 Junxian Fu†
Junxian Fu† Guanglu Yang
Guanglu Yang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr. , 10 February 2021
Sec. Genetics of Common and Rare Diseases
Volume 9 - 2021 | https://doi.org/10.3389/fped.2021.618059
Objective: The aim of the present study is to explore the clinical and genetic characteristics of 3p deletion syndrome to improve clinicians' understanding of the disease.
Methods: The clinical manifestations, process of diagnosis and treatment, and genetic characteristics of an individual case of 3p deletion syndrome were analyzed. CNKI, Wanfang Data, and the Biomedical Literature Database (PubMed) were searched. The search time limit, using “3p deletion syndrome” and “BRPF1” as keywords, was from the creation of the database up to June 2020. Related data were reviewed.
Results: The proband was a male child with general developmental and intellectual disabilities, special facial features and congenital heart disease. The child was the parents' first pregnancy and first born. Gene microarray analysis showed a 10.095 Mb deletion in the 3p26.3-p25.3 region, resulting in a heterozygous mutation of the BRPF1 gene; thus, the patient was diagnosed with 3p deletion syndrome. At the time of diagnosis, the child was 1 year of age and was responding to comprehensive rehabilitation training. A total of 29 well-documented cases were found in the literature, of which 19 cases had an onset within 1 year of birth, and mainly manifested with mental and motor development disabilities and abnormal facial features, with different gene deletions, depending on the size and location of the 3p deletion.
Conclusion: The genetic test results of the child in this study indicated a heterozygous deletion of the BRPF1 gene on the short arm of chromosome 3, which was a unique feature of this study, since it was rarely mentioned in other reports of 3p deletion syndrome. The clinical phenotype of this syndrome is complex as it can include intellectual and motor development backwardness, low muscle tone, certain abnormal facial features (low hairline, bilateral ptosis, widely spaced eyes, a forward nose, left ear auricle deformity, a high-arched palate, a small jaw), and the deformation of systems such as the gastrointestinal tract and the urinary tract malformation or symptoms of epilepsy. As clinical manifestations can be relatively mild, the syndrome is easy to miss or misdiagnose. Clinical workers need to be aware of this disease when they find that children have special features, such as stunted growth, low muscle tone or ptosis, and it needs to be diagnosed through genetic testing. Most children are able to develop certain social skills after rehabilitation treatment.
3p deletion syndrome is a rare autosomal and contiguous genomic disorder characterized by the following: intellectual disability; motor developmental delay; unusual facial features (microcephaly, micrognathia, ptosis, long philtrum, low and deformed ears, polydactyly deformity); hypotonia; and other rarer symptoms, including congenital heart disease (CHD), renal and gastrointestinal malformations, autism, congenital hypothyroidism, epilepsy, and tumors (1). The BRPF1 gene is a chromatin regulator that binds to methylated histone H3 and promotes acetylation. BRPF1 deletion may lead to intellectual disability and abnormal facial features, such as ptosis (2–9). In the present study, the clinical features, the process of diagnosis and treatment, and the genetic findings of a patient with 3p deletion syndrome were reviewed, and a review of the literature was conducted to summarize the clinical features and genetic characteristics of the syndrome to aid in clinical recognition and identification.
The pediatric patient was a 1-year-old male who was the parents' first pregnancy and first born. The patient was born by caesarian section after being unable to enter the pelvic cavity at full term. There was no history of hypoxia or asphyxia. The birth weight was 3.05 kg, and the patient failed to pass the hearing screening at 3, 5, 42 days after birth, and 3 months of age. The parents were non-consanguineous and denied any family history of genetic diseases. There had been no abnormalities in a Down's screening. The mother had a cold during her pregnancy but did not take any medication. There was a history of secondhand smoke exposure but no history of radiation or chemical carcinogen exposure.
Case history: shortly after birth the child started to suffer repeated hiccupping and would vomit more than 10 times a day. The patient was referred to a hospital in Beijing 3 months after birth. A gastrointestinal angiography was performed, which led to the diagnosis of gastroesophageal reflux. Medication was given and the vomiting was relieved at 8 months, but the patient continued to suffer from vomiting when coughing or sneezing. The patient could hold his head erect at 4 months of age, and recognize things and people at 10 months. However, by age 11 months, the patient was no longer able to hold his head erect or sit or climb unassisted. The patient could not walk and could only stand on his toes. The patient could say the words “dad” and “mom” and presented with hyperhidrosis. After being admitted to the hospital, the patient was diagnosed with 3p deletion syndrome after genetic testing. At 1 year of age, the patient was referred to our hospital for rehabilitation training. A physical examination yielded the following observations. The patient was in clear consciousness with average spirit and moderate nutrition. He was 76 cm in height, 8.5 kg in weight, and his head circumference was 44.5 cm. His front hairline was low, and he had bilateral ptosis (drooping eyelids) and wide-set eyes. The pupils were equal in a circle, and the light reflection was sensitive. The patient had a broad nose bridge, forward nostrils, a deformity of the left auricle, a long philtrum, a high-arched palate, and micrognathia (as shown in Figure 1). The teeth were hypoplastic and yellow in color. There was no penetration of the hands. There was no obvious abnormality of the lungs or abdomen and no obvious pathological murmur in the heart valve area. There were no stereotyped movements. The knee reflex and Achilles tendon reflex of the left lower limb were weak, but the tendon reflex of the right lower limb, the bicep reflex of the upper limbs, the triceps reflex, the periosteum reflex of the radius, and the reflex of the abdominal wall were all present, and there was no evident pathological reflex. The patient had hypotonia in the four extremities and uncooperative muscle strength. The patient presented with intellectual disabilities with an inability to recognize strangers or clap hands; further, the patient showed a poor ability to track light and objects.
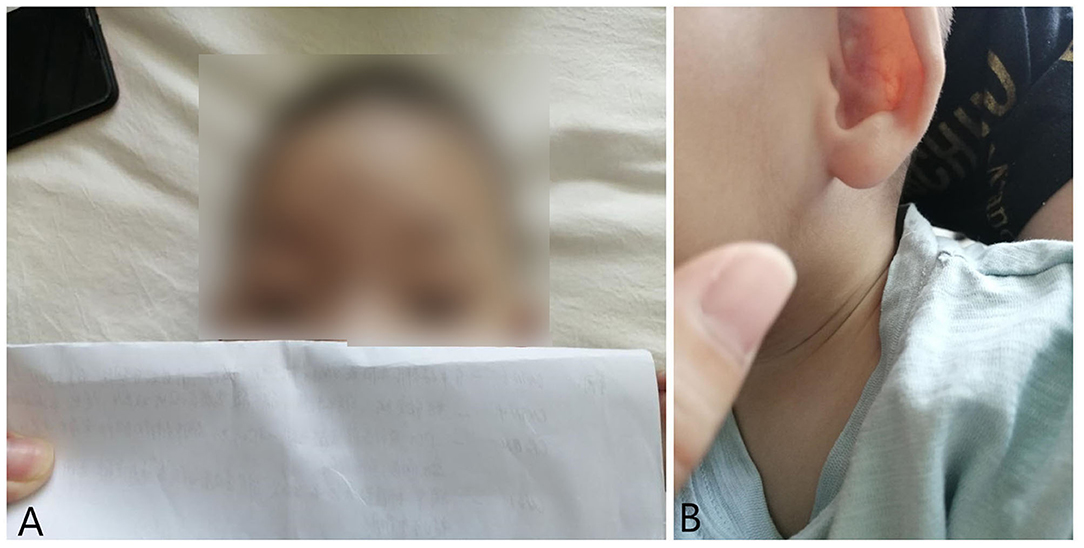
Figure 1. Ptosis (A) and auricular deformity (B) in the case of 1 year old with 3p deletion syndrome.
A number of measurements were made to evaluate the patient's problems. A biochemical analysis showed no obvious abnormalities. The results of the Peabody Developmental Motor Scale were as follows: gesture, three points in the standard score; moving, 1 point in the standard score; physical operation, 0 point in the standard score; grasp, 2 points in the standard score; vision-motion integration, five points in the standard score; for total movement, a development quotient of 51, for coarse movement, a development quotient of 51 and for fine movement, a development quotient of 61. A colored ultrasonography of the heart revealed a congenital heart defect (CHD), namely a ventricular septal defect (peri-membranous) of ~3 mm in size and a left to right shunt at the ventricular level. A video EEG showed an abnormal EEG for a young child with slightly slower background waves during the awake period and spikes and slow waves of the frontal area and midline area (Fz) emitted during sleep. An MRI of the head was normal, but examination of the eyes showed that the lower edge of the pupil was obscured by around 1 mm by both eyelids, giving a diagnosis of bilateral ptosis. With respect to an otolaryngology examination, the patient did not cooperate during the hearing screening, but the child responded to his parents' voices and environmental sounds, indicating that he had no hearing loss. Metabolic screening of the blood and urine revealed no abnormalities.
The patient's clinical manifestations, combined with some well-established auxiliary examinations, indicated a genetic disease, so it was recommended that the family members underwent genetic testing to offer further clarification (Table 1). The genomic DNA of a blood sample was extracted with a Qiagen FlexiGene DNA Kit with approval from the Medical Ethics Committee and family consent. The informed consent forms can be found in the Supplementary Material. An Affymetrix CytoScan750K_Array chromosome microarray chip (Affymetrix, Santa Clara, California, USA) was used to detect the potential copy number variation. DNA extraction and quality control, restrictive enzymatic digestion and labeling, hybridization purification, and scanning were performed, along with other related steps. The genomic variation databases OMIM, HGMD, and ClinVar were used to analyze the array data, and the results were as follows: arr3p26.3-p25.3 (147389-10243084) × 1, 3p26.3-p25.3 was missing 10.095Mb in size. After qPCR verification, heterozygous deletion mutations in exons five and 12 of the BRPF1 gene were found (as shown in Table 2). The parents did not have the above-mentioned variant, so it was considered a novel variant, and the patient was diagnosed with 3p deletion syndrome.
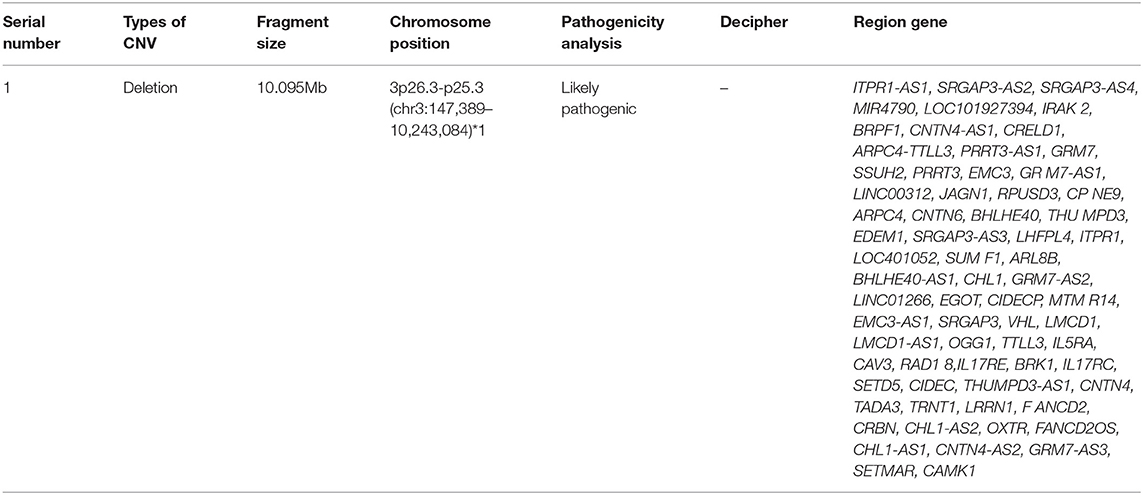
Table 1. Genetic test results.
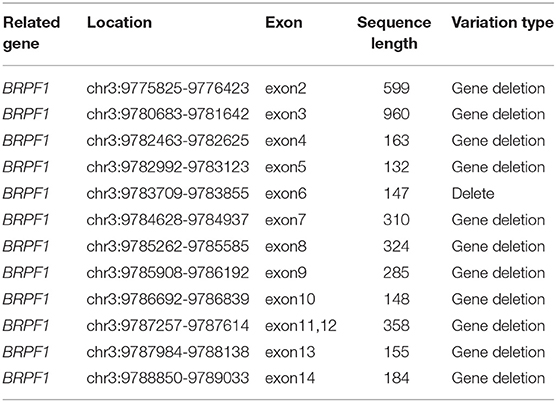
Table 2. Deletion variants of the exons.
The treatment the patient was given consisted of a comprehensive rehabilitation program, which included large joint loosening training, Brunstrom training, exercise system training, head control and climbing strength training, four-point kneeling and one knee kneeling, and sitting training. After more than 12 days of rehabilitation, the responses had improved, and the patient could turn over, clap his hands, chase people and objects, grasp objects, sit up for more than half an hour, and stand for around 3 min.
3p deletion syndrome is a rare contiguous genomic disease. To date, no more than 60 cases have been detected globally. The syndrome is mainly caused by deletion of the short arm of chromosome 3.3p. Currently reported cases mainly show 3p25, 3p25-26, and 3p26 deletions. Both terminal and middle deletions in the short arm of chromosome 3 may cause 3p deletion syndrome, though the syndrome is most common in large terminal de novo deletions. The retrieved clinical data of 29 cases with 3p deletion syndrome reported in China and in other parts of the world are summarized in Table 3. As shown, 19 cases had an onset within 1 year of age, and four pediatric patients died within 3 months of birth. Of these four cases, three died of unknown causes, and one died from complications of influenza.

Table 3. The clinical characteristics and results of the literature review of the 3p deletion syndrome.
3p deletion syndrome was first reported by Verjaal and De Nef in 1978 (10). After more than 40 years of studies, the main clinical phenotypes of the disease have been identified as follows: delayed growth and development, intellectual disability, hypotonia, micrognathia, ptosis, wide nose bridge, long philtrum, low ear position, deformed ears, polydactyly deformity, hearing abnormalities, CHD, renal abnormalities, syndactylism, gastrointestinal abnormalities, and scoliosis (12, 13). The incidence of the main symptoms is 95% for growth retardation, 79% for intellectual disability, 61% for hypotonia, and 42% for ptosis. The incidence of other syndromes is 43% for CHD and 29% for hearing abnormalities. In the present case, the pediatric patient had the main manifestations of delayed growth and development, intellectual disability, hypotonia, micrognathia, ptosis, a wide nose bridge and auricle deformity, and CHD. As the patient did not cooperate with the hearing test, follow-up observation was required to check their hearing.
Exon Slim + CMA chromosome chip testing revealed a 10.095 Mb deletion in 3p26.3-p25.3 and a heterozygous deletion in exons five and 12 in the BRPF1 gene. The patient had most of the main clinical phenotypes, as well as other features, such as CHD. BRPF1 is a chromatin regulator with a PHD domain, bromouracil domain, PWWP domain, and two enhancers (14). PHD is responsible for the activation of three histone lysine acetyltransferases (MOZ, MORF, and HBO1) while bromouracil is used for acetyl-lysine recognition, and the PWWP domain is used for specific interactions with methylated histone H3. BRPF1 forms a complex with MOZ, MORF, and HBO1, and acts as a scaffold to bridge subunit interactions, stimulate acetyltransferase activity, and limit substrate specificity (15). Lysine acetyltransferase 6A (KAT6A) and its secondary chain, KAT6B, form chemical complexes with BRPF1 for the acetylation and propionylation of histone H3 at lysine 23 (H3K23). When missing BRPF1, variant histone H3 acetylation in H3K23 and propionyl defects affect chromatin modification after translation. The lack of BRPF1 can also lead to multiple gene (such as Robo3 and Otx1) transcription reduction (16, 17), the loss of neuronal migration and neural tube closure, the control of transcriptional regulation, and it can cause neurodevelopmental disorders, which can result in intellectual disability. In this case, the child had intellectual disability, and the genetic test results suggested the deletion of BRPF1 gene. Combined with the previous research results, it was proved that the intellectual retardation of this child was caused by the deletion of the BRPF1 gene. In addition, the BRPF1 gene plays a role in craniofacial development, and it has also been confirmed that a lack of BRPF1 can cause ptosis and (or) narrow palpebral fissure (18, 19). According to our statistics, the ptosis occurrence rate is about 40%. In hospitalized cases, during the perfect eye exam, a bilateral palpebral edge of shaded pupil of about 1 mm is considered to be a sign of ptosis. We consider such cases to be BRPF1 ptosis, resulting from gene deletion (20–24). In the present case, the heterozygous deletion of BRPF1 appeared to have led to intellectual disability and ptosis, but not to hearing abnormalities or renal and gastrointestinal malformations. The other clinical phenotypes, such as hypermobility, are thought to have been caused by the absence of large segments of the short arm of chromosome 3.
Previous studies have shown that the size of a deletion can vary, ranging from one to several trillion bases. Furthermore, most deletions are de novo and occur in a family for the first time, though familial cases have also been shown to occur. Not all patients with deficits have the typical features of the syndrome, and some have only mild symptoms or are even completely free of them (25–28, 33). Most patients with 3p deletion syndrome have a moderate or higher intellectual disability. Initially, it was thought that the CHL1 located at 3p26.3 (at the 213649-426090 bp of the p telomere) was associated with neurodevelopment (34), though later genetic studies in familial patients found that a 1.1 Mb deletion covering the CHL1 alone was insufficient to produce a clinical phenotype of the 3p deletion syndrome (35). Genetic analysis of a complex chromosome 3 aberration leading to 3p deletion syndrome conducted by Dijkhuizen and Essen suggested that the main features of 3p deletion syndrome are caused by CNTN4 and CRBN deletions and that CHL1 could be a candidate gene for non-specific developmental delays in the disease (36). As more cases have been discovered, more genes have been found to be linked to intellectual disability. Salwati et al. narrowed down the candidate critical region for intellectual disability to a region ~950 kb in size containing SRGAP3. They therefore concluded that this gene may be the key gene for intellectual disability (37). Peter et al. further reduced the region containing the gene to 485.78 kb (35). However, an SRGAP3 deficiency cannot explain all cases of intellectual disability in 3p deletion syndrome. Gregory reported on a patient with intellectual disability and dysmorphic features and co-analyzed the patient's genes with those of three other reported cases. Here, it was found that the patients shared a missing region, ~124 kb in size, that contained the genes of THUMPD3, SETD5, and LOC440944 (22), which suggested that these genes may correlate with intellectual disability. Xiaofeng et al. reported a case of 3p25.3p25.2 chromosomal heterozygous deletion with gastrointestinal malformation and intellectual disability in a case with 3p deletion syndrome with a missing gene size of 3327 kb containing SETD5, SRGPA3, and SEC13. The deletion of SETD5 and SRGPA3 was associated with intellectual disability, while the deletion of SEC13 may have been responsible for gastrointestinal developmental abnormalities (28). Although recent studies have suggested that SETD5 and SRGPA3 are the key genes involved in intellectual disability in 3p deletion syndrome, the genetic test results in the present case showed that BRPF1 deletion may also cause intellectual disability.
The patient in the present study also had CHD. The candidate genes proposed for CHD in the present study are SEC13R, SLC6A11, and CAV3. Genetic analysis of a further patient with CHD by Helena showed a deletion of SLC6A11, which suggests that it may a candidate gene (13). Cecilia and Cathrine showed that CAV3 haploinsufficiency may cause CHD in 3p deletion syndrome (18). Meanwhile, Timothy et al. proposed locating the cardiac development gene at the junction of D3S1585 and D3S1317, in the region that includes SEC13R, and suggested that it may be a candidate gene for CHD (36–39). However, Elaine and Matthew reported localizing the CHD gene to the D3S1263 to D3S3594 region, in which candidate genes for CHD proposed in previous studies, such as CAV3 and SEC13R, are excluded (37). Salwati analyzed 3p deletion syndrome by microarray and mapped the candidate intervals for CHD susceptibility sites to ~200 kb intervals. On the other hand, some known candidate genes, such as CAV3 and SEC13R, were shown to be outside the target interval. This could be due to the fact that genes beyond the critical interval for CHD may have altered expression due to changes in the deletion of the regulatory regions (29). The CHD-related gene deletions suggested in previous studies were not identified in the present case, which suggested that the development of CHD in the present case may correlate with an absence of large segments of the short arm of chromosome 3.
In addition, some pediatric patients with 3p deletion syndrome may manifest hearing impairment and autistic tendencies, which may correlate with ATP2B2 (38) and CNTN4 deficiency (11, 39), respectively. A small number of patients develop tumors. Sato reported the first case of 3p deletion syndrome combined with cerebellar angioblastoma in a child, which was mainly caused by a VHL gene deletion (30). Some patients with 3p deletion syndrome may also have other chromosomal translocations that lead to a more severe clinical phenotype (31, 32), though genetic microarray analysis can only detect deletions and duplications. The diagnosis of other forms of mutations, including point mutations, inversions, or translocations, requires other tests, such as chromosomal karyotyping.
No abnormalities of the kidney and gastrointestinal tract were found in the present case, but the electroencephalogram showed abnormal discharges. The child did not cooperate with the hearing screening, so auditory disability could not be confirmed. Future follow-up observations are needed to watch for rare manifestations, such as epilepsy.
At present there is still little Chinese research into 3p deletion syndrome, but it is known that the exact location of the deletion can vary and different genes can have different clinical phenotypes. This study describes a case of a typical phenotype of 3 p deletion syndrome, exhibiting intellectual and motor development backwardness, low muscle tone, CHD and ptosis. Genetic test results found a heterozygous mutation of the BRPF1 gene on the short arm of chromosome 3. After more than 2 months of rehabilitation training, the child showed significant growth and progress with their physical responses. Follow-up treatment continued to adhere to the rehabilitation training and was aimed at developing the child's social skills. This case report is expected to provide more references for clinicians to identify the disease. Clinicians should consider 3p deletion syndrome when they are exploring a diagnosis for children with stunting, intellectual disability, ptosis and other special facial manifestations, and the syndrome can be confirmed by means of chromosomal karyotype analysis and gene diagnosis. It has been shown that early rehabilitation therapy and other therapeutic interventions can improve the quality of life of children with this chromosome abnormality.
The original contributions generated for the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by Ethics Committee of The Affiliated Hospital of Inner Mongolia Medical University. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
JF and GY conceived the idea, conceptualized the study, and drafted the manuscript. TW collected the data. ZF and TL analyzed the data. XZ and JS reviewed the manuscript. All authors read and approved the final draft.
This work was supported by Natural Science Foundation of Inner Mongolia Autonomous Region, Project Number: 2015MS08103; Major Project of Inner Mongolia Medical University, Project Number: kjbw2012005; Youth Science and Technology Talents Program-Class A, Project Number: NJYT-17-A19; Major Scientific Research Project of Affiliated Hospital of Inner Mongolia Medical University, Project Number: NTFYZD022; Inner Mongolia Autonomous Region Neurological Disease Clinical Medicine Research Center, Project Number: MYYXT201903.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2021.618059/full#supplementary-material
1. Verjaal M, De Nef MB. A patient with a partial deletion of the short arm of chromosome 3. Am J Dis Child. (1978) 132:43–5. doi: 10.1001/archpedi.1978.02120260045012
2. Narahara K, Kikkawa K, Murakami M, Hiramoto K, Namba H, Tsuji K, et al. Loss of the 3p25.3 band is critical in the manifestation of del(3p) syndrome: karyotype-phenotype correlation in cases with deficiency of the distal portion of the short arm of chromosome 3. Am J Med Genet. (1990) 35:269–73. doi: 10.1002/ajmg.1320350225
3. Phipps ME, Latif F, Prowse A, Payne SJ, Dietz-Band J, Leversha M, et al. Molecular genetic analysis of the 3p- syndrome. Hum Mol Genet. (1994) 3:903–8. doi: 10.1093/hmg/3.6.903
4. Yan K, Rousseau J, Littlejohn RO, Kiss C, Lehman A, Rosenfeld JA, et al. Mutations in the chromatin regulator gene BRPF1 cause syndromic intellectual disability and deficient histone acetylation. Am J Hum Genet. (2017) 100:91–104. doi: 10.1016/j.ajhg.2016.11.011
5. You L, Yan K, Zou J, Zhao H, Bertos NR, Park M, et al. The lysine acetyltransferase activator BRPF1 governs dentate gyrus development through neural stem cells and progenitors. PLoS Genet. (2015) 11:e1005034. doi: 10.1371/journal.pgen.1005034
6. You L, Zou J, Zhao H, Bertos NR, Park M, Wang E, et al. Deficiency of the chromatin regulator BRPF1 causes abnormal brain development. J Biol Chem. (2015) 290:7114–29. doi: 10.1074/jbc.M114.635250
7. Yan K, Rousseau J, Machol K, Cross LA, Agre KE, Gibson CF, et al. Deficient histone H3 propionylation by BRPF1-KAT6 complexes in neurodevelopmental disorders and cancer. Sci Adv. (2020) 6:eaax0021. doi: 10.1126/sciadv.aax0021
8. Naseer MI, Abdulkareem AA, Guzmán-Vega FJ, Arold ST, Pushparaj PN, Chaudhary AG, et al. Novel missense variant in heterozygous state in the BRPF1 gene leading to intellectual developmental disorder with dysmorphic facies and ptosis. Front Genet. (2020) 11:368. doi: 10.3389/fgene.2020.00368
9. Mattioli F, Schaefer E, Magee A, Mark P, Mancini GM, Dieterich K, et al. Mutations in histone acetylase modifier BRPF1 cause an autosomal-dominant form of intellectual disability with associated ptosis. Am J Hum Genet. (2017) 100:105–16. doi: 10.1016/j.ajhg.2016.11.010
10. Ban H, Ren M, Wu L. A infantile patient with 3p deletion syndrome. Chin J Pract Pediatr. (2018) 33:629–30. doi: 10.3760/cma.j.issn.2095-428X.2018.08.020
11. Fernandez T, Morgan T, Davis N, Klin A, Morris A, Farhi A, et al. Disruption of contactin 4 (CNTN4) results in developmental delay and other features of 3p deletion syndrome. Am J Hum Genet. (2004) 74:1286–93. doi: 10.1086/421474
12. Dijkhuizen T, van Essen T, van der Vlies P, Verheij JB, Sikkema-Raddatz B, van der Veen AY, et al. FISH and array-CGH analysis of a complex chromosome 3 aberration suggests that loss of CNTN4 and CRBN contributes to mental retardation in 3pter deletions. Am J Med Genet A. (2006) 140:2482–7. doi: 10.1002/ajmg.a.31487
13. Malmgren H, Sahlén S, Wide K, Lundvall M, Blennow E. Distal 3p deletion syndrome: detailed molecular cytogenetic and clinical characterization of three small distal deletions and review. Am J Med Genet A. (2007) 143A:2143–9. doi: 10.1002/ajmg.a.31902
14. Pohjola P, de Leeuw N, Penttinen M, Kääriäinen H. Terminal 3p deletions in two families–correlation between molecular karyotype and phenotype. Am J Med Genet A. (2010) 152A:441–6. doi: 10.1002/ajmg.a.33215
15. Fernandez TV, García-González IJ, Mason CE, Hernández-Zaragoza G, Ledezma-Rodríguez VC, Anguiano-Alvarez VM, et al. Molecular characterization of a patient with 3p deletion syndrome and a review of the literature. Am J Med Genet A. (2008) 146A:2746–52. doi: 10.1002/ajmg.a.32533
16. Dundar M, Kiraz A, Tasdemir S, Akalin H, Kurtoglu S, Hafo F, et al. Unbalanced 3;22 translocation with 22q11 and 3p deletion syndrome. Am J Med Genet A. (2010) 152A:2791–5. doi: 10.1002/ajmg.a.33249
17. Shuib S, McMullan D, Rattenberry E, Barber RM, Rahman F, Zatyka M, et al. Microarray based analysis of 3p25-p26 deletions (3p- syndrome). Am J Med Genet A. (2009) 149A:2099–105. doi: 10.1002/ajmg.a.32824
18. Gunnarsson C, Foyn Bruun C. Molecular characterization and clinical features of a patient with an interstitial deletion of 3p25.3-p26.1. Am J Med Genet A. (2010) 152A:3110–4. doi: 10.1002/ajmg.a.33353
19. Cuoco C, Ronchetto P, Gimelli S, Béna F, Divizia MT, Lerone M, et al. Microarray based analysis of an inherited terminal 3p26.3 deletion, containing only the CHL1 gene, from a normal father to his two affected children. Orphanet J Rare Dis. (2011) 6:12. doi: 10.1186/1750-1172-6-12
20. Peltekova IT, Macdonald A, Armour CM. Microdeletion on 3p25 in a patient with features of 3p deletion syndrome. Am J Med Genet A. (2012) 158A:2583–6. doi: 10.1002/ajmg.a.35559
22. Kellogg G, Sum J, Wallerstein R. Deletion of 3p25.3 in a patient with intellectual disability and dysmorphic features with further definition of a critical region. Am J Med Genet A. (2013) 161A:1405–8. doi: 10.1002/ajmg.a.35876
23. Omrani MD, Saleh Gargari S, Azizi F, Safavi Naini N, Omrani S. Complex translocation among chromosomes 2, 3, 9, 15, 18, 20 in a patient with 3p-syndrome. Arch Iran Med. (2014) 17:521–2.
24. Suzuki-Muromoto S, Hino-Fukuyo N, Haginoya K, Kikuchi A, Sato H, Sato Y, et al. A case of 3p deletion syndrome associated with cerebellar hemangioblastoma. Brain Dev. (2016) 38:257–60. doi: 10.1016/j.braindev.2015.07.005
25. Zhang K, Song F, Zhang D, Liu Y, Zhang H, Wang Y, et al. Chromosome r(3)(p25.3q29) in a patient with developmental delay and congenital heart defects: a case report and a brief literature review. Cytogenet Genome Res. (2016) 148:6–13. doi: 10.1159/000445273
26. Liu X, Hu Y, Li J, Zhou Y, Li G. A case of 3p deletion and 3q duplication syndrome. Chin J Neonatol. (2018) 33:70. doi: 10.3760/cma.j.issn.2096-2932.2018.01.017
27. Feng L, Cai W, Jiang J, Sun S, Jing C, Yuan L. Clinical phenotype and genetic analysis of a case of a child with 3p26.3-pter deletion and 7q31.33-qter duplication[J]. Chin J Med Genet. (2018) 35:535–9. doi: 10.3760/cma.j.issn.1003-9406.2018.04.017
28. Zhao X, Mao G, Li L, Liu D, Wang N, Qiu W. 3p25.3p25.2 Genetic characteristics and clinical phenotype analysis of a case of chromosomal heterozygous deletion with dwarfism and multiple malformations. J Clin Pediatr. (2020) 38:221–4+8. doi: 10.3969/j.issn.1000-3606.2020.03.015
29. Mowrey PN, Chorney MJ, Venditti CP, Latif F, Modi WS, Lerman MI, et al. Clinical and molecular analyses of deletion 3p25-pter syndrome. Am J Med Genet. (1993) 46:623–9. doi: 10.1002/ajmg.1320460604
30. Moncla A, Philip N, Mattei JF. Blepharophimosis-mental retardation syndrome and terminal deletion of chromosome 3p. J Med Genet. (1995) 32:245–6. doi: 10.1136/jmg.32.3.245-a
31. Cargile CB, Goh DL, Goodman BK, Chen XN, Korenberg JR, Semenza GL, et al. Molecular cytogenetic characterization of a subtle interstitial del(3)(p25.3p26.2) in a patient with deletion 3p syndrome. Am J Med Genet. (2002) 109:133–8. doi: 10.1002/ajmg.10323
32. Riess A, Grasshoff U, Schäferhoff K, Bonin M, Riess O, Horber V, et al. Interstitial 3p25.3-p26.1 deletion in a patient with intellectual disability. Am J Med Genet A. (2012) 158A:2587–90. doi: 10.1002/ajmg.a.35562
33. Iype T, Alakbarzade V, Iype M, Singh R, Sreekantan-Nair A, Chioza BA, et al. A large Indian family with rearrangement of chromosome 4p16 and 3p26.3 and divergent clinical presentations. BMC Med Genet. (2015) 16:104. doi: 10.1186/s12881-015-0251-5
34. Angeloni D, Lindor NM, Pack S, Latif F, Wei MH, Lerman MI. CALL gene is haploinsufficient in a 3p- syndrome patient. Am J Med Genet. (1999) 86:482–5. doi: 10.1002/(SICI)1096-8628(19991029)86:5<482::AID-AJMG15>3.0.CO;2-L
35. Ellery PM, Ellis RJ, Holder SE. Interstitial 3p25 deletion in a patient with features of 3p deletion syndrome: further evidence for the role of SRGAP3 in mental retardation. Clin Dysmorphol. (2014) 23:29–31. doi: 10.1097/MCD.0000000000000017
36. Drumheller T, McGillivray BC, Behrner D, MacLeod P, McFadden DE, Roberson J, et al. Precise localisation of 3p25 breakpoints in four patients with the 3p-syndrome. J Med Genet. (1996) 33:842–7. doi: 10.1136/jmg.33.10.842
37. Green EK, Priestley MD, Waters J, Maliszewska C, Latif F, Maher ER. Detailed mapping of a congenital heart disease gene in chromosome 3p25. J Med Genet. (2000) 37:581–7. doi: 10.1136/jmg.37.8.581
38. McCullough BJ, Adams JC, Shilling DJ, Feeney MP, Sie KC, Tempel BL. 3p– syndrome defines a hearing loss locus in 3p25.3. Hear Res. (2007) 224:51–60. doi: 10.1016/j.heares.2006.11.006
Keywords: 3p deletion syndrome, chromosomal disorder, BRPF1 gene deletion, mental developmental retardation, ptosis
Citation: Fu J, Wang T, Fu Z, Li T, Zhang X, Zhao J and Yang G (2021) Case Report: A Case Report and Literature Review of 3p Deletion Syndrome. Front. Pediatr. 9:618059. doi: 10.3389/fped.2021.618059
Received: 16 October 2020; Accepted: 14 January 2021;
Published: 10 February 2021.
Edited by:
Merlin G. Butler, University of Kansas Medical Center, United StatesReviewed by:
Ana Westenberger, University of Lübeck, GermanyCopyright © 2021 Fu, Wang, Fu, Li, Zhang, Zhao and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guanglu Yang, eWFtZ2d1YW1nbHU0NzFAMTYzLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.