 Debora M. d'Angelo1
Debora M. d'Angelo1 Paola Di Filippo
Paola Di Filippo Francesco Chiarelli
Francesco Chiarelli
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pediatr. , 31 March 2021
Sec. Pediatric Rheumatology
Volume 9 - 2021 | https://doi.org/10.3389/fped.2021.631329
Notable advances in gene sequencing methods in recent years have permitted enormous progress in the phenotypic and genotypic characterization of autoinflammatory syndromes. Interferonopathies are a recent group of inherited autoinflammatory diseases, characterized by a dysregulation of the interferon pathway, leading to constitutive upregulation of its activation mechanisms or downregulation of negative regulatory systems. They are clinically heterogeneous, but some peculiar clinical features may lead to suspicion: a familial “idiopathic” juvenile arthritis resistant to conventional treatments, an early necrotizing vasculitis, a non-infectious interstitial lung disease, and a panniculitis associated or not with a lipodystrophy may represent the “interferon alarm bells.” The awareness of this group of diseases represents a challenge for pediatricians because, despite being rare, a differential diagnosis with the most common childhood rheumatological and immunological disorders is mandatory. Furthermore, the characterization of interferonopathy molecular pathogenetic mechanisms is allowing important steps forward in other immune dysregulation diseases, such as systemic lupus erythematosus and inflammatory myositis, implementing the opportunity of a more effective target therapy.
In the last 20 years, remarkable progress has been made on the phenotypic and genotypic characterization of autoinflammatory disorders (1, 2). The onset of an autoinflammatory phenotype early in infancy is often linked to a monogenic autoinflammatory syndrome, whose diagnosis is currently a challenge for pediatricians (2). Interferonopathies represent the most recent and increasingly featured chapter among these pathologies. The term “interferonopathy” first appeared in 2003, when some authors identified phenotypic overlaps between Aicardi–Goutieres syndrome (AGS) encephalopathy, viral congenital infections, and some autoimmune diseases such as systemic lupus erythematosus (SLE), postulating a common pathological feature as an upregulation of interferon (IFN) α activity (3). Indeed, AGS was originally defined as pseudo-TORCH (toxoplasmosis, rubella, cytomegalovirus, and herpes) syndrome, identifying a group of serologically negative disorders that mimic congenital TORCH infections, suggesting a similar pathogenetic mechanism (3, 4). In the first decade of the 2000s, the genes underlying AGS and monogenic forms of SLE were partially identified and in both cases were associated with type I IFN signaling, confirming the involvement of a common pathway (4–7). In the meantime, the role of nucleic acids and the activation of intracellular receptors for the production of type I IFN by virus-infected cells was identified (4). These considerations led to the definition of a new group of mendelian disorders, characterized by the mutation of single genes involved in the type I IFN signaling: the interferonopathies (8). Further genetic analysis of these patients showed mutations in genes involved in several overlapping pathways: the pathogenesis, still largely to be characterized, lies in alterated response to nucleic acid stimuli or defective regulation of downstream effector molecules (8–10). Despite clinical heterogeneity, some phenotypic characteristics in this group of diseases are shared: early onset of the skin vasculopathy with chilblains, livedo reticularis and panniculitis, involvement of central nervous system (CNS), and interstitial lung disease are the most represented (2, 4, 11). These clinical manifestations represent the “clinical IFN signature.”
In 1957, Isaacs and Lindenmann described a soluble factor in supernatant of cells incubated with influenza virus, able to “interfere” with cell viral infection in vitro; they called this factor “interferon” (12). IFN was subsequently isolated and characterized, showing important biological properties such as inhibition of viral growth and cell multiplication and many immunomodulatory activities (2, 4, 13). Three types of IFNs are described: type I IFN primarily participates in the innate immune system response to viral antigens; type II or IFN-gamma mainly activates Th1 and B lymphocyte adaptive system response; and type III or IFN-lambda has antiviral and modulating properties (13, 14). Precisely, type I IFNs are powerful inflammatory polypeptides, ubiquitously expressed by immune and non-immune cells, including macrophages, lymphocytes, dendritic cells, fibroblasts, and hematopoietic plasmacytoid dendritic cells. This family of cytokines are all induced by microbial and viral nucleic acids, and it consists of the predominantly produced interferon-α (13 variants) and interferon-β and other less frequent variants such as IFN-ε, -κ, -τ, and -ζ (14–20). Indeed, virus infection leads to an intracytoplasmic accumulation of viral double-stranded (ds) RNA or DNA. This surplus of nucleic acids exceeded the activation threshold and is sensed in the cytoplasm or endosomes by different pattern recognition receptors as Toll-like receptors (TLRs), RIG-I-like receptors (RLRs), NOD-like receptors (NLRs), and other cytoplasmic DNA receptors such as the cyclic GMP-AMP synthase (cGAS) (16–21). In particular, intracellular dsDNA interacts with the enzyme cGAS, activating the production of cyclic 2′3′GMP-AMP (cGAMP). In the endoplasmic reticulum (ER), cGAMP binds and activates the STimulator of INterferon Genes (STING), which translocates into the Golgi apparatus. Here, STING C-terminal tail (CTT) recruits TANK-binding kinase 1 (TBK1), which phosphorylates serine 365 of CTT. Phospho-S365 induces the coupling and the subsequent phosphorylation by TBK1 of IFN regulatory factors 3 (IRF3), which translocates to the nucleus and induces the transcription of IFN-β and of IFN regulatory factors 7 (IRF7), which is responsible for IFN-α secretion and autocrine signaling amplification (22).
Another pathway for IFN activation includes three cytosolic RNA helicases: the RIG-I-like helicases (RLHs), the retinoic acid inducible gene-I (RIG-I), and the melanoma differentiation-associated gene 5 (MDA5), which recognize viral dsRNA in the cytoplasm. RIG-I and MDA5 activate downstream IKK epsilon and TBK1, which phosphorylate and activate IRF3 and IRF7, allowing IFNα and β gene transcription (23). Therefore, type I IFNs, produced by the infected cells and released in the extracellular environment, perform an autocrine and paracrine action binding to the heterodimeric interferon receptors (IFNRs), which are constituted by the subunits IFN-α receptor 1 (IFNAR1) and IFNAR2. IFN bound to its receptor induces dimerization of these two subunits, which recruit JAK1 (Janus kinase 1) and TyK2 (tyrosine kinase 2) proteins. This activation further promotes the STAT1–STAT2 dimerization and the recruitment of IFN regulatory factors 9 (IRF9) to assemble the heterotrimeric transcription complex interferon-stimulated gene factor 3 (ISGF3). In the nucleus, ISGF3 binds to IFN-stimulated response elements (ISRE) promoting the expression of interferon-stimulated genes (ISGs) (14, 20, 24). ISG expression controls chemotaxis, cell migration, apoptosis, and cell proliferation, and it regulates immune detection and defense against infections (20). Moreover, a negative feedback mechanism finely regulates signal transduction through the IFN receptor: the ubiquitin-specific peptidase 18 (USP18)–interferon-stimulated gene 15 (ISG15) system. The USP18 binds the IFNAR2 subunit, decoupling it from JAK1 and inhibiting the propagation of the next signal. These events induce a refractory state in the cells with reduced sensitivity to further stimulation. ISG15 is an IFN-induced protein that extracellularly stimulates the production of type II IFN, and intracellularly it binds lysine residues of proteins (ISGylation); this process can be reversed by USP18. In addition, the unconjugated ISG15 form prevents the degradation of UBS18 by the S-phase kinase-associated protein 2 (SPK2), activating a negative regulation of the IFNR signaling (25, 26). Moreover, IFNR activation stimulates the transcription of the suppressor of cytokine signaling 1 (SOCS1), activating another negative feedback mechanism. Indeed, SOCs 1 binds TyK2, reducing IFN signal transduction and, simultaneously, the surface expression of IFNR1, which is stabilized by Tyk2 (27). The IFN pathway described above is illustrated in Figure 1.
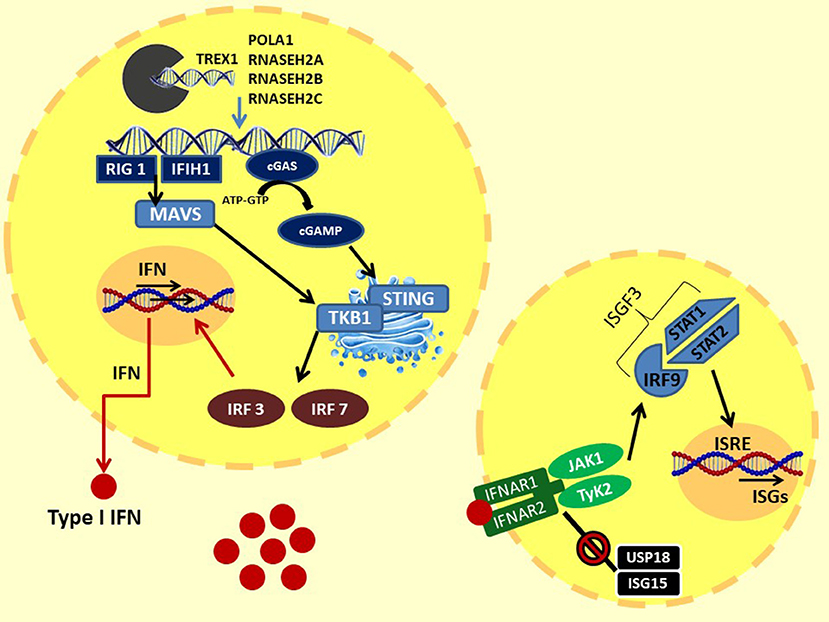
Figure 1. Type I IFN signaling: the intracytoplasmatic accumulation of viral (or endogenous due to loss of function of the enzymes responsible for degradation) nucleic acids is sensed by two systems: (1) the cytoplasmic DNA receptors cyclic GMP-AMP synthase cGAS: intracellular dsDNA activate cGAS, leading to the production of cGAMP. cGAMP binds and activates STING in ER, which translocates into the Golgi apparatus. Here, the STING C-terminal tail recruits TBK1. TBK1 induces the coupling and the phosphorylation of IFR3 and, consequently, IFR7. (2) The cytosolic RNA helicases RIG-I–MDA5 system: intracellular dsRNA activates MDA5 and RIG I, which bind and activate MAVS in the mitochondrial membrane, forming the MAVS signaling complex. MAVS triggers downstream TBK1, than activating IRF3 and IRF7. Finally, IRF3 and IRF7 translocate to the nucleus and induce the transcription of IFN-β and IFN-α, respectively. Type I IFNs, through autocrine and paracrine action, bind to IFN receptors. The IFNR dimerization recruits JAK1 and TyK2 proteins. This activation promotes the STAT1–STAT2 dimerization and the binding of IRF9 to assemble the heterotrimeric transcription complex ISGF3. In the nucleus, ISGF3 binds to IFN-stimulated response elements (ISRE), promoting the expression of interferon-stimulated genes (ISGs). At the same time, IFN signaling is regulated by a negative feedback mechanism by the USP18–ISG15 system. The USP18 binds the IFNAR2 subunit, decoupling it from JAK1 and inhibiting the propagation of the next signal. ISG15 prevents the degradation of UBS18 by SPK2. TREX1, DNA 3′–repair exonuclease 1; RNASEH2, ribonuclease H2; POLA1, polymerase-α; cGAS, GMP-AMP synthase; cGAMP, 2′3′GMP-AMP; STING, STimulator of INterferon Genes; TBK1, TANK-binding kinase 1; MAVS, mitochondrial antiviral-signaling protein; IRF3, IFN regulatory factors 3; IRF7, IFN regulatory factors; RIG-I, retinoic acid-inducible gene-I; MDA5, melanoma differentiation-associated gene 5; JAK1, Janus kinase 1; TyK2, tyrosine kinase 2; IRF9, IFN regulatory factors 9; ISGF3, interferon-stimulated gene factor 3; USP18, ubiquitin-specific peptidase 18; ISG15, interferon-stimulated gene 15; SPK2, S-phase kinase-associated protein 2.
In conclusion, dysregulation of type I IFN signaling can occur through at least five mechanisms: (I) cytosolic accumulation of endogenous nucleic acids by loss-of-function mutations of genes that encode for enzymes responsible for DNA or DNA–RNA hybrid molecule degradation; (II) intracytosolic nucleic acid sensor alteration and following activation of the signal below the threshold; (III) gain-of-function mutations of positive IFN signaling regulators, leading to constitutive activation of the IFN pathway; (IV) loss-of-function mutations of negative IFN signaling regulators; (V) proteasomal dysfunction leading to increased IFN signaling through a mechanism being defined (2, 4). The role of mutations of genes encoding osteopontin activity proteins (TRAP/ACP5) and complement factors in interferonopathies is still not yet clarified (2). These mechanisms are summarized in Figure 2. Very recently, biallelic mutations of two components of the histone pre-mRNA processing complex were implicated: the U7 snRNA-associated Sm-like protein (LSM11) and the RNA U7 Small Nuclear 1 (RNU7-1). Such gene alterations are responsible for a modified stoichiometric conformation of the histones and an altered chromatin formation, with dysregulation of the cGAS-STING activity. These findings highlight further pathways of constitutive activation of IFN, which need to be investigated (28). The main genes involved and their role in the pathogenesis of interferonopathies are summarized in Table 1.
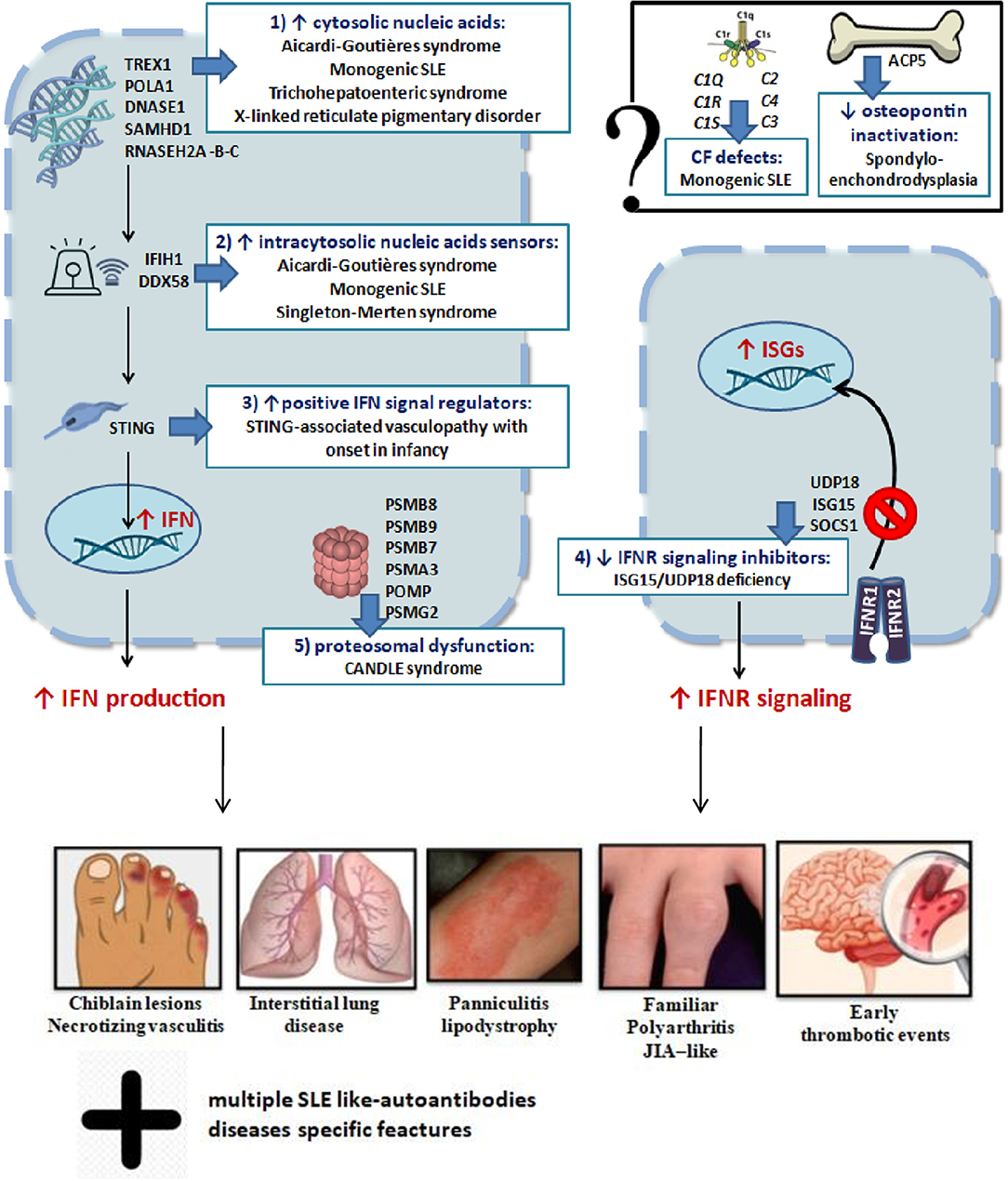
Figure 2. Schematic representation of genetic mechanisms and clinical manifestations in main interferonopathies (see in the text).
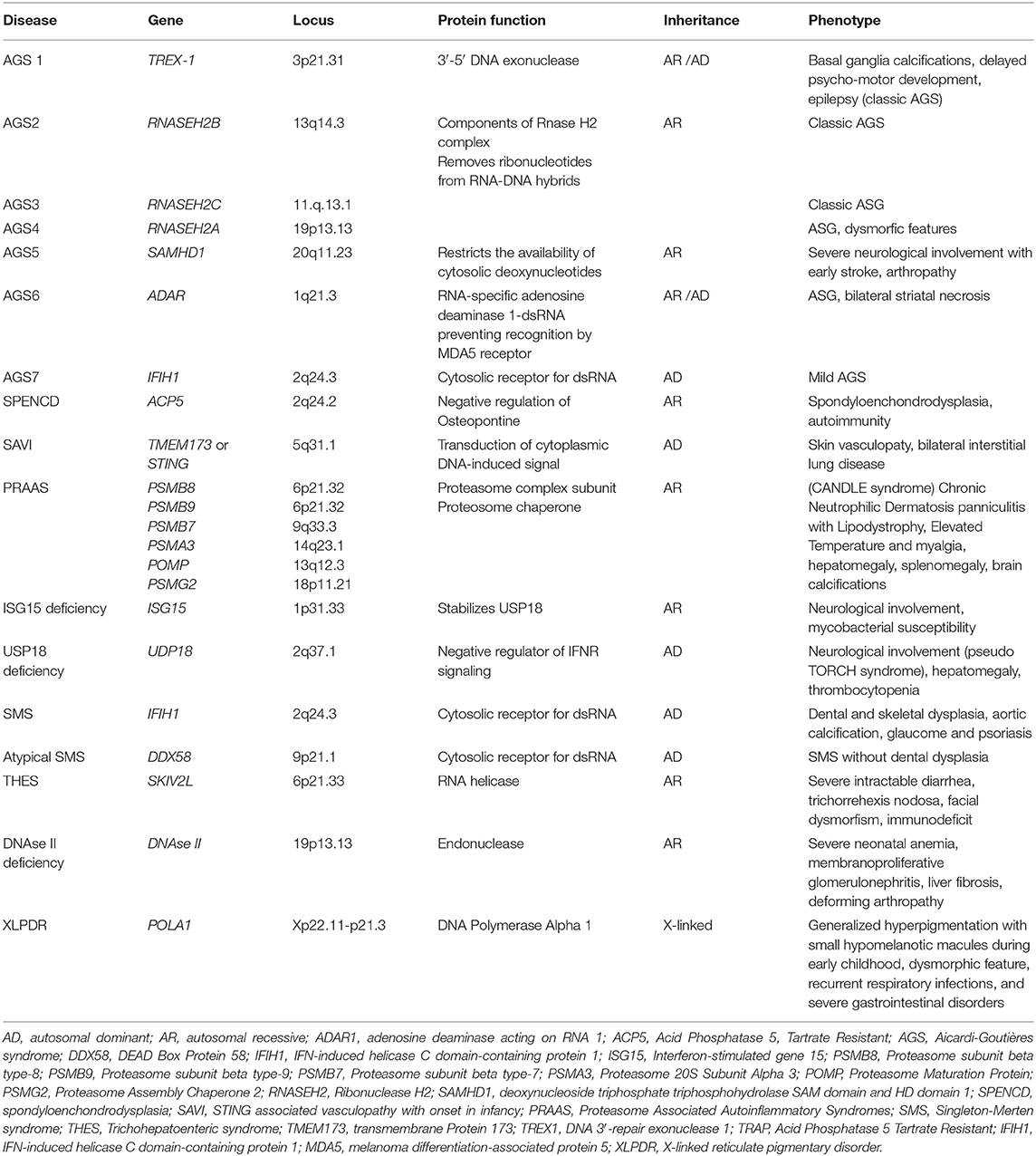
Table 1. Mutated gene and corresponding locus, protein function, pattern of inheritance, and common symptoms of main type I interferonopathies.
Neurological involvement is a prominent feature of most interferonopathies, especially with specific mutations (Table 1) (29, 30). Recent studies showed that IFN type I is implicated in regulating microglial function, both during development and in response to neuroinflammation, ischemia, and neurodegeneration (30, 31). The microglia is the local component of the CNS innate immune system, and it plays a central role in the modulation of CNS inflammatory homeostasis. Microglia, indeed, is essential in phagocytizing myelin debris and providing support to oligodendrocytes and axons, also during synaptogenesis. Disruption in microglial activation may result in detrimental effects within white matter (30–32). Therefore, a disruption in the fine balance between required clearance and excessive phagocytosis is the basis of neurodegenerative pathologies. Type I IFN plays a significant role in regulating this balance.
The effects of IFN on CNS and microglia seem to be dual and opposite (31). Experimental murine models on multiple sclerosis (MS), autoimmune encephalomyelitis, and stroke demonstrated that IFNβ (produced by microglia) has local modulatory effects on myelin inflammation, by increasing phagocytosis (including T cells phagocytosis) and promoting the blood–brain barrier integrity. These data suggest a therapeutic role of IFN in these neurodegenerative diseases (33–35, 35, 36). On the other hand, it was demonstrated that a chronic upregulation of type I IFN signaling by microglia may be injurious and this effect may be greater in white matter (31). Dysregulation of the IFN pathway in mouse knockout for the USP18 gene led to a severe neurological phenotype with seizures, tremor(s), and early mortality (37, 38). Pathological brain evaluation highlighted hydrocephalus with basal ganglia calcifications and clusters of microglia in the white matter which strongly resembled the neuropathological pattern of several human microgliopathies (37, 38). The evaluation of USP18-deficient human patients confirmed the same neuroinflammatory phenotype, with the same astrocyte and microglial activation (25, 39). The susceptibility of nervous tissue to IFN insult seems to be prominent in the fetal CNS, when the process of neurogenesis, synaptogenesis, synaptic pruning, and myelization is paramount (30).
This condition reflects the very early onset of ganglion calcifications, epilepsy, and psychomotor retardation in some interferonopathies, such as AGS (29, 40). Moreover, recent studies suggest that many neuropathies, including Alzheimer's disease and MS, can be considered “microgliopathies,” and the microglia and the IFN signaling pathway may represent a promising therapeutic target (30, 41, 42).
Genetic mutations and clinical syndromes associated with interferonopathies are continuously and rapidly evolving. The first diseases identified as type I interferonopathies were Aicardi–Goutières syndrome (AGS), the spondyloenchondrodysplasia (SPENCD), and the monogenic forms of systemic lupus erythematosus (SLE) (4). According to the 2017 classification of the International Union of Immunological Societies (IUIS), 13 type I interferonopathies were identified. In addition to those initially described, other monogenic syndromes were recognized, including proteasome-associated autoinflammatory syndromes (PRAAS), ISG15 deficiency, Singleton–Merten syndrome (SMS), and STING-associated vasculopathy with onset in infancy (SAVI) (43, 44). Clinical and pathogenetic characteristics of the main interferonopathies are described below.
AGS is a progressive encephalopathy with onset within the first year of life, characterized by basal ganglia calcifications, chronic cerebrospinal fluid (CSF) lymphocytosis, and elevated type I IFN levels in the CSF without evidence of any congenital infection (2, 45). The currently recognized genetic mutations associated with AGS are seven (Table 1), and they led to the identification of seven subgroups of the disease (AGS 1–7). The autosomal recessive forms are caused by mutations of genes encoding for intracellular nucleic acids degradation enzymes: 3′-5′ DNA exonuclease -TREX1 (AGS1), ribonucleases as the RNASEH2B (AGS2), RNASEH2C (AGS3), and RNASEH2A (AGS4), SAM and HD domain-containing deoxynucleoside triphosphate triphosphohydrolase 1-SAMHD1 (AGS5), and adenosine deaminase acting on RNA 1-ADAR1 (AGS6). The autosomal dominant forms are mainly caused by mutations of genes encoding for the intracellular RNA sensor IFN-induced helicase C domain-containing protein 1-IFIH1 (AGS7) (2, 5, 46–52). A less severe phenotype was reported in patients with ISG15 gene mutations (51).
AGS is a clinically heterogeneous disorder. Two forms of AGS were described: in the early-onset form, psychomotor development delay and liver abnormalities are evident from birth; in the later-onset AGS, after the first weeks or months of normal development, symptoms appear as a progressive decline in head growth, spasticity, and cognitive and developmental delay that range from moderate to severe. In addition, abnormal eye movements, nystagmus, glaucoma, and poor visual behavior are often associated (5, 47–52); a peculiar characteristic is a “startle reactions,” in response to even minimal sensory stimuli (53). Intracranial calcifications, white matter destruction, and brain atrophy are evident on neuroimaging, similar to radiological findings seen in congenital infections. Epileptic seizures are present in a variable percentage from 10–30% to 53–75%. In children with the late-onset form, as symptoms lessen, clinical manifestations stabilize with no further worsening of the disease (54). Stroke and cerebral aneurysms are common manifestations in patients affected by mutation of SAMHD1 gene (55). Extraneurological symptoms mainly involve the skin (35% of cases): chilblain-like lesions, characterized by areas of inflammation and necrosis secondary to peripheral inflammatory vasculopathy, are localized mainly in fingers and toes or in the auricles, mostly in the cold months. Other common alterations include thrombocytopenia, hepatosplenomegaly, elevated transaminases, psoriasis, interstitial lung disease, and intermittent fever, which erroneously cause a misdiagnosis with acute CNS infections (meningitis, encephalitis) (45, 46, 48, 50). An autoimmune SLE-like habitus associated with type 1 diabetes mellitus, hypothyroidism, polygammaglobulinemia, and hemolytic anemia has been often described (56). Furthermore, arthropathy with progressive contractures were reported only in children with SAMHD1 gene mutations (57).
SPENCD is a rare autosomic recessive disorder characterized by multiple skeletal dysplasia, neurological involvement, and immune dysfunction with an increased IFN type I signature in peripheral blood and urine, due to mutations of tartrate-resistant acid phosphatase (TRAP) gene (ACP5) (58, 59). TRAP enzyme plays a role in inhibiting the activity of osteopontin (Opn), a protein involved in bone metabolism and immune system function modulation. Opn is expressed by osteoclasts and osteoblasts, and it stimulates bone remodeling and turnover. In addition, it has been shown that secreted Opn acts as a “cytokine” that polarizes lymphocytic differentiation in Th1, promoting macrophage production of IL12. ACP5 gene mutations impair or eliminate TRAP ability to inactivate Opn (58). The role of Opn in modulating type I IFN production is still being defined. Some studies showed that intracellular expression of Opn in plasmacytoid dendritic cells was required for TLR9-dependent expression of IFN-α and colocalization of Opn was essential for efficient nuclear translocation of IRF7 and associated IFNα gene expression. In addition, Opn-deficient mice developed impaired IFNα-dependent natural killer cell responses to tumors and reduced IFN-α responses after infection with herpes simplex virus 1 (HSV-1) (60). Consequently, bone dysplasias with enchondromatosis, platyspondyly and irregularity of vertebral endplates and short stature, brain calcifications with spasticity and autoimmune disorders, including SLE with malar rash, anti-phospholipid syndrome, and glomerulonephritis are the main clinical manifestations (58, 60, 61). Sjögren's syndrome, hemolytic anemia, thrombocytopenia, hypothyroidism, inflammatory myositis, Raynaud's disease, and vitiligo were also described (58, 59).
Increased IFNα serum level in SLE patients was widely demonstrated (62–66). Moreover, increased damage or death of cells and defective nucleic acid metabolism and the consequent production of autoantibodies against nucleic acids are characteristic of SLE. Accordingly, accumulation of nucleic acids may activate the same pathways involved in interferonopathies with an overproduction of IFN, suggesting an overlap between type I interferonopathies and SLE (67, 68). Recently, childhood-onset SLE was linked to single-gene mutations, defining monogenic or familiar SLE. This evolving monogenic mutations can be grouped into four classes: (I) complement factors, (II) enzymes involved in the endogenous metabolism of nucleic acids (extracellular DNASE), (III) proteins directly involved in IFN type I pathway, and (IV) factors involved in the regulation of B and T lymphocyte self-tolerance (69–71). Main genes involved in these forms are summarized in Table 2.
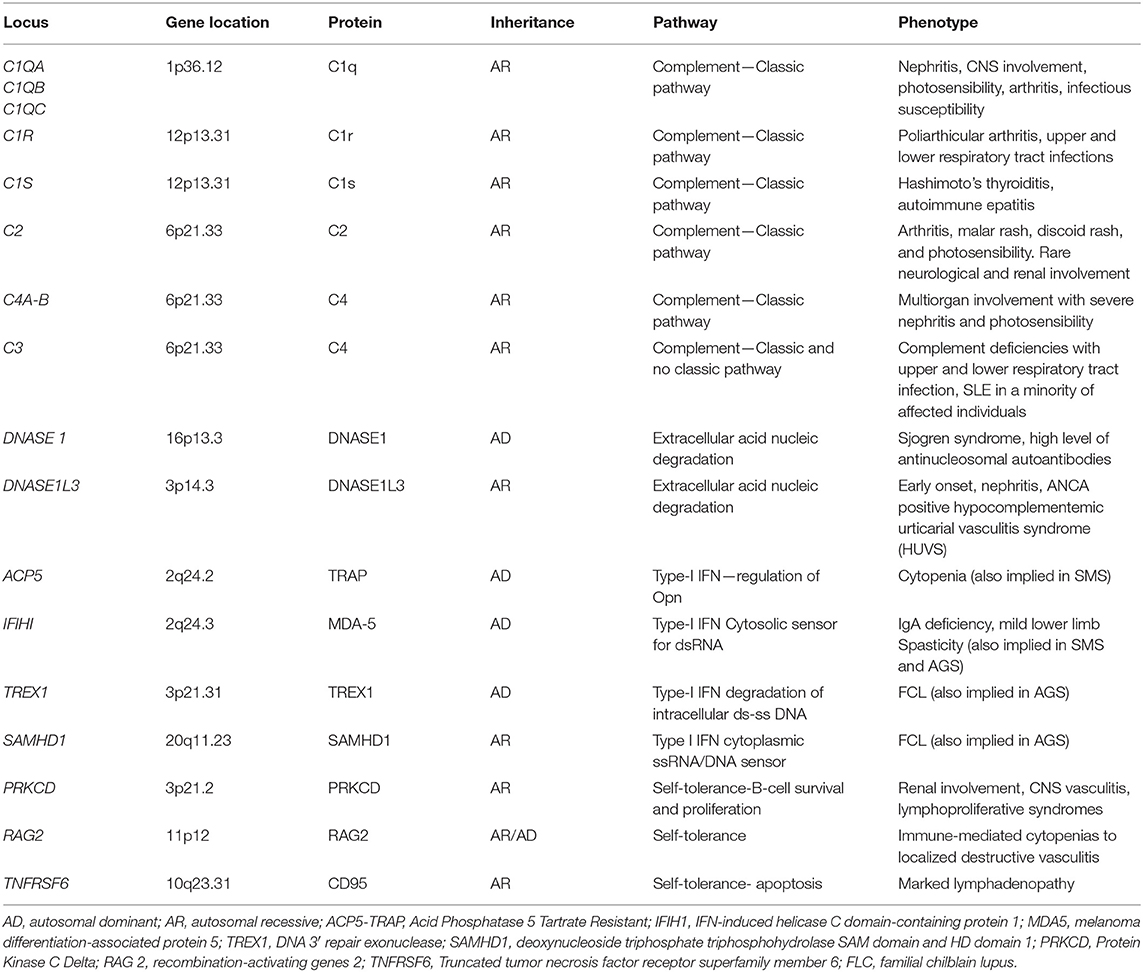
Table 2. The main altered genes described in the monogenic forms of SLE and corresponding phenotype.
Complement protein defects are responsible for autosomal recessive forms of juvenile onset-SLE. C1q deficiency is the strongest risk factor known for the development of SLE, followed by C4A defects. The link between complement protein deficiency and IFN type I upregulation is currently under investigation. A defect of the opsonization of apoptotic self-bodies and, therefore, of the cellular clearance and the “efferocytosis” is considered the main causative factor (69). In addition, it was demonstrated that C1q in vitro inhibits peripheral blood mononuclear cell (PBMCs) and plasmacytoid dendritic cell IFNα production, thus hypothesizing a direct C1q regulatory role (72). The clinical phenotype is represented by an early onset of SLE with frequent early renal glomerular involvement and increased susceptibility to pyogenic infections (especially upper and lower tract respiratory infections) (2, 69). In addition, complement 1r subcomponent (C1r) mutations were described, with a clinical phenotype characterized by prominent neurological involvement and lymphadenopathy (73).
The role of endonuclease gene defects in the development of early SLE was demonstrated. In the early 2000s, some authors highlighted that reduced serum endonuclease DNASE1 activity was linked to the development of autoantibodies (especially anti-nucleosomal autoantibodies) and active SLE disease in patients and mice (74–76). The deficiency of DNASE1L3, a homolog of DNASE1, was first described in 2011 as a pediatric-onset SLE correlated with lupus nephritis (77). Recently, it was demonstrated that DNASE1L3 acts extracellularly and it plays a primary role in digesting genomic DNA circulating microparticles derived from apoptotic cells. Indeed, these DNA microparticles were targeted by autoantibodies in the serum of DNASE1L3-deficient mice and patients and the treatment with exogenous DNASE1L3 inhibited antibody binding. These findings suggest a central role of DNASI1L3 in immune tolerance regulation and in autoantibodies development in SLE (78, 79). Furthermore, the role of another endonuclease, DNAse 2, was recently described. Biallelic mutations in DNASE2 gene were associated with a clinical picture ranging from membrano-proliferative glomerulonephritis and liver fibrosis to severe neonatal anemia (80).
TREX1 is a genetic locus encoding for a form of 3′-5′ DNA exonuclease associated with monogenic SLE (81–85). Single-nucleotide polymorphisms (SNPs) in TREX1 were found associated with common forms of SLE (83–85). Early-onset familial chilblain lupus (FCL) is due to an autosomal dominant mutation of TREX1. It is a rare form of cutaneous SLE characterized by cold-induced severe vasculitic ulcerative lesions of (fingers, toes) and ears (81, 82). In addition, mutations in TREX1 with autosomal dominant inheritance were related to retinal vasculopathy with cerebral leukodystrophy (RVCL). Type I IFN signature was described in these patients. RVCL is characterized by middle-age onset of progressive visual loss due to retinal vasculopathy (telangiectasias, microaneurysms, retinal macular vascular obliteration) and neurologic manifestations including hemiparesis, facial paralysis, aphasia, and hemianopsia up to psychiatric disorders (86). Besides, a homozygous variant of this gene was identified in patients with an early-onset cerebral SLE (87). Also, mutations in SAMHD1 have been reported in patients affected by FCL, with and without vascular CNS involvement (88). Among monogenic SLE associated with mutations of genes involved in the IFN signal regulation, in addition to ISG15 and USP18 gene (discussed in other sections), it is important to mention the role of OTU domain-containing protein 1 (OTUD1). OTUD1 gene encodes for a deubiquitinase that interacts with IRF3, removing the poly-ubiquitin chains on IRF3 and suppressing IFN gene transcription. Patients with loss-of-function missense mutations in OTUD1 present with many different autoimmune disease, including early onset-SLE (71).
PRAAS are a heterogeneous group of interferonopathies caused by inherited loss-of-function mutations in genes encoding proteasome subunits of 20S core particles such as PSMB8, PSMB9, PSMB7, and PSMA3 or proteasome chaperone factors such as POMP and PSMG2 (Table 1). These mutations lead to a failure of proteasome complex formation, causing an alteration of intracellular protein homeostasis and the accumulation of ubiquitinated proteins (2, 89–92). The underlying molecular mechanisms that link this alteration to the activation of the IFN pathway are not yet fully understood. Recently, new insights came from the unfolded protein response (UPR), which originated in response to an accumulation of misfolded proteins in the ER (ER stress). Indeed, defective proteasome compromises the ER-associated protein degradation, resulting in an accumulation of misfolded ER proteins in the lumen. This alteration is determined by three intra ER membrane-resident proteins, in particular the inositol-requiring enzyme 1α/β (IRE1), with subsequent activation of a signal pathway that allows NF-κB activation and IRF3 factor transcription, leading to a IFN-depending inflammation (92, 93).
PRAAS include the Japanese autoinflammatory syndrome with lipodystrophy (JASL) and chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature syndrome (CANDLE) syndromes (2). Common clinical features are the presence of pernio-like purplish nodular lesions (neutrophilic dermatosis), panniculitis with progressive lipodystrophy and muscle atrophy, and joint contractures with extremity deformity. Hepatosplenomegaly and hypochromic or hemolytic anemia were also reported. An early metabolic syndrome, with systemic hypertension and dyslipidemia, occurs in 40–80% of patients (89–92). Proteasome defects were also associated with neurologic diseases. In murine models, loss of proteosome subunits led to development of microcephaly and aberrant rostrocaudal patterning of the branchial arches, suggesting a role of some proteasome components in neuronal development (89–93). Indeed, a mild cognitive delay was also described in some CANDLE/PRAAS patients (89).
ISG15 deficiency consists of two clinical overlapping forms: an immunodeficiency phenotype and a type I interferonopathy (2, 94). It was first described in two families from Turkey and Iran as a Mendelian susceptibility to mycobacterial disease (MSMD) (94). Indeed, these mutations alter the leukocyte secretion of ISG15. Inducing IFNγ, ISG15 plays an essential role in optimizing the anti-mycobacterial immune response by Th1 lymphocytes and macrophages. It also downregulates the IFNR signaling pathway by stabilizing USP18 (2, 14). An upregulation of type IFN I, mostly characterized by a neurological phenotype with basal ganglia calcifications and epilepsy, was described (2, 4). Recently, loss-of-function mutations of ISG15 and signs of IFN activation in keratinocytes, skin endothelial cells, and peripheral blood myeloid cells were reported in five patients affected by skin ulcerative lesions in the perineal, groin, scalp, neck, and axillary areas. These findings may suggest another phenotype with a unique pattern of skin localization of vasculopathic inflammation (95).
In 1973, Singleton et al. described a syndrome characterized by dental dysplasia (periodontitis, root resorption), thoracic and aortic calcifications, osteoporosis, and skeletal radiographic changes (distal limb osteolysis, widened medullary cavities) in two children (96). Later, an atypical form of SMS was identified in two families and it was characterized by abnormal valvular and thoracic calcifications and osteoporosis without dental alterations (97). In addition, psoriatic-like lesions and glaucoma are other clinical features described in SMS. Genetic and pathogenetic bases of this disease were identified about 40 years later. The SMS phenotype results in an upregulation of IFN type I due to gain-of-function mutations of the genes IFIH1 (typical form) and DDX58 (atypical form), encoding for 2 intracytoplasmic nucleic acid sensors (MDM2 and RIG-1, respectively), activating the IFN pathway (97–99).
SAVI was first described in 2014 in six children affected by systemic inflammation, severe cutaneous vasculopathy, and early interstitial lung disease. In these patients, heterozygous gain-of-function mutations in the transmembrane protein 173 (TMEM173) gene result in an upregulation of the IFN gene transcription (100).
Skin and lungs are certainly the most involved systems. Telangiectatic lesions on nose and cheek and violaceous atrophic plaques and nodules on hands are typical clinical signs. Capillary tortuosity with subsequent loss of distal capillary vascularity is responsible for painful ulcerative lesions (especially on fingers and toes, ears, and nose). These lesions evolve into eschars, up to digital amputation, ear cartilage reabsorption, perforation of the nasal septum, or dystrophic nail changes. Skin biopsy revealed capillary wall inflammation with fibrin deposits in the lumen and a dense neutrophilic infiltrate with karyorrhexis throughout the vessel wall, in the absence of granulomatous lesions. Interstitial lung disease with fibrosis together with hilar or paratracheal lymphadenopathy is the main responsible of morbidity and mortality (101). Patients experience a progressive hypoxemic respiratory insufficiency, often asymptomatic in early childhood. Lung biopsy highlighted mixed lymphocytic inflammatory infiltrate (100, 101).
Autoantibodies ANA and cANCA were found in some of these patients, resulting in a difficult differential diagnosis with childhood granulomatosis and polyangiitis (102). Failure to thrive, intermittent fever, polyarthritis, and myositis are other clinical features described in SAVI. Recent reports identified a phenotype mimicking the rheumatoid factor-positive polyarticular juvenile idiopathic arthritis (JIA) associated with an interstitial lung disease (103).
Trichohepatoenteric syndrome (THES) is a very rare disease characterized by typical wooly, brittle, and patchy hair (trichorrhexis nodosa), neonatal-onset intractable diarrhea, facial dysmorphism (“coarse” face feature), and intrauterine growth restriction with short stature. Additional findings include poorly characterized immunodeficiency with recurrent infections and liver disease. Mild intellectual disability was reported in about 50% of affected individuals. Less common findings include congenital heart defects and platelet anomalies.
The disease is caused by loss-of-function mutations of tetratricopeptide repeat domain 37 gene (TTC37) and Ski2-like RNA helicase gene (SKIV2L), which encode for two homologous proteins participating in human RNA exosome homeostasis, with consequent dysregulation of altered mRNA turnover. Moreover, SKIV2L seems to modulate directly RIG-I-like receptors (104).
XLPDR is a rare genodermatosis presenting as an early onset reticulated skin hyperpigmentation associated with recurrent pneumonia and enterocolitis, resembling inflammatory bowel disease. A distinctive face characterized by an upswept frontal hairline and arched eyebrows, severe photophobia with corneal inflammation, and failure to thrive were also described.
XLPDR is caused by an intronic mutation that disrupts the expression of POLA1, which encodes the catalytic subunit of DNA polymerase-α (POLA1). It was highlighted that POLA1 is required for synthesis of RNA–DNA hybrids, which negatively modulate the activation of the IFN regulatory factors (105). Since this is an intronic mutation, it can only be detected by whole genome sequencing (not exone sequencing) (44, 105).
The genetic analysis of patients suffering from a familial form of juvenile “idiopathic” arthritis associated with hemorrhagic interstitial lung disease permitted to identify a new autoinflammatory disease: the COPA syndrome. Indeed, heterozygous mutations were detected in the gene coding for the alpha subunit of the coatomer complex (COPa), which plays a role in the intracytosolic proteins retro-transport between Golgi apparatus-ER. An immune dysregulation of Th17 cells associated with inflammatory cytokine upregulation was described, probably triggered by intracellular ER-stress (106).
A role of IFN signal dysregulation in the pathogenesis was suspected because of the common clinical and pathogenetic features of SAVI syndrome and the finding of IFN-I pathway activation in peripheral blood of COPA syndrome patients. However, the inclusion of COPA syndrome among interferonopathies is still to be defined (44).
For a pediatrician, the first step of the diagnostic workup of these new hereditary diseases is their awareness and knowledge. This is a group of autoinflammatory pathologies that are very different from the more known “inflammasomopathies;” indeed, the typical clinical features are not periodic fever, nor are urticarious skin rash or recurrent serositis. Conversely, an early necrotizing vasculitis, a non-infectious interstitial lung disease in the context of an inflammatory clinical picture, a panniculitis associated with or without lipodystrophy, and early thrombotic events are peculiar elements that should lead to the suspicion of an interferonopathy (2, 44). Type I interferonopathies should also be considered when an autoimmune disease presents a very early onset and/or it is refractory to standard treatment and/or it occurs in several members of the same family (2). For example, a clinical and serological SLE-like syndrome in early life (particularly prepubertal age) should lead to the exclusion of a monogenic form (2). Clinical and laboratory “red flags” for interferonopathies are summarized in Table 3.

Table 3. Red flags of interferonopathies.
In case of suspicion, IFN signaling should always be evaluated. Type I IFN protein dosage is not available in routine clinical practice because of very low circulating levels. However, it was demonstrated that type I IFN pathway upregulation was correlated with increased expression of a subset of six ISG (IFI27, IFI44L, IFIT1, ISG15, RSAD2, and SIGLEC1) by quantitative polymerase chain reaction assays with whole blood samples in AGS patients, the so-called interferon signature. So, they developed the “interferon score” with a high sensitivity for AGS, which may be used both for diagnosis and follow-up and to assess disease activity (107). The IFN signature seems to be also very sensitive to differentiate monogenic and complex type I interferonopathies from inflammasomopathies. However, standardization between different centers could be difficult (44). Therefore, new “interferon scores” were proposed for autoimmune diseases such as SLE or anti-phospholipid antibody syndrome (ALS) using NanoString technology and new standardization methods are being studied to make the reporting homogeneous in research laboratories (108–110).
Since interferonopathies are hereditary genetic diseases, the conclusive diagnosis is the genetic sequencing, with the identification of causative mutations (2, 44).
However, in clinical practice undifferentiated systemic autoinflammatory diseases (USAIDs) are very frequent. More recently, some authors developed a standardized screening for “IFN signature,” using the type-I IFN-response-gene score (IRG-S), cytokine profiling, clinical phenotyping, and genetic evaluation by next-generation sequencing (NGS) (whole-exome sequencing or Sanger sequencing) for diagnosis of new or known interferonopathies, in patients with USAIDs (111). The characterization of the genes and the inflammatory pattern in patients with USAIDs, through modern NGS approaches up to exome or whole-genome sequencing, is currently an important goal, in order to identify the potential appropriate therapeutic targets (1, 2, 44).
Type I interferonopathies are often resistant to conventional immunosuppressive treatment. Intravenous boluses of methylprednisolone or immunoglobulins were used in many cases in the acute phase of the disease, without a conclusive relapse control (2, 44, 112).
A very promising therapeutic strategy is represented by JAK inhibitors. Baricitinib and ruxolitinib are selective JAK1/JAK2 inhibitors; tofacitinib inhibits JAK1/JAK3. Treatment in vitro of peripheral blood mononuclear cells from a patient with SAVI with tofacitinib, ruxolitinib, or baricitinib resulted in an inhibition of constitutive STAT1 activation (100). Later, the clinical efficacy of JAK1 blockade was confirmed by the improvement of vasculitic lesions and lung function in three children suffering from SAVI, treated with ruxolitinib, and in a 9-year-old boy with a SAVI vasculitis treated with tofacitinib (113, 114). The suppression of type I IFN activation during treatment with ruxolitinib was also shown in two children with AGS (115). Recently, the efficacy of baricitinib in the treatment of pediatric interferonopathies was demonstrated: 18 patients were enrolled, 10 with CANDLE, four with SAVI, and four with genetically undefined pathology. Patients treated for a mean of 2.3 years have shown a statistically significant improvement in clinical items and a reduction in the need for glucocorticoids. Additionally, 13 patients had growth potential improved, with catch-up growth observed in nine patients. Bone mineral density also improved, mainly due to steroid-sparing-dependent effects (116). In a study on pharmacokinetics in pediatric and adult patients, a dose-dependent effect of inhibition of the IFN signaling pathway was clearly shown (117). Future studies may clarify other aspects like the efficacy of baricitinib on the neurological involvement (2, 44).
Antiretroviral therapy (RT therapy) usually used for HIV infection is another therapeutic option. In murine studies, TREX-1 (mutated in AGS) plays a role in the degradation of the reverse-transcribed DNA (cDNA) of the HIV virus. Retroelements and mobile genetic elements represent up to half of the human genome; they can move within the genome by a reverse transcription of an RNA intermediate (cDNA) and insertion of its cDNA, a process called retrotransposition (44, 118). Therefore, the human cDNA may be the main substrate of TREX and the cDNA accumulation could be an important cause of IFN pathway activation. Moreover, recent studies have shown that SAMHD1 and ADAR also act on cDNA, confirming this hypothesis. It has been shown that TREX1 knockout mice have a reduction in cardiac inflammation by treatment with a combination of the non-nucleoside RT inhibitor nevirapine together with the nucleoside RT inhibitors tenofovir and emtricitabine (118). The intracellular accumulation of retrotransposomes was also demonstrated, using TREX1 deficiency reprogrammed neuronal cells (an experimental model of the AGS); RT inhibitors stavudine and lamivudine reduced the neurotoxicity of AGS neurons and organoids (119). Recently, the results of a single-center, open-label pilot study involving 11 patients with AGS treated with abacavir, lamivudine, and zidovudine for 12 months were published, demonstrating a reduction in IFN signature and IFN activity in the cerebrospinal fluid and an increase in cerebral blood flow in three of the five patients evaluated. These data support the possible role of HIV RT inhibitors in the treatment of interferonopathies (120).
Another desirable option for treatment of type I interferonopathies are monoclonal antibodies targeting IFN-α (sifalimumab) and IFNAR (anifrolumab). A multicenter, phase 2, open-label study was completed for sifalimumab in patients with SLE, showing good tolerability (121). A recent study confirmed the efficacy against placebo of anifrolumab in the control of skin and joint lesions of 362 SLE patients, showing a significant reduction in the IFN signature of these patients (122). These data highlight their potential use in therapeutic regimens in interferonopathies. Another currently theoretical strategy may be to modulate IFNAR degradation, through regulation of its ubiquitination by histone deacetylase 11 (HDH11) (123).
Finally, future possibility may be allogenic stem cell transplantation and autologous stem cell transplantation after gene correction of a patient's mutation (44).
Type I interferonopathies represent an emerging group of autoinflammatory disorders, with clinical characteristics and therapeutic response very different from previously characterized “inflammasome diseases.” Pediatricians must be aware of the clinical “IFN alarm” signals (such as chilblain-like vasculitis and interstitial lung disease) in order to suspect them and to direct properly the diagnostic workup with eminent prognostic implications. Molecular and biochemical details of IFN signaling permitted the identification of the etiopathological substrate of these rare syndromes, implementing the possibility of a target therapy. Nowadays, thanks to new sequencing techniques such as NGS, many indefinite autoinflammatory syndromes can often assume a genetic and clinical “identity,” offering these children the hope of new therapeutic “tailored” and “customized” options.
All authors contributed equally to this manuscript and approved the final version and its submission to the journal.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank Rose Chilman for helping us with English language.
1. Papa R, Picco P, Gattorno M. The expanding pathways of autoinflammation: a lesson from the first 100 genes related to autoinflammatory manifestations. Adv Protein Chem Struct Biol. (2020) 120:1–44. doi: 10.1016/bs.apcsb.2019.11.001
2. Volpi S, Picco P, Caorsi R, Candotti F, Gattorno M. Type I interferonopathies in pediatric rheumatology. Rheumatology. (2016) 14:35. doi: 10.1186/s12969-016-0094-4
3. Crow YJ, Black DN, Ali M, Bond J, Jackson AP, Lefson M, et al. Cree encephalitis is allelic with Aicardi-Goutières syndrome: implications for the pathogenesis of disorders of interferon alpha metabolism. J Med Genet. (2003) 40:183–7. doi: 10.1136/jmg.40.3.183
4. Rodero MP, Crow YJ. Type I interferon-mediated monogenic autoinflammation: the type I interferonopathies, a conceptual overview. J Exp Med. (2016) 213:2527–38. doi: 10.1084/jem.20161596
5. Rice GI, Bond J, Asipu A, Brunette RL, Manfield IW, Carr IM, et al. Mutations involved in Aicardi-Goutières syndrome implicate SAMHD1 as regulator of the innate immune response. Nat Genet. (2009) 41:829–32. doi: 10.1038/ng.373
6. Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, et al. Mutations in the gene encoding the 3'-5' DNA exonuclease TREX1 cause Aicardi-Goutières syndrome at the AGS1 locus. Nat Genet. (2006) 38:917–20. doi: 10.1038/ng1845
7. Elkon KB, Stone VV. Type I interferon and systemic lupus erythematosus. J Interferon Cytokine Res. (2011) 31:803–12. doi: 10.1089/jir.2011.0045
8. Crow YJ. Type I interferonopathies: a novel set of inborn errors of immunity. Ann N Y Acad Sci. (2011) 1238:91–8. doi: 10.1111/j.1749-6632.2011.06220.x
9. Eleftheriou D, Brogan PA. Genetic interferonopathies: an overview. Best Pract Res Clin Rheumatol. (2017) 31:441–59. doi: 10.1016/j.berh.2017.12.002
10. Crow YJ. Type I interferonopathies: mendelian type I interferon up-regulation. Curr Opin Immunol. (2015) 32:7–12. doi: 10.1016/j.coi.2014.10.005
11. Davidson S, Steiner A, Harapas CR, Masters SL. An update on autoinflammatory diseases: interferonopathies. Curr Rheumatol Rep. (2018) 20:38. doi: 10.1007/s11926-018-0748-y
12. Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci. (1957) 147:258–67. doi: 10.1098/rspb.1957.0048
13. Negishi H, Taniguchi T, Yanai H. The interferon (IFN) class of cytokines and the IFN regulatory factor (IRF) transcription factor family. Cold Spring Harb Perspect Biol. (2018) 10:a028423. doi: 10.1101/cshperspect.a028423
14. Trinchieri G. Type I interferon: friend or foe? J Exp Med. (2010) 207:2053–63. doi: 10.1084/jem.20101664
15. Gough DJ, Messina NL, Clarke CJP, Johnstone RW, Levy DE. Constitutive type I interferon modulates homeostatic balance through tonic signaling. Immunity. (2012) 36:166–74. doi: 10.1016/j.immuni.2012.01.011
16. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. (2014) 14:36–49. doi: 10.1038/nri3581
17. Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. (2004) 75:163–89. doi: 10.1189/jlb.0603252
18. Uddin S, Platanias LC. Mechanisms of type-I interferon signal transduction. J Biochem Mol Biol. (2004) 37:635–41. doi: 10.5483/BMBRep.2004.37.6.635
19. Guiducci C, Coffman RL, Barrat FJ. Signalling pathways leading to IFN-alpha production in human plasmacytoid dendritic cell and the possible use of agonists or antagonists of TLR7 and TLR9 in clinical indications. J Intern Med. (2009) 265:43–57. doi: 10.1111/j.1365-2796.2008.02050.x
20. Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Ann Rev Immunol. (2014) 32:513–45. doi: 10.1146/annurev-immunol-032713-120231
21. Li T, Chen ZJ. The cGAS–cGAMP–STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med. (2018) 215:1287–99. doi: 10.1084/jem.20180139
22. Dobbs N, Burnaevskiy N, Chen D, Gonugunta VK, Alto NM, Yan N. STING activation by translocation from the ER is associated with infection and autoinflammatory disease. Cell Host Microbe. (2015) 18:157–68. doi: 10.1016/j.chom.2015.07.001
23. Reikine S, Nguyen JB, Modis Y. Pattern recognition and signaling mechanisms of RIG-I and MDA5. Front Immunol. (2014) 5:342. doi: 10.3389/fimmu.2014.00342
24. Michalska A, Blaszczyk K, Wesoly J, Bluyssen HAR. A positive feedback amplifier circuit that regulates interferon (IFN)-stimulated gene expression and controls type I and type II IFN responses. Front Immunol. (2018) 28:1135. doi: 10.3389/fimmu.2018.01135
25. Zhang X, Bogunovic D, Payelle-Brogard B, Francois-Newton V, Speer SD, Yuan C, et al. Human intracellular ISG15 prevents interferon-alpha/beta over-amplification and auto-inflammation. Nature. (2015) 517:89–93. doi: 10.1038/nature13801
26. Ketscher L, Knobeloch KP. ISG15 uncut: dissecting enzymatic and non-enzymatic functions of USP18 in vivo. Cytokine. (2015) 76:569–71. doi: 10.1016/j.cyto.2015.03.006
27. Schreiber G. The molecular basis for differential type I interferon signaling. J Biol Chem. (2017) 292:7285–94. doi: 10.1074/jbc.R116.774562
28. Uggenti C, Lepelley A, Depp M, Badrock AP, Rodero MP, El-Daher MT, et al. cGAS-mediated induction of type I interferon due to inborn errors of histone pre-mRNA processing. Nat Genet. (2020) 52:1364–72. doi: 10.1038/s41588-020-00737-3
29. Goldmann T, Blank T, Prinz M. Fine-tuning of type I IFN-signaling in microglia-implications for homeostasis, CNS autoimmunity and interferonopathies. Curr Opin Neurobiol. (2016) 36:38–42. doi: 10.1016/j.conb.2015.09.003
30. McDonough A, Lee RV, Weinstein JR. Microglial interferon signaling and white matter. Neurochem Res. (2017) 42:2625–38. doi: 10.1007/s11064-017-2307-8
31. Hosmane S, Tegenge MA, Rajbhandari L, Uapinyoying P, Kumar NG, Thakor N, et al. Toll/interleukin-1 receptor domain-containing adapter inducing interferon-beta mediates microglial phagocytosis of degenerating axons. J Neurosci. (2012) 32:7745–57. doi: 10.1523/JNEUROSCI.0203-12.2012
32. Anderson SR, Vetter ML. Developmental roles of microglia: a window into mechanisms of disease. Dev Dyn. (2019) 248:98–117. doi: 10.1002/dvdy.1
33. Prinz M, Schmidt H, Mildner A, Knobeloch KP, Hanisch UK, Raasch J, et al. Distinct and nonredundant in vivo functions of IFNAR on myeloid cells limit autoimmunity in the central nervous system. Immunity. (2008) 28:675–86. doi: 10.1016/j.immuni.2008.03.011
34. Veldhuis WB, Derksen JW, Floris S, Van Der Meide PH, De Vries HE, Schepers J, et al. Interferon-beta blocks infiltration of inflammatory cells and reduces infarct volume after ischemic stroke in the rat. J Cereb Blood Flow Metab. (2003) 23:1029–39. doi: 10.1097/01.WCB.0000080703.47016.B6
35. Inacio AR, Liu Y, Clausen BH, Svensson M, Kucharz K, Yang Y, et al. Endogenous IFN-beta signaling exerts anti-inflammatory actions in experimentally induced focal cerebral ischemia. J Neuroinflammation. (2015) 12:211. doi: 10.1186/s12974-015-0427-0
36. Yang C, Hawkins KE, Doré S, Candelario-Jalil E. Neuroinflammatory mechanisms of blood-brain barrier damage in ischemic stroke. Am J Physiol Cell Physiol. (2019) 316:C135–53. doi: 10.1152/ajpcell.00136.2018
37. Knobeloch KP, Utermohlen O, Kisser A, Prinz M, Horak I. Reexamination of the role of ubiquitin-like modifier ISG15 in the phenotype of UBP43-deficient mice. Mol Cell Biol. (2005) 25:11030–4. doi: 10.1128/MCB.25.24.11030-11034.2005
38. Schwabenland M, Mossad O, Peres AG, Kessler F, Maron FJM, Harsan LA, et al. Loss of USP18 in microglia induces white matter pathology. Acta Neuropathol Commun. (2019) 7:106. doi: 10.1186/s40478-019-0757-8
39. Meuwissen ME, Schot R, Buta S, Oudesluijs G, Tinschert S, Speer SD, et al. Human USP18 deficiency underlies type 1 interferonopathy leading to severe pseudo-TORCH syndrome. J Exp Med. (2016) 213:1163–74. doi: 10.1084/jem.20151529
40. Crow YJ, Zaki MS, Abdel-Hamid MS, Abdel-Salam G, BoespflugTanguy O, Cordeiro NJV, et al. Mutations in ADAR1, IFIH1, and RNASEH2B presenting as spastic paraplegia. Neuropediatrics. (2014) 45:386–91. doi: 10.1055/s-0034-1389161
41. Feng X, Bao R, Li L, Deisenhammer F, Arnason BGW, Reder AT. Interferon-β corrects massive gene dysregulation in multiple sclerosis: short-term and long-term effects on immune regulation and neuroprotection. EBioMedicine. (2019) 49:269–83. doi: 10.1016/j.ebiom.2019.09.059
42. Choubey D. Type I interferon (IFN)-inducible Absent in Melanoma 2 proteins in neuroinflammation: implications for Alzheimer's disease. J Neuroinflammation. (2019) 16:236. doi: 10.1186/s12974-019-1639-5
43. Picard C, Bobby Gaspar H, Al-Herz W, Bousfiha A, Casanova JL, Chatila T, et al. International union of immunological societies: 2017 primary immunodeficiency diseases committee report on inborn errors of immunity. J Clin Immunol. (2018) 38:96–128. doi: 10.1007/s10875-017-0464-9
44. Yu ZX, Song HM. Toward a better understanding of type I interferonopathies: a brief summary, update and beyond. World J Pediatr. (2020) 16:44–51. doi: 10.1007/s12519-019-00273-z
45. Crow YJ, Manel N. Aicardi-Goutières syndrome and the type I interferonopathies. Nat Rev Immunol. (2015) 15:429–40. doi: 10.1038/nri3850
46. Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M, Forte GMA, Gornall HL, et al. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAM HD1, ADAR, and IFIH1. Am J Med Genet. (2015) 167:296–312. doi: 10.1055/s-0036-1592307
47. Rice GI, Forte GMA, Szynkiewicz M, Chase DS, Aeby A, Abdel-Hamid MS, et al. Assessment of interferon-related biomarkers in Aicardi-Goutières syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: a case-control study. Lancet Neurol. (2013) 12:1159–69. doi: 10.1016/S1474-4422(13)70258-8
48. Zheng S, Lee PY, Wang J, Wang S, Huang Q, Huang Y, et al. Interstitial lung disease and psoriasis in a child with Aicardi-Goutières syndrome. Front Immunol. (2020) 11:985. doi: 10.3389/fimmu.2020.00985
49. Cuadrado E, Michailidou I, van Bodegraven EJ, Jansen MH, Sluijs JA, Geerts D, et al. Phenotypic variation in Aicardi-Goutières syndrome explained by cell-specific IFN-stimulated gene response and cytokine release. J Immunol. (2015) 194:3623–33. doi: 10.4049/jimmunol.1401334
50. Crow YJ, Rehwinkel J. Aicardi-Goutieres syndrome and related phenotypes: linking nucleic acid metabolism with autoimmunity. Hum Mol Genet. (2009) 18:R130–6. doi: 10.1093/hmg/ddp293
51. Coquel F, Silva MJ, Técher H, Zadorozhny K, Sharma S, Nieminuszczy J, et al. SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature. (2018) 557:57–61. doi: 10.1038/s41586-018-0050-1
52. Rice G, Patrick T, Parmar R, Taylor CF, Aeby A, Aicardi J, et al. Clinical and molecular phenotype of Aicardi-Goutieres syndrome. Am J Hum Genet. (2007) 81:713–25. doi: 10.1086/521373
53. Sun W, Manohar S, Jayaram A, Kumaraguru A, Fu Q, Li J, Allman B. Early age conductive hearing loss causes audiogenic seizure and hyperacusis behavior. Hear Res. (2011) 282:178–83. doi: 10.1016/j.heares.2011.08.004
54. Ramantani G, Maillard LG, Bast T, Husain RA, Niggemann P, Kohlhase J, et al. Epilepsy in Aicardi-Goutières syndrome. Eur J Paediatr Neurol. (2014) 18:30–7. doi: 10.1016/j.ejpn.2013.07.005
55. Thiele H, du Moulin M, Barczyk K, George C, Schwindt W, Nürnberg G, et al. Cerebral arterial stenoses and stroke: novel features of Aicardi-Goutières syndrome caused by the Arg164X mutation in SAMHD1 are associated with altered cytokine expression. Hum Mutat. (2010) 31:1836–50. doi: 10.1002/humu.21357
56. Cattalini M, Galli J, Andreoli L, Olivieri I, Ariaudo G, Fredi M, et al. Exploring autoimmunity in a cohort of children with genetically confirmed Aicardi-Goutières syndrome. J Clin Immunol. (2016) 36:693–9. doi: 10.1007/s10875-016-0325-y
57. Dale RC, Gornall H, Singh-Grewal D, Alcausin M, Rice GI, Crow YJ. Familial Aicardi-Goutières syndrome due to SAMHD1 mutations is associated with chronic arthropathy and contractures. Am J Med Genet A. (2010) 152:938–42. doi: 10.1002/ajmg.a.33359
58. Briggs TA, Rice GI, Daly S, Urquhart J, Gornall H, Bader-Meunier B, et al. Tartrate-resistant acid phosphatase deficiency causes a bone dysplasia with autoimmunity and a type I interferon expression signature. Nat Genet. (2011) 43:127–31. doi: 10.1038/ng.748
59. Lausch E, Janecke A, Bros M, Trojandt S, Alanay Y, De Laet C, et al. Genetic deficiency of tartrate-resistant acid phosphatase associated with skeletal dysplasia, cerebral calcifications and autoimmunity. Nat Genet. (2011) 43:132–7. doi: 10.1038/ng.749
60. Shinohara ML, Lu L, Bu J, Werneck MB, Kobayashi KS, Glimcher LH, et al. Osteopontin expression is essential for interferon-alpha production by plasmacytoid dendritic cells. Nat Immunol. (2006) 7:498–506. doi: 10.1038/ni1327
61. Bilginer Y, Düzova A, Topaloglu R, Batu ED, Boduroglu K, Güçer S, et al. Three cases of spondyloenchondrodysplasia (SPENCD) with systemic lupus erythematosus: a case series and review of the literature. Lupus. (2016) 25:760–5. doi: 10.1177/0961203316629000
62. Kim T, Kanayama Y, Negoro N, Okamura M, Takeda T, Inoue T. Serum levels of interferons in patients with systemic lupus erythematosus. Clin Exp Immunol. (1987) 70:562–9.
63. Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med. (2011) 3:73ra20. doi: 10.1126/scitranslmed.3001201
64. Kirou KA, Lee C, George S, Louca K, Peterson MG, Crow MK. Activation of the interferon-alpha pathway identifies a subgroup of systemic lupus erythematosus patients with distinct serologic features and active disease. Arthritis Rheum. (2005) 52:1491–503. doi: 10.1002/art.21031
65. Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. (2003) 197:711–23. doi: 10.1084/jem.20021553
66. Niewold TB, Hua J, Lehman TJ, Harley JB, Crow MK. High serum IFN-alpha activity is a heritable risk factor for systemic lupus erythematosus. Genes Immun. (2007) 8:492–502. doi: 10.1038/sj.gene.6364408
67. Moulton VR, Suarez-Fueyo A, Meidan E, Li H, Mizui M, Tsokos GC. Pathogenesis of human systemic lupus erythematosus: a cellular perspective. Trends Mol Med. (2017) 23:615–35. doi: 10.1016/j.molmed.2017.05.006
68. Ghodke-Puranik Y, Niewold TB. Immunogenetics of systemic lupus erythematosus: a comprehensive review. J Autoimmun. (2015) 64:125–36. doi: 10.1016/j.jaut.2015.08.004
69. Omarjee O, Picard C, Frachette C, Moreews M, Rieux-Laucat F, Soulas-Sprauel P, et al. Monogenic lupus: dissecting heterogeneity. Autoimmun Rev. (2019) 18:102361. doi: 10.1016/j.autrev.2019.102361
70. Alperin JM, Ortiz-Fernández L, Sawalha AH. Monogenic lupus: a developing paradigm of disease. Front Immunol. (2018) 9:2496. doi: 10.3389/fimmu.2018.02496
71. Demirkaya E, Sahin S, Romano M, Zhou Q, Aksentijevich I. New horizons in the genetic etiology of systemic lupus erythematosus and lupus-like disease: monogenic lupus and beyond. J Clin Med. (2020) 5:712. doi: 10.3390/jcm9030712
72. Lood C, Gullstrand B, Truedsson L, Olin AI, Alm GV, Rönnblom L, et al. C1q inhibits immune complex-induced interferon-alpha production in plasmacytoid dendritic cells: a novel link between C1q deficiency and systemic lupus erythematosus pathogenesis. Arthritis Rheum. (2009) 60:3081–90. doi: 10.1002/art.24852
73. Demirkaya E, Zhou Q, Smith CK, Ombrello MJ, Deuitch N, Tsai WL, et al. Brief report: deficiency of complement 1r subcomponent in early-onset systemic lupus erythematosus: the role of disease-modifying alleles in a monogenic disease. Arthritis Rheumatol. (2017) 69:1832–9. doi: 10.1002/art.40158
74. Napirei M, Karsunky H, Zevnik B, Stephan H, Mannherz HG, Möröy T. Features of systemic lupus erythematosus in Dnase1-deficient mice. Nat Genet. (2000) 25:177–81. doi: 10.1038/76032
75. Yasutomo K, Horiuchi T, Kagami S, Tsukamoto H, Hashimura C, Urushihara M, et al. Mutation of DNASE1 in people with systemic lupus erythematosus. Nat Genet. (2001) 28:313–4. doi: 10.1038/91070
76. Martínez Valle F, Balada E, Ordi-Ros J, Vilardell-Tarres M. DNase 1 and systemic lupus erythematosus. Autoimmun Rev. (2008) 7:359–63. doi: 10.1016/j.autrev.2008.02.002
77. Al-Mayouf S, Sunker A, Abdwani R, Abrawi SA, Almurshedi F, Alhashmi N, et al. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat Genet. (2011) 43:1186–8. doi: 10.1038/ng.975
78. Sisirak V, Sally B, D'Agati V, Martinez-Ortiz W, Özçakar ZB, David J, et al. Digestion of chromatin in apoptotic cell microparticles prevents autoimmunity. Cell. (2016) 166:88–101. doi: 10.1016/j.cell.2016.05.034
79. Wang L, Mi W, Li Q-Z. Targeting the extracellular scavenger DNASE1L3 on SLE. J Xiangya Med. (2017) 2:29. doi: 10.21037/jxym.2017.03.03
80. Rodero MP, Tesser A, Bartok E, Rice GI, Della Mina E, Depp M, et al. Type I interferon-mediated autoinflammation due to DNase II deficiency. Nat Commun. (2017) 8:2176. doi: 10.1038/s41467-017-01932-3
81. Lee-Kirsch MA, Gong M, Schulz H, Ruschendorf F, Stein A, Pfeiffer C, et al. Familial chilblain lupus, a monogenic form of cutaneous lupus erythematosus, maps to chromosome 3p. Am J Hum Genet. (2006) 79:731–7. doi: 10.1086/507848
82. Rice G, Newman WG, Dean J, Patrick T, Parmar R, Flintoff K, et al. Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi- Goutieres syndrome. Am J Hum Genet. (2007) 80:811–5. doi: 10.1086/513443
83. Rice GI, Rodero MP, Crow YJ. Human disease phenotypes associated with mutations in TREX1. J Clin Immunol. (2015) 35:235–43. doi: 10.1007/s10875-015-0147-3
84. Namjou B, Kothari PH, Kelly JA, Glenn SB, Ojwang JO, Adler A, et al. Evaluation of the TREX1 gene in a large multi-ancestral lupus cohort. Genes Immun. (2011) 12:270–9. doi: 10.1038/gene.2010.73
85. Hiraki LT, Silverman ED. Genomics of systemic lupus erythematosus: insights gained by studying monogenic young-onset systemic lupus erythematosus. Rheum Dis Clin North Am. (2017) 43:415–34. doi: 10.1016/j.rdc.2017.04.005
86. Schuh E, Ertl-Wagner B, Lohse P, Wolf W, Mann JF, Lee-Kirsch MA, et al. Multiple sclerosis-like lesions and type I interferon signature in a patient with RVCL. Neurol Neuroimmunol Neuroinflamm. (2015) 2:e55. doi: 10.1212/NXI.0000000000000055
87. Ellyard JI, Jerjen R, Martin JL, Lee AY, Field MA, Jiang SH, et al. Identification of a pathogenic variant in TREX1 in early-onset cerebral systemic lupus erythematosus by Whole-exome sequencing. Arthritis Rheumatol. (2014) 66:3382–6. doi: 10.1002/art.38824
88. Ravenscroft JC, Suri M, Rice GI, Szynkiewicz M, Crow YJ. Autosomal dominant inheritance of a heterozygous mutation in SAMHD1 causing familial chilblain lupus. Am J Med Genet A. (2011) 155A:235–7. doi: 10.1002/ajmg.a.33778
89. de Jesus AA, Brehm A, VanTries R, Pillet P, Parentelli A-S, Montealegre Sanchez GA, et al. Novel proteasome assembly chaperone mutations in PSMG2/PAC2 cause the autoinflammatory interferonopathy CANDLE/PRAAS4. J Allergy Clin Immunol. (2019) 143:1939–43. doi: 10.1016/j.jaci.2018.12.1012
90. Torrelo A, Patel S, Colmenero I, Gurbindo D, Lendinez F, Hernandez A, et al. Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome. J Am Acad Dermatol. (2010) 62:489–95. doi: 10.1016/j.jaad.2009.04.046
91. Garg A, Hernandez MD, Sousa AB, Subramanyam L, Martinez de Villarreal L, dos Santos HG, et al. An autosomal recessive syndrome of joint contractures, muscular atrophy, microcytic anemia, and panniculitis-associated lipodystrophy. J Clin Endocrinol Metab. (2010) 95:58–63. doi: 10.1210/jc.2010-0488
92. Ebstein F, Harlowe MCP, Studencka-Turski M, Kruger E. Contribution of the unfolded protein response (UPR) to the pathogenesis of proteasome-associated autoinflammatory syndromes (PRAAS). Front Immunol. (2019) 10:2756. doi: 10.3389/fimmu.2019.02756
93. Küry S, Besnard T, Ebstein F, Khan TN, Gambin T, Douglas J, et al. De novo disruption of the proteasome regulatory subunit PSMD12 causes a syndromic neurodevelopmental disorder. Am J Hum Genet. (2017) 100:352–63. doi: 10.1016/j.ajhg.2017.01.003
94. Bogunovic D, Byun M, Durfee LA, Abhyankar A, Sanal O, Mansouri D, et al. Mycobacterial disease and impaired IFN-gamma immunity in humans with inherited ISG15 deficiency. Science. (2012) 337:1684–8. doi: 10.1126/science.1224026
95. Fernandez MM, Garcia-Morato MB, Gruber C, Murias Loza S, Malik MNH, Alsohime F, et al. Systemic type I IFN inflammation in human ISG15 deficiency leads to necrotizing skin lesions. Cell Rep. (2020) 31:107633. doi: 10.1016/j.celrep.2020.107633
96. Singleton EB, Merten DF. An unusual syndrome of widened medullary cavities of the metacarpals and phalanges, aortic calcification and abnormal dentition. Pediatr Radiol. (1973) 1:2–7. doi: 10.1007/BF00972817
97. Jang MA, Kim EK, Now H, Nguyen NT, Kim WJ, Yoo JY, et al. Mutations in DDX58, which encodes RIG-I, cause atypical Singleton-Merten syndrome. Am J Hum Genet. (2015) 96:266–74. doi: 10.1016/j.ajhg.2014.11.019
98. Rutsch F, MacDougall M, Lu C, Buers I, Mamaeva O, Nitschke Y, et al. A specific IFIH1 gain-of-function mutation causes Singleton-Merten syndrome. Am J Hum Genet. (2015) 96:275–82. doi: 10.1016/j.ajhg.2014.12.014
99. Pettersson M, Bergendal B, Norderyd J, Nilsson D, Anderlid BM, Nordgren A, et al. Further evidence for specific IFIH1 mutation as a cause of Singleton-Merten syndrome with phenotypic heterogeneity. Am J Med Genet A. (2017) 173:1396–9. doi: 10.1002/ajmg.a.38214
100. Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Sanchez GAM, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med. (2014) 371:507–18. doi: 10.1056/NEJMoa1312625
101. Ozen S, Sag E. Childhood vasculitis. Rheumatology. (2020) 59:iii95–100. doi: 10.1093/rheumatology/kez599
102. Munoz J, Rodiere M, Jeremiah N, Rieux-Laucat F, Oojageer A, Rice GI, et al. Stimulator of interferon genes-associated vasculopathy with onset in infancy: a mimic of childhood granulomatosis with polyangiitis. JAMA Dermatol. (2015) 151:872–7. doi: 10.1001/jamadermatol.2015.0251
103. Clarke SLN, Robertson L, Rice GI, Seabra L, Hilliard TN, Crow YJ, et al. Type 1 interferonopathy presenting as juvenile idiopathic arthritis with interstitial lung disease: report of a new phenotype. Pediatr Rheumatol Online J. (2020) 18:37. doi: 10.1186/s12969-020-00425-w
104. Eckard SC, Rice GI, Fabre A, Badens C, Gray EE, Hartley JL, et al. The SKIV2L RNA exosome limits activation of the RIG-Ilike receptors. Nat Immunol. (2014) 15:839–45. doi: 10.1038/ni.2948
105. Starokadomskyy P, Gemelli T, Rios JJ, Xing C, Wang RC, Li H, et al. DNA polymerase-alpha regulates the activation of type I interferons through cytosolic RNA:DNA synthesis. Nat Immunol. (2016) 17:495–504. doi: 10.1038/ni.3409
106. Vece TJ, Watkin LB, Nicholas S, Canter D, Braun MC, Guillerman RP, et al. Copa syndrome: a novel autosomal dominant immune dysregulatory disease. J Clin Immunol. (2016) 36:377–87. doi: 10.1007/s10875-016-0271-8
107. Rice GI, Melki I, Frémond ML, Briggs TA, Rodero MP, Kitabayashi N, et al. Assessment of type I interferon signaling in pediatric inflammatory disease. J Clin Immunol. (2017) 37:123–32. doi: 10.1007/s10875-016-0359-1
108. El-Sherbiny YM, Psarras A, Yusof MYM, Hensor EMA, Tooze R, Doody G, et al. A novel two-score system for interferon status segregates autoimmune diseases and correlates with clinical features. Sci Rep. (2018) 8: 5793. doi: 10.1038/s41598-018-33062-1
109. Kim H, de Jesus AA, Brooks SR, Liu Y, Huang Y, VanTries R, et al. Development of a validated interferon score using NanoString Technology. J Interferon Cytokine Res. (2018) 38:171–85. doi: 10.1089/jir.2017.0127
110. Pin A, Monasta L, Taddio A, Piscianz E, Tommasini A, Tesser A. An easy and reliable strategy for making type I interferon signature analysis comparable among research centers. Diagnostics. (2019) 9:113. doi: 10.3390/diagnostics9030113
111. de Jesus AA, Hou Y, Brooks S, Malle L, Biancotto A, Huang Y, et al. Distinct interferon signatures and cytokine patterns define additional systemic autoinflammatory diseases. J Clin Invest. (2020) 130:1669–82. doi: 10.1172/JCI129301
112. Bienias M, Brück N, Griep C, Wolf C, Kretschmer S, Kind B, et al. Therapeutic approaches to type I interferonopathies. Curr Rheumatol Rep. (2018) 20:32. doi: 10.1007/s11926-018-0743-3
113. Frémond M-L, Rodero MP, Jeremiah N, Belot A, Jeziorski E, Duffy D, et al. Efficacy of the Janus kinase 1/2 inhibitor ruxolitinib in the treatment of vasculopathy associated with TMEM173-activating mutations in 3 children. J Allergy Clin Immunol. (2016) 138:1752–5. doi: 10.26226/morressier.57bc1755d462b80290b4d6b9
114. Seo J, Kang J-A, Suh DI, Park E-B, Lee C-R, Choi SA, et al. Tofacitinib relieves symptoms of stimulator of interferon genes (STING)-associated vasculopathy with onset in infancy caused by 2 de novo variants in TMEM173. J Allergy Clin Immunol. (2017) 139:1396–9. doi: 10.1016/j.jaci.2016.10.030
115. Meesilpavikkai K, Dik WA, Schrijver B, van Helden-Meeuwsen CG, Versnel MA, van Hagen PM, et al. Efficacy of baricitinib in the treatment of chilblains associated with Aicardi-Goutières syndrome, a type I interferonopathy. Arthritis Rheumatol. (2019) 71:829–31. doi: 10.1002/art.40805
116. Sanchez GAM, Reinhardt A, Ramsey S, Wittkowski H, Hashkes PJ, Berkun Y, et al. JAK1/2 inhibition with baricitinib in the treatment of autoinflammatory interferonopathies. J Clin Invest. (2018) 128:3041–52. doi: 10.1172/JCI98814
117. Kim H, Brooks KM, Tang CC, Wakim P, Blake M, Brooks SR, et al. Pharmacokinetics, pharmacodynamics, and proposed dosing of the oral JAK1 and JAK2 inhibitor baricitinib in pediatric and young adult CANDLE and SAVI patients. Clin Pharmacol Ther. (2017) 104:364–73. doi: 10.1002/cpt.936
118. Beck-Engeser GB, Eilat D, Wabl M. An autoimmune disease prevented by anti-retroviral drugs. Retrovirology. (2011) 8:91. doi: 10.1186/1742-4690-8-91
119. Thomas CA, Tejwani L, Trujillo CA, Negraes PD, Herai RH, Mesci P, et al. Modeling of TREX1-dependent autoimmune disease using human stem cells highlights L1 accumulation as a source of neuroinflammation. Cell Stem Cell. (2017) 21:319–31. doi: 10.1016/j.stem.2017.07.009
120. Rice GI, Meyzer C, Bouazza N, Hully M, Boddaert N, Semeraro M, et al. Reverse-transcriptase inhibitors in the Aicardi-Goutières syndrome. N Engl J Med. (2018) 379:2275–7. doi: 10.1056/NEJMc1810983
121. Takeuchi T, Tanaka Y, Matsumura R, Saito K, Yoshimura M, Amano K, et al. Safety and tolerability of sifalimumab, an anti-interferon-α monoclonal antibody, in Japanese patients with systemic lupus erythematosus: a multicenter, phase 2, open-label study. Mod Rheumatol. (2020) 30:93–100. doi: 10.1080/14397595.2019.1583832
122. Morand EF, Furie R, Tanaka Y, Bruce IN, Askanase AD, Richez C, et al. Trial of anifrolumab in active systemic lupus erythematosus. N Engl J Med. (2020) 382:211–21. doi: 10.1056/NEJMoa1912196
123. Cao J, Sun L, Aramsangtienchai P, Spiegelman NA, Zhang X, Huang W, et al. HDAC11 regulates type I interferon signaling through defatty-acylation of SHMT2. Proc Natl Acad Sci USA. (2019) 116:5487–92. doi: 10.1073/pnas.1815365116
AGS, Aicardi–Goutieres syndrome; SLE, systemic lupus erythematosus; IFN, interferon; toxoplasmosis, rubella, cytomegalovirus, and herpes, TORCH; CNS, central nervous system; TLRs, Toll-like receptors; RLRs, RIG-I-like receptors; NLRs, NOD-like receptors; cGAS, cyclic GMP-AMP synthase; cGAMP, 2′3′GMP-AMP; STING, STimulator of INterferon Genes; ER, endoplasmic reticulum; TBK1, TANK-binding kinase 1; IRF3, IFN regulatory factors 3; IRF7, IFN regulatory factors 7; RLHs, RIG-I-like helicases; RIG-I, retinoic acid-inducible gene-I; MDA5, melanoma differentiation-associated gene 5; IFNRs, interferon receptors; IRF9, IFN regulatory factors 9; ISGs, interferon-stimulated genes; USP18, ubiquitin-specific peptidase 18; ISG15, interferon-stimulated gene 15; SPK2, S-phase kinase-associated protein 2; AD, autosomal dominant; AR, autosomal recessive; ADAR1, adenosine deaminase acting on RNA 1; ACP5, acid phosphatase 5, tartrate resistant; AGS, Aicardi–Goutières syndrome; DDX58, DEAD Box Protein 58; IFIH1, IFN-induced helicase C domain-containing protein 1; ISG15, interferon-stimulated gene 15, PSMB8 (proteasome subunit beta type-8; PSMB9, proteasome subunit beta type-9; PSMB7, proteasome subunit beta type-7; PSMA3, proteasome 20S subunit alpha 3; POMP, proteasome maturation protein; PSMG2, proteasome assembly chaperone 2; RNASEH2, ribonuclease H2; SAMHD1, deoxynucleoside triphosphate triphosphohydrolase SAM domain and HD domain 1; SPENCD, spondyloenchondrodysplasia; SAVI, STING-associated vasculopathy with onset in infancy; PRAAS, proteasome-associated autoinflammatory syndromes; SMS, Singleton–Merten syndrome; THES, trichohepatoenteric syndrome; TMEM173, transmembrane protein 173; TREX1, DNA 3′—repair exonuclease 1; TRAP, acid phosphatase 5 tartrate resistant; IFIH1, IFN-induced helicase C domain-containing protein 1; XLPDR, X-linked reticulate pigmentary disorder; MS, multiple sclerosis; CSF, cerebrospinal fluid; Opn, osteopontin; HSV-1, herpes simplex virus 1; PBMCs, peripheral blood mononuclear cells; SNPs, single-nucleotide polymorphisms; UPR, unfolded protein response; IRE1, inositol requiring enzyme 1α/β; TTC37, tetratricopeptide repeat domain 37 gene; SKIV2L, Ski2-like RNA helicase gene; ALS, anti-phospholipid antibody syndrome; USAIDs, undifferentiated systemic autoinflammatory diseases; NGS, next-generation sequencing; SOCS1, suppressor of cytokine signaling 1; LSM11, U7 snRNA-associated Sm-like protein; RNU7-1, RNA U7 small nuclear 1; OTUD1, OTU domain-containing protein 1; C1r, complement 1r subcomponent.
Keywords: autoinflammatory disease, type I interferon (IFN) signaling, Aicardi-Goutières syndrome, Janus kinase inhibitors, innate immunity, interferon
Citation: d'Angelo DM, Di Filippo P, Breda L and Chiarelli F (2021) Type I Interferonopathies in Children: An Overview. Front. Pediatr. 9:631329. doi: 10.3389/fped.2021.631329
Received: 19 November 2020; Accepted: 05 January 2021;
Published: 31 March 2021.
Edited by:
Erkan Demirkaya, Western University, CanadaReviewed by:
Micol Romano, Istituto Ortopedico Gaetano Pini, ItalyCopyright © 2021 d'Angelo, Di Filippo, Breda and Chiarelli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesco Chiarelli, Y2hpYXJlbGxpQHVuaWNoLml0; orcid.org/0000-0002-3724-1495
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.