RDS. However, SP-A and SP-D (SP-D co-isolates with the surfactant complex) are not included in the formulation, even though a major complication in prematurely born infants with RDS is infection. In addition to its host defense function, SP-A, along with SP-B, is important for the formation of tubular myelin (an extracellular surfactant structure) (22–24). Moreover, SP-A is involved in surfactant-related functions (17, 25) and lung airway function (26).
Multiple genetic variants and single nucleotide polymorphisms (SNP) of the surfactant protein gene (SFTP) have been shown to associate with RDS (10, 27–40). Human SP-A, consisting of SP-A1 and SP-A2 proteins, is encoded by two functional genes SFTPA1 and SFTPA2, respectively (41). The SFTPA1 and SFTPA2 genes share a high degree of sequence similarity but differ at various splice variants at the 5′ untranslated region (UTR) and exhibit sequence variability within coding and non-coding regions (17). Prior studies have also found intragenic and intergenic haplotypes between SFTPA1 and/or SFTPA2 (42) and SFTPB and/or SFTPD haplotypes associated with risk or protective effect in RDS (43).
However, the impact of SNP-SNP interactions on RDS susceptibility has not been addressed before. The synergistic (epistatic) interactions among genetic variants of the surfactant proteins may alter disease susceptibility (44, 45), but this was not possible to study earlier due to the limitation of statistical approaches at the time. However, current more advanced statistical models may help identify the intricate epistatic interaction among multiple gene variants that play a significant role in multifactorial and complex diseases, such as RDS. Such analysis is likely to be beneficial to understand the impact of genetics on complex diseases, especially as we move toward personalized medicine.
In the present study, we studied intergenic and intragenic SNP-SNP interactions of the SFTP genes. We hypothesized that epistatic interactions among SFTP gene variants are associated with RDS susceptibility in preterm infants.
This study used available genotype data and clinical information in the Floros biobank at Penn State University, College of Medicine. These were collected and processed under an approved protocol by the institutional review board from the human subject protection office of the Pennsylvania State University (PSU) College of Medicine as well as the institutional review board of the respective centers where samples were collected in other Institutions other than PSU, as described previously (12, 29, 31, 32, 46, 47). The clinical and demographic data of the study samples are given in Table 1. The patients consisted of 848 preterm infants born <36 weeks of gestation, stratified by RDS, where 458 infants were diagnosed with RDS, and 477 infants did not develop RDS. RDS was diagnosed by clinical features of respiratory distress such as retractions, grunting, and flaring after birth. Chronic lung disease was diagnosed as needing supplemental oxygen at 28 days of life or 36 weeks postmenstrual age (50). Chorioamnionitis was diagnosed by clinical features such as maternal fever. The use of antenatal steroids was variable with betamethasone or dexamethasone.
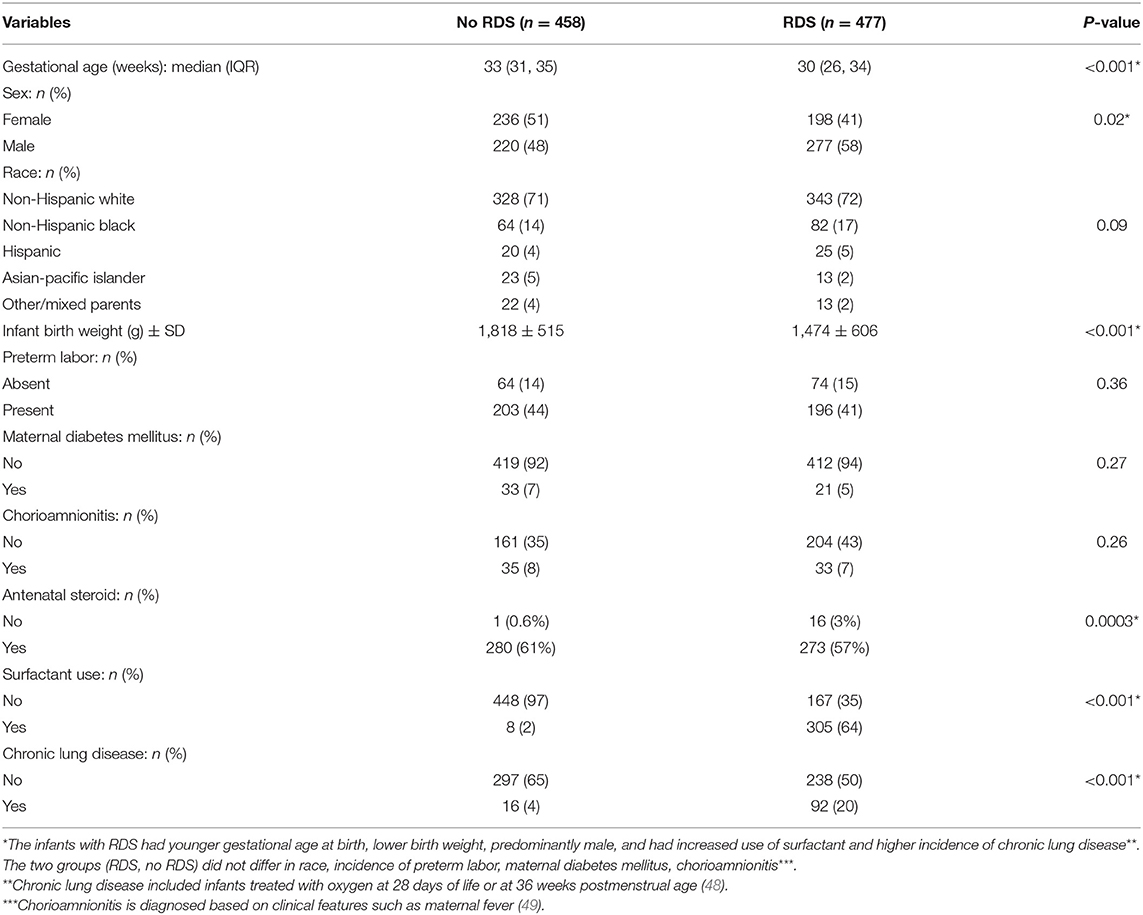
Table 1. Clinical Characteristics of the cohort with and without RDS.
A total of 17 SNPs of the SP genes SFTPA1, SFTPA2, SFTPB, SFTPC, and SFTPD were studied. These included five SNPs from SFTPA1: rs1059047, rs1136450, rs1136451, rs1059057, and rs4253527; four SNPs from SFTPA2: rs1059046, rs17886395, rs1965707, and 1965708; four SNPs from SFTPB: rs1130866, rs7316, rs2077079, and rs3024798; two SNPs from SFTPC: rs4715 and rs1124; and two SNPs from SFTPD: rs721917 and rs2243639. Polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP) was used to analyze the SFTP gene polymorphisms as described (49, 51, 52).
Wang et al. (53) developed a general multi-locus model for analyzing genetic associations in a case-control study. This model has three characteristics. First, it integrates classic quantitative genetic principles into a categorical data analysis framework, allowing epistatic interactions to be interpreted on a solid genetic basis. Second, this model can not only detect the genetic effects of single SNPs and pairwise genetic interactions, but also characterize high-order genetic interactions. That is, the model dissects genotypic differences into additive (a) and dominant (d) genetic effects at individual SNPs: additive × additive (aa), additive × dominant (ad), dominant × additive (da), and dominant × dominant (dd) epistatic effects at a pair of SNPs, and additive × additive × additive (aaa), additive × additive × dominant (aad), additive × dominant × additive (ada), additive × dominant × dominant (add), dominant × additive × additive (daa), dominant × additive × dominant (dad), dominant × dominant × additive (dda), dominant × dominant × dominant (ddd) epistatic effects at a triad of SNPs. Mounting evidence shows that high-order interactions play an important role in mediating complex traits and complex human diseases (54). Third, while the precise detection of a pairwise genetic interaction requires a huge number of samples, such as 5,000 (55), which may be hardly met in general studies, Wang et al.'s model is less sample size-reliant by coalescing case and control samples into a 2 × 2 contingency table for the detection of epistasis at any order. The statistical properties of Wang et al.'s model have been extensively studied through computer simulation, with results, presented in the original article, demonstrating its usefulness and robustness in a small-sample case-control study. Also, a detailed computational procedure of this model was given in the original article, allowing the readers to understand and repeat the model.
For each type of data analysis, case-control genotype observations were sorted into a 2 × 2 contingency table to test each of the genetic effects described above. For example, consider a SNP with three genotypes AA, Aa, and aa. To estimate its dominant effect, the effect size was compared to that of the heterozygote Aa against the average size of each of the two homozygotes AA and aa in cases and controls, respectively. Based on the resulting 2 × 2 contingency table, the logistic regression model was implemented to estimate the dominant effect of this SNP, and the effects were adjusted for age and sex. The odds ratio (OR) was estimated to assess the magnitude of the dominant/additive effect.
To estimate the additive effect, the size was compared as below,
Odds of genotype for cases = number of cases with AA/number of cases with aa
Odds of genotype for controls = number of controls with AA/number of controls with aa
OR = odds for cases/odds for controls
=
For example-
OR = 1: Genotype difference is not associated with the disease;
OR > 1.0: Genotype AA is “more risky” (i.e., associated with higher risk for the disease than genotype aa)
OR < 1.0: Genotype aa is “more risky” for the disease than genotype AA
A similar procedure was applied to analyze all other genetic effects.
The significance of each effect was adjusted for multiple comparisons using the false discovery rate (FDR) controlled at 1%. Wang et al.'s simulation data indicate that a 100 × 100 sample size combination in an epistatic case-control model has a power of > 0.80 to detect significant associations in a 2 × 2 contingency table analysis (53). Thus, our current sample size provides adequate power to detect all the significant epistatic interactions.
Table 1 shows the demographic and clinical characteristics of infants with and without RDS. There were 458 infants without RDS and 477 infants who developed RDS. Infants with RDS were younger as assessed by gestational age at birth (30 vs. 33 weeks) and had lower birth weight (1,474 ± 606 gram vs. 1,818 ± 515 gram) compared to infants without RDS. Infants with RDS were predominantly male (58 vs. 48%, p-value 0.02). The two risk factors for RDS (gestational age and sex) were corrected in the analysis. Gestational age and birth weight are co-linear variables, and only one (gestational age) was chosen to be corrected in the analysis. As expected, infants who developed RDS had increased use of surfactant and a higher incidence of chronic lung disease than infants who did not have RDS. These outcomes are related to RDS rather than predictors (surfactant use and chronic lung disease); therefore, we did not correct them in the SNP-SNP interaction model. The use of antenatal steroids was significantly different between the two groups. However, ~40% of the antenatal steroid data were missing and may have caused bias in estimating this parameter.
The associations of single SNP and intergenic/intragenic two and three SNP interactions with RDS are shown in Tables 2–4, respectively. The tables show the specific SNPs of the SFTP genes and their effect, either additive (a) or dominant (d). The additive effect of the SNP indicates that one of the homozygous alleles (one or two copies) is associated with the disease compared to the other homozygous allele. The dominant effect of the SNP indicates that the heterozygous genotype is associated with the disease compared to the mean of either homozygous genotype. The numbers 1, 2, or 3 are for SNP1, SNP2, or SNP3, respectively. For example, (a) a1d2 (Table 3) interaction means that the presence of any minor allele genotype of SNP1 and the heterozygous genotype of SNP2 is significant. (b) d1d2d3 (Table 4) interaction indicates that the combination of the heterozygous genotype at the first, second, and third SNP is associated with the disease.

Table 2. Single SNP associated with RDS.
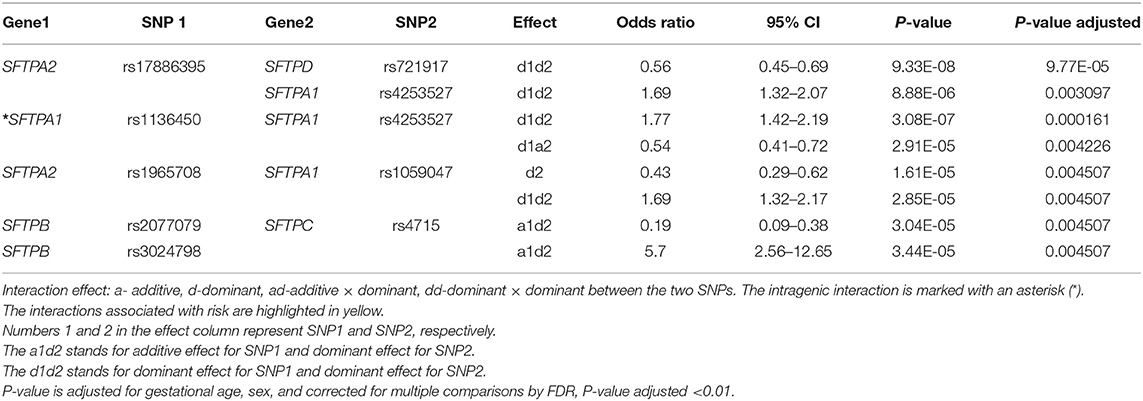
Table 3. The two SNP interactions associated with RDS susceptibility.
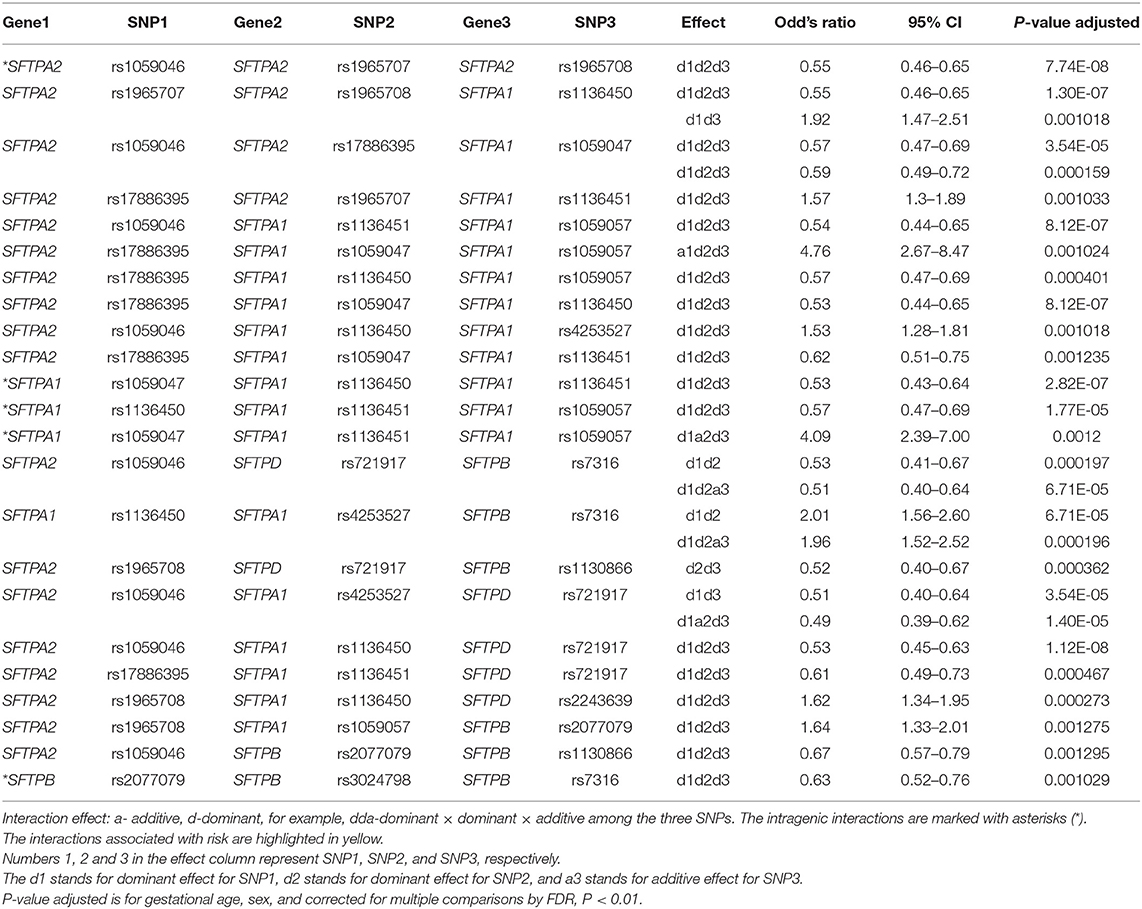
Table 4. Three SNP-SNP-SNP interactions of surfactant protein genes associated with RDS.
Out of the 17 SNPs of the five SFTP genes, only the rs17886395 of the SFTPA2 was associated by itself with RDS (Table 2). This SNP exhibited an additive effect on RDS susceptibility (OR 0.16, 95% CI 0.06–0.43, adjusted p = 0.03). This particular SNP is also noted to interact with other SNPs in the two and three SNP interactions models, as shown in Tables 3, 4. No other SFTP SNP by itself was associated with RDS at the adjusted value p < 0.01.
Among the two SNP interactions, the only intragenic interaction included SFTPA1 SNPs; rs1136450 and rs4253527 (Table 3), and this combination exhibited two effects, where the d1d2 interaction was associated with increased risk for RDS (OR 1.77, 96% CI 1.42–2.19, adjusted P = 0.0001), and the d1a2 was associated with protection for RDS (OR 0.54, 95% CI 0.41–0.72, adjusted P = 0.004) (Figure 1).
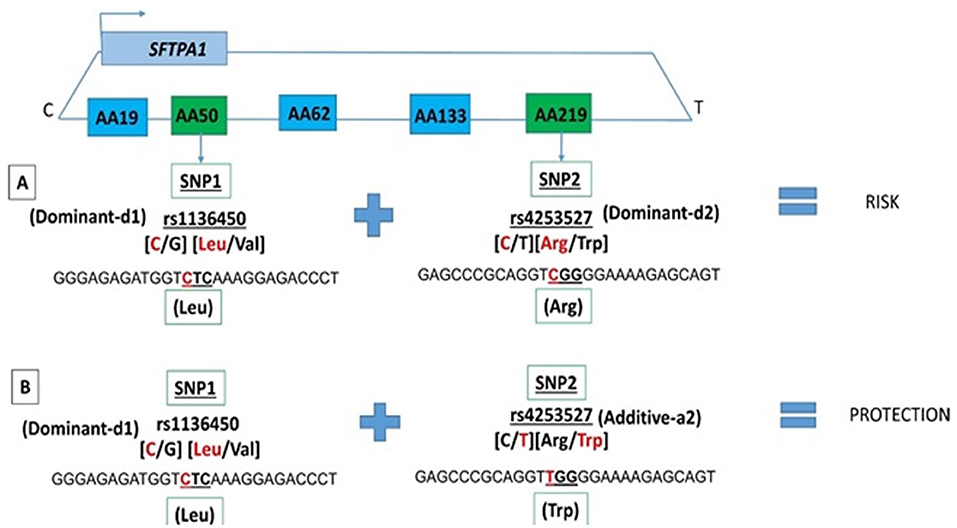
Figure 1. SFTPA1 intragenic two SNP interactions and RDS susceptibility. It shows the schematic presentation of SFTPA1 on the top and the arrow depicts the transcriptional orientation. The relative location of SNPs are shown from centromere (C) to telomere (T) and each box represents the amino acid number that includes the particular SNP. For example, AA50 denotes the rs1136450 SNP and AA219 denotes the rs4253527 SNP. The amino acid numbering is based on the precursor molecule and thus includes the signal peptide (56). In this two SNP intragenic interaction, underneath the green boxes are the SNP ID and the actual SNPs involved. (A) The dominant effect, dl of SNP1, is highlighted by red, as noted for the rs1134650 C variant that codes for leucine. This interacts with the dominant effect of SNP2, rs4253527 C variant that codes for arginine (in red) and increases risk of RDS. (B) The dominant effect, dl of SNP1, is highlighted by red and this SNP interacts with the additive effect of SNP2 rs4253527 T variant that codes for tryptophan and this interaction is protective for RDS.
There were five intragenic interactions associated with RDS. Three interactions were among SNPs of the SFTPA1 and two involved the SFTPA2 and SFTPB genes. The SFTPA2 SNPs: rs1059046, rs1965707, and rs1965708 exhibited an effect, d1d2d3, that was protective for RDS (OR = 0.55, 95% CI 0.46–0.55, adjusted p < 0.01). The SFTPA1 gene variants: rs1059047 (SNP1), rs1136451 (SNP2), rs1059057 (SNP3) in a three-SNP interaction (d1a2d3) increased the risk for RDS (OR 4.09, 95% CI 2.39–7.00, adjusted p = 0.0012) (Table 3). The other intragenic interaction, d1d2d3, was found among SFTPB SNPs: rs2077079 (SNP1), rs3024798 (SNP2), and rs7316 (SNP3), as d1d2d3, and this was protective for RDS (OR = 0.63, 95% CI 0.52–0.76, adjusted P 0.001).
The two SNP interactions are shown in Table 3. The combination of SFTPA2 rs17886395 (SNP1) with (i) SFTPA1 rs4253527 (SNP2) as d1d2, increased risk of RDS (OR 1.69, 95% CI 1.32–2.17, adjusted p = 0.004), and (ii) SFTPA1 rs1059047 (SNP2) as d2 without any epistatic effect from SNP1 was protective (OR 0.43, 95% CI 0.29–0.62, adjusted p = 0.004). The SFTPA2 SNP rs17886395 interaction with the SFTPD SNP rs721917 was protective when both had a dominant effect (OR 0.56, 95% CI 0.45–0.69 adjusted p < 0.01). Intergenic SNP-SNP interactions were also noted between each of the two of the SFTPB SNPs (rs2077079 or rs3024798) and one SFTPC SNP rs4715 associated with protection or risk against RDS, as shown in Table 3.
Table 4 shows the intergenic three SNP interactions of the SFTP genes associated with RDS. There were a total of 28 intergenic interactions. There were four SFTPA2 SNPs studied. Among them, the rs17886395 SNP, found to have an additive effect and be protective for RDS by itself in the single SNP model, was present in 7 out of the 28 intergenic interactions and in 5 out of the 7 interactions were noted to be protective.
The five SFTPA1 gene SNPs exhibited mainly a dominant effect. The rs1136450 was involved in the highest number of interactions (10 intergenic interactions), and the other SFTPA1 SNPs had fewer than 5 interactions showing either protective or risk effect. An example of a three intergenic SNP interaction is shown diagrammatically in Figure 2. This figure depicts an interaction among three SNPs of SFTPA1 and SFTPA2. In this intergenic interaction, the additive effect of SNP1, rs17886395, G variant that codes for alanine interacts with SNP2 (rs1059047) and SNP3 (rs1059057) of SFTPA1 in a dominant effect. This interaction, based on odd's ratios, is associated with increased disease susceptibility. It has the highest odd's ratio (OR 4.76, 95% CI 2.67–8.47) compared to the odd's ratios of the other three SNP interactions.

Figure 2. Intergenic three SNP interaction and RDS susceptibility. It shows the schematic presentation of SFTPA2 and SFTPA1 on the top and the arrows depict the opposite transcriptional orientation. The relative location of SNPs is shown from centromere (C) to telomere (T) and each box represents the amino acid number that includes the particular SNP. For example, AA91 denotes the rs17886395 SNP, AA19 denotes the rs1059047 SNP, and AA133 denotes the rs1059057 SNP. In this three SNP intergenic interaction, underneath the green boxes are the SNP ID and the SNPs involved. The additive effect of SNP1, rs17886395 G variant that codes for alanine (highlighted in red) interacts with SNP2 and SNP3 of SFTPA1 in a dominant effect and increases risk of RDS.
The SFTPB SNPs (rs7316, rs1130866, rs2077079) were involved in 5 intergenic interactions, and the SFTPD SNPs (rs721917, rs2243639) were involved in a total of 6 intergenic interactions, and they were mainly in a dominant effect.
Figure 3 shows that the SNPs of the hydrophobic SFTPB and SFTPC interacted with each other in the two-SNP model, and the SNPs of the hydrophilic SFTPA1, SFTPA2, and SFTPD SNPs also interacted with each other. There was no interaction between any of the hydrophobic and the hydrophilic SPs SNPs. The three-SNP model (Figure 4) depicted an intricate network of interactions among all the SFTP genes, except for SFTPC. A total of 28 three SNP interactions were identified. The SFTPA1 and SFTPA2 have the maximum number of interactions and, along with SFTPD, interacted with SFTPB. All three SNP interactions, except for one intragenic interaction of SFTPB (rs2077079-SNP1, rs3024798-SNP2, rs7316-SNP3 as d1d2d3), involved either SFTPA1 or SFTPA2. This highlights the impact and importance of SFTPA1 and SFTPA2 in RDS.
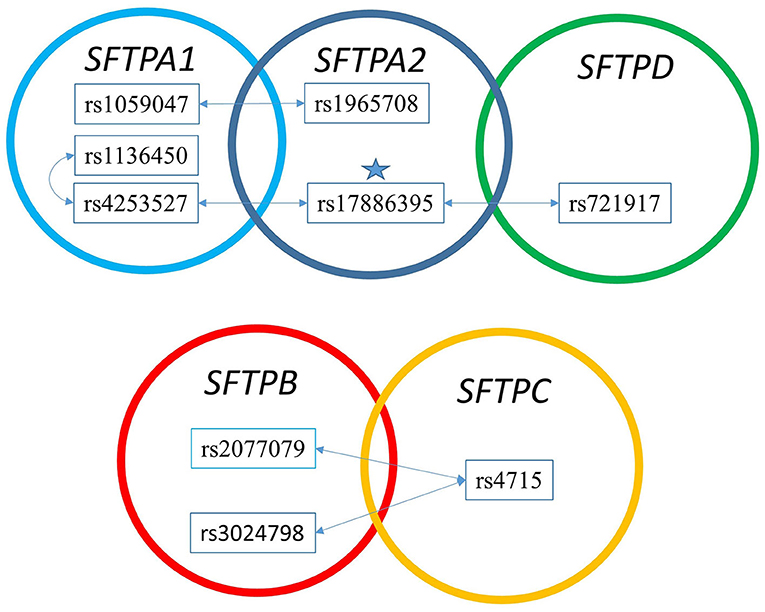
Figure 3. The two SNP interaction in RDS susceptibility. Associations between RDS and the two SNP-SNP interactions are shown. The star marks the SFTPA2 SNP shown to associate with RDS by itself. (A) depicts the two SNP-SNP intergenic and intragenic interactions of the hydrophilic SP genes associated with RDS. (B) depicts the two SNP-SNP intergenic and intragenic interactions of the hydrophobic SP genes associated with RDS.
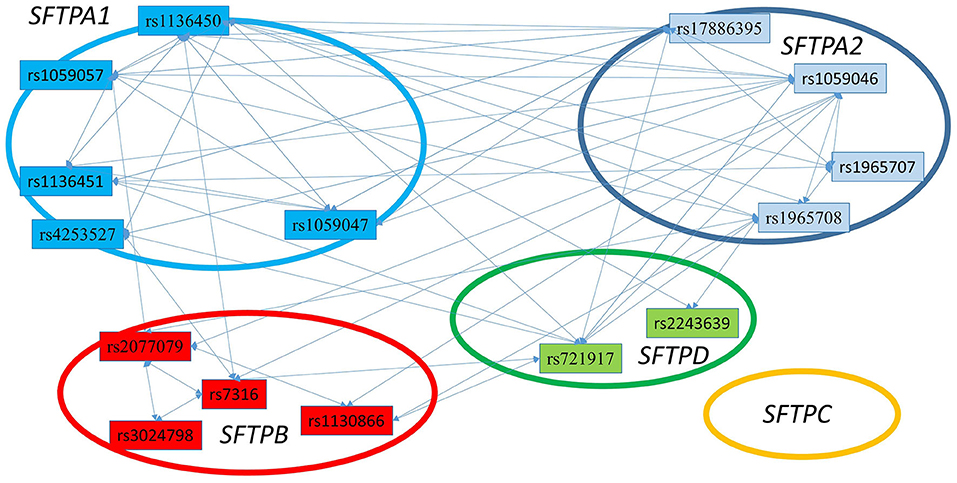
Figure 4. The three SNP interactions associated with RDS susceptibility. The figure depicts intergenic and intragenic interactions of SFTPA1, SFTPA2, SFTPD, and SFTPB genes. No three SNP interactions were observed that involved SFTPC SNPs. There are a total of 28 three SNP interactions. All interactions (but one) involved SFTPA1 and/or SFTPA2.
Although SFTP variants have been implicated in RDS (10, 27, 39), the statistical method used at the time had a limited ability to detect complex epistatic interactions among multiple SNPs. However, a more recent methodology by Wang et al. (53) enables investigation of complex SNP-SNP interactions by employing SNP interaction models. As one of very few statistical models that can analyze high-order interactions, Wang et al.'s (53) model has been used in a variety of case-control studies, showing its elegance and robustness. For example, using this model, interactions among SFTP SNPs were detected to impact cystic fibrosis (57), pediatric acute respiratory failure (58), and hypersensitivity pneumonitis (59). In this study, we used 17 well-studied SNPs of SFTPA1, SFTPA2, SFTPB, SFTPC, and SFTPD to investigate the impact of individual SNPs and SNP interactions among SNPs in the same gene or between two or among three different genes. This approach revealed that (a) the highest number of the two and three SNP interactions were among SNPs of the SFTPA1 and SFTPA2, and these were associated with risk or protection for RDS. (b) Only the rs17886395 (C allele) of the SFTPA2 was protective for RDS in a single-SNP model. (c) In the two SNP models, there was no interaction between the hydrophilic SFTPA1, SFTPA2, SFTPD SNPs, and the hydrophobic SFTPB or SFTPC SNPs. (d) the three SNP models showed intricate intergenic and intragenic interactions among SNPs of the SFTPA1, SFTPA2, SFTPB, and SFTPD; however, SFTPC did not interact with any of the other SFTP SNPs. Thus, in the present study we show not only association of a single SNP but also of two and three SNP interactions to associate with RDS susceptibility.
Using the stringent criteria of FDR correction with 1% (p < 0.01), none of the single SFTP SNPs was associated with RDS. When the FDR correction was set at 5% (p < 0.05), the rs17886395 G allele of the SFTPA2 gene exhibited an additive effect and increased risk for neonatal RDS compared to the C allele. The 1A3 haplotype that includes the G allele increased the risk of TB in Mexicans (60). However, the C allele of the same SNP, found to be protective of RDS (present study), has also been protective against infection, such as RSV in Finnish infants (61). In contrast, in an Ethiopian study group, the C allele was associated with increased risk of TB (62), and this allele as part of 6A/1A genotype was associated with risk in community-acquired pneumonia in a Spanish study group (63). Several haplotypes of SFTPA1 and SFTPA2 have been well-characterized (39, 64) and the most common haplotype, 6A2/1A0, has been associated with low SP-A protein expression in a study of patients with sudden infant death syndrome (65). It is of interest that the C allele of the rs17886395 SNP in pediatric diseases (i.e., RDS, RSV) is associated with protection, but in diseases likely to occur in adults (i.e., TB, community-acquired pneumonia) is associated with risk. Whether disease susceptibility by the C allele of the rs17886395 SNP is influenced by the lung environment in an age-dependent manner remains to be determined. The association of this particular SNP (rs17886395) in RDS susceptibility in the current study may not be surprising. Infection is a common complication of RDS and prematurity, and therefore the alleles of this SNP may differentially affect disease susceptibility.
The rs17886395 (C/G) is located in the collagen-like domain of SFTPA2 and changes the encoded amino acid Pro/Ala at codon 91 (41). It has been shown that proline normally stabilizes collagen triple helices due to conformational restrictions of the pyrrolidine ring and the presence of tertiary amides, while alanine substitutions tend to destabilize the triple helix (66). Thus, the G allele/GCT encoding alanine may destabilize the structure and explain the risk susceptibility.
We observed an association of the intragenic interaction between two SNPs (rs1136450 and rs4253527) of the SFTPA1 with RDS susceptibility in the two-SNP model. The susceptibility of RDS changes based on the effect of rs4253527 in that interaction, i.e., dominant and additive effect of rs4253527 is associated with increased and decreased risk of RDS, respectively (Figure 1). This indicates that an additive or a dominant effect of the same SNP may change the susceptibility of an individual to a particular disease based on interactions with other SNPs. The rs4253527 (C/T) is located within the carbohydrate recognition domain (CRD) of the SFTPA1 and changes the amino acid arginine (CGG) to tryptophan (TGG) at amino acid 219. This change may differentially affect innate immune processes under various conditions, including oxidative stress, because tryptophan is more sensitive to oxidation than arginine (56, 67). SFTPA1 variants that differ in CRD at rs4253527 have been shown to differ in their ability to enhance phagocytosis (68) and cytokine production (69). Moreover, the CRD of surfactant proteins A and D are known to mediate binding to infectious agents such as Pneumocystis carinii (70, 71) and therefore the susceptibility to RDS may be interconnected with response to infection. The rs1136450 (C/G) SNP has a leucine (CTC) to valine (CTC) substitution at amino acid 50 and together with rs4253527, may impact protein function, but direct experimental evidence is lacking. Moreover, SFTPA1 has been shown to more efficiently affect surfactant reorganization (than SFTPA2) in the alveolar space and inhibit surfactant inactivation by serum proteins (25). However, considering the complexity of SFTPA variants and their potential contribution to health and disease status, it is conceivable that the activity of a gene product in a given microenvironment, such as that in prematurity, is altered, and this may variably affect the health of the individual.
There were no significant interactions observed between SNPs of the hydrophilic and hydrophobic SPs. In contrast, previous observations have shown an association of SFTPB and SFTPA1 and/or SFTPA2 with increased risk of neonatal RDS in case-control studies (32, 34, 36). These apparent contrasting findings could be due to differences in the patient population, sample size, and/or statistical approaches used in previously reported studies and the present study.
This study, to our knowledge, is the first to show that interactions among three SNPs of the SP genes and their epistatic effect associate with RDS susceptibility. The majority of prior studies have at most reported interactions between two SNPs of the SP genes. The three SNP models in the present study showed that the highest number of intergenic and intragenic interactions involved SFTPA1 and SFTPA2, indicating perhaps the importance of these genes in RDS.
The SNP rs1136450 with a dominant effect had the highest number of interactions (n = 9), and these were associated with either risk or protection for RDS. The rs1136450 (C/G) results in an amino acid change, Leu/Val (CTC/GTC) at codon 50 (39, 41). This SNP is located in the N-terminal collagen region and the change in amino acid may affect the binding to receptors such as calreticulin/CD91 on phagocytes (72–74). The G allele (valine) of this SNP is associated with risk of interstitial pulmonary fibrosis (IPF) in a Mexican study group (49). On the other hand, the same allele was protective in community-acquired pneumonia (63). In prior studies, this allele has been associated with risk for RDS in Finnish, whites, and blacks (29, 30); however, this was not seen in a Korean study group (75). The current study showed that this SNP had a risk or protective effect based on interactions with SNPs of other SFTP genes. The various interactions may change the qualitative and/or quantitative function of SFTPA, and this could explain the variable outcome.
The rs1059046 SNP of SFTPA2 was also found to have a high number of interactions (n = 8), and all of the interactions with a dominant effect were shown to be protective for RDS. This SNP changes the amino acid Asn/Thr at codon 9 (AAC/ACC). This amino acid is part of the signal peptide and may affect the processing of SP-A2. The A allele of this SNP of SFTPA2 was also noted to have a protective role in community-acquired pneumonia (63). Of note, prior studies have shown the A allele, either in its homozygous or heterozygous form to be associated with risk for the respiratory syncytial virus (RSV) (61, 76) as well as influenza (77). The rs17886395 of SFTPA2, which was described in detail above, was also noted to have a high number of three SNP interactions (n = 7), five of them had a dominant effect with a protective role and the remaining two (dominant or additive) were associated with risk in RDS. These together highlight the complexity of SNP interactions and their important effect on disease susceptibility.
There was one significant intragenic interaction (rs2077079, rs3024798, and rs7316). Each SNP exhibited a dominant effect and this interaction was associated with decreased risk of RDS. The rs2077079 (C/A) is located 10 nt downstream of the TATAA box, 5′ regulatory region and may affect gene transcription. The rs3024798 (A/C) is located at the splice sequence of intron 2-exon 3 and may affect splicing. The rs7316 (A/G) is located in the 3′UTR, at 4 nt upstream of the TAATAAA polyadenylation signal and may affect polyadenylation (78). The location of these SNPs indicates that these may affect the processing and/or regulation of SP-B. Whether any of these mechanisms are negatively affected in RDS remains to be determined. However, each of these three SNPs has been previously shown to associate with various lung diseases (29, 57, 79, 80). The A allele of rs2077079 is associated with risk of RDS in blacks, whereas the A allele of rs3024798 is associated with protection of RDS (29). The A allele of rs7316 is associated with risk of RDS (79) and acute lung injury in African-American children (80). However, the dominant effect of rs7316 is associated with mild CF (57). It is interesting that these SNPs by themselves have been associated with risk or protection of RDS; however, the present study highlights the importance of SNP interactions, as these could mediate a differential epistatic effect compared to individual SNPs and that this may have a significant effect on the actual health/disease outcome of an individual under certain conditions.
None of the SNPs were identified in the three SNP model, even though single SFTPC SNPs have been associated with RDS (38, 81) and other pulmonary diseases such as interstitial lung disease (82). The hydrophobic SFTPB and SFTPC SNPs showed significant interactions in the two SNP model but not in the three SNP model. Furthermore, the two SFTPC SNPs rs1124 and rs4715 change amino acids 186 and 138, respectively. Although their effect on the functional or structural integrity of SP-C is not known, these likely affect processing of the precursor SP-C molecule rather than the mature SP-C, because these amino acids are part of the SP-C precursor and not of the mature SP-C.
The SFTPD SNPs were involved in intergenic interactions associated with RDS susceptibility. The SFTPD rs721917 (C/T) SNP changes Threonine (C) to Methionine (T) at position 11 in the mature protein. The C allele of the rs721917 SNP, is associated with O- linked glycosylation of threonine leading to a partial posttranslational modification and this may alter the tendency to form multimers (83, 84). Moreover, this SNP is associated with SP-D levels, with the T allele (methionine) being correlated with increased levels (83–85). The T allele of this SNP was protective for RDS (86, 87), whereas some studies reported no association with RDS (88). The current study also supports previous observations where SFTPA2 and SFTPD haplotypes were shown to be protective against RDS (42).
Although the present study has a relatively large sample size, one limitation is that the patient population differs from that of the controls in terms of age, birth weight, and sex. However, the analyses were adjusted for age and sex (birth weight was not corrected due to collinearity with gestational age). Another study limitation may be reduced generalizability as both study groups were predominantly whites. It is also possible that we have missed some significant interactions due to the use of stringent criteria such as those imposed by the FDR correction, set at 1% to avoid spurious associations. Nonetheless, the present findings need to be replicated. The SNP interactions and their association with the disease phenotype may be affected by the severity of RDS, which was not captured in this study. Around 40% of the data on important parameters such as antenatal use of steroids were missing and that may have introduced bias in the estimation of the difference between groups. The diagnosis of chronic lung disease included oxygen use at 28 days or oxygen at 36 weeks postmenstrual age. The definition for BPD has evolved over time and hence the study characteristic does not capture the current definition of BPD, consistently, as per NICHD 2019 (89).
Despite the above limitations, this study indicates a greater role of SFTPA1 and SFTPA2 in RDS susceptibility as they had the most interactions with SNPs of other SFTPs in the two and three-SNP models. Furthermore, the concern for infection in the setting of prematurity and chorioamnionitis sets up the SFTPA1 and SFTPA2 gene products, SP-A1 and SP-A2, as very important molecules for the first line of defense and regulation of various processes of the alveolar macrophage (17). Our animal studies, among others, have shown that SP-A1 and SP-A2 regulate the miRNome of the alveolar macrophage (90) and the alveolar epithelial type II cells in response to ozone exposure (91). Most importantly, these differentially affect survival in response to infection in young and old mice (92, 93) and lung function (26). Of interest, the commercially available exogenous surfactant preparations used to treat RDS, lack SP-A (94) (they only have SP-B and S-PC), but yet infection is a major comorbidity with RDS.
Furthermore, surfactant lipids and SP-A exhibit anti- and pro-inflammatory effects, respectively, on immune cells under baseline conditions, and surfactant lipids have been shown to attenuate the SP-A effect (13, 95, 96). Thus, the absence of SP-A in the exogenous surfactant preparations and the additional surfactant lipids provided by the exogenous preparation may negatively contribute to a further imbalance of pro and anti-inflammatory processes (95) in the premature lungs. With ongoing trials of SP-A peptides to treat asthma and the use of SP-A peptides to treat RSV (97–99) the present findings point to a future need to investigate SP-A as adjunct therapeutic modality for RDS as well.
The data analyzed in this study are subject to the following licenses/restrictions: the de-identified dataset is part of the FLOROS biobank at the Penn State University, College of Medicine. Requests to access these datasets should be directed to Joanna FLoros, SmZsb3Jvc0Bwc3UuZWR1.
The studies involving human participants were reviewed and approved by Institutional Research Board (IRB) at Penn State University, College of Medicine. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
SA: data curation. MY, LY, and RW: formal analysis. JF: funding acquisition. BN and JF: resources. RW and JF: supervision and writing—review and editing. SA, CG, and JF: writing—original draft. All authors read and approved the final manuscript.
This study was supported by NIH grant R37 HL34788 to JF.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors would like to acknowledge all the collaborators associated with the different institutions as they have been mentioned in previously published papers and Dr. R. Auten for contributing nine samples and Dr. T. Weaver and P. Ballard for contributing one specimen each.
SNP, Single nucleotide polymorphism; RDS, respiratory distress syndrome; SFTP, surfactant protein gene; BW, birth weight; SP, surfactant protein.
1. Avery ME, Mead J. Surface properties in relation to atelectasis and hyaline membrane disease. AMA J Dis Children. (1959) 97:517–23. doi: 10.1001/archpedi.1959.02070010519001
2. March of Dimes. Infant Deaths Due to Respiratory Distress Syndrome United States | PeriStats | March Of Dimes. (2017). Available online at: www.marchofdimes.org
3. Crowley PA. Antenatal corticosteroid therapy: a meta-analysis of the randomized trials, 1972 to 1994. Am J Obstet Gynecol. (1995) 173:322–35. doi: 10.1016/0002-9378(95)90222-8
4. Fanaroff AA, Stoll BJ, Wright LL, Carlo WA, Ehrenkranz RA, Stark AR, et al. Trends in neonatal morbidity and mortality for very low birthweight infants. Am J Obstet Gynecol. (2007) 196:147–e1. doi: 10.1016/j.ajog.2006.09.014
5. Usher RH, Allen AC, McLean FH. Risk of respiratory distress syndrome related to gestational age, route of delivery, and maternal diabetes. Am J Obstet Gynecol. (1971) 111:826–32. doi: 10.1016/0002-9378(71)90495-9
7. Richardson DK, Torday JS. Racial differences in predictive value of the lecithin/sphingomyelin ratio. Am J Obstet Gynecol. (1994) 170:1273–8. doi: 10.1016/S0002-9378(13)90449-X
8. Myrianthopoulos NC, Churchill JA, Baszynski AJ. Respiratory distress syndrome in twins. Acta Geneticae Med Gemellologiae Twin Res. (1971) 20:199–204. doi: 10.1017/S1120962300011628
9. Lankenau HM. A genetic and statistical study of the respiratory distress syndrome. Eur J Pediatr. (1976) 123:167–77. doi: 10.1007/BF00452094
10. Floros J, Kala P. Surfactant proteins: molecular genetics of neonatal pulmonary diseases. Annu Rev Physiol. (1998) 60:365–84. doi: 10.1146/annurev.physiol.60.1.365
11. DeMello DE, Phelps DS, Patel G, Floros J, Lagunoff D. Expression of the 35kDa and low molecular weight surfactant-associated proteins in the lungs of infants dying with respiratory distress syndrome. Am J Pathol. (1989) 134:1285.
12. deMello DE, Heyman S, Phelps DS, Floros J. Immunogold localization of SP-A in lungs of infants dying from respiratory distress syndrome. Am J Pathol. (1993) 142:1631.
13. Phelps DS. Surfactant regulation of host defense function in the lung: a question of balance. Pediatric Pathol Mol Med. (2001) 20:269–92. doi: 10.1080/152279501750412225
14. Kishore U, Greenhough TJ, Waters P, Shrive AK, Ghai R, Kamran MF, et al. Surfactant proteins SP-A and SP-D: structure, function and receptors. Mol Immunol. (2006) 43:1293–315. doi: 10.1016/j.molimm.2005.08.004
15. Floros J, Wang G, Mikerov AN. Genetic complexity of the human innate host defense molecules, surfactant protein A1 (SP-A1) and SP-A2—impact on function. Crit Reviews™ Eukaryotic Gene Expression. (2009) 19:125–37. doi: 10.1615/CritRevEukarGeneExpr.v19.i2.30
16. Crouch EC. Surfactant protein-D and pulmonary host defense. Respir Res. (2000) 1:1–16. doi: 10.1186/rr19
17. Floros J, Thorenoor N, Tsotakos N, Phelps DS. Human surfactant protein SP-A1 and SP-A2 variants differentially affect the alveolar microenvironment, surfactant structure, regulation and function of the alveolar macrophage, and animal and human survival under various conditions. Front Immunol. (2021) 12:2889. doi: 10.3389/fimmu.2021.681639
18. Weaver TE, Conkright JJ. Function of surfactant proteins B and C. Annu Rev Physiol. (2001) 63:555–78. doi: 10.1146/annurev.physiol.63.1.555
19. Cochrane CG, Revak SD. Pulmonary surfactant protein B (SP-B): structure-function relationships. Science. (1991) 254:566–8. doi: 10.1126/science.1948032
20. Pérez-Gil J. Structure of pulmonary surfactant membranes and films: the role of proteins and lipid–protein interactions. Biochimica Biophysica Acta Biomembranes. (2008) 1778:1676–95. doi: 10.1016/j.bbamem.2008.05.003
21. Cañadas O, Olmeda B, Alonso A, Pérez-Gil J. Lipid–protein and protein–protein interactions in the pulmonary surfactant system and their role in lung homeostasis. Int J Mol Sci. (2020) 21:3708. doi: 10.3390/ijms21103708
22. Williams MC, Hawgood S. Hamilton RL. Changes in lipid structure produced by surfactant proteins SP-A, SP-B, and SP-C. Am J Respir Cell Mol Biol. (1991) 5:41. doi: 10.1165/ajrcmb/5.1.41
23. Poulain FR, Allen L, Williams MC, Hamilton RL, Hawgood S. Effects of surfactant apolipoproteins on liposome structure: implications for tubular myelin formation. Am J Physiol Lung Cell Mol Physiol. (1992) 262:L730–9. doi: 10.1152/ajplung.1992.262.6.L730
24. Korfhagen TR, Bruno MD, Ross GF, Huelsman KM, Ikegami M, Jobe AH, et al. Altered surfactant function and structure in SP-A gene targeted mice. Proc Nat Acad Sci USA. (1996) 93:9594–9. doi: 10.1073/pnas.93.18.9594
25. Lopez-Rodriguez E, Pascual A, Arroyo R, Floros J, Perez-Gil J. Human pulmonary surfactant protein SP-A1 provides maximal efficiency of lung interfacial films. Biophys J. (2016) 111:524–36. doi: 10.1016/j.bpj.2016.06.025
26. Thorenoor N, Zhang X, Umstead TM, Halstead ES, Phelps DS, Floros J. Differential effects of innate immune variants of surfactant protein-A1 (SFTPA1) and SP-A2 (SFTPA2) in airway function after Klebsiella pneumoniae infection and sex differences. Respir Res. (2018) 19:1–14. doi: 10.1186/s12931-018-0723-1
27. Tsitoura MEI, Stavrou EF, Maraziotis IA, Sarafidis K, Athanassiadou A, Dimitriou G. Surfactant protein A and B gene polymorphisms and risk of respiratory distress syndrome in late-preterm neonates. PLoS ONE. (2016) 11:e0166516. doi: 10.1371/journal.pone.0166516
28. Somaschini M, Presi S, Ferrari M, Vergani B, Carrera P. Surfactant proteins gene variants in premature newborn infants with severe respiratory distress syndrome. J Perinatol. (2018) 38:337–44. doi: 10.1038/s41372-017-0018-2
29. Floros J, Fan R, Matthews A, DiAngelo S, Luo J, Nielsen H, et al. Family-based transmission disequilibrium test (TDT) and case–control association studies reveal surfactant protein A (SP-A) susceptibility alleles for respiratory distress syndrome (RDS) and possible race differences. Clin Genet. (2001) 60:178–87. doi: 10.1034/j.1399-0004.2001.600303.x
30. Rämet M, Haataja R, Marttila R, Floros J, Hallman M. Association between the surfactant protein A (SP-A) gene locus and respiratory-distress syndrome in the Finnish population. Am J Human Genet. (2000) 66:1569–79. doi: 10.1086/302906
31. Floros J, Veletza SV, Kotikalapudi P, Krizkova L, Karinch AM, Friedman C, et al. Dinucleotide repeats in the human surfactant protein-B gene and respiratory-distress syndrome. Biochem J. (1995) 305:583–90. doi: 10.1042/bj3050583
32. Kala P, Ten Have T, Nielsen H, Dunn M, Floros J. Association of pulmonary surfactant protein A (SP-A) gene and respiratory distress syndrome: interaction with SP-B. Pediatr Res. (1998) 43:169–77. doi: 10.1203/00006450-199802000-00003
33. Wambach JA, Yang P, Wegner DJ, An P, Hackett BP, Cole FS, et al. Surfactant protein-C promoter variants associated with neonatal respiratory distress syndrome reduce transcription. Pediatr Res. (2010) 68:216–20. doi: 10.1203/PDR.0b013e3181eb5d68
34. Marttila R, Haataja R, Guttentag S, Hallman M. Surfactant protein A and B genetic variants in respiratory distress syndrome in singletons and twins. Am J Respir Crit Care Med. (2003) 168:1216–22. doi: 10.1164/rccm.200304-524OC
35. Marttila R, Haataja R, Rämet M, Pokela ML, Tammela O, Hallman M. Surfactant protein A gene locus and respiratory distress syndrome in Finnish premature twin pairs. Ann Med. (2003) 35:344–52. doi: 10.1080/07853890310006389
36. Floros J, Fan R. Surfactant protein A and B genetic variants and respiratory distress syndrome: allele interactions. Neonatology. (2001) 80:22–5. doi: 10.1159/000047173
37. Hilgendorff A, Heidinger K, Bohnert A, Kleinsteiber A, König IR, Ziegler A, et al. Association of polymorphisms in the human surfactant protein-D (SFTPD) gene and postnatal pulmonary adaptation in the preterm infant. Acta Paediatr. (2009) 98:112–7. doi: 10.1111/j.1651-2227.2008.01014.x
38. Lahti M, Marttila R, Hallman M. Surfactant protein C gene variation in the Finnish population–association with perinatal respiratory disease. Euro J Human Genet. (2004) 12:312–20. doi: 10.1038/sj.ejhg.5201137
39. Silveyra P, Floros J. Genetic variant associations of human SP-A and SP-D with acute and chronic lung injury. Front Biosci. (2012) 17:407. doi: 10.2741/3935
40. Floros J., Thomas N. (2009). Genetic variations of surfactant proteins and lung injury. Surfactant Pathogenesis and Treatment of Lung Disease, edited by Nakos G, Papathanasiou A. Kerala, India: Research Signpost 25–48.
41. Karinch AM, Floros J. 5'splicing and allelic variants of the human pulmonary surfactant protein A genes. Am J Respir Cell Mol Biol. (1995) 12:77–88. doi: 10.1165/ajrcmb.12.1.7811473
42. Thomas NJ, Fan R, Diangelo S, Hess JC, Floros J. Haplotypes of the surfactant protein genes A and D as susceptibility factors for the development of respiratory distress syndrome. Acta paediatrica. (2007) 96:985–9. doi: 10.1111/j.1651-2227.2007.00319.x
43. Haataja R, Rämet M, Marttila R, Hallman M. Surfactant proteins A and B as interactive genetic determinants of neonatal respiratory distress syndrome. Hum Mol Genet. (2000) 9:2751–60. doi: 10.1093/hmg/9.18.2751
44. Frankel WN, Schork NJ. Who's afraid of epistasis?. Nat Genet. (1996) 14:371–3. doi: 10.1038/ng1296-371
45. Moore JH. The ubiquitous nature of epistasis in determining susceptibility to common human diseases. Hum Hered. (2003) 56:73–82. doi: 10.1159/000073735
46. Floros J, DiAngelo S, Koptides M, Karinch AM, Rogan PK, Nielsen H, et al. Human SP-A locus: allele frequencies and linkage disequilibrium between the two surfactant protein A genes. Am J Respir Cell Mol Biol. (1996) 15:489–98. doi: 10.1165/ajrcmb.15.4.8879183
47. Floros J, Thomas NJ, Liu W, Papagaroufalis C, Xanthou M, Pereira S, et al. Family-based association tests suggest linkage between surfactant protein B (SP-B) (and flanking region) and respiratory distress syndrome (RDS): SP-B haplotypes and alleles from SP-B–linked loci are risk factors for RDS. Pediatr Res. (2006) 59:616–21. doi: 10.1203/01.pdr.0000203145.48585.2c
48. Newton ER. Chorioamnionitis and intraamniotic infection. Clin Obstet Gynecol. (1993). 36:795–808. doi: 10.1097/00003081-199312000-00004
49. Selman M, Lin HM, Montaño M, Jenkins AL, Estrada A, Lin Z, et al. Surfactant protein A and B genetic variants predispose to idiopathic pulmonary fibrosis. Hum Genet. (2003) 113:542–50. doi: 10.1007/s00439-003-1015-4
50. Jobe AH, Bancalari E. Bronchopulmonary dysplasia. Am J Respir Crit Care Med. (2001) 163:1723–9. doi: 10.1164/ajrccm.163.7.2011060
51. Lin Z, Pearson C, Chinchilli V, Pietschmann SM, Luo J, Pison U, et al. Polymorphisms of human SP-A, SP-B, and SP-D genes: association of SP-B Thr131Ile with ARDS. Clin Genet. (2000) 58:181–91. doi: 10.1034/j.1399-0004.2000.580305.x
52. DiAngelo S, Lin Z, Wang G, Phillips S, Ramet M, Luo J, et al. Novel, non-radioactive, simple and multiplex PCR-cRFLP methods for genotyping human SP-A and SP-D marker alleles. Dis Markers. (1999) 15:269–81. doi: 10.1155/1999/961430
53. Wang Z, Liu T, Lin Z, Hegarty J, Koltun WA, Wu R. A general model for multilocus epistatic interactions in case-control studies. PLoS ONE. (2010) 5:e11384. doi: 10.1371/journal.pone.0011384
54. Taylor MB, Ehrenreich IM. Higher-order genetic interactions and their contribution to complex traits. Trends Genet. (2015) 31:34–40. doi: 10.1016/j.tig.2014.09.001
55. Zuk O, Hechter E, Sunyaev SR, Lander ES. The mystery of missing heritability: Genetic interactions create phantom heritability. Proc Nat Acad Sci USA. (2012) 109:1193–8. doi: 10.1073/pnas.1119675109
56. Wang G, Bates-Kenney SR, Tao JQ, Phelps DS, Floros J. Differences in biochemical properties and in biological function between human SP-A1 and SP-A2 variants, and the impact of ozone-induced oxidation. Biochemistry. (2004) 43:4227–39. doi: 10.1021/bi036023i
57. Lin Z, Thorenoor N, Wu R, DiAngelo SL, Ye M, Thomas NJ, et al. Genetic association of pulmonary surfactant protein genes, SFTPA1, SFTPA2, SFTPB, SFTPC, and SFTPD with cystic fibrosis. Front Immunol. (2018) 9:2256. doi: 10.3389/fimmu.2018.02256
58. Gandhi CK, Chen C, Wu R, Yang L, Thorenoor N, Thomas NJ, et al. Association of SNP–SNP interactions of surfactant protein genes with pediatric acute respiratory failure. J Clin Med. (2020) 9:1183. doi: 10.3390/jcm9041183
59. Gandhi CK, Chen C, Amatya S, Yang L, Fu C, Zhou S, et al. SNP and haplotype interaction models reveal association of surfactant protein gene polymorphisms with hypersensitivity pneumonitis of Mexican population. Frontiers in medicine. (2020) 7. doi: 10.3389/fmed.2020.588404
60. Floros J, Lin HM, García A, Salazar MA, Guo X, DiAngelo S, et al. Surfactant protein genetic marker alleles identify a subgroup of tuberculosis in a Mexican population. J Infect Dis. (2000) 182:1473–8. doi: 10.1086/315866
61. Lüfgren J, Rämet M, Renko M, Marttila R, Hallman M. Association between surfactant protein A gene locus and severe respiratory syncytial virus infection in infants. J Infect Dis. (2002) 185:283–9. doi: 10.1086/338473
62. Malik S, Greenwood CMT, Eguale T, Kifle A, Beyene J, Habte A, et al. Variants of the SFTPA1 and SFTPA2 genes and susceptibility to tuberculosis in Ethiopia. Hum Genet. (2006) 118:752–9. doi: 10.1007/s00439-005-0092-y
63. García-Laorden M, de Castro FR, Solé-Violán J, Rajas O, Blanquer J, Borderías L, et al. Influence of genetic variability at the surfactant proteins A and D in community-acquired pneumonia: a prospective, observational, genetic study. Crit Care. (2011) 15:1–12. doi: 10.1186/cc10030
64. Floros J, Wang G, Lin Z. Genetic diversity of human SP-A, a molecule with innate host defense and surfactant-related functions; characteristics, primary function, and significance. Curr Pharmacogenomics. (2005) 3:87–95. doi: 10.2174/1570160054022935
65. Stray-Pedersen A, Vege A, Opdal SH, Moberg S, Rognum TO. Surfactant protein A and D gene polymorphisms and protein expression in victims of sudden infant death. Acta paediatrica. (2009) 98:62–8. doi: 10.1111/j.1651-2227.2008.01090.x
66. Kersteen EA, Raines RT. Contribution of tertiary amides to the conformational stability of collagen triple helices. Biopolymers. (2001) 59:24–8. doi: 10.1002/1097-0282(200107)59:1<24::AID-BIP1002>3.0.CO;2-N
67. Floros J, Wang G. A point of view: quantitative and qualitative imbalance in disease pathogenesis; pulmonary surfactant protein A genetic variants as a model. Comparative Biochem Physiol Part A Mol Integrative Physiol. (2001) 129:295–303. doi: 10.1016/S1095-6433(01)00325-7
68. Mikerov AN, Umstead TM, Gan X, Huang W, Guo X, Wang G, et al. Impact of ozone exposure on the phagocytic activity of human surfactant protein A (SP-A) and SP-A variants. Am J Physiol Lung Cell Mol Physiol. (2008) 294:L121–L130. doi: 10.1152/ajplung.00288.2007
69. Wang G, Umstead TM, Phelps DS, Al-Mondhiry H, Floros J. The effect of ozone exposure on the ability of human surfactant protein a variants to stimulate cytokine production. Environ Health Perspect. (2002) 110:79–84. doi: 10.1289/ehp.0211079
70. McCormack FX, Festa AL, Andrews RP, Linke M, Walzer PD. The carbohydrate recognition domain of surfactant protein A mediates binding to the major surface glycoprotein of Pneumocystis carinii. Biochemistry. (1997) 36:8092–9. doi: 10.1021/bi970313f
71. Vuk-Pavlovic Z, Standing JE, Crouch EC, Limper AH. Carbohydrate recognition domain of surfactant protein D mediates interactions with Pneumocystis carinii glycoprotein A. Am J Respir Cell Mol Biol. (2001) 24:475–84. doi: 10.1165/ajrcmb.24.4.3504
72. Hickling TP, Malhotra R, Sim RB. Human lung surfactant protein A exists in several different oligomeric states: oligomer size distribution varies between patient groups. Mol Med. (1998) 4:266–75. doi: 10.1007/BF03401923
73. Crouch EC. Collectins and pulmonary host defense. Am J Respir Cell Mol Biol. (1998) 19:177–201. doi: 10.1165/ajrcmb.19.2.140
74. Palaniyar N, Ikegami M, Korfhagen T, Whitsett J, McCormack FX. Domains of surfactant protein A that affect protein oligomerization, lipid structure and surface tension. Compar Biochem Physiol Part A Mol Integrative Physiol. (2001) 129:109–27. doi: 10.1016/S1095-6433(01)00309-9
75. Jo HS, Cho SI, Chang YH, Kim BI, Choi JH. Surfactant protein A associated with respiratory distress syndrome in Korean preterm infants: evidence of ethnic difference. Neonatology. (2013) 103:44–7. doi: 10.1159/000342498
76. El Saleeby CM, Li R, Somes GW, Dahmer MK, Quasney MW, DeVincenzo JP. Surfactant protein A2 polymorphisms and disease severity in a respiratory syncytial virus-infected population. J Pediatr. (2010) 156:409–14. doi: 10.1016/j.jpeds.2009.09.043
77. Herrera-Ramos E, López-Rodríguez M, Ruíz-Hernández JJ, Horcajada JP, Borderías L, Lerma E, et al. Surfactant protein A genetic variants associate with severe respiratory insufficiency in pandemic influenza A virus infection. Crit Care. (2014) 18:1–12. doi: 10.1186/cc13934
78. Lin Z, Demello DE, Batanian JR, Khammash HM, DiAngelo S, Luo J, et al. Aberrant SP-B mRNA in lung tissue of patients with congenital alveolar proteinosis (CAP). Clin Genet. (2000) 57:359–69. doi: 10.1034/j.1399-0004.2000.570506.x
79. Fatahi N, Niknafs N, Kalani M, Dalili H, Shariat M, Amini E, et al. Association of SP-B gene 9306 A/G polymorphism (rs7316) and risk of RDS. J Maternal Fetal Neonatal Med. (2018) 31:2965–70. doi: 10.1080/14767058.2017.1359829
80. Dahmer MK, O'cain P, Patwari PP, Simpson P, Li SH, Halligan N, et al. The influence of genetic variation in surfactant protein B on severe lung injury in African American children. Crit. Care Med. (2011) 39:1138–44. doi: 10.1097/CCM.0b013e31820a9416
81. Fatahi N, Dalili H, Kalani M, Niknafs N, Shariat M, Tavakkoly-Bazzaz J, et al. Association of SP-C gene codon 186 polymorphism (rs1124) and risk of RDS. The J Maternal Fetal Neonatal Med. (2017) 30:2585–9. doi: 10.1080/14767058.2016.1256994
82. Nogee LM, Dunbar AE, Wert SE, Askin F, Hamvas A, Whitsett JA. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N Engl J Med. (2001) 344:573–9. doi: 10.1056/NEJM200102223440805
83. Leth-Larsen R, Garred P, Jensenius H, Meschi J, Hartshorn K, Madsen J, et al. A common polymorphism in the SFTPD gene influences assembly, function, and concentration of surfactant protein D. J Immunol. (2005) 174:1532–8. doi: 10.4049/jimmunol.174.3.1532
84. Sorensen GL. Surfactant protein D in respiratory and non-respiratory diseases. Front Med. (2018) 5:18. doi: 10.3389/fmed.2018.00018
85. Heidinger K, König IR, Bohnert A, Kleinsteiber A, Hilgendorff A, Gortner L, et al. Polymorphisms in the human surfactant protein-D (SFTPD) gene: strong evidence that serum levels of surfactant protein-D (SP-D) are genetically influenced. Immunogenetics. (2005) 57:1–7. doi: 10.1007/s00251-005-0775-5
86. Chang HY, Li F, Li FS, Zheng CZ, Lei YZ, Wang J. Genetic polymorphisms of SP-A, SP-B, and SP-D and risk of respiratory distress syndrome in preterm neonates. Med Sci Monit Int Med J Exp Clin Res. (2016) 22:5091. doi: 10.12659/MSM.898553
87. Sorensen GL, Dahl M, Tan Q, Bendixen C, Holmskov U, Husby S. Surfactant protein-D–encoding gene variant polymorphisms are linked to respiratory outcome in premature infants. J Pediatr. (2014) 165:683–9. doi: 10.1016/j.jpeds.2014.05.042
88. Gower WA, Nogee LM. Candidate gene analysis of the surfactant protein D gene in pediatric diffuse lung disease. J Pediatr. (2013) 163:1778–80. doi: 10.1016/j.jpeds.2013.06.063
89. Jensen EA, Dysart K, Gantz MG, McDonald S, Bamat NA, Keszler M, et al. The diagnosis of bronchopulmonary dysplasia in very preterm infants. An evidence-based approach. Am J Respir Crit Care Med. (2019) 200:751–9. doi: 10.1164/rccm.201812-2348OC
90. Noutsios GT, Thorenoor N, Zhang X, Phelps DS, Umstead TM, Durrani F, et al. SP-A2 contributes to miRNA-mediated sex differences in response to oxidative stress: pro-inflammatory, anti-apoptotic, and anti-oxidant pathways are involved. Biol Sex Differ. (2017) 8:1–15. doi: 10.1186/s13293-017-0158-2
91. Noutsios GT, Thorenoor N, Zhang X, Phelps DS, Umstead TM, Durrani F, et al. Major effect of oxidative stress on the male, but not female, SP-A1 type II cell miRNome. Front Immunol. (2019) 10:1514. doi: 10.3389/fimmu.2019.01514
92. Thorenoor N, Umstead TM, Zhang X, Phelps DS, Floros J. Survival of surfactant protein-A1 and SP-A2 transgenic mice after Klebsiella pneumoniae infection, exhibits sex-, gene-, and variant specific differences; treatment with surfactant protein improves survival. Front Immunol. (2018) 9:2404. doi: 10.3389/fimmu.2018.02404
93. Thorenoor N, Phelps SD, Kala P, Ravi R, Floros Phelps A, Umstead MT, et al. Impact of surfactant protein-A variants on survival in aged mice in response to Klebsiella pneumoniae infection and ozone: serendipity in action. Microorganisms. (2020) 8:1276. doi: 10.3390/microorganisms8091276
94. Echaide M, Autilio C, Arroyo R, Perez-Gil J. Restoring pulmonary surfactant membranes and films at the respiratory surface. Biochimica Biophysica Acta Biomembranes. (2017) 1859:1725–39. doi: 10.1016/j.bbamem.2017.03.015
95. Phelps D. Pulmonary surfactant modulation of host-defense function. Appl Cardiopulmonary Pathophysiol. (1995) 5:221–9.
96. Koptides M, Umstead TM, Floros J, Phelps DS. Surfactant protein A activates NF-kappa B in the THP-1 monocytic cell line. Am J Physiol Lung Cell Mol Physiol. (1997) 273:L382–8. doi: 10.1152/ajplung.1997.273.2.L382
97. Watson A, Kronqvist N, Spalluto CM, Griffiths M, Staples KJ, Wilkinson T, et al. Novel expression of a functional trimeric fragment of human SP-A with efficacy in neutralisation of RSV. Immunobiology. (2017) 222:111–8. doi: 10.1016/j.imbio.2016.10.015
98. Watson A, Sørensen GL, Holmskov U, Whitwell HJ, Madsen J, Clark H. Generation of novel trimeric fragments of human SP-A and SP-D after recombinant soluble expression in E. coli. Immunobiology. (2020) 225:151953. doi: 10.1016/j.imbio.2020.151953
Keywords: epistasis, neonatal, genetic variants, pulmonary, allele
Citation: Amatya S, Ye M, Yang L, Gandhi CK, Wu R, Nagourney B and Floros J (2021) Single Nucleotide Polymorphisms Interactions of the Surfactant Protein Genes Associated With Respiratory Distress Syndrome Susceptibility in Preterm Infants. Front. Pediatr. 9:682160. doi: 10.3389/fped.2021.682160
Received: 17 March 2021; Accepted: 06 September 2021;
Published: 04 October 2021.
Edited by:
Maria Elisabetta Baldassarre, University of Bari Aldo Moro, ItalyReviewed by:
Uday Kishore, Brunel University London, United KingdomCopyright © 2021 Amatya, Ye, Yang, Gandhi, Wu, Nagourney and Floros. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joanna Floros, amZsb3Jvc0BwZW5uc3RhdGVoZWFsdGgucHN1LmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.