 Yu Hu
Yu Hu XinDong Xue
XinDong Xue JianHua Fu
JianHua Fu- Department of Pediatrics, Shengjing Hospital of China Medical University, Shenyang, China
Objective: The clinical symptoms of neonatal Prader-Willi syndrome (PWS) are not typical and are easy to miss. The aim of the study was to investigate the clinical features and genetic characteristics of seven cases of neonatal PWS from northern China, and to improve the understanding of PWS in neonates.
Methods: We retrospectively analyzed seven infants diagnosed by methylation specific multiplex ligation probe amplification technology (MS-MLPA) in the Neonatology Unit of Shengjing Hospital of China Medical University from September 2016 to July 2020.
Results: All seven cases involved full term or nearly full-term infants born to mothers without a history of abnormal pregnancy or delivery. Difficulty in feeding occurred immediately after birth in infants with decreased hypotonia. Five patients had characteristic craniofacial morphology, such as a prominent forehead, narrow face, almond-shaped eyes, small mouth, and downturned mouth. Further, three of the seven infants had patent ductus arteriosus (PDA). In addition, three neonates had hyperammonemia, hypoglycemia, and idiopathic edema, respectively. PWS could be effectively diagnosed and genotyped by MS-MLPA.
Conclusion: Neonates with PWS have hypotonia and feeding difficulty. Characteristic facial features and genital hypoplasia are common in neonatal PWS. Infants with PWS may be predisposed to PDA, hypoglycemia, hyperammonemia, and edema.
Introduction
Prader-Willi syndrome (PWS) is a complex neuroendocrine hereditary syndrome first described by John Langdon Down in 1887 and named by Swiss doctors Andrea Prader, Heinrich Willi, and Alexis Labhartin in 1956, based on the clinical characteristics of nine pediatric cases (1). The incidence of PWS varies in the range of 1/10,000–1/30,000 across ethnicities (2). The epidemiological data of PWS is lacking in China. The National Health Commission, the Ministry of Science and Technology, the Ministry of Industry and Information Technology, the State Drug Administration, and the State Administration of Traditional Chinese Medicine jointly issued a list of rare diseases in China. The list included 121 diseases, 18 of which have a neonatal onset. PWS is one of the 18 rare neonatal diseases in the list. The causes and genetic mechanisms in PWS are complex (3). Most cases are due to new mutations, meaning both parents are normal, and only a small proportion of PWS cases are inherited. It occurs due to a genetic defect during the stage of egg (sperm) or embryo formation, which results in the deletion or disruption of more than 10 genes on the long arm of chromosome 15. If the defect is inherited from the father, or if the patient has two maternal copies of chromosome 15 with the defect, the result is PWS.
Prader-Willi syndrome results from the paternally inherited loss of the chromosome region 15q11.2-13 (3), which causes complex influences in appetite, development, metabolism, cognition, and behavior (4). PWS is also the most common cause of genetic obesity (5). Additionally, difficulty with public transport and hyperlexia are two of the lesser known manifestations of PWS (6). There is a continuum in the clinical presentation and physical characteristics of PWS, and variations exist among different age groups (7). In the neonatal stage, marked hypotonia and feeding difficulties are characteristic clinical features. In the infantile stage, patients gradually show developmental delay. In the childhood stage, hyperphagia for high carbohydrate food with an evident satiety is typically observed. Later, in the adolescence stage, the patient usually shows weight gain and several behavioral problems. In fact, most patients seek medical advice only after they have developed obesity (7).
It is important to recognize the features of PWS as soon as possible because of its long-term implications. Since PWS is a complex genetic disorder caused by the lack of expression of paternally active genes on chromosome region 15q11.2-q13, there are three different genetic subtypes of PWS (8): deletion of the paternal copy of 15q11-q13 (70–75% of cases), maternal uniparental disomy (UPD) for chromosome 15 (25–30%), and imprinting defect because of the silencing of paternal alleles (1–3%). Methylation specific multiplex ligation probe amplification (MS-MLPA) is an effective molecular detection method for PWS based on differential methylation status. In this study, we report seven neonatal patients from northern China diagnosed with PWS by MS-MLPA technology and analyzed their clinical features and genetic characteristics. The aim of this study was to strengthen the understanding of PWS in neonates.
Materials and Methods
Subjects
We retrospectively analyzed the medical records of seven neonates with PWS (three boys and four girls) who were genetically diagnosed using MS-MLPA. All of them were hospitalized in the Department of Neonatology, Shengjing Hospital of China Medical University from September 2016 to July 2020. The study protocols were approved by the Hospital Ethics Committee for Human Research of Shengjing Hospital of China Medical University.
Methods
All the seven neonates were diagnosed by MS-MLPA after blood sampling. Abnormal methylation pattern, chromosome microdeletions, and maternal uniparental disomy were analyzed to aid the genetic diagnosis.
Statistical Analysis
Clinical data on sex, birth weight, gestational age (GA), age of onset, age at hospital admission, first symptoms, clinical magnifications, genotype, age at genetic diagnosis, treatment, and prognosis were collected. All clinical data were descriptively analyzed. Birth weights were presented as means ± standard deviations.
Results
General Data
In total, seven neonates (three boys and four girls) were included in the study. The GA ranged from 36 weeks and 6 days to 40 weeks. Birth weight varied from 2.4 to 3.4 kg with an average weight of 2.79 ± 0.35 kg. Maternal ages ranged from 23 to 37 years old. All cases were appropriate for GA. Interestingly none of the seven neonates was a firstborn and all had a maternal history of multiple pregnancies. Breech presentation was observed in one patient. Of note, 71.4% (5/7) were delivered by cesarean section; one of them was delivered in breech position, and another suffered from intrauterine distress and other symptoms. Four patients were second-born children, three of whom had healthy families, and one had a brother suffering from ventricular septal defect. They were all full term or nearly full-term infants without maternal history of abnormal pregnancy or asphyxia at birth. Age at hospital admission varied from 2 days to 16 days. The results are summarized in Table 1.
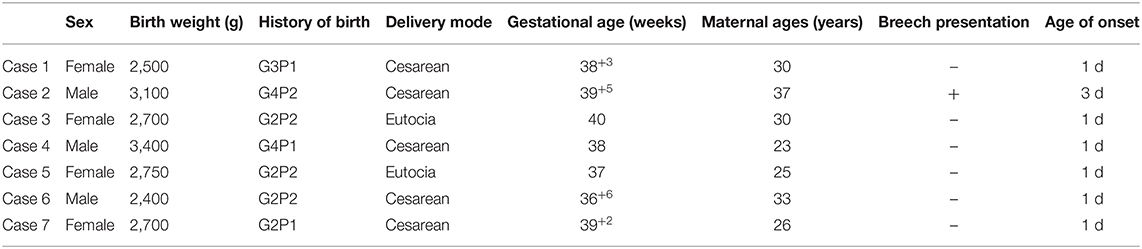
Table 1. Demographical characteristics of the neonates with Prader-Willi syndrome (n = 7).
Clinical Features
All the seven cases were Han Chinese. As shown in Table 2, five of them had decreased fetal movement during pregnancy. The first symptom in all cases was a difficulty in feeding immediately after birth. On admission, the physical examination showed a weak crying, poor rooting, sucking and swallowing reflexes in all cases. All the seven neonates had central hypotonia and poor feeding ability. Five cases (71%) had characteristic craniofacial morphology including prominent forehead, narrow face, almond-shaped eyes, small mouth, and downturned mouth. Further, two of the three boys (67%) had bilateral cryptorchidism. The genitalia of one girl (25%) had the appearance of labia minora. Three cases (43%) were diagnosed with patent ductus arteriosus (PDA) by cardiac ultrasonography with a diameter range from 1.5 to 3.3 mm. In addition, three neonates had hyperammonemia, hypoglycemia, and idiopathic edema, respectively. All of them completed the head magnetic resonance and electromyography examination without abnormalities. Another girl unfortunately suffered from nemaline myopathy caused by a mutation in the NEB gene.
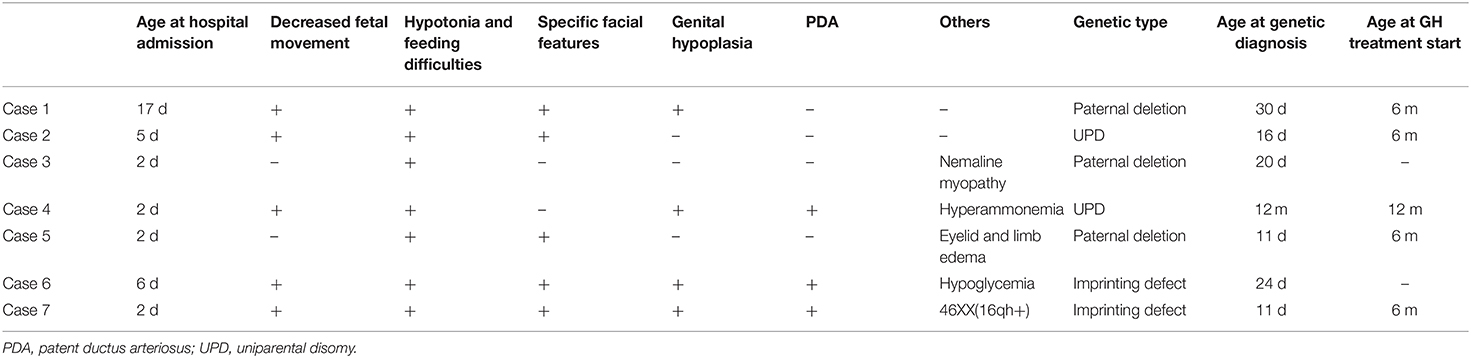
Table 2. Clinical characteristics of the neonates with Prader-Willi syndrome (n = 7).
Genetics
Using the MS-MLPA method, a deletion was detected in three patients (43%). Two patients had a maternal uniparental disomy (29%), and an abnormal methylation pattern without deletion was observed in other two patients (29%). Further, one girl exhibited chromosomal polymorphism, 46XX(16qh+). The results are summarized in Table 2.
Diagnosis, Treatment, and Outcome
As shown in Table 2, five neonates with PWS were diagnosed between postnatal days 11 and 30, and were discharged when their feeding problems started to improve at 12 to 47 days (average 22 days) of age. Four of them initiated growth hormone (GH) treatment from the age of 6 months. A girl of the five neonates gave up the treatment because of comorbid nemaline myopathy disease. Apart from the five neonates, one case was discharged before laboratory examination and received a genetic diagnosis and GH treatment in a follow-up visit at 1 year old. Another patient was discharged just after genetic diagnosis at postnatal days 24 and died shortly after leaving the hospital because of poor sucking.
Discussion
Recently, an MS-MLPA kit equipped with suitable discharge was introduced. It can detect gene duplication or deletion, distinguish types of deletion, and estimate the methylation status. MS-MLPA allows to detect large, abnormal, or subtle deletions of chromosomes, but not balanced translocations. MS-MLPA cannot detect any changes that lie outside the target critical region on chromosome 15. The positive detection rate can be as high as 99% (9). In this article, the genetic analysis, which was performed using the MS-MLPA method, showed a deletion of the paternal chromosome 15 in three patients (43%), maternal uniparental disomy in two patients (29%), and imprinting defect in two other patients (29%). Furthermore, their parents and relatives were all healthy. Deletion of the paternal chromosome was the main genetic feature. Patients with paternal deficit in PWS are more common than those with maternal UPD. This result was in line with those of other studies (10, 11). Intrauterine hypotonia causes polyhydramnios, malpresentation, and reduced fetal movements, leading to a higher probability of cesarean. All these features were found in this study, in accord with other reports (12).
In the fetal period, PWS may manifest as fetal movement reduction, excessive amniotic fluid, and breech presentation. In the neonatal stage, marked hypotonia, weak cry, and feeding difficulties are characteristic clinical features (12). PWS is one of the leading causes of neonatal hypotonia (13). We collected the most accurate and detailed clinical information possible and found that all the seven cases showed poor sucking ability and central hypotonia. Therefore, if neonates present these two symptoms, especially in the presence of reduced intrauterine fetal movement, PWS should be considered. Beyhan Tuysuz and Gillessen-Kaesbach et al. found that PWS is commonly the cause of neonatal hypotonia, with a prevalence of 10.7–45% among infants with hypotonia (14, 15).
In this study, the results indicate that all seven cases were (nearly) full term and appropriate for GA, which was in accord with another Chinese study (11). It was reported that the full-term infants accounted for 80.4% of all 102 PWS patients. However, other studies have reported on the prevalence of preterm births and small for GA newborns (12). The discrepancies may be explained by the differences in sample size and ethnic differences. Specific genetic testing for PWS should be taken into account for all neonates with unknown central hypotonia, even in the absence of other major features of this syndrome. In this study, one PWS infant had a complication of mitochondrial myopathy, which had not been reported previously.
Patients with PWS often show a characteristic craniofacial morphology, including prominent forehead, narrow face, almond-shaped eyes, small mouth, and downturned mouth. However, craniofacial morphology are not obvious at birth. It become more typical with age (7). These symptoms are easy to miss if proper attention is not paid. Of the seven children in this study, we noted that five had one or more of these characteristic features. In addition, white skin and light hair are more conspicuous in patients of Asian origin. Small hands and feet and hypogonadism are also characteristic features of PWS (16). In fact, very few PWS patients are diagnosed in the neonatal period, and most of them are ignored and missed. The poor muscle tone gradually improves but persists in adulthood. Developmental delay, hyperphagia, rapid weight gain, and many behavioral problems gradually arise (17). PWS is commonly found after the onset of hyperphagia and obesity (18). It is important for clinicians to recognize the clinical continuum at different ages in the early diagnosis of this condition.
The relationship between PWS and PDA has not been well recognized until now. Abduljawad et al. (19) reported that PDA was the second most prevalent congenital heart disease, accounting for 25.5% of a total of 212 syndromic children. However, in their study, there was only one subject diagnosed with PWS. Olander et al. (20) reported a patient with maternal UPD and mosaic trisomy and 15 patients who had a more severe PWS phenotype with high morbidity of congenital heart disease. In this study, we found that three out of seven infants had PDA. Interestingly, there was one PDA infant of the two UPD patients, and the two infants with imprinting defect had PDA.
Infants with PWS are in a bad metabolic condition, often ignored in the early period. Harrington et al. reported that about 12.6% (12/95) of children with PWS develop hypoglycemia, possibly because of adrenal and GH deficiency (21). Ma et al. found five cases with hyperammonemia and two cases with hypoglycemia among nine PWS patients aged 10 days to 11 months (22). In this study, we found two cases of PWS with hypoglycemia and two with hyperammonemia. We speculated that the pathogenesis of hypoglycemia and hyperammonemia may be associated with endocrine metabolic abnormalities.
In this study, eyelid and limb edema were observed in one infant with PWS. Temporary eyelid and pedal edema were also previously reported in an infant case of PWS by Schmeling et al. (23). In fact, adult cases with PWS are often complicated with edema. The (U.S.) Prader-Willi Syndrome Association monitors revealed that edema is present in 16.78% of a total of 1,067 adult cases (24). The cause of the edema is mostly unclear but has frequently been attributed to obesity.
Since obesity-related complications are important causes of death in PWS cases, early diagnosis and careful follow-up are essential for the management of these patients. PWS has no cure so far. Prevention of obesity and appetite control are important therapeutic strategies. Early intervention, strict diet control, and GH therapy can improve quality of life (25). GH is a safe and effective drug to use for PWS treatment, as it promotes carbohydrate and fat metabolism, ensures the normal growth and development of children with PWS, controls body mass index, and improves cognitive function. It is often prescribed from 4 to 6 months old to adulthood (26). The annual mortality of PWS was about 3%, which is much higher compared with the general population. Respiratory failure caused by obesity has been reported to be the main cause of death in PWS patients (27). The treatment of PWS requires a multidisciplinary management involving rehabilitation, as well as endocrine, genetic, metabolic, psychological, and nutrition therapies. Different treatments have been adopted depending on the age group and endocrine disorders (28). These mainly include dietary behavior, nutrition management, management of cryptorchidism, sex hormone replacement therapy in adolescent patients, and use of GH (29). In our study, GH treatment started when the infant was 6 months old in order to increase the growth rate, reduce fat accumulation, improve muscle mass and strength, promote caloric consumption and protein synthesis, and increase bone density. GH is beneficial to improve the prognosis of PWS (30). Recently, it was found that topiramate may be helpful in the treatment of behavioral disorders. Topiramate had a significant effect on eating disorders, with a dose-effect relationship (31). Excitingly, CRISPR (clustered regularly interspaced short palindromic repeats) therapy and oligonucleotide therapy are now in the clinical validation phase of the American Food and Drug Administration (32).
The clinical features of PWS change with age. It is necessary for pediatricians to properly identify this disease. PWS is often misdiagnosed in neonates because of its nonspecific clinical manifestations. Pediatricians should recognize PWS in neonates with central hypotonia, feeding difficulty, and weak crying. It is advised to use a genetic method such as MS-MLPA in neonates with central hypotonia and feeding difficulties. Characteristic facial features and genital hypoplasia are also important features of PWS. Patent ductus arteriosus, hypoglycemia, hyperammonemia, and idiopathic edema may also be present in PWS patients. PWS can be promptly diagnosed in the neonatal period if proper attention is given to neonatal clinical features. Early diagnosis contributes to improve prognosis, and genetic testing is an effective diagnostic tool. The limitations of the study include the limited number of cases and the retrospective observational study design; therefore, further investigation are needed to confirm our results.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of the Shengjing Hospital of China Medical University (Protocol no. KYCS2019404). Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
YH collected and analyzed the clinical data. XX searched literature. JF designed the study and was the major contributor in writing the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Prader A. Adrenogenital syndrome, adrenogenital salt deficiency syndrome and Cushing's syndrome in childhood. Schweiz Med Wochenschr. (1956) 12:289–99.
2. Crinò A, Fintini D, Bocchini S, Grugni G. Obesity management in Prader-Willi syndrome: current perspectives. Diabetes Metab Syndr Obes. (2018) 11:579–93. doi: 10.2147/DMSO.S141352
3. Cheon CK. Genetics of Prader-Willi syndrome and Prader-Will-Like syndrome. Ann Pediatr Endocrinol Metab. (2016) 3:126–35. doi: 10.6065/apem.2016.21.3.126
4. Hurren BJ, Flack NA. Prader-Willi syndrome: a spectrum of anatomical and clinical features. Clin Anat. (2016) 5:590–605. doi: 10.1002/ca.22686
5. Khan MJ, Gerasimidis K, Edwards CA, Shaikh MG. Mechanisms of obesity in Prader-Willi syndrome. Pediatr Obes. (2018) 1:3–13. doi: 10.1111/ijpo.12177
6. Cassidy SB, Schwartz S, Miller JL, Driscoll DJ. Prader-Willi syndrome. Genet Med. (2012) 14:10–26. doi: 10.1038/gim.0b013e31822bead0
7. Angulo MA, Butler MG, Cataletto ME. Prader-Willi syndrome: a review of clinical, genetic, and endocrine findings. J Endocrinol Invest. (2015) 12:1249–63. doi: 10.1007/s40618-015-0312-9
8. Butler MG, Manzardo AM, Forster JL. Prader-Willi syndrome: clinical genetics and diagnostic aspects with treatment approaches. Curr Pediatr Rev. (2016) 2:136–66. doi: 10.2174/1573396312666151123115250
9. Botezatu A, Puiu M, Cucu N, Diaconu CC, Badiu C, Arsene C, et al. Comparative molecular approaches in Prader-Willi syndrome diagnosis. Gene. (2016) 2 (Pt 1):353–8. doi: 10.1016/j.gene.2015.08.058
10. Cassidy SB, Forsythe M, Heeger S, Nicholls RD, Schork N, Benn P, et al. Comparison of phenotype between patients with Prader-Willi syndrome due to deletion 15q and uniparental disomy 15. Am J Med Genet. (1997) 4:433–40.
11. Ge MM, Gao YY, Wu BB, Yan K, Qin Q, Wang HJ, et al. Relationship between phenotype and genotype of 102 Chinese newborns with Prader-Willi syndrome. Mol Biol Rep. (2019) 46:4717–24. doi: 10.1007/s11033-019-04916-2
12. Çizmecioglu FM, Jones JH, Paterson WF, Kherra S, Kourime M, McGowan R, et al. Neonatal features of the Prader-Willi syndrome; the case for making the diagnosis during the first week of life. J Clin Res Pediatr Endocrinol. (2018) 3:264–73. doi: 10.4274/jcrpe.0029
13. Harris SR. Congenital hypotonia: clinical and developmental assessment. Dev Med Child Neurol. (2008) 12:889–92. doi: 10.1111/j.1469-8749.2008.03097.x
14. Gillessen-Kaesbach G, Gross S, Kaya-Westerloh S, Passarge E, Horsthemke B. DNA methylation based testing of 450 patients suspected of having Prader-Willi syndrome. J Med Genet. (1995) 2:88–92. doi: 10.1136/jmg.32.2.88
15. Tuysuz B, Kartal N, Erener-Ercan T, Guclu-Geyik F, Vural M, Perk Y, et al. Prevalence of Prader-Willi syndrome among infants with hypotonia. J Pediatr. (2014) 5:1064–7. doi: 10.1016/j.jpeds.2014.01.039
16. Belengeanu D, Bratu C, Stoian M, Motoc A, Ormerod E, Podariu AC, et al. The heterogeneity of craniofacial morphology in Prader-Willi patients. Rom J Morphol Embryol. (2012) 3:527–32.
17. Sanjeeva GN, Maganthi M, Kodishala H, Marol RKR, Kulshreshtha PS, Lorenzetto E, et al. Clinical and molecular characterization of Prader-Willi syndrome. Indian J Pediatr. (2017) 11:815–21. doi: 10.1007/s12098-017-2386-1
18. Muscogiuri G, Formoso G, Pugliese G, Ruggeri RM, Scarano E, Colao A, et al. Prader-Willi syndrome: an uptodate on endocrine and metabolic complications. Rev Endocr Metab Disord. (2019) 2:239–50. doi: 10.1007/s11154-019-09502-2
19. Abduljawad EM, AlHarthi A, AlMatrafi SA, Hussain M, Shawli A, Waggass R. The prevalence of congenital heart diseases in syndromic children at King Khalid National Guard Hospital from 2005 to 2016. Cureus. (2020) 4:e7891. doi: 10.7759/cureus.7891
20. Olander E, Stamberg J, Steinberg L, Wulfsberg EA. Third Prader-Willi syndrome phenotype due to maternal uniparental disomy 15 with mosaic trisomy 15. Am J Med Genet. (2000) 3:215–8. doi: 10.1002/1096-8628(20000731)93:3<215::AID-AJMG11>3.0.CO;2-K
21. Harrington RA, Weinstein DA, Miller JL. Hypoglycemia in Prader-Willi syndrome. Am J Med Genet A. (2014) 5:1127–9. doi: 10.1002/ajmg.a.36405
22. Ma YY, Wu TF, Liu YP, Wang Q, Song JQ, Song F, et al. Nutritional and metabolic findings in patients with Prader-Willi syndrome diagnosed in early infancy. J Pediatr Endocrinol Metab. (2012) 11–12:1103–9. doi: 10.1515/jpem-2012-0167
23. Schmeling H, Gillessen-Kaesbach G, Schulte-Mattler U, Burdach S, Horneff G. Prader-Labhart-Willi syndrome in infants. Klin Padiatr. (2002) 2:51–3. doi: 10.1055/s-2002-25265
24. Manzardo AM, Heinemann J, McManus B, Loker C, Loker J, Butler MG. Venous thromboembolism in Prader-Willi syndrome: a questionnaire survey. Genes. (2019) 7:550. doi: 10.3390/genes10070550
25. Passone CBG, Pasqualucci PL, Franco RR, Ito SS, Mattar LBF, Koiffmann CP, et al. Prader-willi syndrome: what is the general pediatrician supposed to do? -A review. Rev Paul Pediatr. (2018) 3:345–52. doi: 10.1515/JPEM.1991.4.3.167
26. Stipančić G, PoŽgaj Šepec M, La Grasta Sabolić L. Effect of growth hormone therapy in children with prader-willi sndrom - our first experimences. Acta Clin Croat. (2018) 4:744–55. doi: 10.20471/acc.2018.57.04.17
27. Butler MG, Manzardo AM, Heinemann J, Loker C, Loker J. Causes of death in Prader-Willi syndrome: Prader-Willi Syndrome Association (USA) 40-year mortality survey. Genet Med. (2017) 6:635–42. doi: 10.1038/gim.2016.178
28. Luo F, Luo X. The future perspectives of clinical diagnosis and management of genetic diseases: lessons learned from the current status of Prader-Willi syndrome in China. Zhonghua Er Ke Za Zhi. (2015) 6:409–11.
29. East N, Maroney M. Topiramate in the treatment of Prader-Willi syndrome: a case report. Ment Health Clin. (2018) 1:7–9. doi: 10.9740/mhc.2017.01.007
30. Bridges N. What is the value of growth hormone therapy in Prader Willi syndrome? Arch Dis Child. (2014) 2:166–70. doi: 10.1136/archdischild-2013-303760
31. Consoli A, Çabal Berthoumieu S, Raffin M, Thuilleaux D, Poitou C, Coupaye M, et al. Effect of topiramate on eating behaviours in Prader-Willi syndrome: TOPRADER double-blind randomised placebo-controlled study. Transl Psychiatry. (2019) 1:274. doi: 10.1038/s41398-019-0597-0
Keywords: clinical features, genetic testing, infant, neonate, Prader-Willi syndrome
Citation: Hu Y, Xue X and Fu J (2021) Case Report: Clinical Analysis of Seven Neonates With Prader-Willi Syndrome and Review of the Literature. Front. Pediatr. 9:633532. doi: 10.3389/fped.2021.633532
Received: 25 November 2020; Accepted: 01 February 2021;
Published: 18 February 2021.
Edited by:
Anup C. Katheria, Sharp Mary Birch Hospital for Women & Newborns, United StatesReviewed by:
Rachel Kai-Xin Yim, University of California, San Diego, United StatesPhoebe Jen, Sharp Mary Birch Hospital for Women & Newborns, United States
Copyright © 2021 Hu, Xue and Fu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: JianHua Fu, ZnVqaDE4MzBAMTYzLmNvbQ==