 Federica Buonocore
Federica Buonocore
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Pediatr. , 14 December 2020
Sec. Pediatric Endocrinology
Volume 8 - 2020 | https://doi.org/10.3389/fped.2020.619041
This article is part of the Research Topic Endocrine Diseases of Newborn: Epidemiology, Pathogenesis, Therapeutic Option and Outcome View all 14 articles
Adrenal insufficiency (AI) is a potentially life-threatening condition that can be difficult to diagnose, especially if it is not considered as a potential cause of a child's clinical presentation or unexpected deterioration. Children who present with AI in early life can have signs of glucocorticoid deficiency (hyperpigmentation, hypoglycemia, prolonged jaundice, poor weight gain), mineralocorticoid deficiency (hypotension, salt loss, collapse), adrenal androgen excess (atypical genitalia), or associated features linked to a specific underlying condition. Here, we provide an overview of causes of childhood AI, with a focus on genetic conditions that present in the first few months of life. Reaching a specific diagnosis can have lifelong implications for focusing management in an individual, and for counseling the family about inheritance and the risk of recurrence.
Adrenal insufficiency (AI) is a potentially life-threatening condition that needs urgent diagnosis and treatment (1–4). AI is relatively rare in early life, affecting approximately 1:5,000–10,000 children, and its features can be non-specific. Children can be initially mis-diagnosed as having sepsis, metabolic conditions, or cardiovascular disease, highlighting the need to consider adrenal dysfunction as a differential diagnosis for an unwell or deteriorating infant. Prompt recognition allows the correct investigations to be undertaken urgently and definitive management to be established.
AI can be broadly divided into secondary causes, due to disruption of hypothalamic or pituitary (corticotrope) ACTH release, and primary causes, which affect the adrenal gland itself. Although some conditions have fairly typical presentation patterns and ages of onset, there is often a spectrum of features, and milder variants may produce partial or delayed onset forms of classic conditions (5, 6). Associated features can sometimes give a clue to the diagnosis.
Here, we provide a brief summary of the genetic causes of AI that tend to present in the neonatal period or first few months of life, and the implications of making a specific genetic diagnosis for management. While the focus of this minireview is very much on genetic causes, physical causes (such as adrenal hemorrhage or infiltration) should not be overlooked.
Secondary AI is caused by impaired ACTH synthesis and release from pituitary corticotrope cells (Figure 1A). ACTH deficiency can be isolated or can occur as part of a combined (multiple) pituitary hormone deficiency (CPHD) due to defects in hypothalamo-pituitary function (Table 1). Usually glucocorticoid release is affected, whereas disturbances in mineralocorticoid function and salt balance are unusual as aldosterone synthesis is primarily under the control of the renin-angiotensin system.
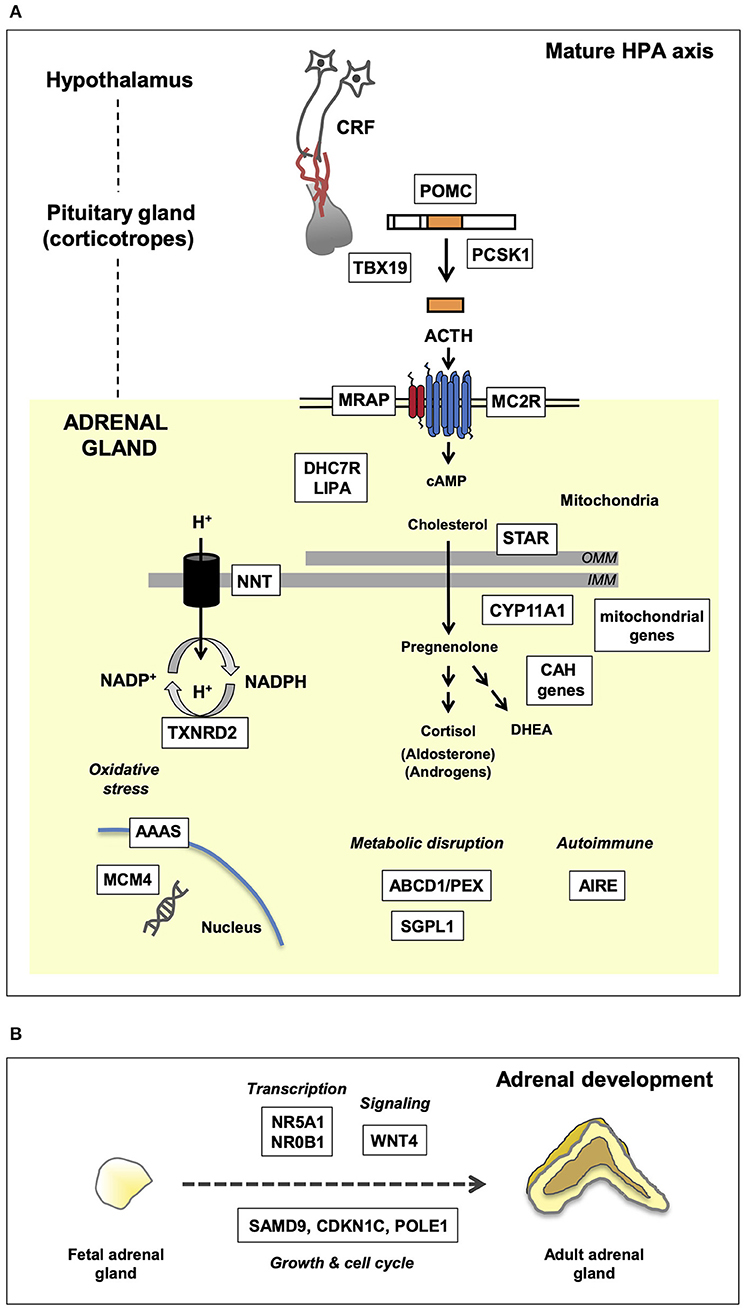
Figure 1. Genetic mechanisms of pediatric adrenal insufficiency (AI) along the hypothalamo-pituitary-adrenal (HPA) axis. Key genes are shown in white boxes. (A) Genetic causes of AI as they relate to the mature HPA axis. These genes are required for a multitude of key enzymatic and biochemical processes occurring within the nucleus, mitochondria, and cytoplasm. Disruption of these genes gives rise to the clinical phenotypes discussed in the text. OMM, outer mitochondrial membrane; IMM, inner mitochondrial membrane. (B) Overview of adrenal development and key genes associated with adrenal hypoplasia. Adrenal hypoplasia is mediated by disruption of key genes required for normal fetal adrenal development. These genes are involved in transcription, signaling, and growth/cell cycle processes.
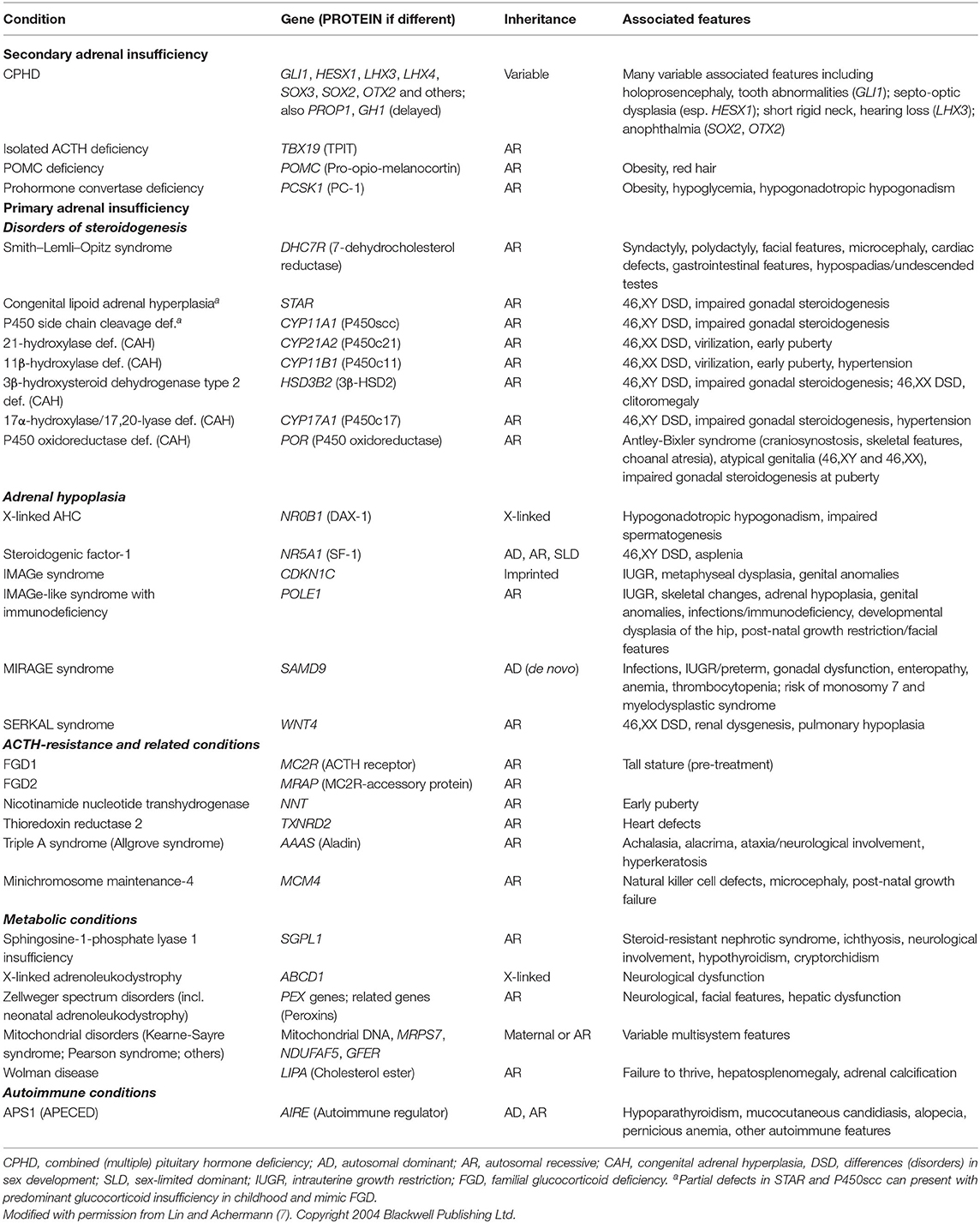
Table 1. Selected monogenic causes of adrenal insufficiency in children.
Several genetic causes of CPHD are reported (e.g., GLI1, HESX1, LHX3, LHX4, SOX3, SOX2 and others) (8). Pituitary ACTH insufficiency usually occurs together with loss of other anterior pituitary hormones (GH, TSH, LH/FSH). Concomitant GH and ACTH insufficiency often causes hypoglycemia in young children, and a small penis and undescended testes may be a sign of congenital gonadotropin insufficiency in boys (9). Other associated features include septo-optic dysplasia or specific associations such as micro-ophthalmia (SOX2, OTX2). Disruption of PROP1 or GH1 can cause ACTH insufficiency in later life (10).
Isolated ACTH deficiency can occur due to disruption of TPIT (TBX19), or with associated features due to defects in pro-opiomelanocortin (POMC) or pro-hormone convertase-1 (PC-1/PCSK1).
TPIT is a transcription factor that regulates synthesis of POMC in pituitary corticotrope cells, but not in other POMC producing cells of the body (e.g., skin, hypothalamus) (11). POMC is a precursor molecule that is cleaved to release ACTH along with other peptides (e.g., alpha-MSH, beta-endorphin) (Figure 1A). Children with severe disruption of TPIT usually present with evidence of glucocorticoid insufficiency, such as hypoglycemia or hypoglycemic seizures, and prolonged conjugated hyperbilirubinemia in the first few weeks of life (12, 13). This contrasts to late-onset isolated ACTH insufficiency, where the molecular basis is currently unknown.
Defects in POMC itself also result in ACTH insufficiency and adrenal dysfunction in early infancy (14). Children have red (or auburn) hair and pale skin due to MSH deficiency, and profound hyperphagia and weight gain from later infancy due to hypothalamic POMC disruption (15). MC4R agonists, which mimic MSH, have had promising results in suppressing hyperphagia in this condition, so it is an important diagnosis to make (16).
Disruption of the cleavage enzyme prohormone convertase-1 (PC-1, PCSK1) also presents with ACTH insufficiency, together with hypoglycemia, malabsorptive diarrhea, obesity, and hypogonadism (17, 18). This diagnosis is rare.
An overview of monogenic causes of primary adrenal insufficiency (PAI) in childhood is shown in Table 1 and Figures 1A,B, together with inheritance patterns and associated features. Here, we focus primarily on key genetic causes of PAI that present in the first few months of life. Disorders of salt-balance (e.g., aldosterone synthase deficiency) are not included.
Smith–Lemli–Opitz syndrome is a defect in cholesterol biosynthesis due to disruption of the enzyme 7-dehydrocholesterol reductase (DHCR7) (19). Common findings in infancy are microcephaly, cleft palate, syndactyly of the second and third toes, post-axial polydactyly, congenital heart defects, gastrointestinal issues (e.g., pyloric stenosis), atypical genital and undescended testes (46,XY) and characteristic facial features (20). AI or impaired stress response can occur, but are surprisingly rare (21, 22). Elevated 7-dehydrocholesterol is diagnostic, coupled with genetic testing.
Steroidogenic acute regulatory protein (STAR) plays a key role in cholesterol transport into the mitochondria, whereas the P450 cholesterol side-change cleave enzyme (P450scc, encoded by CYP11A1) catalyzes the conversion of cholesterol to pregnenolone (Figure 1) (23, 24). These proteins are both required for adrenal (glucocorticoid, mineralocorticoid) and gonadal (testosterone, estrogen) steroid synthesis. Severe disruption causes salt-losing AI in all children and female-typical external genitalia in 46,XY infants. The onset of PAI usually occurs at around 3–4 weeks of age in complete STAR deficiency (also known as “congenital lipoid adrenal hyperplasia”), as build-up of intracellular cholesterol takes time to cause cellular damage (“two-hit hypothesis”) (25). In contrast, infants with a severe P450scc deficiency usually present with salt-losing PAI at around 7-10 days (26, 27). Partial disruption of these proteins results in predominant glucocorticoid insufficiency in childhood (27–31).
The most common cause of PAI in the first month of life is congenital adrenal hyperplasia (CAH) (23). In virtually all populations, 21-hydroxylase deficiency (21-OHD, CYP21A2) is most prevalent, with an incidence of 1:10,000–1:20,000 (although geographical hotspots occur) (32, 33). Other rare forms of CAH include 11 beta-hydroxylase deficiency (CYP11B1) (especially in Jewish populations originating from Morocco), 3 beta-hydroxysteroid dehydrogenase deficiency (HSD3B2), 17 alpha-hydroxylase deficiency (CYP17A1) and P450 oxidoreductase deficiency (POR), all of which have specific presentations and biochemical profiles (Table 1) (23).
Approximately 80% of 46,XX girls with confirmed 21-OHD have atypical genitalia at birth, so any baby with genital differences and non-palpable gonads should be considered as having 21-OHD until proven otherwise (33, 34). Progressive salt loss usually results in hyperkalemia and hyponatremia at around 5–7 days of life, mandating urgent monitoring and treatment once the diagnosis is made. Boys (46,XY) with 21-OHD have no obvious signs at birth and usually present in a salt-losing adrenal crisis between 1 and 2 weeks of age, so many countries include 17-hydroxyprogesterone in their newborn screening program. More detailed reviews of CAH and its management are presented elsewhere (32, 33).
Adrenal hypoplasia is an underdevelopment of the adrenal glands, which often presents with PAI in early life (Figure 1B). Usually this is an X-linked condition, or sometimes associated with intrauterine growth restriction (IUGR) (fetal growth restriction, FGR) syndromes.
X-linked congenital adrenal hypoplasia (adrenal hypoplasia congenita, AHC) primarily affects boys and is associated with disruption of the nuclear receptor, DAX-1 (encoded by NR0B1) (35, 36). This condition presents with salt-losing PAI in the first 2 months of life (40%), or more insidiously with AI in childhood (37). Late-onset forms of the condition have also been described (38–40).
X-linked AHC has three main features: PAI, hypogonadotropic hypogonadism (HH) in adolescence, and impaired spermatogenesis (41). Some boys may paradoxically have macrophallia at birth, and initial presentation with either isolated mineralocorticoid insufficiency or isolated glucocorticoid insufficiency is reported (42, 43). Growth hormone insufficiency has also been diagnosed in a small subset of boys (37, 44–46).
Boys with PAI and HH in adolescence almost invariably have X-linked AHC, especially if there is a family history of X-linked adrenal dysfunction. Even without such a history, we found that approximately 40% of boys presenting with salt-losing AI in the first two months of life had X-linked AHC, once more common conditions such as CAH had been excluded (47). Approximately two-thirds of boys have pathogenic missense or loss-of-function (stop gain, frameshift) variants in DAX-1/NR0B1, around one-sixth have a localized deletion of this gene on the X-chromosome (Xp21), and one-sixth have a larger Xp contiguous gene deletion syndrome that can involve genes causing glycerol kinase deficiency (GKD), ornithine transcarbamylase deficiency (OTC) and Duchenne Muscular Dystrophy (DMD) (47). Very rarely, girls have X-linked AHC due to skewed X-inactivation (48). Establishing this diagnosis early allows prompt recognition and management of both the PAI and potential associated conditions (39). Families can be counseled about risk in brothers or in the maternal family, and presymptomatic boys diagnosed (49).
Steroidogenic factor-1 (SF-1/NR5A1) is a related nuclear receptor considered as a “master-regulator” of adrenal and reproductive development (36). Severe disruption of SF-1 has very rarely been associated with early-onset PAI in 46,XX girls and 46,XY phenotypic female babies with testicular dysgenesis, usually due to disruption of key DNA-binding elements of this transcription factor (50, 51). In contrast, more than 200 individuals and families with heterozygous pathogenic variants in SF-1/NR5A1 have been reported, having a wide spectrum of reproductive phenotypes (from gonadal dysgenesis through to male factor infertility or primary ovarian insufficiency and ovotesticular DSD) (36, 52–54). To date, adrenal function is normal in most of these individuals.
AI associated with IUGR/FGR can occur as part of IMAGe syndrome (intrauterine growth restriction, metaphyseal dysplasia, adrenal hypoplasia, genitourinary anomalies) (55, 56). Children usually present with salt-losing PAI in early life. Other variable features include frontal bossing, impaired glucose tolerance, and hearing loss.
Classic IMAGe syndrome is associated with gain-of-function variants in the cell-cycle repressor, CDKN1C (56). This is a paternally-imprinted (maternally-expressed gene), so is usually inherited from the mother, but can occur de novo. IMAGe-associated pathogenic variants are localized within a very specific motif in the PCNA-binding motif of CDKN1C, causing impaired cell cycle S-phase progression (57). Variants neighboring this motif can cause IUGR/Russell-Silver Syndrome phenotypes with normal adrenal function (58, 59). Of note, loss-of-function of CDKN1C is associated with Beckwith-Wiedemann syndrome, an “overgrowth” syndrome, highlighting how different changes in one gene can have opposing phenotypes (58).
Recently, an “IMAGe-like” syndrome with AI and immunodeficiency (infections, lymphopenia, hypogammaglobulinemia) has been reported (60). These children have profound postnatal growth restriction, distinctive facial features, hip dysplasia and hypoplastic patellae. This condition results from pathogenic biallelic variants in polymerase epsilon-1 (POLE1, Pol ε), often involving a heterozygous intronic variant (c.1686 + 32C>G). POLE1 is a DNA polymerase that interacts with PCNA in S-phase DNA replication.
Another multisystem growth restriction syndrome associated with adrenal hypoplasia is MIRAGE syndrome (myelodysplasia, infections, restriction of growth, adrenal hypoplasia, genital phenotypes, enteropathy) (61, 62). Infants with severe forms of MIRAGE are born preterm and develop salt-losing PAI in early life. Recurrent infections (viral, bacterial and fungal), anemia/thrombocytopenia, nephropathy, severe enteropathy, esophageal reflux and aspiration are common, and 46,XY children have penoscrotal hypospadias or gonadal (testicular) dysfunction with a female phenotype. Mortality is high and children who survive show long-term growth restriction. As many of these features are found in sick, preterm, growth-restricted babies, it is likely that this condition is under-diagnosed.
MIRAGE syndrome results from heterozygous gain-of-function missense mutations in the growth repressor, sterile alpha domain containing 9 (SAMD9) (61, 62). These changes usually occur de novo and restrict cell growth and division, potentially through reduced recycling of growth factor receptors. SAMD9 also plays a role in innate viral immunity and host defense.
One interesting aspect of MIRAGE syndrome is how secondary genetic events can dynamically modify the phenotype through “revertant mosaicism.” For example, development of progressive, somatic monosomy 7 in cis (i.e., on the same allele) “removes” the deleterious gain-of-function mutation in SAMD9 allowing a clonal growth advantage of these affected cells, especially in the hematopoietic system, and reversal of the postnatal anemia and thrombocytopenia (61, 62). However, monosomy 7 is linked to the development of myelodysplastic syndrome, which can lead to leukemia if other genetic changes occur. Interestingly, somatic loss-of-function (nonsense or frameshift) changes or uniparental disomy in cis can also “remove” the mutant allele and ameliorate the phenotype (5, 62–64). Increasingly, children with milder MIRAGE-like features are being reported, many with normal adrenal function (65).
SeRKAL syndrome (female sex reversal and dysgenesis of kidneys, adrenals, and lungs) has been reported in a single family with homozygous disruptive mutations in WNT4, a signaling molecule implicated in adrenal development (66). Other historic reports have rarely described AI with Pena–Shokeir syndrome type I (DOK7, RAPSN), pseudotrisomy 13, Galloway–Mowat syndrome (WDR73), Pallister–Hall syndrome (GLI3, with pituitary defects) and Meckel–Gruber syndrome (MKS1) (67).
Another important group of conditions causing PAI in childhood are ACTH-resistance conditions (also known as Familial Glucocorticoid Deficiency, FGD) and related disorders (68). Some of these may present in early infancy.
Familial Glucocorticoid Deficiency Type 1 (FGD1) is a recessive condition that results from pathogenic variants in the ACTH receptor (melanocortin 2 receptor, MC2R) (68, 69). Children sometimes present in the first weeks of life with signs of cortisol insufficiency (hypoglycemia/convulsions, prolonged jaundice) and marked hyperpigmentation. Genuine mineralocorticoid insufficiency is very rare, but transient salt loss or dilutional hyponatremia can occur, sometimes leading to a misdiagnosis of adrenal hypoplasia (70, 71). FGD1 can also present later in childhood with recurrent infections, hyperpigmentation, and lethargy. Generally, children respond very well to glucocorticoid replacement, but ACTH concentrations can be difficult to suppress.
A similar form of ACTH-resistance results from disruption of the melanocortin 2 receptor accessory protein, MRAP (72). MRAP traffics MC2R to the adrenal cell membrane surface, so disruption of its function (usually due to splicing defects in exon 3) impairs ACTH signaling (68, 73, 74). Affected children usually present with severe glucocorticoid insufficiency and hyperpigmentation in the first few months of life.
Defects in nicotinamide nucleotide transhydrogenase (NNT) are a well-established cause of isolated PAI in children, and occasionally additional features such as early puberty have been reported (68, 75, 76). This condition mostly presents after 1 year of age but has been reported as early as 4 months of age. To date, defects in thioredoxin reductase 2 (TNXRD2) are reported in a single family (sometimes with cardiac defects), and present in mid- or later childhood (77).
Triple A syndrome is a well-established condition linking PAI (“Addison disease”), with alacrima and achalasia of the esophagus (78, 79). This condition results from disruption of the protein aladin (encoded by AAAS), a potential nucleoporin component that may also influence cellular stress (80–82). Alacrima is often present from birth but is difficult to diagnose. Other features usually develop in childhood, or in the second decade of life (83, 84). Progressive neurological and autonomic dysfunction can also co-occur, so this is an important diagnosis to consider.
Disruption of minichromosome maintenance 4 (MCM4) causes mild PAI together with short stature and immunodeficiency (85). To date, this is only reported in individuals of Irish Traveller ancestry, and typically manifests in mid-childhood. As noted above, partial (non-classic) high steroidogenic blocks (STAR and CYP11A1) can present in childhood with PAI. Non-classic STAR defects are sometimes termed FGD3 (27–31).
Several metabolic conditions are associated with PAI but the presentation and features can be variable (86).
SGPL1 deficiency is a novel sphingolipidosis that results from impaired breakdown of sphingosine 1-phosphate (87–89). Key features are PAI (sometimes with adrenal calcifications) and steroid-resistant nephrotic syndrome (SRNS), as well as ichthyosis, neurological dysfunction, primary hypothyroidism, lymphopenia and undescended testes. Many children present with PAI in the first year of life (hyperpigmentation, or adrenal crisis), although some first present with SRNS and the use of steroid treatment may delay the adrenal phenotype. Other features are variable and can appear or progress with time.
Adrenoleukodystrophy (ALD) is a very important cause of PAI because of associated progressive neurological features (90). The X-linked form of ALD due to defects in ABCD1 usually presents in childhood, and sometimes with adrenal-only features. Thus, all boys with undiagnosed causes of PAI should have long-chain fatty acids measured and there are some calls for newborn screening to increase early detection, since allogenic hematopoietic stem cell transplantation may reduce the progression of cerebral X-ALD in patients with early stages of disease, and hematopoietic stem cell gene therapy has been investigated (91–94). In contrast, “neonatal adrenal leukodystrophy” is now classified as part of the “Zellweger Spectrum Disorders” (with Zellweger syndrome/cerebrohepatorenal syndrome, infantile Refsum disease and rhizomelic chondrodysplasia punctata type 1) (95). This spectrum of disorders results from defects in peroxisomal function (13 different PEX genes and others) and has many features including hypotonia, seizures, hepatic dysfunction and renal cysts. PAI has been reported, usually in childhood or with an impaired stress response (96), so screening after 1 year of age has been recommended (95).
Mitochondrial defects have a range of causes and presenting features. Adrenal dysfunction occurs in rare cases, more often associated with large scale mitochondrial DNA deletions (e.g., Kearns-Sayre and Pearson syndromes), but also pathogenic variants in other related genes (e.g., MK-TK, MRPS7, QRSL1, NDUFAF5, GFER) (86, 97).
Wolman disease (primary xanthomatosis) results from disruption of lysosomal acid lipase (LIPA), and is associated with AI (often with adrenal calcifications), failure to thrive, hepatosplenomegaly and anemia in the first few months of life. It is a lysosomal storage disorder that is usually fatal, although improvements with enzyme replacement treatment (sebelipase alfa) are reported (98–100).
Although autoimmune “Addison disease” is the most common cause of AI in adolescents and adults, autoimmune PAI is rare in children (101). The best-established condition is Autoimmune Polyglandular Syndrome type 1 (APS1, also known as APECED), due to defects in autoimmune regulator (AIRE). Early features can include mucocutaneous candidiasis and rarely hypoparathyroidism (hypocalcemia). PAI and other associations usually occur in childhood or later life.
Physical causes of AI such as hemorrhage or infiltration (e.g., neuroblastoma) should not be overlooked. Unilateral adrenal hemorrhages detected by imaging are common (1:200–500 newborn), but usually asymptomatic (102). Symptomatic bilateral hemorrhages are rare but can cause profound AI. As in older children, prolonged administration of glucocorticoids for other conditions can suppress the hypothalamo-pituitary adrenal (HPA) axis and cause AI if withdrawal is rapid.
Transient, relative AI has been described in some very preterm babies, or in sick newborn children under stress. The physiological basis of this is unclear, but steroid supplements have been used in some situations (103).
Making a specific genetic diagnosis has several benefits. It allows tailored treatment of the specific underlying hormonal defect (such as the need for ongoing mineralocorticoid replacement or not) and permits the surveillance, early recognition, and prompt treatment of associated extra-adrenal features (16, 61, 62, 70, 71, 87, 98).
Reaching a specific genetic diagnosis also has wider implications for the family, especially as these conditions have a range of inheritance patterns (e.g., autosomal recessive, dominant/de novo; X-linked, imprinted). This information guides genetic counseling during future pregnancies, and potentially allows pre-symptomatic diagnosis and treatment in relatives with subclinical disease (49).
Traditionally, genetic testing has relied on Sanger sequencing of candidate genes one at a time. This approach may still have a role in common conditions such as 21-OHD (CYP21A2), where there is a specific biochemical profile, well-established pathogenic variants, and a pseudogene that can complicate analysis, or in X-linked AHC (DAX-1/NR0B1) when well-established associations (e.g., HH) or inheritance patterns (e.g., X-linked) are present.
However, associated features or pathognomonic biochemical patterns are often not present when an infant presents with PAI, so “next generation” sequencing (NGS) approaches are increasingly time- and cost-effective.
Access to services varies from country to country, but targeted “panels” to analyze all key PAI genes at once are increasingly available as a clinical service. In addition, several studies have shown how “trio” whole exome sequencing (WES) can help diagnose sick infants and children, especially when there are complex, multisystem features, and exome analysis has been reported to help in the diagnosis of children with PAI (104). Whole genome sequencing (WGS) will become increasingly available and has potential advantages and disadvantages at present compared to panels/WES.
In general, genetic testing for PAI has a high diagnosis rate, certainly when compared to other pediatric endocrine conditions such as congenital hypothyroidism and hypothalamo-pituitary hormone deficiencies. For example, in a national cohort study of rare, undiagnosed PAI in Turkey (with CAH and obvious metabolic causes excluded), a specific genetic diagnosis was reached in 80–90% of children (105), although the diagnostic yield of autosomal recessive conditions was high due to high consanguinity rates. New genetic causes may still emerge as our understanding of human adrenal development expands (106).
Finally, founder effects and genetic “hotspots” can be very important in identifying a specific genetic cause of PAI and taking a history of family ancestry is key. For example, in Turkey the MRAP splice variant cIVS3ds + 1delG is found in the West, whereas P450scc/CYP11A1 p.R451W occurs in Eastern regions (105). As families migrate around the world, founder effects are seen in children born in other countries. Knowing a specific hotspot can allow focused and cost-effective screening of “at risk” family members before the onset of PAI.
AI is an important diagnosis to consider in any sick newborn infant and prompt investigation and treatment is essential. Genetic testing is increasingly useful for finding a specific cause, predicting associated features, counseling families and, in some situations, for modifying treatments.
All authors were involved in the writing, editing, and final approval of the manuscript.
JA is a Wellcome Trust Senior Research Fellow in Clinical Science (209328/Z/17/Z) with research support from Great Ormond Street Hospital Children's Charity (Grant V2518) and the National Institute for Health Research, Great Ormond Street Hospital Biomedical Research Centre (Grant IS-BRC-1215-20012). The views expressed are those of the authors and not necessarily those of the National Health Service, National Institute for Health Research, or Department of Health. SMcG-B holds a Wellcome Trust Clinical PhD Fellowship (216362/Z/19/Z).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are grateful to the children and families involved in our studies over the years, and to Louise Metherell and colleagues at Queen Mary University London for their work on genetic glucocorticoid insufficiency. This article is dedicated to the late Dr. Paolo Ghirri, whose collaborations helped us to understand the role of SAMD9 in MIRAGE syndrome.
1. Bornstein SR, Allolio B, Arlt W, Barthel A, Don-Wauchope A, Hammer GD, et al. Diagnosis and treatment of primary adrenal insufficiency: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. (2016) 101:364–89. doi: 10.1210/jc.2015-1710
2. Park J, Didi M, Blair J. The diagnosis and treatment of adrenal insufficiency during childhood and adolescence. Arch Dis Child. (2016) 101:860–5. doi: 10.1136/archdischild-2015-308799
3. Bowden SA, Henry R. Pediatric adrenal insufficiency: diagnosis, management, and new therapies. Int J Pediatr. (2018) 2018:1–8. doi: 10.1155/2018/1739831
4. Kirkgoz T, Guran T. Primary adrenal insufficiency in children: diagnosis and management. Best Pract Res Clin Endocrinol Metab. (2018) 32:397–424. doi: 10.1016/j.beem.2018.05.010
5. Buonocore F, Achermann JC. Primary adrenal insufficiency: new genetic causes and their long-term consequences. Clin Endocrinol. (2020) 92:11–20. doi: 10.1111/cen.14109
6. Flück CE. Mechanisms in endocrinology: update on pathogenesis of primary adrenal insufficiency: beyond steroid enzyme deficiency and autoimmune adrenal destruction. Eur J Endocrinol. (2017) 177:R99–111. doi: 10.1530/EJE-17-0128
7. Lin L, Achermann JC. Inherited adrenal hypoplasia: not just for kids! Clin Endocrinol. (2004) 60:529–37. doi: 10.1111/j.1365-2265.2004.01988.x
8. Kelberman D, Rizzoti K, Lovell-Badge R, Robinson ICAF, Dattani MT. Genetic regulation of pituitary gland development in human and mouse. Endocr Rev. (2009) 30:790–829. doi: 10.1210/er.2009-0008
9. Alatzoglou KS, Dattani MT. Genetic forms of hypopituitarism and their manifestation in the neonatal period. Early Hum Dev. (2009) 85:705–12. doi: 10.1016/j.earlhumdev.2009.08.057
10. Cerbone M, Dattani MT. Progression from isolated growth hormone deficiency to combined pituitary hormone deficiency. Growth Horm IGF Res. (2017) 37:19–25. doi: 10.1016/j.ghir.2017.10.005
11. Pulichino A-M. Human and mouse TPIT gene mutations cause early onset pituitary ACTH deficiency. Genes Dev. (2003) 17:711–6. doi: 10.1101/gad.1065603
12. Vallette-Kasic S, Brue T, Pulichino A-M, Gueydan M, Barlier A, David M, et al. Congenital isolated adrenocorticotropin deficiency: an underestimated cause of neonatal death, explained by TPIT gene mutations. J Clin Endocrinol Metab. (2005) 90:1323–31. doi: 10.1210/jc.2004-1300
13. Couture C, Saveanu A, Barlier A, Carel JC, Fassnacht M, Flück CE, et al. Phenotypic homogeneity and genotypic variability in a large series of congenital isolated ACTH-deficiency patients with TPIT gene mutations. J Clin Endocrinol Metab. (2012) 97:E486–95. doi: 10.1210/jc.2011-1659
14. Krude H, Biebermann H, Luck W, Horn R, Brabant G, Grüters A. Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat Genet. (1998) 19:155–7. doi: 10.1038/509
15. Çetinkaya S, Güran T, Kurnaz E, Keskin M, Sagsak E, Savaş Erdeve S, et al. A patient with proopiomelanocortin deficiency: an increasingly important diagnosis to make. J Clin Res Pediatr Endocrinol. (2018) 10:68–73. doi: 10.4274/jcrpe.4638
16. Kühnen P, Clément K, Wiegand S, Blankenstein O, Gottesdiener K, Martini LL, et al. Proopiomelanocortin deficiency treated with a melanocortin-4 receptor agonist. N Engl J Med. (2016) 375:240–6. doi: 10.1056/NEJMoa1512693
17. Jackson RS, Creemers JWM, Farooqi IS, Raffin-Sanson M-L, Varro A, Dockray GJ . Small-intestinal dysfunction accompanies the complex endocrinopathy of human proprotein convertase 1 deficiency. J Clin Invest. (2003) 112:1550–60. doi: 10.1172/JCI18784
18. Stijnen P, Ramos-Molina B, O'Rahilly S, Creemers JWM. PCSK1 mutations and human endocrinopathies: from obesity to gastrointestinal disorders. Endocr Rev. (2016) 37:347–71. doi: 10.1210/er.2015-1117
19. Nowaczyk MJ, Wassif CA. Smith–Lemli–Opitz syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al., editors. GeneReviews®. Seattle, WA: University of Washington (1998 [updated 2020]). p. 1–38.
20. Nowaczyk MJM, Irons MB. Smith–Lemli–Opitz syndrome: phenotype, natural history, and epidemiology. Am J Med Genet Part C Semin Med Genet. (2012) 160C:250–62. doi: 10.1002/ajmg.c.31343
21. Bianconi SE, Conley SK, Keil MF, Sinaii N, Rother KI, Porter FD, et al. Adrenal function in Smith–Lemli–Opitz syndrome. Am J Med Genet Part A. (2011) 155:2732–8. doi: 10.1002/ajmg.a.34271
22. Jayamanne C, Sandamal S, Jayasundara K, Saranavabavananthan M, Mettananda S. Smith–Lemli–Opitz syndrome presenting as acute adrenal crisis in a child: a case report. J Med Case Rep. (2018) 12:217. doi: 10.1186/s13256-018-1738-4
23. Miller WL, Auchus RJ. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr Rev. (2011) 32:81–151. doi: 10.1210/er.2010-0013
24. Miller WL. Disorders in the initial steps of steroid hormone synthesis. J Steroid Biochem Mol Biol. (2017) 165:18–37. doi: 10.1016/j.jsbmb.2016.03.009
25. Bose HS, Sugawara T, Strauss JF, Miller WL. The pathophysiology and genetics of congenital lipoid adrenal hyperplasia. N Engl J Med. (1996) 335:1870–9. doi: 10.1056/NEJM199612193352503
26. Hiort O, Holterhus PM, Werner R, Marschke C, Hoppe U, Partsch CJ, et al. Homozygous disruption of P450 side-chain cleavage (CYP11A1) is associated with prematurity, complete 46,XY reversal, and severe adrenal failure. J Clin Endocrinol Metab. (2005) 90:538–41. doi: 10.1210/jc.2004-1059
27. Tee MK, Abramsohn M, Loewenthal N, Harris M, Siwach S, Kaplinsky A, et al. Varied clinical presentations of seven patients with mutations in CYP11A1 encoding the cholesterol side-chain cleavage enzyme, P450scc. J Clin Endocrinol Metab. (2013) 98:713–20. doi: 10.1210/jc.2012-2828
28. Baker BY, Lin L, Kim CJ, Raza J, Smith CP, Miller WL, et al. Nonclassic congenital lipoid adrenal hyperplasia: a new disorder of the steroidogenic acute regulatory protein with very late presentation and normal male genitalia. J Clin Endocrinol Metab. (2006) 91:4781–5. doi: 10.1210/jc.2006-1565
29. Metherell LA, Naville D, Halaby G, Begeot M, Huebner A, Nürnberg G, et al. Nonclassic lipoid congenital adrenal hyperplasia masquerading as familial glucocorticoid deficiency. J Clin Endocrinol Metab. (2009) 94:3865–71. doi: 10.1210/jc.2009-0467
30. Parajes S, Kamrath C, Rose IT, Taylor AE, Mooij CF, Dhir V . A novel entity of clinically isolated adrenal insufficiency caused by a partially inactivating mutation of the gene encoding for P450 side chain cleavage enzyme (CYP11A1). J Clin Endocrinol Metab. (2011) 96:E1798–806. doi: 10.1210/jc.2011-1277
31. Maharaj A, Buonocore F, Meimaridou E, Ruiz-Babot G, Guasti L, Peng H-M, et al. Predicted benign and synonymous variants in CYP11A1 cause primary adrenal insufficiency through missplicing. J Endocr Soc. (2019) 3:201–21. doi: 10.1210/js.2018-00130
32. Speiser PW, Arlt W, Auchus RJ, Baskin LS, Conway GS, Merke DP, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. (2018) 103:4043–88. doi: 10.1210/jc.2018-01865
33. Merke DP, Auchus RJ. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. N Engl J Med. (2020) 383:1248–61. doi: 10.1056/NEJMra1909786
34. Khalid JM, Oerton JM, Dezateux C, Hindmarsh PC, Kelnar CJ, Knowles RL. Incidence and clinical features of congenital adrenal hyperplasia in Great Britain. Arch Dis Child. (2012) 97:101–6. doi: 10.1136/archdischild-2011-300234
35. Muscatelli F, Strom TM, Walker AP, Zanaria E, Récan D, Meindl A, et al. Mutations in the DAX-1 gene give rise to both X-linked adrenal hypoplasia congenita and hypogonadotropic hypogonadism. Nature. (1994) 372:672–6. doi: 10.1038/372672a0
36. Suntharalingham JP, Buonocore F, Duncan AJ, Achermann JC. DAX-1 (NR0B1) and steroidogenic factor-1 (SF-1, NR5A1) in human disease. Best Pract Res Clin Endocrinol Metab. (2015) 29:607–19. doi: 10.1016/j.beem.2015.07.004
37. Reutens AT. Clinical and functional effects of mutations in the DAX-1 gene in patients with adrenal hypoplasia congenita. J Clin Endocrinol Metab. (1999) 84:504–11. doi: 10.1210/jc.84.2.504
38. Tabarin A, Achermann JC, Recan D, Bex V, Bertagna X, Christin-Maitre S, et al. A novel mutation in DAX1 causes delayed-onset adrenal insufficiency and incomplete hypogonadotropic hypogonadism. J Clin Invest. (2000) 105:321–8. doi: 10.1172/JCI7212
39. Guclu M, Lin L, Erturk E, Achermann JC, Cangul H. Puberty, stress, and sudden death. Lancet. (2010) 376:1512. doi: 10.1016/S0140-6736(10)61153-1
40. Kyriakakis N, Shonibare T, Kyaw-Tun J, Lynch J, Lagos CF, Achermann JC, et al. Late-onset X-linked adrenal hypoplasia (DAX-1, NR0B1): two new adult-onset cases from a single center. Pituitary. (2017) 20:585–93. doi: 10.1007/s11102-017-0822-x
41. Achermann JC, Vilain EJ. NR0B1-related adrenal hypoplasia congenita. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al., editors. GeneReviews®. Seattle, WA: University of Washington (2001 [updated 2018]), 1–41.
42. Wiltshire E, Couper JJ, Rodda C, Jameson JL, Achermann JC. Variable presentation of X-linked adrenal hypoplasia congenita. J Pediatr Endocrinol Metab. (2001) 14:1093–6. doi: 10.1515/jpem-2001-0804
43. Landau Z, Hanukoglu A, Sack J, Goldstein N, Weintrob N, Eliakim A, et al. Clinical and genetic heterogeneity of congenital adrenal hypoplasia due to NR0B1 gene mutations. Clin Endocrinol. (2010) 72:448–54. doi: 10.1111/j.1365-2265.2009.03652.x
44. Rojek A, Obara-Moszynska M, Malecka E, Slomko-Jozwiak M, Niedziela M. NR0B1 (DAX1) mutations in patients affected by congenital adrenal hypoplasia with growth hormone deficiency as a new finding. J Appl Genet. (2013) 54:225–30. doi: 10.1007/s13353-013-0135-3
45. Chung ST, Chi CH, Haymond MW, Jeha GS. Infantile growth hormone deficiency and X-linked adrenal hypoplasia congenita. Jacobs J Pediatr. (2015) 1:003.
46. Suthiworachai C, Tammachote R, Srichomthong C, Ittiwut R, Suphapeetiporn K, Sahakitrungruang T, et al. Identification and functional analysis of six DAX1 mutations in patients with X-linked adrenal hypoplasia congenita. J Endocr Soc. (2019) 3:171–80. doi: 10.1210/js.2018-00270
47. Lin L, Gu W-X, Ozisik G, To WS, Owen CJ, Jameson JL, et al. Analysis of DAX1 (NR0B1) and steroidogenic factor-1 (NR5A1) in children and adults with primary adrenal failure: ten years' experience. J Clin Endocrinol Metab. (2006) 91:3048–54. doi: 10.1210/jc.2006-0603
48. Shaikh MG, Boyes L, Kingston H, Collins R, Besley GTN, Padmakumar B, et al. Skewed X inactivation is associated with phenotype in a female with adrenal hypoplasia congenita. J Med Genet. (2008) 45:e1–e1. doi: 10.1136/jmg.2007.055129
49. Achermann JC, Silverman BL, Habiby RL, Jameson JL. Presymptomatic diagnosis of X-linked adrenal hypoplasia congenita by analysis of DAX1. J Pediatr. (2000) 137:878–81. doi: 10.1067/mpd.2000.108567
50. Achermann JC, Ito M, Ito M, Hindmarsh PC, Jameson JL. A mutation in the gene encoding steroidogenic factor-1 causes XY sex reversal and adrenal failure in humans. Nat Genet. (1999) 22:125–6. doi: 10.1038/9629
51. Achermann JC, Ozisik G, Ito M, Orun UA, Harmanci K, Gurakan B, et al. Gonadal determination and adrenal development are regulated by the orphan nuclear receptor steroidogenic factor-1, in a dose-dependent manner. J Clin Endocrinol Metab. (2002) 87:1829–33. doi: 10.1210/jcem.87.4.8376
52. Lourenço D, Brauner R, Lin L, De Perdigo A, Weryha G, Muresan M, et al. Mutations in NR5A1 associated with ovarian insufficiency. Obstet Gynecol Surv. (2009) 64:665–6. doi: 10.1097/01.ogx.0000359268.91477.36
53. Bashamboo A, Ferraz-de-Souza B, Lourenço D, Lin L, Sebire NJ, Montjean D, et al. Human male infertility associated with mutations in NR5A1 encoding steroidogenic factor 1. Am J Hum Genet. (2010) 87:505–12. doi: 10.1016/j.ajhg.2010.09.009
54. Bashamboo A, Donohoue PA, Vilain E, Rojo S, Calvel P, Seneviratne SN, et al. A recurrent p.Arg92Trp variant in steroidogenic factor-1 (NR5A1) can act as a molecular switch in human sex development. Hum Mol Genet. (2016) 25:3446–53. doi: 10.1093/hmg/ddw186
55. Vilain E, Merrer MLE, Lecointre C, Desangles F, Kay MA, Maroteaux P, et al. IMAGe, a new clinical association of Intrauterine growth retardation, metaphyseal dysplasia, adrenal hypoplasia congenita, and Genital anomalies. J Clin Endocrinol Metab. (1999) 84:4335–40. doi: 10.1210/jcem.84.12.6186
56. Arboleda VA, Lee H, Parnaik R, Fleming A, Banerjee A, Ferraz-de-Souza B, et al. Mutations in the PCNA-binding domain of CDKN1C cause IMAGe syndrome. Nat Genet. (2012) 44:788–92. doi: 10.1038/ng.2275
57. Borges KS, Arboleda VA, Vilain E. Mutations in the PCNA-binding site of CDKN1C inhibit cell proliferation by impairing the entry into S phase. Cell Div. (2015) 10:2. doi: 10.1186/s13008-015-0008-8
58. Eggermann T, Binder G, Brioude F, Maher ER, Lapunzina P, Cubellis MV, et al. CDKN1C mutations: two sides of the same coin. Trends Mol Med. (2014) 20:614–22. doi: 10.1016/j.molmed.2014.09.001
59. Suntharalingham JP, Ishida M, Buonocore F, del Valle I, Solanky N, Demetriou C, et al. Analysis of CDKN1C in fetal growth restriction and pregnancy loss. F1000Research. (2019) 8:90. doi: 10.12688/f1000research.15016.1
60. Logan C V, Murray JE, Parry DA, Robertson A, Bellelli R, Tarnauskaite Ž, et al. DNA polymerase epsilon deficiency causes IMAGe syndrome with variable immunodeficiency. Am J Hum Genet. (2018) 103:1038–44. doi: 10.1016/j.ajhg.2018.10.024
61. Narumi S, Amano N, Ishii T, Katsumata N, Muroya K, Adachi M, et al. SAMD9 mutations cause a novel multisystem disorder, MIRAGE syndrome, and are associated with loss of chromosome 7. Nat Genet. (2016) 48:792–7. doi: 10.1038/ng.3569
62. Buonocore F, Kühnen P, Suntharalingham JP, Del Valle I, Digweed M, Stachelscheid H, et al. Somatic mutations and progressive monosomy modify SAMD9-related phenotypes in humans. J Clin Invest. (2017) 127:1700–13. doi: 10.1172/JCI91913
63. Csillag B, Ilencikova D, Meissl M, Webersinke G, Laccone F, Narumi S, et al. Somatic mosaic monosomy 7 and UPD7q in a child with MIRAGE syndrome caused by a novel SAMD9 mutation. Pediatr Blood Cancer. (2019) 66:e27589. doi: 10.1002/pbc.27589
64. Roucher-Boulez F, Mallet D, Chatron N, Dijoud F, Gorduza DB, Bretones P, et al. Reversion SAMD9 mutations modifying phenotypic expression of MIRAGE syndrome and allowing inheritance in a usually de novo disorder. Front Endocrinol. (2019) 10:625. doi: 10.3389/fendo.2019.00625
65. Shima H, Hayashi M, Tachibana T, Oshiro M, Amano N, Ishii T, et al. MIRAGE syndrome is a rare cause of 46,XY DSD born SGA without adrenal insufficiency. PLoS ONE. (2018) 13:e0206184. doi: 10.1371/journal.pone.0206184
66. Mandel H, Shemer R, Borochowitz ZU, Okopnik M, Knopf C, Indelman M, et al. SERKAL syndrome: an autosomal-recessive disorder caused by a loss-of-function mutation in WNT4. Am J Hum Genet. (2008) 82:39–47. doi: 10.1016/j.ajhg.2007.08.005
67. Kyritsi EM, Sertedaki A, Charmandari E, Chrousos GP. Familial or sporadic adrenal hypoplasia syndromes. Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dungan K, et al., editors. Endotext. South Dartmouth, MA: MDText.com, Inc. (2018). p. 1-77.
68. Maharaj A, Maudhoo A, Chan LF, Novoselova T, Prasad R, Metherell LA, et al. Isolated glucocorticoid deficiency: genetic causes and animal models. J Steroid Biochem Mol Biol. (2019) 189:73–80. doi: 10.1016/j.jsbmb.2019.02.012
69. Clark AJL, Grossman A, McLoughlin L. Familial glucocorticoid deficiency associated with point mutation in the adrenocorticotropin receptor. Lancet. (1993) 341:461–2. doi: 10.1016/0140-6736(93)90208-X
70. Lin L, Hindmarsh PC, Metherell LA, Alzyoud M, Al-Ali M, Brain CE, et al. Severe loss-of-function mutations in the adrenocorticotropin receptor (ACTHR, MC2R) can be found in patients diagnosed with salt-losing adrenal hypoplasia. Clin Endocrinol. (2007) 66:205–10. doi: 10.1111/j.1365-2265.2006.02709.x
71. Chan LF, Metherell LA, Krude H, Ball C, O'Riordan SMP, Costigan C, et al. Homozygous nonsense and frameshift mutations of the ACTH receptor in children with familial glucocorticoid deficiency (FGD) are not associated with long-term mineralocorticoid deficiency. Clin Endocrinol. (2009) 71:171–5. doi: 10.1111/j.1365-2265.2008.03511.x
72. Metherell LA, Chapple JP, Cooray S, David A, Becker C, Rüschendorf F, et al. Mutations in MRAP, encoding a new interacting partner of the ACTH receptor, cause familial glucocorticoid deficiency type 2. Nat Genet. (2005) 37:166–70. doi: 10.1038/ng1501
73. Chung TT, Webb TR, Chan LF, Cooray SN, Metherell LA, King PJ, et al. The majority of adrenocorticotropin receptor (melanocortin 2 receptor) mutations found in familial glucocorticoid deficiency type 1 lead to defective trafficking of the receptor to the cell surface. J Clin Endocrinol Metab. (2008) 93:4948–54. doi: 10.1210/jc.2008-1744
74. Novoselova T V, Hussain M, King PJ, Guasti L, Metherell LA, Charalambous M, et al. MRAP deficiency impairs adrenal progenitor cell differentiation and gland zonation. FASEB J. (2018) 32:6186–96. doi: 10.1096/fj.201701274RR
75. Meimaridou E, Kowalczyk J, Guasti L, Hughes CR, Wagner F, Frommolt P, et al. Mutations in NNT encoding nicotinamide nucleotide transhydrogenase cause familial glucocorticoid deficiency. Nat Genet. (2012) 44:740–2. doi: 10.1038/ng.2299
76. Roucher-Boulez F, Mallet-Motak D, Samara-Boustani D, Jilani H, Ladjouze A, Souchon P-F, et al. NNT mutations: a cause of primary adrenal insufficiency, oxidative stress and extra-adrenal defects. Eur J Endocrinol. (2016) 175:73–84. doi: 10.1530/EJE-16-0056
77. Prasad R, Chan LF, Hughes CR, Kaski JP, Kowalczyk JC, Savage MO, et al. Thioredoxin reductase 2 (TXNRD2) mutation associated with familial glucocorticoid deficiency (FGD). J Clin Endocrinol Metab. (2014) 99:E1556–63. doi: 10.1210/jc.2013-3844
78. Allgrove J, Clayden GS, Grant DB, Macaulay JC. Familial glucocorticoid deficiency with achalasia of the cardia and deficient tear production. Lancet. (1978) 311:1284–6. doi: 10.1016/S0140-6736(78)91268-0
79. Sarathi V, Shah NS. Triple-A Syndrome. Adv Exp Med Biol. (2010) 685:1–8. doi: 10.1007/978-1-4419-6448-9_1
80. Tullio-Pelet A, Salomon R, Hadj-Rabia S, Mugnier C, de Laet M-H, Chaouachi B, et al. Mutant WD-repeat protein in triple-A syndrome. Nat Genet. (2000) 26:332–5. doi: 10.1038/81642
81. Handschug K. Triple A syndrome is caused by mutations in AAAS, a new WD-repeat protein gene. Hum Mol Genet. (2001) 10:283–90. doi: 10.1093/hmg/10.3.283
82. Prasad R, Metherell LA, Clark AJ, Storr HL. Deficiency of ALADIN impairs redox homeostasis in human adrenal cells and inhibits steroidogenesis. Endocrinology. (2013) 154:3209–18. doi: 10.1210/en.2013-1241
83. Milenkovic T, Zdravkovic D, Savic N, Todorovic S, Mitrovic K, Koehler K, et al. Triple A syndrome: 32 years experience of a single centre (1977–2008). Eur J Pediatr. (2010) 169:1323–8. doi: 10.1007/s00431-010-1222-7
84. Patt H, Koehler K, Lodha S, Jadhav S, Yerawar C, Huebner A, et al. Phenotype–genotype spectrum of AAA syndrome from Western India and systematic review of literature. Endocr Connect. (2017) 6:901–13. doi: 10.1530/EC-17-0255
85. Hughes CR, Guasti L, Meimaridou E, Chuang C-H, Schimenti JC, King PJ, et al. MCM4 mutation causes adrenal failure, short stature, and natural killer cell deficiency in humans. J Clin Invest. (2012) 122:814–20. doi: 10.1172/JCI60224
86. Hannah-Shmouni F, Stratakis CA. An overview of inborn errors of metabolism manifesting with primary adrenal insufficiency. Rev Endocr Metab Disord. (2018) 19:53–67. doi: 10.1007/s11154-018-9447-2
87. Prasad R, Hadjidemetriou I, Maharaj A, Meimaridou E, Buonocore F, Saleem M, et al. Sphingosine-1-phosphate lyase mutations cause primary adrenal insufficiency and steroid-resistant nephrotic syndrome. J Clin Invest. (2017) 127:942–53. doi: 10.1172/JCI90171
88. Lovric S, Goncalves S, Gee HY, Oskouian B, Srinivas H, Choi W, et al. Mutations in sphingosine-1-phosphate lyase cause nephrosis with ichthyosis and adrenal insufficiency. J Clin Invest. (2017) 127:912–28. doi: 10.1172/JCI89626
89. Janecke AR, Xu R, Steichen-Gersdorf E, Waldegger S, Entenmann A, Giner T, et al. Deficiency of the sphingosine-1-phosphate lyase SGPL1 is associated with congenital nephrotic syndrome and congenital adrenal calcifications. Hum Mutat. (2017) 38:365–72. doi: 10.1002/humu.23192
90. Turk BR, Theda C, Fatemi A, Moser AB. X-linked adrenoleukodystrophy: pathology, pathophysiology, diagnostic testing, newborn screening, and therapies. Int J Dev Neurosci. (2019) 80:52–72 doi: 10.1016/j.ijdevneu.2019.11.002
91. Eng L, Regelmann MO. Early onset primary adrenal insufficiency in males with adrenoleukodystrophy: case series and literature review. J Pediatr. (2019) 211:211–4. doi: 10.1016/j.jpeds.2019.04.021
92. Mahmood A, Raymond G V, Dubey P, Peters C, Moser HW. Survival analysis of haematopoietic cell transplantation for childhood cerebral X-linked adrenoleukodystrophy: a comparison study. Lancet Neurol. (2007) 6:687–92. doi: 10.1016/S1474-4422(07)70177-1
93. Eichler F, Duncan C, Musolino PL, Orchard PJ, De Oliveira S, Thrasher AJ, et al. Hematopoietic stem-cell gene therapy for cerebral adrenoleukodystrophy. N Engl J Med. (2017) 377:1630–8. doi: 10.1056/nejmoa1700554
94. Zhu J, Eichler F, Biffi A, Duncan CN, Williams DA, Majzoub JA. The changing face of adrenoleukodystrophy. Endocr Rev. (2020) 41:577–93 doi: 10.1210/endrev/bnaa013
95. Braverman NE, Raymond G V, Rizzo WB, Moser AB, Wilkinson ME, Stone EM, et al. Peroxisome biogenesis disorders in the Zellweger spectrum: an overview of current diagnosis, clinical manifestations, and treatment guidelines. Mol Genet Metab. (2016) 117:313–21. doi: 10.1016/j.ymgme.2015.12.009
96. Berendse K, Engelen M, Linthorst GE, van Trotsenburg AP, Poll-The BT. High prevalence of primary adrenal insufficiency in Zellweger spectrum disorders. Orphanet J Rare Dis. (2014) 9:133. doi: 10.1186/s13023-014-0133-5
97. Chow J, Rahman J, Achermann JC, Dattani MT, Rahman S. Mitochondrial disease and endocrine dysfunction. Nat Rev Endocrinol. (2017) 13:92–104. doi: 10.1038/nrendo.2016.151
98. Burton BK, Balwani M, Feillet F, Barić I, Burrow TA, Camarena Grande C, et al. A phase 3 trial of sebelipase alfa in lysosomal acid lipase deficiency. N Engl J Med. (2015) 373:1010–20. doi: 10.1056/NEJMoa1501365
99. Jones SA, Rojas-Caro S, Quinn AG, Friedman M, Marulkar S, Ezgu F, et al. Survival in infants treated with sebelipase Alfa for lysosomal acid lipase deficiency: an open-label, multicenter, dose-escalation study. Orphanet J Rare Dis. (2017) 12:25. doi: 10.1186/s13023-017-0587-3
100. Pastores GM, Hughes DA. Lysosomal acid lipase deficiency: therapeutic options. Drug Des Devel Ther. (2020) 14:591–601. doi: 10.2147/DDDT.S149264
101. Frommer L, Kahaly GJ. Autoimmune polyendocrinopathy. J Clin Endocrinol Metab. (2019) 104:4769–82. doi: 10.1210/jc.2019-00602
102. Toti MS, Ghirri P, Bartoli A, Caputo C, Laudani E, Masoni F, et al. Adrenal hemorrhage in newborn: how, when and why—from case report to literature review. Ital J Pediatr. (2019) 45:58. doi: 10.1186/s13052-019-0651-9
103. Shaffer ML, Baud O, Lacaze-Masmonteil T, Peltoniemi OM, Bonsante F, Watterberg KL. Effect of prophylaxis for early adrenal insufficiency using low-dose hydrocortisone in very preterm infants: an individual patient data meta-analysis. J Pediatr. (2019) 207:136-142.e5. doi: 10.1016/j.jpeds.2018.10.004
104. Chan LF, Campbell DC, Novoselova T V, Clark AJL, Metherell LA. Whole-Exome Sequencing in the differential diagnosis of primary adrenal insufficiency in children. Front Endocrinol. (2015) 6. doi: 10.3389/fendo.2015.00113
105. Guran T, Buonocore F, Saka N, Ozbek MN, Aycan Z, Bereket A, et al. Rare causes of primary adrenal insufficiency: genetic and clinical characterization of a large nationwide cohort. J Clin Endocrinol Metab. (2016) 101:284–92. doi: 10.1210/jc.2015-3250
Keywords: adrenal insufficiency, Addison's disease, adrenal hypoplasia, congenital adrenal hyperplasia, glucocorticoid, DAX-1, MIRAGE syndrome, genetic testing
Citation: Buonocore F, McGlacken-Byrne SM, del Valle I and Achermann JC (2020) Current Insights Into Adrenal Insufficiency in the Newborn and Young Infant. Front. Pediatr. 8:619041. doi: 10.3389/fped.2020.619041
Received: 19 October 2020; Accepted: 25 November 2020;
Published: 14 December 2020.
Edited by:
Paula Caroline Midgley, University of Edinburgh, United KingdomReviewed by:
Jarmo Jääskeläinen, Kuopio University Hospital, FinlandCopyright © 2020 Buonocore, McGlacken-Byrne, del Valle and Achermann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John C. Achermann, j.achermann@ucl.ac.uk
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.