 Alison B. Nair
Alison B. Nair Robert I. Parker
Robert I. Parker- 1Pediatric Critical Care Medicine, University of California, San Francisco, San Francisco, CA, United States
- 2Pediatric Hematology/Oncology, Renaissance School of Medicine, Stony Brook University, Stony Brook, NY, United States
Children with critical illness frequently manifest imbalances in hemostasis with risk of consequent bleeding or pathologic thrombosis. Traditionally, plasma-based tests measuring clot formation by time to fibrin clot generation have been the “gold standard” in hemostasis testing. However, these tests are not sensitive to abnormalities in fibrinolysis or in conditions of enhanced clot formation that may lead to thrombosis. Additionally, they do not measure the critical roles played by platelets and endothelial cells. An added factor in the evaluation of these plasma-based tests is that in infants and young children plasma levels of many procoagulant and anticoagulant proteins are lower than in older children and adults resulting in prolonged clot generation times in spite of maintaining a normal hemostatic “balance.” Consequently, newer assays directly measuring thrombin generation in plasma and others assessing the stages hemostasis including clot initiation, propagation, and fibrinolysis in whole blood by viscoelastic methods are now available and may allow for a global measurement of the hemostatic system. In this manuscript, we will review the processes by which clots are formed and by which hemostasis is regulated, and the rationale and limitations for the more commonly utilized tests. We will also discuss selected newer tests available for the assessment of hemostasis, their “pros” and “cons,” and how they compare to the traditional tests of coagulation in the assessment and management of critically ill children.
Introduction
Hemostatic dysfunction and resultant pathologic bleeding and/or thrombosis is a common complication of critical illness in children. There are several important considerations of this dysfunction that are imperative for accurate diagnosis and clinical management in the pediatric population. First, the hemostatic system changes and develops from birth to infancy, childhood, adolescence, and ultimately adulthood. Understanding this evolution is important in the interpretation of physiologic and pathologic hemostasis. Second, traditional tests of hemostasis have focused on platelet count and plasma-based measurements of clot formation. There are limitations in this traditional approach including lack of consideration for (1) platelet function and activity, (2) the imperfect sensitivity of plasma-based tests, particularly to abnormalities in fibrinolysis and conditions of enhanced clot formation, (3) the critical role of the endothelium in hemostasis and crosstalk between the endothelial, inflammatory, and coagulation systems, and (4) the stages of clot formation and lysis over time. Newer studies address some of these challenges by directly measuring thrombin generation in plasma and assessing the stages of hemostasis in whole blood. Successful diagnosis and management of hemostatic dysfunction in critically ill children requires both an age-dependent understanding of clot formation and regulation of hemostasis as well as of the strengths and weaknesses of the tests employed.
Hemostasis and the Regulation of Clot Formation
Hemostasis is the normal physiologic process by which blood is maintained in fluid state while also allowing for blood clot formation at the site of injury maintaining the integrity of the closed circulatory system after vascular damage (1). It is a highly complex series of interwoven processes that result in a rapid, localized, and highly regulated response. Key elements of hemostasis include (1) formation of the platelet plug (referred to as primary hemostasis), (2) soluble phase coagulation with propagation of clotting through the clotting cascade, (3) termination of clot formation, and (4) clot dissolution (fibrinolysis). Under physiologic conditions, a clot is formed at the site of injury to stop bleeding locally with clot lysis and tissue remodeling to follow (1). Abnormalities in any part of these processes can result in dysfunctional hemostasis manifest as abnormal bleeding or thrombosis.
The hemostatic process starts with injury to the vascular endothelium (2). The undisturbed endothelium has multiple guards against undesired coagulation by functioning to counteract platelet activation and aggregation, and to maintain blood fluidity (2). When the integrity of the endothelium is compromised, exposure of subendothelial elements trigger thrombus formation (3). The endothelium also responds to injury through vasoconstriction which results from impairment of the vasodilatory action of damaged endothelial cells as well as direct access of smooth muscle cells to locally generated vasoconstrictive agents (3).
Platelet activation and adhesion upon exposure to vascular injury occurs in a highly coordinated method involving tethering, rolling, activation, and firm adhesion (4, 5). The initial interactions between platelets and the extracellular matrix are highly dictated by local rheological conditions (4). Platelet activation is triggered by several highly adhesive macromolecules including collagen and von Willebrand factor (vWf) along with the weaker agonists adenosine diphosphate (ADP) and epinephrine (4–6). This activation is also triggered by thrombin (4, 5). As part of the platelet activation process, platelets undergo shape change and develop elongated pseudopods that aid in the subsequent adhesion process (7). Platelet adhesion is primarily mediated by attachment of the platelet surface receptor GPIb/IX/V complex to vWf in plasma and the subendothelial matrix (7). Further, platelet activation results in exposure and conformational change of the GPIIb/IIIa receptor on the platelet surface. The GPIIb/IIIa receptor then binds vWf and fibrinogen ultimately resulting in platelet-platelet cohesion (8, 9).
After platelet binding to subendothelial structures and subsequent signaling to the platelet cytoplasm, platelet granules fuse with the open canalicular system of the platelet membrane and empty their contents into the local environment. The paracrine and autocrine nature of these bioactive contents causes increased activation of nearby platelets in both number and degree (4). This results in secondary secretion and significant amplification of the platelet activation and adhesion processes. Alpha (α)granules contain a heterogeneous complement of proteins that effect multiple biologic systems beyond primary hemostasis and coagulation including inflammation, angiogenesis, wound healing, and others (4). Important to formation of the platelet plug is the α-granule release of vWf which further increases formation of the platelet scaffold via GPIIb/IIIa and GPIb/IX/V as well as release of fibrinogen which crosslinks with GPIIb/IIIa and provides an additional source of fibrinogen to what is present in the plasma (4, 10). Dense (δ)-granules secrete ADP and serotonin which stimulate and recruit additional platelets (11). Finally, platelet procoagulant activity is an important facet of platelet activation. This procoagulant activity includes both exposure of procoagulant phospholipids such as phosphatidylserine and the ensuing formation of enzyme complexes on the platelet surface essential to the clotting cascade (12).
The soluble phase of clotting primarily involves the clotting cascade (Figures 1, 2) (13). In general, the clotting cascade involves successive activation of a series of proenzymes or zymogens into active enzymes resulting in an incremental but amplified clotting response. The clotting cascade is classically described by the intrinsic, extrinsic, and common pathway, though further investigation has revealed these pathways to be much more complex and interrelated (Figure 1) (13).
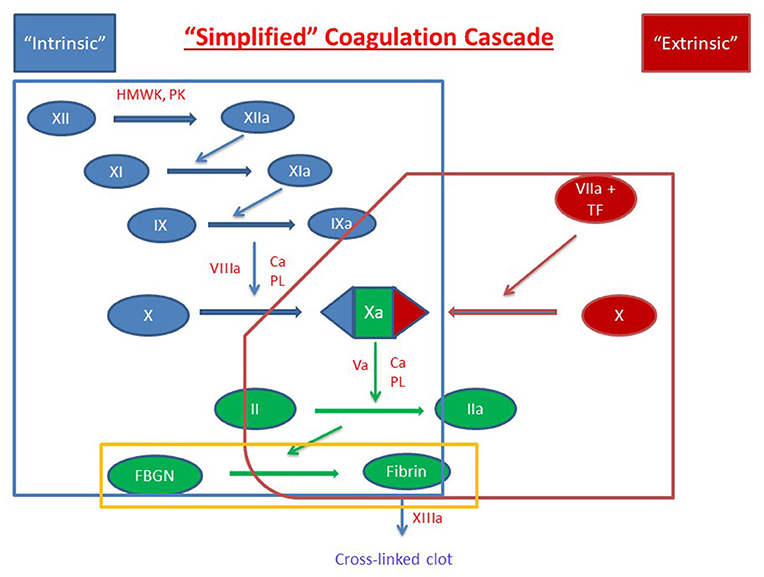
Figure 1. Simplified Coagulation Cascade. Clot formation as activated through the extrinsic pathway (red), the intrinsic pathway (blue) and the common pathway (green). Components of these pathways are measured by prothrombin time, activated partial thromboplastin time, and thrombin time are circled in red, blue, and yellow, respectively. Factors are indicated with a roman numeral and an “a” if activated. Abbreviations: Ca2+, calcium ions; FBGN, fibrinogen; HMWK, high-molecular-weight kininogen; PL, platelet membrane phospholipid; PK, prekallikrein; TF, tissue factor.
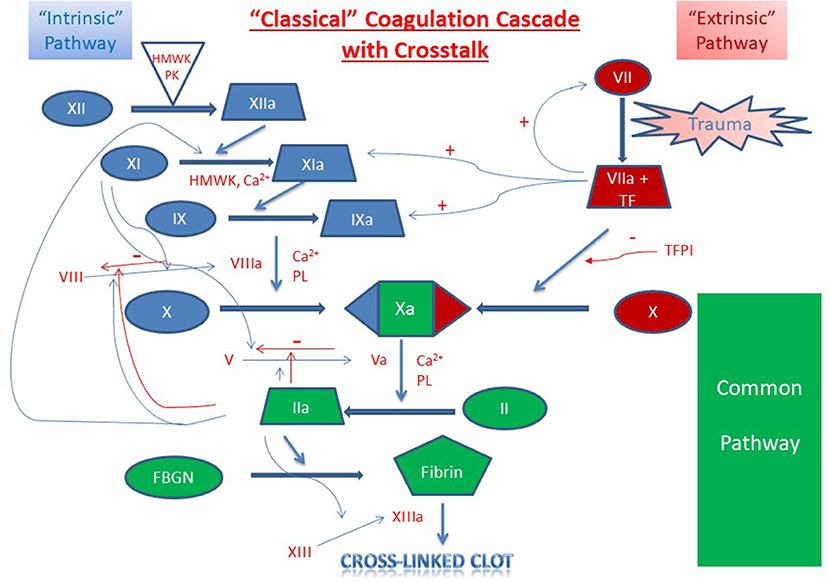
Figure 2. Classical Coagulation Cascade with Crosstalk. Clot formation as activated through the extrinsic pathway (red), intrinsic pathway (blue) and the common pathway (green). Crosstalk and feedback between pathways is shown in blue if positive and red if negative. Factors are indicated with a roman numeral and an “a” if activated. Abbreviations: Ca2+, calcium ions; FBGN, fibrinogen; HMWK, high-molecular-weight kininogen; PL, platelet membrane phospholipid; PK, prekallikrein; TF, tissue factor; TFPI, tissue factor pathway inhibitor.
The clotting cascade is initiated through the extrinsic pathway and starts with exposure of tissue factor (TF) at the site of injury. Blood is exposed to TF either directly by the subendothelial matrix or by cytokine-induced expression on endothelial cells or activated monocytes (14, 15). Platelets may also generate their own TF in proximity to the injured vessel (16). TF operates as a cofactor in the activation of factor VII and together these two factors form the extrinsic tenase multimeric complex which goes on to activate factors X and IX to factors Xa and IXa, respectively (17, 18).
The intrinsic or contact activation pathway is initiated through the interaction of negatively charged surfaces resulting in the activation of factor XII, high-molecular-weight kininogen, and plasma kallikrein among others (13). Activated factor XII in conjunction with high-molecular-weight kininogen activate factor XI which then activates factor IX (19). Activated factor IX forms a multimeric complex with activated factor VIII, which has been activated by factor X and thrombin produced in the extrinsic pathway. This multimeric complex, referred to as intrinsic tenase, subsequently activates sufficient factor X for clot formation (13, 19). This process is amplified because (1) factor VIII is activated by both activated factor X and thrombin and (2) activated factor IX is further activated by the thrombin-induced activation of factor XI (19, 20). As a result, there is a progressive increase in factor VIII and factor IX activation as factor Xa and thrombin are formed. Through these mechanisms, sustained and amplified generation of thrombin is achieved through the intrinsic pathway. Of note, while more activated factor X is generated through the intrinsic pathway due to amplifying steps in the cascade, the extrinsic pathway is physiologically more important clinically (21).
From there, both the extrinsic and intrinsic pathway proceed to the common pathway. Factor V, which is released from platelet α-granules, is cleaved by thrombin to form activated factor V (22, 23). Activated factors X and V bind on the platelet phospholipid surface to form the prothrombinase complex which converts prothrombin (factor II) to thrombin (activated factor II, factor IIa) (24). Thrombin then converts fibrinogen to fibrin and activated factor XIII crosslinks overlapping fibrin stands which leads to stabilization of the clot (25).
Intrinsic to the built-in amplification systems of formation of the platelet plug and the coagulation cascade, hemostasis also requires highly regulated mechanisms of control of clot extension and termination of clot formation (Figure 2). Systemic control of this localized response is modulated through several mechanisms including (1) dilution of procoagulants in the bloodstream, (2) elimination of activated factors through the reticuloendothelial system, and (3) control through antithrombotic pathways anchored on vascular endothelial cells (1). Physiologic inhibitors of coagulation include tissue factor pathway inhibitor (TFPI) which inhibits TF-mediated and factor VIIa-mediated factor X activation, and C1 esterase inhibitor which inhibits activated factor XII, plasma kallikrein, and several complement proteases (26, 27).
Cessation of clot formation is critical in maintaining a targeted response and mediating the extent of the clot. Clot termination involves two key components, antithrombin (antithrombin-III, AT) and the protein C pathway. AT is a serine protease inhibitor that neutralizes many enzymes in the clotting cascade by irreversibly binding to them (28). Its function is enhanced by endogenous heparins and heparan sulfate, which induce a conformational change and increase its affinity for thrombin approximately 300-fold (28–30). Vascular endothelial cells are coated with activated AT and therefore equipped to rapidly inactivate any excess generated thrombin (29, 30). The protein C pathway is initiated through the binding of thrombin to the endothelial membrane-expressed thrombomodulin which induces the ability of the thrombomodulin-thrombin complex to activate protein C (31, 32). In association with protein S, activated protein C then proteolytically inactivates activated factors V and VIII (33, 34).
Also important to the limitation of clot formation is the regulation of vascular and platelet reactivity which is primarily modulated through prostacyclin, thromboxane, and nitric oxide. Specifically, undisturbed endothelial cells adjacent to endothelial injury release arachidonic acid which is subsequently converted to thromboxane A2 by cyclooxygenase-1 in platelets and prostacyclin via cyclooxygenase-1 on the endothelium (35). Prostacyclin blocks platelet aggregation and counteracts thromboxane A2-mediated vasoconstriction (36). In addition to its vasodilatory effects, nitric oxide inhibits platelet adhesion and aggregation (37). In fact, platelets may enhance their synthesis of nitric oxide in the setting of platelet adhesion to collagen providing negative feedback to limit excessive platelet adhesion and vasoconstriction at the site of injury (38).
Clot removal following hemostasis is achieved through fibrinolysis. Central to this process, plasmin is formed from the cleavage of plasminogen by bound fibrin and tissue-type plasminogen activator (38, 39). Urokinase is a secondary plasminogen activator, primarily acting in the extravascular compartment (39). Once formed, plasmin cleaves fibrinogen, fibrin, activated factor XIII, as well as a variety of plasma proteins and other clotting factors (39–41). Fibrinolytic activity can be generated either on the surface of the fibrin-containing thrombus or on cells that express profibrinolytic receptors (38). Plasmin activity is regulated by vascular endothelial cells that secrete both serine protease plasminogen activators and plasminogen activator inhibitors (42). Fibrinolysis works in conjunction with wound healing and tissue remodeling to restore vessel patency following injury.
The above discussion focuses primarily on plasma proteins with only limited mention of the role cellular components play in hemostasis. Indeed, a major limitation of plasma-based coagulation tests is that they do not take into consideration the myriad roles platelets, endothelial and leukocytes play in this process (43). Platelets are recruited to areas of vascular injury and adhere to endothelial cells and subendothelial structures (e.g., collagen) via specific receptors on these structures, a process augmented through binding to von Willebrand factor (vWf), thereby forming a platelet plug (Figure 3A). As noted, endothelial cells are critical in modulating the balance between activation of coagulation and fibrinolysis mediated through the binding of thrombin to thrombomodulin (Figure 3B). Additionally, platelet-neutrophil interactions are important in the formation of Neutrophil Extracellular Traps (NETs) and the process of NETosis which can induce a process referred to as immunothrombosis (Figure 4) (44, 45).
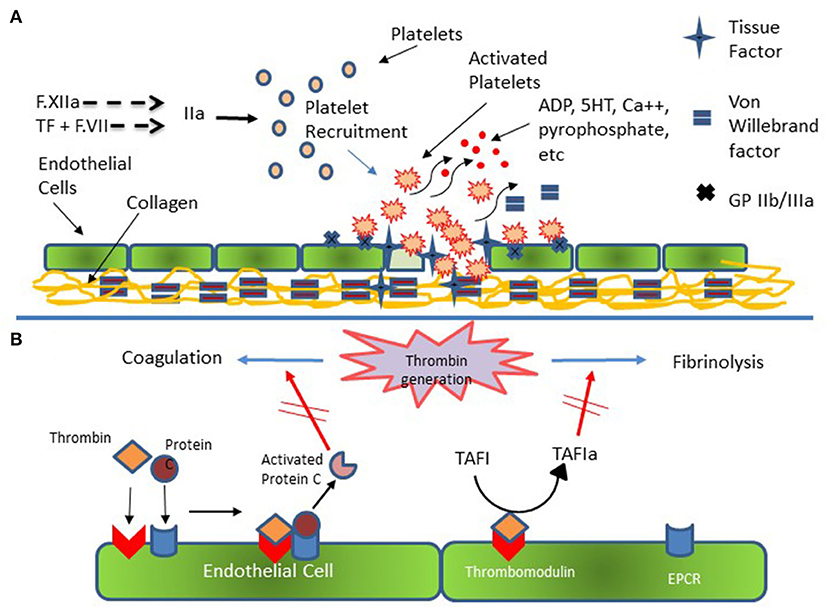
Figure 3. Platelet and Endothelial Cell Interactions in Hemostasis. (A) Injured endothelial cells elaborate tissue factor (TF) with consequent activation of the coagulation cascade and generation of thrombin (F.IIa). Thrombin activates platelets which subsequently adhere to injured endothelial cells and to subendothelial collagen via specific receptors on endothelial and platelet membrane and via von Willebrand [vWf]. Additional platelets are recruited to the area of injury with subsequent formation of platelet plug. (B) Thrombin generation results in activation of coagulation and fibrinolysis. Thrombin bound to endothelial cell surface thrombomodulin catalyzes the conversion of Protein C bound to endothelial cell protein C receptor (EPCR). Activated Protein C then down regulates further thrombin formation by inactivating activated F.VIII and activated F.V. Additionally, Thrombomodulin bound thrombin catalyzes Thrombin Activatable Fibrinolysis Inhibitor (TAFI) to its active form (TAFIa) which damps the activation of fibrinolysis thereby promoting clot formation and stability.
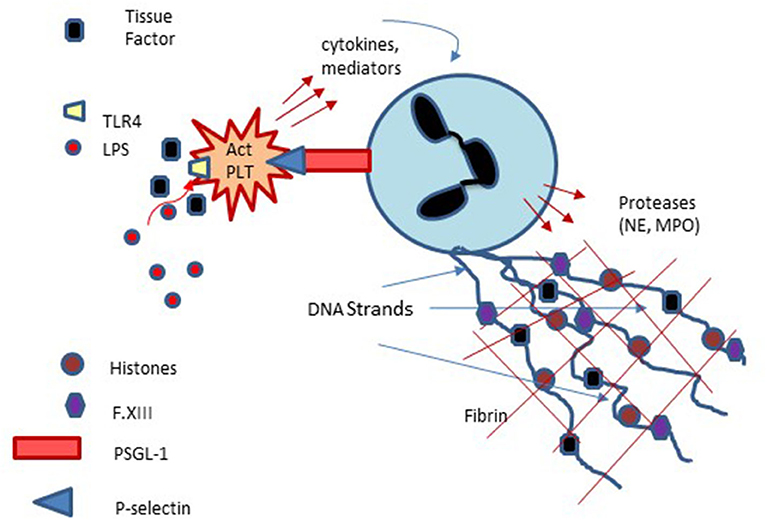
Figure 4. Platelet involvement in NETosis and Immunothrombosis: Platelets interact with neutrophils via platelet membrane expressed P-selectin and P-selectin glycoprotein ligand-1 (PSGL-1) expressed on neutrophils with subsequent enhancement of neutrophils, extrusion of neutrophil DNA and formation of Neutrophil Extracellular Traps (NETs) which can serve as a nidus of thrombus formation. Platelet activation via lipopolysaccharide (LPS) binding to platelet Toll-like receptors may also participate in NET formation and NETosis.
Developmental Hemostasis
Developmental hemostasis describes the physiologic changes that occur with an increase in age. These changes are particularly relevant in the pediatric population from birth through infancy when plasma levels of several important procoagulant and anticoagulant factors rapidly increase or decrease to normal adult levels. Because of the dynamic development of the hemostatic system, an understanding of age-specific physiology and quantification of normal reference ranges for coagulation parameters is of particular importance in pediatric medicine.
Early study of developmental hemostasis included the establishment of reference ranges for basic global measures of coagulation in infants using cord blood (46, 47). Andrew et al. were the first to define the field of developmental hemostasis in a series of articles describing reference values for coagulation parameters in (1) healthy term infants from birth to 6 months of age, (2) healthy preterm infants from birth to 6 months of age, and (3) healthy children and adolescents between 1 and 16 years of age (Table 1) (48–50). This work defined normal values of global measures of coagulation, individual factors that facilitate clot formation, and inhibitors of coagulation (Table 2).
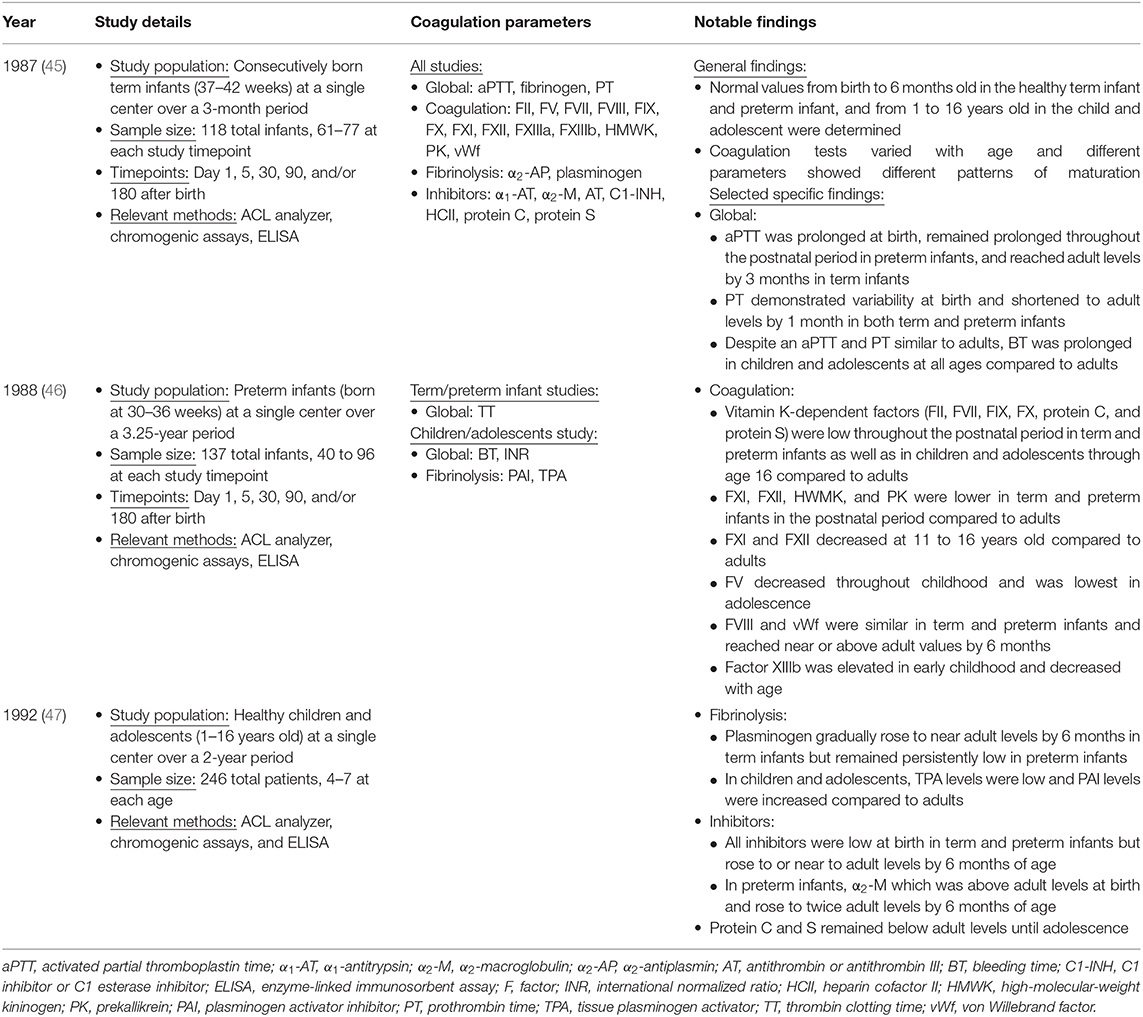
Table 1. Study details of the Andrew et al. series describing the developing hemostatic system during infancy, childhood, and adolescence (45–47).
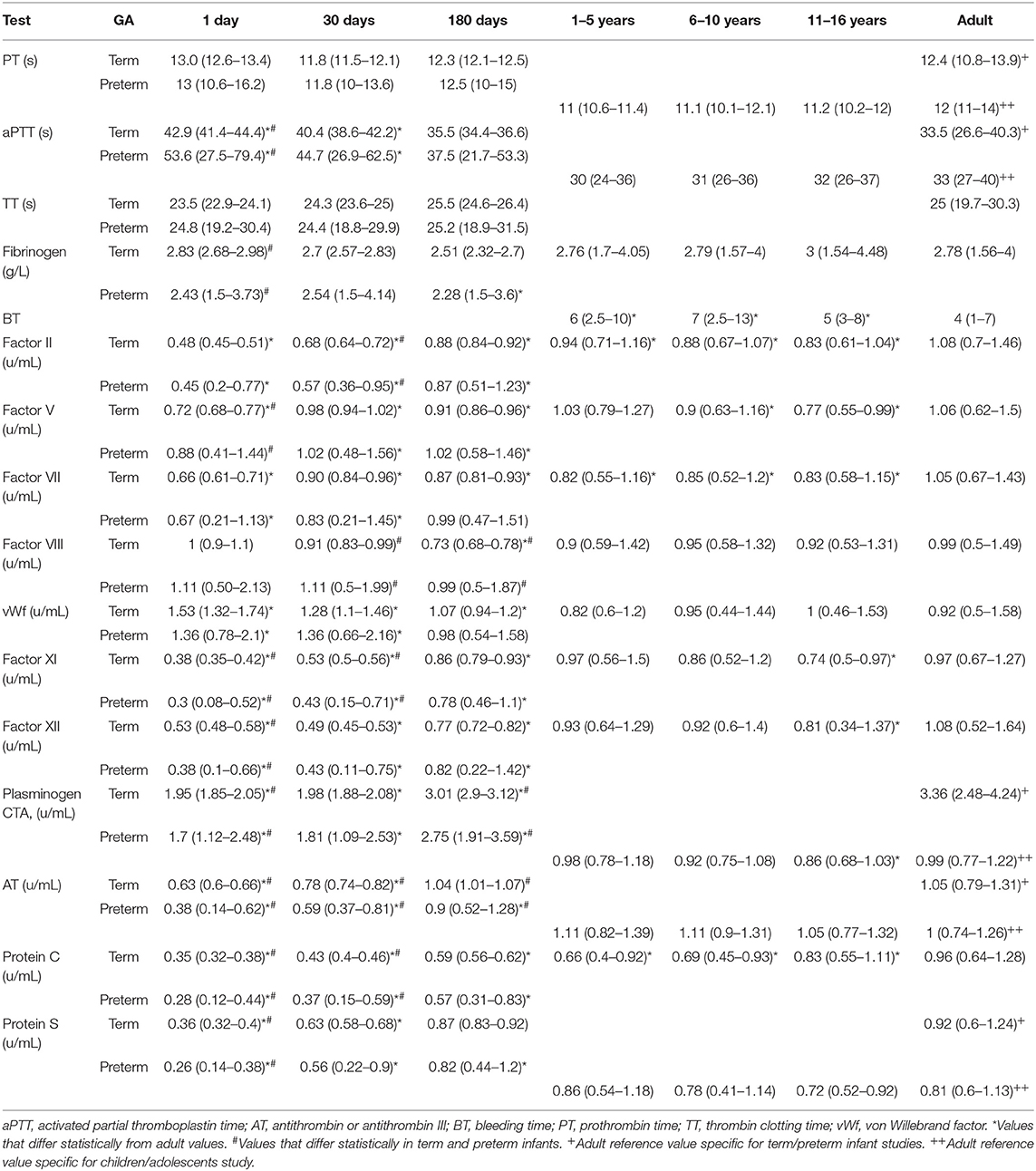
Table 2. Mean values and 95% confidence intervals of key hemostasis measures in term infants, preterm infants, children, and adolescents compared to normal adult values based on the Andrew et al. series (46–48).
Overall, Andrew et al. found that coagulation tests varied with age and different parameters showed different patterns of maturation (Tables 1, 2) (48–50). At birth, plasma levels of many important coagulation factors were found to be around half of that found in adults. Full maturation to adult levels of these factors varied between a few months to 16 years of age (48–50). While there were continued increases in some factors through later childhood, most of these factors reached near normal adult levels by 6 months of age. Specifically, in both term and preterm infants the mean values for the vitamin K-dependent factors, contact factors, and selected inhibitors including antithrombin (AT), protein C and protein S were well-below adult values. In contrast, fibrinogen, factors V, VIII, and XIII, vWf and the inhibitors α1-antitrypsin, α2-antiplasmin, α2-macroglobulin, and C1 esterase inhibitor were noted to be at normal or somewhat supra-normal adult levels at birth. Globally, both prothrombin time (PT) and activated partial thromboplastin time (aPTT) were prolonged in neonates. While there was wide variation in normal PT in infancy, mean PT normalized to adult levels by 1 month of age whereas aPTT normalized by 6 months of age (48, 49). Differences between preterm and term infants tend to be present but relatively small (48, 49). By 6 months of age both preterm and full term infants exhibit equivalent levels of all but four components of the coagulation system, namely factor VIII (mean 0.99 in preterm compared to 0.73 in term), plasminogen (mean 2.75 in preterm compared to 3.01 in term), antithrombin (mean 0.9 in preterm compared to 1.04 in term), and heparin cofactor II (mean 0.89 in preterm compared to 1.2 in term) (49).
Subsequent studies went on to confirm the highly selective pattern of maturation of the coagulation system described by Andrew et al. using different analyzer and reagent combinations (Table 3) (51–55). Compared to Andrew et al. these studies report the same trends by age and value relative to adult values but establish different reference intervals due to use of different equipment and reagents instead of the manual methods employed by Andrew (51–55). In addition to updated analysis techniques, several studies have expanded the panel hemostatic markers (Table 3) (51–53). Flanders et al. described differences by sex in protein S levels with females having higher than adult values at all ages and males having higher than adult values only in the 16–17 year old age group (101–103% in females 7–17 years old vs. 98% in female adults; 116% in males 16–17 years old vs. 108% in male adults) (51).

Monagle et al. described elevated D-dimer, reduced TFPI, and reduced endogenous thrombin potential in healthy neonates and children compared to established adult values (52). Appel et al. described variation in development of vWf and factor VIII across blood groups, specifically (1) a higher median vWf antigen in non-O compared to O groups at 1–5 years old (97 vs. 71%) and >19 years old (118 vs. 99%), (2) a higher median ristocetin cofactor activity in non-O compared to O groups at 1–5 years old (82 vs. 66%) and >19 years old (106 vs. 82%), and (3) a higher median factor VIII in non-O compared to O groups at 1–5 years old (121 vs. 100%) and >19 years old (131 vs. 116%) (53). Attard et al. combined the results of several cohorts of infants at different ages and found maturation patterns that mirrored prior studies along with a high level of interindividual variability in protein levels (54). Finally, Toulon et al. looked across seven centers and found values that were not significantly different between centers when using the same analysis techniques and reagents (55). In contrast to other reports in which primarily activity of clotting proteins is measured, Attard tracks changes in antigenic levels of hemostatic factors over age (54). It is assumed that the relationship between antigen and activity is (relatively) fixed and that a decrease in antigen implies a commensurate decrease in activity with clinical decisions being made accordingly. However, one cannot rule out the possibility that changes in antigenic level or protein activity noted with age reflect an element of post-translational protein modification.
Taken together, these studies show that most coagulation parameters are highly dependent on age and undergo the greatest changes during the 1st year of life. These changes require the use of age-specific reference intervals in the clinical assessment of hemostasis and the diagnosis of pediatric bleeding and thrombotic disorders. Despite these age-dependent differences, the changing coagulation system throughout infancy, childhood, and adolescence should be viewed as physiologic, i.e., “balanced,” with decreased factor levels and prolonged PT and aPTT in infants compared to normal adolescents and adults not necessarily reflecting an increased risk for hemorrhage. In fact, the pediatric hemostatic system may favor a reduced risk of thrombosis without an increased risk of bleeding (52, 56). While generation of thrombin in infants has been shown to be less than that in adults, this is counterbalanced by the reduction in AT and the fact that that the major physiologic inhibitor of thrombin in infants, α2-macroglobulin, is less efficient than is AT. However, other studies have shown that thrombin generation (endogenous thrombin potential; ETP) is “normal” in infants if measured in the presence of added thrombomodulin and that thrombin generation in very pre-term newborns is not different than that in near term infants (57, 58). That infants exhibit a “balanced” hemostatic system is supported by clinical findings such as decreased thromboembolic disease following surgery or prolonged immobilization in children compared to adults with similar risk factors (52, 59, 60). That said, the molecular basis underlying age-related numerical changes in coagulation measures as well as the clinical manifestation of these changes is not fully understood. Animal studies have demonstrated structural differences in coagulation proteins in newborn compared to adult models but this work has yet to be translated to humans (61). Further translational study is necessary to elucidate the important connections between coagulation parameters and clinical thrombotic and bleeding risk.
Key Laboratory Tests of Hemostasis
Evaluation of abnormalities in pediatric hemostasis requires (1) knowledge of different hemostatic testing options, (2) an understanding of the strengths and weaknesses of each test, and (3) an awareness of utility in the pediatric population, particularly in the context of the developmental hemostasis consideration discussed above. The following are key tests of pediatric hemostasis with specific focus on platelet evaluation, clotting assays, and global measures. Logistics of testing as well as strengths and weaknesses are summarized in Table 4.
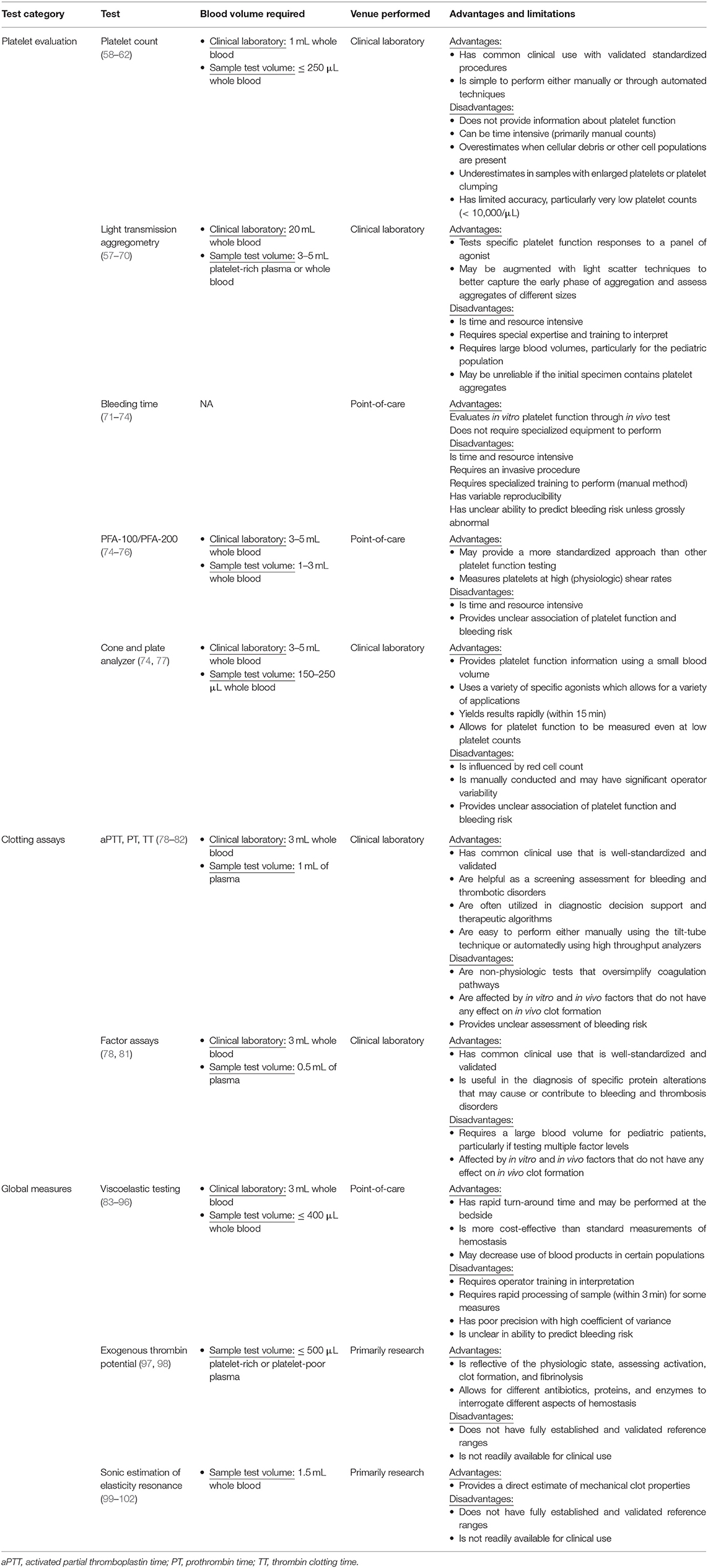
Table 4. Overview of selected testing in hemostasis.
Platelet Evaluation
While the important role platelets play in the initiation and regulation of clot formation has been recognized for over 100 years, the ability to measure how well-platelets function in these processes remains limited (59). Initial evaluations were confined to determining platelet number and morphology in peripheral blood and did not provide any specific information regarding platelet function (62). However, with the development of light transmission aggregometry (LTA), direct assessment of how platelets respond and aggregate to agonists ex vivo was possible (63). While admittedly non-physiologic, this methodology has remained the cornerstone of platelet function assessment for the past 60 years (discussed further below).
Platelet enumeration can be performed in a variety of ways and remains a first-line screening test in platelet evaluation. In manual methods, platelets are observed under light microscopy and counted by hand, a process which is time consuming and carries a coefficient of variation between 15 and 25% (64). Automated counts are more typically utilized as they are rapid, reproduceable and require less tech time. However, while they are highly accurate in most circumstances, cellular debris may result in overestimation or underestimation of platelet number in samples with platelet clumping or in patients with enlarged platelets (6, 66). Optical counting methods, in which platelets are recognized by light scattering or fluorescent dye, increase accuracy by better measurement of different sized platelets and better discrimination of platelets from cellular debris and other cell populations (66). In flow cytometric methods, platelets are incubated with fluorescent monoclonal antibodies directed against an antigen on the platelet membrane. While determination of platelet number may be different depending on the analyzer, all have been shown to be accurate with platelet counts as low as 10,000/μL (64, 67, 68).
Despite these advances, all platelet counting methods may be problematic in severely thrombocytopenic patients, an inaccuracy that may have substantial clinical impact around bleeding assessment and transfusion. A common cause of artifactual underestimation of platelet count occurs when patient plasm contains EDTA reactive antibodies causing in vitro platelet clumping. Most clinical laboratories will identify the presence of platelet clumps or cellular debris and not report an actual platelet number due to the possibility of a spurious result. The clinician must be aware of this possibility when an unexpected high or low platelet count is reported.
Assessment of platelet function through measurement of in vitro platelet aggregation, developed in the 1960's, significantly improved the ability to identify alterations in platelet function (63). In this technique, referred to as Light Transmission Aggregation (LTA), platelet-rich plasma is stirred in a cuvette between a light source and a measuring photocell. Agonist-induced platelet aggregation results in increased transmission of light through plasma over time. Multiple agonists are commonly employed and trigger a highly characterized response for each agonist in normal platelets. Deviation from that agonist-specific response enables diagnosis of platelet disorders based on the profile of aggregation (69). In addition to aggregation, shape change, adenosine triphosphate secretion, and (with special equipment) an increase in cytoplasmic calcium ions can also be measured. While much information can be derived from this classical method, turbidimetric platelet aggregometry has several important barriers that limit its practical use. First, it is labor intensive and requires a significant degree of technical expertise to carry out (70, 71). Second, this method is restricted in its ability to assess small aggregates and therefore may have limited sensitivity to both preexisting aggregates or the early phase of aggregation (72). Third, platelet adhesion and change in surface expression of cytoadhesion receptors are also not assessed (70), and fourth, it requires large volume blood sample and (generally) a platelet count over 100,000–200,000/υL limiting its use in small infants and in those with severe thrombocytopenia.
Whole blood aggregometry is another type of LTA and variation of turbidimetric aggregometry. In this technique, whole blood is stirred between two platinum wire electrodes set at a fixed distance (73). The electrode becomes covered with platelets and further adhesion of platelet aggregates is induced after the addition of agonists all of which change the impedance with time (73). Whole blood aggregometry may be superior to turbidimetric aggregometry when monitoring anti-platelet therapy, but is similarly insensitive to smaller platelet aggregates (74). Light scattering methods analyze particle size by flow cytometry and increases the accuracy for counting platelets and for assessing aggregates of all sizes during the early aggregation phase (103). Light scattering may be combined with aggregometry to monitor the continued formation of platelet microaggregates (73). This combined technique accurately assesses primary and secondary aggregation responses and may be of specific use in platelet hyperreactivity disease states (104).
Bleeding time (BT) was the first, and remains one of the only, test to evaluate in vivo platelet function. Via the Duke, Ivy, or Mielke method, a “standardized” cut is created and the time until bleeding flow arrests is assessed (75, 76). For the pediatric population, there are commercially available devices based on the Mielke method with age-appropriate templates that take into account the thinner epidermis and size of newborns and infants. While still occasionally used as first line screening for severe hemostatic defects, the perioperative use of the BT has largely disappeared because it, along with many other platelet function tests, does not accurately predict the risk related to bleeding for surgical procedures unless grossly abnormal (77, 105). This suggests that either the test is insensitive or mild platelet defects are not clinically important. Advantages of bleeding time include study of platelet function in situ and the ability to perform the test without complex equipment. Disadvantages include its manual nature with variable reproducibility, the invasiveness of the procedure, and that it is time consuming (77, 105).
In addition to bleeding time, there are number of other tests that have been developed to mimic the processes that occur in vivo following vessel injury. The hemostatometer, a commercially available clot signature analyzer, punches holes within a tube containing flowing anticoagulated blood. Under controlled conditions, the punched holes stimulate formation of the primary platelet plug and allow for the assessment of platelet function and of involvement of GPIb, GPIIb/IIIa, and vWf (19, 106). The thrombotic status analyzer pulls whole blood through a capillary tube and the resulting force induces platelet activation, formation of the platelet plug, and eventually capillary tube occlusion (107). The platelet function analyzer (PFA-100 or updated PFA-200) exposes citrated whole blood to high shear in a capillary tube with a center membrane coated with an agonist. As platelets are activated and aggregate, the device measures the drop in flow rate between the capillary tube and center membrane and ultimately measures tube occlusion or closing time (78, 105). While referred to as an in vitro bleeding time, its ability to identify platelet disorders is poor and currently this test may serve as a test for screening and monitoring of von Willebrand disease in the pediatric population (78, 79). The cone and plate analyzer exposes a whole blood sample to a plate under arterial flow conditions and assesses platelet adhesion and aggregation on the surface of the plate (80, 105). For the pediatric population, the main advantage of this system is that it requires only a small volume of blood to perform (150–250 μL) (80). Platelet mapping can be performed using thromboelastography (TEG) though its clinical utility and correlation with other platelet function assays has not been determined (81). In general, none of these tests have been shown to have clinical utility in the routine assessment of platelet function.
Finally, platelet function may be assessed through the identification of markers of platelet activation and reactivity in plasma and/or on circulating platelets. This may be evaluated through measurement of platelet release products such as platelet factor 4 and β-thromboglobulin in platelet-poor plasma. Whole blood flow cytometry can be used to measure activated platelets, platelet hypo-reactivity or hyper-reactivity, platelet-leukocyte aggregates, platelet microparticles, and platelet turnover which is inferred through the measurement of reticulated (i.e., young) platelets (82). Nucleic acid-specific dyes can be used to measure young platelets that contain residual RNA and aid in the assessment of platelet turnover or thrombopoiesis (83).
Clotting Assays
The most commonly utilized tests measuring the effectiveness of in vivo clotting measure the conversion of fibrinogen to fibrin and utilize the time to formation of a fibrin clot as determined by one of several methodologies each of which have a different degree of sensitivity. The most common of these tests are PT, aPTT, and thrombin time (TT) (84). A few important aspects of nomenclature for these tests should be noted. First, in contrast to the older PTT, aPTT is an updated, modified, test that includes the addition of an activator to accelerate clotting time resulting in a narrower reference range and increased sensitivity (84, 85). Second, because it is calculated from PT, International Normalized Ratio (INR) is frequently employed as a surrogate PT. However, INR has only been validated as a measure of intensity of vitamin K antagonist induced anti-coagulation and has not been validated as a measure of bleeding risk (86). While the Activated Clotting Time (ACT) is generally performed to monitor anticoagulation during cardiopulmonary bypass surgery and during ExtraCorporeal Membrane Oxygenation (ECMO), outside of these clinical settings it has not gained widespread use.
PT, aPTT, and TT are reported as time to clot formation while fibrinogen is reported as mg/dL determined by a lab-specific calibration curve (84, 87). In regards to assays utilized for the quantification of protein levels of specific clotting assays, time to clot formation is measured and then reported as a percent in relation to time to clot formation using pooled normal adult plasma. By convention, the amount of a specific clotting factor contained in normal plasma is defined as 100%, equivalent to 1 unit/mL in normal plasma. From this assumption, time to clot formation is converted to a U/mL concentration. All of these assays where the end point is the time to a fibrin clot are referred to as “functional assays.” An advantage of these assays is that they are relatively simple and easy to standardize, but they can be affected by in vitro and in vivo factors that do not have any effect on in vivo clot formation (84, 87). One such circumstance is common in neonates, particularly premature neonates, and in infants with cyanotic heart disease who manifest a reactive polycythemia with hematocrit >55%. Under this condition, blood collected into standard coagulation testing vacuum tubes (for example, “blue top tubes”) may produce PT or aPTT with values that are spuriously prolonged as these samples contain less plasma and are therefore over-anticoagulated (88). This can be remedied by collecting blood in tubes that contain a reduced amount of anticoagulant.
Global Measures of Hemostasis
There are several important global measures of hemostasis that are gaining use and clinical application. This includes viscoelastic testing, a method of assessing clot formation dynamically in whole blood. Viscoelastic testing is designed to imitate sluggish venous blood flow and triggers clot formation to derive measurements of hemostasis (89). Developed in 1948, but not adopted in the clinical setting until the 1980's, viscoelastic testing now has more broad use primarily in trauma, surgery, and cardiopulmonary bypass (89–91).
Currently available methods for viscoelastic testing include thromboelastography (TEG) and the related rapid thromboelastography (r-TEG) as well as rotational thromboelastography (ROTEM) (89). In both, a small sample of patient blood is placed into a cup and a pin that serves as a sensor is inserted. In TEG and r-TEG, the cup is oscillated with a specified rotational cycle to precipitate clot formation. With clot formation, the torque of the rotating cup is transmitted to the pin and the degree of pin rotation is converted to an electrical signal and recorded. The device tracing reflects this change in electrical signal reflecting change in clot strength resulting from clot formation or fibrinolysis. ROTEM uses similar methods but the sensor pin instead rotates within the cup (91).
Viscoelastic testing measures clot initiation or time to initial clot formation, kinetics or growth of the clot, clot strength, and clot lysis. All components are identified by specific measurements on a tracing produced by the device over time. Clot initiation or clot time provides an assessment of early activity of the clotting cascade before clot amplification and the burst of thrombin production. Clot kinetics or clot formation time assesses clot potentiation by activation of platelets and thrombin-mediated cleavage of soluble fibrinogen. Clot strength describes maximal clot strength achieved via GPIIb/IIIa-mediated platelet-fibrin interactions. Finally, clot lysis provides an assessment of activation of the fibrinolytic system (89, 92, 93). In addition to standard viscoelastic testing, specific elements of the clotting cascade can be interrogated by the addition of specific clotting activators or inhibitors (94).
The diagnostic and prognostic role of viscoelastic testing is being studied in a range of clinical areas (89, 92, 93). The majority of these studies have been in adult perioperative cardiac surgery patients as well as other surgical patients at risk for hemorrhage (89). A recent Cochrane review of 17 studies in a largely cardiac surgery population, only 2 of which included pediatric patients, addressed the utility of thromboelastic testing in bleeding patients (95). They found that while TEG/ROTEM-guided transfusion strategies might decrease transfusion requirements and support a tendency toward improved mortality outcomes, conclusive recommendations could not be made because of limitations in study design and power (88). Subsequent studies in pediactric patients (largely neonates) has continued to investigate the utility of viscoelastic testing as a modality to better manage patients with acquired coagulopathies or requiring anticoagulation (96, 97). Reference range values have been established in term, preterm and very low birth weight infants (98, 99). In the trauma population, TEG-guided resuscitation is growing in use, though current studies are insufficient to determine if thromboelastography-based transfusion is better than existing transfusion practices (100, 101). Other populations of potential interest include liver transplantation, sepsis, obstetric hemorrhage, perioperative thromboembolism, and management of anticoagulation on mechanical circulator support, though evidence from large trials required to formulate consensus recommendations in these areas is lacking (89). This work is even more limited in pediatric specific populations, though studies have suggested a benefit in similar disease populations as to adults (102, 108). That said, viscoelastic testing may be particularly advantageous in pediatric populations since only a small volume of blood is required for sample analysis. Further, in limited use TEG has been shown to be reproducible in some infant populations, and cost-effectiveness studies seem to clearly favor viscoelastic testing compared to standard laboratory run measures of hemostasis (102, 109–113). Taken together, viscoelastic testing remains a novel method that may be advantageous in certain populations including children.
Endogenous thrombin generation is a measure of the global efficiency of coagulation as thrombin sits at the key intersection of endothelial cell, platelet, and coagulation activation, clot formation, and fibrinolysis. Thrombin generated can be quantified in platelet-rich or platelet-poor plasma using the calibrated automated thrombogram (CAT) method. Specifically, CAT monitors the cleavage of a fluorogenic substrate that is compared to the known thrombin activity in a non-clotting plasma sample (100). The CAT system is open, allowing for different antibiotics, proteins, and enzymes to be introduced to evaluate specific aspects of hemostatic process (114). Validation of reference ranges are just now being established with preliminary studies suggesting children and adolescents have lower exogenous thrombin potential compared to adults (115); although as previously stated, low thrombin generation potential noted in infants may reflect in part how the assay is performed (58). While these assays have great clinical potential, standardization and validation need refining as does a more rapid technical process.
Another potentially promising method is sonic estimation of elasticity via resonance. This technology applies acoustic radiation force to induce shear wave resonance within a rigid test chamber. The characteristics of this resonance are compared to numerical or analytical models to quantify the sample filling the chamber (116, 117). The technique may be performed rapidly and shows promise as a point-of-care test and may rival TEG/ROTEM in assessment of clot stiffness and fibrinogen contribution (117–119).
Special Considerations
There are a number of additional considerations in the assessment of hemostasis in the pediatric population. First, there are numerous technical challenges. This includes practical difficulty drawing sufficient blood volume from small infants and children given their small vessels for certain tests (e.g., LTA) and the overall smaller total blood volume infants and children which limits the quantity of blood that can safely be obtained to perform comprehensive diagnostic testing. Consequently, tests that use small volumes may be preferred and careful consideration is necessary to parsimoniously select appropriate tests given the clinical situation. While central lines help with the technical difficulties of obtaining adequate testing volumes, samples can be contaminated by anticoagulation or other medications running through the line itself. In fact, samples collected through central lines may require 10 mL of blood be withdrawn prior to sample collection for the result to be considered valid. Techniques to mitigate this waste include using the initial sample collection for other labs or reinfusing blood that would otherwise be discarded.
There are also a number of infant and pediatric conditions and diseases that require special testing considerations. Pediatric trauma patients, particularly infants, demonstrate significant endothelial injury, present with acute traumatic coagulopathy and/or coagulopathy related to iatrogenic causes, and, while they may be at less risk of bleeding than their adult counterparts, they are also at risk of thrombosis during recovery from their injury (120–123). While there have been several pediatric-focused studies around damage control resuscitation and thromboelastographic-guided transfusion, there are still gaps in how these tests account for the developmental changes in hemostasis noted in infants and children (99, 116). Pediatric patients with liver dysfunction may have significant hemostatic abnormalities from impaired synthetic function including coagulation factor defects, thrombocytopenia and platelet dysfunction, alterations in the fibrinolytic system, and alterations in endogenous inhibitors of coagulation. While clotting times may be significantly elevated in these patients, these abnormal results often do not correlate with bleeding risk (124, 125). Indeed, assessment of hemostatic balance in patients with liver disease generally demonstrates a neutral or mildly pro-thrombotic hemostatic system (126, 127). The concept of “hemostatic balance” has also been conceptually addressed as alternatively being a condition of a loss in “hemostatic reserve” by some hematologists, a concept that carries some merit (128). In the oncologic population, patients may be at risk of hyperviscosity, thrombosis, and cytopenias that may increase bleeding risk depending on the type of cancer and therapies. The hemostatic system is frequently monitored in these patients, but transfusion and other therapeutic decisions necessitate an understanding of the limitations and normal variations of these standard hematologic measurements (129).
Patients on extracorporeal life support present with unique hemostatic risks as both the extracorporeal circuit and the underlying disease process induce important dysregulation of the hemostatic system that may favor either hemorrhage or thrombosis (119, 120). Hematologic monitoring of these patients may be one of the most active parts of their management and typically includes frequent evaluation for bleeding, consumptive coagulopathy, hemolysis, and adequacy of anticoagulation. In addition to aPTT to assess anticoagulant effect of unfractionated heparin (UFH), anti-Xa assay (assessing effect of UFH or low molecular weight heparin [LMWH] or some of the new direct acting oral anticoagulants [DOACs]) or the Activated Clotting Time (ACT) are utilized to assess bleeding/thrombosis risks (130, 131) Increasingly, the anti-Xa assay is becoming the preferred test to monitor UFH as well as anti-Xa inhibitors due to its insensitivity to low levels of AT. However, therapeutic range for anti-Xa has been extrapolated from those for UFH by matching aPTT result to anti-Xa level in plasma samples to which heparin was added ex vivo. This practice does not account for the primarily anti-IIa action of UFH potentially introducing a degree of uncertainty regarding the extrapolated therapeutic threshold (132, 133). ACT is the preferred monitoring test for UFH given during cardiopulmonary bypass surgery with “therapeutic” (i.e., desired) values being surgeon and device specific (134, 135).
Finally, trauma patients often present in the field or to community hospitals and must be transported to the regional trauma center for care. The use of point of care (POC) tests of hemostasis have only recently been investigated in this patient population (136, 137). While some studies suggest the feasibility of these devices, there are as of yet insufficient data to adequately assess their use.
Conclusions
Understanding of hemostatic dysfunction remains a unique challenge in critically ill infants and children. It requires knowledge of physiologic hemostatic pathways, comprehension of the development of the hemostatic system by age, an understanding of logistics, advantages, and limitations in hematologic testing, and an awareness of the specifics of application to pediatric populations. Taken together, this knowledge will enable the practitioner to more accurately assess hemostatic dysfunction and determine bleeding and thrombosis risk resulting in more successful diagnosis and management of this vulnerable population.
Author Contributions
AN drafted all tables included. RP created all figures included in manuscript. Both authors participated in reviewing appropriate published literature, writing all drafts, and the final submitted draft manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Furie B, Furie BC. Mechanisms of thrombus formation. N Engl J Med. (2008) 359:938–49. 359:938–49. doi: 10.1056/NEJMra0801082
2. Kazmi RS, Boyce S, Lwaleed BA. Homeostasis of hemostasis: the role of endothelium. Semin Thromb Hemos. (2015) 41:549–55. 41:549–55. doi: 10.1055/s-0035-1556586
3. Hinsbergh VWM. Endothelium – role in regulation of coagulation and inflammation. Semin Immunopatho. (2012) 34:93–106. 34:93–106. doi: 10.1007/s00281-011-0285-5
4. Broos K, Feys HB, De Meyer SF, Vanhoorelbeke K, Deckmyn H. Platelets at work in primary hemostasis. Blood Rev. (2011) 25:155–67. 25:155–67. doi: 10.1016/j.blre.2011.03.002
5. Swieringa F, Spronk HMH, Heemskerk JWM, van der Meijden PEJ. Integrating platelet and coagulation activation in fibrin clot formation. Res Pract Thromb Haemost. (2018) 2:450–60. doi: 10.1002/rth2.12107
6. Shen J, Sampietro S, Wu J, Tang J, Gupta S, Matzko CN, et al. Coordination of platelet agonist signaling during the hemostatic response in vivo. Blood Adv. (2017) 1:2767–75. 1:2767–75. doi: 10.1182/bloodadvances.2017009498
7. Clemetson KJ. Platelet GPIb-V-IX complex. Thromb Haemost. (1997) 78:266–70. doi: 10.1055/s-0038-1657537
8. Savage B, Saldívar E, Ruggeri ZM. Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von willebrand factor. Cell. (1996) 84:289–97. 84:289–97. doi: 10.1016/S0092-8674(00)80983-6
9. Savage B, Shattil SJ, Ruggeri ZM. Modulation of platelet function through adhesion receptors. A dual role for glycoprotein IIb-IIIa (integrin alpha IIb beta 3) mediated by fibrinogen and glycoprotein Ib-von willebrand factor. J Biol Chem. (1992) 267:11300–6.
10. Harrison P, Savidge GF, Cramer EM. The origin and physiological relevance of alpha-granule adhesive proteins. Review Br J Haematol. (1990) 74:125–30. 74:125–30. doi: 10.1111/j.1365-2141.1990.tb02554.x
11. Kroll MH, Schafer AI. Biochemical mechanisms of platelet activation. Blood. (1989) 74:1181–95. 74:1181–95. doi: 10.1182/blood.V74.4.1181.1181
12. Monroe DM, Hoffman M, Roberts HR. Platelets and thrombin generation. Arterioscler Thromb Vasc Biol. (2002) 22:1381–9. 22:1381–9. doi: 10.1161/01.ATV.0000031340.68494.34
13. Mann KG, Brummel-Ziedins K, Orfeo T, Butenas S. Models of blood coagulation. Blood Cells Mol Dis. (2006) 36:108–17. 36:108–17. doi: 10.1016/j.bcmd.2005.12.034
14. Rapaport SI, Rao LV. The tissue factor pathway: how it has become a “prima ballerina”. Thromb Haemost. (1995) 74:7–17. 74:7–17. doi: 10.1055/s-0038-1642646
15. Levi M, van der Poll T, Schultz M. Systemic versus localized coagulation activation contributing to organ failure in critically ill patients. Semin Immunopathol. (2012) 34:167–79. 34:167–79. doi: 10.1007/s00281-011-0283-7
16. Panes O, Matus V, Sáez CG, Quiroga T, Pereira J, Mezzano D. Human platelets synthesize and express functional tissue factor. Blood. (2007) 109:5242–50. doi: 10.1182/blood-2006-06-030619
17. Konigsberg W, Kirchhofer D, Riederer MA, Nemerson Y. The TF:VIIa complex: clinical significance, structure-function relationships and its role in signaling and metastasis. Thromb Haemost. (2001) 86:757–71. 86:757–71. doi: 10.1055/s-0037-1616129
18. Morrison SA, Jesty J. Tissue factor-dependent activation of tritium-labeled factor IX and factor X in human plasma. Blood. (1984) 63:1338–47. doi: 10.1182/blood.V63.6.1338.1338
19. Butenas S, van 't Veer C, Mann KG. Evaluation of the initiation phase of blood coagulation using ultrasensitive assays for serine proteases. J Biol Chem. (1997) 272:21527–33. 272:21527–33. doi: 10.1074/jbc.272.34.21527
20. Von dem Borne PA, Meijers JC, Bouma BN. Feedback activation of factor XI by thrombin in plasma results in additional formation of thrombin that protects fibrin clots from fibrinolysis. Blood. (1995) 86:3035–42. 86:3035–42. doi: 10.1182/blood.V86.8.3035.3035
21. O'Donnell JS, O'Sullivan JM, Preston RJS. Advances in understanding the molecular mechanisms that maintain normal haemostasis. Br J Haematol. (2019) 186:24–36. doi: 10.1111/bjh.15872
22. Mann KG, Krishnaswamy S, Lawson JH. Surface-dependent hemostasis. Semin Hematol. (1992) 29:213–26.
23. Suzuki K, Dahlbäck B, Stenflo J. Thrombin-catalyzed activation of human coagulation factor V. J Biol Chem. (1982) 257:6556–64.
24. Tracy PB, Eide LL, Mann KG. Human prothrombinase complex assembly and function on isolated peripheral blood cell populations. J Biol Chem. (1985) 260:2119–24.
25. Mosesson MW. The roles of fibrinogen and fibrin in hemostasis and thrombosis. Semin Hematol. (1992) 29:177–88.
26. Baugh RJ, Broze GJ, Krishnaswamy S. Regulation of extrinsic pathway factor Xa formation by tissue factor pathway inhibitor. J Biol Chem. (1998) 273:4378–86. doi: 10.1074/jbc.273.8.4378
27. Cicardi M, Zingale L, Zanichelli A, Pappalardo E, Cicardi B. C1 inhibitor: molecular and clinical aspects. Springer Semin Immunopathol. (2005) 27:286–98. doi: 10.1007/s00281-005-0001-4
28. Perry DJ. Antithrombin and its inherited deficiencies. Blood Rev. (1994) 8:37–55. doi: 10.1016/0268-960X(94)90006-X
29. Marcum JA, McKenney JB, Rosenberg RD. Acceleration of thrombin-antithrombin complex formation in rat hindquarters via heparinlike molecules bound to the endothelium. J Clin Invest. (1984) 74:341–50. 74:341–50. doi: 10.1172/JCI111429
30. Mammen EF. Antithrombin: its physiological importance and role in DIC. Semin Thromb Hemost. (1998) 24:19–25. 24:19–25. doi: 10.1055/s-2007-995819
31. Esmon CT. The roles of protein C and thrombomodulin in the regulation of blood coagulation. J Biol Chem. (1989) 264:4743–6.
32. Esmon CT. The protein C pathway. Chest. (2003) 124(3 Suppl):26S−32. doi: 10.1378/chest.124.3_suppl.26S
33. Fulcher CA, Gardiner JE, Griffin JH, Zimmerman TS. Proteolytic inactivation of human factor VIII procoagulant protein by activated human protein C and its analogy with factor V. Blood. (1984) 63:486–9. 63:486–9. doi: 10.1182/blood.V63.2.486.486
34. O'Brien LM, Mastri M, Fay PJ. Regulation of factor VIIIa by human activated protein C and protein S: inactivation of cofactor in the intrinsic factor Xase. Blood. (2000) 95:1714–20. 95:1714–20. doi: 10.1182/blood.V95.5.1714.005k40_1714_1720
35. Smith WL. The eicosanoids and their biochemical mechanisms of action. Biochem J. (1989) 259:315–24. 259:315–24. doi: 10.1042/bj2590315
36. Moncada S, Higgs A. The L-arginine-nitric oxide pathway. N Engl J Med. (1993) 329:2002–12. 329:2002–12. doi: 10.1056/NEJM199312303292706
37. Cozzi MR, Guglielmini G, Battiston M, Momi S, Lombardi E, Miller EC, et al. Visualization of nitric oxide production by individual platelets during adhesion in flowing blood. Blood. (2015) 125:697–705. 125:697–705. doi: 10.1182/blood-2014-06-579474
38. Hoylaerts M, Rijken DC, Lijnen HR, Collen D. Kinetics of the activation of plasminogen by human tissue plasminogen activator. Role of fibrin. J Biol Chem. (1982) 257:2912–9. doi: 10.1055/s-0038-1652446
39. Chapin JC, Hajjar KA. Fibrinolysis and the control of blood coagulation. Blood Rev. (2015) 29:17–24. 29:17–24. doi: 10.1016/j.blre.2014.09.003
40. Samis JA, Ramsey GD, Walker JB, Nesheim ME, Giles AR. Proteolytic processing of human coagulation factor IX by plasmin. Blood. (2000) 95:943–51. doi: 10.1182/blood.V95.3.943.003k34_943_951
41. Hur WS, Mazinani N, Lu XD, Britton HM, Byrnes JR, Wolberg AS, et al. Coagulation factor XIIIa is inactivated by plasmin. Blood. (2015) 126:2329–37. doi: 10.1182/blood-2015-07-650713
42. Kolev K, Machovich R. Molecular and cellular modulation of fibrinolysis. Thromb Haemost. (2003) 89:610–21. 89:610–21. doi: 10.1055/s-0037-1613567
43. Van der Poll T, Parker RI. Platelet activation and endothelial cell dysfunction. Crit Care Clin. (2020) 36:233–53. 36:233–53. doi: 10.1016/j.ccc.2019.11.002
44. Kimball AS, Obi AT, Diaz JA, Henke PK. The emerging role of NETs in venous thrombosis and immunothrombosis. Front Immunol. (2016) 7:236. 7:236. doi: 10.3389/fimmu.2016.00236
45. Franchi T, Eaton S, De Coppi P, Giuliani S. The emerging role of immunothrombosis in paediatric conditions. Pediatr Res. (2019) 86:19–27. 86:19–27. doi: 10.1038/s41390-019-0343-6
46. Bleyer WA, Hakami N, Shepard TH. The development of hemostasis in the human fetus and newborn infant. J Pediatr. (1971) 79:838–53. 79:838–53. doi: 10.1016/S0022-3476(71)80405-5
47. Foley ME, Clayton JK, McNicol GP. Hemostatic mechanisms in maternal umbilical vein and umbilical artery blood at the time delivery. Br J Obstet Gynaecol. (1977) 84:81–7. 84:81–7. doi: 10.1111/j.1471-0528.1977.tb12532.x
48. Andrew M, Paes B, Milner R, Johnson M, Mitchell L, Tollefsen DM, et al. Development of the human coagulation system in the full-term infant. Blood. (1987) 70:165–72. doi: 10.1182/blood.V70.1.165.165
49. Andrew M, Paes B, Milner R, Johnson M, Mitchell L, Tollefsen DM, et al. Development of the human coagulation system in the healthy premature infant. Blood. (1998) 72:1651–7. 72:1651–7. doi: 10.1182/blood.V72.5.1651.1651
50. Andrew M, Vegh P, Johnston M, Bowker J, Frederick O, Mitchell L. Maturation of the hemostatic system during childhood. Blood. (1992) 80:1998–2005. doi: 10.1182/blood.V80.8.1998.1998
51. Flanders MM, Phansalkar R, Crist A, Robers WL, Rogers GM. Pediatric reference intervals for uncommon bleeding and thrombotic disorders. J Pediatr. (2006) 149:275–7. 149:275–7. doi: 10.1016/j.jpeds.2006.04.008
52. Monagle P, Barnes C, Ignjatovic V, Furmedge J, Newall F, Chan A, et al. Developmental haemostasis. Impact for clinical haemostasis laboratories. Thromb Haemost. (2006) 95:362–72. 95:362–72. doi: 10.1160/TH05-01-0047
53. Appel I, Grimminck B, Geerts J, Stigter R, Cnossen MH, Beishuizen A. Age dependency of coagulation parameters during childhood and puberty. J Thromb Haemost. (2012) 10:2254–63. 10:2254–63. doi: 10.1111/j.1538-7836.2012.04905.x
54. Attard C, van der Straaten T, Karlaftis V, Monagle P, Ignjatovic V. Developmental hemostasis: age-specific differences in the levels of hemostatic proteins. J Thromb Haemost. (2013) 11:1850–4. 11:1850–4. doi: 10.1111/jth.12372
55. Toulon P, Berruyer M, Brionne-François M, Grand F, Lasne D, Telion C, et al. Age dependency for coagulation parameters in paediatric populations. Results of a multicentre study aimed at defining the age-specific reference ranges. Thromb Haemost. (2016) 116:9–16. 116:9–16. doi: 10.1160/TH15-12-0964
56. Tripodi A, Raffaeli G, Scalambrino E, Padovan L, Clerici M, Chantarangkul V, et al. Procoagulant imbalance in preterm neonates detected by thrombin generation procedures. Thromb Res. (2020) 185:96–101. 185:96–101. doi: 10.1016/j.thromres.2019.11.013
57. Tripodi A, Ramenghi LA, Chantarangkul V, De Carli A, Clerici M, Groppo M, et al. Normal thrombin generation in neonates in spite of prolonged conventional coagulation tests. Haematologica. (2008) 93:1256–9. 93:1256–9. doi: 10.3324/haematol.12566
58. Neary E, McCallion N, Kevane B, Cotter M, Egan K, Regan I, et al. Coagulation indices in very preterm infants from cord blood and postnatal samples. J Thromb Haemost. (2015) 13:2021–30. 13:2021–30. doi: 10.1111/jth.13130
59. Monagle P, Chan A, Massicotte P, Chalmers E, Michelson AD, Chan A. Antithrombotic therapy in children: the seventh ACCP conference on antithrombotic and thrombolytic therapy. Chest. (2004) 126(3 Suppl):645S−87S. Suppl):645S−87S. doi: 10.1378/chest.126.3_suppl.645S
60. Andrew M. Developmental hemostasis: relevance to thromboembolic complications in pediatric patients. Thromb Haemost. (1995) 74:415–25. 74:415–25. doi: 10.1055/s-0038-1642714
61. Monagle P, Hagstrom JN. Developmental hemostasis. In: Abman PF, editor. Fetal and Neonatal Physiology. 3rd edn. St. Louis: Elsevier (2003). p. 1435–46. doi: 10.1016/B978-0-7216-9654-6.50151-X
62. Bizzozero J. On a new blood particle and its role in thrombosis and blood coagulation. Virchows Arch B Cell Pathol Incl Mol Pathol. (1982) 90:261. 90:261. doi: 10.1007/BF01931360
63. Born GV. Aggregation of blood platelets by adenosine diphosphate and its reversal. Nature. (1962) 194:927–9. 194:927–9. doi: 10.1038/194927b0
64. Harrison P, Horton A, Grant D, Briggs C, MacHin S. Immunoplatelet counting: a proposed new reference procedure. Br J Haematol. (2000) 108:228–35. doi: 10.1046/j.1365-2141.2000.01846.x
66. Kunz D. Possibilities and limitations of automated platelet counting procedures in the thrombocytopenic range. Semin Thromb Hemost. (2001) 27:229–35. doi: 10.1055/s-2001-15252
67. Davis B, Bigelow N. Indirect immunoplatelet counting by flow cytometry as a reference method for platelet count calibration. Lab Hematol. (1999) 5:15.
68. Segal HC, Briggs C, Kunka S, Casbard A, Harrison P, Machin SJ, et al. Accuracy of platelet counting haematology analysers in severe thrombocytopenia and potential impact on platelet transfusion. Br J Haematol. (2005) 128:520–5. 128:520–5. doi: 10.1111/j.1365-2141.2004.05352.x
69. Hayward CPM, Pai M, Liu Y, Moffat KA, Seecharan J, Webert KE, et al. Diagnostic utility of light transmission platelet aggregometry: results from a prospective study of individuals referred for bleeding disorder assessments. J Thromb Haemost. (2009) 7:676–84. 7:676–84. doi: 10.1111/j.1538-7836.2009.03273.x
70. Cattaneo M, Hayward CPM, Moffat KA, Pugliano MT, Liu Y, Michelson AD. Results of a worldwide survey on the assessment of platelet function by light transmission aggregometry: a report from the platelet physiology subcommittee of the SSC of the ISTH. J Thromb Haemost. (2009) 7:1029. 7:1029. doi: 10.1111/j.1538-7836.2009.03458.x
71. Quiroga T, Goycoolea M, Matus V, Zúñiga P, Martínez C, Garrido M, et al. Diagnosis of mild platelet function disorders. Reliability and usefulness of light transmission platelet aggregation and serotonin secretion assays. Br J Haematol. (2009) 147:729–36. 147:729–36. doi: 10.1111/j.1365-2141.2009.07890.x
72. Thompson NT, Scrutton MC, Wallis RB. Particle volume changes associated with light transmittance changes in the platelet aggregometer: dependence upon aggregating agent and effectiveness of stimulus. Thromb Res. (1986) 41:615–26. doi: 10.1016/0049-3848(86)90358-0
73. Cardinal DC, Flower RJ. The electronic aggregometer: a novel device for assessing platelet behavior in blood. J Pharmacol Methods. (1980) 3:135–58. 3:135–58. doi: 10.1016/0160-5402(80)90024-8
74. Nicholson NS, Panzer-Knodle SG, Haas NF, Taite BB, Szalony JA, Page JD, et al. Assessment of platelet function assays. Am Heart J. (1998) 135:S170–8. doi: 10.1016/S0002-8703(98)70245-5
75. Mielke CH, Kaneshiro MM, Maher IA, Weiner JM, Rapaport SI. The standardized normal Ivy bleeding time and its prolongation by aspirin. Blood. (1969) 34:204–15. doi: 10.1182/blood.V34.2.204.204
76. Harker LA, Slicther SJ. The bleeding time as a screening test for evaluation of platelet function. N Engl J Med. (1972) 287:155–9. 287:155–9. doi: 10.1056/NEJM197207272870401
77. Rodgers RP, Levin J. A critical reappraisal of the bleeding time. Semin Thromb Hemost. (1990) 16:1–20. 16:1–20. doi: 10.1055/s-2007-1002658
78. Kundu SK, Heilmann EJ, Sio R, Garcia, Davidson CRM, Ostgaard RA. Description of an in vitro platelet function analyzer–PFA-100. Semin Thromb Hemost. (1995) 21(Suppl. 2):106–12. 2):106–12. doi: 10.1055/s-0032-1313612
79. Carcao MD, Blanchette VS, Dean JA, He L, Kern MA, Stain AM, et al. The platelet function analyzer (PFA-100): a novel in-vitro system for evaluation of primary haemostasis in children. Br J Haematol. (1998) 101:70–3. 101:70–3. doi: 10.1046/j.1365-2141.1998.00656.x
80. Varon D, Dardik R, Shenkman B, Kotev-Emeth S, Farzame N, Tamarin I, et al. A new method for quantitative analysis of whole blood platelet interaction with extracellular matrix under flow conditions. Thromb Res. (1997) 85:283–94. 85:283–94. doi: 10.1016/S0049-3848(97)00014-5
81. Chen A, Teruya J. Global hemostasis testing thromboelastography: old technology, new applications. Clin Lab Med. (2009) 29:391–407. 29:391–407. doi: 10.1016/j.cll.2009.04.003
82. Michelson AD. Flow cytometry: a clinical test of platelet function. Blood. (1996) 87:4925–36. 87:4925–36. doi: 10.1182/blood.V87.12.4925.bloodjournal87124925
83. Ault KA, Knowles C. In vivo biotinylation demonstrates that reticulated platelets are the youngest platelets in circulation. Exp Hematol. (1995) 23:996–1001.
84. Winter WE, Flax SD, Harris NS. Coagulation testing in the core laboratory. Lab Med. (2017) 48:295–313. 48:295–313. doi: 10.1093/labmed/lmx050
85. Poller L. Standardization of the aPTT test. Current status. Scand J Haematol Suppl. (1980) 37:49–63. 37:49–63. doi: 10.1111/j.1600-0609.1980.tb01341.x
86. Gatt A, Chen D, Pruthi RK, Kamath PS, Leise MD, Ashrani AA, et al. From vitamin K antagonists to liver international normalized ratio: a historical journal and critical perspective. Semin Thromb Hemost. (2014) 40:845–51. 40:845–51. doi: 10.1055/s-0034-1395160
87. McCraw A, Hillarp A, Echenagucia M. Considerations in the laboratory assessment of haemostasis. Haemophilia. (2010) 16 (Suppl. 5):74–8. 5):74–8. doi: 10.1111/j.1365-2516.2010.02302.x
88. Pal S, Curley A, Stanworth S. Interpretation of clotting tests in the neonate. Arch Dis Child Fetal Neonatal Ed. (2015) 100:F270–4. 100:F270–4. doi: 10.1136/archdischild-2014-306196
89. Shen L, Tabaie S, Ivascu N. Viscoelastic testing inside and beyond the operating room. J Thorac Dis. (2017) 9(Suppl. 4):S299–308. 4):S299–308. doi: 10.21037/jtd.2017.03.85
90. Hartert H. Blutgerinnungsstudien mit der thrombelastographie; einem neuen untersuchungs verfahren. Klin Wochenschr. (1948) 26:577–83. 26:577–83. doi: 10.1007/BF01697545
91. Kang YG, Martin DJ, Marquez J, Lewis JH, Bontempo FA, Shaw BW, et al. Intraoperative changes in blood coagulation and thrombelastographic monitoring in liver transplantation. Anesth Analg. (1985) 64:888–96. 64:888–96. doi: 10.1213/00000539-198509000-00008
92. Whiting D, DiNardo JA. TEG and ROTEM: technology and clinical applications. Am J Hematol. (2014) 89:228–32. 89:228–32. doi: 10.1002/ajh.23599
93. Hans GA, Besser MW. The place of viscoelastic testing in clinical practice. Br J Haematol. (2016) 173:37–48. 173:37–48. doi: 10.1111/bjh.13930
94. Chow JH, Richards JE, Morrison JJ, Galvagno SM, Tanaka KA, Madurska MJ, et al. Viscoelastic signals for optimal resuscitation in trauma: kaolin thrombelastography cutoffs for diagnosing hypofibrinogenemia (VISOR Study). Anesth Analg. (2019) 129:1482–91. 129:1482–91. doi: 10.1213/ANE.0000000000004315
95. Wikkelsø A, Wetterslev J, Ann Merete Møller AM, Arash Afshari A. Thromboelastography (TEG) or thromboelastometry (ROTEM) to monitor haemostatic treatment versus usual care in adults or children with bleeding. Cochrane Database Syst Rev. (2016) 2016:CD007871. 2016:CD007871. doi: 10.1002/14651858.CD007871.pub3
96. Sewell EK, Forman KR, Wong EC, Gallagher M, Luban NL, Massaro AN. Thromboelastography in term neonates: an alternative approach to evaluating coagulopathy. Arch Dis Child Fetal Neonatal Ed. (2017) 102:F79–84. 102:F79–84. doi: 10.1136/archdischild-2016-310545
97. Raffaeli G, Tripodi A, Cavallaro G, Cortesi V, Scalambrino E, Pesenti N, et al. Thromboelastographic profiles of healthy very low birthweight infants serially during their first month. Arch Dis Child Fetal Neonatal Ed. (2020) 105:412–18. doi: 10.1136/archdischild-2019-317860
98. Motta M, Guaragni B, Pezzotti E, Rodriguez-Perez C, Chirico G. Reference intervals of citrated-native whole blood thromboelastography in premature neonates. Early Hum Dev. (2017) 115:60–63. 115:60–63. doi: 10.1016/j.earlhumdev.2017.09.014
99. Sokou R, Foudoulaki-Paparizos L, Lytras T, Konstantinidi A, Theodoraki M, Lambadaridis I, et al. Reference ranges of thromboelastometry in healthy full-term and pre-term neonates. Clin Chem Lab Med. (2017) 55:1592–7. 55:1592–7. doi: 10.1515/cclm-2016-0931
100. Hunt H, Stanworth S, Curry N, Woolley T, Cooper C, Ukoumunne O, et al. Thromboelastometry (TEG) and rotational thromboelastometry (ROTEM for trauma induced coagulopathy in adult trauma patient with bleeding. Cochrane Syst Rev. (2015) 2015:CD10438. doi: 10.1002/14651858.CD010438.pub2
101. Whiting P, Al M, Westwood M, Ramos IC, Ryder S, Armstrong N, et al. Viscoelastic point-of-care testing to assist with the diagnosis, management and monitoring of haemostasis: a systematic review and cost-effectiveness analysis. Health Technol Assess. (2015) 19:1–228. 19:1–228. doi: 10.3310/hta19580
102. Henderson N, Sullivan JE, Myers J, Wells T, Calhoun A, Berkenbosch J, et al. Use of thromboelastography to predict thrombotic complications in pediatric and neonatal extracorporeal membranous oxygenation. J Extra Corpor Technol. (2018) 50:149–154.
103. Solis RT, Wright CB, Gibbs MB. Electronic particle size measurements of platelet aggregates formed in vitro. J Appl Physiol. (1975) 38:739–44. doi: 10.1152/jappl.1975.38.4.739
104. Ozaki Y, Satoh K, Yatomi Y, Yamamoto T, Shirasawa Y, Kume S. Detection of platelet aggregates with a particle counting method using light scattering. Anal Biochem. (1994) 218:284–94. 218:284–94. doi: 10.1006/abio.1994.1180
105. Panzer S, Jilma P. Methods for testing platelet function for transfusion medicine. Vox Sang. (2011) 101:1–9. 101:1–9. doi: 10.1111/j.1423-0410.2011.01467.x
106. Görög P, Ahmed A. Haemostatometer: a new in vitro technique for assessing haemostatic activity of blood. Thromb Res. (1984) 34:341–57. doi: 10.1016/0049-3848(84)90391-8
107. Gorog DA, Kovacs IB. Thrombotic status analyser. Measurement of platelet-rich thrombus formation and lysis in native blood. Thromb Haemost. (1995) 73:514–20. doi: 10.1055/s-0038-1653806
108. Phillips RC, Shahi N, Leopold D, Levek C, Shirek G, Hilton S, et al. Thromboelastography-guided management of coagulopathy in neonates with congenital diaphragmatic hernia supported by extracorporeal membrane oxygenation. Pediatr Surg Int. (2020) 36:1027–33. 36:1027–33. doi: 10.1007/s00383-020-04694-0
109. Ghirardello S, Raffaeli G, Scalambrino E, Chantarangkul V, Cavallaro G, Artoni A, et al. The intra-assay reproducibility of thromboelastography in very low birth weight infants. Early Hum Dev. (2018) 127:48–52. 127:48–52. doi: 10.1016/j.earlhumdev.2018.10.004
110. Wikkelsø A, Wetterslev J, Møller AM, Afshari A. Thromboelastography (TEG) or rotational thromboelastometry (ROTEM) to monitor haemostatic treatment in bleeding patients: a systematic review with meta-analysis and trial sequential analysis. Anaesthesia. (2017) 72:519–31. 72:519–31. doi: 10.1111/anae.13765
111. Emani S, Sleeper LA, Faraoni D, Mulone M, Diallo F, DiNardo JA, et al. Thromboelastography is associated with surrogates for bleeding after pediatric cardiac operations. Ann Thorac Surg. (2018) 106:799–806. 106:799–806. doi: 10.1016/j.athoracsur.2018.04.023
112. Sujka J, Gonzalez KW, Curiel KL, Daniel J, Fischer RT, Andrews WS, et al. The impact of thromboelastography on resuscitation in pediatric liver transplantation. Pediatr Transplant. (2018) 22:e13176112. 22:e13176112. doi: 10.1111/petr.13176
113. Gilley M, Beno S. Damage control resuscitation in pediatric trauma. Curr Opin Pediatr. (2018) 30:338–43. 30:338–43. doi: 10.1097/MOP.0000000000000617
114. Duarte RCF, Ferreira CN, Rios DRA, dos Reis HJ, Carvalho MG. Thrombin generation assays for global evaluation of the hemostatic system: perspectives and limitations. Rev Bras Hematol Hemoter. (2017) 39:259–65. 39:259–65. doi: 10.1016/j.bjhh.2017.03.009
115. Haidl H, Cimenti C, Leschnik B, Zach D, Muntean W. Age-dependency of thrombin generation measured by means of calibrated automated thrombography (CAT). Thromb Haemost. (2006) 95:772–5. 95:772–5. doi: 10.1160/TH05-10-0685
116. Corey FS, Walker WF. Sonic estimation of elasticity via resonance: a new method of assessing hemostasis. Ann Biomed Eng. (2016) 44:1405–24. 44:1405–24. doi: 10.1007/s10439-015-1460-y
117. Ferrante EA, Blasier KR, Givens TB, Lloyd CA, Fischer TJ, Viola F. A novel device for the evaluation of hemostatic function in critical care settings. Anesth Analg. (2016) 123:1372–9. 123:1372–9. doi: 10.1213/ANE.0000000000001413
118. Penny S Reynolds PS, Paul Middleton P, Harry McCarthy H, Bruce D Spiess BD. A comparison of a new ultrasound-based whole blood viscoelastic test (SEER sonorheometry) versus thromboelastography in cardiac surgery. Anesth Analg. (2016) 123:1400–7. 123:1400–7. doi: 10.1213/ANE.0000000000001362
119. Huffmyer JL, Fernandez LG, Haghighian C, Terkawi AS, Groves DS. Comparison of SEER sonorheometry with rotational thromboelastometry and laboratory parameters in cardiac surgery. Anesth Analg. (2016) 123:1390–9. 123:1390–9. doi: 10.1213/ANE.0000000000001507
120. Christiaans SC, Duhachek-Stapelman AL, Russell RT, Lisco SJ, Kerby JD, Pittet JF. Coagulopathy after severe pediatric trauma. Shock. (2014) 41:476–90. doi: 10.1097/SHK.0000000000000151
121. Nair A, Flori H, Cohen MJ. Characterization of organ dysfunction and mortality in pediatric patients with trauma with acute traumatic coagulopathy. Trauma Surg Acute Care Open. (2020) 5:e000382. 5:e000382. doi: 10.1136/tsaco-2019-000382
122. Cohen MJ, Christie SA. Coagulopathy of trauma. Crit Care Clin. (2017) 33:101–18. 33:101–18. doi: 10.1016/j.ccc.2016.08.003
123. Leeper CM, Neal MD, McKenna C, Sperry JL, Gaines BA. Abnormalities in fibrinolysis at the time of admission are associated with deep vein thrombosis, mortality, and disability in a pediatric trauma population. Trauma Acute Care Surg. (2017) 82:27–34. 82:27–34. doi: 10.1097/TA.0000000000001308
124. Leeper CM, Gaines BA. Viscoelastic hemostatic assays in the management of the pediatric trauma patient. Semin Pediatr Surg. (2017) 26:8–13. 26:8–13. doi: 10.1053/j.sempedsurg.2017.01.004
125. Bhatt H, Rao GS. Management of acute liver failure: a pediatric perspective. Curr Pediatr Rep. (2018) 6:246–57. 6:246–57. doi: 10.1007/s40124-018-0174-7
126. Sollazzi L, Perilli V. End stage liver disease: a delicate balance of bleeding and thrombosis. Minerva Anestesiol. (2019) 85:712–4. 85:712–4. doi: 10.23736/S0375-9393.19.13859-X
127. Labidi A, Baccouche H, Fekih M, Mahioub S, BenMustapha N, Serghini M, et al. The relationship between coagulation disorders and the risk of bleeding in cirrhotic patients. Ann Hepatol. (2019) 18:627–32. 18:627–32. doi: 10.1016/j.aohep.2018.12.007
128. Kenet G, Barg AA, Nowak-Göttl U. Hemostasis in the very young. Semin Thromb Hemost. (2018) 44:617–23. 44:617–23. doi: 10.1055/s-0038-1660852
129. Nellis ME, Goel R, Karam O. Transfusion management in pediatric oncology patients. Hematol Oncol Clin North Am. (2019) 33:903–13. 33:903–13. doi: 10.1016/j.hoc.2019.05.011
130. Dalton HJ, Garcia-Filion P, Holubkov R, Moler FW, Shanley T, Heidemann S, et al. Association of bleeding and thrombosis with outcome in extracorporeal life support. Pediatr Crit Care Med. (2015) 16:167–74. 16:167–74. doi: 10.1097/PCC.0000000000000317
131. Dalton HJ, Reeder R, Garcia-Filion P, Holubkov R, Berg RA, Zuppa A, et al. Factors associated with bleeding and thrombosis in children receiving extracorporeal membrane oxygenation. Am J Respir Crit Care Med. (2017) 196:762–71. doi: 10.1164/rccm.201609-1945OC
132. Ignjatovic V, Summerhayes R, Gan A, Than J, Chan A, Cochrane A, et al. Monitoring unfractionated heparin (UFH) therapy: which anti-factor Xa assay is appropriate? Thromb Res. (2007) 120:347–51. 120:347–51. doi: 10.1016/j.thromres.2006.10.006
133. Arachchillage DRJ, Kamani F, Deplano S, Banya W, Laffan M. Should we abandon the APTT for monitoring unfractionated heparin? Thromb Res. (2017) 157:157–61. doi: 10.1016/j.thromres.2017.07.006
134. Moynihan K, Johnson K, Straney L, Stocker C, Anderson B, Venugopal P, et al. Coagulation monitoring correlation with heparin dose in pediatric extracorporeal life support. Perfusion. (2017) 32:675–85. 32:675–85. doi: 10.1177/0267659117720494
135. Thompson TZ, Kunak RL, Savage NM, Agarwal S, Chazelle J, Singh G. Intraoperative monitoring of heparin: comparison of activated coagulation time and whole blood heparin measurements by different point-of-care devices with heparin concentration by laboratory-performed plasma anti-Xa assay. Lab Med. (2019) 50:348–56. doi: 10.1093/labmed/lmz014
136. Levi M, Hunt BJ. A critical appraisal of point-of-care coagulation testing in critically ill patients. J Thromb Haemost. (2015) 13:1960–7. 13:1960–7. doi: 10.1111/jth.13126
Keywords: pediatric, developmental hemostasis, diagnosis, coagulopathy, viscoelastic
Citation: Nair AB and Parker RI (2021) Hemostatic Testing in Critically Ill Infants and Children. Front. Pediatr. 8:606643. doi: 10.3389/fped.2020.606643
Received: 15 September 2020; Accepted: 10 December 2020;
Published: 08 January 2021.
Edited by:
Gemma Crighton, Royal Children's Hospital, AustraliaReviewed by:
Genny Raffaeli, IRCCS Ca 'Granda Foundation Maggiore Policlinico Hospital, ItalyPaul Monagle, The University of Melbourne, Australia
Copyright © 2021 Nair and Parker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert I. Parker, cm9iZXJ0LnBhcmtlckBzdG9ueWJyb29rLmVkdQ==