
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pediatr., 10 May 2021
Sec. Pediatric Neurology
Volume 9 - 2021 | https://doi.org/10.3389/fped.2021.676616
 Han-yu Luo1,2†Ling-ling Xie1,2†Si-qi Hong1,2Xiu-juan Li1,2Mei Li1,2Yue Hu1,2Jian-nan Ma1,2Peng Wu1,2Min Zhong1,2Min Cheng1,2Ting-song Li1,2Li Jiang1,2*
Han-yu Luo1,2†Ling-ling Xie1,2†Si-qi Hong1,2Xiu-juan Li1,2Mei Li1,2Yue Hu1,2Jian-nan Ma1,2Peng Wu1,2Min Zhong1,2Min Cheng1,2Ting-song Li1,2Li Jiang1,2*Objectives: To study the genetic and clinical characteristics of Chinese children with pathogenic proline-rich transmembrane protein 2 (PRRT2) gene-associated disorders.
Methods: Targeted next generation sequencing (NGS) was used to identify pathogenic PRRT2 variations in Chinese children with epilepsy and/or kinesigenic dyskinesia. Patients with confirmed PRRT2-associated disorders were monitored and their clinical data were analyzed.
Results: Forty-four patients with pathogenic PRRT2 variants were recruited. Thirty-five of them (79.5%) had heterozygous mutations, including 30 frameshifts, three missenses, one nonsense, and one splice site variant. The c.649dupC was the most common variant (56.8%). Eight patients (18.2%) showed whole gene deletions, and one patient (2.3%) had 16p11.2 microdeletion. Thirty-four cases (97.1%) were inherited and one case (2.9%) was de novo. Forty patients were diagnosed with benign familial infantile epilepsy (BFIE), two patients had paroxysmal kinesigenic dyskinesia (PKD) and two had infantile convulsions and choreoathetosis (ICCA). Patients with whole gene deletions had a later remission than patients with heterozygous mutations (13.9 vs. 7.1 months, P = 0.001). Forty-two patients were treated with antiseizure medications (ASMs). At last follow-up, 35 patients, including one who did not receive therapy, were asymptomatic, and one patient without ASMs died of status epilepticus at 12 months of age. One patient developed autism, and one patient showed mild developmental delay/intellectual disability.
Conclusion: Our data suggested that patients with whole gene deletions could have more severe manifestations in PRRT2-associated disorders. Conventional ASMs, especially Oxcarbazepine, showed a good treatment response.
Pathogenic mutations in the proline-rich transmembrane protein 2 (PRRT2) gene have recently been identified in a series of disorders. Among these PRRT2-associated disorders, a majority of patients have benign familial infantile epilepsy (BFIE), characterized by self-limiting seizures, which usually onset before the age of 6 months, and normal psychomotor development (1), paroxysmal kinesigenic dyskinesia (PKD), characterized by short attacks of dyskinesia triggered by sudden voluntary movements (2), or infantile convulsions with choreoathetosis (ICCA), presenting paroxysmal movement disorders during childhood after remission of infantile convulsions (3). Other rare cases, including hemiplegic migraine and episodic ataxia, have rapidly expanded the boundary of this spectrum (4). Most patients have a tendency to natural remission and favorable outcome. The diverse clinical manifestations caused by variants in the same gene indicate a conventional spectrum that share similar clinical and genetic features, although the pathophysiology of variants in PRRT2 is not entirely understood.
In this study, we aimed to describe clinical and genetic features of a selected cohort of Chinese children with PRRT2-associated disorders and discuss the treatment strategies and prognosis of these patients.
A total of 44 children who were diagnosed and treated at the Children's Hospital of Chongqing Medical University were recruited between April 2016 and May 2019. Inclusion criteria for the study were: (I) children (age < 18 years) receiving a new diagnosis of epilepsy or children presenting with paroxysmal movement disorders; (II) patients with pathogenic PRRT2 variants. Clinical data, including demographic information, family history, clinical manifestation, electroencephalogram (EEG), brain magnetic resonance imaging (MRI), treatment, and follow-up were collected and analyzed. BFIE was defined when patients met the following conditions: (I) onset of seizures between the age of 3 and 12 months; (II) focal seizures with or without secondary generalization; (III) normal interictal EEGs and brain MRI; (IV) normal psychomotor development; (V) positive family history; (VI) seizure-free before 2 years of age; and (VII) exclusion of other metabolic disorders (5). The diagnostic criteria of PKD were revised from Bruno et al. (6) as follows: (I) an identified kinesigenic trigger for attacks; (II) duration of attacks <1 minute; (III) attacks without loss of consciousness or pain; (IV) response to antiseizure medications (ASMs); (V) age of onset 1-20 years old; and (VI) exclusion of other organic diseases. ICCA was diagnosed when patients presented with both BFIE and PKD. All patients participated in a follow up at least 3 months after inclusion in the study, including number of seizure episodes, developmental milestones, mental conditions, and therapeutic regimes.
Written informed consent was obtained from legal guardians of patient. This study was approved by the Children's Hospital of Chongqing Medical University Ethics Committee. Genomic DNA from each individual and 50 normal controls without epilepsy or any related history were extracted from peripheral blood leukocytes. A custom-designed panel that captured the coding exons of 535 genes associated with epilepsy, including PRRT2, was synthetized using the Agilent SureSelect Target Enrichment System which contained a total of 12,000 probes covering 1.285 Mbp. Then, targeted next generation sequencing (NGS) was performed following previously reported experimental procedures (7). Sequencing results were aligned to the Genome Reference Consortium Homo sapiens (human) genome assembly GRCh37 (GRCh37/hg19) and compared with the established human PRRT2 sequences (NM_145239). Sanger sequencing was performed to validate variants identified by NGS and for segregation analysis. We used PolyPhen-2, SIFT, MutationTaster, and FATHMM-MKL to predict the pathogenicity of the missense variants.
Continuous variables were expressed as mean ± SD. Differences were tested using Student's t-test or the Mann–Whitney U-test. Categorical variables were summarized as percentages, and compared byχ2 test or by Fisher's exact test. A P-value ≤ 0.05 in a two-tailed test was considered statistically significant. Statistical analyses were performed using SPSS version 23.0 (IBM, Armonk, NY).
Forty-four patients (29 males and 15 females) with pathogenic PRRT2 variants were identified (Table 1). Nine patients (20.5%) showed PRRT2 deletions (eight patients with PRRT2 whole gene deletions, and one with 16p11.2 microdeletion). The remaining 35 patients (79.5%) were PRRT2 heterozygous variants, including 30 frameshift mutations, three missense mutations, one nonsense mutation, and one splice site change. Of the 35 variants, 32 (91.4%) coded the proline-rich domain and extracellular domain, including 25 c.649dupC mutations, three c.649delC mutations, one c.224C>T mutation, one c.2484C>G mutation, one c.439G>C mutation, and one c.615dupA mutation (Figure 1). Based on the Human Gene Mutation Database (HGMD), four patients displayed novel variants (c.284C>G, c.883_884insGGAA, c.879+4A>G, and c.914G>A). The nonsense variant c.284C>G (p.S95X) resulted in a premature stop codon. The c.883_884insGGAA (p.N296Kfs*45) produced a frameshift variant with a premature stop codon 45 amino acids downstream. Both of them were classified as “pathogenetic” according to the ACMG criteria. The c.879+4A>G variant affected the splice site. According to the ACMG criteria, it was classified as “variant of unknown significance.” The missense variant c.914G>A (p.G305E) affected highly conserved residues of the cytoplasmic domain toward the C terminus of PRRT2, which was considered “variant of unknown significance” according to the ACMG criteria. All missense variants were predicted to be damaging to protein function by prediction tools used (Table 2). Thirty-four (97.1%) of the frameshift mutations were inherited, and only one c.649dupC mutation (2.9%) was de novo.
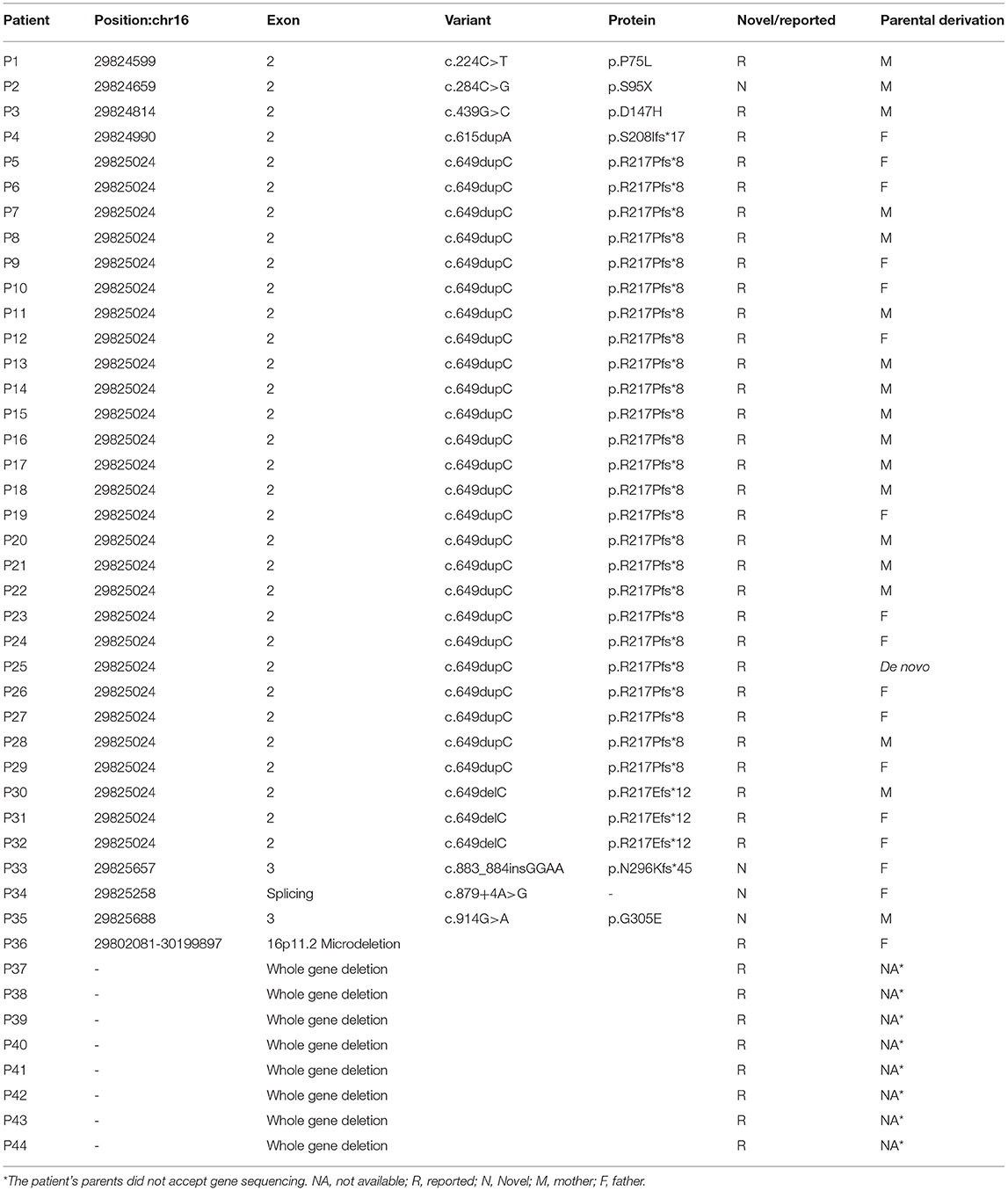
Table 1. Genotypes of 44 patients with pathogenic PRRT2 mutations.
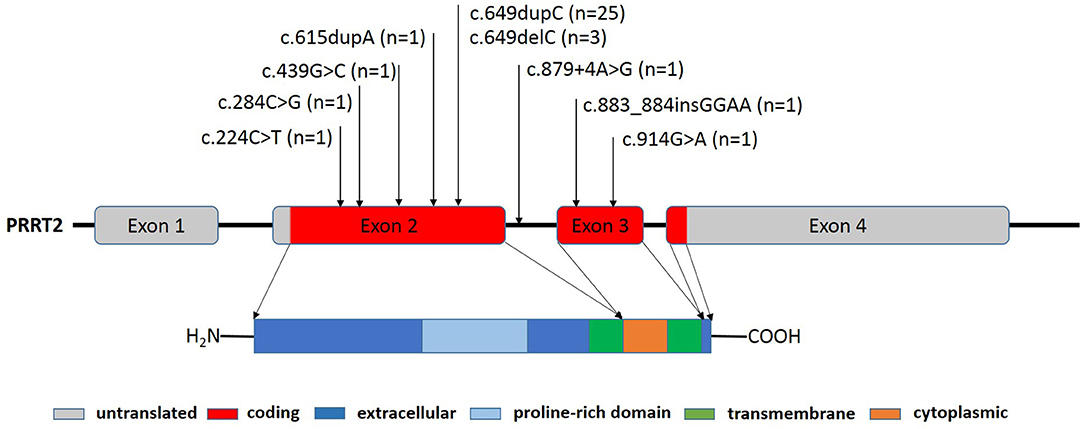
Figure 1. PRRT2 gene structure and locations of PRRT2 mutations in the PRRT2 gene sequence. The PRRT2 gene contains four exons that encode a 340 amino acid protein, including a protein-rich domain (amino acids 131-216) in its extracellular region (amino acids 1-268), one cytoplasmic region (amino acids 290-317), and two transmembrane regions (amino acids 269-289 and 318-338). The positions of the 35 heterozygous mutations identified in this study are shown.

Table 2. Pathogenicity prediction of three missense mutations.
Patients' detailed clinical information was gathered from 3 months to 2 years following PRRT2 mutation diagnosis (Table 3). No specific perinatal history or abnormal physical examination were noted in any patients when admitted. Figure 2 presented three patients' pedigrees (Patients 8, 9, and 35) that showed segregation of PRRT2 variants in more than two generations.
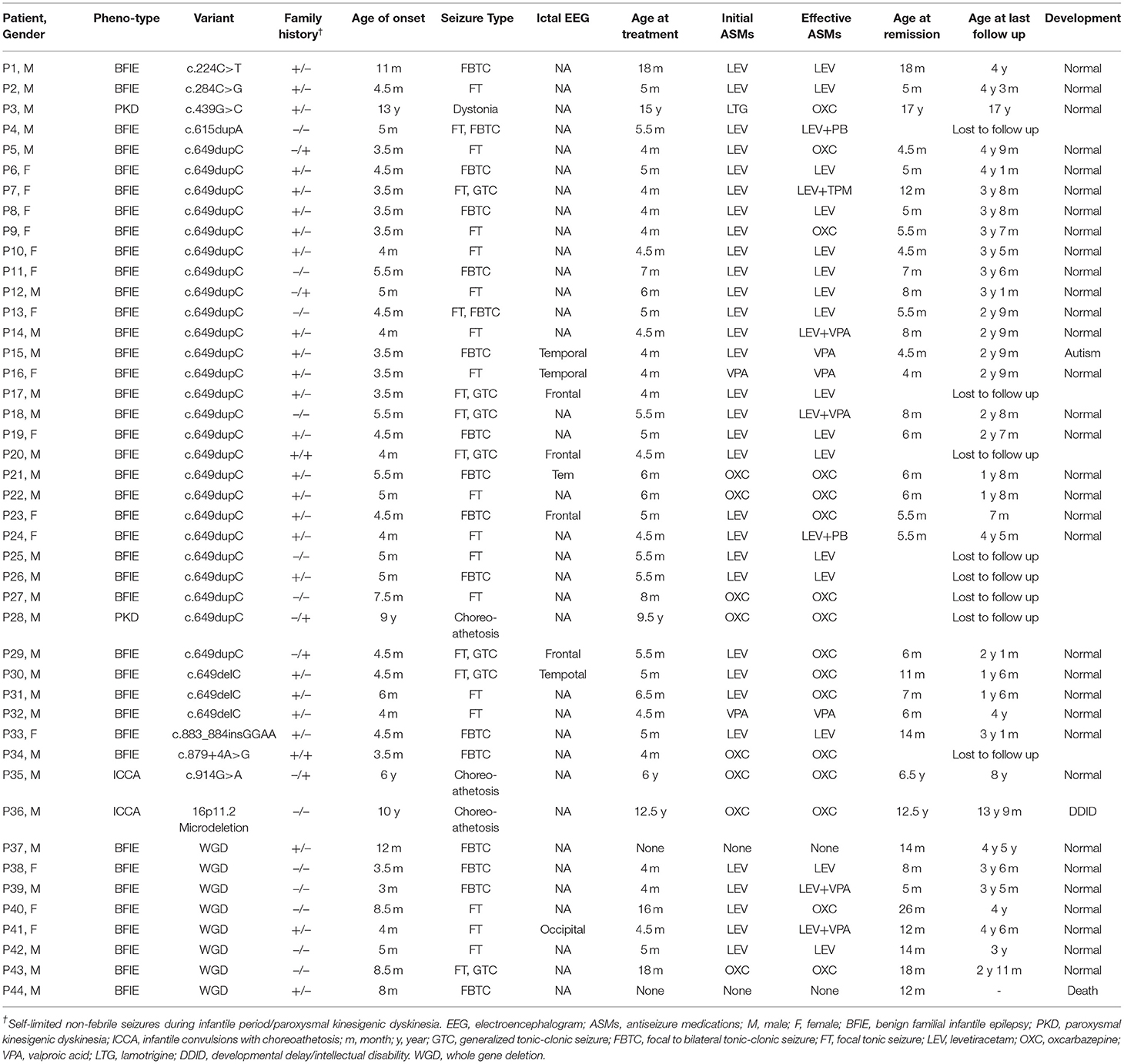
Table 3. Clinical characteristics of patients harboring pathogenic PRRT2 mutations.
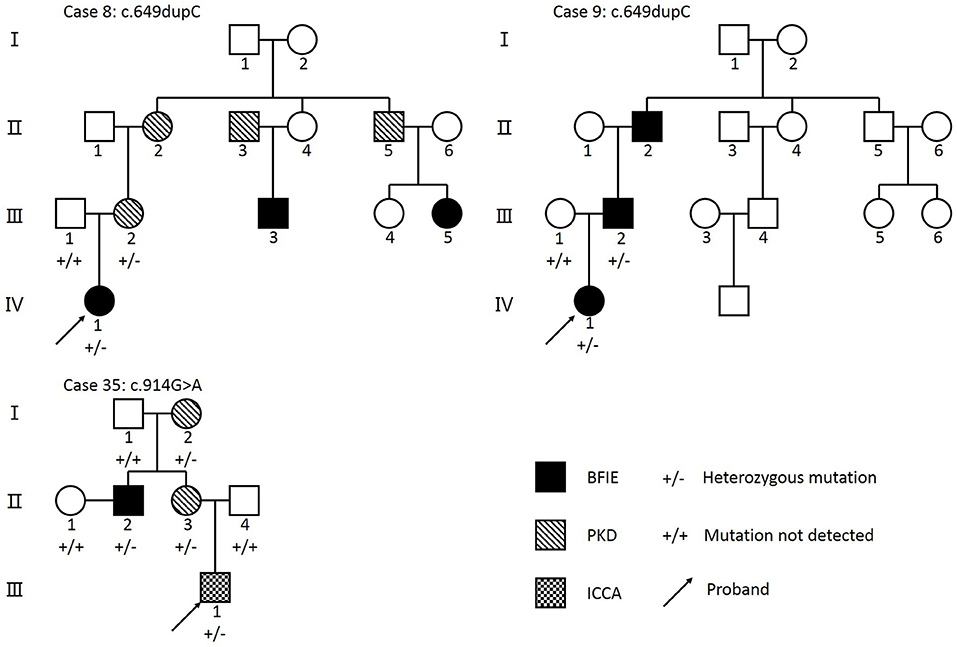
Figure 2. Pedigrees from three families with PRRT2 mutations. Arrow, proband; BFIE, benign familial infantile epilepsy; PKD, paroxysmal kinesigenic dyskinesia; ICCA, infantile convulsions with choreoathetosis. Individuals who underwent genetic sequencing are indicated by + and –. Individuals with a PRRT2 heterozygous mutation are shown using +/–, and individuals that tested negative for a PRRT2 mutation are shown using +/+. The c.649dupC mutation was identified in family 8 and family 9. The c.914G>A mutation was identified in Family 35.
Forty patients (25 males and 15 females) with pathogenic PRRT2 variants were diagnosed with BFIE, and 29 of them had a family history of seizures or PKD. The average age of onset was 5.1 ± 2.0 months. There was no significant difference (4.7 vs. 6.6 months, P = 0.143) in age of onset between patients with heterozygous mutations (n = 32) and patients with whole gene deletions (n = 8). All the patients presented with focal motor seizures. Twenty-three patients had focal to bilateral tonic-clonic seizures. Twenty-eight patients presented seizures in clusters (2-20 attacks per day). Status epilepticus was noted in one patient (Patient 44). Ictal EEG showed that discharges originated from temporal lobe (Patients 15, 16, 21, and 30), frontal lobe (Patients 17, 20, 23, and 29) and occipital lobe (Patient 41).The interictal EEGs were normal in 35 patients, whereas 4 patients showed interictal spike waves that originated from the frontal lobe (Patients 38 and 42), temporal lobe (Patient 33), or occipital lobe (Patient 14) during frequent attacks, which disappeared during subsequent follow-up. All patients demonstrated a normal brain MRI.
Two patients were diagnosed with PKD (Patients 3 and 28). Both of them had a family history of movement disorders. The age of symptoms onset was 9 and 13 years old, respectively. Patient 3 presented with dystonia triggered by standing up suddenly, and patient 28 presented with bilateral choreoathetosis triggered by sudden movement. The attacks occurred several times per day and usually lasted <1 min without loss of consciousness. Both patients showed normal interictal EEG and brain MRI.
Two patients (Patients 35 and 36) were diagnosed with ICCA. Patient 35 had manifested self-limited clusters of non-febrile focal motor seizures since the age of 5 months and developed bilateral dyskinesia at the age of 6 years. Patient 36 had focal to bilateral tonic-clonic seizures from 8 to 18 months of age, and presented with unilateral choreoathetosis at 10 years old. These two ICCA patients had normal brain MRI and EEG.
A total of 38 patients with BFIE initiated ASMs at the average age of 5.9 ± 3.5 months (Table 4). Thirty-one patients received levetiracetam (LEV; 10 mg/kg/day), and 16 (51.6%) patients were seizure free while using a dose between 15 and 40 mg/kg/d. However, the remaining 15 (48.4%) patients still experienced seizures after the LEV dosage was gradually increased to 30-40 mg/kg/d. Of these 15 patients, seven reached seizure freedom after the addition of phenobarbital (PB), topiramate (TPM), or valproic acid (VPA), respectively. Another seven patients were switched to oxcarbazepine (OXC; 10-20 mg/kg) and the seizures were completely controlled. The remaining one patient became seizure free after switching to VPA. Meanwhile, five patients were initially treated with OXC (10 mg/kg/d), and all of them reached seizure freedom at 15-20 mg/kg/d OXC. Two patients with BFIE initially received VPA (20 mg/kg/d), and their symptoms were controlled.
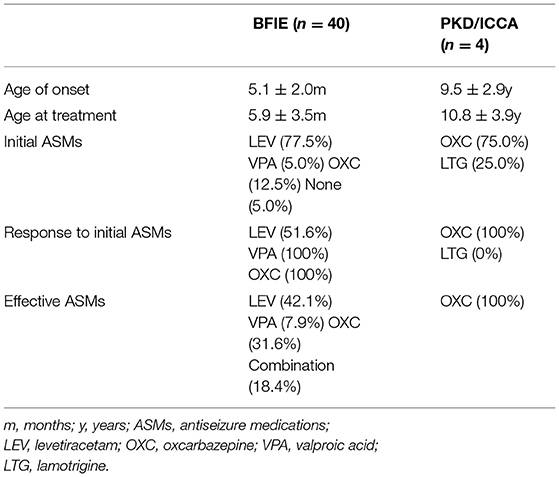
Table 4. Treatment of PRRT2-associated disorders.
Two patients (Patient 37 and 44) with BFIE did not receive treatment due to their parents' personal decision. Patient 37 was seizure free 2 months after the first seizure. Patient 44 presented with focal to bilateral tonic-clonic seizures in clusters since the age of 8 months. Interictal EEG during hospitalization once showed bilateral centro-frontal epileptic activity. Unfortunately, he died of status epilepticus (duration unknown) at home without any emergency aids at the age of 12 months.
The average age of seizure remission in the 32 patients with BFIE that received follow-up was 8.5 ± 5.0 months. Compared with patients with heterozygous mutations (n = 25), patients with whole gene deletions (n = 7) had a significantly later remission (13.9 vs. 7.1 months, P = 0.001). All patients had normal developmental milestones during follow-up except Patient 15 who developed autism at 2 years old.
One patient with PKD and two patients with ICCA were initially administered OXC. Patient 28 was lost to follow up, and the remaining two patients remitted from dyskinesia at a dose of 10-20 mg/kg/d. Patient 3 with PKD was initially administered lamotrigine (LTG, dosage unavailable) with no response. The attacks were controlled after switching to OXC (10 mg/kg/day). The average age at last episode was 12 ± 5.3 years. One patient with 16p11.2 microdeletion (Patient 36) showed mild developmental delay/intellectual disability. The remaining patients had normal cognitive outcome.
PRRT2 gene locates on chromosome 16p11.2, and encodes the proline-rich transmembrane protein 2 that is mainly expressed in the cerebral cortex, basal ganglia, and cerebellum (Figure 1). The function of the PRRT2 gene remains unclear, but current findings indicate that it is involved in the process of Ca2+-related presynaptic neurotransmitter release (8). A recent study demonstrated that PRRT2 also interacts with the voltage-gated Na+ channels Nav1.2 and Nav1.6 to negatively modulate neural activity (9). Thus, pathogenic mutations in the PRRT2 gene may lead to a state of neuronal hyper-excitability, which clinically presents a series of paroxysmal disorders. In this study, we reported 44 children with PRRT2 pathogenic mutations. In line with current data, the most common variant was the c.649dupC frameshift mutation, which was identified in 56.8% of patients in our study (4). We also reported four novel variants. The c.284C>G (p.S95X) and the c.883_884insGGAA (p.N296Kfs*45) led to a truncated PRRT2 protein and were classified as “pathogenetic” according to the ACMG criteria. The splice site variant (c.879+4A>G) was inherited from patient's father with cosegregation consistence which suggested its pathogenicity, although it was classified as “variant of unknown significance” according to the ACMG criteria. But further studies are needed to confirm its pathogenicity. The missense variant c.914G>A (p.G305E) was predicted to be damaging by Mutation Taster, Polyphen2, SIFT and FATHMM-MKL (Table 2). According to the study from Tsai et al., pathogenic missense variants at the C terminus often lead to failure of protein targeting to the plasma membrane that could be an important mechanism for PRRT2-associated disorders (10).
Several studies have reported the existence of homozygous mutations, compound heterozygous mutations, and 16p11.2 microdeletion of PRRT2 in a small group of cases (11–14). In 2015, Ebrahimi-Fakhari et al. summarized that these rare cases accounted for 1.5% (21/1,444) of all PRRT2-associated cases (4). Meanwhile, only 3.7% of patients were identified with a whole deletion of PRRT2 gene in a recent Italian cohort study (15). In this study, the proportion (20.5%) of patients with whole gene deletions or 16p11.2 microdeletion was significantly higher than previous data. This could be explained by the selective bias due to different standards for accepting genetic testing in clinical practice. The biallelic mutation or complete deletion of the PRRT2 gene led to total impairment of function of the PRRT2 protein, possibly resulting in the patient's severe clinical presentation. In our cohort, one patient suffered status epilepticus, and one patient showed mild developmental delay/intellectual disability. Both of them had complete deletions of the PRRT2 gene. In addition, we found that patients with whole gene deletions had a later remission in PRRT2-associated BFIE. Previous study have revealed that patients with homozygous, compound heterozygous and microdeletion mutation more frequently presented with more severe phenotypes including intellectual/development disorders when comparing with heterozygous mutations cases (16–18). However, evidences remain insufficient to prove the existence of genotype-phenotype correlations among PRRT2-associated disorders. The “gene-dosage effect” that causes severe impairment or total loss of the PRRT2 protein that leads to clinical presentations still require further validation.
Yang et al. found that the whole PRRT2 gene mutation and the 16p11.2 microdeletion were more likely be de novo (19). Unfortunately, the parental derivation of eight patients with whole gene deletions was not available. Five of them had no family history, which could possibly serve as potential indirect evidence. However, seven family members carried a pathogenic mutation without clinical symptoms although PRRT2-associated disorders are autosomal dominant inherited, which suggested the phenomenon of incomplete penetrance. Balagura et al. reported that the penetrance of PRRT2 pathogenic mutation was 89% (15). Schubert et al. estimated an 82% penetrance of PRRT2 mutations in BFIE (20). Meanwhile, the penetrance of PRRT2 mutations in PKD was estimated to be 61%, which was raised to nearly 90% when ICCA cases were taken into account (21).
All patients in this study were diagnosed with BFIE, PKD, or ICCA due to inclusion criteria, and their clinical characteristics were similar with previous studies. BFIE, PKD, and ICCA are main phenotypes of PRRT2-associated disorders, accounting for more than 90% of all cases (4). In addition, several studies have reported that other disorders, including hemiplegic migraine and episodic ataxia, could be a PRRT2-associated phenotype (4). In a recent study, migraine occurred as a concomitant diagnosis of BFIE in 10% of PRRT2-associated patients, suggesting an increased risk for migraine in younger patients with PRRT2-associated BFIE (22). PRRT2-associated disorders are relatively benign. However, recent studies suggested that pathogenic PRRT2 mutation could be responsible for some severe epileptic syndromes. Döring et al. reported a case who presented BFIE at the age of 4 months and developed continuous spikes and waves during sleep at the age of 4 years (22). Only an inherited c.649dupC variant was identified through whole gene sequencing. Furthermore, Jafarpour and Desai reported that infantile spams could also be a PRRT2-associated phenotype (23). In addition, PRRT2 mutations could be responsible for early childhood myoclonic epilepsy according to a recent research (18). These findings suggested that this evolving spectrum has a more widely boundary. However, further studies are needed to establish whether PPRT2 mutations play an important role in these situations.
Symptoms of PRRT2-associated disorders are readily controlled by conventional ASMs (4). Monotherapy was effective in a majority of patients with BFIE in our study. Considering both the efficacy and safety, LEV is one of the first-line choices for clinical treatment of pediatric epilepsy, especially for infantile patients. Most patients in our study were initially treated with LEV. Experiences from Zhao et al. suggested that OXC seems to be more effective than LEV (24). Similarly, half of patients in our study had no response to LEV, whereas all patients treated with OXC quickly became seizure free. These results revealed that OXC seems to be superior to LEV for patients with PRRT2-associated BFIE. OXC is a derivative of carbamazepine (CBZ) that has fewer side effects and drug interactions (25). Pan et al. suggested that OXC had a significant effect on pediatric PKD (26). Moreover, compared with non-PRRT2 mutations cases, CBZ seemed to be more effective in patients with PRRT2-associated PKD (27). A recent study revealed that PRRT2 could serve as a negative modulator of sodium channels (9). Therefore, as sodium channel blockers, CBZ and OXC probably have specific mechanisms for controlling symptoms in PRRT2-associated disorders. In addition, Symonds et al. recently reported that PRRT2-associated BFIE was the most frequent single-gene epilepsy, with an incidence of 1 per 9,970 live births (28). Thus, early genetic testing for patients with suspected BFIE is helpful to clinical treatment. Nevertheless, considering the self-limited course of PRRT2-associated diseases, whether ASMs play an essential role is ambiguous. However, one patient without ASMs suffered status epilepticus and died of delayed treatment, suggesting that the appropriate treatment is still necessary. Thus, comprehensive evaluation should be individualized when planning the treatment regime for patients with PRRT2-associated disorders.
Outcome of patients with PRRT2-associated disorders is usually favorable. In the present study, the average age of remission of patients with BFIE was 8.5 ± 5.0 months, which was similar to previously published data (29). Most patients had normal psychomotor development at last follow-up. Interestingly, one patient carrying the c.649dupC variant showed language retardation that was diagnosed with autism after seizure remission. Although microdeletions and duplications at 16p11.2, where the PRRT2 gene is located, have been observed in patients with autism (30, 31). Current studies have found no clear relationship between PRRT2 pathogenic mutation and autism (32). Thus, we postulated that PRRT2 mutations play no pathological role in the development of autism in this patient. However, the function of the PRRT2 gene during the psychomotor development period needs further study.
This study has several limitations. Firstly, patients with BFIE may not yet develop movement disorders during the relatively short follow-up. Meanwhile, considering the self-limited course of PRRT2-associated disorders, some patients did not accept gene tests. Moreover, we did not include patients with other rare phenotypes. Thus, our results may not reflect the accurate phenotype distribution. Secondly, pedigree analysis was limited due to insufficient data of family members. Thus, continuous follow-up and detailed information of probands and family members are needed to better understand the characteristics of PRRT2-associated disorders.
In conclusion, pathogenic PRRT2 mutations are responsible for a series of paroxysmal diseases, mainly including BFIE, PKD, and ICCA. Patients with whole gene deletions could have more severe phenotypes. Conventional antiseizure medications, especially OXC, could be the first-line choice. Further long-term cohort studies and pedigree analysis are needed to fully illuminate characteristics of PRRT2-associated diseases.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
The studies involving human participants were reviewed and approved by the ethics committee of Children's Hospital of Chongqing Medical University. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
LJ and L-lX designed the study. S-qH, X-jL, ML, YH, J-nM, PW, MZ, MC, and T-sL collected the patients' information. H-yL performed the statistical analysis. H-yL and L-lX drafted first version of the manuscript. All authors participated in the critical review of the manuscript, contributed to the article, and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank the patients and their families for participating in this study.
1. Caraballo RH, Cersosimo RO, Amartino H, Szepetowski P, Fejerman N. Benign familial infantile seizures: further delineation of the syndrome. J Child Neurol. (2002) 17:696-9. doi: 10.1177/088307380201700909
2. Bennett LB, Roach ES, Bowcock AM. A locus for paroxysmal kinesigenic dyskinesia maps to human chromosome 16. Neurology. (2000) 54:125-30. doi: 10.1212/WNL.54.1.125
3. Szepetowski P, Rochette J, Berquin P, Piussan C, Lathrop GM, Monaco AP. Familial infantile convulsions and paroxysmal choreoathetosis: a new neurological syndrome linked to the pericentromeric region of human chromosome 16. Am J Hum Genet. (1997) 61:889-98. doi: 10.1086/514877
4. Ebrahimi-Fakhari D, Saffari A, Westenberger A, Klein C. The evolving spectrum of PRRT2-associated paroxysmal diseases. Brain. (2015) 138:3476-95. doi: 10.1093/brain/awv317
5. Vigevano F. Benign familial infantile seizures. Brain Dev. (2005) 27:172-7. doi: 10.1016/j.braindev.2003.12.012
6. Bruno MK, Hallett M, Gwinn-Hardy K, Sorensen B, Considine E, Tucker S, et al. Clinical evaluation of idiopathic paroxysmal kinesigenic dyskinesia: new diagnostic criteria. Neurology. (2004) 63:2280-7. doi: 10.1212/01.WNL.0000147298.05983.50
7. Fang ZX, Zhang M, Xie LL, Jiang L, Hong SQ, Li XJ, et al. KCNQ2 related early-onset epileptic encephalopathies in Chinese children. J Neurol. (2019) 266:2224-32. doi: 10.1007/s00415-019-09404-y
8. Valtorta F, Benfenati F, Zara F, Meldolesi J. PRRT2: from paroxysmal disorders to regulation of synaptic function. Trends Neurosci. (2016) 39:668-79. doi: 10.1016/j.tins.2016.08.005
9. Fruscione F, Valente P, Sterlini B, Romei A, Baldassari S, Fadda M, et al. PRRT2 controls neuronal excitability by negatively modulating Na+ channel 1.2/1.6 activity. Brain. (2018) 141:1000-16. doi: 10.1093/brain/awy051
10. Tsai MH, Nian FS, Hsu MH, Liu WS, Liu YT, Liu C, et al. PRRT2 missense mutations cluster near C-terminus and frequently lead to protein mislocalization. Epilepsia. (2019) 60:807-17. doi: 10.1111/epi.14725
11. Delcourt M, Riant F, Mancini J, Milh M, Navarro V, Roze E, et al. Severe phenotypic spectrum of biallelic mutations in PRRT2 gene. J Neurol Neurosurg Psychiatry. (2015) 86:782-5.
12. Chen YP, Song W, Yang J, Zheng ZZ, Huang R, Chen K, et al. PRRT2 mutation screening in patients with paroxysmal kinesigenic dyskinesia from Southwest China. Eur J Neurol. (2014) 21:174-6. doi: 10.1111/ene.12122
13. Labate A, Tarantino P, Viri M, Mumoli L, Gagliardi M, Romeo A, et al. Homozygous c.649dupC mutation in PRRT2 worsens the BFIS/PKD phenotype with mental retardation, episodic ataxia, and absences. Epilepsia. (2012) 53:e196-9. doi: 10.1111/epi.12009
14. Dale RC, Grattan-Smith P, Nicholson M, Peters GB. Microdeletions detected using chromosome microarray in children with suspected genetic movement disorders: a single-centre study. Dev Med Child Neurol. (2012) 54:618-23. doi: 10.1111/j.1469-8749.2012.04287.x
15. Balagura G, Riva A, Marchese F, Iacomino M, Madia F, Giacomini T, et al. Clinical spectrum and genotype-phenotype correlations in PRRT2 Italian patients. Eur J Paediatr Neurol. (2020) 28:193-7. doi: 10.1016/j.ejpn.2020.06.005
16. Guerrero-Lopez R, Ortega-Moreno L, Giraldez BG, Alarcon-Morcillo C, Sanchez-Martin G, Nieto-Barrera M, et al. Atypical course in individuals from Spanish families with benign familial infantile seizures and mutations in the PRRT2 gene. Epilepsy Res. (2014) 108:1274-8. doi: 10.1016/j.eplepsyres.2014.06.011
17. Weber A, Kohler A, Hahn A, Neubauer B, Muller U. Benign infantile convulsions (IC) and subsequent paroxysmal kinesigenic dyskinesia (PKD) in a patient with 16p11.2 microdeletion syndrome. Neurogenetics. (2013) 14:251-3. doi: 10.1007/s10048-013-0376-7
18. Vlaskamp DRM, Callenbach PMC, Rump P, Giannini LAA, Brilstra EH, Dijkhuizen T, et al. PRRT2-related phenotypes in patients with a 16p11.2 deletion. Eur J Med Genet. (2019) 62:265-9. doi: 10.1016/j.ejmg.2018.08.002
19. Yang L, You C, Qiu S, Yang X, Li Y, Liu F, et al. Novel and de novo point and large microdeletion mutation in PRRT2-related epilepsy. Brain Behav. (2020) 10:e01597. doi: 10.1002/brb3.1597
20. Schubert J, Paravidino R, Becker F, Berger A, Bebek N, Bianchi A, et al. PRRT2 mutations are the major cause of benign familial infantile seizures. Hum Mutat. (2012) 33:1439-43. doi: 10.1002/humu.22126
21. van Vliet R, Breedveld G, de Rijk-van Andel J, Brilstra E, Verbeek N, Verschuuren-Bemelmans C, et al. PRRT2 phenotypes and penetrance of paroxysmal kinesigenic dyskinesia and infantile convulsions. Neurology. (2012) 79:777-84. doi: 10.1212/WNL.0b013e3182661fe3
22. Döring JH, Saffari A, Bast T, Brockmann K, Ehrhardt L, Fazeli W, et al. The phenotypic spectrum of PRRT2-associated paroxysmal neurologic disorders in childhood. Biomedicines. (2020) 8:456. doi: 10.3390/biomedicines8110456
23. Jafarpour S, Desai J. Infantile spasms associated with a pathogenic PRRT2 variant. Pediatr Neurol. (2021) 115:41. doi: 10.1016/j.pediatrneurol.2020.10.010
24. Zhao Q, Liu Z, Hu Y, Fang S, Zheng F, Li X, et al. Different experiences of two PRRT2-associated self-limited familial infantile epilepsy. Acta Neurol Belg. (2020) 120:1025-8. doi: 10.1007/s13760-020-01348-9
25. Schmidt D, Elger CE. What is the evidence that oxcarbazepine and carbamazepine are distinctly different antiepileptic drugs? Epilepsy Behav. (2004) 5:627-35. doi: 10.1016/j.yebeh.2004.07.004
26. Pan G, Zhang L, Zhou S. Clinical features of patients with paroxysmal kinesigenic dyskinesia, mutation screening of PRRT2 and the effects of morning draughts of oxcarbazepine. BMC Pediatr. (2019) 19:439. doi: 10.1186/s12887-019-1798-7
27. Li HF, Chen WJ, Ni W, Wang KY, Liu GL, Wang N, et al. PRRT2 mutation correlated with phenotype of paroxysmal kinesigenic dyskinesia and drug response. Neurology. (2013) 80:1534-5. doi: 10.1212/WNL.0b013e31828cf7e1
28. Symonds JD, Zuberi SM, Stewart K, McLellan A, O'Regan M, MacLeod S, et al. Incidence and phenotypes of childhood-onset genetic epilepsies: a prospective population-based national cohort. Brain. (2019) 142:2303-18. doi: 10.1093/brain/awz195
29. Okumura A, Shimojima K, Kurahashi H, Numoto S, Shimada S, Ishii A, et al. PRRT2 mutations in Japanese patients with benign infantile epilepsy and paroxysmal kinesigenic dyskinesia. Seizure. (2019) 71:1-5. doi: 10.1016/j.seizure.2019.05.017
30. Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, et al. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med. (2008) 358:667-75. doi: 10.1056/NEJMoa075974
31. Kumar RA, KaraMohamed S, Sudi J, Conrad DF, Brune C, Badner JA, et al. Recurrent 16p11.2 microdeletions in autism. Hum Mol Genet. (2008) 17:628-38. doi: 10.1093/hmg/ddm376
Keywords: PRRT2, genotype, phenotype, benign familial infantile epilepsy, treatment, prognosis
Citation: Luo H-y, Xie L-l, Hong S-q, Li X-j, Li M, Hu Y, Ma J-n, Wu P, Zhong M, Cheng M, Li T-s and Jiang L (2021) The Genotype and Phenotype of Proline-Rich Transmembrane Protein 2 Associated Disorders in Chinese Children. Front. Pediatr. 9:676616. doi: 10.3389/fped.2021.676616
Received: 05 March 2021; Accepted: 13 April 2021;
Published: 10 May 2021.
Edited by:
Pasquale Striano, University of Genoa, ItalyReviewed by:
Ganna Balagura, Giannina Gaslini Institute (IRCCS), ItalyCopyright © 2021 Luo, Xie, Hong, Li, Li, Hu, Ma, Wu, Zhong, Cheng, Li and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Jiang, dr_jiangcqmu@163.com
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.