 Hassan Abolhassani
Hassan Abolhassani
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pediatr., 30 April 2021
Sec. Pediatric Immunology
Volume 9 - 2021 | https://doi.org/10.3389/fped.2021.615724
This article is part of the Research TopicMeaningful Cases of Primary Immunodeficiencies, Volume IIIView all 15 articles
Collective clinical and immunologic findings of defects in the CD27–CD70 axis indicate a primary immunodeficiency associated with terminal B-cell development defect and immune dysregulation leading to autoimmunity, uncontrolled viral infection, and lymphoma. Since the molecular mechanism underlying this entity of primary immunodeficiency has been recently described, more insight regarding the function and profile of immunity is required. Therefore, this study aimed to investigate stimulated antibody production, polyclonal vs. virus-specific T-cell response, and cytokine production of a CD70-deficient patient reported previously with early-onset antibody deficiency suffering from chronic viral infections and B-cell lymphoma. The patient and her family members were subjected to clinical evaluation, immunological assays, and functional analyses. The findings of this study indicate an impaired ability of B cells to produce immunoglobulins, and a poor effector function of T cells was also associated with the severity of clinical phenotype. Reduced proportions of cells expressing the memory marker CD45RO, as well as T-bet and Eomes, were observed in CD70-deficient T cells. The proportion of 2B4+ and PD-1+ virus-specific CD8+ T cells was also reduced in the patient. Although the CD70-mutated individuals presented with early-onset clinical manifestations that were well-controlled by using conventional immunological and anticancer chemotherapies, with better prognosis as compared with CD27-deficient patients, targeted treatment toward specific disturbed immune profile may improve the management and even prevent secondary complications.
Common variable immunodeficiency (CVID) is the most common symptomatic primary immune deficiency (PID) characterized by reduced production of immunoglobulins (Igs), predisposing affected individuals to recurrent infections (1, 2). Selected patients may also suffer from autoimmunity, chronic enteropathy, lymphoproliferative disorders, and malignancy (1, 3).
The genetic defects underlying the disease have only been identified in a minority of CVID patients (4, 5). Approximately 5–10% of patients harbor mutations in the TNFRSF13B gene (TACI) (6). Disease-causing mutations in TNFRSF13C (BAFFR), encoding one of the ligands for TACI (7); T-cell co-stimulation gene encoding inducible co-stimulator (ICOS) (8); B-cell co-receptor complex genes including CD19, CD81, CD20, and CD21 (9–12); genes encoding the lipopolysaccharide-responsive vesicle trafficking beach and anchor containing protein (LRBA) (13); cytotoxic T-lymphocyte-associated protein 4 (CTLA4) (14); Ras-related C3 botulinum toxin substrate 2 (RAC2) (15); phosphatidylinositol 3-kinase (PI3K) receptor 1 (PIK3R1) (16); PI3K catalytic subunit delta (PIK3CD); phosphatase and tensin homolog (PTEN); protein kinase C delta (PRKCD) (17); tumor necrosis factor-like weak inducer of apoptosis (TWEAK/TNFSF12) (18); nuclear factor-kappa B1 (NFKB1) (19); NFKB2; and interleukin 21 (IL21) (14) and its receptor IL21R (20) have also been described in a few patients presenting with features of CVID. Furthermore, mutations in tRNA nucleotidyltransferase 1 (TRNT1), IKAROS Family Zinc Finger 1 (IKZF1), interferon regulatory factor 2 binding protein 2 (IRF2BP2), ATPase proton transporting accessory protein 1 (ATP6AP1), Rho guanine nucleotide exchange factor 1 (ARHGEF1), SH3 domain-containing kinase-binding protein 1 (SH3KBP1), SEC61 translocon Subunit alpha 1 (SEC61A1), and mannosyl-oligosaccharide glucosidase (MOGS) have been shown to be associated with the development of the disease (2).
We have recently shown the association of defects in the CD70–CD27 signaling pathway with clinical presentation resembling the CVID phenotype (21–23). CD70 is a co-stimulatory molecule expressed on several types of immune cells including T cells, B cells, and dendritic (DC) cells. Interaction with its ligand, CD27, leads to the signaling cascade in CD70-positive cells via anti-apoptotic kinases, in particular PI3K (24–26). On the other hand, CD70 is essential for triggering the survival and proliferation of CD27-positive immune cells through the IL-2-dependent activation of the NFκB pathway (25, 27, 28). Previous animal studies have also implicated the CD70/CD27 pathway in the regulation of immunity vs. tolerance by several mechanisms including T-cell expansion and survival, co-stimulation of antigen presentation, germinal center formation, B-cell activation, and antibody production (29–31). Although several correlations have been observed between deficient patients and animal models, there are still several immune functions and profiling that should be investigated in these newly discovered genetic defects.
The index family recruited to this study is the first identified family with CD70 deficiency [with confirmed homozygous mutation c.250delT, p.S84Pfs27X (22)]. Written informed consent for this study was obtained from the patients and their relatives, following the principles of the Ethics Committee of Tehran University of Medical Sciences. The immune profile of the index family including complete blood count, lymphocyte subpopulations, serum Ig levels, specific antibody levels, and autoantibodies was documented in our previous report (22, 23, 32). Updated clinical follow-up of the family members was provided, and for more detailed lymphocyte phenotyping, peripheral venous blood was collected both in ethylenediaminetetraacetic acid (EDTA) and later in Streck cell preservative vials (Streck, La Vista, USA) to preserve cells during transport. The samples in the preservative containing vials were processed for flow cytometric immunophenotyping within 36 h. Antibody combinations used for polychromatic 8-color surface staining of lymphocytes have been described previously (33).
Peripheral blood mononuclear cells (PBMCs) were also isolated in parallel from whole blood by Hypaque-Ficoll (GE Healthcare) density gradient centrifugation and cryopreserved in freezing medium (Synth-a-Freeze CTS, Life Technologies). Frozen PBMCs were thawed and cultured at 1 × 106 cells per well in 1 ml complete RPMI 1640 (InvivoGen, USA) supplemented with 10% FBS (HyClone Laboratories), either with medium only, or with T-cell-independent stimulation (2.5 μg/ml human CpG oligodeoxynucleotides, InvivoGen + 1 μg/ml Anti-IgM MP Biomedicals, USA) or T-cell-dependent stimulation (10 ng/ml IL-10, R&D, USA + 300 ng/ml CD40L, Immunokemi, Sweden). Cell proliferation was measured on days 0 and 5 using the CellTiter 96 Aqueous One Solution Kit (Promega, USA), following the instructions from the manufacturer. The Ig concentration in the cell culture supernatant was measured at day 7 by standard enzyme-linked immunosorbent assay (ELISA) as described previously (6). Rabbit anti-human IgA and IgM (1:4,000, Dako, UK) and IgG (1:4,000, Jackson ImmunoResearch, USA) were used as capture antibodies, and peroxidase-conjugated goat anti-human IgA, IgG, and IgM were used as secondary antibodies (1:5,000, Dako, UK).
The protocol for intracellular staining of transcription factors and functional markers after polyclonal and virus-specific stimulation has been described elsewhere (34). Antibodies used for the functional and phenotypic panels of T cells are shown in Supplementary Table 1. LIVE/DEAD Aqua amine dye (Life Technologies) was used to discriminate dead cells or debris. PBMCs were analyzed on a four-laser LSR Fortessa (BD Biosciences). Antibody capture beads (BD Biosciences) were stained individually with all antibodies used in the experiments for compensation setup. Gating analysis was performed using FlowJo 8.8.7 (TreeStar, Ashland, USA). Manual gates were based on unstained cells or fluorescence minus one (FMO) gating strategies as previously described (34, 35). A T-cell response was considered positive if the frequency of cytokine-producing cells were twice the negative background and >0.1% of total CD4+ or CD8+ T cells after background reduction.
The manual gating of the flow cytometry data produced CD4+ and CD8+ T-cell gated data, and the fluorescence shift between the samples was normalized using Gauss norm (36). The visual interactive stochastic neighbor embedding (viSNE) (37) tool was used to map the high-dimensional cytometry data onto a two-dimensional scatterplot. In parallel, the data were analyzed by the PhenoGraph algorithm (38) to allow automated multidimensional clustering of the cells. The subpopulations determined by PhenoGraph were relayed back on to the viSNE map. Heat maps and unsupervised hierarchical clustering of the average fluorescence of each PhenoGraph population were used for visualization. In summary, the flow cytometry data were analyzed using cyt (37), PhenoGraph algorithm (38), SPICE (39), and R environment (40).
The clinical manifestations [recurrent upper respiratory tract infections, varicella pneumonia, a viral infection of the central nervous system, Behçet's syndrome, alopecia areata, and Epstein–Barr virus (EBV)-mediated Hodgkin lymphoma] of two patients from this index family with shared homozygous mutation of c.250delT, p.S84Pfs27X have been reported (22). At the most recent follow-up, the proband (age 33) was clinically stable on monthly immunoglobulin replacement therapy and prophylactic antimicrobial therapy; she only experienced only one admission during the last 5 years due to pneumonia and no sign of lymphoma relapse was observed; P2 (age 37) was intellectually disabled but did not present with other clinical manifestations and without progression of his humoral immunodeficiency (specific antibody deficiency). Moreover, basic humoral immunologic assays (hypogammaglobulinemia, increased percentage of CD38+IgM+ transitional B cells and CD27− naive B cells, and a diminished IgM−IgD−CD27+ class-switched memory B cells) have been identified in the previous report of these CD70-deficient patients (22), resembling B-cell phenotype that is most similar to CVID patients who are classified as B+smB−Trhi according to the EURO classification (41). Our further investigation on the B-cell subset of the proband indicated a slightly increased frequency of plasmablasts and CD21low B cells (Table 1).
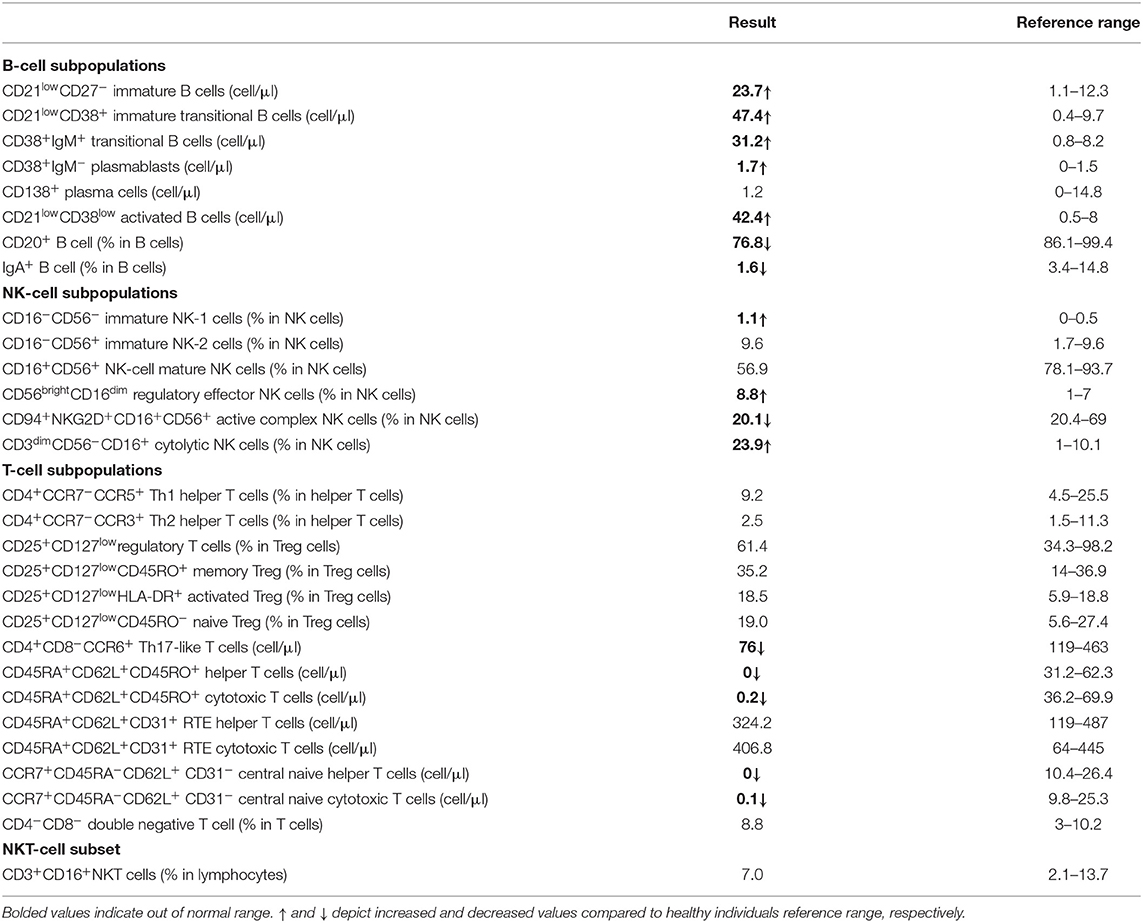
Table 1. Extended B-, T-, and NK-cell subsets within the lymphocytes of the proband with homozygous CD70 mutation.
We have also reported increased frequencies of naive CD4+ and CD8+ T cells, decreases in memory T-cell subsets, and almost normal proportions of NK cells and NK subsets in CD70-deficient patients (22). In line with our previous observation, a more detailed T-cell immunophenotyping was performed for the proband to evaluated T-stem cell memory thymic emigrated (CD45RA+CD62L+CD45RO+) helper and cytotoxic T cells, which showed a reduced number as well as decreased Th17-like T cells. No obvious differences in the proportion of the invariant natural killer T cells (iNKT, TCR Vα24-Jα18) between the proband and normal controls were detected (Table 1).
PBMCs from the proband, the homozygous sibling, a heterozygous relative, a healthy blood donor, and a healthy travel control were tested in vitro for their ability to be induced and produce immunoglobulins. The proliferation response to the T-dependent stimuli was normal in both the proband and the homozygous brother (Supplementary Figure 1). Cells from the proband (with severe phenotype), but not the homozygous brother (with milder phenotype), had a reduced ability to respond to T-independent (anti-IgM+CpG) and T-dependent stimuli (IL-10+CD40L) to produce IgA and IgG, especially the latter (Figure 1).
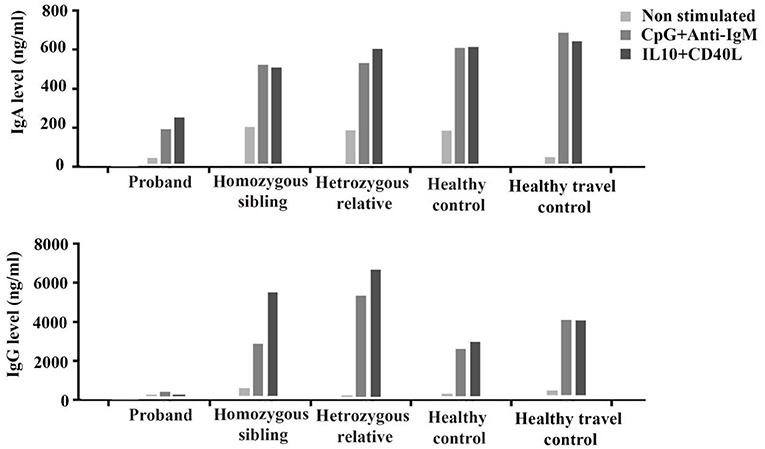
Figure 1. In vitro production of IgA and IgG before (non-stimulated) and after stimulation with T-independent (CpG+anti-IgM) and T-dependent (IL-10+CD40L) cytokines in the proband, the homozygous sibling, a heterozygous relative, and two healthy controls.
To further analyze the T-cell profile in CD70-deficient subjects, we developed polychromatic flow cytometry panels to simultaneously distinguish the expression levels of multiple phenotypic markers (CD45RO, CD27, CCR7, CD57, CD127, CD70) and key transcription factors (T-bet and Eomes) on CD4+ and CD8+ T cells. To achieve a high-dimensional view of the phenotypic and transcriptional heterogeneity of CD4+ and CD8+ T cells between the subjects, CD4+ and CD8+ T cells from each subject were analyzed using the PhenoGraph algorithm. PhenoGraph is an automated clustering approach that defines biologically relevant cell populations from multiparametric flow cytometry datasets without any subjective gating. The hierarchical clustering identified CD70 as the main outlier, not clustering well with other markers of T-cell differentiation. Seventeen CD4+ T-cell and 14 CD8+ T-cell subpopulations were identified based on the combination of the phenotypic and transcriptional flow dataset. By employing hierarchical clustering of the average fluorescence of each PhenoGraph population, we were able to identify T-cell subpopulations that clustered together into naive-, central/transitional memory (CM/TM)-, effector memory (EM)-, and effector (Eff)-like CD4+ and CD8+ T-cell groups (Figures 2A–D). Detailed expression levels of all T-cell markers for each population are listed in Supplementary Tables 2, 3.
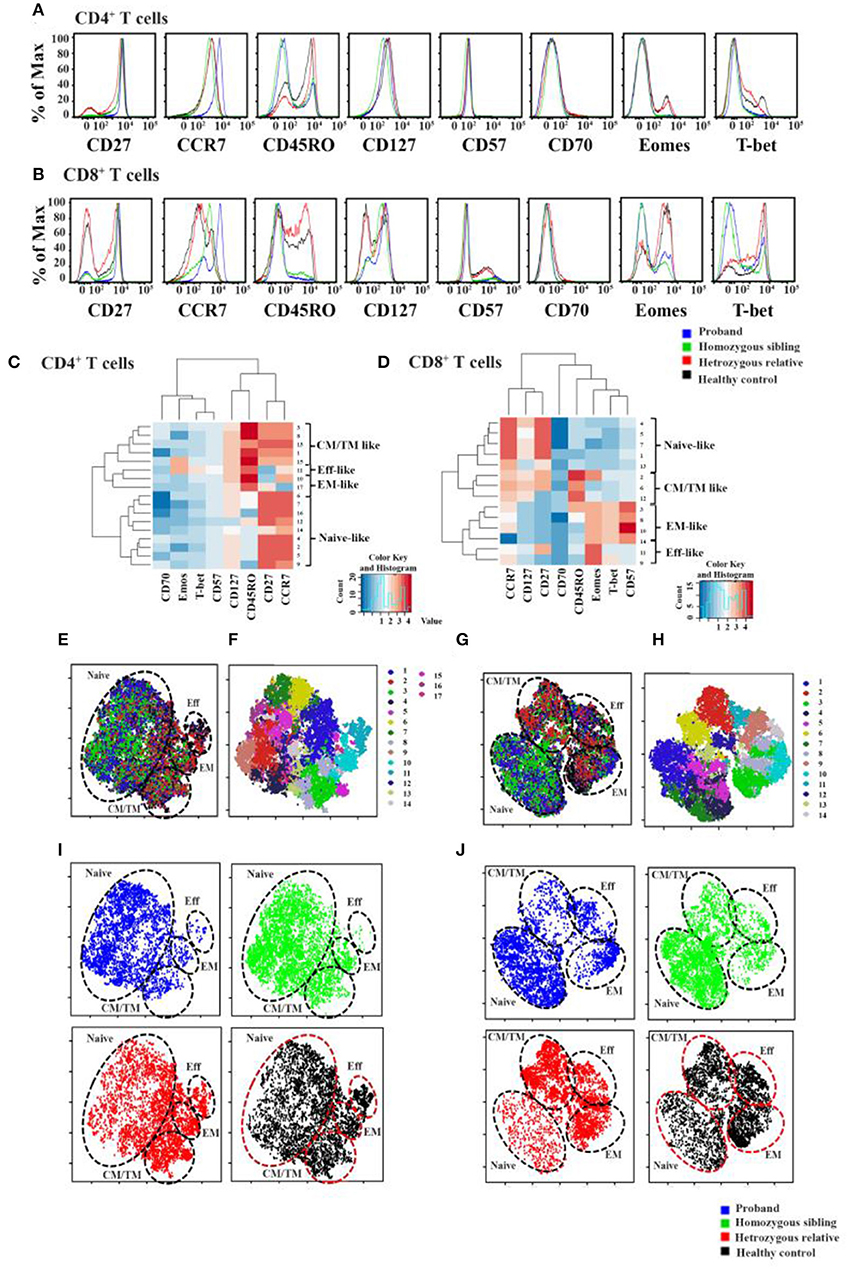
Figure 2. Immunologic markers of cells in CD70-deficient family. Histogram overlays of all differentiation markers (CCR7, CD27, CD57, CD70, CD127, CD45RO, Eomes, and T-bet) for CD4+ (A) and CD8+ cells (B) in two homozygous patients, a heterozygous relative father, and a healthy control. Using an automated density-based algorithm called PhenoGraph, 17 CD4+ T-cell (C) and 14 CD8+ T-cell (D) subclusters were identified based on the intensity of CCR7, CD27, CD57, CD70, CD127, CD45RO, Eomes, and T-bet expression. Hierarchical Spearman clustering was performed to delineate the relationship between the different T-cell clusters using dendograms and heat maps. The relationships between different clusters were identified as naive-, central/transitional memory- (CM/TM-), effector memory- (EM-), and effector (Eff-) like T-cell populations. Localization of each PhenoGraph-derived subcluster and overlapped plotted viSNE map of individuals within the viSNE map for CD4+ and CD8+ T cells was shown in (E,F) and (G,H), respectively (for more information about each PhenoGraph population, see Supplementary Tables 4, 5). In (I,J), each subject's phenotypic data were plotted on the viSNE map, represented with different colors (blue, proband; green, homozygous brother; red, heterozygous father; black, healthy control). Naive-, CM/TM-, EM-, and Eff-like T-cell populations were marked within the viSNE plot based on the subclusters that were determined using PhenoGraph in sections (E–H).
To generate comparisons of the identified T-cell clusters between the different subjects, we applied the viSNE tool in our analysis (37, 42). This generated a representative map of individual cells similar to biaxial plots, but uses pairwise distances that reflect each cell's proximity in a high-dimensional, rather than a two-dimensional, space (Figures 2E–J). Figure 3 illustrates the distribution of CD27 and CD70-positive CD4+ and CD8+ T cells on the representative viSNE plot. A general absence of CD70 expression and increased intensity of CD27 in subclusters enriched with cells from the homozygous patients were observed. Notably, reduced proportions of cells expressing the memory marker CD45RO, as well as T-bet and Eomes, were observed in the proband and her homozygous brother, as compared with the heterozygous relative and healthy control. Furthermore, the homozygous patients demonstrated elevated levels of naive and early differentiated T-cell markers (CCR7 and CD127), particularly for CD8+ T cells. The viSNE approach demonstrated that approximately 70% of all CD4+ T cells for both homozygous individuals were present within different subclusters of naive-like CD4+ T cells, equaling twice as many in comparison with the heterozygous relative and the healthy control. Conversely, fewer cells were present in different EM- and Eff-like clusters in both homozygous patients, especially for the Eff-like cluster 11 expressing both T-bet and Eomes (Figure 2E and Supplementary Table 2). In addition, both homozygous patients showed a general lack of CD45RO+ cells and possessed 3–6 times higher frequencies of naive-like CD8+ T cells than the heterozygous relative and the control. Although some variation was noted between the two homozygous patients, they possessed low frequencies of mainly EM-like (such as clusters 3 and 8) or Eff-like cells co-expressing both T-bet and Eomes (cluster 9) (Figure 2G and Supplementary Table 3).
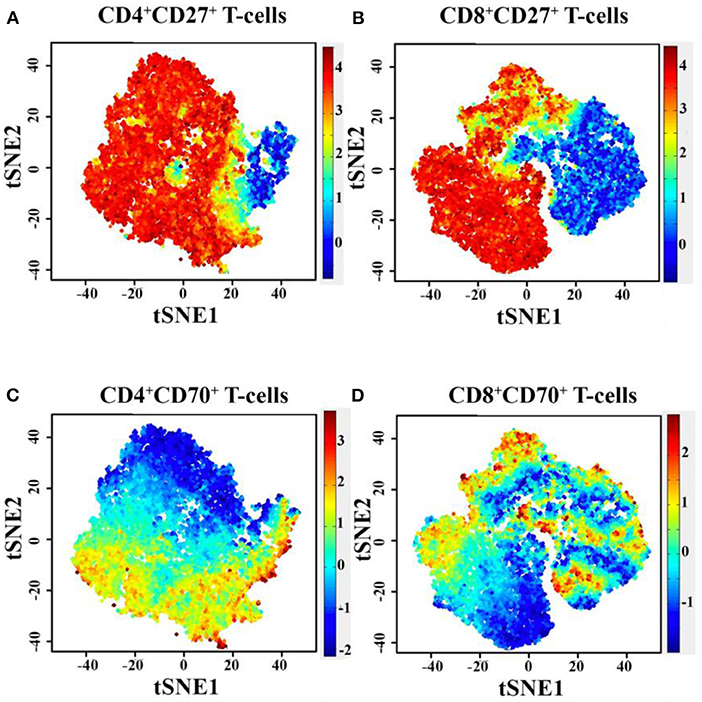
Figure 3. Diagram of PhenoGraph-derived subcluster within the viSNE map, showing the level of CD27 (A,B) and CD70 (C,D) protein expression on CD4+ and CD8+ T cells. t-SNE, t-distributed stochastic neighbor embedding.
Based on the multidimensional phenotypic and transcriptional T-cell profiling data, we next sought to determine the functional characteristics of T cells in CD70-deficient patients. Polyclonal stimulation using Staphylococcal enterotoxin B (SEB) further confirmed that particularly CD8+, but also CD4+, T cells contained low frequencies of responding cells. The lower proportion of SEB-polyclonal CD8+ T cells was strongly associated with a higher frequency of naive T cells in both homozygous individuals (Figures 4A,B). The expression pattern of several effector molecules (IFNγ, TNF, CD107a, and granzyme B) for polyclonal (SEB)-specific T cells was subsequently assessed (Figures 4C,D and Supplementary Tables 4, 5). The SEB stimulations confirmed that particularly CD8+, but also CD4+, T cells possessed low frequencies of responding cells (Figures 4C,D). By employing SPICE analysis to determine the co-expression pattern of all functional markers (Figures 4E,F), we observed that for both of the homozygous patients, there was a clear lack of cells producing two or more cytokines, indicating that Th1 and cytolytic CD4+ T-cell polarization is severely affected by the lack of CD70 (Figure 4E and Supplementary Table 4), as previously demonstrated in murine models (43). In line with the CD4+ T-cell data, polyclonal stimulations revealed that both homozygous cases had very low frequencies of CD8+ T cells capable of producing multiple cytokines after polyclonal stimulations (Figure 4F and Supplementary Table 5). The proliferative response (Ki-67 expression) for a healthy control and proband was also assessed following a 3-day stimulation with the polyclonal antigen SEB. The homozygous proband possessed fewer CD8+ T cells (23.5%) that proliferated in response against SEB than the healthy subject (37.5%, Figure 5).
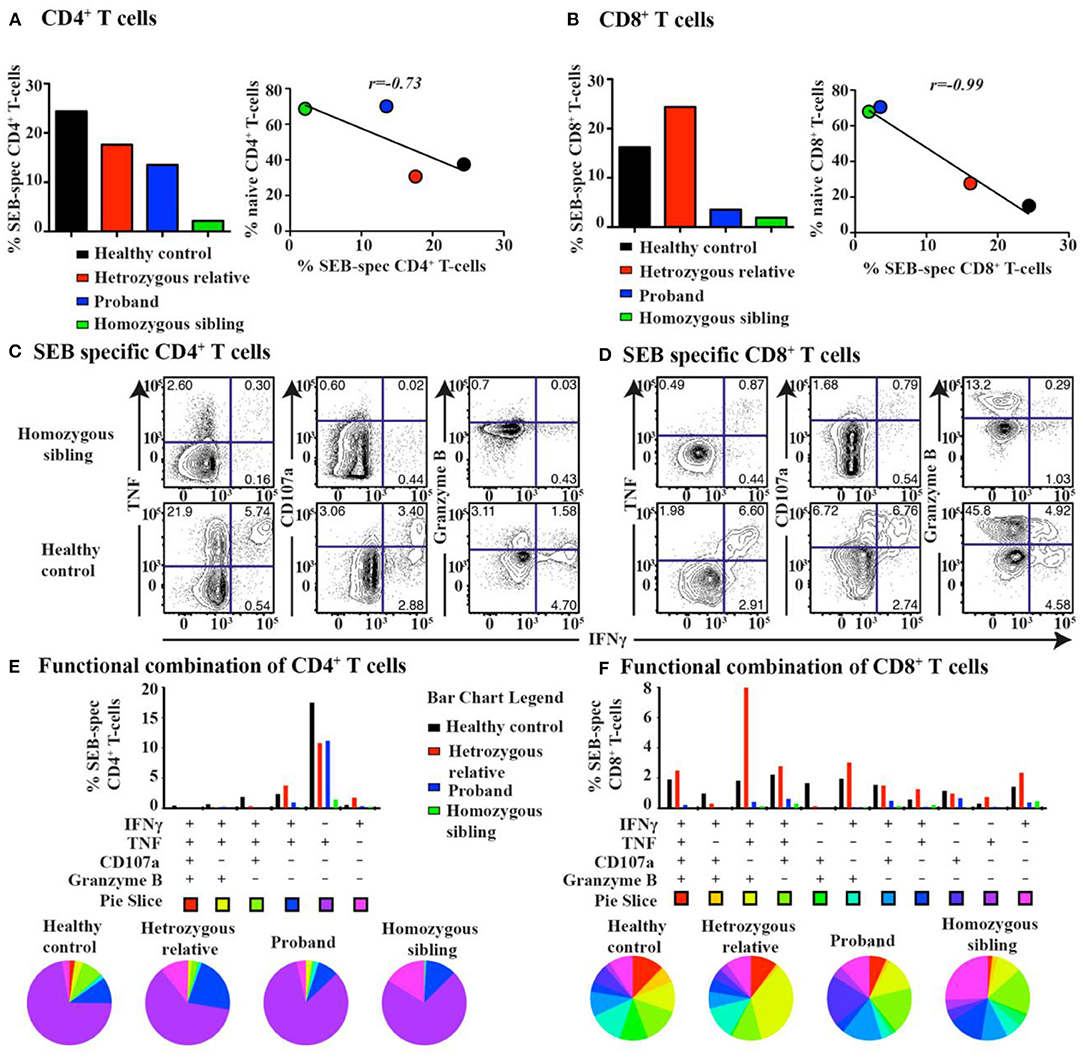
Figure 4. T-cell response and cytokine production after super-antigen simulation in CD70 deficiency. The total frequency of super-antigen (SEB)-specific (A) CD4+ and (B) CD8+ T cells (left). Pearson correlation analysis between the frequency of SEB-specific T cells and naive T cells for all subjects (right). Representative biaxial plots of SEB-specific (C) CD4+ and (D) CD8+ T-cell expression of IFNγ together with TNF, CD107a, or granzyme B for a homozygous patient and a healthy control. Corresponding SPICE analysis of all functional combinations (based on Boolean gating) for each patient's SEB-specific (E) CD4+ and (F) CD8+ T-cell response (below). Functional combinations with no detectable response (<0.05%) are not depicted in the graph.
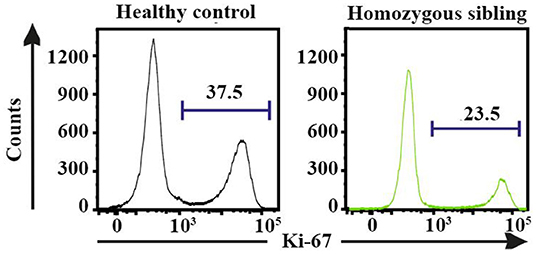
Figure 5. The proliferative (Ki-67+) response for staphylococcal enterotoxin B-specific CD8+ T cells following 3-day stimulations in the homozygous patient and a healthy control.
In our previous report, we presented impaired specific immunity against Epstein–Barr virus (EBV) aligned with high viral and antibody titers for EBV. However, we also observed increased antibody titers against cytomegalovirus (CMV), herpes simplex virus 1 (HSV-1), and varicella-zoster virus (VZV) in the homozygous patients (22), suggesting CD70 deficiency is associated with increased susceptibility to infections by the Herpesviridae family. We, therefore, extended our functional T-cell analyses to assess the CMV-specific T-cell response. A barely detectable response against HCMV-pp65 was observed both in the homozygous patients and heterozygous individuals. The healthy control, however, clearly showed a positive cytolytic response against HCMV-pp65 (Figures 6A–C and Supplementary Tables 4, 5). Intriguingly, the proportion of 2B4+ and PD-1+ CMV-specific CD8+ T cells was reduced in the proband and the homozygous sibling compared with a heterozygous relative and a healthy control after HCMV-pp65 stimulation (Figure 6D).
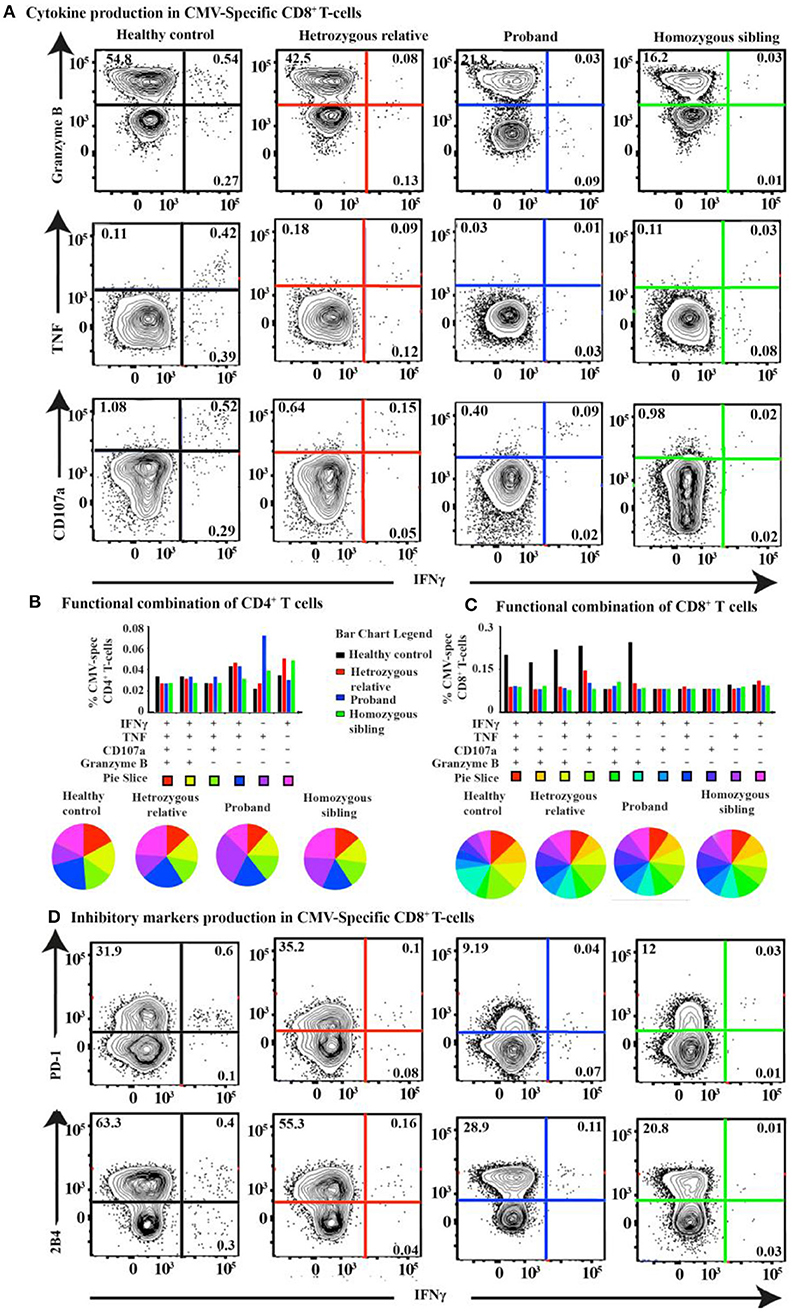
Figure 6. T-cell response and cytokine production after CMV-pp65 antigen simulation in CD70 deficiency. (A) FACS plots of the IFNγ vs. granzyme B, TNF, and CD107a CMV-pp65-specific CD8+ T-cell response for the proband, the homozygous sibling, a heterozygous relative, and a healthy control. Corresponding SPICE analysis of all functional combinations (based on Boolean gating) for each patient's CMV-specific (B) CD4+ and (C) CD8+ T-cell response. (D) FACS plots of the IFNγ vs. PD-1 and 2B4 CMV-pp65-specific CD8+ T-cell responses.
In our previous study, we have shown that the clinical and immunologic findings on the first two CD70-deficient individuals indicate a terminal B-cell developmental defect and a reduced T-cell effector function underlying their antibody deficiency, susceptibility to viral infection, autoimmunity, and lymphoid malignancy. CD27–CD70 interaction is not essential for B-cell development but has been suggested to be important for B-cell activation, germinal center (GC) formation, generation of plasma cells, and Ig production (24, 25, 44). This was in line with the current observations in our CD70-deficient proband that besides hypogammaglobulinemia and reduced proportion of switched memory B cells, the patient had an impaired ability of B cells to secrete IgG and IgA in vitro. In CD27-deficient mice, the levels of virus-specific Igs in the serum and the frequency and pattern of somatic hypermutation (SHM) of GC B cells in response to conjugated chicken γ globulin seem to be normal (45). In a CD70-deficient mouse model, only T-cell function has been thoroughly studied (43, 46). Nevertheless, concerning B-cell function, these mouse models may not fully reflect the role of CD27–CD70 in humans, as CD27 is expressed on a much higher frequency of human GC B cells and may have a more profound impact on GC reactions such as class-switching recombination and SHM in human cells (25). In support of this view, antibody deficiency is also one of the main clinical features in CD27-deficient patients (21, 23). One question remains, however, if the hypogammaglobulinemia is primarily due to intrinsic B- and T-cell defects and/or whether it is secondary to EBV or other viral infections. In several CD27-deficient patients described previously, the serum level of Igs was normal/high initially but subsequently declined dramatically in the months following documented EBV infections (47, 48). The mechanism linking hypogammaglobulinemia and viral infections, however, remains elusive.
Of note, almost 25% of the B cells did not express CD20 which was associated with <2% frequency of plasma cells/plasmablast and the remission phase of lymphoma and absence of treatment with anti-CD20 monoclonal antibodies. Since no mutation was observed in negative or positive regulators of CD20 (data not shown), defective CD27/CD70 signaling might indirectly impact the CD20 regulators, a notion which has been suggested by previous studies (25, 49–51). Moreover, reflecting the discrepancy in the humoral immunity in these homozygous siblings (one with CVID phenotype and another with specific antibody deficiency), the in vitro study showed the impaired ability of B cells to secrete IgG and IgA in proband but not in the homozygous brother. Intriguingly, the high percentage of CD21lowCD38low B-cell subset (10% of total B cells) might be the reason underlying low B-cell response and also probably the specific autoimmune response in the proband but not the homozygous brother, a notion which was supported by the normal count of this specific subset in the latter patient (CD21lowCD38low B cell: 2.4%).
We have identified in our previous report that CD27–CD70 co-stimulation is essential for T-cell activation and CD8+ memory effector functions as illustrated by in vitro analyses (25) and data derived from CD70 knockout mouse models (43, 46, 52). Investigations in our patients provided evidence for a reduced proliferative capacity and cytokine production in both CD4+ and CD8+ T cells in response to either polyclonal stimulation or CMV-specific antigens. Furthermore, the patients' T cells presented with a more “naive”-like phenotype, with reduced expression of the memory T-cell marker (CD45RO) as well as the transcription factors T-bet and Eomes. These new findings support our previous observation on EBV-specific CD8+ T cells from the patients that exhibited a naive-like phenotype (22). Thus, although the T-cell number and subsets were largely unaffected, the T-cell effector function and differentiation of naive precursors into memory cells were abnormal in CD70-deficient patients. As we have shown in our previous study that stem cell-like memory (CD45RA+CCR7+CD95−) has been decreased in association with increased naive markers on CD4 and CD8 T cells, our new observation supports that stem cell-like memory emigrant T cells (CD45RA+CD62L+CD45RO+) are reduced, but total recent thymic emigrant T-cell (CD45RA+CD62L+CD31+) subsets were normal both in helper and cytotoxic T cells in the proband with CD70 deficiency. Considering the recurrent viral (particularly EBV) infection in the proband and distinct functional role of stem cell-like memory compared to other naive emigrant T cells (despite shared similar recirculation patterns and distribution), the CD70–CD27 axis is critical for the development of the former subset with minimally differentiated T cells (53).
Moreover, although NK-cell effector function was normal in CD70−/− mice (46), a reduction of several activation-regulating markers on NK cells was observed in the CD70-deficient proband. In this context, failure to trigger CD27 on NK cells through CD70 directly affects NK activity by decreasing effector–target conjugate formation and IFNγ production of freshly isolated CD27hiCD56bright NK cells (54, 55). The spectrum of viral infections seen in CD70-deficient patients is reminiscent of those in other patients with NK-cell deficiencies (e.g., GATA2 and FCGR3A mutations) presenting with susceptibility to Herpesviridae virus infection due to defect in targeted NK-cell-mediated defenses by downmodulating major histocompatibility complex (MHC) class 1 (56). Taken together, reduced T-cell function and a subtle defect in NK cells may thus underlie the susceptibility to Herpesviridae virus infection in these patients. This also suggests that although several other T-cell co-stimulation pathways exist, such as the OX40/OX40L (57) and 4-1BB/4-1BB-L pathways, the CD27/CD70 interaction has a unique functional role in fine-tuning T-cell-mediated immunity to virus infections.
Behçet's syndrome and alopecia areata were diagnosed in the proband, thereby providing a potential link between CD70 deficiency and a higher risk for autoimmunity. Behçet's syndrome is a rare, systemic inflammatory vascular disorder with unknown etiology, although associations with HLA-B*51 and several non-MHC loci including IL23R-IL12RB2 and IL-10 have been described (58). Besides, environmental factors such as infections, especially viral infections, have been considered to trigger the disease in genetically predisposed individuals (59). Notably, although not diagnosed as Behçet's syndrome, several CD27-deficient patients also suffer from uveitis and EBV-induced oral/perianal ulcers, which are typical signs of this syndrome (21). Furthermore, there have also been some indications that the onset of alopecia areata is associated with EBV infection (60). Our data would therefore suggest that uncontrolled or chronic viral infections, especially with EBV, due to lack of CD27–CD70 co-stimulation, trigger the autoimmunity observed in the index patient. The CD27–CD70 axis has also been suggested to play a role in central tolerance and the development of regulatory T cells (Tregs) (61, 62) and Th17 cells (63). Moreover, overexpression of CD27 and its soluble form and surprisingly also a higher expression of CD70 have been documented in several types of autoimmune disorders including systemic lupus erythematosus, rheumatoid arthritis, multiple sclerosis, and psoriasis (64, 65). Thus, additional mechanisms may explain the phenotype of our patient, and dysregulation of the CD27–CD70 pathway can contribute to various forms of inflammatory and autoimmune disorders, although additional investigations are required to fully understand the mechanisms involved (66).
The lymphoid malignancy observed in the CD70-deficient proband and various other cancers in the family members suggest that CD70 has a potential role in preventing tumorigenesis. There are two major, mutually nonexclusive hypotheses (67): [1] CD27–CD70 interaction is important for generating NK- and T-cell-mediated antivirus responses; thus, the CD70 mutations cause a defective response to the Herpesviridae family, leading to lymphoid proliferation and malignancy; and [2] CD27–CD70 interaction is important for generating NK- and T-cell-mediated antitumor responses, and thus, mutations in CD70 could be considered as a mechanism of immune evasion where malignant cells escape circumvent NK- and T-cell surveillance. In line with these notions, somatic mutations and deletions affecting both alleles of CD70 have been observed in diffuse large B-cell lymphomas (DLBCLs), supporting its potential role as a tumor suppressor (68, 69). However, paradoxically, a higher expression of CD70 in DLBCL is associated with an unfavorable prognosis, and constitutive expression of CD70 has also been described in other types of lymphomas including Hodgkin's lymphoma and mantle cell lymphoma, as well as solid tumors such as clear cell, ovarian, and nasopharyngeal carcinomas (70). Thus, several oncogenic roles of CD70 have been proposed, including recruitment of CD27+ Treg, induction of apoptosis of lymphocytes, and skewing toward T-cell exhaustion (71–73). Therapies targeting the CD27/CD70 axis have furthermore been shown to have some, though not dramatic, clinical effects in kidney cancer patients (70). Our patient lacked CD70 expression, had an EBV+ lymphoma, and showed impaired CD8+ T-cell memory and effector functions. Furthermore, the heterozygous family members presented with various types of cancers. Thus, our result would argue for a tumor suppressor role for CD70, although further analyses would be required to dissect the function of the CD27–CD70 axis in tumorigenesis.
In conclusion, we have extended our previous observation of the first cases of CD70 deficiency in humans (Table 2). Based on the current study, although CD70 appears not to be essential for T- and B-cell development, together with its ligand CD27, it plays a unique role in T-cell-mediated (T-cell activation and CD8+ memory effector function) and B-cell-mediated (B-cell activation, GC formation, generation of plasma cells) immunity. Impaired B-cell Ig induction, poor effector, and memory function of T cells with reduced proportions of CD45RO, T-bet, and Eomes were documented in these patients and need to be evaluated in the future on other patients reported with this group of primary immunodeficiency.
Table 2. A summary of new findings of patients with CD70 deficiency.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
The studies involving human participants were reviewed and approved by the Regional Ethical Review Board (EPN) in Tehran University of Medical Sciences, Tehran, Iran. Written informed consent to participate in this study was provided by the participants.
HA performed the conception and design of the study, acquisition of data, analysis and interpretation of data, drafting the article, and has approved it for publication.
This work was supported by grants provided by the Jonas Söderquist scholarship and Åke Wibergs stiftelse.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank all the donors and their guardians for their willingness to support this work. We also thank support from colleagues at the Department of Laboratory Medicine, Qiang Pan Hammarstrom, Lennart Hammarström, Asghar Aghamohammadi, Likun Du, Marcus Buggert, and Juliet Frederiksen.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2021.615724/full#supplementary-material
1. Hammarstrom L, Abolhassani H, Baldanti F, Marcotte H, Pan-Hammarstrom Q. Development of passive immunity against SARS-CoV-2 for management of immunodeficient patients-a perspective. J Allergy Clin Immunol. (2020) 146:58–60. doi: 10.1016/j.jaci.2020.04.043
2. Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human inborn errors of immunity: 2019 update on the classification from the international union of immunological societies expert committee. J Clin Immunol. (2020) 40:24–64. doi: 10.1007/s10875-019-00737-x
3. Abolhassani H, Aghamohammadi A, Fang M, Rezaei N, Jiang C, Liu X, et al. Clinical implications of systematic phenotyping and exome sequencing in patients with primary antibody deficiency. Genet Med. (2019) 21:243–51. doi: 10.1038/s41436-018-0012-x
4. Yazdani R, Habibi S, Sharifi L, Azizi G, Abolhassani H, Olbrich P, et al. Common variable immunodeficiency: epidemiology, pathogenesis, clinical manifestations, diagnosis, classification, and management. J Investig Allergol Clin Immunol. (2020) 30:14–34. doi: 10.18176/jiaci.0388
5. Yazdani R, Abolhassani H, Kiaee F, Habibi S, Azizi G, Tavakol M, et al. Comparison of common monogenic defects in a large predominantly antibody deficiency cohort. J Allergy Clin Immunol Pract. (2019) 7:864–78.e9. doi: 10.1016/j.jaip.2018.09.004
6. Salzer U, Chapel HM, Webster AD, Pan-Hammarstrom Q, Schmitt-Graeff A, Schlesier M, et al. Mutations in TNFRSF13B encoding TACI are associated with common variable immunodeficiency in humans. Nat Genet. (2005) 37:820–8. doi: 10.1038/ng1600
7. Warnatz K, Salzer U, Rizzi M, Fischer B, Gutenberger S, Bohm J, et al. B-cell activating factor receptor deficiency is associated with an adult-onset antibody deficiency syndrome in humans. Proc Natl Acad Sci USA. (2009) 106:13945–50. doi: 10.1073/pnas.0903543106
8. Grimbacher B, Hutloff A, Schlesier M, Glocker E, Warnatz K, Drager R, et al. Homozygous loss of ICOS is associated with adult-onset common variable immunodeficiency. Nat Immunol. (2003) 4:261–8. doi: 10.1038/ni902
9. Kuijpers TW, Bende RJ, Baars PA, Grummels A, Derks IA, Dolman KM, et al. CD20 deficiency in humans results in impaired T cell-independent antibody responses. J Clin Invest. (2010) 120:214–22. doi: 10.1172/JCI40231
10. van Zelm MC, Reisli I, van der Burg M, Castano D, van Noesel CJ, van Tol MJ, et al. An antibody-deficiency syndrome due to mutations in the CD19 gene. N Engl J Med. (2006) 354:1901–12. doi: 10.1056/NEJMoa051568
11. Thiel J, Kimmig L, Salzer U, Grudzien M, Lebrecht D, Hagena T, et al. Genetic CD21 deficiency is associated with hypogammaglobulinemia. J Allergy Clin Immunol. (2012) 129:801–10.e6. doi: 10.1016/j.jaci.2011.09.027
12. van Zelm MC, Smet J, Adams B, Mascart F, Schandene L, Janssen F, et al. CD81 gene defect in humans disrupts CD19 complex formation and leads to antibody deficiency. J Clin Invest. (2010) 120:1265–74. doi: 10.1172/JCI39748
13. Lopez-Herrera G, Tampella G, Pan-Hammarstrom Q, Herholz P, Trujillo-Vargas CM, Phadwal K, et al. Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity. Am J Hum Genet. (2012) 90:986–1001. doi: 10.1016/j.ajhg.2012.04.015
14. Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med. (2014) 20:1410–6. doi: 10.1038/nm.3746
15. Alkhairy OK, Rezaei N, Graham RR, Abolhassani H, Borte S, Hultenby K, et al. RAC2 loss-of-function mutation in 2 siblings with characteristics of common variable immunodeficiency. J Allergy Clin Immunol. (2014) 135:1380–4. doi: 10.1016/j.jaci.2014.10.039
16. Lucas CL, Zhang Y, Venida A, Wang Y, Hughes J, McElwee J, et al. Heterozygous splice mutation in PIK3R1 causes human immunodeficiency with lymphoproliferation due to dominant activation of PI3K. J Exp Med. (2014) 211:2537–47. doi: 10.1084/jem.20141759
17. Salzer E, Santos-Valente E, Klaver S, Ban SA, Emminger W, Prengemann NK, et al. B-cell deficiency and severe autoimmunity caused by deficiency of protein kinase C delta. Blood. (2013) 121:3112–6. doi: 10.1182/blood-2012-10-460741
18. Wang HY, Ma CA, Zhao Y, Fan X, Zhou Q, Edmonds P, et al. Antibody deficiency associated with an inherited autosomal dominant mutation in TWEAK. Proc Natl Acad Sci USA. (2013) 110:5127–32. doi: 10.1073/pnas.1221211110
19. Abolhassani H, El-Sherbiny YM, Arumugakani G, Carter C, Richards S, Lawless D, et al. Expanding clinical phenotype and novel insights into the pathogenesis of ICOS deficiency. J Clin Immunol. (2020) 40:277–88. doi: 10.1007/s10875-019-00735-z
20. Kotlarz D, Zietara N, Milner JD, Klein C. Human IL-21 and IL-21R deficiencies: two novel entities of primary immunodeficiency. Curr Opin Pediatr. (2014) 26:704–12. doi: 10.1097/MOP.0000000000000160
21. Alkhairy OK, Perez-Becker R, Driessen GJ, Abolhassani H, van Montfrans J, Borte S, et al. Novel mutations in TNFRSF7/CD27: clinical, immunologic, and genetic characterization of human CD27 deficiency. J Allergy Clin Immunol. (2015) 136:703–12.e10. doi: 10.1016/j.jaci.2015.02.022
22. Abolhassani H, Edwards ES, Ikinciogullari A, Jing H, Borte S, Buggert M, et al. Combined immunodeficiency and Epstein-Barr virus-induced B cell malignancy in humans with inherited CD70 deficiency. J Exp Med. (2017) 214:91–106. doi: 10.1084/jem.20160849
23. Ghosh S, Kostel Bal S, Edwards ESJ, Pillay B, Jimenez-Heredia R G, Rao, et al. Extended clinical and immunological phenotype and transplant outcome in CD27 and CD70 deficiency. Blood. (2020) 136:2638–55. doi: 10.1182/blood.2020006738
24. Arens R, Nolte MA, Tesselaar K, Heemskerk B, Reedquist KA, van Lier RA, et al. Signaling through CD70 regulates B cell activation and IgG production. J Immunol. (2004) 173:3901–8. doi: 10.4049/jimmunol.173.6.3901
25. Borst J, Hendriks J, Xiao Y. CD27 and CD70 in T cell and B cell activation. Curr Opin Immunol. (2005) 17:275–81. doi: 10.1016/j.coi.2005.04.004
26. Nolte MA, van Olffen RW, van Gisbergen KP, van Lier RA. Timing and tuning of CD27-CD70 interactions: the impact of signal strength in setting the balance between adaptive responses and immunopathology. Immunol Rev. (2009) 229:216–31. doi: 10.1111/j.1600-065X.2009.00774.x
27. Tesselaar K, Arens R, van Schijndel GM, Baars PA, van der Valk MA, Borst J, et al. Lethal T cell immunodeficiency induced by chronic costimulation via CD27-CD70 interactions. Nat Immunol. (2003) 4:49–54. doi: 10.1038/ni869
28. Jacquot S. CD27/CD70 interactions regulate T dependent B cell differentiation. Immunol Res. (2000) 21:23–30. doi: 10.1385/IR:21:1:23
29. Francosalinas G, Cantaert T, Nolte MA, Tak PP, van Lier RA, Baeten DL. Enhanced costimulation by CD70+ B cells aggravates experimental autoimmune encephalomyelitis in autoimmune mice. J Neuroimmunol. (2013) 255:8–17. doi: 10.1016/j.jneuroim.2012.10.010
30. Denoeud J, Moser M. Role of CD27/CD70 pathway of activation in immunity and tolerance. J Leukoc Biol. (2011) 89:195–203. doi: 10.1189/jlb.0610351
31. Riether C, Schurch C, Ochsenbein AF. Modulating CD27 signaling to treat cancer. Oncoimmunology. (2012) 1:1604–6. doi: 10.4161/onci.21425
32. Abolhassani H, Chou J, Bainter W, Platt CD, Tavassoli M, Momen T, et al. Clinical, immunologic, and genetic spectrum of 696 patients with combined immunodeficiency. J Allergy Clin Immunol. (2018) 141:1450–8. doi: 10.1016/j.jaci.2017.06.049
33. Boldt A, Borte S, Fricke S, Kentouche K, Emmrich F, Borte M, et al. Eight-color immunophenotyping of T-, B-, and NK-cell subpopulations for characterization of chronic immunodeficiencies. Cytometry B Clin Cytom. (2014) 86:191–206. doi: 10.1002/cyto.b.21162
34. Buggert M, Norstrom MM, Salemi M, Hecht FM, Karlsson AC. Functional avidity and IL-2/perforin production is linked to the emergence of mutations within HLA-B*5701-restricted epitopes and HIV-1 disease progression. J Immunol. (2014) 192:4685–96. doi: 10.4049/jimmunol.1302253
35. Norstrom MM, Buggert M, Tauriainen J, Hartogensis W, Prosperi MC, Wallet MA, et al. Combination of immune and viral factors distinguishes low-risk versus high-risk HIV-1 disease progression in HLA-B*5701 subjects. J Virol. (2012) 86:9802–16. doi: 10.1128/JVI.01165-12
36. Hahne F, Khodabakhshi AH, Bashashati A, Wong CJ, Gascoyne RD, Weng AP, et al. Per-channel basis normalization methods for flow cytometry data. Cytometry A. (2010) 77:121–31. doi: 10.1002/cyto.a.20823
37. Amir el AD, Davis KL, Tadmor MD, Simonds EF, Levine JH, Bendall SC, et al. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat Biotechnol. (2013) 31:545–52. doi: 10.1038/nbt.2594
38. Levine JH, Simonds EF, Bendall SC, Davis KL, Amir el AD, Tadmor MD, et al. Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell. (2015) 162:184–97. doi: 10.1016/j.cell.2015.05.047
39. Roederer M, Nozzi JL, Nason MC. SPICE: exploration and analysis of post-cytometric complex multivariate datasets. Cytometry A. (2011) 79:167–74. doi: 10.1002/cyto.a.21015
40. RRDCT. R: A Language and Environment for Statistical Computing. R Found Stat Comput 1: 409. (2011). Available online at: http://www.r-project.org.
41. Wehr C, Kivioja T, Schmitt C, Ferry B, Witte T, Eren E, et al. The EUROclass trial: defining subgroups in common variable immunodeficiency. Blood. (2008) 111:77–85. doi: 10.1182/blood-2007-06-091744
42. DiGiuseppe JA, Tadmor MD, Pe'er D. Detection of minimal residual disease in B lymphoblastic leukemia using viSNE. Cytometry B Clin Cytom. (2015) 10:294–304. doi: 10.1002/cyto.b.21252
43. Munitic I, Kuka M, Allam A, Scoville JP, Ashwell JD. CD70 deficiency impairs effector CD8 T cell generation and viral clearance but is dispensable for the recall response to lymphocytic choriomeningitis virus. J Immunol. (2013) 190:1169–79. doi: 10.4049/jimmunol.1202353
44. Agematsu K, Hokibara S, Nagumo H, Shinozaki K, Yamada S, Komiyama A. Plasma cell generation from B-lymphocytes via CD27/CD70 interaction. Leuk Lymphoma. (1999) 35:219–25. doi: 10.3109/10428199909145724
45. Xiao Y, Hendriks J, Langerak P, Jacobs H, Borst J. CD27 is acquired by primed B cells at the centroblast stage and promotes germinal center formation. J Immunol. (2004) 172:7432–41. doi: 10.4049/jimmunol.172.12.7432
46. Allam A, Swiecki M, Vermi W, Ashwell JD, Colonna M. Dual function of CD70 in viral infection: modulator of early cytokine responses and activator of adaptive responses. J Immunol. (2014) 193:871–8. doi: 10.4049/jimmunol.1302429
47. Salzer E, Daschkey S, Choo S, Gombert M, Santos-Valente E, Ginzel S, et al. Combined immunodeficiency with life-threatening EBV-associated lymphoproliferative disorder in patients lacking functional CD27. Haematologica. (2013) 98:473–8. doi: 10.3324/haematol.2012.068791
48. van Montfrans JM, Hoepelman AI, Otto S, van Gijn M, van de Corput L, de Weger RA, et al. CD27 deficiency is associated with combined immunodeficiency and persistent symptomatic EBV viremia. J Allergy Clin Immunol. (2012) 129:787–9.e6. doi: 10.1016/j.jaci.2011.11.013
49. Wojciech L, Ignatowicz L. Tregs strip dendritic cells of CD70 to regulate Th1 differentiation. EMBO J. (2015) 34:1290–2. doi: 10.15252/embj.201591547
50. Yamamoto H, Kishimoto T, Minamoto S. NF-kappaB activation in CD27 signaling: involvement of TNF receptor-associated factors in its signaling and identification of functional region of CD27. J Immunol. (1998) 161:4753–9.
51. Riether C, Schürch CM, Bührer ED, Hinterbrandner M, Huguenin AL, Hoepner S, et al. CD70/CD27 signaling promotes blast stemness and is a viable therapeutic target in acute myeloid leukemia. J Exp Med. (2017) 214:359–80. doi: 10.1084/jem.20152008
52. Welten, Redeker SP, Franken KL, Benedict CA, Yagita H, Wensveen FM, et al. CD27-CD70 costimulation controls T cell immunity during acute and persistent cytomegalovirus infection. J Virol. (2013) 87:6851–65. doi: 10.1128/JVI.03305-12
53. Gattinoni L, Speiser DE, Lichterfeld M, Bonini C. T memory stem cells in health and disease. Nat Med. (2017) 23:18–27. doi: 10.1038/nm.4241
54. Vossen MT, Matmati M, Hertoghs KM, Baars PA, Gent MR G, Leclercq, et al. CD27 defines phenotypically and functionally different human NK cell subsets. J Immunol. (2008) 180:3739–45. doi: 10.4049/jimmunol.180.6.3739
55. Hayakawa Y, Smyth MJ. CD27 dissects mature NK cells into two subsets with distinct responsiveness and migratory capacity. J Immunol. (2006) 176:1517–24. doi: 10.4049/jimmunol.176.3.1517
56. Orange JS. Natural killer cell deficiency. J Allergy Clin Immunol. (2013) 132:515–25; quiz 526. doi: 10.1016/j.jaci.2013.07.020
57. Byun M, Ma CS, Akcay A, Pedergnana V, Palendira U, Myoung J, et al. Inherited human OX40 deficiency underlying classic Kaposi sarcoma of childhood. J Exp Med. (2013) 210:1743–59. doi: 10.1084/jem.20130592
58. Mizuki N, Meguro A, Ota M, Ohno S, Shiota T T, Kawagoe, et al. Genome-wide association studies identify IL23R-IL12RB2 and IL10 as Behcet's disease susceptibility loci. Nat Genet. (2010) 42:703–6. doi: 10.1038/ng.624
59. Seoudi N, Bergmeier LA, Hagi-Pavli E, Bibby D, Fortune F. The seroprevalence and salivary shedding of herpesviruses in Behcet's syndrome and recurrent aphthous stomatitis. J Oral Microbiol. (2015) 7:27156. doi: 10.3402/jom.v7.27156
60. Rodriguez TA, Duvic M, Areata R. National Alopecia Onset of alopecia areata after Epstein-Barr virus infectious mononucleosis. J Am Acad Dermatol. (2008) 59:137–9. doi: 10.1016/j.jaad.2008.02.005
61. Nolte MA, Arens R, van Os R, van Oosterwijk M, Hooibrink B, van Lier RA, et al. Immune activation modulates hematopoiesis through interactions between CD27 and CD70. Nat Immunol. (2005) 6:412–8. doi: 10.1038/ni1174
62. Appay V, Dunbar PR, Callan M, Klenerman P, Gillespie GM, Papagno L, et al. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat Med. (2002) 8:379–85. doi: 10.1038/nm0402-379
63. Coquet JM, Middendorp S, van der Horst G, Kind J, Veraar EA, Xiao Y, et al. The CD27 and CD70 costimulatory pathway inhibits effector function of T helper 17 cells and attenuates associated autoimmunity. Immunity. (2013) 38:53–65. doi: 10.1016/j.immuni.2012.09.009
64. Wei C, Anolik J, Cappione A, Zheng B, Pugh-Bernard A, Brooks J, et al. A new population of cells lacking expression of CD27 represents a notable component of the B cell memory compartment in systemic lupus erythematosus. J Immunol. (2007) 178:6624–33. doi: 10.4049/jimmunol.178.10.6624
65. Han BK, Olsen NJ, Bottaro A. The CD27-CD70 pathway and pathogenesis of autoimmune disease. Semin Arthritis Rheum. (2015) 45:496–501. doi: 10.1016/j.semarthrit.2015.08.001
66. Asgardoon MH, Azizi G, Yazdani R, Sohani M, Pashangzadeh S, Kalantari A, et al. Monogenic primary immunodeficiency disorder associated with common variable immunodeficiency and autoimmunity. Int Arch Allergy Immunol. (2020) 181:706–14. doi: 10.1159/000508817
67. Kiaee F, Azizi G, Rafiemanesh H, Zainaldain H, Sadaat Rizvi F, Alizadeh M, et al. Malignancy in common variable immunodeficiency: a systematic review and meta-analysis. Exp Rev Clin Immunol. (2019) 15:1105–13. doi: 10.1080/1744666X.2019.1658523
68. de Miranda NF, Georgiou K, Chen L, Wu C, Gao Z, Zaravinos A, et al. Exome sequencing reveals novel mutation targets in diffuse large B-cell lymphomas derived from Chinese patients. Blood. (2014) 124:2544–53. doi: 10.1182/blood-2013-12-546309
69. Bertrand P, Maingonnat C, Penther D, Guney S, Ruminy P, Picquenot JM, et al. The costimulatory molecule CD70 is regulated by distinct molecular mechanisms and is associated with overall survival in diffuse large B-cell lymphoma. Genes Chromosomes Cancer. (2013) 52:764–74. doi: 10.1002/gcc.22072
70. Jacobs J, Deschoolmeester V, Zwaenepoel K, Rolfo C, Silence K, Rottey S, et al. CD70: an emerging target in cancer immunotherapy. Pharmacol Ther. (2015) 155:1–10. doi: 10.1016/j.pharmthera.2015.07.007
71. Claus C, Riether C, Schurch C, Matter MS, Hilmenyuk T, Ochsenbein AF. CD27 signaling increases the frequency of regulatory T cells and promotes tumor growth. Cancer Res. (2012) 72:3664–76. doi: 10.1158/0008-5472.CAN-11-2791
72. Chahlavi A, Rayman P, Richmond AL, Biswas K, Zhang R, Vogelbaum M, et al. Glioblastomas induce T-lymphocyte death by two distinct pathways involving gangliosides and CD70. Cancer Res. (2005) 65:5428–38. doi: 10.1158/0008-5472.CAN-04-4395
Keywords: primary immunodeficiency, inborn errors of immunity, CD70 deficiency, class-switching recombination, T-bet, Eomes, PD-1
Citation: Abolhassani H (2021) Specific Immune Response and Cytokine Production in CD70 Deficiency. Front. Pediatr. 9:615724. doi: 10.3389/fped.2021.615724
Received: 09 October 2020; Accepted: 10 March 2021;
Published: 30 April 2021.
Edited by:
Ivan K. Chinn, Baylor College of Medicine, United StatesReviewed by:
Henner Morbach, University Hospital Würzburg, GermanyCopyright © 2021 Abolhassani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hassan Abolhassani, SGFzc2FuLmFib2xoYXNzYW5pQGtpLnNl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.