
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr., 24 June 2021
Sec. Pediatric Immunology
Volume 9 - 2021 | https://doi.org/10.3389/fped.2021.633996
This article is part of the Research TopicMeaningful Cases of Primary Immunodeficiencies, Volume IIIView all 15 articles
 Vera Maria Dantas1*
Vera Maria Dantas1* Cassandra Teixeira Valle2Roberta Piccin de Oliveira3Mylena Taíse Azevedo L. Bezerra4Cleia Teixeira do Amaral5Raissa Anielle S. Brandão5
Cassandra Teixeira Valle2Roberta Piccin de Oliveira3Mylena Taíse Azevedo L. Bezerra4Cleia Teixeira do Amaral5Raissa Anielle S. Brandão5 Jussara M. Cerqueira Maia6Tirzah Braz Petta7
Jussara M. Cerqueira Maia6Tirzah Braz Petta7Familial hemophagocytic lymphohistiocytosis (FHL) is a rare, potentially fatal autosomal-recessive immunodeficiency, and STXBP2 mutations have been associated with FHL type 5 (FHL-5). Here, we report a case of a 2-year-old boy who presented with recurrent fever, hepatosplenomegaly, pancytopenia, hyperferritinemia, and hypofibrinogenemia since 4 months of age. His genetic analysis revealed a compound heterozygosity of the STXBP2 gene with a described pathogenic mutation, c.1247-1G>C (splicing acceptor site), harbored by his father and a likely pathogenic variant of uncertain significance (VUS), c.704G>A (p.Arg235Gln), harbored by his mother. He was diagnosed as compound heterozygous for FHL-5 and was treated with the HLH-2004 protocol. Since treatment, this patient has been in remission, and he is being evaluated for a hematopoietic stem cell transplantation (HSCT).
Familial hemophagocytic lymphohistiocytosis (FHL) is a rare disease with an autosomal-recessive inheritance pattern. It is caused by defects in immune regulation, such as mutations in genes controlling cytotoxicity and the killing effect of natural killer (NK) and T cells (1, 2). FHL is included in inherited hemophagocytic lymphohistiocytosis disorders, also known as primary hemophagocytic lymphohistiocytosis (HLH) (2).
There are several mutations associated with congenital immunodeficiency syndromes that can lead to episodes of immune dysregulation and HLH: Chédiak–Higashi syndrome (LYST), Griscelli syndrome type 2 (RAB27A), Hermansky–Pudlak syndrome (AP3B1), and X-linked lymphoproliferative syndrome (XLP)-1 (SH2D1A) and XLP-2 (XIAP) (3).
Secondary HLH includes patients without a known familial mutation. These patients can also develop acute HLH in the context of infections, malignancies, autoinflammatory or metabolic diseases, and acquired immunodeficiencies (4). Viral infections are frequently implicated in the onset of active HLH episodes, both in primary, genetic HLH, as well as in the secondary acquired form. The most common infections are those of herpes viruses, such as Epstein–Barr virus and cytomegalovirus (4).
According to Janka and Stadt, as cited by George (3), FHL syndromes are subclassified into five subtypes (FHL-1 to FHL-5) based on the functional protein anomalies and the prerequisite genetic mutations. Viñas-Giménez et al. (5) related four genes (PRF1, UNC13D, STX11, and STXBP2) in the 9q21.3–22 region that were identified as possible causes of FHL-2, FHL-3, FHL-4, and FHL-5, respectively. The mutated gene for the FHL-1 subtype is currently undefined. Sieni et al. (6) have reported that variants in these four genes comprise 240 missense, 69 frameshift, 51 non-sense, 51 splicing, 10 in-frame indel, 7 deep intronic, and 5 large rearrangement variants. FHL-5 is caused by homozygous or compound heterozygous mutations in the STXBP2 gene, which has 19 exons and is located in the 19p13 region. This gene encodes the 593-amino acid protein Munc18-2, and its functional alterations may impair the secretion of cytotoxic granules by NK cells (7).
It is estimated that the incidence of FHL is ~0.12–0.15 per 100,000 children per year, and FHL-5 only accounts for 10% of all cases of FHL (8). Generally, the first episodes of the disease occur during infancy, with a peak incidence between 1 and 6 months of age (6). Clinical manifestations of HLH are mainly a result of tissue infiltration by T cells and macrophages as well as an inappropriate pro-inflammatory cytokine release (4). The clinical findings are fever, hepatosplenomegaly, cytopenia, hypofibrinogenemia, hypertriglyceridemia, hyperferritinemia, less frequently, and central nervous system involvement (2, 4).
HLH, whether primary or secondary, can have a fulminant and fatal course or, in some circumstances, may have a non-severe picture with a transient course. Treatment of HLH should be guided by both the severity of the disease and its underlying etiology (3).
In 1994, the Histiocyte Society proposed the first protocol for the treatment of HLH (HLH-94). In 2004, a revised protocol (HLH-2004) was proposed (2). These protocols have greatly improved the outcomes of this frequently life-threatening disease. Both protocols are primarily based on the administration of steroids and etoposide to control the inflammation and its end-organ damage. However, in the case of relapse or that with primary etiology, hematopoietic stem cell transplantation (HSCT) may be required (2, 3).
Herein, we report a case of a patient diagnosed with FHL-5 and canonical phenotype found to be compound heterozygous in the STXBP2 gene of one known pathogenic mutation and one variant of uncertain significance (VUS) not previously associated with FHL.
A 2-year-old boy with recurrent hospitalizations since 4 months of age was admitted with fever, diarrhea, and abdominal distension. His weight and height were in the 50th and fifth percentiles, respectively. His physical examination was noticeable for splenomegaly 8 cm below the left costal margin and hepatomegaly 4 cm below the right costal margin, but there was no palpable lymphadenopathy. His laboratory workup revealed pancytopenia, hypofibrinogenemia, and hyperferritinemia (Table 1). Serological investigations for rubella, cytomegalovirus, herpes simplex virus, hepatitis B, human immunodeficiency virus, toxoplasmosis, visceral leishmaniosis, and autoimmune diseases were negative. His immunologic evaluation showed normal immunoglobulin levels (IgG, IgM, IgA, and IgE) and normal lymphocyte subset counts (CD3, CD4, CD8, CD19, and NK cells). An estimate of neutrophil function using the dihydrorhodamine test was normal and therefore not suggestive of chronic granulomatous disease (CGD). Bone marrow aspiration and biopsy did not show hemophagocytic pictures, malignancy, or infection. Of note is that the patient's parents were not consanguineous, and there was no family history of similar presentations, including in two maternal half-siblings, a 10-year-old boy, and a 13-year-old girl.
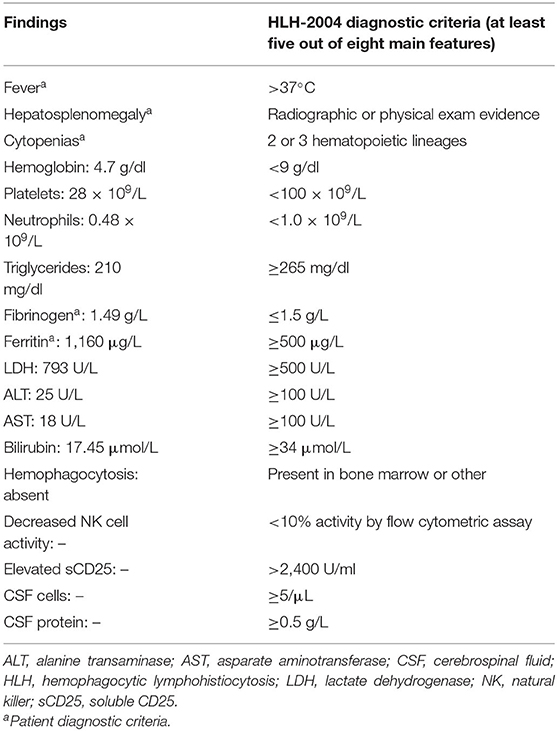
Table 1. Patient's clinical laboratory findings and HLH diagnostic criteria.
The patient was empirically treated with corticosteroids, intravenous immunoglobulins, and antibiotics. This resulted in gradual clinical and laboratorial improvement over the following 3 weeks. The patient had recurrent similar presentations over the following year, but was always responsive to steroids. Then, primary immunodeficiency next-generation sequencing panels of 207 genes (Invitae, San Francisco, CA, USA) were performed on the blood of the patient and his parents. This revealed that the patient had biallelic mutations in the STXBP2 gene: one pathogenic variant, c.1247-1G>C (splicing acceptor site), and one VUS, c.704G>A (p.Arg235Gln) (Figure 1). Genetic analyses of the patient's parents showed that both were heterozygous for the STXBP2 gene; the father harbored a pathogenic mutation, c.1247-1G>C (splicing acceptor site), and the mother harbored a VUS, c.704G>A (p.Arg235Gln).

Figure 1. (A) Pathogenic variant and a variant of uncertain significance (VUS) for the STXBP2 gene from ClinVar. The pathogenic variant c.1247-1G>C found in the patient is in a splicing site and therefore is not illustrated in this figure (variation ID: 330555; dbSNP: rs140148806). The VUS found in the patient is indicated by a black arrow (c.704>A; p. Arg235gln). There are 10 pathogenic variants and 81 VUS described for STXBP2 in ClinVar, as shown in the figure. The color legend is indicated in the corner of the figure. (B) Protein alignment for the residue Arg235 (VUS) among five organisms: Homo sapiens, Rattus norvegicus, Mus musculus, Canis lupus familiaris, and Bos taurus. Conservation of the arginine (R) is highlighted in blue. Alignment position among the five species: 257; sequence position: 235.
The patient was diagnosed with FHL-5 and was started on treatment with the HLH-2004 protocol. Since treatment with this protocol, the patient has been in remission (Figure 2). His family states that he is improving and has had only one relapse of disease (which was associated with the public health service's lack of one of the protocol medications for 2 months). The patient is currently under evaluation for HSCT.
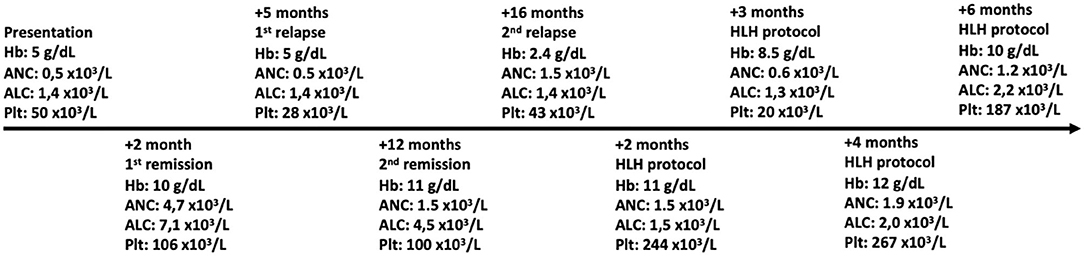
Figure 2. Peripheral blood counts over time. Hb, hemoglobin; ANC, absolute neutrophil count; ALC, absolute lymphocyte count; Plt, platelet count.
In this case report, we describe a 2-year-old boy from Brazil with five of the eight clinical laboratory criteria for hemophagocytic lymphohistiocytosis (2). He presented with recurrent fever, hepatosplenomegaly, pancytopenia, hypofibrinogenemia, and hyperferritinemia (Table 1) and intercalated with short remission periods after empirical treatment with corticosteroids (Figure 2). Of note is that the bone marrow aspirate did not reveal hemophagocytosis, but this does not rule out the diagnosis of HLH (6).
HLH is often triggered by viral infection in a genetically predisposed child (2). but an extensive viral workup was negative, as described above. Visceral leishmaniasis is a differential diagnosis of HLH (9). particularly in a patient's endemic area, but this was ruled out by a normal rK39 (a sensitivity test) (10) and the bone marrow evaluation.
Primary immunodeficiencies that confer impaired killing of infectious pathogens can cause patients to display the HLH phenotype (e.g., the inappropriate cytokine release in patients with CGD) (11). However, an evaluation for primary immunodeficiencies (including CGD) was negative, as reported above.
The child presented a molecular diagnosis consistent with HLH-5 (MedGen ID: 416514), with heterozygous compound mutations in STXBP2, the pathogenic variant c.1247-1G>C, and the VUS c.704G>A (p.Arg235Gln) (Figure 1).
STXBP2 encodes for the 593-amino acid protein Munc18-2, whose deficiency causes familial hemophagocytic lymphohistiocytosis type 5 (OMIM: 613101) and impairs cytotoxic granule exocytosis by NK cells (7, 12). Studies have reported that individuals with HLH may be compound heterozygotes and may also have monoallelic mutations of known familial HLH genes (13). According to Spessott et al., as cited by Chinn (14), the possibility that some of these monogenic variants may cause dominant-negative loss of function of the corresponding protein cannot be fully excluded.
The patient's reported STXBP2 VUS c.704G>A (p.Arg235Gln) is presented in a population database (rs757488006, 0.002%; ExAC) and the Invitae database. A VUS in the same amino acid has been reported previously, c.704G>C p.Arg235Pro, with a high likelihood score for pathogenicity (5). Pathogenicity prediction algorithms are controversial for the impact of this missense change (SIFT: “Deleterious”; PolyPhen-2: “Benign”; Align-GVGD: “Class C0”). We have applied a decision tree based on a machine learning technique for data classification, and the algorithm predicted the VUS as pathogenic or damaging (15).
Studies of FHL-5 patients with biallelic mutations in STXBP2, homozygous or compound heterozygous mutations, and one allele carrying an exon 15 c.1247-1G>C splice-site mutation showed later-onset, long-lasting remission and mild relapses (16, 17). This supports the assumption of a residual function of exon 15 splice-site mutations with a less complete or partial defect in this pathway. This could explain why our patient survived recurrent disease flare-ups that transiently responded to therapy with only steroids.
The FHL-5 phenotype of this patient was associated with the compound heterozygosity of one known pathogenic STXBP2 mutation and the VUS c.704>A (p.Arg235gln), highlighting the possible association of this VUS with FHL-5.
The original contributions presented in the study are included in the article /supplementary material, further inquiries can be directed to the corresponding author/s.
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
VD provided clinical expertise and conceived and drafted the manuscript. CV provided clinical expertise and provided patient care. CA, RB, and RO collected the case history and additional clinical data. MB and JC provided patient care. TP interpreted genetic data and assisted with preparing the figures and supervising the project. All authors were involved in the critical revision of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors are grateful to the patient's parents for their help and consent to the reporting of this case, to Prof. Antonio Condino Neto at the University of São Paulo for the insightful discussions, and to the Jeffrey Modell Foundation for supporting the genetic study.
1. Nagai K, Yamamoto K, Fujiwara H, An J, Ochi T, Suemori K, et al. Subtypes of familial hemophagocytic lymphohistiocytosis in Japan based on genetic and functional analyses of cytotoxic T lymphocytes. PLoS ONE. (2010) 5:e14173. doi: 10.1371/journal.pone.0014173
2. Henter JI, Horne A, Aricò M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. (2007) 48:124–31. doi: 10.1002/pbc.21039
3. George, MR. Hemophagocytic lymphohistiocytosis: review of etiologies and management. J Blood Med. (2014) 5:69–86. doi: 10.2147/JBM.S46255
4. Brisse E, Wouters CH, Andrei G, Matthys P. How viruses contribute to the pathogenesis of hemophagocytic lymphohistiocytosis. Front Immunol. (2017) 8:1102. doi: 10.3389/fimmu.2017.01102
5. Viñas-Giménez L, Padilla N, Batlle-Masó L, Casals F, Rivière JG, Martínez-Gallo, et al. FHLdb: a comprehensive database on the molecular basis of familial hemophagocytic lymphohistiocytosis. Front Immunol. (2020) 11:107. doi: 10.3389/fimmu.2020.00107
6. Sieni E, Cetica V, Heckmann Y, Coniglio ML, Da Ros M, Ciambotti B, et al. Familial hemophagocytic lymphohistiocytosis: when rare diseases shed light on immune system functioning. Front Immunol. (2014) 5:167. doi: 10.3389/fimmu.2014.00167
7. zur Stadt U, Rohr J, Seifert W, Koch F, Grieve S, Pagel J, et al. Familial hemophagocytic lymphohistiocytosis type 5 (FHL-5) is caused by mutations in Munc18-2 and impaired binding to syntaxin 11. Am J Hum Genet. (2009) 85:482–92. doi: 10.1016/j.ajhg.2009.09.005
8. Tang X, Guo X, Li Q, Huang Z. Familial hemophagocytic lymphohistiocytosis type 5 in a Chinese Tibetan patient caused by a novel compound heterozygous mutation in STXBP2. Medicine. (2019) 98:43. doi: 10.1097/MD.0000000000017674
9. Rajagopala S, Dutta U, Chandra KS, Bhatia P, Varma N, Kochhar R. Visceral leishmaniasis associated hemophagocytic lymphohistiocytosis – case report and systematic review. J Infect. (2008) 56:381–8. doi: 10.1016/j.jinf.2008.02.013
10. Braz R, Nascimento ET, Martins DA, Wilson ME, Pearson RD, Reed SG, et al. The sensitivity and specificity of Leishmania chagasi recombinant K39 antigen in the diagnosis of American visceral leishmaniasis and in differentiating active from subclinical infection. Am J Trop Med Hyg. (2002) 67:344–8. doi: 10.4269/ajtmh.2002.67.344
11. Parekh C, Hofstra T, Church JA, Coates TD. Hemophagocytic lymphohistiocytosis in children with chronic granulomatous disease. Pediatr Blood Cancer. (2011) 56:46. doi: 10.1002/pbc.22830
12. Côte M, Ménager MM, Burgess A, Mahlaoui N, Picard C, Schaffner C, et al. Munc18-2 deficiency causes familial hemophagocytic lymphohistiocytosis type 5 and impairs cytotoxic granule exocytosis in patient NK cells. J Clin Invest. (2009) 119:3765–73. doi: 10.1172/JCI40732
13. Cetica V, Santoro A, Gilmour KC, Sieni E, Beutel K, Pende D, et al. STXBP2 mutations in children with familial haemophagocytic lymphohistiocytosis type 5. J Med Genet. (2010) 47:595–60. doi: 10.1136/jmg.2009.075341
14. Chinn IK, Eckstein OS, Peckham-Gregory EC, Goldberg BR, Forbes LR, Nicholas SK, et al. Genetic and mechanistic diversity in pediatric hemophagocytic lymphohistiocytosis. Blood. (2018) 132:89–100. doi: 10.1182/blood-2017-11-814244
15. Nascimento PM, Medeiros IG, Falcão RM, Stransky B, Souza JE. A decision tree to improve identification of pathogenic mutations in clinical practice. BMC Med Inform Decis Mak. (2020) 20:52. doi: 10.1186/s12911-020-1060-0
16. Pagel J, Beutel K, Lehmberg K, Koch F, Maul-Pavicic A, Rohlfs AK, et al. Distinct mutations in STXBP2 are associated with variable clinical presentations in patients with familial hemophagocytic lymphohistiocytosis type 5 (FHL5). Blood. (2012) 119:6016–24. doi: 10.1182/blood-2011-12-398958
Keywords: pancytopenia, hepatosplenomegaly, hyperferritinemia, STXBP2, familial hemophagocytic lymphohistiocytosis type 5
Citation: Dantas VM, Valle CT, de Oliveira RP, Bezerra MTAL, do Amaral CT, Brandão RAS, Cerqueira Maia JM and Petta TB (2021) Germline Compound Heterozygous Variants Identified in the STXBP2 Gene Leading to a Familial Hemophagocytic Lymphohistiocytosis Type 5: A Case Report. Front. Pediatr. 9:633996. doi: 10.3389/fped.2021.633996
Received: 26 November 2020; Accepted: 11 May 2021;
Published: 24 June 2021.
Edited by:
Samuel Cern Cher Chiang, Cincinnati Children's Hospital Medical Center, United StatesReviewed by:
Tiphanie Phillips Vogel, Baylor College of Medicine, United StatesCopyright © 2021 Dantas, Valle, de Oliveira, Bezerra, do Amaral, Brandão, Cerqueira Maia and Petta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vera Maria Dantas, dmVyYW1kYW50YXNAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.