
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Pediatr. , 19 February 2021
Sec. Genetics of Common and Rare Diseases
Volume 9 - 2021 | https://doi.org/10.3389/fped.2021.633822
This article is part of the Research Topic New Therapeutic Options for Rare Diseases View all 21 articles
 Min Liu1,2
Min Liu1,2 Li-Ying Sun1,2,3*
Li-Ying Sun1,2,3*Glycogen storage disease type IV (GSD IV) is a rare autosomal recessive disorder caused by glycogen–branching enzyme (GBE) deficiency, leading to accumulation of amylopectin–like glycogen that may damage affected tissues. The clinical manifestations of GSD IV are heterogeneous; one of which is the classic manifestation of progressive hepatic fibrosis. There is no specific treatment available for GSD IV. Currently, liver transplantation is an option. It is crucial to evaluate long–term outcomes of liver transplantation. We reviewed the published literature for GSD IV patients undergoing liver transplantation. To date, some successful liver transplantations have increased the quantity and quality of life in patients. Although the extrahepatic manifestations of GSD IV may still progress after transplantation, especially cardiomyopathy. Patients with cardiac involvement are candidates for cardiac transplantation. Liver transplantation remains the only effective therapeutic option for treatment of GSD IV. However, liver transplantation may not alter the extrahepatic progression of GSD IV. Patients should be carefully assessed before liver transplantation.
Glycogen storage disease type IV (GSD IV, Andersen disease, amylopectinosis, OMIM:232500) is an autosomal recessive disorder of glycogen metabolism (1). GSD IV is estimated to occur in 1 in 600 000 to 800 000 individuals worldwide (2). GSD IV is caused by deficiency of glycogen-branching enzyme (GBE), encoded by GBE1 gene, leading to accumulation of amylopectin-like glycogen (polyglucosan) in affected tissues (3–6). More than 40 mutations in the GBE1 gene have been found to cause GSD IV. GBE1 is located on chromosome 3p14 and encodes a 702-amino-acid protein (7–9).
GSD IV is a rare form of glycogen storage disease that accounts for ~3% of glycogenosis (2). The accumulation of abnormal branched glycogen results in tissue damage. The clinical manifestations of GSD IV are heterogeneous with variable age of onset, severity and involvement of the liver, cardiac muscle neuromuscular system (3). The progressive hepatic type is the most common and classic form of GSD IV and is characterized by hepatomegaly that rapidly progresses to liver cirrhosis. Patients ultimately die from liver failure by 5 years of age, unless liver transplantation is performed (10, 11). Patients with a rare non-progressive hepatic form do not develop cirrhosis and survive to adulthood without liver transplantation (12–14). The neuromuscular form of GSD IV varies in onset (from fetal to adult age) and severity (4, 7, 15), which present with symptoms of myopathy, cardiomyopathy, and central and peripheral nervous system dysfunction (7, 16).The fatal infantile neuromuscular type is the most severe form of GSD IV (17, 18). Patients with the congenital neuromuscular form have severe muscle wasting, hypotonia, cardiomyopathy, respiratory distress, neuronal involvement and death in early infancy (19, 20). Neurological adult forms can present as isolated myopathy or as widespread upper and lower motor neuron lesions (adult polyglucosan body disease, APBD) (21). Liver biopsies have confirmed signs of cirrhosis and cells with periodic acid-Schiff (PAS) –stained, diastase-resistant inclusions, consistent with GSD IV. There is no specific treatment for GSD IV. Prognosis is unfavorable for patients with severe cardiomyopathy, associated neurological dysfunction and classic forms who do not undergo liver transplantation.
As there is no treatment to compensate for the deficient enzyme activity, additionally, the classic form of GSD IV usually leads to liver failure, and liver transplantation is the only effective treatment presently available for GSD IV with progressive liver disease (22). Since the first successful liver transplant for GSD IV in September 1984 (23), it has been increasingly used to treat GSD IV cases with liver failure and has improved survival. It is important to know the long–term outcomes of this therapeutic option. In this article, we review all GSD IV patients who underwent liver transplantation to explore the effectiveness and outcomes of liver transplantation for GSD IV.
The English–language literature about GSD IV patients undergoing liver transplantation was systematically reviewed in PubMed and in the references of relevant publications. The key words used included glycogen storage disease type IV, Andersen disease, amylopectinosis, type IV glycogenosis, glycogen-branching enzyme deficiency, and liver transplantation. Indications for liver transplantation, age at transplantation, follow-up period and complications were reviewed. Liver function post–transplantation, extrahepatic manifestations after transplantations, and clinical outcomes were analyzed to better clarify the clinical value of liver transplantation for GSD IV.
We summarized the characteristics and results of 24 patients with GSD IV who underwent liver transplantation between 1984 and 2017 (Table 1). There were 19 male patients with two sets of brothers (cases 7 and12, 8 and 11) and five female patients. The youngest was transplanted at 10 months of age and the oldest at 23 years. Two patients required a second liver transplantation: one for nonfunction of the graft 2 d after the first liver transplant, and one for antibody–mediated rejection due to ABO–incompatible live donor graft 6.2 mo after the first transplant. A living related transplantation was performed in seven patients. The indication for liver transplantation was based on the occurrence of progressive liver cirrhosis with evidence of portal hypertension in most cases; for one patient, liver cirrhosis was associated with liver adenoma, and for another, it was heart failure.
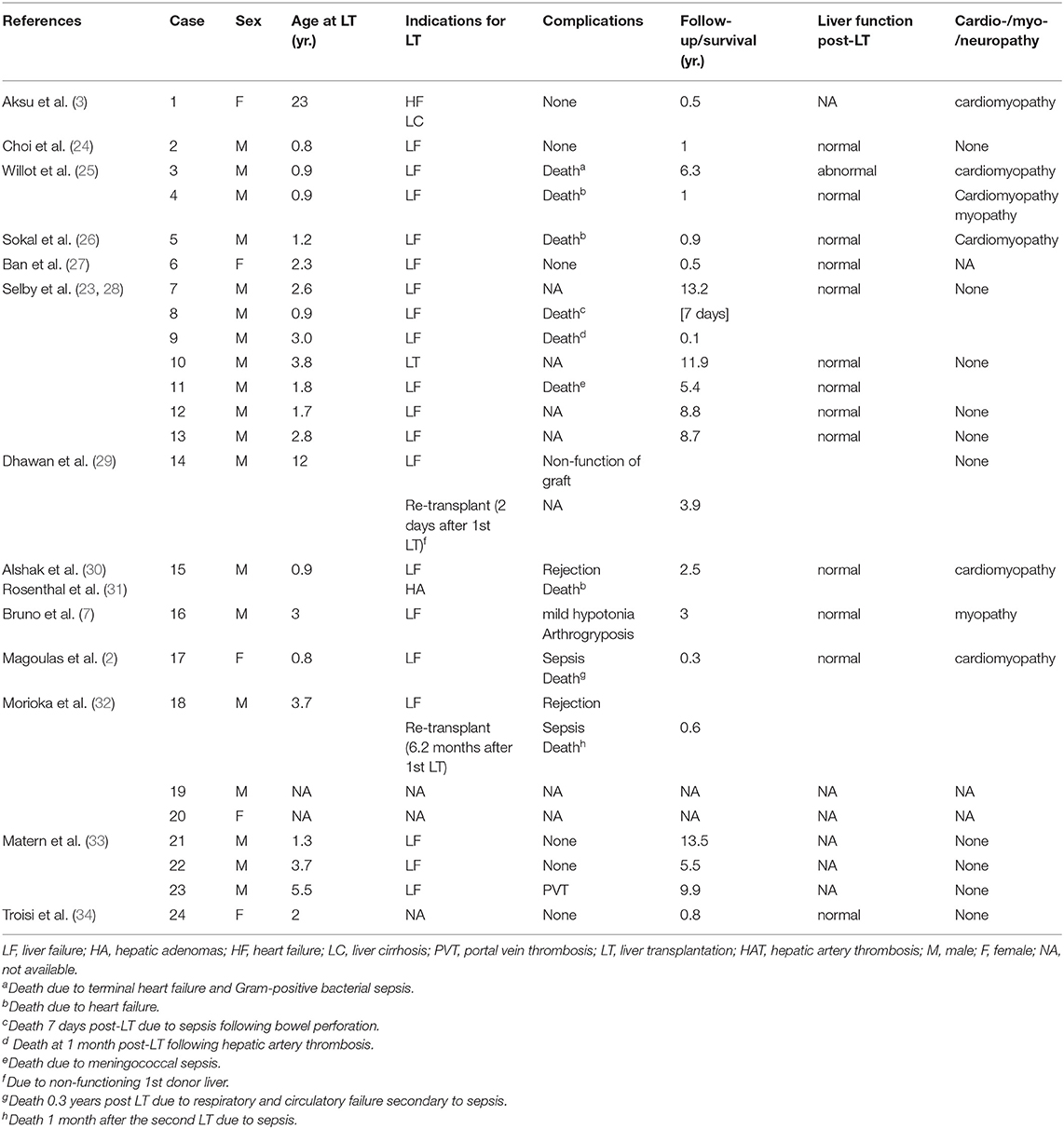
Table 1. Patients with GSD IV who underwent liver transplantation.
On myocardial biopsy, PAS–positive deposits in the myocardium were found in 10 patients with GSD IV, including five postmortem specimens (Table 2). All myocardial biopsies were performed postoperatively, except for biopsy of the first patient. Death was reported in nine (37.5%) patients because of: sepsis following bowel perforation 7 d after transplantation (n = 1); meningococcal sepsis (n = 1); sepsis 1 mo after second liver transplantation (n = 1); respiratory and circulatory failure secondary to sepsis 0.3 years post-transplantation (n = 1); terminal heart failure and Gram-positive bacterial sepsis 76 mo after liver transplantation (n = 1); cardiomyopathy (n = 3); and hepatic artery thrombosis 1 mo post–liver transplantation (n = 1). Five patients died <1 year after transplantation;two of whom died <30 d after liver transplantation. Four patients died > 1-year post–transplant. All other patients were alive at time of follow–up (range several months to 13.5 years after transplantation). In all identified survivors, liver function was normal and portal hypertension was decompressed via the new liver. Catch–up growth was reported in eight patients.

Table 2. PAS-positive inclusion deposits in the myocardium of ten patients with GSD IV pre-or-post liver transplantation.
Cardiomyopathy was the most frequent and serious complication, eventually leading to heart failure. Cardiomyopathy developed in six (25%) patients, including one with pre-transplantation heart failure. None of these individuals had undergone combined liver–heart transplantation. Transplant–related complications were seen in eight (33.3%) patients and included portal vein thrombosis (n = 1), hepatic artery thrombosis (n = 1), rejection (n = 2), sepsis (n = 4) and non-function of graft (n = 1). One of the eight patients had both rejection and sepsis. Of the 15 (62.5%) survivors, one presented at birth with severe hypotonia and flexion contractures of the hips and articulus. He was reported to have symptomatic improvement with mild hypotonia and arthrogryposis 3 years post–transplantation. Thirteen survivors had no neuromuscular or heart complications during follow-up for as long as 13.5 years. Notably, there was a marked reduction in the amount of amylopectin on myocardial biopsy in one of these patients after transplantation (case 12, Table 1). The patient who underwent liver transplantation for heart failure had not experienced any symptoms of cardiac failure at 6 mo after transplantation (case 1, Table 1).
Our review shows that liver transplantation corrects the primary hepatic enzyme defect, so the graft does not accumulate polyglucosan. Liver transplantation normalized the liver function of all the patients with GSD IV except case 3, thereby improving the survival rate and quality of life of patients. Since then, liver transplantation has improved patients' outcome because metabolic sequelae, such as cardiomyopathy and skeletal myopathy, have not developed post–transplantation in most cases. Most remarkable, eight patients were reported to had retarded growth pre–transplantation, but developed well with normal growth, after liver transplantation (25, 27, 28). As there is no way to compensate for the insufficient enzyme activity, liver transplantation remains the only effective treatment for patients with the progressive hepatic subtype of GSD IV who develop liver failure.
The clinical onset frequently in patients with classical GSD IV is earlier than in atypical individuals (23). We identified that most patients did not develop cardiomyopathy or neuromuscular complications during follow–up of up to 13.5 years (11). Seven of these patients received liver transplantation from living donors, which included five heterozygous and three ABO–incompatible donors, and no mortality or morbidity associated with heterozygosity has yet been observed (3, 24, 27, 32, 34). One of these patents was a 3.7-year-old boy with GSD IV (case 18, Table 1), who underwent first liver transplantation using an ABO–incompatible liver graft from his mother, which led to graft failure due to ABO-incompatibility and was replaced by an ABO–incompatible liver graft from his father 6.2 mo after the first liver transplant. In one patient who underwent liver transplantation for heart failure due to cardiomyopathy, and her heart failure was restored during 6 mo follow–up (3). Remarkably, there was resorption of amylopectin on myocardial biopsy in one patient after liver transplantation (35). The mechanism of this decrease of abnormal glycogen remains unclear (11). It may have been due to migration of donor cells from the liver allograft to the recipient heart and microchimerism (5, 35).
However, the prognosis is grave when extrahepatic manifestations of GSD IV develop, especially cardiomyopathy (36). Accordingly, it is a major concern that liver transplantation may only improve the hepatic function of individuals with GSD IV, and extrahepatic manifestations might develop post–transplantation and result in poor prognosis. Amylopectin is not soluble, and the enzyme defect is present in other affected organs such as muscle, heart or nervous system. Amylopectin accumulation in other affected tissues might progress after transplantation. Postoperative extrahepatic progression of the disease caused by amylopectin accumulation in other affected tissues might be a potential risk for these individuals. Cardiac amylopectionosis was the most common postoperative complication in GSD IV patients (6/24) and four of those died from heart failure. We identified only one patient with heart failure before transplantation. The evaluation of pretransplant cardiac function is not a predicting factor of poor outcome in this situation. Of the four patients who died of cardiac failure after liver transplantation, all preoperative heart function was normal, but they developed cardiac amylopectionosis, attributed to accumulation of amylopectin in cardiac muscle. The patients died 2.55 years (range: 0.9–6.3years) after transplantation (25, 26, 31). In three of those four patients, autopsy showed that cardiomyocytes contained massive PAS–positive, diastase-resistant inclusions (25, 31). In the fourth patient, postoperative myocardial biopsy showed PAS–positive, amylase–resistant deposits in cardiomyocytes up to 9 mo following liver transplantation (26). In another patient, cardiomyopathy was discovered by postmortem examination after he died from respiratory and circulatory failure secondary to sepsis (2). Myocardial biopsy is a potential predictor of cardiac functional prognosis after transplantation, but the number of amylopectin–like deposits related to progressive fatal cardiac failure needs to be defined. Further, long-term follow-up is necessary to evaluate possible cardiac or neuromuscular complications (24). To further evaluate this risk, patients with GSD IV need careful assessment of heart, liver, and muscle before and after liver transplantation.
Heart transplantation has been suggested in cases with severe cardiac involvement. Patients with progressive cardiomyopathy and myocardial involvement confirmed by myocardial biopsy secondary to GSD IV may be candidates for cardiac transplantation. The experience with cardiac transplantation for GSD IV is insufficient. Only three patients are known to have undergone cardiac transplantation for extrahepatic progression related to GSD IV (37–39). Two patients were transplanted successfully and in good condition during follow–up. The third one died due to infectious complications after orthoptic heart transplantation. Evaluation of the genotype–phenotype correlation in GSD IV may be helpful, which may provide valuable information in decision-making and help us to better understand the outcome of liver transplantation (6, 33).
Liver transplantation remains the only therapeutic option for treatment of hepatic manifestation of GSD IV. Our review shows that all GSD IV patients who survived had normal liver function after liver transplantation. Selection of patients with GSD IV for liver transplantation should be alert to extrahepatic progression, as the cardiomyopathy may lead to fatal complications. Consideration of combined liver–heart transplantation and careful assessment of cardiac function even in the absence of evidence of clinical decompensation appears warranted for patients with GSD IV. Histopathological studies of myocardial tissues and evaluation of the correlation between genotype–phenotype and the condition may predict the degree of severity and assist with treatment decisions.
ML identified all the cases to be included, analyzed and interpreted the data and drafted the manuscript. L-YS reviewed the manuscript. Both authors read and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Andersen DH. Familial cirrhosis of the liver with storage of abnormal glycogen. Lab Invest. (1956)5:11–20.
2. Magoulas PL, El-Hattab AW, Roy A, Bali DS, Finegold MJ, Craigen WJ. Diffuse reticuloendothelial system involvement in type IV glycogen storage disease with a novel GBE1 mutation: a case report and review. Hum Pathol. (2012) 43:943–51. doi: 10.1016/j.humpath.2011.10.001
3. Aksu T, Colak A, Tufekcioglu O. Cardiac involvement in glycogen storage disease type iv: two cases and the two ends of a spectrum. Case Rep Med. (2012)2012:764286. doi: 10.1155/2012/764286
4. Fernandez C, Halbert C, De Paula AM, Lacroze V, Froissart R, Figarella-Branger D, et al. Non-lethal neonatal neuromuscular variant of glycogenosis type IV with novel GBE1 mutations. Muscle Nerve. (2010) 41:269–71. doi: 10.1002/mus.21499
5. Dainese L, Adam N, Boudjemaa S, Hadid K, Rosenblatt J, Jouannic JM, et al. Glycogen storage disease type IV and early implantation defect: early trophoblastic involvement associated with a New GBE1 mutation. Pediatr Dev Pathol. (2016) 19:512–5. doi: 10.2350/14-09-1557-CR.1
6. Moses SW, Parvari R. The variable presentations of glycogen storage disease type IV: a review of clinical, enzymatic and molecular studies. Curr Mol Med. (2002) 2:177–88. doi: 10.2174/1566524024605815
7. Bruno C, van Diggelen OP, Cassandrini D, Gimpelev M, Giuffre B, Donati MA, et al. Clinical and genetic heterogeneity of branching enzyme deficiency (glycogenosis type IV). Neurology. (2004) 63:1053–8. doi: 10.1212/01.wnl.0000138429.11433.0d
8. Lamperti C, Salani S, Lucchiari S, Bordoni A, Ripolone M, Fagiolari G, et al. Neuropathological study of skeletal muscle, heart, liver, and brain in a neonatal form of glycogen storage disease type IV associated with a new mutation in GBE1 gene. J Inherit Metab Dis. (2009) 32(Suppl 1):S161–8. doi: 10.1007/s10545-009-1134-8
9. Shin YS. Glycogen storage disease: clinical, biochemical, and molecular heterogeneity. Semin Pediatr Neurol. (2006) 13:115–20. doi: 10.1016/j.spen.2006.06.007
10. Cenacchi G, Papa V, Costa R, Pegoraro V, Marozzo R, Fanin M, et al. Update on polyglucosan storage diseases. Virchows Arch. (2019) 475:671–86. doi: 10.1007/s00428-019-02633-6
11. Davis MK, Weinstein DA. Liver transplantation in children with glycogen storage disease: controversies and evaluation of the risk/benefit of this procedure. Pediatr Transplant. (2008) 12:137–45. doi: 10.1111/j.1399-3046.2007.00803.x
12. Bao Y, Kishnani P, Wu JY, Chen YT. Hepatic and neuromuscular forms of glycogen storage disease type IV caused by mutations in the same glycogen-branching enzyme gene. J Clin Invest. (1996) 97:941–8. doi: 10.1172/JCI118517
13. Greene HL, Brown BI, McClenathan DT, Agostini RJ, Taylor SR. A new variant of type IV glycogenosis: deficiency of branching enzyme activity without apparent progressive liver disease. Hepatology. (1988) 8:302–6. doi: 10.1002/hep.1840080219
14. McConkie-Rosell A, Wilson C, Piccoli DA, Boyle J, DeClue T, Kishnani P, et al. Clinical and laboratory findings in four patients with the non-progressive hepatic form of type IV glycogen storage disease. J Inherit Metab Dis. (1996) 19:51–8. doi: 10.1007/BF01799348
15. Radhakrishnan P, Moirangthem A, Nayak SS, Shukla A, Mathew M, Girisha KM. Novel pathogenic variants in GBE1 causing fetal akinesia deformation sequence and severe neuromuscular form of glycogen storage disease type IV. Clin Dysmorphol. (2019) 28:17–21. doi: 10.1097/MCD.0000000000000248
16. Tay SK, Akman HO, Chung WK, Pike MG, Muntoni F, Hays AP, et al. Fatal infantile neuromuscular presentation of glycogen storage disease type IV. Neuromuscul Disord. (2004) 14:253–60. doi: 10.1016/j.nmd.2003.12.006
17. Assereto S, van Diggelen OP, Diogo L, Morava E, Cassandrini D, Carreira I, et al. Null mutations and lethal congenital form of glycogen storage disease type IV. Biochem Biophys Res Commun. (2007) 361:445–50. doi: 10.1016/j.bbrc.2007.07.074
18. Yu W, Brundler MA, Wright JJ. Polyglucosan bodies in placental extravillious trophoblast for the diagnosis of fatal perinatal neuromuscular-type glycogen storage disease type IV. Pediatr Dev Pathol. (2018) 21:423–7. doi: 10.1177/1093526617707852
19. Nolte KW, Janecke AR, Vorgerd M, Weis J, Schroder JM. Congenital type IV glycogenosis: the spectrum of pleomorphic polyglucosan bodies in muscle, nerve, and spinal cord with two novel mutations in the GBE1 gene. Acta Neuropathol. (2008) 116:491–506. doi: 10.1007/s00401-008-0417-8
20. Taratuto AL, Akman HO, Saccoliti M, Riudavets M, Arakaki N, Mesa L, et al. Branching enzyme deficiency/glycogenosis storage disease type IV presenting as a severe congenital hypotonia: muscle biopsy and autopsy findings, biochemical and molecular genetic studies. Neuromuscul Disord. (2010) 20:783–90. doi: 10.1016/j.nmd.2010.07.275
21. Bruno C, Cassandrini D, Assereto S, Akman HO, Minetti C, Di Mauro S. Neuromuscular forms of glycogen branching enzyme deficiency. Acta Myol. (2007)26:75–8.
22. Kido J, Nakamura K, Matsumoto S, Mitsubuchi H, Ohura T, Shigematsu Y, et al. Current status of hepatic glycogen storage disease in Japan: clinical manifestations, treatments and long-term outcomes. J Hum Genet. (2013) 58:285–92. doi: 10.1038/jhg.2013.17
23. Selby R, Starzl TE, Yunis E, Todo S, Tzakis AG, Brown BI, et al. Liver transplantation for type I and type IV glycogen storage disease. Eur J Pediatr. (1993) 152(Suppl 1):S71–6. doi: 10.1007/BF02072093
24. Choi SY, Kang B, Choe JY, Lee Y, Jang HJ, Park HD, et al. A case of glycogen storage disease iv with rare homozygous mutations in the glycogen branching enzyme gene. Pediatr Gastroenterol Hepatol Nutr. (2018) 21:365–8. doi: 10.5223/pghn.2018.21.4.365
25. Willot S, Marchand V, Rasquin A, Alvarez F, Martin SR. Systemic progression of type IV glycogen storage disease after liver transplantation. J Pediatr Gastroenterol Nutr. (2010) 51:661–4. doi: 10.1097/MPG.0b013e3181d29780
26. Sokal EM, Van Hoof F, Alberti D, de Ville DGJ, de Barsy T, Otte JB. Progressive cardiac failure following orthotopic liver transplantation for type IV glycogenosis. Eur J Pediatr. (1992) 151:200–3. doi: 10.1007/BF01954384
27. Ban HR, Kim KM, Jang JY, Kim GH, You HW, Kim K, et al. Living donor liver transplantation in a korean child with glycogen storage disease type IV and a GBE1 mutation. Gut Liver. (2009)3:60–3. doi: 10.5009/gnl.2009.3.1.60
28. Selby R, Starzl TE, Yunis E, Brown BI, Kendall RS, Tzakis A. Liver transplantation for type IV glycogen storage disease. N Engl J Med. (1991) 324:39–42. doi: 10.1056/NEJM199101033240107
29. Dhawan A, Tan KC, Portmann B, Mowat AP. Glycogenosis type IV: liver transplant at 12 years. Arch Dis Child. (1994) 71:450–1. doi: 10.1136/adc.71.5.450
30. Alshak NS, Cocjin J, Podesta L, van de Velde R, Makowka L, Rosenthal P, et al. Hepatocellular adenoma in glycogen storage disease type IV. Arch Pathol Lab Med. (1994)118:88–91.
31. Rosenthal P, Podesta L, Grier R, Said JW, Sher L, Cocjin J, et al. Failure of liver transplantation to diminish cardiac deposits of amylopectin and leukocyte inclusions in type IV glycogen storage disease. Liver Transpl Surg. (1995) 1:373–6. doi: 10.1002/lt.500010607
32. Morioka D, Kasahara M, Takada Y, Corrales JP, Yoshizawa A, Sakamoto S, et al. Living donor liver transplantation for pediatric patients with inheritable metabolic disorders. Am J Transplant. (2005) 5:2754–63. doi: 10.1111/j.1600-6143.2005.01084.x
33. Matern D, Starzl TE, Arnaout W, Barnard J, Bynon JS, Dhawan A, et al. Liver transplantation for glycogen storage disease types I, III, and IV. Eur J Pediatr. (1999) 158(Suppl 2):S43–8. doi: 10.1007/pl00014320
34. Troisi RI, Elsheikh YM, Shagrani MA, Broering D. First fully laparoscopic donor hepatectomy for pediatric liver transplantation using the indocyanine green near-infrared fluorescence imaging in the Middle East: a case report. Ann Saudi Med. (2014) 34:354–7. doi: 10.5144/0256-4947.2014.354
35. Starzl TE, Demetris AJ, Trucco M, Ricordi C, Ildstad S, Terasaki PI, et al. Chimerism after liver transplantation for type IV glycogen storage disease and type 1 Gaucher's disease. N Engl J Med. (1993) 328:745–9. doi: 10.1056/NEJM199303183281101
36. Maheshwari A, Rankin R, Segev DL, Thuluvath PJ. Outcomes of liver transplantation for glycogen storage disease: a matched-control study and a review of literature. Clin Transplant. (2012) 26:432–6. doi: 10.1111/j.1399-0012.2011.01549.x
37. de La Blanchardière A, Vayssier C, Duboc D, Jacquemin IE, Eymard B, Fardeau M, et al. [Severe cardiomyopathy revealing amylopectinosis. Two cases in adolescents from the same family]. Presse Med. (1994) 23:1124–7.
38. Ewert R, Gulijew A, Wensel R, Dandel M, Hummel M, Vogel M, et al. [Glycogenosis type IV as a seldom cause of cardiomyopathy - report about a successful heart transplantation]. Z Kardiol. (1999) 88:850–6. doi: 10.1007/s003920050361
Keywords: glycogen storage disease type IV, glycogen branching enzyme, Andersen disease, liver transplantation, metabolism
Citation: Liu M and Sun L-Y (2021) Liver Transplantation for Glycogen Storage Disease Type IV. Front. Pediatr. 9:633822. doi: 10.3389/fped.2021.633822
Received: 26 November 2020; Accepted: 02 February 2021;
Published: 19 February 2021.
Edited by:
Andrea Gropman, Children's National Hospital, United StatesReviewed by:
Priya Kishnani, Duke University, United StatesCopyright © 2021 Liu and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li-Ying Sun, c3VueGx4QG91dGxvb2suY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.