 Yu-qing Pan
Yu-qing Pan- Department of Pediatrics, Shengjing Hospital of China Medical University, Shenyang, China
Partial deletion of 10p chromosome is a rare chromosomal aberration. Submicroscopic deletion of 10p15.3 is mainly related to cognitive deficits, speech disorders, motor delay, and hypotonia with the deleted region ranging from 0.15 to 4 Mb. The clinical phenotype is mainly determined by the ZMYND11 and DIP2C genes. Here, we report a rare case of feeding difficulties, hypocalcemia, and psychomotor retardation. Our patient has a 12.48 Mb deletion in 10p15.3–10p13, which is the second case of large 10p deletion among reported cases thus far.
Introduction
Partial deletion of chromosome 10p is a rare chromosomal aberration that is associated with different syndromes (1). It involves a known monogenic syndrome; the hypoparathyroidism, sensorineural deafness, and renal dysplasia (HDR or Barakat) syndrome (OMIM #146255); and DiGeorge syndrome 2 (DGS2), as well as other syndromes (2). HDR is a rare autosomal dominant disorder, which is characterized by varying degrees of HDR. It is caused by the dysfunction of the glutamyl-amidotransferase-subunit A (GATA3) gene located on 10p14 (3). GATA3 plays an important role in the embryonic development of the parathyroid gland, inner ear, kidney, and central nervous system (4). However, haploinsufficiency of a more centromeric region on 10p13–10p14 was previously found to be related to DGS2 (velocardiofacial syndrome complex three), which also includes as a feature congenital heart defects and thymus hypoplasia/aplasia or T cell defects (5). Additional features include renal anomalies, eye anomalies, hypoparathyroidism, skeletal defects, and developmental delay (6). Importantly, hypocalcemia is one of the key characteristics present in DGS2 and HDR. Thus, this characteristic can aid in the diagnosis of the possible genetic disorders (7) (the causes of hypocalcemia are summarized in Supplementary Table 1).
In 10p13–10p15.3 microdeletions, zinc finger MYND-type containing 11 (ZMYND11), disco-interacting protein 2 homolog C (DIP2C), La ribonucleoprotein 4B (LARP4B), and other genes have been reported to be responsible for DGS2, HDR syndrome, or other similar phenotypes (8). The protein encoded by the ZMYND11 gene (also called BS69 or BRAM1), a cellular nuclear protein containing PHD, Bromo, PWWP, and MYND domains, was originally identified as an adenovirus E1A-binding protein that inhibits the transactivation function of E1A (9). It is associated with autosomal dominant non-syndromic intellectual disability (or autosomal dominant mental retardation type 30, OMIM #616083) involving complex cognitive, behavioral, and developmental difficulties (10, 11). The DIP2C gene encodes a member of the disco-interacting protein homolog two family with two other isoforms, DIP2A and DIP2B. It is a candidate for developmental dyslexia and autism (12). The LARP4B gene encodes a member of an evolutionarily conserved protein family implicated in RNA metabolism and translation (13).
Recently, Kim et al. reported the so far largest deletion (16 Mb) in chromosome 10p in a Korean neonate (8). The clinical manifestations included hypoparathyroidism, hearing loss, genitourinary and cardiac anomalies, thymus hypoplasia, and neural system abnormalities and limb deformities.
In the present study, a rare case of feeding difficulties, hypocalcemia, and psychomotor retardation is reported in which our patient harbors a 12.48 Mb deletion in 10p15.3–10p13, which is the second case of large 10p deletion among reported cases so far.
Clinical Report
Our patient was admitted when she was 6 days old to the Department of Neonatology, Shengjing Hospital of China Medical University. She was born at 39 weeks of gestation from a 34-year-old G4P2 mother without asphyxia. Her parents were non-consanguineous, and there was one healthy older female sibling in the family. The patient was spontaneously conceived, and her prenatal history was reportedly uneventful with no exposure to known teratogens. Birth weight, length, and head circumference were within normal ranges. Facial dysmorphism and feeding difficulties were noted soon after birth. Accordingly, the following features were noted: brachycephaly, round face, down-ward slanting palpebral fissures, hypertelorism, curled eye lashes, a broad and low nasal root, micrognathia, high arched palate, low-set ears, muscular hypertonia, irritability, cyanosis during feeding, weak sucking, and severe swallowing dysfunction (Figure 1). In addition, the patient had multiple daily episodes of apnea, desaturation, and cyanosis for a few seconds with spontaneous resolution.
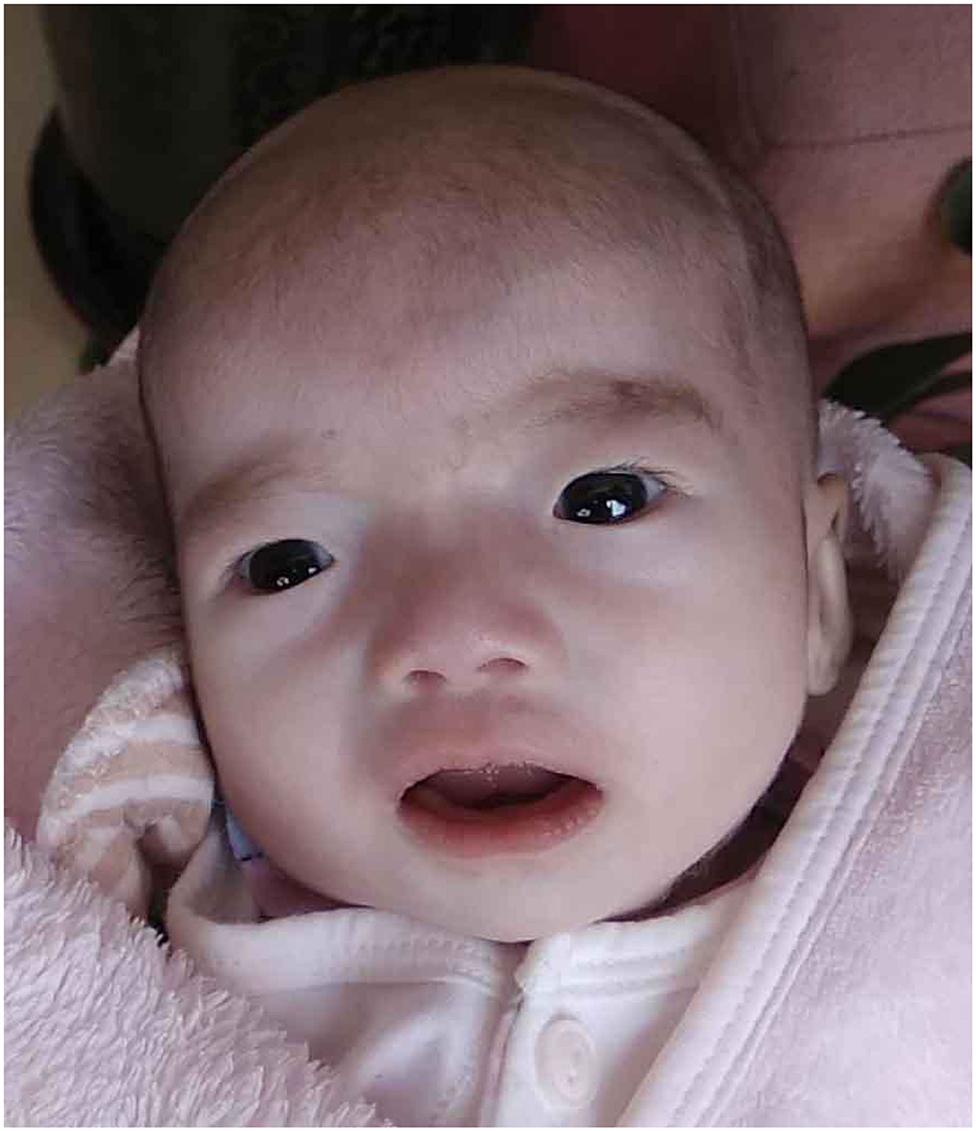
Figure 1. Our patient at the age of 45 days showed brachycephaly, round face, down-ward slanting palpebral fissures, hypertelorism, curled eye lashes, a broad and low nasal root, micrognathia, high arched palate, low-set ears.
She presented with hypoparathyroidism, and her persistent hypocalcemia was difficult to correct despite treatment with calcium gluconate solution and calcitriol. Additionally, parathyroid ultrasound did not demonstrate any anomalies, whereas a chest CT scan showed normal thymus but diffuse bilateral bronchopneumonia, and laryngotracheal CT was normal. The patient's 25-hydroxyvitamin D blood level was low (8.96 ng/ml, normal: 30–70 ng/ml), and immunological studies demonstrated normal counts and a ratio of T and B cells. Myocardial enzyme spectrum was abnormal: creatine kinase (CK) 1,335 U/L (normal: <171 U/L), CK-MB 67 U/L (normal: <24 U/L), lactate dehydrogenase (LDH) 612 U/L (normal: 80–285 U/L), and troponin I: 0.657 μg/L (normal: 0–0.04 μg/L). Myoglobin was normal. Serum creatinine and urine routine test were normal. Thus, laboratory results did not disclose renal functional abnormality. Abdominal ultrasound was planned to be performed on control examination to rule out any urogenital developmental disorder. Brain MRIs performed during hospitalization were found to be normal; however, the electroencephalogram (EEG) was severely abnormal: (1) the sleep–wake cycle was disturbed and not consistent with the corresponding gestational age; (2) EEG activity in the QS phase was abnormal with multifocal spikes and sharp waves released asynchronously; and (3) multiple θ rhythms were issued in the central midline and parietal regions. The infant's clinical manifestations and the abnormal EEG pointed toward a severe neurological dysfunction in the patient. Moreover, upper gastrointestinal radiography exhibited severe swallowing dysfunction of the epiglottis. Visual and brainstem auditory evoked potentials were observed to be normal. Fundoscopy, screening for inborn errors of metabolism, and echocardiography were found to be without a pathological sign.
Materials and Methods
Karyotype Analysis
G-band karyotype analysis from the patient's peripheral blood was performed (14). Karyotypes were reviewed according to the International System for Human Cytogenetics Nomenclature (ISCN 2013).
Detection of Chromosome Copy Number Variation
Genomic DNA was extracted from the patient's peripheral blood using the DNeasy blood and tissue kit (Qiagen GmbH, Hilden, Germany). The quality and concentration of DNA were measured using the NanoDrop spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, USA). Next-generation sequencing (NGS) and copy number variation (CNV) detection was performed by Berry Genomics Corporation (Beijing, China) according to previous studies (15). Briefly, 50 ng of amniocyte DNA was fragmented to an average size of 300 bp. DNA libraries were constructed by end filling, adapter ligation, and PCR amplification as previously described (16). DNA libraries were then subjected to massively parallel sequencing on the NextSeq 500 platform (Illumina, San Diego, CA, USA) in order to generate approximately five million 36-bp single-end reads, representing 0.06–0.1-fold genome coverage. All the sequences were aligned to the unmasked hg19 genome using the Burrows–Wheeler algorithm (17). Mapped reads were allocated progressively to 20 kb bin sizes from the p to q arms of the 24 chromosomes.
Results
The routine karyotype analysis already pointed toward a deletion in one of the short arms of chromosome 10. Subsequent NGS and CNV analyses of the data found a heterozygous 10p15.3–10p13 deletion [arr 10p15.3–p13 (120,000–12,600,000) × 1], illustrating that the patient had an ~12.48 Mb deletion in the short arm of chromosome 10 (Figure 2). The genes located in the deleted region were checked using the UCSC Genome Browser (www.genome.ucsc.edu) (Figure 3). The ZMYND11, DIP2C, LARP4B, and GATA3 were included in the deleted region.
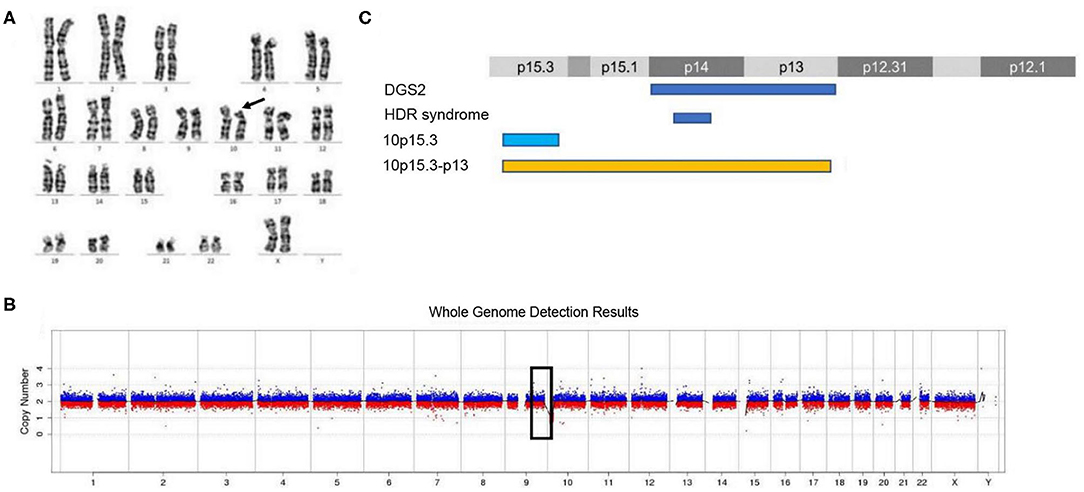
Figure 2. Karyotype analysis and CNV analysis showed ~12.48 Mb deletion at 10p15.3–10p13. (A) Karyotype analysis; (B) CNV sequence; (C) the published deletions in this region. (C) Showed the four deletion types in the 10p15–10p13 region. The deletion of 10p15.3 has been reported by Descipio et al. (11), Vargiami et al. (12), Eggert et al. (18), and Poluha et al. (19). The deletion of 10p15.3–p13 has been reported by Kim et al. (8) and our study.
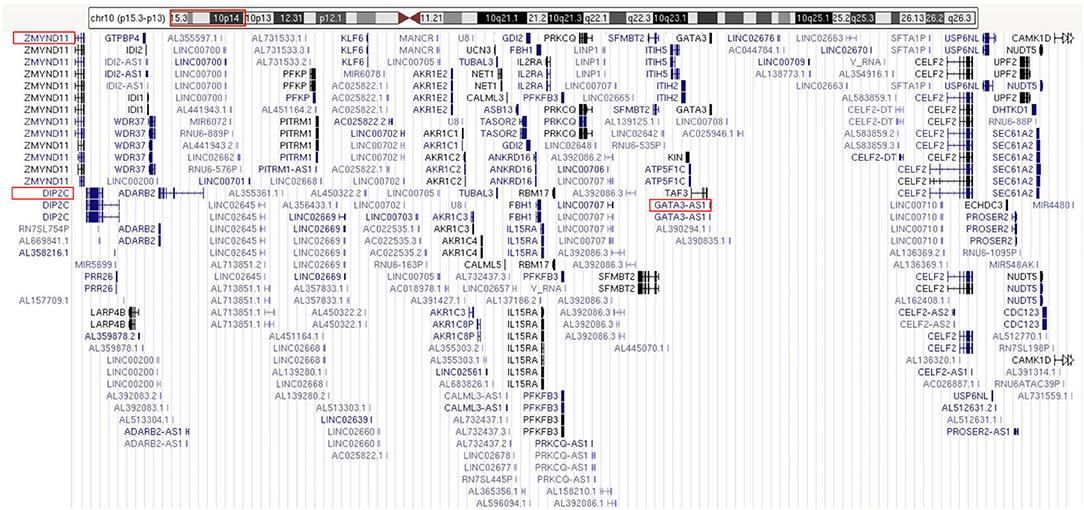
Figure 3. The genes included in the deleted region (chr10: 120,000–12,600,000).
Discussion
The clinical characteristics of reported patients with a microdeletion in the 10p13–15 region are summarized in Table 1. The present patient who has a 12.48 Mb deletion in 10p15.3–10p13 shows clinical features of facial dysmorphism, swallowing dysfunction, hypoparathyroidism, and neurological abnormalities. These phenotypes are not typical of HDR and DGS2, though the patient is only the second reported case with a large deletion.
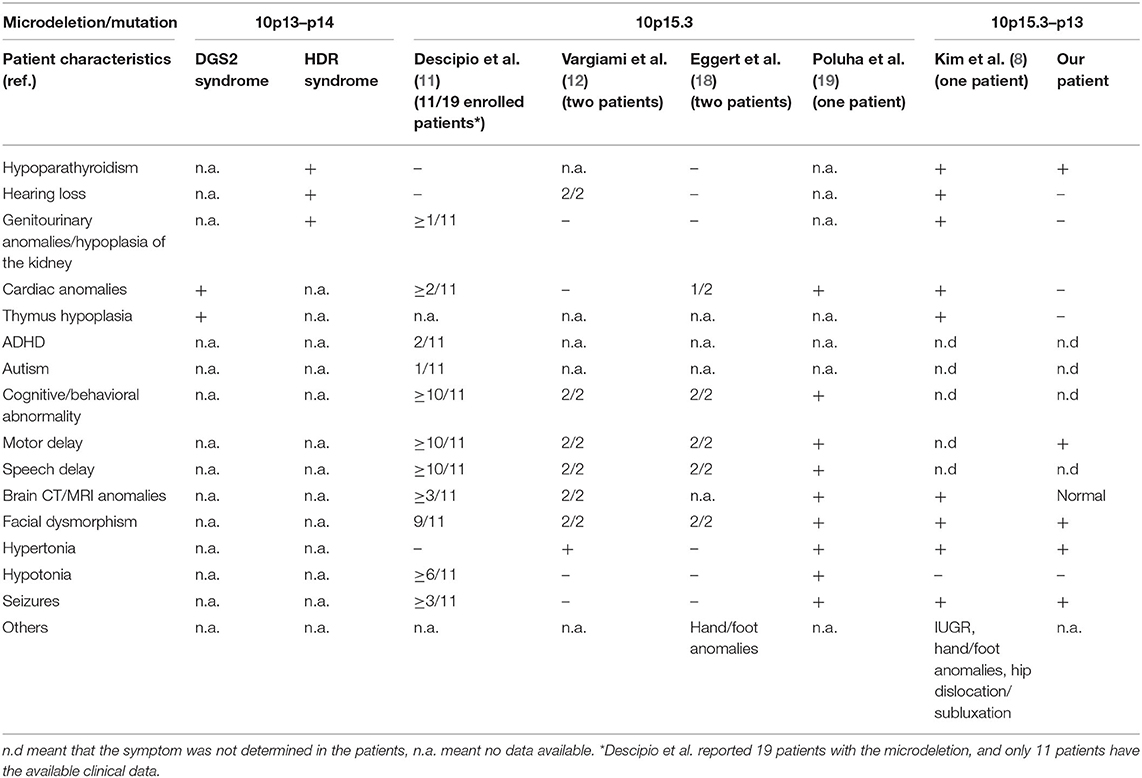
Table 1. Clinical features of patients with a microdeletion/mutation involving the 10p15–10p13 region.
Previously, Tremblay et al. described that the deletion of chromosome 10p15.3–10p15.2 was associated with ventricular septal defects/septal aneurysms among 18 family members in three generations (20). Moreover, 19 unrelated individuals were characterized with submicroscopic deletions involving 10p15.3 (21). Their common clinical features (12 of the 19 individuals) included cognitive/behavioral/developmental difficulties, speech delay/language disorder, motor delay (10/10), craniofacial dysmorphism, hypotonia, brain anomalies, and seizures. A monozygotic female twin pair with a de novo 2.7 Mb deletion of 10p15.3 presented with severe developmental delay, including severe visual and sensorineural hearing impairment. Additionally, both showed generalized dystonia, microcephaly, complete absence of voluntary movements, and visual/auditory unresponsiveness (22). Submicroscopic deletion of 10p15.3 is mainly related to cognitive deficits, speech disorders, motor delay, and hypotonia with a deleted region from 0.15 to 4 Mb (21). However, Gamba et al. reported a male child with a 5.6 Mb deletion at 10p15.3–10p14 who exhibited short stature, cleft lip/palate, and feeding problems (23). In view of the aforementioned studies, the microdeletion of 10p15.3 was inferred to give rise to different features and is seemingly not related to the deletion size.
Tumiene et al. suggested that the clinical features of 10p15.3 microdeletion included neurodevelopmental disorders, characteristic dysmorphic features, and some other more frequent symptoms, and that the ZMYND11 gene was responsible for the above phenotype (24). As a transcriptional repressor, mutations of ZMYND11 have been associated with autosomal dominant mental retardation type 30 leading to intellectual disability, behavioral abnormalities, and seizures (25). Pathogenic single nucleotide variants (SNVs) of ZMYND11 were also associated with Cornelia de Lange syndrome in a large study (26). In addition, the DIP2C gene is located on the minimal region of the overlap of the deletions (24). It is highly expressed in the brain and encompasses various neurological functions, such as “memory,” “neuropeptide signaling pathway,” and “response to amphetamine” according to a gene ontology analysis in the DIP2C gene knock-out mice (12). The deletion of DIP2C could induce the cell enlargement and growth retardation by stimulating DNA methylation (27). However, no pathogenic point mutations or gene deletions of the DIP2C gene have been described so far with a human phenotype. Although the ZMYND11 and DIP2C genes are located on the minimal region of the overlap of the deletions (24), it seems that they serve as key genes for 10p15.3 microdeletion syndrome. However, the deletion is so large that it is impossible to correlate the entire manifestation with the function of single genes. The exact role of the ZMYND11 and DIP2C genes in regard to their clinical features requires further investigation.
In summary, this study reports a patient with a 12.48 Mb deletion in 10p15.3–10p13. The phenotype was not found to be typical of HDR and DGS2, though the patient is only the second reported case having a large deletion thus far.
Data Availability Statement
The original contributions generated for the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee of Shengjing hospital of China Medical University. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
Y-qP performed the data collection and analysis and wrote the original draft. J-hF performed the supervision and edited the writing.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2021.603666/full#supplementary-material
References
1. Melis D, Genesio R, Boemio P, Del Giudice E, Cappuccio G, Mormile A, et al. Clinical description of a patient carrying the smallest reported deletion involving 10p14 region. Am J Med Genet A. (2012) 158A:832–5. doi: 10.1002/ajmg.a.34133
2. Bilous RW, Murty G, Parkinson DB, Thakker RV, Coulthard MG, Burn J, et al. Brief report: autosomal dominant familial hypoparathyroidism, sensorineural deafness, and renal dysplasia. N Engl J Med. (1992) 327:1069–74. doi: 10.1056/NEJM199210083271506
3. Van Esch H, Groenen P, Nesbit MA, Schuffenhauer S, Lichtner P, Vanderlinden G, et al. GATA3 haplo-insufficiency causes human HDR syndrome. Nature. (2000) 406:419–22. doi: 10.1038/35019088
4. Nesbit MA, Bowl MR, Harding B, Ali A, Ayala A, Crowe C, et al. Characterization of GATA3 mutations in the hypoparathyroidism, deafness, and renal dysplasia (HDR) syndrome. J Biol Chem. (2004) 279:22624–34. doi: 10.1074/jbc.M401797200
5. Lichtner P, Konig R, Hasegawa T, Van Esch H, Meitinger T, Schuffenhauer S. An HDR (hypoparathyroidism, deafness, renal dysplasia) syndrome locus maps distal to the DiGeorge syndrome region on 10p13/14. J Med Genet. (2000) 37:33–7. doi: 10.1136/jmg.37.1.33
6. McDonald-McGinn DM, Sullivan KE. Chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Medicine. (2011) 90:1–18. doi: 10.1097/MD.0b013e3182060469
7. Kita M, Kuwata Y, Usui T. Familial congenital choanal atresia with GATA3 associated hypoparathyroidism-deafness-renal dysplasia syndrome unidentified on auditory brainstem response. Auris Nasus Larynx. (2019) 46:808–12. doi: 10.1016/j.anl.2018.10.005
8. Kim SB, Kim YE, Jung JM, Jin HY, Lim YJ, Chung ML. Clinical description of a neonate carrying the largest reported deletion involving the 10p15.3p13 region. Clin Case Reports. (2017) 5:1369–75. doi: 10.1002/ccr3.1070
9. Yang JP, Yang JK, Li C, Cui ZQ, Liu HJ, Sun XF, et al. Downregulation of ZMYND11 induced by miR-196a-5p promotes the progression and growth of GBM. Biochem Biophys Res Commun. (2017) 494:674–80. doi: 10.1016/j.bbrc.2017.10.098
10. Moskowitz AM, Belnap N, Siniard AL, Szelinger S, Claasen AM, Richholt RF, et al. A de novo missense mutation in ZMYND11 is associated with global developmental delay, seizures, and hypotonia. Cold Spring Harbor Mol Case Stud. (2016) 2:a000851. doi: 10.1101/mcs.a000851
11. Coe BP, Witherspoon K, Rosenfeld JA, van Bon BW, Vulto-van Silfhout AT, Bosco P, et al. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat Genetics. (2014) 46:1063–71. doi: 10.1038/ng.3092
12. Oo ZM, Adlat S, Sah RK, Myint MZZ, Hayel F, Chen Y, et al. Brain transcriptome study through CRISPR/Cas9 mediated mouse Dip2c gene knock-out. Gene. (2020) 758:144975. doi: 10.1016/j.gene.2020.144975
13. Kuspert M, Murakawa Y, Schaffler K, Vanselow JT, Wolf E, Juranek S, et al. LARP4B is an AU-rich sequence associated factor that promotes mRNA accumulation and translation. RNA. (2015) 21:1294–305. doi: 10.1261/rna.051441.115
14. Shimono J, Miyoshi H, Kiyasu J, Kamimura T, Eto T, Miyagishima T, et al. Clinicopathological analysis of polyploid diffuse large B-cell lymphoma. PLoS ONE. (2018) 13:e0194525. doi: 10.1371/journal.pone.0194525
15. Liang D, Peng Y, Lv W, Deng L, Zhang Y, Li H, et al. Copy number variation sequencing for comprehensive diagnosis of chromosome disease syndromes. J Mol Diagn. (2014) 16:519–26. doi: 10.1016/j.jmoldx.2014.05.002
16. Liang D, Lv W, Wang H, Xu L, Liu J, Li H, et al. Non-invasive prenatal testing of fetal whole chromosome aneuploidy by massively parallel sequencing. Prenat Diagn. (2013) 33:409–15. doi: 10.1002/pd.4033
17. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. (2009) 25:1754–60. doi: 10.1093/bioinformatics/btp324
18. Eggert M, Muller S, Heinrich U, Mehraein Y. A new familial case of microdeletion syndrome 10p15.3. Eur J Med Genet. (2016) 59:179–82. doi: 10.1016/j.ejmg.2016.02.008
19. Poluha A, Bernaciak J, Jaszczuk I, Kedzior M, Nowakowska BA. Molecular and clinical characterization of new patient with 1.08 Mb deletion in 10p15.3 region. Mol Cytogenet. (2017) 10:34. doi: 10.1186/s13039-017-0336-2
20. Tremblay N, Yang SW, Hitz MP, Asselin G, Ginns J, Riopel K, et al. Familial ventricular aneurysms and septal defects map to chromosome 10p15. Eur Heart J. (2011) 32:568–73. doi: 10.1093/eurheartj/ehq447
21. DeScipio C, Conlin L, Rosenfeld J, Tepperberg J, Pasion R, Patel A, et al. Subtelomeric deletion of chromosome 10p15.3: clinical findings and molecular cytogenetic characterization. Am J Med Genet A. (2012) 158A:2152–61. doi: 10.1002/ajmg.a.35574
22. Vargiami E, Ververi A, Kyriazi M, Papathanasiou E, Gioula G, Gerou S, et al. Severe clinical presentation in monozygotic twins with 10p15.3 microdeletion syndrome. Am J Med Genet A. (2014) 164A:764–8. doi: 10.1002/ajmg.a.36329
23. Gamba BF, Rosenberg C, Costa S, Richieri-Costa A, Ribeiro-Bicudo LA. Cleft lip/palate, short stature, and developmental delay in a boy with a 5.6-mb interstitial deletion involving 10p15.3p14. Mol Syndromol. (2015) 6:39–43. doi: 10.1159/000371404
24. Tumiene B, Ciuladaite Z, Preiksaitiene E, Mameniskiene R, Utkus A, Kucinskas V. Phenotype comparison confirms ZMYND11 as a critical gene for 10p15.3 microdeletion syndrome. J Appl Genet. (2017) 58:467–74. doi: 10.1007/s13353-017-0408-3
25. Yates TM, Drucker M, Barnicoat A, Low K, Gerkes EH, Fry AE, et al. ZMYND11-related syndromic intellectual disability: 16 patients delineating and expanding the phenotypic spectrum. Hum Mutat. (2020) 41:1042–50. doi: 10.1002/humu.24001
26. Aoi H, Mizuguchi T, Ceroni JR, Kim VEH, Furquim I, Honjo RS, et al. Comprehensive genetic analysis of 57 families with clinically suspected Cornelia de Lange syndrome. J Hum Genet. (2019) 64:967–78. doi: 10.1038/s10038-019-0643-z
Keywords: 10p deletion, copy number variation, feeding difficulty, hypocalcemia, psychomotor retardation
Citation: Pan Y-q and Fu J-h (2021) Case Report: Clinical Description of a Patient Carrying a 12.48 Mb Microdeletion Involving the 10p13–15.3 Region. Front. Pediatr. 9:603666. doi: 10.3389/fped.2021.603666
Received: 07 September 2020; Accepted: 13 January 2021;
Published: 25 February 2021.
Edited by:
Katalin Komlosi, Medical Center University of Freiburg, GermanyReviewed by:
Rita Selvatici, University of Ferrara, ItalyJudit Kárteszi, Zala Megyei Szent Raphael Kórház, Hungary
Copyright © 2021 Pan and Fu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jian-hua Fu, fujh77@163.com