
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pediatr., 29 April 2021
Sec. Pediatric Nephrology
Volume 9 - 2021 | https://doi.org/10.3389/fped.2021.544925
This article is part of the Research TopicRecent Advances in Pediatric NephrologyView all 5 articles
 Lingxia Zhang1†
Lingxia Zhang1† Ke Huang2†Shugang Wang3†Haidong Fu1
Ke Huang2†Shugang Wang3†Haidong Fu1 Jingjing Wang1Huijun Shen1Zhihong Lu1Junyi Chen1Yu Bao1
Jingjing Wang1Huijun Shen1Zhihong Lu1Junyi Chen1Yu Bao1 Chunyue Feng1*Guanping Dong2*
Chunyue Feng1*Guanping Dong2* Jianhua Mao1*
Jianhua Mao1*Gitelman syndrome (GS, OMIM 263800) is a genetic congenital tubulopathy associated with salt loss, which is characterized by hypokalemic metabolic toxicity, hypocalciuria, and hypomagnesemia. GS, which is typically detected in adolescence or adulthood, has long been considered a benign tubular lesion; however, the disease is associated with a significant decrease in the quality of life. In this study, we assessed the genotype–phenotype correlations based on the medical histories, clinical symptoms, laboratory test results, and whole-exome sequencing profiles from pediatric patients with GS. Between January 2014 and December 2020, all 31 consecutively enrolled patients complained of fatigue, salt craving, and muscle weakness. Sixteen patients demonstrated growth retardation, and five patients presented with nocturia and constipation. All patients presented with hypokalemic metabolic alkalosis, normal blood pressure, hyperaldosteronism, and a preserved glomerular filtration rate, and 24 of the 31 (77.4%) patients had hypomagnesemia. Homozygous, compound heterozygous, and heterozygous mutations in SLC12A3 were detected in 4, 24, and 3 patients, respectively. GS patients often present with muscle weakness and fatigue caused by hypokalemia and hypomagnesemia. Therefore, early diagnosis of GS is important in young children to reduce the possibility of growth retardation, tetany, and seizures. Next-generation sequencing such as whole-exome or whole-genome sequencing provides a practical tool for the early diagnosis and improvement of GS prognosis. Further whole-genome sequencing is expected to reveal more variants in SLC123A among GS patients with single heterozygous mutations.
Gitelman syndrome (GS, OMIM 263800) is a genetic congenital tubulopathy associated with salt loss that is caused by defects in the sodium chloride cotransporter (NCCT, encoded by SLC12A3) with an autosomal recessive inheritance pattern (1). Epidemiological studies have shown that GS is the most frequent inherited tubulopathy with an estimated prevalence ranging from 1:40,000 in Europeans to 10.3:10,000 in the Japanese population (2), although the proportion of heterozygote carriers can reach up to 1% (3). However, the incidence of GS in the Chinese population is currently unclear (4).
The majority of GS patients present with mild and non-specific symptoms during adolescence or adulthood (1). The main characteristics of GS include hypokalemic metabolic toxicity, hypocalciuria, and hypomagnesemia. These electrolyte abnormalities lead to the common clinical manifestations of muscular weakness, salt craving tetany, and growth retardation. However, the phenotype of GS is highly variable and is linked to quality of life. Currently, a diagnosis of GS is based on clinical symptoms, biochemical abnormalities (i.e., normal/lower blood pressure, increased activity of the renin–angiotensin–aldosterone system, metabolic alkalosis, hypomagnesemia, and hypocalciuria), and genetic testing (5). Recently, next-generation sequencing (NGS) has begun to play an increasingly important role in the diagnosis of GS.
In the present study, we aimed to obtain a clearer picture of the genotype–phenotype relationships in Chinese patients with GS based on laboratory test data, clinical features, and medical histories, along with whole-exome sequencing profiles.
Between January 2014 and December 2020, 31 probands (18 male and 13 female) with a clinical diagnosis of GS were recruited from the Children's Hospital of Zhejiang University School of Medicine. Patients were selected for inclusion in this study according to the classical GS standard: hyperaldosteronism, hypomagnesemia, hypocalciuria, renal hypokalemia, secondary hyperreninism, and metabolic alkalosis. However, plasma renin and aldosterone were not detected in every patient at diagnosis. In addition, hypomagnesemia or hypocalciuria was not described in the records of some genetically confirmed cases; therefore, we did not confirm all of the classical GS diagnostic criteria before genetic testing. Estimated glomerular filtration rates (eGFRs) were calculated using the Schwartz equation (6) that was developed for patients <18 years old. A total of 100 healthy children were selected as control subjects.
Patients with metastatic hypokalemia, potassium loss in the digestive tract, renal tubular acidosis, or medication history with cascara, diuretics, or ethanol were excluded from this study (7).
This study protocol was approved by the Ethics Committee of the Children's Hospital of Zhejiang University School of Medicine. All study participants or their guardians provided written informed consent before enrollment. The collected clinical data were statistically analyzed using an independent samples t-test.
An automatic biochemical analyzer was used to determine the blood concentration of electrolytes. A radioimmunoassay was used to determine plasma aldosterone, plasma renin activity, and plasma angiotensin.
Genomic DNA was extracted from the peripheral blood of the patients using QIAamp Blood DNA Mini Kit (Qiagen, Milano, Italy) according to the manufacturer's instructions. DNA concentrations were determined using a NanoDrop spectrophotometer (Thermo Scientific, Waltham, MA, USA). The DNA samples were then stored at −20°C until analysis.
The whole-exome library was constructed using the Roche Nimble Gen Seq EZ Exome Enrichment Kit with capturing probes V2.0 (Roche, USA), and the DNA of total exons and their flanking introns were enriched. High-throughput sequencing was performed on an Illumina NovaSeq 6000 series sequencer (Illumina, USA), with at least 99% of target sequences sequenced at a 150 × reading depth.
Mutation analysis was performed as previously described (8, 9). For variant annotation and prediction of pathogenicity, the raw data were cleaned and then aligned to the National Center for Biotechnology Information human reference genome (hg18) using the BWA software package. GATK software was used to call single nucleotide polymorphisms and indels (<50 bp), and non-synonymous variations with a minor allele frequency <5% were screened using SIFT. All variations and related diseases were annotated using dbSNP, 1000 Genomes Project, ExAC, ESP, OMIM, Swiss-var, HGMD, ClinVar SNP, and/or disease databases. In addition, the Provean, SIFT, Polyphen2-HVAR, Polyphen2-HDIV, and Mutationtster software packages were used to predict functional changes in all detected variations, and MaxEntScan software was used to assess the impact of splice site variants. No significant difference was observed in the year at diagnosis, serum potassium, serum magnesium, blood pH value, epidermal growth factor receptor (eGFR), standard base excess, plasma renin activity, and plasma aldosterone concentration between patients under or above 7 years according to age of onset (Table 1).
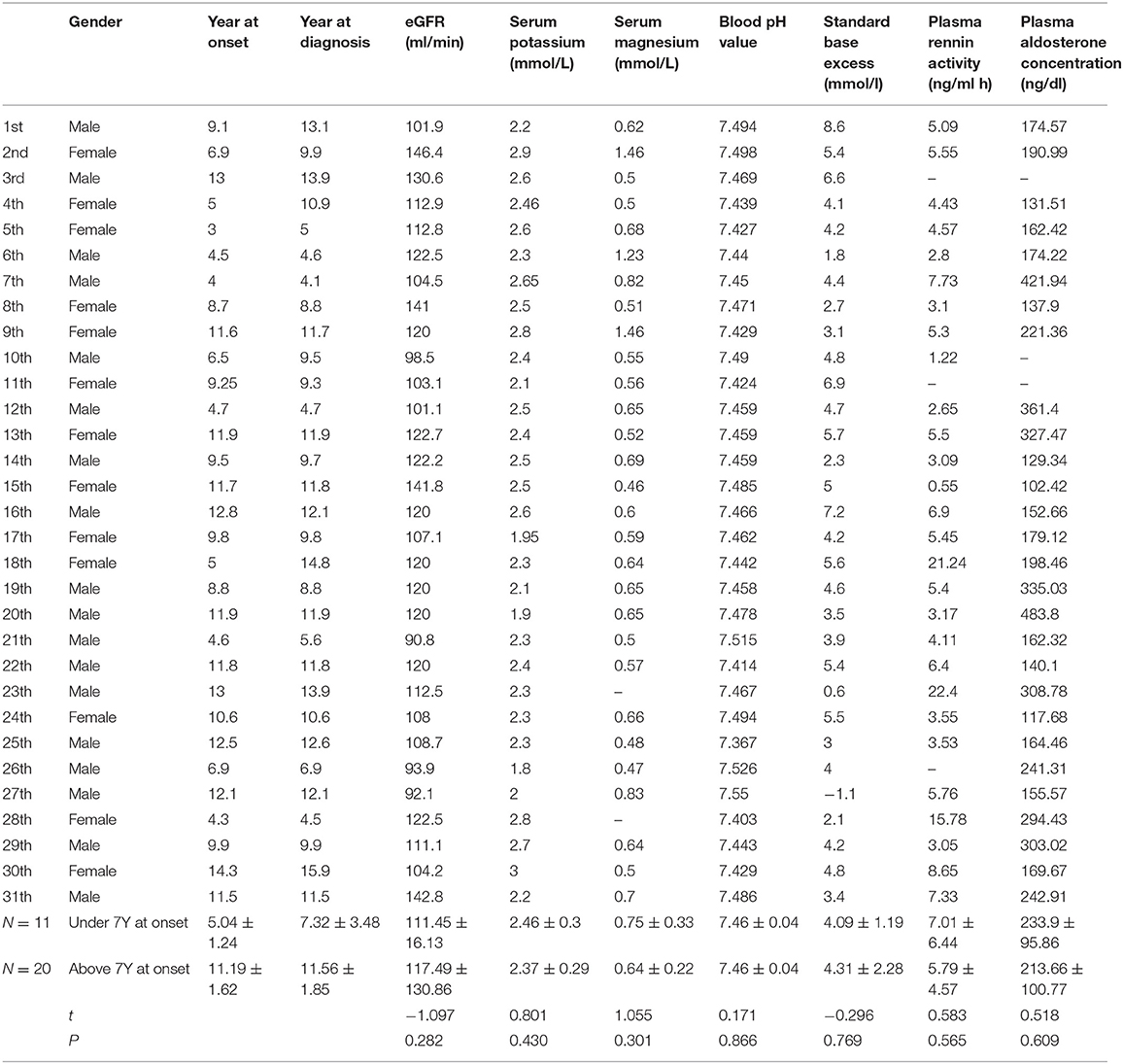
Table 1. Fundamental data and laboratory findings in 31 consecutive children with Gitelman syndrome.
Sanger sequencing was used to confirm the candidate variations associated with GS. DNA extraction was performed as described above. Primer6 software was used for primer design, and polymerase chain reaction was performed using the QIAquick PCR Purification Kit (Qiagen, Milano, Italy). Following ABI BigDye sequencing protocols (cat. no. 4376484; Applied Biosystems; Thermo Fisher Scientific, Inc.), standard Sanger sequencing was performed on an ABI 3130XL genetic analyzer (Applied Biosystems; Thermo Fisher Scientific, Inc.). Sequencher DNA Sequence Analysis Software (http://www.genecodes.com) was used for data analysis.
Between January 2014 and December 2020, 31 consecutive patients (18 boys and 13 girls) with a clinical diagnosis of GS were recruited and sequenced from the Children's Hospital of Zhejiang University School of Medicine.
The clinical and biochemical features of the 31 patients with GS are shown in Table 1. All patients complained of fatigue, salt craving, and muscle weakness. Sixteen patients demonstrated growth retardation, and five patients presented with nocturia and constipation.
All patients presented with hypokalemic metabolic alkalosis, normal blood pressure, hyperaldosteronism, and a preserved glomerular filtration rate, and 24/31 (77.4%) patients had hypomagnesemia (normal range, 0.73–1.06 mmol/L). Twelve of the 31 patients presented with hypocalciuria. No significant difference was observed in the year of onset, year at diagnosis, serum potassium, serum magnesium, blood pH value, eGFR, standard base excess, plasma renin activity, and plasma aldosterone concentration according to age (Table 1).
A total of 36 different mutations were identified in 18 of the 26 exons of SLC12A3 from the 31 consecutive patients, 25 of which were previously reported and 11 of which were novel (Supplementary Table 1).
Furthermore, among the 36 different mutations detected, the most common mutation types were missense mutations (23/36, 63.9%), splicing mutations (7/36, 19.4%), frameshift mutations (4/36, 11.1%), non-frameshift deletion (1/36, 2.8%), and non-sense mutation (1/36, 2.8%).
Among them, 21 mutation loci were located in the transmembrane coding region of the encoded protein NCCT, and 11 mutation loci were located in the coding region of the membrane.
Only 3 of the 36 cases exhibited a single heterozygous mutation, which were all frameshift or splicing mutations (cases 13, 18, and 26). Homozygous mutations were found in four patients with non-sense or missense mutations (cases 5, 8, 9, and 28). All remaining children carried compound heterozygous mutations (24/31, 77.4%) (Table 2 and Figure 1).
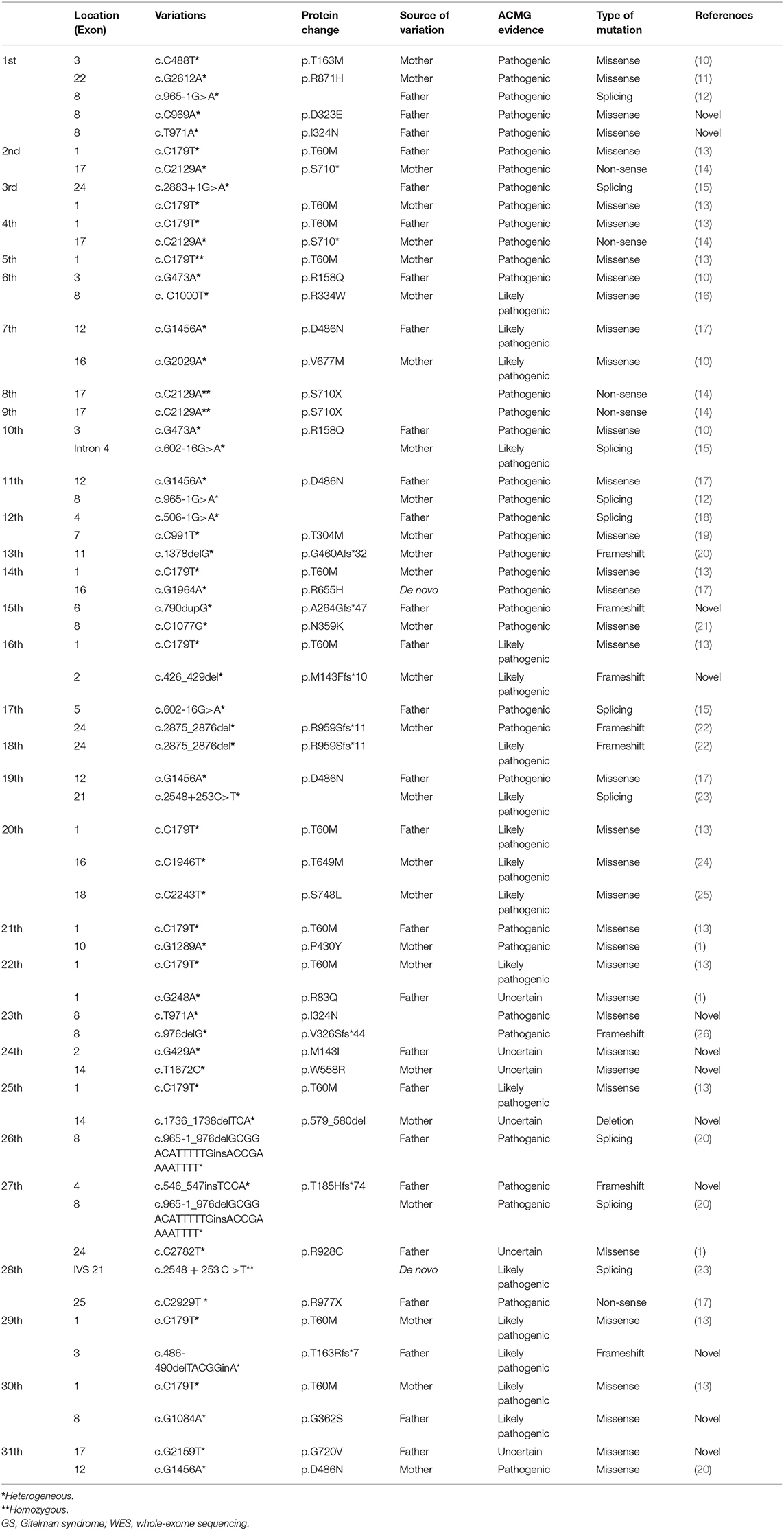
Table 2. SLC12A3 mutations by WES and corroborated sanger sequencing in patients with clinically diagnosed GS.
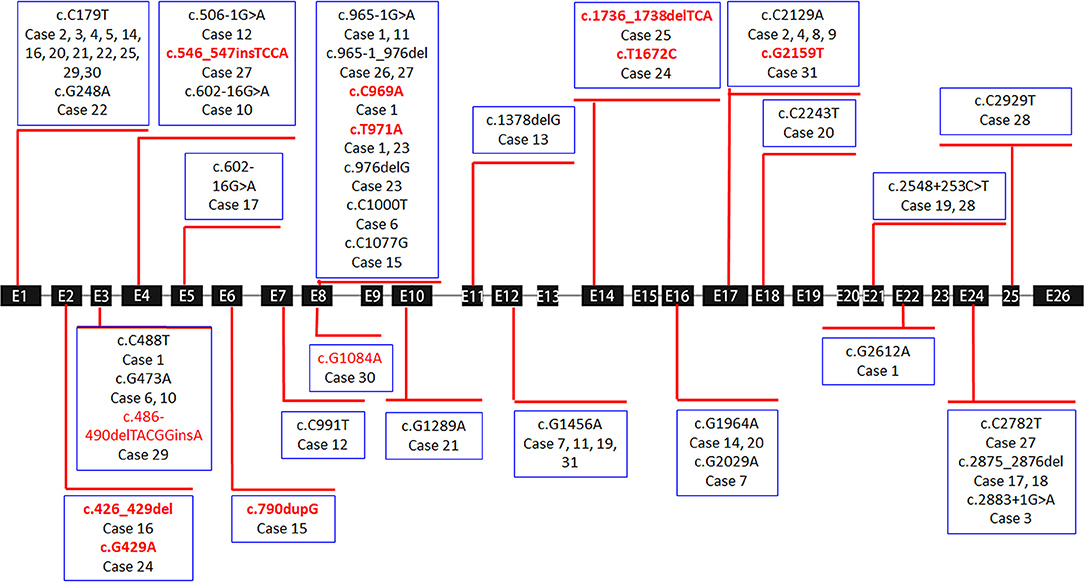
Figure 1. Exon structure of the SLC12A3 gene with geometric shapes indicating the relative positions of different types of mutations. The 11 different types of novel mutations identified in this study are marked in red.
The predicted protein changes caused by the 11 novel mutations are summarized in Figure 2: p.D323E, p.G362S, p.M143I, p.M143Ffs*10, p.W558R, p.579_580del, p.A264Gfs*47, p.T163Rfs*7, p.G720V, and p.T185Hfs*74 are indicated in hot pink, and p.I324N is marked in light pink. These mutations were all found in the amino acid permeases domain (amino acids 141–647; http://pfam.xfam.org/protein/P55017) of NCCT protein.
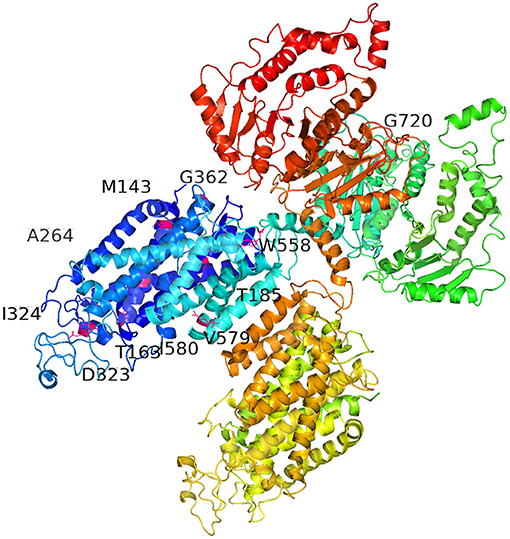
Figure 2. Three-dimensional model of sodium chloride cotransporter (NCCT) showing the locations of the 11 novel SLC12A3 mutations, including missense, non-sense, deletion, and frameshift mutations, which resulted in the following amino acid variations: p.D323E, p.G362S, p.M143I, p.M143Ffs*10, p.W558R, p.579_580del, p.A264Gfs*47, p.T163Rfs*7, p.G720V, and p.T185Hfs*74 (hot pink), and p.I324N (light pink). These mutations are found in the amino acid permeases domain (amino acids 141–647; http://pfam.xfam.org/protein/P55017). Full protein sequences of the SLC12A3 mutations were submitted to SWISS MODEL (http://swissmodel.expasy.org/) servers. The figure was generated using Pymol 2.3.2.
All patients harboring the novel mutations presented with hypokalemic metabolic alkalosis, hypomagnesemia, salt craving, and muscle weakness. Increased plasma renin appeared in some patients (cases 1, 16, 23, 25, 27, 30, and 32). Growth retardation was present in case 25, and tetany and hyperuricemia were found in case 30.
In this study, 31 children with newly diagnosed GS were subjected to clinical testing and genetic sequencing. All patients presented with hypokalemic metabolic alkalosis, normal blood pressure, hyperaldosteronism, and a preserved glomerular filtration rate, and 24/31 (77.4%) patients had hypomagnesemia. Whole-exome sequencing identified 4 cases with homozygous mutations, 3 cases with a single heterozygous mutation, and 24 cases with compound heterogeneous mutations in the causal gene SLC12A3.
GS is an autosomal recessive disorder caused by loss-of-function mutations in SLC12A3 (24), and the majority of patients exhibit homozygous or compound heterozygous mutations of SLC12A3. However, based on SLC12A3 mutation screening, 18–40% of GS patients are typically found to carry only one mutant allele (1, 14, 24, 27). Because GS is evidently recessive, and heterozygous relatives of patients with GS are clinically and metabolically asymptomatic, the reason for this phenomenon is unclear (28). Before the clinical introduction of NGS, the single-strand conformation polymorphism approach was most commonly used for mutation screening; however, this method is limited in resolution power. Moreover, traditionally, only exon and exon-boundary single nucleotide variants have been screened for GS, which may exclude relevant variations in the promoter region, introns, signaling poly(A) region, and both the 5′ and 3′ untranslated regions, which may explain why these patients were identified as single heterozygous carriers (24). In the present study, whole-exome sequencing and subsequent Sanger sequencing were applied in clinically diagnosed GS patients, revealing that only 3/31 (9.7%) carried heterozygous mutations (frameshift mutation in cases 13 and 18 and a splicing mutation in case 26). Thus, the incidence of heterozygous mutation in our study is much lower than that reported previously (18–40%) (1, 14, 24, 27). Although the reason for this discrepancy is unknown, we recommend whole-exome or whole-genome sequencing for patients with GS to identify undiscovered variations in SLC12A3, which may increase the identification rate for genetic diseases such as GS.
GS has long been deemed a benign disorder, which is generally diagnosed during adolescence or adulthood. However, with the introduction of NGS, the establishment of a GS diagnosis is becoming easier in young children. This can be meaningful as GS is associated with a significant reduction in quality of life and is not always a benign tubulopathy, as mentioned by Cruz et al. (16).
GS often manifests with symptoms of cramps/muscle weakness, dizziness, fatigue, salt craving, thirst, nocturia/nocturnal enuresis, low blood pressure, constipation, carpopedal spasms, or tetanic episodes triggered by hypomagnesemia. Treatment of hypomagnesemia is difficult. In GS patients with hypomagnesemia, magnesium supplementation should be considered first, which can accelerate potassium repletion to reduce the risk of tetany and other complications (29, 30). Oral magnesium supplements are appropriate to correct hypomagnesemia, which deteriorates to hypokalemia and makes it difficult to supply potassium. Although all types of magnesium salts are effective (31), their bioavailability is highly variable, resulting in osmotic diarrhea at high doses (32). For patients with acute and severe complications of hypomagnesemia (such as tetany, arrhythmia) or digestive intolerance to oral magnesium supplements, intravenous infusion of magnesium should be used (3). Furthermore, a series of drugs should be avoided for patients with GS, including drugs that slow down the sinus rhythm or influence the QT interval, drugs that can potentially exacerbate hypomagnesemia, and acetazolamide.
Above all, GS patients often present with muscle weakness and fatigue caused by hypokalemia and hypomagnesemia, which contribute to a significant reduction in quality of life. Therefore, early diagnosis of GS is necessary in young children to reduce the possibility of growth retardation, tetany, and seizures. NGS such as whole-exome or whole-genome sequencing provides a practical tool for the early diagnosis and improvement of the prognosis of GS. Importantly, further whole-genome sequencing is expected to reveal more variants in SLC123A among GS patients with single heterozygous mutations.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: NCBI, SRA repository with accession number SUB7270931.
The studies involving human participants were reviewed and approved by The Ethics Committee of the Children Hospital of Zhejiang University School of Medicine. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
LZ, KH, and SW contributed to patient service and data collection. CF, GD, and JM contributed to study conception and design. HF, JW, HS, and ZL collected the data. JC, YB, and SW contributed to data analysis and interpretation. All authors participated in drafting of the article or key modifications of important content and approved the final version to be published.
This study was supported by the National Natural Foundation of China (U20A20351, 81770710), Key Research and Development Plan of Zhejiang Province (2021C03079, 2019C03028), Major projects jointly constructed by Zhejiang province and the National Health Commission (WKJ-ZJ-1908), and Natural Science Foundation of Zhejiang Province (LQ18H050001).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We acknowledge all patients for participating in the present study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2021.544925/full#supplementary-material
1. Vargas-Poussou R, Dahan K, Kahila D, Venisse A, Riveira-Munoz E, Debaix H, et al. Spectrum of mutations in Gitelman syndrome. J Am Soc Nephrol. (2011) 22:693–703. doi: 10.1681/ASN.2010090907
2. Tago N, Kokubo Y, Inamoto N, Naraba H, Tomoike H, Iwai N. A high prevalence of Gitelman's syndrome mutations in Japanese. Hypertens Res. (2004) 27:327–31. doi: 10.1291/hypres.27.327
3. Knoers NVAM, Levtchenko EN. Gitelman syndrome. Orphanet J Rare Dis. (2008) 3:22. doi: 10.1186/1750-1172-3-22
4. Wang F, Shi C, Cui Y, Li C, Tong A. Mutation profile and treatment of Gitelman syndrome in Chinese patients. Clin Exp Nephrol. (2017) 21:293–9. doi: 10.1007/s10157-016-1284-6
5. Urwin S, Willows J, Sayer JA. The challenges of diagnosis and management of Gitelman syndrome. Clin Endocrinol. (2020) 92:3–10. doi: 10.1111/cen.14104
6. Schwartz GJ, Gauthier B. A simple estimate of glomerular filtration rate in adolescent boys. J Pediatr. (1985) 106:522–6. doi: 10.1016/S0022-3476(85)80697-1
7. Sinha A, Lněnička P, Basu B, Gulati A, Hari P, Bagga A. Gitelman syndrome: novel mutation and long-term follow-up. Clin Exp Nephrol. (2012) 16:306–9. doi: 10.1007/s10157-011-0542-x
8. Ye Q, Shen Q, Rao J, Zhang A, Zheng B, Liu X, et al. Multicenter study of the clinical features and mutation gene spectrum of Chinese children with dent disease. Clin Genet. (2020) 97:407–17. doi: 10.1111/cge.13663
9. Zhou F, Mao J, Ye Q, Zhu X, Zhang Y, Ye Y, et al. Clinical features and genetic findings in Chinese children with distal renal tubular acidosis. Int J Clin Exp Pathol. (2018) 11:3523–32.
10. Syrén M-L, Tedeschi S, Cesareo L, Bellantuono R, Colussi G, Procaccio M, et al. Identification of fifteen novel mutations in the SLC12A3 gene encoding the Na-Cl Co-transporter in Italian patients with Gitelman syndrome. Hum Mutat. (2002) 20:78. doi: 10.1002/humu.9045
11. Lin S-H, Shiang J-C, Huang C-C, Yang S-S, Hsu Y-J, Cheng C-J. Phenotype and genotype analysis in Chinese patients with Gitelman's syndrome. J Clin Endocrinol Metab. (2005) 90:2500–7. doi: 10.1210/jc.2004-1905
12. Huang K, Dai Y-L, Zhang J-W, Zhang L, Wu W, Dong G-P, Ullah R, Fei Y, Fu J-F: Gitelman syndrome combined with growth hormone deficiency: three cases report. Medicine (2019) 98:e17244. doi: 10.1097/MD.0000000000017244
13. Maki N, Komatsuda A, Wakui H, Ohtani H, Kigawa A, Aiba N, et al. Four novel mutations in the thiazide-sensitive Na-Cl co-transporter gene in Japanese patients with Gitelman's syndrome. Nephrol Dial Transplant. (2004) 19:1761–6. doi: 10.1093/ndt/gfh239
14. Lemmink HH, Knoers NV, Károlyi L, van Dijk H, Niaudet P, Antignac C, et al. Novel mutations in the thiazide-sensitive NaCl cotransporter gene in patients with Gitelman syndrome with predominant localization to the C-terminal domain. Kidney Int. (1998) 54:720–30. doi: 10.1046/j.1523-1755.1998.00070.x
15. Glaudemans B, Yntema HG, San-Cristobal P, Schoots J, Pfundt R, Kamsteeg E-J, et al. Novel NCC mutants and functional analysis in a new cohort of patients with Gitelman syndrome. Eur J Hum Genet. (2012) 20:263–70. doi: 10.1038/ejhg.2011.189
16. Cruz DN, Shaer AJ, Bia MJ, Lifton RP, Simon DB, Yale GS, et al. Gitelman's syndrome revisited: an evaluation of symptoms and health-related quality of life. Kidney Int. (2001) 59:710–7. doi: 10.1046/j.1523-1755.2001.059002710.x
17. Simon DB, Nelson-Williams C, Bia MJ, Ellison D, Karet FE, Molina AM, et al. Gitelman's variant of Bartter's syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat Genet. (1996) 12:24–30. doi: 10.1038/ng0196-24
18. Abuladze N, Yanagawa N, Lee I, Jo OD, Newman D, Hwang J, et al. Peripheral blood mononuclear cells express mutated NCCT mRNA in Gitelman's syndrome: evidence for abnormal thiazide-sensitive NaCl cotransport. J Am Soc Nephrol. (1998) 9:819–26.
19. Miao Z, Gao Y, Bindels RJM, Yu W, Lang Y, Chen N, et al. Coexistence of normotensive primary aldosteronism in two patients with Gitelman's syndrome and novel thiazide-sensitive Na-Cl cotransporter mutations. Eur J Endocrinol. (2009) 161:275–83. doi: 10.1530/EJE-09-0271
20. Shao L, Liu L, Miao Z, Ren H, Wang W, Lang Y, Yue S, Chen N: A novel SLC12A3 splicing mutation skipping of two exons and preliminary screening for alternative splice variants in human kidney. Am J Nephrol. (2008) 28:900–7. doi: 10.1159/000141932
21. Qin L, Shao L, Ren H, Wang W, Pan X, Zhang W, et al. Identification of five novel variants in the thiazide-sensitive NaCl co-transporter gene in Chinese patients with Gitelman syndrome. Nephrology. (2009) 14:52–8. doi: 10.1111/j.1440-1797.2008.01042.x
22. Lin S-H, Cheng N-L, Hsu Y-J, Halperin ML. Intrafamilial phenotype variability in patients with Gitelman syndrome having the same mutations in their thiazide-sensitive sodium/chloride cotransporter. Am J Kidney Dis. (2004) 43:304–12. doi: 10.1053/j.ajkd.2003.10.018
23. Lo Y-F, Nozu K, Iijima K, Morishita T, Huang C-C, Yang S-S, et al. Recurrent deep intronic mutations in the SLC12A3 gene responsible for Gitelman's syndrome. Clin J Am Soc Nephrol. (2011) 6:630–9. doi: 10.2215/CJN.06730810
24. Ji W, Foo JN, O'Roak BJ, Zhao H, Larson MG, Simon DB, et al. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat Genet. (2008) 40:592–9. doi: 10.1038/ng.118
25. Lee JW, Lee J, Heo NJ, Cheong HI, Han JS. Mutations in SLC12A3 and CLCNKB and their correlation with clinical phenotype in patients with Gitelman and Gitelman-like syndrome. J Korean Med Sci. (2016) 31:47–54. doi: 10.3346/jkms.2016.31.1.47
26. Shi RR, Li CC, Fang L, Xu J, Guan QB, Zhou XL, et al. Clinical and genetic characteristics of Gitelman syndrome in 5 pedigrees. Zhonghua Nei Ke Za Zhi. (2017) 56:104–11. doi: 10.3760/cma.j.issn.0578-1426.2017.02.005
27. Riveira-Munoz E, Chang Q, Godefroid N, Hoenderop JG, Bindels RJ, Dahan K, et al. Transcriptional and functional analyses of SLC12A3 mutations: new clues for the pathogenesis of Gitelman syndrome. J Am Soc Nephrol. (2007) 18:1271–83. doi: 10.1681/ASN.2006101095
28. Gamba G. Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiol Rev. (2005) 85:423–93. doi: 10.1152/physrev.00011.2004
29. Knoers NVAM. Gitelman syndrome. Adv Chronic Kidney Dis. (2006) 13:148–54. doi: 10.1053/j.ackd.2006.01.014
30. Blanchard A, Bockenhauer D, Bolignano D, Calò LA, Cosyns E, Devuyst O, et al. Gitelman syndrome: consensus and guidance from a Kidney Disease: Improving Global Outcomes (KDIGO) controversies conference. Kidney Int. (2017) 91:24–33. doi: 10.1016/j.kint.2016.09.046
31. Huang C-L, Kuo E. Mechanism of hypokalemia in magnesium deficiency. J Am Soc Nephrol. (2007) 18:2649–52. doi: 10.1681/ASN.2007070792
Keywords: Gitelman's syndrome, congenital tubulopathy, genotype, mutation-genetics, phenotype [mesh]
Citation: Zhang L, Huang K, Wang S, Fu H, Wang J, Shen H, Lu Z, Chen J, Bao Y, Feng C, Dong G and Mao J (2021) Clinical and Genetic Features in 31 Serial Chinese Children With Gitelman Syndrome. Front. Pediatr. 9:544925. doi: 10.3389/fped.2021.544925
Received: 23 March 2020; Accepted: 19 March 2021;
Published: 29 April 2021.
Edited by:
Richard Nisan Fine, Stony Brook Children's Hospital, United StatesReviewed by:
Se Jin Park, Eulji University, South KoreaCopyright © 2021 Zhang, Huang, Wang, Fu, Wang, Shen, Lu, Chen, Bao, Feng, Dong and Mao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianhua Mao, bWFvamg4OEB6anUuZWR1LmNu; Guanping Dong, ZGdweGx4QHpqdS5lZHUuY24=; Chunyue Feng, c3ByaW5nbW9vbmZlbmdAMTYzLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.