 Juliann M. Savatt
Juliann M. Savatt Scott M. Myers
Scott M. Myers
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pediatr. , 19 February 2021
Sec. Genetics of Common and Rare Diseases
Volume 9 - 2021 | https://doi.org/10.3389/fped.2021.526779
This article is part of the Research Topic Genetic Testing in Pediatric Disorders View all 35 articles
Neurodevelopmental disorders are the most prevalent chronic medical conditions encountered in pediatric primary care. In addition to identifying appropriate descriptive diagnoses and guiding families to evidence-based treatments and supports, comprehensive care for individuals with neurodevelopmental disorders includes a search for an underlying etiologic diagnosis, primarily through a genetic evaluation. Identification of an underlying genetic etiology can inform prognosis, clarify recurrence risk, shape clinical management, and direct patients and families to condition-specific resources and supports. Here we review the utility of genetic testing in patients with neurodevelopmental disorders and describe the three major testing modalities and their yields – chromosomal microarray, exome sequencing (with/without copy number variant calling), and FMR1 CGG repeat analysis for fragile X syndrome. Given the diagnostic yield of genetic testing and the potential for clinical and personal utility, there is consensus that genetic testing should be offered to all patients with global developmental delay, intellectual disability, and/or autism spectrum disorder. Despite this recommendation, data suggest that a minority of children with autism spectrum disorder and intellectual disability have undergone genetic testing. To address this gap in care, we describe a structured but flexible approach to facilitate integration of genetic testing into clinical practice across pediatric specialties and discuss future considerations for genetic testing in neurodevelopmental disorders to prepare pediatric providers to care for patients with such diagnoses today and tomorrow.
With a combined prevalence of ~17% among 3- to 17-year-old children in the United States, neurodevelopmental disabilities are the most prevalent chronic medical conditions encountered in pediatric primary care (1). The vast majority of these individuals have diagnoses captured within the Diagnostic and Statistical Manual of Mental Disorders, 5th Edition (DSM-5) neurodevelopmental disorders category that includes intellectual disability (ID), global developmental delay (GDD), communication disorders (language disorder, speech sound disorder, childhood onset fluency disorder, and social/pragmatic communication disorder), autism spectrum disorder (ASD), attention deficit/hyperactivity disorder (ADHD), specific learning disorder (involving reading, written expression, and/or mathematics), and motor disorders (developmental coordination disorder, stereotypic movement disorder, and tic disorders) (2). Broader conceptualizations of neurodevelopmental disorders include conditions outside of the realm of the DSM-5, such as cerebral palsy (CP) and epilepsy, and sometimes neuropsychiatric disorders for which there is strong clinical and biological evidence of developmental origins, such as schizophrenia (3–6).
Neurodevelopmental disorders are characterized by developmental deficits in cognition, language, behavior, and/or motor skills that cause impairment of personal, social, academic, and/or occupational functioning (2, 7). These clinically and etiologically heterogeneous disorders represent manifestations of altered neural development and, as such, are typically diagnosed during infancy, childhood, or adolescence. Although subject to maturational changes, neurodevelopmental disorders are non-progressive and tend to follow a relatively steady trajectory rather than a pattern of remitting and relapsing (7). Tic disorders are an exception to the latter, since there is typically waxing and waning of core symptoms rather than a steady course. Because there is insufficient information to allow systematic classification of neurodevelopmental conditions based on etiology and pathophysiology, a descriptive (phenomenological) categorical nosology based on groups of signs and symptoms that define disorders or syndromes (e.g., ASD, ADHD, CP, etc.), has been adopted (8, 9). Descriptive categorical diagnoses are useful heuristics for improving interrater reliability and enhancing information exchange, but their limited alignment with many clinical and biological findings is well-described (8, 10–12). Pearn (9) noted that “syndromic diagnosis is a concise shorthand for describing a constellation of clinical symptoms and signs - but is an acknowledgment of causal ignorance, which in turn demands differential reappraisal as new biochemical, genetic, or ultimately molecular causes of the syndrome are defined.” Comprehensive developmental care, in addition to identifying appropriate descriptive diagnoses and guiding families to evidence-based treatments and supports, includes a search for an underlying etiologic diagnosis (13–16).
The scope of this review is limited to the genetic etiologic evaluation of children and adolescents with neurodevelopmental disorders. The focus is on individuals with diagnoses including GDD, ID, or ASD, for which there are existing guidelines for genetic testing, but we discuss the prospect of future genetic etiologic testing across broader neurodevelopmental disorders. Because of the focus on determining etiology using diagnostic genetic testing, the review does not cover pharmacogenomics, carrier testing, newborn screening, or pre-symptomatic/predictive testing. We provide an overview of the value of clinical genetic testing; the most commonly performed tests and the yield of these tests for determining a genetic diagnosis (focusing on established indications, ID/GDD and ASD); approaches to service delivery and implementation of genetic testing in clinical practice; and future directions, including emerging indications for genetic testing and forthcoming technologies.
Identification of an underlying genetic etiology for a child's categorical neurodevelopmental diagnosis (or diagnoses) can provide both clinical and personal utility to patients and their families. Table 1 provides several examples of genetic causes of neurodevelopmental disorders and the potential utility of identifying such a diagnosis. Establishing a genetic basis for a child's neurodevelopmental phenotype can provide additional information about their prognosis and enable caretakers to understand potential areas of need and opportunities for increased support (39). For example, a genetic diagnosis, and the prognostic information it provides, may facilitate acquisition of educational, disability, and employment services (40–45). Although currently infrequent in the case of neurodevelopmental disorders, a genetic diagnosis might also provide access to etiology-specific treatments (45–49). Given the number of treatments that are currently being explored in animal models and clinical trials, such gene-specific therapies are likely to become more ubiquitous (50). In the interim, as new therapies are being developed, a genetic diagnosis can provide access to condition-specific research protocols enrolling human subjects (51, 52). It is also important to note that elucidating etiologies and pathophysiologic processes is a necessary step toward developing animal models and human cell lines to use in pathogenesis studies, clinical trials, and, ultimately, mechanism-based targeted treatments for neurodevelopmental disorders (13, 53). In addition to facilitating medical management and access to services, a genetic diagnosis can also enable families to avoid unnecessary diagnostic tests (42, 45, 49, 52, 54) and, with an etiologic diagnosis in hand, families may be more empowered to avoid therapeutic interventions that are based on unfounded etiologic theories and are potentially harmful (e.g., chelation therapy) (55). Additionally, genetic testing can afford patients and their caregivers the ability to identify, treat, and/or prevent medical comorbidities at the time of diagnosis, as well as conditions that may develop later in life (42, 45, 49, 52, 56–65).
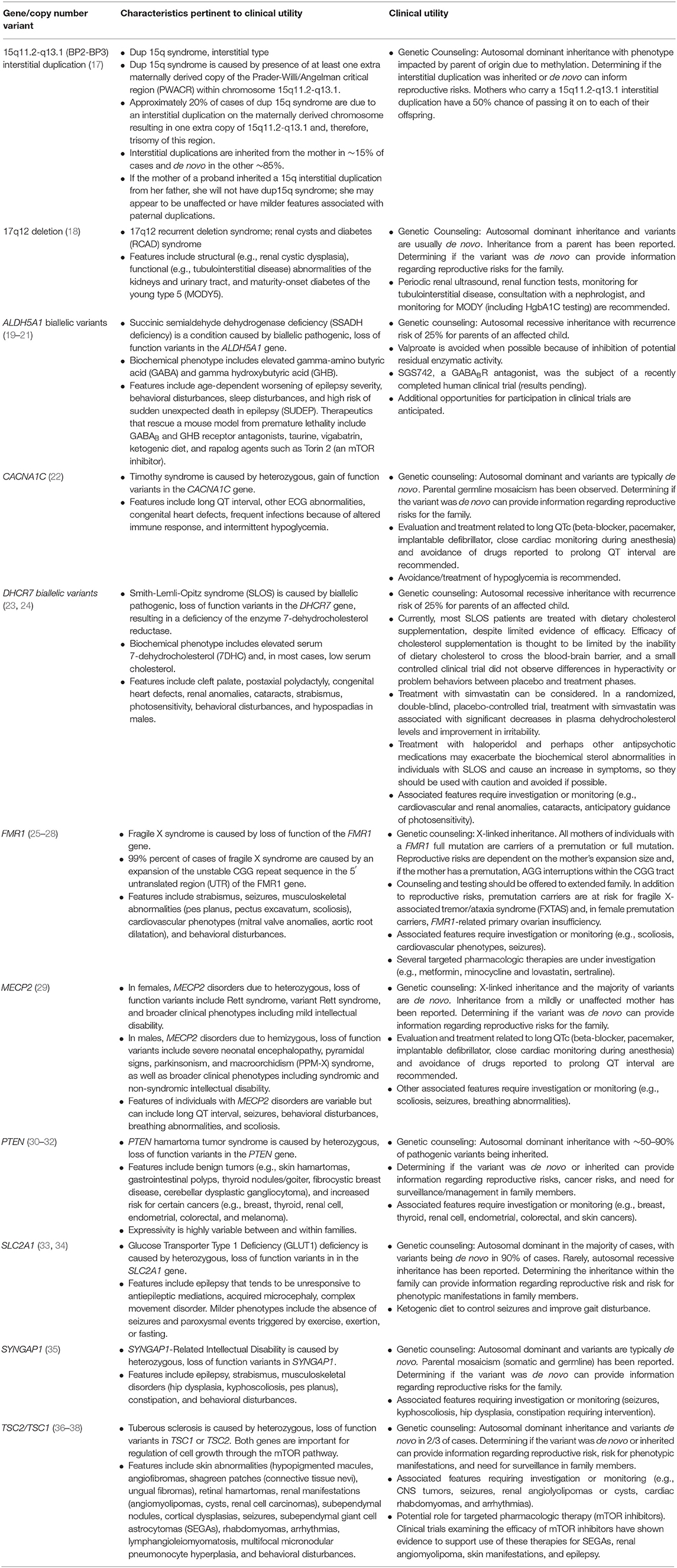
Table 1. Examples of clinical utility of specific genetic findings.
Furthermore, genetic testing allows healthcare providers to refine recurrence risk counseling for the family to inform reproductive decision making (39, 42, 45, 49, 52, 58, 59, 63, 65–67). When an etiology is determined, the risk of recurrence for an individual family varies depending on the specific genomic variant(s) identified. For parents of a child with a neurodevelopmental disorder, recurrence may be 50%, for example, in the case of an inherited, maternally-derived chromosome 15q11–q13 interstitial duplication, 25% in the case of an autosomal recessive disorder such as Smith-Lemli-Opitz syndrome, or approximately the prevalence rate of the particular disorder in the general population (e.g., ~1.5% for ASD) if the child has a de novo explanatory variant. This refined recurrence risk information offers patients, parents, and family members an understanding of their reproductive risks and informs reproductive decision making, restores reproductive confidence, and enables prenatal diagnosis (45, 48, 68). Furthermore, an understanding of recurrence risk in subsequent children and generations can enable families and their medical providers to identify neurodevelopmental disorders and initiate beneficial behavioral treatments and therapies at earlier ages (69). In the case of inherited etiologies, identifying a genetic diagnosis for the proband may also provide diagnoses to other family members with a history of a neurodevelopmental disorder (70).
In addition to the clinical utility conferred through a genetic diagnosis, identifying an etiology can also provide psychosocial benefit to families. A genetic diagnosis can provide families with an explanation for their child's developmental history and, therefore, bring an end to the child's “diagnostic odyssey” that may have included years of uncertainty, anxiety, and evaluations (49, 58, 60, 71), and can also guide patients and families to condition-specific resources and supports (45). Receiving a diagnosis has also been shown to increase knowledge, provide a sense of empowerment (45), result in peace of mind (41), increase parental quality of life (72), decrease parental guilt (59, 73), and foster increased acceptance (74).
In addition to wanting to know their child's clinical diagnosis and prognosis, families typically want to know the cause of the child's developmental disability. In the last two decades, rapid advances in the development of genetic testing technologies and application of these technologies to well-characterized patient cohorts have revolutionized our ability to make specific genetic diagnoses in patients presenting with neurodevelopmental disorders. New genes are being implicated in neurodevelopment at a rapid pace. Online resources such as the Geisinger Developmental Brain Disorder Genes Database (https://dbd.geisingeradmi.org/), the Clinical Genome Resource's Gene Validity Curations and Dosage Sensitivity Map (https://search.clinicalgenome.org/kb/gene-validity/; https://dosage.clinicalgenome.org/), and DECIPHER's Development Disorder Genotype - Phenotype Database (https://decipher.sanger.ac.uk/ddd#ddgenes) can provide up to date information about genes' and genomic variants' relationship to neurodevelopmental disorders (75–78).
Genetic testing is now the standard of care for several neurodevelopmental disorders and the indications for testing will almost certainly broaden in the future. In practice, a genetic etiologic diagnosis may be suspected clinically and confirmed by gene-specific genetic testing or, more commonly, it may be revealed by chromosomal microarray (CMA) analysis, exome sequencing (ES), or FMR1 CGG repeat analysis for fragile X syndrome completed as a routine part of the evaluation of a patient with ID/GDD or ASD in the absence of a clinically recognizable syndrome. It is important for pediatric clinicians to understand these common tests and their role in the care of children with neurodevelopmental disorders.
Genome-wide CMA has been endorsed as a first-tier test for several indications including in patients with ASD, ID, GDD, and/or multiple congenital anomalies (79, 80). CMA technologies (Comparative Genomic Hybridization (CGH) array CGH and Single Nucleotide Polymorphism (SNP)-based testing) detect copy number variants (CNVs) – gains or losses of chromosomal material (81). In some cases, such gains or losses affect gene function and impact health and development. The resolution, or size of gains and losses that can be detected by CMA, varies and is determined by the specific technology used and the genomic distance between DNA probes.
CNVs detected through CMA should be categorized into the following categories in accordance with the American College of Medical Genetics and Genomics (ACMG) and Clinical Genome Resource (ClinGen) guidelines (82).
1) Pathogenic – CNVs that are thought to be associated with disease. Pathogenic CNVs may include those that explain the patient's phenotype, those that are associated with carrier status for a recessive condition, and those that indicate disease risk for an unrelated phenotype. Pathogenic CNVs that explain a patient's neurodevelopmental history can include recurrent deletions or duplications such as 22q11.2 deletion syndrome or the 17p11.2 recurrent microdeletion that causes Smith-Magenis syndrome (83, 84), or novel gains or losses that impact dosage sensitive gene(s).
2) Likely pathogenic – CNVs that have considerable evidence to suggest that they are associated with disease but where additional evidence could further clarify the variant's pathogenicity.
3) Uncertain Significance – CNVs that do not have enough information to determine if they are pathogenic or benign. When a CNV with uncertain significance is identified, parental studies might be used to provide additional information to clarify pathogenicity.
4) Likely Benign – CNVs that have considerable evidence to suggest that they are not associated with disease but where additional evidence could further clarify this.
5) Benign – CNVs that are not thought to be associated with disease. These are often present in phenotypically normal individuals or in the general population.
Interpretations may change over time as new evidence emerges informing a variants' pathogenicity. Because CMA employs genome-wide testing, incidental findings, or those that are unrelated to the primary indication, may be identified (82). CNVs often include multiple genes and, in rare instances, a CNV might explain a child's neurodevelopmental history, but given the gene content, may also confer risk for an unrelated condition (61). In other cases, a CNV that is unrelated to a patient's phenotype but that has implications for care may be identified and reported through testing (85).
In addition to CNVs, laboratories that employ SNP-based CMA can also detect regions of homozygosity (chromosomal segments that are identical to one another). Identification of regions of homozygosity enables the potential detection of conditions that can be caused by uniparental isodisomy (UPD) such as Silver-Russell (maternal UPD chromosome 7), Angelman (paternal UPD chromosome 15), and Prader-Willi syndromes (maternal UPD chromosome 15) (86). In other instances, regions of homozygosity may be indicative of ancestral homozygosity or parental consanguinity (86–88). Regions of homozygosity can also be indicative of an increased risk for an autosomal recessive condition due to the potential for homozygous variation in single gene (88, 89).
Although CMA is able to detect CNVs, this technology has limited ability to detect balanced chromosomal rearrangements (translocations, inversions), trinucleotide repeat expansions, imbalances in the mitochondrial genome, epigenetic abnormalities (e.g., methylation abnormalities), sequence level variants, or low-level mosaicism for CNVs (Table 2) (65, 90). As stated above, the size of deletions and/or duplications that can be detected by CMA varies. Current clinical CMA platforms can detect CNVs ~400 kb in size (80) with many laboratories detecting those >250 kilobases. Certain regions may be more specifically targeted enabling even smaller CNV detection (Table 2).
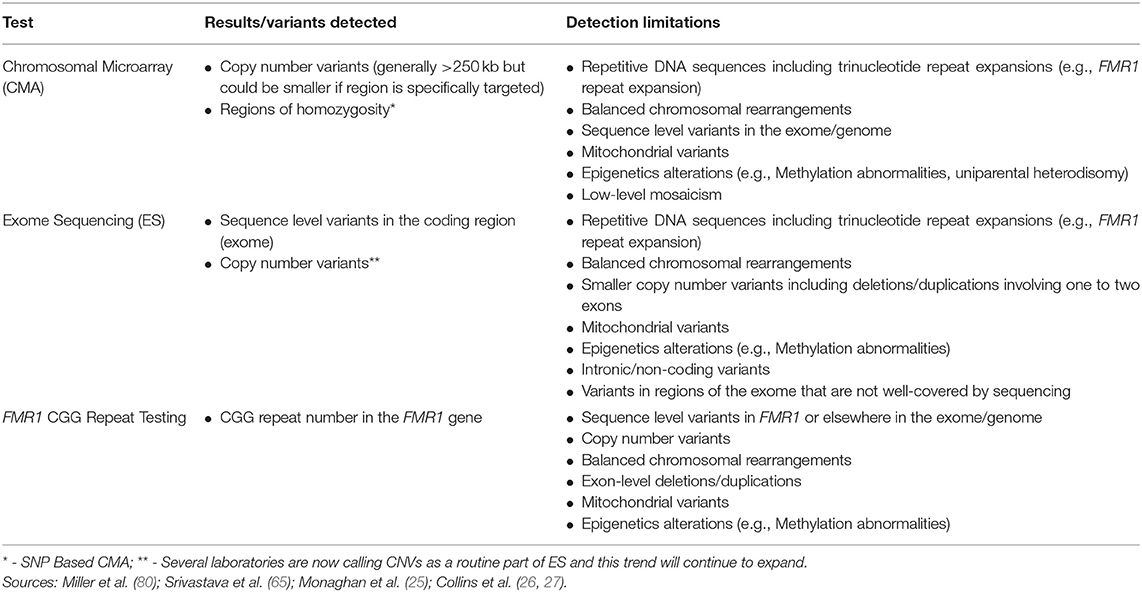
Table 2. Genetic tests commonly used in evaluation of neurodevelopmental disorders.
Additional testing discussed in this review can aid in detection of some variants that CMA is unable to detect, including FMR1 CGG repeat analysis for the trinucleotide repeat expansion that causes fragile X syndrome and ES for detection of exonic sequence variants. Cytogenetic testing, including G-banded karyotype and/or florescence in situ hybridization (FISH) as well as methylation testing, can detect additional variants or clarify results from CMA. As reviewed elsewhere, such testing should be considered on a case by case basis (e.g., for detection of balanced rearrangements and other complex rearrangements or mosaicism for partial or whole chromosome aneuploidy, or for clarification of the location of a duplication) (90).
Authors reporting CMA yield use variable nomenclature to describe variants' pathogenicity and diverse interpretation practices, thus limiting between-study comparisons and pooling of data across studies of CMA diagnostic yield. The diagnostic yield of CMA is the proportion of tests performed that identify a variant that is considered causative for a patient's phenotype. Taken together, a causative result can be identified by CMA in 15–20% of individuals with ID/DD, ASD, and/or multiple congenital anomalies (54, 79, 80). For the purpose of trying to capture yield specific to neurodevelopmental disorders, one can examine 19 studies that report on the yield of CMA in more than 150 individuals with ID/GDD where the diagnostic yield ranged from 4.5 to 28.0% (median 13.7%) (91–109). Additionally, one can look to 11 studies limited to patients with ASD (each with a sample size ≥50), in which CMA identified a causative variant in 1.5 to 20.5% of subjects (median 8.1%) (96, 98, 99, 104, 109–115). Because karyotype was the standard etiologic approach before the clinical integration of CMA, many publications summarizing diagnostic yields of CMA excluded patients with an abnormal karyotype. Since the majority of pathogenic results identified by karyotype would be detected by CMA, a number of publications may be reporting yields ~3.7% percent lower, on average, than if CMA had been applied as a first-tier evaluation (116). Overall, the diagnostic yield of CMA in patients with ID, GDD, and/or ASD suggests that CNVs comprise a substantial proportion of underlying genetic etiologies.
Although the diagnostic yield of CMA has been well-examined in the literature, there are few studies exploring the frequency that incidental findings are identified with this testing technology. In two studies, 0.15–0.48% of patients had a CNV that included a gene associated with hereditary cancer pre-disposition (117, 118), and in a third study, 1.2% had a CNV that included a gene associated with an adult-onset condition (119) The pathogenicity of these CNVs were not reported by the study authors, so the true rate of pathogenic incidental findings in these study populations is somewhat unclear (117–119). Taken together, however, these studies suggest that clinically significant incidental findings likely occur in <1% of patients undergoing CMA.
In the last decade, the arrival of high-throughput sequencing technologies, collectively referred to as next generation sequencing (NGS) or massively parallel sequencing, has reduced the cost and increased the speed of sequencing, enabling laboratories to sequence large amounts of DNA. Rather than testing a single gene or several genes, laboratories can now offer sequencing of extensive gene panels, the exome, or the genome.
In ES, protein coding regions, or exons, of the genome are sequenced. The exome makes up ~1.5–2% of the genome, but the vast majority of alleles underlying Mendelian disorders impact coding sequences (120). ES allows detection of sequence-level, single gene variants across almost all of the exome and a small number of intronic nucleotides at the boundaries of each exon. In addition to sequence-level variant detection, several clinical laboratories are now incorporating CNV calling into ES and are able to detect multi-exon deletions and duplications (65, 121, 122) and more laboratories will likely add this to their exome analyses moving forward. Shorter CNVs including those that involve one to two exons are less reliabily detected using ES (121). Given the breadth of information that is generated using ES, parental samples are recommended for analysis (ideally trio analysis or duo analysis if only one parent is available) since this can reduce the number of candidate variants that require review and facilitate variant interpretation (78, 123).
As with CMA, variants detected by ES are categorized as pathogenic, likely pathogenic, uncertain significance, likely benign, or benign in accordance with ACMG/ Association for Molecular Pathology (AMP) sequence variant interpretation guidelines (123). Due to the rapid increase in our understanding of genes and genomic variants, improved variant annotation and filtering, and evolving patient phenotypes (65, 124–129), an iterative reanalysis of ES data may enable additional diagnoses. Over time, a variant's pathogenicity may be updated as new information emerges. ES also includes analysis of genes not yet associated with a specific phenotype. As a result, iterative reanalysis might also identify a novel variant.
ES, like CMA, may identify results that are clinically relevant but unrelated to the patient's neurodevelopmental disorder. ES includes almost all of the coding portions of known genes, providing an opportunity to examine sequence data for pathogenic/likely pathogenic variants in medically actionable genes that are unrelated to the indication for testing (secondary findings). The ACMG has recommended that clinicians notify their patient if a variant known or expected to increase disease risk was identified in a list of, currently 59, clinically actionable genes (130, 131). ACMG has specified that patients and families have the ability to opt out of such findings (132) and will continue to update this list of clinically actionable genes as additional evidence emerges (130). In addition to secondary findings, the use of parental samples in ES can result in incidental identification of possible misattributed parentage; reporting and disclosure of this information varies between laboratories and clinicians (133, 134).
Although ES captures the majority of the exome, the exome is not covered in its entirety and coverage may differ across platforms and laboratories (135). As a result, ES may not detect all coding, sequence variants. ES also has lower sensitivity for detection of mosaicism and exon-level deletions and/or duplications compared to gene panels that include deletion/duplication analysis (65). An increasing number of laboratories are calling CNVs from ES data; laboratories that are detecting and reporting CNVs have limited ability to detect deletions or duplications involving only one to two exons, however (Table 2) (121). Furthermore, ES is unable to detect repetitive DNA sequences including trinucleotide repeats (e.g., the CGG repeat expansion that causes fragile X syndrome), intronic/non-coding variants, mitochondrial variants, epigenetic variants (e.g., methylation abnormalities), or balanced chromosomal rearrangements.
The diagnostic yield is the proportion of exome analyses with variants that are determined to be pathogenic or likely pathogenic and explain the patient's phenotype. A recent meta-analysis of 21 ES studies that focused on isolated neurodevelopmental disorders (GDD, ID, and/or ASD; n = 3,173) identified a diagnostic yield of 31% (95% CI 25–38%) (65). When nine additional studies of individuals with these neurodevelopmental disorders plus associated neurological or syndromic conditions or clinical characteristics were included, increasing the total number of participants to 3,350, the yield was 36% (95% CI 30–43%). In a large, laboratory-based study not included in the meta-analysis because the cohort included a potentially broader group of neurodevelopmental phenotypes, the diagnostic yield among individuals undergoing ES due to neurodevelopmental disorders (ID, ASD, developmental delay, or speech delay) with or without involvement of other organ systems was 25.4% (425 of 1,673) (136).
Delineation of the diagnostic yield of ES for cohorts ascertained for ID/GDD vs. ASD is limited by the phenotype data reported in the literature. Srivastava et al. (65) reported in their meta-analysis that among studies including individuals with primarily ID (n = 10), the diagnostic yield was 39% (95% CI 29–50%), whereas studies including individuals with primarily ASD (n = 5) had a yield of 16% (95% CI 11–24%) and those with a more heterogenous mix of ID and/or ASD (n = 6) identified a diagnosis in 37% of participants (95% CI 29–46%) (65). Among seven ES studies each involving more than 50 individuals with ID/GDD, a diagnosis was established in 34% of patients (range 28–43%) (128, 137–142). Studies restricted to clinically ascertained samples of patients with ASD and analyses of ASD subgroups within the large, laboratory-based samples have reported lower yields of 8–26% (median 15%) (139, 140, 143–146). There is substantial variability among these studies regarding the information provided about cognitive status and the stringency of ASD diagnosis. In most cases the patients included had previously had genetic testing including fragile X analysis and CMA that did not result in a molecular diagnosis. ES reveals two or, rarely, three molecular diagnoses in ~1% of individuals undergoing clinical testing (136, 140). When this occurs, the patient typically has a “blended phenotype,” with features that are accounted for by each of the pathogenic variants and not by a single molecular diagnosis.
Several studies suggest that the diagnostic yield of ES is higher when trios (probands and parents) are tested than when only the DNA of probands is sequenced (139, 140, 147). Because of the rapid advances in gene and variant curation and factors such as evolution of the phenotype in an individual over time, periodic reanalysis of ES data may enhance the diagnostic yield considerably (48, 124, 127, 138, 148, 149). There is wide variability in the rate of secondary findings among individuals who undergo ES; several recent large studies have reported rates of 2–6% (136, 140, 150, 151). These reportable secondary findings, which are most commonly related to cardiomyopathies, cardiac conduction disorders, hereditary cancer pre-disposition, or familial hypercholesterolemia, often result in additional testing and/or surveillance of the proband and relatives (140).
In addition to ES, single gene testing and targeted next-generation sequencing panels have been used in evaluation of patients with neurodevelopmental disorders including ID, GDD, and ASD historically, and a number of clinical laboratories currently offer such testing. Compared to ES, more targeted sequencing allows for increased read depth and sequence coverage thus increasing detection of mosaicism (152). Moreover, single gene testing and gene panels are better able to detect indels and those that include deletion/duplication analysis are better able to detect exon-level deletions and/or duplications (65, 153). While ES identifies 100–200 potentially deleterious sequence variants on average, more targeted analysis identifies a smaller number of variants requiring less analysis and reducing the potential for variants of uncertain significance (81, 120, 154).
Although there are some advantages to more targeted testing and clinical scenarios that warrant such testing, the genetic heterogeneity of ID, GDD, and ASD and frequency of causative de novo variants, ES is an appropriate approach for many patients with these diagnoses (129). Studies have found that ES can detect more than 98% of pathogenic variants identified on gene panels (155). Additionally, the gene content of targeted gene panels vary significantly between laboratories meaning that diagnostic yields also vary. In Hoang et al. (156), comparison of ASD gene panels across 21 laboratories found that the number of genes included on ASD-related panels ranged from 11–2,562 and only a single gene (MECP2) was included on all panels. In a simulation study comparing ES to gene panels, providers were asked to choose a commercially available gene panel as an alternative when ordering ES for a patient; of the patients receiving a diagnosis through ES, 23% would not have had their variant identified through the provider-chosen gene panel (157). Additionally, as an increasing number of genes are being implicated in Mendelian conditions (158) including neurodevelopmental disorders, panels quickly can become out of date and updating panels can be a time-consuming process (159). After undergoing gene panel testing, 11% of panel-negative cases in one cohort received a diagnosis via ES; many of these diagnoses were attributed to a gene-disease relationship being identified after the panel assay was established (154). In addition to being able to more readily report recently described genes implicated in neurodevelopmental disorders, ES also enables increasing diagnoses over time due to reanalysis. Unlike gene panels, ES includes analysis of genes not yet associated with a specific phenotype. As a result, iterative reanalysis enables reporting of novel variants recently implicated in neurodevelopment and can allow for diagnoses into the future while gene panels typically only enable reanalysis of the sequenced genes. Finally, more targeted testing has been viewed as a less expensive testing option; however, more recent publications have suggested that ES is cost-effective (142, 157, 160–163).
Although ES and CMA have revolutionized our ability to detect genomic variants, there are limitations in our ability to interpret such variants and determine their impact on health and development. Copy number and sequence variants identified through CMA and ES might be interpreted as variants of uncertain significance (VUS) based on available evidence. VUS include variants in known disease genes that have insufficient evidence to be classified as benign or pathogenic as well as sequence and copy number variants that involve genes that are not yet associated with a disease or phenotype (also referred to as genes of uncertain significance/ candidate genes) (82, 123). VUS in patients undergoing CMA due to a history of developmental delay, ID, ASD, and/or multiple congenital anomalies have been reported to carry at least one VUS 7.9–19% of the time (94–96, 104, 105, 107, 164). The frequency of VUS in patients undergoing ES depends on a number of factors including the reporting laboratory's reporting practices (165), phenotypic features provided by the ordering clinician (166), and inclusion of parental samples. Several studies examined VUS rates among patients undergoing ES for a variety of indications and found VUS rates of 25.3–86% (63, 166, 167).
Uncertain results, although not unique to genetics (168), can pose challenges for patients and providers alike. Compared to other potential results, providers are least comfortable explaining VUS results to families and report that additional preparation is required for such results (169–171). Non-genetics providers express a need for additional education and access to genetics professionals' expertise to facilitate understanding and disclosure of such results (169). Despite expressing less comfort with VUS results and calls for more support and education, non-genetics, pediatric providers that regularly order testing appear to be able to understand and interpret VUS results accurately (172).
Studies of parents have suggested that they are interested in receiving uncertain results and those that receive such results post-natally see the them as important, often demonstrate an understanding that the result is uncertain, and acknowledge that future advancements can increase understanding (73, 171, 173–178). Although parents seem to accurately recall the concept of uncertainty, parents report difficulty understanding how the result impacts them and their child, and some over interpret the variant as being causative (73, 170, 173, 174). Emotional reactions to VUS vary between families and over time (174, 176).
Given the potential for misunderstandings and variable emotional reactions, providers need to be equipped to discuss uncertain results or refer on to providers with genetics expertise (73). The option for in-person, timely discussions with empathic and honest providers and access to supplemental information including documentation of the result have been suggested to improve understanding of VUS (73, 173, 176, 177). In contrast, internet searches have been shown to increase uncertainty; therefore, anticipatory guidance about misleading or irrelevant online information should be provided to families receiving VUS results (73). In addition to providing families with appropriate information, support, and resources to facilitate understanding of uncertain results, providers returning VUS should consider if additional evaluations that could inform the pathogenicity of the variant are indicated such as parental or familial testing, imaging, or specialist referrals (82, 123, 137, 179). Furthermore, knowledge of genomic variants and their relationship to health and development will continue to improve. As such, VUS will be updated to benign or pathogenic over time. For example, in one study of 2,250 patients undergoing ES reanalysis, 23 had variants initially reported as VUS upgraded to diagnostic (125). Patients receiving VUS results need to be informed of the potential for interpretation updates and ordering providers should discuss the process for reassessing variants.
Fragile X syndrome, caused by loss of function of the FMR1 gene, is thought to be one of the most common inherited genetic causes of ID and ASD (54, 180, 181). Ninety-nine percent of cases of fragile X syndrome are caused by an expansion of the unstable CGG repeat sequence in the 5′ untranslated region (UTR) of the FMR1 gene (25–27). Most individuals in the general population have 44 or fewer CGG repeats, while more than 200 CGG repeats in the FMR1 gene results in hypermethylation and, consequently, transcriptional silencing of the gene (182, 183). The FMR1 gene is located on the X chromosome (Xq27.3) and, consequently, mutations can cause variable phenotypes in males and females (54, 184–186). FMR1 CGG repeat analysis is usually completed using polymerase chain reaction (PCR) analysis and Southern Blot Analysis (25). FMR1 CGG repeat analysis can yield four main categories of results (25):
1) Normal: ≤44 repeats.
2) Intermediate (Inconclusive, Borderline, Gray Zone): 45–54 repeats. Alleles with 45–54 repeats have not been observed to expand to a full mutation in one generation. Because minor increases or decreases in repeat size can occur, alleles of this size could be associated with fragile X syndrome in future generations.
3) Pre-mutation: 55–200 repeats. Expansions in FMR1 of this size are unstably passed from parent to child and, when passed from the mother, expansion from the pre-mutation to a full mutation may occur. The risk for expansion is greatest in those with larger repeat sizes (187). Furthermore, the presence of AGG interruptions within the CGG repeat tract is associated with decreased risk for expansion (187–189).
4) Full Mutation: >200 repeats (typically several hundred to several thousand repeats).
In addition to the risk of expansion to a full mutation in offspring, female pre-mutation carriers are at risk for hypergonadotropic hypogonadism (fragile X pre-mature ovarian insufficiency) before age 40 years. Although more prevalent in males, both male and female pre-mutation carriers are also at an increased risk for fragile X-Associated Tremor/Ataxia Syndrome (FXTAS), a late-onset neurodegenerative condition characterized by cerebellar ataxia, intention tremor, cognitive impairment with increased penetrance observed in males (190). When offering FMR1 CGG repeat testing, pre-mutation carrier status may be identified in the proband and/or relatives and incidentally identify risks for these adult-onset conditions (181).
FMR1 CGG repeat testing cannot detect other variants causing loss of function of the FMR1 gene (e.g., deletions, sequence variants that result in protein truncation), which are rare. Testing does not detect other genomic causes of neurodevelopmental disorders (e.g., sequence variants in other genes, copy number variants, epigenetic abnormalities) (Table 2).
The yield of fragile X testing among individuals with ID/GDD and/or ASD varies widely based on study design and characteristics of the population being tested, such as severity of cognitive impairment, whether both males and females are included, and whether clinical judgement or phenotypic feature checklists were used to exclude some potential participants (191, 192). For example, the diagnostic yield varied from 0.5 to 6% among 14 studies reviewed by Peprah (192) that included at least 200 individuals with ID who were ascertained either through clinical referral or special needs service utilization (192). Hunter et al. (191) identified 15 studies that estimated the frequency of the full mutation in populations with ID before extrapolating to the total population, with the goal of including them in a meta-analysis, but it was not possible to combine the data and calculate a valid mean prevalence due to variability in study methods and lack of reported measures of uncertainty (191).
Several studies that attempted to capture populations of males with neurodevelopmental disorders for assessment of the rate of fragile X syndrome reported full mutations in 8/611 (1.3%) with unexplained ID, 20/3,738 (0.5%) with special education needs related to cognitive deficiencies, and 7/2,471 (0.28%) receiving special education services (excluding isolated speech therapy or gifted services) (193–195). An epidemiological study of a representative sample of 3,313 people with ID (56% male) in the Netherlands included 1,143 individuals (55% male) who were not eligible for testing because they had other etiologic diagnoses, including fragile X syndrome (30 males and 2 females; 4.75 and 0.39%, respectively), or had previous negative clinical testing, and 2,170 (57% male) who were eligible for testing as part of the study (196). Among those who were eligible, 1,520 individuals (57% male) consented and were tested. Full mutations were identified in 9/866 males (1.0%) and 2/654 females (0.31%) (196). Allowing the unknown diagnostic yield for those who were eligible but did not consent to testing to vary from 0.5 to 2.0 times the sex-specific diagnostic yield of those who were tested and including the ineligible individuals, the overall prevalence estimates are 2.2–2.5% for males with ID and 1.3–1.6% for females with ID. The rates of full FMR1 mutations identified among clinical ASD cohorts are generally lower than those identified in clinical ID cohorts. Although early studies suggested higher rates, the larger studies published in the last decade (n = 142 to n = 861) have identified fragile X full mutations in only 0.23 to 1.2% of individuals ascertained for ASD diagnosis (111, 197–201). The combined yield of these studies was nine full mutations in 1,984 individuals tested (0.45%), including seven males and two females. Fragile X syndrome has been identified in females with ASD with and without ID (202–204).
The diagnostic yield of fragile X testing has also been described by several clinical laboratories, typically with little information available about phenotype. For example, a large-volume commercial diagnostic laboratory reported FMR1 full mutation alleles in 1.4% of 59,707 males and 0.61% of 59,525 females tested post-natally over a 14-year period (1992–2006) (205). More recently, two university hospital diagnostic laboratories reported yields of 0% (0/654) and 0.9% (11/1,177) in males under age 22 and 19 years, respectively (206, 207). A similar yield of 0.8% (43/5,401) was reported by another hospital-based genetics laboratory that did not describe the diagnostic rate in males and females separately (208). Borch et al. (209) found a diagnostic yield of 1.2% (30/2,486) among pediatric patients who had fragile X testing, including 1.3% of males (25/1,919) and 0.9% of females (5/567). The vast majority, 96% (29/30), had clinical features and/or family history that were suggestive of fragile X syndrome.
Although studies vary in the level of phenotypic detail provided, several authors reporting on yields of CMA, ES, and FMR1 CGG repeat analysis in patients with neurodevelopmental disorders suggest that specific characteristics and additional diagnoses are associated with increased diagnostic yields (95, 111, 113, 140, 146, 198, 201, 210, 211). Lower IQ, dysmorphic features, and congenital anomalies have been found to be associated with higher diagnostic yield of CMA and ES in some studies examining yield in patients with ASD (111, 113, 146, 198, 201). Similarly, in Fan et al. (95), co-occurring congenital heart defects, facial dysmorphisms, microcephaly, and hypotonia were associated with increased diagnostic yield of CMA in patients with developmental delay or ID (95). A higher diagnostic yield has also been reported for moderate to severe ID than for mild ID. Among individuals with severe ID, it is estimated that more than 60% harbor causative CNVs or exonic sequence variants (211, 212). Finally, in a meta-analysis of published studies using clinical checklists among patients with ID undergoing testing for fragile X, soft and velvety skin on the palms with redundancy on the dorsum of the hand, large prominent ears, pale blue eyes, family history of ID, autistic behavior, flat feet, and plantar crease all are correlated with an increased likelihood of identifying a FMR1 full mutation (210). Although specific features and additional diagnoses have been correlated with increased diagnostic yield, the yield in patients with isolated ID/GDD and/or ASD is high enough to warrant an etiologic genetic investigation following the application of these categorical diagnoses.
Although heterogeneous study designs and methodologies limit conclusions that can be drawn regarding diagnosis-specific diagnostic yields, current evidence suggests that among patients with ID or GDD, the diagnostic yields are at least 15% for CMA, 35% for ES, and 1% (in females) to 2% (in males) for FMR1 CGG repeat analysis. For those with primarily ASD, the diagnostic yields are at least 10% for CMA and 15% for ES, and 0.5% or less for FMR1 CGG repeat analysis. Across all of these tests and clinical diagnoses, the diagnostic yield rates are higher in the presence of lower IQ, neurological comorbidities, dysmorphic features, and congenital anomalies. The cumulative diagnostic yield for all three tests, therefore, is over 50% for individuals with ID/GDD and over 25% for individuals with primarily ASD.
Due to the yields of genetic testing and corresponding potential for clinical and personal utility, there is consensus that genetic testing should be offered to patients with GDD, ID, and/or ASD diagnoses (13–15, 65, 80, 213). The specific testing algorithm varies between guidelines and consensus statements (214), and many recommendations predate the broad integration of exome sequencing into clinical care (Table 3).
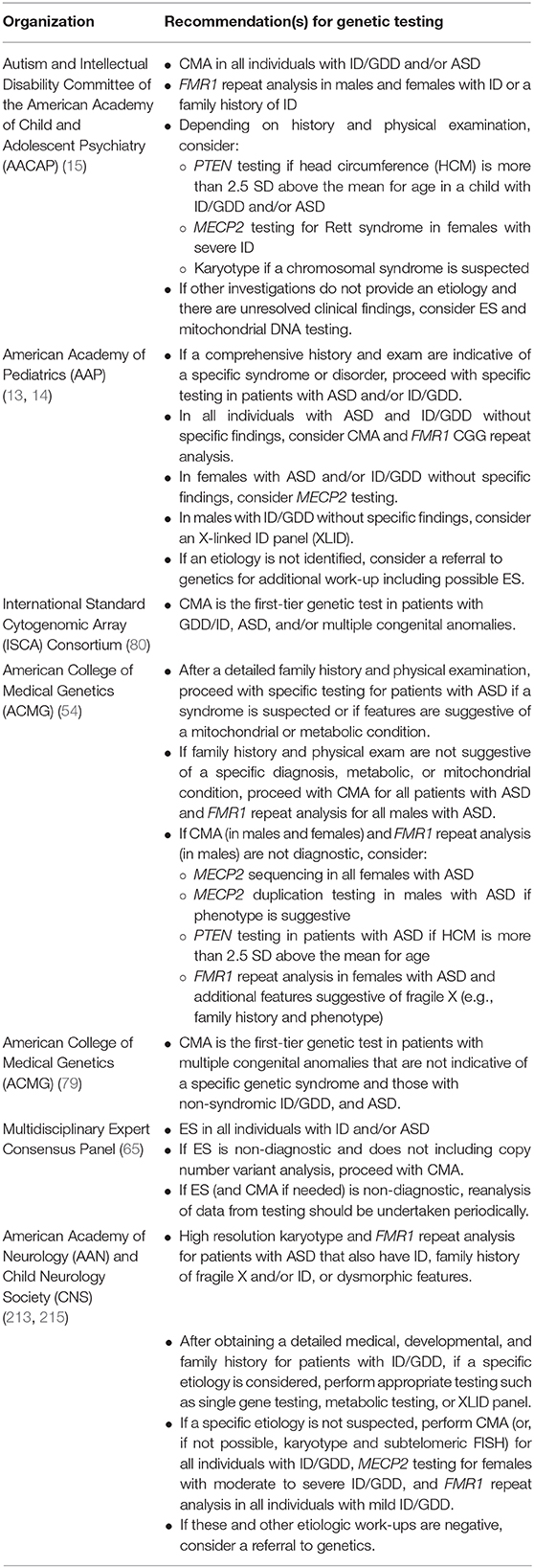
Table 3. Genetic testing guidelines for ID/GDD and/or ASD.
Despite widespread recommendations for genetic testing for neurodevelopmental disorders in pediatric patients, surveys, interviews (69, 214, 216–221) and retrospective studies of clinical cohorts (111, 198) suggest that that a minority of children with ASD (16.5%-45%) and ID (43%) in the United States have undergone any genetic testing. Furthermore, a study examining pediatric providers' use of genetic testing in the care of simulated patients with ID, GDD, and/or ASD suggest that genetic testing is underutilized (222). In Europe, rates of testing vary from country to country with 61.7% French parents reporting that their child with ASD underwent some diagnostic genetic test compared to 13% of children with ASD in Spain (216, 223). Although there is consensus agreement that a genetic evaluation should be offered to individuals with GDD, ID, and/or ASD, this does not appear to be occurring in practice. Pediatric providers caring for patients with neurodevelopmental disorders should work to address this gap in care.
Pediatric providers, from general practitioners to neurodevelopmental pediatricians, are in a position to empower themselves to consent for and order genetic testing. Given the increasing availability of genetic testing and limited number of genetics professionals, genetics providers cannot and need not be involved in all instances of consent for genetic testing and disclosure of results (224–227). Genetic testing for patients with neurodevelopmental disorders has shifted to be within the purview of non-genetics providers and is something that pediatric providers can be equipped to undertake (214, 228–233). Below we describe a structured but flexible approach to facilitate integration of genetic testing into clinical practice across pediatric specialties (Table 4).

Table 4. Approach to genetic etiologic evaluation in GDD, ID, and/or ASD.
In most cases of patients with ID, GDD, and/or ASD, developmental, medical, and family histories do not point to a specific genetic etiology. In these cases, the approach to genetic testing should include broad analysis for copy number and exonic sequence variants (Table 4). This can be achieved by ordering a single genetic test - ES with CNV analysis (65). Diagnostic yields of ES and emerging evidence that ES is cost-efficient in the etiologic evaluation of children with neurodevelopmental disorders (142, 160–163) support this assertion that ES is the most appropriate first test to pursue (65). Payers are even altering their policies to support increasing efficiency through such testing algorithms (234).
It is important to note that, at present, some genetic testing laboratories may not include CNV analysis as part of their ES and some payers may require completion of CMA prior to coverage of ES. As a result, ES (without CNV analysis) and CMA may need to be pursued in a stepwise manner (65, 141). In cases where both CMA and ES are pursued independently, the order of testing will be dictated by individual factors such as insurance requirements (e.g., payer may only cover ES after a negative CMA) and additional phenotypic features (e.g., non-specific epilepsy phenotype in addition to ASD increasing the likelihood of a sequence variant) (235).
Less often, a patient presenting with ID, GDD, and/or ASD may have a constellation of features and/or history that are suggestive of a particular genetic etiology. In these instances, if a specific disorder is suspected, specific genetic evaluation(s) should be pursued (Table 4). For example, in a male patient with developmental delay, head circumference that is more than 2.5 standard deviations above the mean for age, and penile freckling, PTEN gene analysis may be pursued as an initial evaluation given that these features are highly suggestive of PTEN hamartoma tumor syndrome (13, 30). Similarly, in the case of a female with severe developmental delay, acquired microcephaly, seizures, and stereotypic hand movements, MECP2 gene analysis may be pursued given that these features are suggestive of Rett syndrome (13, 79).
Recommendations for when to pursue FMR1 repeat analysis are diverse with some publications recommending that all individuals with ASD and/or ID/GDD undergo testing (13, 14, 213) while others make a distinction based on sex and recommend testing for all males (54). Finally, some authors suggest ordering FMR1 repeat analysis as a second-tier test (209) or restricting testing to those with the highest likelihood of testing positive for an FMR1 full mutation based on specific clinical findings could reduce the number of individuals tested while maintaining high sensitivity (236–238). With the relatively low cost of FMR1 repeat analysis and reproductive risks for family members should a child have fragile X, there should be a low threshold for offering this testing if testing is not offered to all patients with ID, GDD. and/or ASD.
Once a testing approach is determined, the ordering provider should engage families in a conversation prior to testing so they understand the nature and scope of the test(s) (e.g., purpose and potential results including uncertain results), benefits, limitations (e.g., does not detect all genetic causes of neurodevelopmental disorders), risks (e.g., uncertain results and familial implications), and costs (228, 233). Following receipt of results, providers will need to review any variants identified and put them in the context of their patient's clinical features and what is known about the gene, communicate results to the family, establish a follow-up plan, and collaborate with and refer to subspecialists (e.g., medical geneticist) as needed (233).
Some non-genetics providers express a lack of knowledge and/or discomfort in ordering and interpreting genetic testing (239–243), but genomics education (244), relationships with genetic testing laboratory staff (245), and the ability to refer to genetics (241) once results are received can afford providers with knowledge and support to integrate genomic medicine into their practice. In addition to consenting for and ordering testing independently, pediatric providers can integrate genetic counselors into their clinics for additional support and expertise and a multidisciplinary approach to care. Genetic counselors provide care in at least 28 countries and are typically graduate level providers that have training in both genetics and counseling enabling them to interpret genetic test results and support patients and families in their care (246–249). Increasingly, genetic counselors are working as experts in genetic testing alongside non-genetics providers in various clinical settings, including pediatric neurology and neurodevelopmental clinics (250–252). With the ability to foster and maintain interdisciplinary relationships included in their code of ethics and practice-based competencies, genetic counselors can educate other providers about genetic testing and the genetics of neurodevelopmental disorders (249, 253–256). Furthermore, genetic counselors' expertise makes them well-suited to work alongside pediatric providers to obtain consent for genetic testing, aid in ordering of appropriate testing, interpret genetic test results, and facilitate a family's understanding of and adjustment to genetic information (179, 249, 253–259). Genetic counselors' skill sets can enable them to be important members of a multidisciplinary neurodevelopmental care team along with other specialists such as neurodevelopmental pediatricians (16).
Pediatric healthcare providers may also elect to refer a subset of patients with neurodevelopmental disorders on to an external genetics provider (260). Medical geneticists have expertise in genetic diagnostics, management of genetic conditions, and counseling services (261). Pediatric healthcare providers might choose to refer to a genetics provider in cases where a specific genetic condition is suspected, additional etiological work-up is indicated after non-diagnostic testing, the provider is not equipped to facilitate discussion and ordering of specific testing, or where insurance requires a genetics evaluation before coverage of genetic testing (137, 234). Following receipt of results, pediatric healthcare providers could also refer to medical geneticists given that medical geneticists are well-poised to facilitate clinical correlation of genomic variants, evaluation of VUS, and management of patients with genetic diagnoses requiring ongoing surveillance and care (14, 137, 224, 262, 263).
Given the diagnostic yields and implications of genetic test results, genetic testing should be considered for any patient with ID, GDD, or ASD across the globe (Table 4). With that said, approaches to testing might be influenced by country and/or regional-specific factors. The healthcare system (universal health coverage vs. primarily private, for profit insurers) (216), recognition of genetic professionals (223, 247), access to testing due to availability and cost (247, 264–266), and genetics education for non-genetics professionals (265–267) all affect integration of genetics into the care of patients with neurodevelopmental disorders.
Although the majority of the literature concerning genetic testing for patients with neurodevelopmental disorders focuses on genetic testing for three neurodevelopmental conditions – ASD, ID, and GDD; the rapid evolution of genomic medicine and expansion of the literature will continue to change the landscape of genetic testing shifting who is offered genetic testing, the test offerings available, and our understanding of genomic variants.
In addition to ASD, ID, and GDD, increasing evidence supports consideration of genetic testing for other neurodevelopmental disorders. In epilepsy, between 4 and 78% of selected patients have genomic variants that are probable or definitive explanations for their epilepsy phenotypes (235). In a recent meta-analysis, of studies reporting on the yield of CMA, epilepsy gene panels, and ES, the diagnostic yield was 8, 23, and 45%, respectively (235). Specific epilepsy phenotypes, such as epileptic encephalopathies and comorbid ID have been associated with increased yields of testing (268, 269). Importantly, an increasing number of genetic conditions associated with seizure phenotypes influence therapeutic decision making; therefore, identification of a genetic etiology increasingly can enable personalized care (270–274). Despite genetic testing yields similar to those if ID, GDD, and ASD, no formal practice guidelines regarding genetic testing for individuals with epilepsy have been published by major medical societies; although various approaches to etiologic genetic testing in this patient population have been suggested by independent authors (273, 275).
In CP, studies have suggested a yield of 9.6–31.0% from CMA (276–279) and 10.6–65.0% using ES (280–283). In addition to emerging evidence that CP has a sizable genetic underpinning, genetic testing in CP has already resulted in changes to care (282, 284). Although a practice parameter published more than 15 years ago, the 2004 joint practice parameter from the American Academy of Neurology and Child Neurology Society, indicates that genetic testing should not be pursued in children with CP (285), no guidelines have been published since the emergence of data that demonstrates a significant diagnostic yield of genetic testing in CP patients.
Additional neurodevelopmental disorders, including speech disorders (286–290), Tourette syndrome (291–294) ADHD (295–298), and schizophrenia (299) have emerging evidence that genetic testing can provide an etiology to a portion of patients. Further studies are needed to understand the diagnostic yields and inform testing algorithms for these conditions.
The overlapping symptoms (300, 301), shared genetic underpinnings (4, 81, 211, 229, 302), and comorbidity of neurodevelopmental and neuropsychiatric diagnoses (303–307), have prompted many to advocate that neurodevelopmental and neuropsychiatric conditions collectively be considered as a continuum of developmental brain dysfunction (4, 229, 308). Familial aggregation and genetic studies have further supported the heterogenous nature of neurodevelopment and neuropsychiatric disorders (309–314). Given this and the emerging evidence of utility of genetic testing in a number of neurodevelopmental disorders, in the future, evidence may support broad implementation of genetic testing across neurodevelopmental and neuropsychiatric conditions.
In addition to considering genetic testing for additional neurodevelopmental diagnoses, current testing technology will continue to improve, and new testing offerings will emerge. For example, ES analysis did not routinely detect copy number variants and uniparental disomy, but now some laboratories are identifying and reporting these potentially causative variants (121, 140). With the rapid improvement of testing technologies and bioinformatics pipelines, ES may one day be able to detect additional genetic causes of neurodevelopmental disorders, including trinucleotide repeat expansions (315).
Not only will existing testing continue to improve, the clinical integration of other testing modalities such as genome sequencing, that increases coverage of exons, improves detection of structural variants, and interrogates non-coding regions; RNA-sequencing that analyzes gene expression and mRNA splicing; and genome-wide methylation studies that assesses epigenetic modification of the genome that can impact gene expression will increase diagnostic yield (212, 316–319). It is estimated that up to 3% of all patients with negative exomes could be explained by variants in the non-coding, regulatory regions that would be detectable by genome sequencing (316) and could be further assessed through technologies such as RNA-sequencing (317) and genome-wide methylation studies (319, 320). In addition to informing pathogenicity of non-coding variants, RNA-sequencing and genome-wide methylation studies could also detect epigenetic changes that impact gene expression but do not alter the DNA sequence and are thus undetectable through sequencing (319, 321).
Furthermore, genome sequencing and single-cell sequencing will also enable increased detection of somatic mosaicism that, using current technologies, appear to constitute 5–10% of de novo variants (211, 322). Because somatic variants are not always detectable in the blood, future testing that includes additional cell types will also increase the identification of genetic etiologies for patients and families (211). Availability of relevant tissues, such as single neurons, may be limited, however.
In addition to advancements in testing technology, the understanding of genes and variants and their relationships to disease will continue to improve. Currently, only a fraction of the 20,000 human coding genes have an established disease relationship, and variants in known disease genes may be uncertain given the current knowledge base. As reference databases are bolstered (317), data sharing of variants and candidate genes increases (317, 323, 324), and additional technologies such RNA-sequencing and genome-wide methylation studies are integrated into testing algorithms (317, 319), our understanding of genes' relationship to neurodevelopment disorders and variants' pathogenicity will improve, thus increasing diagnostic yield of testing.
As new tests emerge and previous test results are reanalyzed or updated, recontacting of patients should be a shared responsibility among health-care providers, the clinical testing laboratories, and the patients (325, 326). As such, pediatric providers ordering genetic testing and those with an ongoing relationship with the patient and family, should remain informed of advancements that might provide additional information about a reported variant or indicate that additional testing or evaluation is warranted (82, 325, 327). Providers should bear in mind that, even if an individual has had genetic testing, there may be additional testing (e.g., exome reanalysis, CMA if ES does not include adequate CNV detection, genome sequencing, and novel testing such as genome sequencing) to consider over time and it is important that clinicians continue to consider the utility of genetic testing in individuals with neurodevelopmental diagnoses.
Currently, diagnostic genetic testing in neurodevelopmental disorders is focused on identifying rare variants of large effect size. As our ability to detect and interpret genetic causes of NDD increases, including those of more modest effect size, more families will be afforded the clinical and personal utility of genetic diagnoses. Many genetic underpinnings of NDD are characterized by variable expressivity, however, often leaving families with questions about recurrence and prognosis. In addition to the important impact of environmental and stochastic variation, recent advances suggest that the observed variability in phenotypic expression of large-effect rare variants may be explained, in part, by the additive effects of additional high-impact variants (328, 329) and background polygenic risk conferred by a large number of common variants of small individual effect size (330–336). Larger sample sizes and improved study methodologies are expected to lead to identification of rare variants of smaller effect size and elucidate the potential clinical role for polygenic scores in the near future. Increased understanding of additional genetic and other factors that influence phenotypes will enable tailored counseling and care for families receiving a genetic etiology for their child's NDD (17, 334).
Neurodevelopmental disorders are common, and a significant proportion are caused by rare copy number and exonic sequence variants of large effect size that can be identified by genetic testing. Identifying a genetic etiologic diagnosis can allow clinicians to provide more accurate prognostication and recurrence risk counseling, identify and treat or prevent medical comorbidities, guide patients and families to condition-specific resources and supports and, in some cases, refine treatment options. Genetic testing is now the standard of care for individuals with ID/GDD and/or ASD, and the indications for testing will almost certainly broaden to include other neurodevelopmental disorders in the future. Most genetic etiologic diagnoses in patients with ID/GDD and/or ASD can be made with broad examination of copy number and exonic sequence variants that is most efficiently achieved using ES with CNV calling. It is important for clinicians who provide healthcare to children and adolescents with neurodevelopmental disorders to gain an understanding of these common tests and their role in providing the best medical care for patients and to work to facilitate the genetic etiologic evaluation of these patients by ordering testing or partnering with genetic providers.
JS and SM contributed to the literature review and synthesis of this review article.
Research reported in this publication was supported by the National Institute of Mental Health and the Eunice Kennedy Shriver National Institute of Child Health and Human Development of the National Institutes of Health under award numbers R01MH074090, R01MH107431, and U01MH119705.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank David H. Ledbetter and Christa Lese Martin for their thoughtful review and comments that helped shape this review.
1. Zablotsky B, Black LI, Maenner MJ, Schieve LA, Danielson ML, Bitsko RH, et al. Prevalence and trends of developmental disabilities among children in the United States: 2009–2017. Pediatrics. (2019) 144:e20190811. doi: 10.1542/peds.2019-0811
2. American Psychiatric Association (2013). Diagnostic and Statistical Manual of Mental Disorders: DSM-5. 5th ed. Arlington, VA: American Psychiatric Association. doi: 10.1176/appi.books.9780890425596
3. Ismail FY, Shapiro BK. What are neurodevelopmental disorders? Curr Opin Neurol. (2019) 32:611–6. doi: 10.1097/WCO.0000000000000710
4. Moreno-De-Luca A, Myers SM, Challman TD, Moreno-De-Luca D, Evans DW, Ledbetter DH. Developmental brain dysfunction: revival and expansion of old concepts based on new genetic evidence. Lancet Neurol. (2013) 12:406–14. doi: 10.1016/S1474-4422(13)70011-5
5. Owen MJ, O'Donovan MC, Thapar A, Craddock N. Neurodevelopmental hypothesis of schizophrenia. Br J Psychiatry. (2011) 198:173–5. doi: 10.1192/bjp.bp.110.084384
6. Rapoport JL, Giedd JN, Gogtay N. Neurodevelopmental model of schizophrenia: update 2012. Mol Psychiatr. (2012) 17:1228–38. doi: 10.1038/mp.2012.23
7. Thapar A, Cooper M, Rutter M. Neurodevelopmental disorders. Lancet Psychiat. (2017) 4:339–46. doi: 10.1016/S2215-0366(16)30376-5
8. Clark LA, Cuthbert B, Lewis-Fernandez R, Narrow WE, Reed GM. Three approaches to understanding and classifying mental disorder: ICD-11, DSM-5, and the National Institute of Mental Health's Research Domain Criteria (RDoC). Psychol Sci Public Interest. (2017) 18:72–145. doi: 10.1177/1529100617727266
9. Pearn J. Differentiating diseases: the centrum of differential diagnosis in the evolution of Oslerian medicine. Fetal Pediatr Pathol. (2011) 30:1–15. doi: 10.3109/15513815.2011.520252
10. Hyman SE. The diagnosis of mental disorders: the problem of reification. Annu Rev Clin Psychol. (2010) 6:155–79. doi: 10.1146/annurev.clinpsy.3.022806.091532
11. Insel TR, Cuthbert BN. Medicine. Brain disorders? Precisely Sci. (2015) 348:499–500. doi: 10.1126/science.aab2358
12. Lilienfeld SO, Treadway MT. Clashing diagnostic approaches: DSM-ICD versus RDoC. Annu Rev Clin Psychol. (2016) 12:435–63. doi: 10.1146/annurev-clinpsy-021815-093122
13. Hyman SL, Levy SE, Myers SM. Identification, evaluation, and management of children with autism spectrum disorder. Pediatrics. (2020) 145:e20193447. doi: 10.1542/peds.2019-3447
14. Moeschler JB, Shevell M. Comprehensive evaluation of the child with intellectual disability or global developmental delays. Pediatrics. (2014) 134:e903–18. doi: 10.1542/peds.2014-1839
15. Muhle RA, Reed HE, Vo LC, Mehta S, McGuire K, Veenstra-VanderWeele J, et al. Clinical diagnostic genetic testing for individuals with developmental disorders. J Am Acad Child Adolesc Psychiatry. (2017) 56:910–3. doi: 10.1016/j.jaac.2017.09.418
16. Myers SM. Diagnosing developmental disabilities. In: Batshaw ML, Roizen NJ, Pellegrino L, editors. Children with Disabilities. 8th ed. Baltimore: Brookes (2019). p. 199–223.
17. Finucane BM, Lusk L, Arkilo D, Chamberlain S, Devinsky O, Dindot S, et al. 15q duplication syndrome and related disorders. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle. Available online at: https://www.ncbi.nlm.nih.gov/books/NBK367946/ (accessed March 2, 2020).
18. Mitchel MW, Moreno-De-Luca D, Myers SM, Levy RV, Turner S, Ledbetter DH. 17q12 recurrent deletion syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle (2016). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK401562/ (accessed January 25, 2021).
19. DiBacco ML, Roullet JB, Kapur K, Brown MN, Walters DC, Gibson KM, et al. Age-related phenotype and biomarker changes in SSADH deficiency. Ann Clin Transl Neurol. (2018) 6:114–20. doi: 10.1002/acn3.696
20. Vogel KR, Ainslie GR, Walters DC, McConnell A, Dhamne SC, Rotenberg A, et al. Succinic semialdehyde dehydrogenase deficiency, a disorder of GABA metabolism: an update on pharmacological and enzyme-replacement therapeutic strategies. J Inherit Metab Dis. (2018) 41:699–708. doi: 10.1007/s10545-018-0153-8
21. Pearl PL, Wiwattanadittakul N, Roullet JB, Gibson KM. Succinic semialdehyde dehydrogenase deficiency. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle (2004). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK1195/ (accessed March 2, 2020).
22. Napolitano C, Splawski I, Timothy KW, Bloise R, Priori SG. Timothy syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle (2006). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK1403/ (accessed March 2, 2020).
23. Nowaczyk MJM, Wassif CA. Smith-lemli-opitz syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle (1998). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK1143/ (accesed January 26, 2021).
24. Wassif CA, Kratz L, Sparks SE, Wheeler C, Bianconi S, Gropman A, et al. A placebo-controlled trial of simvastatin therapy in Smith-Lemli-Opitz syndrome. Genet Med. (2017) 19:297–305. doi: 10.1038/gim.2016.102
25. Monaghan KG, Lyon E, Spector EB, erican College of Medical Genetics and Genomics. ACMG Standards and Guidelines for fragile X testing: a revision to the disease-specific supplements to the Standards and Guidelines for Clinical Genetics Laboratories of the American College of Medical Genetics and Genomics. Genet Med. (2013) 15:575–86. doi: 10.1038/gim.2013.61
26. Collins SC, Bray SM, Suhl JA, Cutler DJ, Coffee B, Zwick ME, et al. Identification of novel FMR1 variants by massively parallel sequencing in developmentally delayed males. Am J Med Genet A. (2010) 152A:2512–20. doi: 10.1002/ajmg.a.33626
27. Collins SC, Coffee B, Benke PJ, Berry-Kravis E, Gilbert F, Oostra B, et al. Array-based FMR1 sequencing and deletion analysis in patients with a fragile X syndrome-like phenotype. PLoS ONE. (2010) 5:e9476. doi: 10.1371/journal.pone.0009476
28. Hunter JE, Berry-Kravis E, Hipp H, Gilbert F, Oostra B, et al. FMR1 disorders. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle (1998). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK1384/ (accessed March 2, 2020).
29. Kaur S, Christodoulou J. MECP2 disorders. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle (2001). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK1497/ (accessed January 25, 2020).
30. Pilarski R. PTEN hamartoma tumor syndrome: a clinical overview. Cancers. (2019) 11:844. doi: 10.3390/cancers11060844
31. Eng C. PTEN hamartoma tumor syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle (2001). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK1488/ (accessed March 2, 2020).
32. National Comprehensive Cancer Network (NCCN). Clinical Practice Guidelines in Oncology – Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic Version 2.2021. (2021). Available online at: https://www.nccn.org/professionals/physician_gls/pdf/genetics_bop.pdf (accessed January 25, 2021).
33. Klepper J. GLUT1 deficiency syndrome in clinical practice. Epilepsy Res. (2012) 100:272–7. doi: 10.1016/j.eplepsyres.2011.02.007
34. Wang D, Pascual JM, De Vivo D. Glucose transporter type 1 deficiency syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle (2002). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK1430/ (accessed March 2, 2020).
35. Holder JL Jr, Hamdan FF, Michaud JL. SYNGAP1-related intellectual disability. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle (2019). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK537721/ (accessed March 2, 2020).
36. Curatolo P, Moavero R. mTOR inhibitors in tuberous sclerosis complex. Curr Neuropharmacol. (2012) 10:404–15. doi: 10.2174/157015912804143595
37. Northrup H, Koenig MK, Pearson DA, Au KS. Tuberous sclerosis complex. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle (1999). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK1220/ (accessed January 26, 2021).
38. Krueger DA, Northrup H, International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex surveillance and management: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. (2013) 49:255–65. doi: 10.1016/j.pediatrneurol.2013.08.002
39. Schaefer GB, Bodensteiner JB. Evaluation of the child with idiopathic mental retardation. Pediatr Clin North Am. (1992) 39:929–43. doi: 10.1016/S0031-3955(16)38381-X
40. Bilkey GA, Burns BL, Coles EP, Bowman FL, Beilby JP, Pachter NS, et al. Genomic testing for human health and disease across the life cycle: applications and ethical, legal, and social challenges. Front Public Health. (2019) 7:40. doi: 10.3389/fpubh.2019.00040
41. Hayeems RZ, Babul-Hirji R, Hoang N, Weksberg R, Shuman C. Parents' experience with pediatric microarray: transferrable lessons in the era of genomic counseling. J Genet Couns. (2016) 25:298–304. doi: 10.1007/s10897-015-9871-3
42. Iglesias A, Anyane-Yeboa K, Wynn J, Wilson A, Truitt Cho M, Guzman E, et al. The usefulness of whole-exome sequencing in routine clinical practice. Genet Med. (2014) 16:922–31. doi: 10.1038/gim.2014.58
43. Reiff M, Giarelli E, Bernhardt BA, Easley E, Spinner NB, Sankar PL, et al. Parents' perceptions of the usefulness of chromosomal microarray analysis for children with autism spectrum disorders. J Autism Dev Disord. (2015) 45:3262–75. doi: 10.1007/s10803-015-2489-3
44. Stivers T, Timmermans S. The actionability of exome sequencing testing results. Sociol Health Illn. (2017) 39:1542–56. doi: 10.1111/1467-9566.12614
45. Sun F, Oristaglio J, Levy SE, Hakonarson H, Sullivan N, Fontanarosa J, et al. In Genetic Testing for Developmental Disabilities, Intellectual Disability, and Autism Spectrum Disorder [Internet]. Rockville, MD: Agency for Healthcare Research and Quality (US) (2015). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK304462/ (accessed December 10, 2019).
46. Dixon-Salazar TJ, Silhavy JL, Udpa N, Schroth J, Bielas S, Schaffer AE, et al. Exome sequencing can improve diagnosis and alter patient management. Sci Transl Med. (2012) 4:138ra178. doi: 10.1126/scitranslmed.3003544
47. van Karnebeek CDM, Stockler S. Treatable inborn errors of metabolism causing intellectual disability: a systematic literature review. Mol Genet. (2012) 105:368–81. doi: 10.1016/j.ymgme.2011.11.191
48. Fung JLF, Yu MHC, Huang S, Chung CCY, Chan MCY, Pajusalu S, et al. A three-year follow-up study evaluating clinical utility of exome sequencing and diagnostic potential of reanalysis. npj Genom Med. (2020) 5:37. doi: 10.1038/s41525-020-00144-x
49. American College of Medical Genetics and Genomics (ACMG). Clinical utility of genetic and genomic services: a position statement of the American College of Medical Genetics and Genomics. Genet Med. (2015) 17:505–7. doi: 10.1038/gim.2015.41
50. Tărlungeanu DC, Novarino G. Genomics in neurodevelopmental disorders: an avenue to personalized medicine. Exp Mol Med. (2018) 50:100. doi: 10.1038/s12276-018-0129-7
51. Saul RA editor. Medical Genetics in Pediatric Practice. Itasca, IL: American Academy of Pediatrics (2013).
52. Niguidula N, Alamillo C, Shahmirzadi Mowlavi L, Powis Z, Cohen JS, Farwell Hagmen KD. Clinical whole-exome sequencing results impact medical management. Mol Genet Genomic Med. (2018) 6:1068–78. doi: 10.1002/mgg3.484
53. Zoghbi HY, Bear MF. Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb Perspect Biol. (2012) 4:a009886. doi: 10.1101/cshperspect.a009886
54. Schaefer GB, Mendelsohn NJ, the Professional Practive and Guidelines Committeee 29 of the American College of Medical Genetics. Clinical genetics evaluation in identifying the etiology of autism spectrum disorders: 2013 guideline revisions. Genet Med. (2013) 15:399–407. doi: 10.1038/gim.2013.32
55. Medavarapu S, Marella LL, Sangem A, Kairam R. Where is the evidence? a narrative literature review of the treatment modalities for autism spectrum disorders. Cureus. (2019) 11:e3901. doi: 10.7759/cureus.3901
56. Coulter ME, Miller DT, Harris DJ, Hawley P, Picker J, Roberts AE, et al. Chromosomal microarray testing influences medical management. Genet Med. (2011) 13:770–6. doi: 10.1097/GIM.0b013e31821dd54a
57. Ellison JW, Ravnan JB, Rosenfeld JA, Morton SA, Neill NJ, Williams MS, et al. Clinical utility of chromosomal microarray analysis. Pediatrics. (2012) 130:e1085–95. doi: 10.1542/peds.2012-0568
58. Henderson LB, Applegate CD, Wohler E, Sheridan MB, Hoover-Fong J, Batista DA. The impact of chromosomal microarray on clinical management: a retrospective analysis. Genet Med. (2014) 16:657–64. doi: 10.1038/gim.2014.18
59. Hens K, Peeters H, Dierickx K. Genetic testing and counseling in the case of an autism diagnosis: a caregivers perspective. Eur J Med Genet. (2016) 59:452–8. doi: 10.1016/j.ejmg.2016.08.007
60. Mroch AR, Flanagan JD, Stein QP. Solving the puzzle: case examples of array comparative genomic hybridization as a tool to end the diagnostic odyssey. Curr Probl Pediatr Adolesc Health Care. (2012) 42:74–8. doi: 10.1016/j.cppeds.2011.10.003
61. Riggs ER, Wain KE, Riethmaier D, Smith-Packard B, Faucett WA, Hoppman N, et al. Chromosomal microarray impacts clinical management. Clin Genet. (2014) 85:147–53. doi: 10.1111/cge.12107
62. Saam J, Gudgeon J, Aston E, Brothman AR. How physicians use array comparative genomic hybridization results to guide patient management in children with developmental delay. Genet Med. (2008) 10:181–6. doi: 10.1097/GIM.0b013e3181634eca
63. Srivastava S, Cohen JS, Vernon H, Baranano K, McClellan R, Jamal L, et al. Clinical whole exome sequencing in child neurology practice. Ann Neurol. (2014) 76:473–83. doi: 10.1002/ana.24251
64. Tao VQ, Chan KY, Chu YW, Mok GT, Tan TY, Yang W, et al. The clinical impact of chromosomal microarray on paediatric care in Hong Kong. PLoS ONE. (2014) 9:e109629. doi: 10.1371/journal.pone.0109629
65. Srivastava S, Love-Nichols JA, Dies KA, Ledbetter DH, Martin CL, Chung WK, et al. Meta-analysis and multidisciplinary consensus statement: exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders. Genet Med. (2019) 21:2413–21. doi: 10.1038/s41436-019-0554-6
66. Hayeems RZ, Hoang N, Chenier S, Stavropoulos DJ, Pu S, Weksberg R, et al. Capturing the clinical utility of genomic testing: medical recommendations following pediatric microarray. Eur J Hum Genet. (2015) 23:1135–41. doi: 10.1038/ejhg.2014.260
67. Shevell MI, Majnemer A, Rosenbaum P, Abrahamowicz M. Etiologic yield of subspecialists' evaluation of young children with global developmental delay. J Pediatr. (2000) 136:593–8. doi: 10.1067/mpd.2000.104817
68. Turner G, Boyle J, Partington MW, Kerr B, Raymond FL, Gécz J. Restoring reproductive confidence in families with X-linked mental retardation by finding the causal mutation. Clin Genet. (2008) 73:188–90. doi: 10.1111/j.1399-0004.2007.00929.x
69. Narcisa V, Discenza M, Vaccari E, Rosen-Sheidley B, Hardan AY, Couchon E. Parental interest in a genetic risk assessment test for autism spectrum disorders. Clin Pediat. (2012) 52:139–46. doi: 10.1177/0009922812466583
70. Trosman JR, Weldon CB, Slavotinek A, Norton ME, Douglas MP, Phillips KA. Perspectives of US private payers on insurance coverage for pediatric and prenatal exome sequencing: results of a study from the Program in Prenatal and Pediatric Genomic Sequencing (P3EGS). Genet Med. (2020) 22:283–91. doi: 10.1038/s41436-019-0650-7
71. Mollison L, O'Daniel JM, Henderson GE, Berg JS, Skinner D. Parents' perceptions of personal utility of exome sequencing results. Genet. Med. (2020) 22:752–7. doi: 10.1038/s41436-019-0730-8
72. Lingen M, Albers L, Borchers M, Haass S, Gärtner J, Schröder S, et al. Obtaining a genetic diagnosis in a child with disability: impact on parental quality of life. Clin Genet. (2016) 89:258–66. doi: 10.1111/cge.12629
73. Reiff M, Bernhardt BA, Mulchandani S, Soucier D, Cornell D, Pyeritz RE, Spinner NB. “What does it mean?”: uncertainties in understanding results of chromosomal microarray testing. Genet Med. (2012) 14:250–8. doi: 10.1038/gim.2011.52
74. Krabbenborg L, Vissers LELM, Schieving J, Kleefstra T, Kamsteeg EJ, Veltman JA, et al. Understanding the psychosocial effects of WES test results on parents of children with rare diseases. J Genet Couns. (2016) 25:1207–14. doi: 10.1007/s10897-016-9958-5
75. Gonzalez-Mantilla AJ, Moreno-De-Luca A, Ledbetter DH, Martin CL. A cross-disorder method to identify novel candidate genes for developmental brain disorders. JAMA Psychiatry. (2016) 73:275–83. doi: 10.1001/jamapsychiatry.2015.2692
76. Strande NT, Riggs ER, Buchanan AH, Ceyhan-Bisroy O, DiStefano M, Dwight SS, et al. Evaluating the clinical validity of gene-disease associations: an evidence-based framework developed by the clinical genome resource. Am J Hum Genet. (2017) 100:895–906. doi: 10.1016/j.ajhg.2017.04.015
77. Riggs ER, Church DM, Hanson K, Horner VL, Kaminsky EB, Kuhn RM, et al. Towards an evidence-based process for the clinical interpretation of copy number variation. Clin Genet. (2012) 81:403–12. doi: 10.1111/j.1399-0004.2011.01818.x
78. Wright CF, Fitzgerald TW, Jones WD, Clayton S, McRae JF, van Kogelenberg M, et al. Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. Lancet. (2015) 385:1305–14. doi: 10.1016/S0140-6736(14)61705-0
79. Manning M, Hudgins L. Array-based technology and recommendations for utilization in medical genetics practice for detection of chromosomal abnormalities. Genet Med. (2010) 12:742–5. doi: 10.1097/GIM.0b013e3181f8baad
80. Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. (2010) 86:749–64. doi: 10.1016/j.ajhg.2010.04.006
81. Mefford HC, Batshaw ML, Hoffman EP. Genomics, intellectual disability, and autism. New Engl J Med. (2012) 366:733–43. doi: 10.1056/NEJMra1114194
82. Riggs ER, Andersen EF, Cherry AM, Kantarci S, Kearney H, Patel A, et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med. (2020) 22:245–57. doi: 10.1038/s41436-019-0686-8
83. Kaminsky EB, Kaul V, Paschall J, Church DM, Bunke B, Kunig D, et al. An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities. Genet Med. (2011) 13:777–84. doi: 10.1097/GIM.0b013e31822c79f9
84. Watson CT, Marques-Bonet T, Sharp AJ, Mefford HC. The genetics of microdeletion and microduplication syndromes: an update. Annu Rev Genomics Hum Genet. (2014) 15:215–44. doi: 10.1146/annurev-genom-091212-153408
85. Hogan J, Turner A, Tucker K, Warwick L. Unintended diagnosis of Von Hippel Lindau syndrome using Array Comparative Genomic Hybridization (CGH): counseling challenges arising from unexpected information. J Genet Couns. (2013) 22:22–6. doi: 10.1007/s10897-012-9520-z
86. Kearney HM, Kearney JB, Conlin LK. Diagnostic implications of excessive homozygosity detected by SNP-based microarrays: consanguinity, uniparental disomy, and recessive single-gene mutations. Clin Lab Med. (2011) 31:595–613. doi: 10.1016/j.cll.2011.08.003
87. Gibson J, Morton NE, Collins A. Extended tracts of homozygosity in outbred human populations. Hum Mol Genet. (2006) 15:789–95. doi: 10.1093/hmg/ddi493
88. Rehder CW, David KL, Hirsch B, Toriello HV, Wilson CM, Kearney HM. American College of Medical Genetics and Genomics: standards and guidelines for documenting suspected consanguinity as an incidental finding of genomic testing. Genet Med. (2013) 15:150–2. doi: 10.1038/gim.2012.169
89. Sund KL, Zimmerman SL, Thomas C, Mitchell AL, Prada CE, Grote L, et al. Regions of homozygosity identified by SNP microarray analysis aid in the diagnosis of autosomal recessive disease and incidentally detect parental blood relationships. Genet Med. (2013) 15:70–8. doi: 10.1038/gim.2012.94
90. Waggoner D, Wain KE, Dubuc AM, Conlin L, Hickey SE, Lamb AN, et al. Yield of additional genetic testing after chromosomal microarray for diagnosis of neurodevelopmental disability and congenital anomalies: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med. (2018) 20:1105–13. doi: 10.1038/s41436-018-0040-6
91. Baris HN, Tan WH, Kimonis VE, Irons MB. Diagnostic utility of array-based comparative genomic hybridization in a clinical setting. Am J Med Genet A. (2007)143A:2523–33. doi: 10.1002/ajmg.a.31988
92. Bartnik M, Nowakowska B, Derwińska K, Wiśniowiecka-Kowalnik B, Kedzior M, Bernaciak J, et al. Application of array comparative genomic hybridization in 256 patients with developmental delay or intellectual disability. J Appl Genet. (2014) 55:125–44. doi: 10.1007/s13353-013-0181-x
93. Battaglia A, Doccini V, Bernardini L, Novelli A, Loddo S, Capalbo A, et al. Confirmation of chromosomal microarray as a first-tier clinical diagnostic test for individuals with developmental delay, intellectual disability, autism spectrum disorders and dysmorphic features. Eur J Paediatr Neurol. (2013) 17:589–99. doi: 10.1016/j.ejpn.2013.04.010
94. Doherty E, O'Connor R, Zhang A, Lim C, Love JM, Ashton F, et al. Developmental delay referrals and the roles of Fragile X testing and molecular karyotyping: a New Zealand perspective. Mol Med Rep. (2013) 7:1710–4. doi: 10.3892/mmr.2013.1386
95. Fan Y, Wu Y, Wang L, Wang Y, Gong Z, Qiu W, et al. Chromosomal microarray analysis in developmental delay and intellectual disability with comorbid conditions. BMC MedGenomics. (2018) 11:49. doi: 10.1186/s12920-018-0368-4
96. Ho KS, Wassman ER, Baxter AL, Hensel CH, Martin MM, Prasad A, et al. Chromosomal microarray analysis of consecutive individuals with autism spectrum disorders using an ultra-high resolution chromosomal microarray optimized for neurodevelopmental disorders. Int J Mol Sci. (2016) 17:2070. doi: 10.3390/ijms17122070
97. Lee CG, Park SJ, Yun JN, Ko JM, Kim HJ, Yim SY, et al. Array-based comparative genomic hybridization in 190 Korean patients with developmental delay and/or intellectual disability: a single tertiary care university center study. Yonsei Med J. (2013) 54:1463–70. doi: 10.3349/ymj.2013.54.6.1463
98. Lu X, Shaw CA, Patel A, Li J, Cooper ML, Wells WR, et al. Clinical implementation of chromosomal microarray analysis: summary of 2513 postnatal cases. PloS ONE. (2007) 2:e327. doi: 10.1371/journal.pone.0000327
99. Marano RM, Mercurio L, Kanter R, Doyle R, Abuelo D, Morrow EM, et al. Risk assessment models in genetics clinic for array comparative genomic hybridization: clinical information can be used to predict the likelihood of an abnormal result in patients. J Pediatr Genet. (2013) 2:25–31. doi: 10.3233/PGE-13044
100. Nicholl J, Waters W, Mulley JC, Suwalski S, Brown S, Hull Y, et al. Cognitive deficit and autism spectrum disorders: prospective diagnosis by array CGH. Pathology. (2014) 46:41–5. doi: 10.1097/PAT.0000000000000043
101. Pereira RR, Pinto IP, Minasi LB, de Melo AV, da Cruz e Cunha DM, Cruz AS, et al. Screening for intellectual disability using high-resolution CMA technology in a retrospective cohort from Central Brazil. PLoS ONE. (2014) 9:e103117. doi: 10.1371/journal.pone.0103117
102. Preiksaitiene E, Molyte A, Kasnauskiene J, Ciuladaite Z, Utkus A, Patsalis PC, et al. Considering specific clinical features as evidence of pathogenic copy number variants. J Appl Genet. (2014) 55:189–96. doi: 10.1007/s13353-014-0197-x
103. Qiao Y, Harvard C, Tyson C, Liu X, Fawcett C, Pavlidis P, et al. Outcome of array CGH analysis for 255 subjects with intellectual disability and search for candidate genes using bioinformatics. Hum Genet. (2010) 128:179–94. doi: 10.1007/s00439-010-0837-0
104. Roberts JL, Hovanes K, Dasouki M, Manzardo AM, Butler MG. Chromosomal microarray analysis of consecutive individuals with autism spectrum disorders or learning disability presenting for genetic services. Gene. (2014) 535:70–8. doi: 10.1016/j.gene.2013.10.020
105. Sansovic I, Ivankov AM, Bobinec A, Kero M, Barisic I. Chromosomal microarray in clinical diagnosis: a study of 337 patients with congenital anomalies and developmental delays or intellectual disability. Croat Med J. (2017) 58:231–8. doi: 10.3325/cmj.2017.58.231
106. Sharp AJ, Hansen S, Selzer RR, Cheng Z, Regan R, Hurst JA, et al. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat Genet. (2006) 38:1038–42. doi: 10.1038/ng1862
107. Shoukier M, Klein N, Auber B, Wickert J, Schroder J, Zoll B, et al. Array CGH in patients with developmental delay or intellectual disability: are there phenotypic clues to pathogenic copy number variants? Clin Genet. (2013) 83:53–65. doi: 10.1111/j.1399-0004.2012.01850.x
108. Xiang B, Zhu H, Shen Y, Miller DT, Lu K, Hu X, et al. Genome-wide oligonucleotide array comparative genomic hybridization for etiological diagnosis of mental retardation: a multicenter experience of 1499 clinical cases. J Mol Diagn. (2010) 12:204–12. doi: 10.2353/jmoldx.2010.090115
109. Xu M, Ji Y, Zhang T, Jiang X, Fan Y, Geng J, et al. Clinical application of chromosome microarray analysis in Han Chinese children with neurodevelopmental disorders. Neurosci Bull. (2018) 34:981–91. doi: 10.1007/s12264-018-0238-2
110. Ho KS, Twede H, Vanzo R, Harward E, Hensel CH, Martin MM, et al. Clinical performance of an ultrahigh resolution chromosomal microarray optimized for neurodevelopmental disorders. BioMed Res Int. (2016) 2016:3284534. doi: 10.1155/2016/3284534
111. McGrew SG, Peters BR, Crittendon JA, Veenstra-Vanderweele J. Diagnostic yield of chromosomal microarray analysis in an autism primary care practice: which guidelines to implement? J Autism Dev Disord. (2012) 42:1582–91. doi: 10.1007/s10803-011-1398-3
112. Rosenfeld JA, Ballif BC, Torchia BS, Sahoo T, Ravnan JB, Schultz R, et al. Copy number variations associated with autism spectrum disorders contribute to a spectrum of neurodevelopmental disorders. Genet Med. (2010) 12:694–702. doi: 10.1097/GIM.0b013e3181f0c5f3
113. Schaefer GB, Starr L, Pickering D, Skar G, Dehaai K, Sanger WG. Array comparative genomic hybridization findings in a cohort referred for an autism evaluation. J Child Neurol. (2010) 25:1498–503. doi: 10.1177/0883073810370479
114. Nava C, Keren B, Mignot C, Rastetter A, Chantot-Bastaraud S, Faudet A, et al. Prospective diagnostic analysis of copy number variants using SNP microarrays in individuals with autism spectrum disorders. Eur J Hum Genet. (2014) 22:71–8. doi: 10.1038/ejhg.2013.88
115. Bremer A, Giacobini M, Eriksson M, Gustavsson P, Nordin V, Fernell E, et al. Copy number variation characteristics in subpopulations of patients with autism spectrum disorders. Am J Med Genet B Neuropsychiatr Genet. (2011) 115–24. doi: 10.1002/ajmg.b.31142
116. Shevell M, Ashwal S, Donley D, Flint J, Gingold M, Hirtz D, et al. Practice parameter: evaluation of the child with global developmental delay: report of the Quality Standards Subcommittee of the American Academy of Neurology and The Practice Committee of the Child Neurology Society. Neurology. (2003) 60:367–80. doi: 10.1212/01.WNL.0000031431.81555.16
117. Pichert G, Mohammed SN, Ahn JW, Ogilvie CM, Izatt L. Unexpected findings in cancer predisposition genes detected by array comparative genomic hybridisation: what are the issues? J Med Genet. (2011) 48:535–9. doi: 10.1136/jmg.2010.087593
118. Adams SA, Coppinger J, Saitta SC, Stroud T, Manikum K, Fan Z, et al. Impact of genotype-first diagnosis: the detection of microdeletion and microduplication syndromes with cancer predisposition by aCGH. Genet Med. (2009) 11:314–22. doi: 10.1097/GIM.0b013e3181a028a5
119. Boone PM, Soens ZT, Campbell IM, Stankiewicz P, Cheung SW, Patel A, et al. Incidental copy-number variants identified by routine genome testing in a clinical population. Genet Med. (2013) 15:45–54. doi: 10.1038/gim.2012.95
120. Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. (2011) 12:745–55. doi: 10.1038/nrg3031
121. Retterer K, Scuffins J, Schmidt D, Lewis R, Pineda-Alvarez D, Stafford A, et al. Assessing copy number from exome sequencing and exome array CGH based on CNV spectrum in a large clinical cohort. Genet Med. (2015) 17:623–9. doi: 10.1038/gim.2014.160
122. Marchuk DS, Crooks K, Strande N, Kaiser-Rogers K, Milko LV, Brandt A, et al. Increasing the diagnostic yield of exome sequencing by copy number variant analysis. PLoS ONE. (2018) 13:e0209185. doi: 10.1371/journal.pone.0209185
123. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
124. Ewans LJ, Schofield D, Shrestha R, Zhu Y, Gayevskiy V, Ying K, et al. Whole-exome sequencing reanalysis at 12 months boosts diagnosis and is cost-effective when applied early in Mendelian disorders. Genet Med. (2018) 20:1564–74. doi: 10.1038/gim.2018.39
125. Liu P, Meng L, Normand EA, Xia F, Song X, Ghazi A, et al. Reanalysis of Clinical Exome Sequencing Data. New Engl J Med. (2019) 380:2478–80. doi: 10.1056/NEJMc1812033
126. Smith ED, Radtke K, Rossi M, Shinde DN, Darabi S, El-Khechen D, et al. Classification of genes: standardized clinical validity assessment of gene-disease associations aids diagnostic exome analysis and reclassifications. Hum Mutat. (2017) 38:600–8. doi: 10.1002/humu.23183
127. Wenger AM, Guturu H, Bernstein JA, Bejerano G. Systematic reanalysis of clinical exome data yields additional diagnoses: implications for providers. Genet Med. (2017) 19:209–14. doi: 10.1038/gim.2016.88
128. Wright CF, McRae JF, Clayton S, Gallone G, Aitken S, FitzGerald TW, et al. Making new genetic diagnoses with old data: iterative reanalysis and reporting from genome-wide data in 1,133 families with developmental disorders. Genet Med. (2018) 20:1216–23. doi: 10.1038/gim.2017.246
129. Xue Y, Ankala A, Wilcox W, Madhuri H. Solving the molecular diagnostic testing conundrum for Mendelian disorders in the era of next-generation sequencing: single-gene, gene panel, or exome/genome sequencing. Genet Med. (2015) 17:444–51. doi: 10.1038/gim.2014.122
130. Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. (2013) 15:565–74. doi: 10.1038/gim.2013.73
131. Kalia SS, Adelman K, Bale SJ, Chung WK, Eng C, Evans JP, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med. (2017) 19:249–55. doi: 10.1038/gim.2016.190
132. American College of Medical Genetics and Genomics (ACMG). Incidental findings in clinical genomics: a clarificatoin. Genet Med. (2013) 15:664–6. doi: 10.1038/gim.2013.82
133. Eno C, Bayrak-Toydemir P, Bean L, Braxton A, Chao EC, El-Khechen D, et al. Misattributed parentage as an unanticipated finding during exome/genome sequencing: current clinical laboratory practices and an opportunity for standardization. Genet Med. (2019) 21:861–6. doi: 10.1038/s41436-018-0265-4
134. Hercher L, Jamal L. An old problem in a new age: revisiting the clinical dilemma of misattributed paternity. Appl Transl Genom. (2016) 8:36–9. doi: 10.1016/j.atg.2016.01.004
135. Kong SW, Lee I-H, Liu X, Hirschhorn JN, Mandl KD. Measuring coverage and accuracy of whole-exome sequencing in clinical context. Genet Med. (2018) 20:1617–26. doi: 10.1038/gim.2018.51
136. Yang Y, Muzny DM, Xia F, Niu Z, Person R, Ding Y, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. (2014) 312:1870–9. doi: 10.1001/jama.2014.14601
137. Baldridge D, Heeley J, Vineyard M, Manwaring L, Toler TL, Fassi E, et al. The Exome Clinic and the role of medical genetics expertise in the interpretation of exome sequencing results. Genet Med. (2017) 19:1040–8. doi: 10.1038/gim.2016.224
138. Bowling KM, Thompson ML, Amaral MD, Finnila CR, Hiatt SM, Engel KL, et al. Genomic diagnosis for children with intellectual disability and/or developmental delay. Genome Med. (2017) 9:43. doi: 10.1186/s13073-017-0433-1
139. Lee H, Deignan JL, Dorrani N, Strom SP, Kantarci S, Quintero-Rivera F, et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA. (2014) 312:1880–7. doi: 10.1001/jama.2014.14604
140. Retterer K, Juusola J, Cho MT, Vitazka P, Millan F, Gibellini F, et al. Clinical application of whole-exome sequencing across clinical indications. Genet Med. (2016) 18:696–704. doi: 10.1038/gim.2015.148
141. Vissers LELM, van Nimwegen KJM, Schieving JH, Kamsteeg E-J, Kleefstra T, Yntema HG, et al. A clinical utility study of exome sequencing versus conventional genetic testing in pediatric neurology. Genet Med. (2017) 19:1055–63. doi: 10.1038/gim.2017.1
142. Vrijenhoek T, Middelburg EM, Monroe GR, van Gassen KLI, Geenen JW, Hövels AM, et al. Whole-exome sequencing in intellectual disability; cost before and after a diagnosis. Eur J Hum Genet. (2018) 26:1566–71. doi: 10.1038/s41431-018-0203-6
143. Butler MG, Rafi SK, Hossain W, Stephan DA, Manzardo AM. Whole exome sequencing in females with autism implicates novel and candidate genes. Int J Mol Sci. (2015) 16:1312–35. doi: 10.3390/ijms16011312
144. Codina-Solà M, Rodríguez-Santiago B, Homs A, Santoyo J, Rigau M, Aznar-Laín G, et al. Integrated analysis of whole-exome sequencing and transcriptome profiling in males with autism spectrum disorders. Molec Autism. (2015) 6:21. doi: 10.1186/s13229-015-0017-0
145. Rossi M, El-Khechen D, Black MH, Farwell Hagman KD, Tang S, Powis Z. Outcomes of diagnostic exome sequencing in patients with diagnosed or suspected autism spectrum disorders. Pediatr Neurol. (2017) 70:34–43. doi: 10.1016/j.pediatrneurol.2017.01.033
146. Tammimies K, Marshall CR, Walker S, Kaur G, Thiruvahindrapuram B, Lionel AC, et al. Molecular diagnostic yield of chromosomal microarray analysis and whole-exome sequencing in children with autism spectrum disorder. JAMA. (2015) 314:895–903. doi: 10.1001/jama.2015.10078
147. Clark MM, Stark Z, Farnaes L, Tan TY, White SM, Dimmock D, et al. Meta-analysis of the diagnostic and clinical utility of genome and exome sequencing and chromosomal microarray in children with suspected genetic diseases. NPJ Gen Med. (2018) 3:16. doi: 10.1038/s41525-018-0053-8
148. Al-Nabhani M, Al-Rashdi S, Al-Murshedi F, Al-Kindi A, Al-Thihli K, Al-Saegh A, et al. Reanalysis of exome sequencing data of intellectual disability samples: yields and benefits. Clin Genet. (2018) 94:495–501. doi: 10.1111/cge.13438
149. Basel-Salmon L, Orenstein N, Markus-Bustani K, Ruhrman-Shahar N, Kilim Y, Magal N, et al. Improved diagnostics by exome sequencing following raw datareevaluation by clinical geneticists involved in the medical care of the individualstested. Genet Med. (2019) 21:1443–51. doi: 10.1038/s41436-018-0343-7
150. Green RC, Goddard KAB, Jarvik GP, Amendola LM, Appelbaum PS, Berg JS, et al. Clinical sequencing exploratory research consortium: accelerating evidence-based practice of genomic medicine. Am J Hum Genet. (2016) 98:1051–66. doi: 10.1016/j.ajhg.2016.04.011
151. Haer-Wigman L, van der Schoot V, Feenstra I, Vulto-van Silfhout AT, Gilissen C, Brunner HG, et al. 1 in 38 individuals at risk of a dominant medically actionable disease. Eur J Hum Genet. (2019) 27:325–30. doi: 10.1038/s41431-018-0284-2
152. Sun Y, Ruivenkamp CA, Hoffer MJ, Vrijenhoek T, Kriek M, van Asperen CJ, et al. Next-generation diagnostics: gene panel, exome, or whole genome? Hum Mutat. (2015) 36:648–55. doi: 10.1002/humu.22783
153. Brett M, McPherson J, Zang ZJ, Lai A, Tan ES, Ng I, et al. Massively parallel sequencing of patients with intellectual disability, congenital anomalies and/or autism spectrum disorders with a targeted gene panel. PLoS ONE. (2014) 9:e93409. doi: 10.1371/journal.pone.0093409
154. Saudi Mendeliome Group. Comprehensive gene panels provide advantages over clinical exome sequencing for Mendelian diseases [published correction appears in Genome Biol. 2015;16:226. Genome Biol. (2015) 16:134. doi: 10.1186/s13059-015-0693-2
155. LaDuca H, Farwell KD, Vuong H, Lu HM, Mu W, Shahmirzadi L, et al. Exome sequencing covers >98% of mutations identified on targeted next generation sequencing panels. PloS ONE. (2017) 12:e0170843. doi: 10.1371/journal.pone.0170843
156. Hoang N, Buchanan JA, Scherer SW. Heterogeneity in clinical sequencing tests marketed for autism spectrum disorders. NPJ Genom Med. (2018) 3:27. doi: 10.1038/s41525-018-0066-3
157. Dillon OJ Lunke S Stark Z Yeung A Thorne N Melbourne Genomics Health Alliance. Exome sequencing has higher diagnostic yield compared to simulated disease-specific panels in children with suspected monogenic disorders. Eur J Hum Genet. (2018) 26:644–51. doi: 10.1038/s41431-018-0099-1
158. Chong JX, Buckingham KJ, Jhangiani SN, Boehm C, Sobreira N, Smith JD, et al. The genetic basis of mendelian phenotypes: discoveries, challenges, and opportunities. Am J Hum Genet. (2015) 97:199–215. doi: 10.1016/j.ajhg.2015.06.009
159. Check W. In next-gen sequencing, panel versus exome. CAP Today (2016, March 7). Available online at: http://www.captodayonline.com/next-gen-sequencing-panel-versus-exome/ (accessed March 3, 2020).
160. Soden SE, Saunders CJ, Willig LK, Farrow EG, Smith LD, Petrikin JE, et al. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci Transl Med. (2014) 6:265ra168. doi: 10.1126/scitranslmed.3010076
161. Valencia CA, Husami A, Holle J, Johnson JA, Qian Y, Mathur A, et al. Clinical impact and cost-effectiveness of whole exome sequencing as a diagnostic tool: a pediatric center's experience. Front Pediatr. (2015) 3:67. doi: 10.3389/fped.2015.00067
162. Monroe GR, Frederix GW, Savelberg SM, de Vries TI, Duran KJ, et al. Effectiveness of whole-exome sequencing and costs of the traditional diagnostic trajectory in children with intellectual disability. Genet Med. (2016) 18:949–56. doi: 10.1038/gim.2015.200
163. Nolan D, Carlson M. Whole exome sequencing in pediatric neurology patients: clinical implications and estimated cost analysis. J Child Neurol. (2016) 31:887–94. doi: 10.1177/0883073815627880
164. Jang W, Kim Y, Han E, Park J, Chae H, Kwon A, et al. Chromosomal microarray analysis as a first-tier clinical diagnostic test in patients with developmental delay/intellectual disability, autism spectrum disorders, and multiple congenital anomalies: a prospective multicenter study in Korea. Ann Lab Med. (2019) 39:299–310. doi: 10.3343/alm.2019.39.3.299
165. Vears DF, Niemiec E, Howard HC, Borry P. Analysis of VUS reporting, variant reinterpretation and recontact policies in clinical genomic sequencing consent forms. Eur J Hum Genet. (2018) 26:1743–51. doi: 10.1038/s41431-018-0239-7
166. Trujillano D, Bertoli-Avella AM, Kumar Kandaswamy K, Weiss ME, Köster J, Marais A, et al. Clinical exome sequencing: results from 2819 samples reflecting 1000 families. EJHG. (2017) 25:176–82. doi: 10.1038/ejhg.2016.146
167. Shashi V, McConkie-Rosell A, Schoch K, Katsuri V, Rehder C, Jiang YH, et al. Practical considerations in the clinical application of whole-exome sequencing. Clin Genet. (2016) 89:173–81. doi: 10.1111/cge.12569
168. Han PK, Klein WM, Arora NK. Varieties of uncertainty in health care: a conceptual taxonomy. Med Decis Making. (2011) 31:828–38. doi: 10.1177/0272989X10393976
169. Reiff M, Ross K, Mulchandani S, Propert KJ, Pyeritz RE, Spinner NB, et al. Physicians' perspectives on the uncertainties and implications of chromosomal microarray testing of children and families. Clin Genet. (2013) 83:23–30. doi: 10.1111/cge.12004
170. Wynn J, Lewis K, Amendola LM, Bernhardt BA, Biswas S, Joshi M, et al. Clinical providers' experiences with returning results from genomic sequencing: an interview study. BMC Med Genomics. (2018) 11:45. doi: 10.1186/s12920-018-0360-z
171. Turbitt E, Halliday J, Amor D, Metcalfe S. Preferences for results from genomic microarrays: comparing parents and health care providers. Clin Genet. (2015) 87:21–9. doi: 10.1111/cge.12398
172. Menke C. Non-Genetics Pediatric Providers' Understanding and Interpretation of a VUS Result [Master's Thesis]. [Abstract]. Cincinnati, OH: University of Cincinnati (2019).
173. Jez S, Martin M, South S, Vanzo R, Rothwell E. Variants of unknown significance on chromosomal microarray analysis: parental perspectives. J Communit Genet. (2015) 6:343–9. doi: 10.1007/s12687-015-0218-4
174. Kiedrowski LA, Owens KM, Yashar BM, Schuette JL. Parents' perspectives on variants of uncertain significance from chromosome microarray analysis. J Genet Couns. (2016) 25:101–11. doi: 10.1007/s10897-015-9847-3
175. Daack-Hirsch S, Driessnack M, Hanish A, Johnson VA, Shah LL, Simon CM, et al. 'Information is information': a public perspective on incidental findings in clinical and research genome-based testing. Clin Genet. (2013) 84:11–8. doi: 10.1111/cge.12167
176. Li X, Nusbaum R, Smith-Hicks C, Jamal L, Dixon S, Mahida S. Caregivers' perception of and experience with variants of uncertain significance from whole exome sequencing for children with undiagnosed conditions. J Genet Couns. (2019) 28:304–12. doi: 10.1002/jgc4.1093
177. Wilkins EJ, Archibald AD, Sahhar MA, White SM. “It wasn't a disaster or anything”: parents' experiences of their child's uncertain chromosomal microarray result. Am J Med Genet A. (2016) 170:2895–904. doi: 10.1002/ajmg.a.37838
178. McConkie-Rosell A, Hooper SR, Pena LDM, Schoch K, Spillmann RC, Jiang YH, et al. Psychosocial profiles of parents of children with undiagnosed diseases: managing well or just managing? J Genet Couns. (2018) 27:935–46. doi: 10.1007/s10897-017-0193-5
179. Wain KE, Azzariti DR, Goldstein JL, Johnson AK, Krautscheid P, Lepore B, et al. Variant interpretation is a component of clinical practice among genetic counselors in multiple specialties. Genet Med. (2020) 22:785–92. doi: 10.1038/s41436-019-0705-9
180. Gallagher A, Hallahan B. Fragile X-associated disorders: a clinical overview. J Neurol. (2012) 259:401–13. doi: 10.1007/s00415-011-6161-3
181. Sherman S, Pletcher BA, Driscoll DA. Fragile X syndrome: diagnostic and carrier testing. Genet Med. (2005) 7:584–7. doi: 10.1097/01.GIM.0000182468.22666.dd
182. Fu YH, Kuhl DP, Pizzuti A, Pieretti M, Sutcliffe JS, Richards S, et al. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell. (1991) 67:1047–58. doi: 10.1016/0092-8674(91)90283-5
183. Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. (1991) 65:905–14. doi: 10.1016/0092-8674(91)90397-H
184. Alanay Y, Unal F, Turanli G, Alikaşifoglu M, Alehan D, Akyol U, et al. A multidisciplinary approach to the management of individuals with fragile X syndrome. J Intellect Disabil Res. (2007) 51:151–61. doi: 10.1111/j.1365-2788.2006.00942.x
185. Musumeci SA, Hagerman RJ, Ferri R, Bosco P, Dalla Bernardina B, Tassinari CA, et al. Epilepsy and EEG findings in males with fragile X syndrome. Epilepsia. (1999) 40:1092–9. doi: 10.1111/j.1528-1157.1999.tb00824.x
186. Sansone SM, Schneider A, Bickel E, Berry-Kravis E, Prescott C, Hessl D. Improving IQ measurement in intellectual disabilities using true deviation from population norms. J Neurodev Disord. (2014) 6:16. doi: 10.1186/1866-1955-6-16
187. Nolin SL, Glicksman A, Tortora N, Allen E, Macpherson J, Mila M, et al. Expansions and contractions of the FMR1 CGG repeat in 5,508 transmissions of normal, intermediate, and premutation alleles. Am J Med Genet. (2019) 179:1148–56. doi: 10.1002/ajmg.a.61165
188. Latham GJ, Coppinger J, Hadd AG, Nolin SL. The role of AGG interruptions in fragile X repeat expansions: a twenty-year perspective. Front Genet. (2014) 5:244. doi: 10.3389/fgene.2014.00244
189. Nolin SL, Sah S, Glicksman A, Sherman SL, Allen E, Berry-Kravis E. Fragile X AGG analysis provides new risk predictions for 45–69 repeat alleles. Am J Med Genet. (2013) 161A:771–8. doi: 10.1002/ajmg.a.35833
190. Hagerman RJ, Hagerman P. Fragile X-associated tremor/ataxia syndrome - features, mechanisms and management. Nat Rev Neurol. (2016) 12:403–12. doi: 10.1038/nrneurol.2016.82
191. Hunter J, Rivero-Arias O, Angelov A, Kim E, Fotheringham I, Leal J. Epidemiology of fragile X syndrome: a systematic review and meta-analysis. Am J Med Genet A. (2014) 164A:1648–58. doi: 10.1002/ajmg.a.36511
192. Peprah E. Fragile X syndrome: the FMR1 CGG repeat distribution among world populations. Ann Hum Genet. (2012) 76:178–91. doi: 10.1111/j.1469-1809.2011.00694.x
193. Patsalis PC, Sismani C, Hettinger JA, Boumba I, Georgiou I, Stylianidou G, et al. Molecular screening of fragile X (FRAXA) and FRAXE mental retardation syndromes in the Hellenic population of Greece and Cyprus: incidence, genetic variation, and stability. Am J Med Genet. (1999) 84:184–90. doi: 10.1002/(sici)1096-8628(19990528)84:3<184::aid-ajmg2>3.0.co;2-b
194. Youings SA, Murray A, Dennis N, Ennis S, Lewis C, McKechnie N, et al. FRAXA and FRAXE: the results of a five year survey. J Med Genet. (2000) 37:415–21. doi: 10.1136/jmg.37.6.415
195. Crawford DC, Meadows KL, Newman JL, Taft LF, Scott E, Leslie M, et al. Prevalence of the fragile X syndrome in African-Americans. Am J Med Genet. (2002) 110:226–33. doi: 10.1002/ajmg.10427
196. de Vries BB, van den Ouweland AM, Mohkamsing S, Duivenvoorden HJ, Mol E, Gelsema K, et al. Screening and diagnosis for the fragile X syndrome among the mentally retarded: an epidemiological and psychological survey. Collaborative Fragile X Study Group. Am J Hum Genet. (1997) 61:660–7. doi: 10.1086/515496
197. Shen Y, Dies KA, Holm IA, Bridgemohan C, Sobeih MM, Caronna EB, et al. Clinical genetic testing for patients with autism spectrum disorders. Pediatrics. (2010) 125:727–35. doi: 10.1542/peds.2009-1684
198. Roesser J. Diagnostic yield of genetic testing in children diagnosed with autism spectrum disorders at a regional referral center. Clin Pediatr (Phila). (2011) 50:834–43. doi: 10.1177/0009922811406261
199. Tassone F, Choudhary NS, Tassone F, Durbin-Johnson B, Hansen R, Hertz-Picciotto I, et al. Identification of expanded alleles of the FMR1 gene in the Childhood Autism Risks from Genes and Environment (CHARGE) study. J Autism Dev Disord. (2013) 43:530–9. doi: 10.1007/s10803-012-1580-2
200. Mordaunt D, Gabbett M, Waugh M, O'Brien K, Heussler H. Uptake and diagnostic yield of chromosomal microarray in an Australian Child Development Clinic. Children. (2014) 1:21–30. doi: 10.3390/children1010021
201. Eriksson MA, Liedén A, Westerlund J, Bremer A, Wincent J, Sahlin E, et al. Rare copy number variants are common in young children with autism spectrum disorder. Acta Paediatr. (2015) 104:610–8. doi: 10.1111/apa.12969
202. Mazzocco MMM, Kates WR, Baumgardner TL, Freund LS, Reiss AL. Autistic behaviors among girls with fragile X syndrome. J Autism Dev Disord. (1997) 27:415–35. doi: 10.1023/A:1025857422026
203. Clifford S, Dissanayake C, Bui QM, Huggins R, Taylor AK, Loesch DZ. Autism spectrum phenotype in males and females with fragile X full mutation and premutation. J Autism Dev Disord. (2007) 37:738–47. doi: 10.1007/s10803-006-0205-z
204. Chaste P, Betabcur C, Gérard-Blanluet M, Bargiacchi A, Kuzbari S, Drunat S, et al. High-functioning autism spectrum disorder and fragile X syndrome: report of two affected sisters. Molec Autism. (2012) 3:5. doi: 10.1186/2040-2392-3-5
205. Strom CM, Crossley B, Redman JB, Buller A, Quan F, Peng M, et al. Molecular testing for Fragile X Syndrome: lessons learned from 119,232 tests performed in a clinical laboratory. Genet Med. (2007) 9:46–51. doi: 10.1097/GIM.0b013e31802d833c
206. Mullegama SV, Klein SD, Nguyen DC, Kim A, Signer R, Fox M, et al. Is it time to retire fragile X testing as a first-tier test for developmental delay, intellectual disability, and autism spectrum disorder? Genet Med. (2017) 19:1380. doi: 10.1038/gim.2017.146
207. Hartley T, Potter R, Badalato L, Smith AC, Jarinova O, Boycott KM. Fragile X testing as a second-tier test. Genet Med. (2017) 19:147. doi: 10.1038/gim.2017.147
208. Smith K, Chandler K, Hindley D, Ramsden SC. Fragile X syndrome testing in the North West. Arch Dis Child. (2013) 98:239. doi: 10.1136/archdischild-2012-302934
209. Borch LA, Parboosingh J, Thomas MA, Veale P. Re-evaluating the first-tier status of fragile X testing in neurodevelopmental disorders. Genet Med. (2020) 22:1036–9. doi: 10.1038/s41436-020-0773-x
210. Lubala TK, Lumaka A, Kanteng G, Mutesa L, Mukuku O, Wembonyama S, et al. Fragile X checklists: a meta-analysis and development of a simplified universal clinical checklist. Mol Genet Genom Med. (2018) 6:526–32. doi: 10.1002/mgg3.398
211. Vissers L, Gilissen C, Veltman J. Genetic studies in intellectual disability and related disorders. Nat Rev Genet. (2016) 17:9–18. doi: 10.1038/nrg3999
212. Gilissen C, Hehir-Kwa JY, Thung DT, van de Vorst M, van Bon BW, Willemsen MH, et al. Genome sequencing identifies major causes of severe intellectual disability. Nature. (2014) 511:344–7. doi: 10.1038/nature13394
213. Michelson DJ, Shevell MI, Sherr EH, Moeschler JB, Gropman AL, Ashwal S. Evidence Report: genetic and metabolic testing on children with global developmental delay. Report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. (2011) 77:1629–35. doi: 10.1212/WNL.0b013e3182345896
214. Barton KS, Tabor HK, Starks H, Garrison NA, Laurino M, Burke W. Pathways from autism spectrum disorder diagnosis to genetic testing. Genet Med. (2018) 20:737–44. doi: 10.1038/gim.2017.166
215. Filipek PA, Accardo PJ, Ashwal S, Baranek GT, Cook EH Jr, et al. Practice parameter: screening and diagnosis of autism: report of the Quality Standards Subcommittee of the American Academy of Neurology and the Child Neurology Society. Neurology. (2000) 55:468–79. doi: 10.1212/wnl.55.4.468
216. Amiet C, Couchon E, Carr K, Carayol J, Cohen D. Are there cultural differences in parental interest in early diagnosis and genetic risk assessment for autism spectrum disorder? Front Pediatr. (2014) 2:32. doi: 10.3389/fped.2014.00032
217. Chen LS, Xu L, Huang TY, Dhar SU. Autism genetic testing: a qualitative study of awareness, attitudes, and experiences among parents of children with autism spectrum disorders. Genet Med. (2013) 15:274–81. doi: 10.1038/gim.2012.145
218. Kiely B, Vettam S, Adesman A. Utilization of genetic testing among children with developmental disabilities in the United States. App Clin Genet. (2016) 9:93–100. doi: 10.2147/TACG.S103975
219. Vande Wydeven K, Kwan A, Hardan AY, Bernstein JA. Underutilization of genetics services for autism: the importance of parental awareness and provider recommendation. J Genet Couns. (2012) 21:803–13. doi: 10.1007/s10897-012-9494-x
220. Cuccaro ML, Czape K, Alessandri M, Lee J, Deppen AR, Bendik E, et al. Genetic testing and corresponding services among individuals with autism spectrum disorder (ASD). Am J Med Genet A. (2014) 164A:2592–600. doi: 10.1002/ajmg.a.36698
221. Moreno-De-Luca D, Kavanaugh BC, Best CR, Sheinkopf SJ, Phornphutkul C, Morrow EM. Clinical genetic testing in autism spectrum disorder in a large community-based population sample. JAMA Psychiatry. (2020) 77:979–81. doi: 10.1001/jamapsychiatry.2020.0950
222. Peabody J, DeMaria L, Tamandong-LaChica D, Florentino J, Acelajado MC, Burgon T. Low rates of genetic testing in children with developmental delays, intellectual disability, and autism spectrum disorders. Glob Pediatr Health. (2015) 2:2333794X15623717. doi: 10.1177/2333794X15623717
223. Codina-Solà M, Pérez-Jurado LA, Cuscó I, Serra-Juhé C. Provision of genetic services for autism and its impact on Spanish families. J Autism Dev Disord. (2017) 47:2947–56. doi: 10.1007/s10803-017-3203-4
224. Korf BR, Ledbetter D, Murray MF. Report of the Banbury Summit Meeting on the evolving role of the medical geneticist, February 12–14, 2006. Genet Med. (2008) 10:502–7. doi: 10.1097/GIM.0b013e31817701fe
225. Ormond KE, Hallquist MLG, Buchanan AH, Dondanville D, Cho MK, Smith M, et al. Developing a conceptual, reproducible, rubric-based approach to consent and result disclosure for genetic testing by clinicians with minimal genetics background. Genet Med. (2019) 21:727–35. doi: 10.1038/s41436-018-0093-6
226. Vassy JL, Korf BR, Green RC. How to know when physicians are ready for genomic medicine. Sci Transl Med. (2015) 7:287fs19. doi: 10.1126/scitranslmed.aaa2401
227. Bowdin S, Gilbert A, Bedoukian E, Carew C, Adam MP, Belmont J, et al. Recommendations for the integration of genomics into clinical practice. Genet Med. (2016) 18:1075–84. doi: 10.1038/gim.2016.17
228. Cohen J, Hoon A, Floet AMW. Providing family guidance in rapidly shifting sand: informed consent for genetic testing. Dev Med Child Neurol. (2013) 55:766–8. doi: 10.1111/dmcn.12102
229. Finucane B, Myers SM. Genetic counseling for autism spectrum disorder in an evolving theoretical landscape. Curr Genet Med Rep. (2016) 4:147–53. doi: 10.1007/s40142-016-0099-9
230. Reiff M, Mueller R, Mulchandani S, Spinner NB, Pyeritz RE, Bernhardt BA. A qualitative study of healthcare providers' perspectives on the implications of genome-wide testing in pediatric clinical practice. J Genet Couns. (2014) 23:474–88. doi: 10.1007/s10897-013-9653-8
231. Wolfe K, Stueber K, McQuillin A, Jichi F, Patch C, Flinter F, et al. Genetic testing in intellectual disability psychiatry: opinions and practices of UK child and intellectual disability psychiatrists. J Appl Res Intellect Disabil. (2017) 31:273–84. doi: 10.1111/jar.12391
232. Sanderson SC, Hill M, Patch C, Searle B, Lewis C, Chitty LS. Delivering genome sequencing in clinical practice: an interview study with healthcare professionals involved in the 100 000 Genomes Project. BMJ Open. (2019) 9:e029699. doi: 10.1136/bmjopen-2019-029699
233. Biesecker LG, Biesecker BB. An approach to pediatric exome and genome sequencing. Curr Opin Pediatr. (2014) 26:639–45. doi: 10.1097/MOP.0000000000000150
234. Aetna(2019). Medical Clinical Policy Bulletins – Number:0140 Genetic Testing. Available online at: http://www.aetna.com/cpb/medical/data/100_199/0140.html (accessed January 12, 2020).
235. Sánchez Fernández I, Loddenkemper T, Gaínza-Lein M, Sheidley BR, Poduri A. Diagnostic yield of genetic tests in epilepsy: A meta-analysis and cost-effectiveness study. Neurology. (2019) 92:e418–28. doi: 10.1212/WNL.0000000000006850
236. Giangreco CA, Steele MW, Aston CE, Cummins JH, Wenger SL. A simplified six item checklist for screening for fragile X syndrome in the pediatric population. J Pediatr. (1996) 129:611–4. doi: 10.1016/S0022-3476(96)70130-0
237. Christofolini DM, Abbud EM, Lipay MV, Costa SS, Vianna-Morgante AM, Bellucco FT, et al. Evaluation of clinical checklists for fragile X syndrome screening in Brazilian intellectually disabled males: proposal for a new screening tool. J Intellect Disabil. (2009) 13:239–48. doi: 10.1177/1744629509348429
238. de Vries BB, Mohkamsing S, van den Ouweland AM, Mol E, Gelsema K, van Rijn M, et al. Screening for the fragile X syndrome among the mentally retarded: a clinical study. The Collaborative Fragile X Study Group. J Med Genet. (1999) 36:467–70.
239. Baars MJ, Henneman L, Ten Kate LP. Deficiency of knowledge of genetics and genetic tests among general practitioners, gynecologists, and pediatricians: a global problem. Genet Med. (2005) 7:605–10. doi: 10.1097/01.gim.0000182895.28432.c7
240. Burke S, Stone A, Bedward J, Thomas H, Farndon P. A “neglected part of the curriculum” or “of limited use?” Views on genetics training by nongenetics medical trainees and implications for delivery. Genet Med. (2006) 8:109–15. doi: 10.1097/01.gim.0000200159.19920.b5
241. Klitzman R, Chung W, Marder K, Shanmugham A, Chin LJ, Stark M, et al. Attitudes and practices among internists concerning genetic testing. J Genet Couns. (2013) 22:90–100. doi: 10.1007/s10897-012-9504-z
242. Owusu Obeng A, Fei K, Levy KD, Elsey AR, Pollin TI, Ramirez AH, et al. Physician-reported benefits and barriers to clinical implementation of genomic medicine: a multi-site IGNITE-network survey. J Pers Med. (2018) 8:24. doi: 10.3390/jpm8030024
243. Selkirk CG, Weissman SM, Anderson A, Hulick PJ. Physicians' preparedness for integration of genomic and pharmacogenetic testing into practice within a major healthcare system. Genet Test Mol Biomarkers. (2013) 17:219–25. doi: 10.1089/gtmb.2012.0165
244. Reed EK, Johansen Taber KA, Ingram Nissen T, Schott S, Dowling LO, O'Leary JC, et al. What works in genomics education: outcomes of an evidenced-based instructional model for community-based physicians. Genet Med. (2016) 18:737–45. doi: 10.1038/gim.2015.144
245. Scacheri C, Redman JB, Pike-Buchanan L, Steenblock K. Molecular testing: improving patient care through partnering with laboratory genetic counselors. Genet Med. (2008) 10:337–42. doi: 10.1097/GIM.0b013e31817283a5
246. Abacan M, Alsubaie L, Barlow-Stewart K, Caanen B, Cordier C, Courtney E, et al. The global state of the genetic counseling profession. Eur J Hum Genet. (2019) 27:183–97. doi: 10.1038/s41431-018-0252-x
247. Ormond KE, Laurino MY, Barlow-Stewart K, Wessels TM, Macaulay S, Austin J, et al. Genetic counseling globally: Where are we now? Am J Med Genet C Semin Med Genet. (2018) 178:98–107. doi: 10.1002/ajmg.c.31607
248. Patch C, Middleton A. Genetic counselling in the era of genomic medicine. Br Med Bull. (2018) 126:27–36. doi: 10.1093/bmb/ldy008
249. Accreditation Council for Genetic Counseling (ACGC). Practice Based Competencies. (2019). Available online at: https://www.gceducation.org/wp-content/uploads/2019/06/ACGC-Core-Competencies-Brochure_15_Web_REV-6-2019.pdf (accessed November 15, 2019).
250. Ormond KE. From genetic counseling to “genomic counseling”. Mol Genet Genom Med. (2013) 1:189–93. doi: 10.1002/mgg3.45
251. Stein Q, Loman R, Zuck T. Genetic counseling in pediatrics. Pediatr Rev. (2018) 39:323–31. doi: 10.1542/pir.2017-0194
252. Morad Y, Sutherland J, DaSilva L, Ulster A, Shik J, Gallie B, et al. Ocular Genetics Program: multidisciplinary care of patients with ocular genetic eye disease. Can J Ophthalmol. (2007) 42:734–8. doi: 10.3129/i07-144
253. Bernhardt B. Genetic counselors and the future of clinical genomics. Genome Med. (2014) 6:49. doi: 10.1186/gm565
254. Doyle DL, Awwad RI, Austin JC, Baty BJ, Bergner AL, Brewster SJ, et al. 2013 review and update of the genetic counseling practice based competencies by a task force of the accreditation council for genetic counseling. J Genet Couns. (2016) 25:868–79. doi: 10.1007/s10897-016-9984-3
255. Middleton A, Marks P, Bruce A, Protheroe-Davies LK, King C, Claber O, et al. The role of genetic counsellors in genomic healthcare in the United Kingdom: a statement by the Association of Genetic Nurses and Counsellors. Eur J Hum Genet. (2017) 25:659–61. doi: 10.1038/ejhg.2017.28
256. National Society of Genetic Counselors (NSGC). NSGC Code of Ethics. (2017). Available online at: https://www.nsgc.org/p/cm/ld/fid=12#intro (accessed November 15, 2019).
257. Wilkes MS, Day FC, Fancher TL, McDermott H, Lehman E, Bell RA, et al. Increasing confidence and changing behaviors in primary care providers engaged in genetic counselling. BMC Med Educ. (2017) 17:163. doi: 10.1186/s12909-017-0982-4
258. Stoll K, Kubendran S, Cohen SA. The past, present and future of service delivery in genetic counseling: keeping up in the era of precision medicine. Am J Med Genet C Semin Med Genet. (2018) 178:24–37. doi: 10.1002/ajmg.c.31602
259. Conta J, Sternen D, Stasi S, Candadai S, Wellner M, Dickerson J. An integrated approach to genetic test stewardship: the role of laboratory genetic counselors in test preauthorization and ordering. Am J Clin Pathol. (2019) 152:S148. doi: 10.1093/ajcp/aqz130.015
260. Pletcher BA, Toriello HV, Noblin SJ, Seaver LH, Driscoll DA, Bennett RL, et al. Indications for genetic referral: a guide for healthcare providers. Genet Med. (2007) 9:385–9. doi: 10.1097/GIM.0b013e318064e70c
261. American Board of Medical Genetics and Genomics (ABMGG). ABMGG: Training Options. (2017). Available online at: http://www.abmgg.org/pages/training_options.shtml (accessed November 15, 2019).
262. Hennekam RC, Biesecker LG. Next-generation sequencing demands next-generation phenotyping. Hum Mutat. (2012) 33:884–6. doi: 10.1002/humu.22048
263. Mak CC, Leung GK, Mok GT, Yeung KS, Yang W, Fung CW, et al. Exome sequencing for paediatric-onset diseases: impact of the extensive involvement of medical geneticists in the diagnostic odyssey. NPJ Genom Med. (2018) 3:19. doi: 10.1038/s41525-018-0056-5
264. Margarit SB, Alvarado M, Alvarez K, Lay-Son G. Medical genetics and genetic counseling in Chile. J Genet Couns. (2013) 22:869–74. doi: 10.1007/s10897-013-9607-1
265. Bucio D, Ormond KE, Hernandez D, Bustamante CD, Lopez Pineda A. A genetic counseling needs assessment of Mexico. Mol Genet Genomics. (2019) 7:e668. doi: 10.1002/mgg3.668
266. Horovitz DD, de Faria Ferraz VE, Dain S, Marques-de-Faria AP. Genetic services and testing in Brazil. J Community Genet. (2013) 4:355–75. doi: 10.1007/s12687-012-0096-y
267. Rodas-Pérez C, Clarke A, Powell J, Thorogood M. Challenges for providing genetic counselling in Colombian genetic clinics: the viewpoint of the physicians providing genetic consultations. J Community Genet. (2015) 6:301–11. doi: 10.1007/s12687-015-0237-1
268. Mullen SA, Carvill GL, Bellows S, Bayly MA, Trucks H, Lal D, et al. Copy number variants are frequent in genetic generalized epilepsy with intellectual disability. Neurology. (2013) 81:1507–14. doi: 10.1212/WNL.0b013e3182a95829
269. Møller RS, Larsen LH, Johannesen KM, Talvik I, Talvik T, Vaher U, et al. Gene panel testing in epileptic encephalopathies and familial epilepsies. Mol Syndromol. (2016) 7:210–9. doi: 10.1159/000448369
270. Dang LT, Silverstein FS. Drug treatment of seizures and epilepsy in newborns and children. Pediatr Clin North Am. (2017) 64:1291–308. doi: 10.1016/j.pcl.2017.08.007
271. Poduri A. When should genetic testing be performed in epilepsy patients? Epilepsy Curr. (2017) 17:16–22. doi: 10.5698/1535-7511-17.1.16
272. Ream MA, Patel AD. Obtaining genetic testing in pediatric epilepsy. Epilepsia. (2015) 56:1505–14. doi: 10.1111/epi.13122
273. Thodeson DM, Park JY. Genomic testing in pediatric epilepsy. Cold Spring Harb Mol Case Stud. (2019) 5:a004135. doi: 10.1101/mcs.a004135
274. Snoeijen-Schouwenaars FM, van Ool JS, Verhoeven JS, van Mierlo P, Braakman HMH, Smeets EE, et al. Diagnostic exome sequencing in 100 consecutive patients with both epilepsy and intellectual disability. Epilepsia. (2019) 60:155–64. doi: 10.1111/epi.14618
275. Mefford HC. Clinical genetic testing in epilepsy. Epilepsy Curr. (2015) 15:197–201. doi: 10.5698/1535-7511-15.4.197
276. Oskoui M, Gazzellone M, Thiruvahindrapuram B, Zarrei M, Anderson J, Wei J, et al. Clinically relevant copy number variations detected in cerebral palsy. Nat Commun. (2015) 6:7949. doi: 10.1038/ncomms8949
277. Segel R, Ben-Pazi H, Zeligson S, Fatal-Valevski A, Aran A, Gross-Tsur V, et al. Copy number variations in cryptogenic cerebral palsy. Neurology. (2015) 84:1660–8. doi: 10.1212/WNL.0000000000001494
278. McMichael G, Girirajan S, Moreno-De-Luca A, Gecz J, Shard C, Nguyen LS, et al. Rare copy number variation in cerebral palsy. Eur J Hum Genet. (2014) 22:40–5. doi: 10.1038/ejhg.2013.93
279. Zarrei M, Fehlings DL, Mawjee K, Switzer L, Thiruvahindrapuram B, Walker S, et al. De novo and rare inherited copy-number variations in the hemiplegic form of cerebral palsy. Genet Med. (2018) 20:172–80. doi: 10.1038/gim.2017.83
280. McMichael G, Bainbridge MN, Haan E, Corbett M, Gardner A, Thompson S, et al. Whole-exome sequencing points to considerable genetic heterogeneity of cerebral palsy. Mol Psychiatry. (2015) 20:176–82. doi: 10.1038/mp.2014.189
281. Corbett MA, van Eyk CL, Webber DL, Bent SJ, Newman M, Harper K, et al. Pathogenic copy number variants that affect gene expression contribute to genomic burden in cerebral palsy. NPJ Genom Med. (2018) 3:33. doi: 10.1038/s41525-018-0073-4
282. Matthews AM, Blydt-Hansen I, Al-Jabri B, Andersen J, Tarailo-Graovac M, Price M, et al. Atypical cerebral palsy: genomics analysis enables precision medicine. Genet Med. (2019) 21:1621–8. doi: 10.1038/s41436-018-0376-y
283. Moreno-De-Luca A, Millan F, Pesacreta DR, Oetjens MT, Teigen C, Wain KE, et al. Molecular diagnostic yield of exome sequencing in patients with cerebral palsy. JAMA. (2021) 325:467–75. doi: 10.1001/jama.2020.26148
284. Jin SC, Lewis SA, Bakhtiari S, Zeng X, Sierant MC, Shetty S, et al. Mutations disrupting neuritogenesis genes confer risk for cerebral palsy. Nat Genet. (2020) 52:1046–56. doi: 10.1038/s41588-020-0695-1
285. Ashwal S, Russman BS, Blasco PA, Miller G, Sandler A, Shevell M, et al. Practice parameter: diagnostic assessment of the child with cerebral palsy: report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. (2004) 62:851–63. doi: 10.1212/01.WNL.0000117981.35364.1B
286. Barnett CP, van Bon BW. Monogenic and chromosomal causes of isolated speech and language impairment. J Med Genet. (2015) 52:719–29. doi: 10.1136/jmedgenet-2015-103161
287. Chen XS, Reader RH, Hoischen A, Veltman JA, Simpson NH, Francks C, et al. Next-generation DNA sequencing identifies novel gene variants and pathways involved in specific language impairment. Sci Rep. (2017) 7:46105. doi: 10.1038/srep46105
288. Eising E, Carrion-Castillo A, Vino A, Strand EA, Jakielski KJ, Scerri TS, et al. A set of regulatory genes co-expressed in embryonic human brain is implicated in disrupted speech development. Mol Psychiatr. (2019) 24:1065–78. doi: 10.1038/s41380-018-0020-x
289. Laffin JJS, Raca G, Jackson CA, Strand EA, Jakielski KJ, Shriberg LD. Novel candidate genes and regions for childhood apraxia of speech identified by array comparative genomic hybridization. Genet Med. (2012) 14:928–36. doi: 10.1038/gim.2012.72
290. Worthey EA, Raca G, Laffin JJ, Wilk BM, Harris JM, Jakielski KJ, et al. Whole-exome sequencing supports genetic heterogeneity in childhood apraxia of speech. J Neurodev Disord. (2013) 5:29. doi: 10.1186/1866-1955-5-29
291. Willsey JA, Fernandez TV, Yu D, King RA, Dietrich A, Xing J. De novo coding variants are strongly associated with tourette disoder. Neuron. (2017) 94:486–99. doi: 10.1016/j.neuron.2017.04.024
292. Huang AY, Yu D, Davis LK, Sul JH, Tsestos F, Ramensky V, et al. Rare copy number variants in NRXN1 and CNTN6 increase risk for tourette syndrome. Neuron. (2017) 94:1101–11. doi: 10.1016/j.neuron.2017.06.010
293. Bassett AS, Scherer SW. Copy number variation in tourette syndrome. Neuron. (2017) 94:1041–3. doi: 10.1016/j.neuron.2017.06.017
294. Wang S, Mandell JD, Kumar Y, Sun N, Morris MT, Arbelaez J, et al. De novo sequence and copy number variants are strongly associated with tourette disorder and implicate cell polarity in pathogenesis. Cell Rep. (2018) 24, 344–345. doi: 10.1016/j.celrep.2018.12.024
295. Williams NM, Zaharieva I, Martin A, Langley K, Mantripragada K, Fossdal R, et al. Rare chromosomal deletions and duplications in attention-deficit hyperactivity disorder: a genome-wide analysis. Lancet. (2010) 376:1401–8. doi: 10.1016/S0140-6736(10)61109-9
296. Elia J, Gai X, Xie HM, Perin JC, Geiger E, Glessner JT, et al. Rare structural variants found in attention-deficit hyperactivity disorder are preferentially associated with neurodevelopmental genes. Mol Psychiatr. (2010) 15:637–46. doi: 10.1038/mp.2009.57
297. Lesch KP, Selch S, Renner TJ, Jacob C, Nguyen TT, Hahn T, et al. Genome-wide copy number variation analysis in attention-deficit/hyperactivity disorder: association with neuropeptide Y gene dosage in an extended pedigree. Mol Psychiatry. (2011) 16:491–503. doi: 10.1038/mp.2010.29
298. Gudmundsson OO, Walters GB, Ingason A, Johansson S, Zayats T, Athanasiu L, et al. Attention-deficit hyperactivity disorder shares copy number variant risk with schizophrenia and autism spectrum disorder. Transl Psychiatry. (2019) 9:258. doi: 10.1038/s41398-019-0599-y
299. Singh T, Kurki MI, Curtis D, Purcell SM, Crooks L, McRae J, et al. Rare loss-of-function variants in SETD1A are associated with schizophrenia and developmental disorders. Nature Neurosci. (2016) 19:571–7. doi: 10.1038/nn.4267
300. Reiss AL. Childhood developmental disorders: an academic and clinical convergence point for psychiatry, neurology, psychology and pediatrics. J Child Psychol Psychiatry. (2009) 50:87–98. doi: 10.1111/j.1469-7610.2008.02046.x
301. Thurm A, Farmer C, Salzman E, Lord C, Bishop S. State of the field: differentiating intellectual disability from autism spectrum disorder. Front Psychiatry. (2019) 10:526. doi: 10.3389/fpsyt.2019.00526
302. Zhu X, Need AC, Petrovski S, Goldstein DB. One gene, many neuropsychiatric disorders: lessons from Mendelian diseases. Nat Neurosci. (2014) 17:773–81. doi: 10.1038/nn.3713
303. Einfeld SL, Ellis LA, Emerson E. Comorbidity of intellectual disability and mental disorder in children and adolescents: a systematic review. J Intellect Dev Disabil. (2011) 36:137–43. doi: 10.1080/13668250.2011.572548
304. Franke B, Michelini G, Asherson P, Banaschewski T, Bilbow A, Buitelaar JK. Live fast, die young? A review on the developmental trajectories of ADHD across the lifespan. Eur Neuropsychopharmacol. (2018) 28:1059–88. doi: 10.1016/j.euroneuro.2018.08.001
305. Johnson MR, Shorvon SD. Heredity in epilepsy: neurodevelopment, comorbidity, and the neurological trait. Epilepsy Behav. (2011) 22:421–7. doi: 10.1016/j.yebeh.2011.07.031
306. Mazefsky CA, Oswald DP, Day TN, Eack SM, Minshew NJ, Lainhart JE. ASD, a psychiatric disorder, or both? Psychiatric diagnoses in adolescents with high-functioning ASD. J Clin Child Adolesc Psychol. (2012) 41:516–23. doi: 10.1080/15374416.2012.686102
307. Taurines R, Schmitt J, Renner T, Conner AC, Warnke A, Romanos M. Developmental comorbidity in attention-deficit/hyperactivity disorder. Atten Defic Hyperact Disord. (2010) 2:267–89. doi: 10.1007/s12402-010-0040-0
308. Owen MJ, O'Donovan MC. Schizophrenia and the neurodevelopmental continuum:evidence from genomics. World Psychiatry. (2017) 16:227–35. doi: 10.1002/wps.20440
309. de la Serna E, Baeza I, Toro J, Andrés S, Puig O, Sánchez-Guistau V, et al. Relationship between clinical and neuropsychological characteristics in child and adolescent first degree relatives of subjects with schizophrenia. Schizophr Res. (2010) 116:159–67. doi: 10.1016/j.schres.2009.09.001
310. Gur RE, Nimgaonkar VL, Almasy L, Calkins ME, Ragland JD, Pogue-Geile MF, et al. Neurocognitive endophenotypes in a multiplex multigenerational family study of schizophrenia. Am J Psychiatry. (2007) 164:813–9. doi: 10.1176/ajp.2007.164.5.813
311. Larsson H, Asherson P, Chang Z, Ljung T, Friedrichs B, Larsson JO, et al. Genetic and environmental influences on adult attention deficit hyperactivity disorder symptoms: a large Swedish population-based study of twins. Psychol Med. (2013) 43:197–207. doi: 10.1017/S0033291712001067
312. Sullivan PF, Daly MJ, O'Donovan M. Genetic architectures of psychiatric disorders: the emerging picture and its implications. Nat Rev Genet. (2012) 13:537–51. doi: 10.1038/nrg3240
313. Tollanes MC, Wilcox AJ, Lie RT, Moster D. Familial risk of cerebral palsy: population based cohort study. BMJ. (2014) 349:g4294. doi: 10.1136/bmj.g4294
314. Toulopoulou T, Goldberg TE, Mesa IR, Picchioni M, Rijsdijk F, Stahl D, et al. Impaired intellect and memory: a missing link between genetic risk and schizophrenia? Arch Gen Psychiatry. (2010) 67:905–13. doi: 10.1001/archgenpsychiatry.2010.99
315. Tankard RM, Bennett MF, Degorski P, Delatycki MB, Lockhart PJ, Bahlo M. Detecting expansions of tandem repeats in cohorts sequenced with short-read sequencing data. Am J Hum Genet. (2018) 103:858–73. doi: 10.1016/j.ajhg.2018.10.015
316. Short PJ, McRae JF, Gallone G, Sifrim A, Won H, Geschwind DH, et al. De novo mutations in regulatory elements in neurodevelopmental disorders. Nature. (2018) 555:611. doi: 10.1038/nature25983
317. Frésard L, Montgomery SB. Diagnosing rare diseases after the exome. Cold Spring Harb Mol Case Stud. (2018) 4:a003392. doi: 10.1101/mcs.a003392
318. Costain G, Walker S, Marano M, Veenma D, Snell M, Curtis M, et al. Genome sequencing as a diagnostic test in children with unexplained medical complexity. JAMA Netw Open. (2020) 3:e2018109. doi: 10.1001/jamanetworkopen.2020.18109
319. Aref-Eshghi E, Bend EG, Colaiacovo S, Caudle M, Chakrabarti R, Napier M, et al. Diagnostic utility of genome-wide DNA methylation testing in genetically unsolved individuals with suspected hereditary conditions. Am J Hum Genet. (2019) 104:685–700. doi: 10.1016/j.ajhg.2019.03.008
320. Aref-Eshghi E, Rodenhiser DI, Schenkel LC, Lin H, Skinner C, Ainsworth P, et al. Genomic DNA methylation signatures enable concurrent diagnosis and clinical genetic variant classification in neurodevelopmental syndromes. Am J Hum Genet. (2018) 102:156–74. doi: 10.1016/j.ajhg.2017.12.008
321. Barbosa M, Joshi RS, Garg P, Martin-Trujillo A, Patel N, Jadhav B, et al. Identification of rare de novo epigenetic variations in congenital disorders. Nat Commun. (2018) 9:2064. doi: 10.1038/s41467-018-04540-x
322. D'Gama AM, Walsh CA. Somatic mosaicism and neurodevelopmental disease. Nat Neurosci. (2018) 21:1504–14. doi: 10.1038/s41593-018-0257-3
323. Rehm HL, Berg JS, Brooks LD, Bustamante CD, Evans JP, Landrum MJ, et al. ClinGen–the Clinical Genome Resource. N Engl J Med. (2015) 372:2235–42. doi: 10.1056/NEJMsr1406261
324. Philippakis AA, Azzariti DR, Beltran S, Brookes AJ, Brownstein CA, Brudno M, et al. The Matchmaker Exchange: a platform for rare disease gene discovery. Hum Mutat. (2015) 36:915–21. doi: 10.1002/humu.22858
325. David KL, Best RG, Brenman LM, Bush L, Deignan JL, Flannery D, et al. Patient re-contact after revision of genomic test results: points to consider-a statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med. (2019) 21:769–71. doi: 10.1038/s41436-018-0391-z
326. Carrieri D, Howard HC, Benjamin C, Clarke AJ, Dheensa S, Doheny S, et al. Recontacting patients in clinical genetics services: recommendations of the European Society of Human Genetics. Eur J Hum Genet. (2019) 27:169–82. doi: 10.1038/s41431-018-0285-1
327. Hirschhorn K, Fleisher LD, Godmilow L, Howell RR, Lebel RR, McCabe ER, et al. Duty to re-contact [published correction appears in Genet Med 1999 Jul-Aug;1, 186]. Genet Med. (1999) 1:171–2. doi: 10.1097/00125817-199905000-00010
328. Girirajan S, Rosenfeld JA, Cooper GM, Antonacci F, Siswara P, Itsara A, et al. A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat Genet. (2010) 42:203–9. doi: 10.1038/ng.534
329. Girirajan S, Rosenfeld JA, Coe BP, Parikh S, Friedman N, Goldstein A. Phenotypic heterogeneity of genomic disorders and rare copy-number variants. N Engl J Med. (2012) 367:1321–31. doi: 10.1056/NEJMoa1200395
330. Turner TN, Coe BP, Dickel DE, Hoekzema K, Nelson BJ, Zody MC, et al. Genomic patterns of de novo mutation in simplex autism. Cell. (2017) 171:710–22. doi: 10.1016/j.cell.2017.08.047
331. Cleynen I, Engchuan W, Hestand MS, Heung T, Holleman AM, Johnston HR, et al. Genetic contributors to risk of schizophrenia in the presence of a 22q11.2 deletion. Mol Psychiatry. (2020) 3:10.1038/s41380-020-0654-3. doi: 10.1038/s41380-020-0654-3
332. Davies RW, Fiksinski AM, Breetvelt EJ, Williams NM, Hooper SR, Monfeuga T. Using common genetic variation to examine phenotypic expression and risk prediction in 22q11.2 deletion syndrome. Nat Med. (2020) 26:1912–8. doi: 10.1038/s41591-020-1103-1
333. Niemi M, Martin HC, Rice DL, Gallone G, Gordon S, Kelemen M, et al. Common genetic variants contribute to risk of rare severe neurodevelopmental disorders. Nature. (2018) 562:268–71. doi: 10.1038/s41586-018-0566-4
334. Pizzo L, Jensen M, Polyak A, Rosenfeld JA, Mannik K, Krishan A, et al. Rare variants in the genetic background modulate cognitive and developmental phenotypes in individuals carrying disease-associated variants. Genet Med. (2019) 21:816–25. doi: 10.1038/s41436-018-0266-3
335. Moreno-De-Luca A, Evans DW, Boomer KB, Hanson E, Bernier R, Goin-Kochel RP, et al. The role of parental cognitive, behavioral, and motor profiles in clinical variability in individuals with chromosome 16p11.2 deletions. JAMA Psychiatry. (2015) 72:119–26. doi: 10.1001/jamapsychiatry.2014.2147
336. Olszewski AK, Radoeva PD, Fremont W, Kates WR, Antshel KM. Is child intelligence associated with parent and sibling intelligence in individuals with developmental disorders? An investigation in youth with 22q11.2 deletion (velo-cardio-facial) syndrome. Res Dev Disabil. (2014) 35:3582–90. doi: 10.1016/j.ridd.2014.08.034
Keywords: autism, intellectual disability, global developmental delay, genetic testing, neurodevelopment, exome sequencing, chromosomal microarray, fragile x
Citation: Savatt JM and Myers SM (2021) Genetic Testing in Neurodevelopmental Disorders. Front. Pediatr. 9:526779. doi: 10.3389/fped.2021.526779
Received: 14 January 2020; Accepted: 18 January 2021;
Published: 19 February 2021.
Edited by:
Merlin G. Butler, University of Kansas Medical Center, United StatesReviewed by:
Flora Tassone, University of California, Davis, United StatesCopyright © 2021 Savatt and Myers. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juliann M. Savatt, am1zYXZhdHRAZ2Vpc2luZ2VyLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.