 Yu-Cheng Lin
Yu-Cheng Lin Chi-Chien Wu
Chi-Chien Wu Yen-Hsuan Ni
Yen-Hsuan Ni- 1Department of Pediatrics, Far Eastern Memorial Hospital, New Taipei City, Taiwan
- 2Department of Healthcare Administration, Oriental Institute of Technology, New Taipei City, Taiwan
- 3Departments of Pediatrics, National Taiwan University Hospital, Taipei, Taiwan
Non-alcoholic or recently re-defined metabolic associated fatty liver disease (MAFLD), a spectrum of progressive hepatic disease, has become a public health issue in obese children and adolescents. MAFLD is a complex metabolic disease strongly associated with obesity and insulin resistance. It is not known why not every obese subject will develop MAFLD. Different ethnic/racial groups display differences in MAFLD prevalence, indicating genetic factor plays a role. In the past two decades, sequence variations in genetic loci, including PNPLA3, TM6SF2, GCKR, MBOAT7, HSD17B13, etc. have been shown to confer susceptibility to MAFLD in children and adults. This review article provides an updated viewpoint of genetic predictors related to pediatric MAFLD. We discuss whether these susceptible genes can be clinically used for risk stratification and personalized care. Understanding human genetics and molecular mechanisms can give important information not only for prediction of risk but also on how to design drugs. In view of current epidemic of MAFLD worldwide, it is necessary to identify which children with MAFLD progress rapidly and need earlier intervention. In the future, a comprehensive analysis of individualized genetic and environmental factors may help assess the risk of children with MAFLD and personalize their treatment.
Introduction
There is increasing interest in metabolic associated fatty liver disease (MAFLD), defined as excessive deposition of fat in the liver in the absence of significant alcohol consumption. The progression of MAFLD encompasses a spectrum of conditions ranging from fat in the liver—simple steatosis, fat with inflammation and/or fibrosis-steatohepatitis to advanced fibrosis and cirrhosis over time (1). MAFLD is one of the most common chronic liver disease in the whole world (2, 3), becoming a global health burden (4). The long-term follow-up study revealed adults with MAFLD had increased liver related and non-liver related mortalities (5). MAFLD is now a serious health condition not only for adults, but also for children (6).
Pathogenesis of MAFLD is complicated, multifactorial (7), and strongly associated with obesity related comorbidities such as insulin resistance, cardiac dysfunction, and kidney disease, etc. (8–11). Nowadays, MAFLD is no longer regarded as a primary hepatic disease, but rather a component of metabolic syndrome. Therefore, a recent expert consensus group suggested the metabolic associated fatty liver disease “MAFLD” as a more appropriate term than the nomenclature of non-alcoholic fatty liver disease (NAFLD). Since children drink less alcohol, we believe that the term MAFLD is more suitable for children and should replace NAFLD (12).
Due to a continuum from obesity to metabolic syndrome, patients with MAFLD may benefit from early identification of the disease risk and individually targeted treatment (13). Establishing clinical predictors is necessary for MAFLD diagnosis and risk stratification. Previously reported biochemical factors include elevated total cholesterol, triglycerides, fasting insulin, increased fasting glucose and insulin concentrations, homeostatic model assessment for insulin resistance (HOMA-IR) index, and aspartate aminotransferase (AST)/alanine aminotransferase (ALT) ratio (14, 15).
There are several differences between pediatric and adult MAFLD in prevalence, risk propensity, and liver histology (Table 1) (20, 21). Risk factors such as alcohol abuse, drug abuse and comorbidities among children are much less than those in adults. Understanding the differences between pediatric and adult MAFLD can help assess how NAFLD progresses from childhood toward adulthood. The histological pattern of pediatric MAFLD is different from that of adults. The classic histological findings that represent MAFLD are: steatosis, swelling, inflammation, and fibrosis. In adults, steatosis, inflammation, and accumulation of collagen start in the perivenular area (zone 3), while in children, it usually starts in the periportal area (zone 1) with lack of ballooning (22). However, it is not clear whether patients with pediatric MAFLD pattern differ in pathogenesis, prognosis, or response to treatment (23). In addition, due to lack of long-term follow-up from children to adults, the natural history and prognosis of MAFLD in children are still uncertain. Compared with adults with MAFLD, children with MAFLD have a much longer course of disease. Reversing the course of MAFLD in childhood is indeed an unmet need.
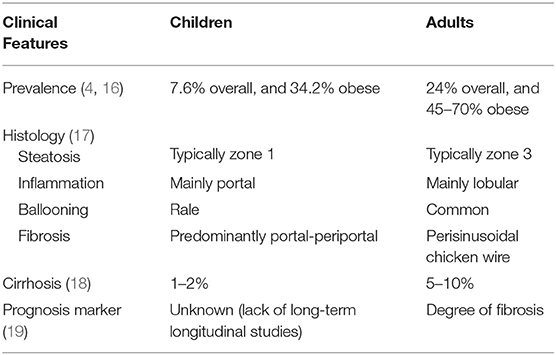
Table 1. Differences between adult and pediatric MAFLD.
Epidemiology
The increasing epidemic of obesity and sedentary lifestyle continues to raise the prevalence of MAFLD (24). As the global obesity epidemic worsens metabolic disorders, the health burden of children's MAFLD has become huge (16, 25). Recent research indicated in obese children, MAFLD is present in nearly one-third of boys and one-fourth of girls (26). Not every obese subject will suffer from MAFLD, which suggests that genetic and/or environmental factors contribute to each individual's susceptibility. In fact, MAFLD can occur in non-obese individuals (27, 28).
Different ethnic/racial groups display differences in MAFLD prevalence (29). It is recognized the highest prevalence is in the American Hispanic population followed by the Caucasian and the African-American (30). In pediatric population, obese Hispanic adolescents are more likely to develop MAFLD than obese non-Hispanic adolescents (31). Compared with the West, the East has a lower MAFLD incidence and prevalence (32).
Different lifestyle and nutrition status may partially account for the differences among ethnic groups. However, the Western diet and sedentary lifestyle have led to the emergence of obesity and MAFLD in Asia over the last decade (33). On the other hand, growing evidence reveals the importance of genetic factors in the development and progression of MAFLD. Since the frequency of genetic variants differs among ethnic groups, increasing understanding of the genetic predisposition to MAFLD may help us to decipher the reasons for its occurrence (34).
Heritability of MAFLD
There is growing awareness of the role of genetic factors in the etiology and prognosis of MAFLD. The differences in disease distribution observed in adults and children with MAFLD indicate genetic susceptibility plays a crucial role in the development of MAFLD (6). In a twin study, Loomba et al. revealed that MAFLD-associated hepatic steatosis and fibrosis are heritable traits (35). Genome-wide association studies (GWAS) have identified several important single nucleotide polymorphisms (SNPs) affecting the severity and progression of MAFLD (36). Overall, the dynamic interactions between genetic and environmental factors further modulate the disease phenotype, susceptibility, development, and progression (37–39).
Till now, MAFLD susceptible genes have been reported to be involved in a wide spectrum of pathogenic mechanisms, including lipid metabolism, insulin signaling, oxidative stress, inflammation and fibrogenesis, etc. Genetic factors do not have the same effects across studies due to different study populations and designs. For example, people with PNPLA3 variant are usually considered to have more hepatic steatosis, inflammation, and fibrosis. However, Kotronen et al. reported PNPLA3 SNP did not improve the prediction of liver fat content by using their liver fat score equation (40). In fact, MAFLD is a polygenetic disease and we need more genetic information, rather than just one SNP, to establish the predictive model.
Major Common Genetic Variations of MAFLD
Herein, we provide a comprehensive update on genetic variations related to pediatric MAFLD (Table 2). The genetic determinants of MAFLD not only can predict the progression of MAFLD, but also are the possible targets for therapy.
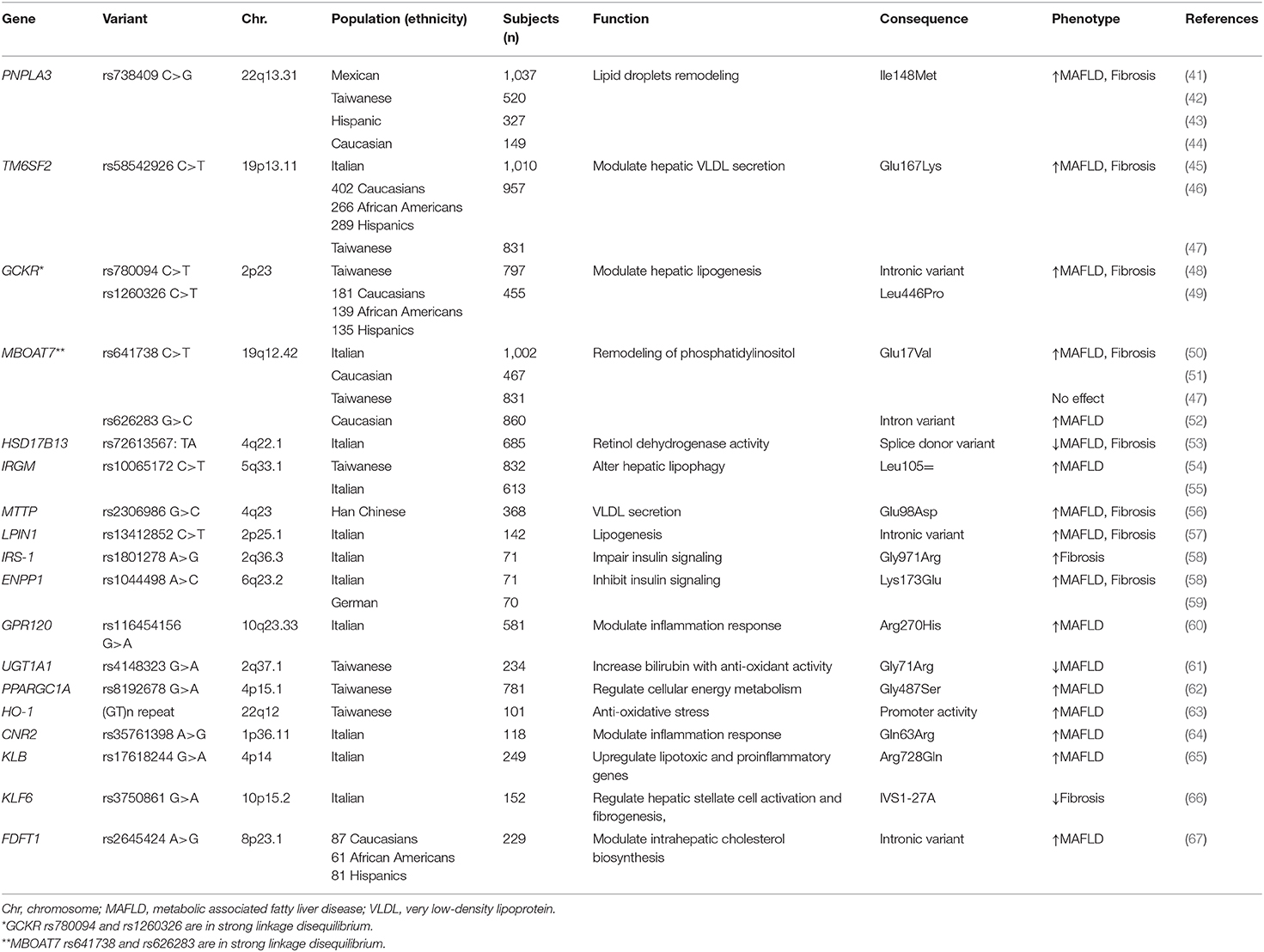
Table 2. Genetic variations associated with pediatric MAFLD.
To date, several major common MAFLD susceptible genes reported in adults have been replicated in pediatric studies, such as PNPLA3, TM6SF2, GCKR, MBOAT7, and HSD17B13. The effects and presumptive functions of these susceptible genes are discussed as follows:
PNPLA3
Patatin-like phospholipase domain containing 3 (PNPLA3) gene encodes a transmembrane protein “adiponutrin,” which is expressed predominantly in the liver, retina, skin, and adipose tissue (68, 69). In 2008, Romeo et al. first identified the association of a PNPLA3 gene I148M variant with hepatic fat content in adults by GWAS (70). Subsequently, Romeo et al. reported this variant was associated with increased levels of ALT/AST in obese children, which suggests that it confers a genetic susceptibility to liver damage since childhood (71). Furthermore, PNPLA3 gene I148M variant modulates the progression and liver-related outcomes in patients with MAFLD (72). The effects of PNPLA3 variants on pediatric MAFLD have been validated in different ethnicities, including Han Chinese, Hispanic, and Caucasian (41–44). Till now, PNPLA3 is regarded as the most robust susceptible gene for MAFLD across different ethnicities.
The wild type PNPLA3 protein facilitates triglyceride hydrolysis. Impaired hydrolysis of triglycerides/lipid droplet (LD) remodeling in hepatocytes leads to hepatic steatosis (73, 74). PNPLA3 I148M promotes the accumulation of intracellular lipids in the liver by reducing the lipidation and secretion of low-density lipoprotein (VLDL) particles (75). Further studies showed the PNPLA3 variant not only increases the odds of developing fatty liver itself, but it also determines the degree of hepatic injury and the full spectrum of histopathologic consequences of MAFLD (76). Several studies have shown that PNPLA3 increases the stimulation of hepatic stellate cells by affecting the metabolism of retinoids, leading to liver fibrosis (77–79). Hence, PNPLA3 SNP has emerged as the key genetic determinant of MAFLD severity in both adults and pediatric patients (80). A recent study showed that silencing Pnpla3 with antisense oligonucleotides improved liver steatosis and fibrosis in Pnpla3 I148M knock-in mice, indicating that PNPAL3 could be a potential target for treatment in human (81).
In addition to the effect on liver, PNPLA3 rs738409 variant has been reported to be associate with reduced glomerular filtration rate (GFR) in children with obesity, indicating the variant PNPLA3 genotype may be related to kidney dysfunction in children independent of MAFLD status (82, 83).
TM6SF2
The human transmembrane 6 superfamily member 2 (TM6SF2) gene encodes a protein of 351 amino acids with 7–10 predicted transmembrane domains. TM6SF2 protein facilitates the transfer of neutral lipids from cytoplasmic to luminal LDs and VLDL particles. Overexpression of TM6SF2 decreases the number and size of LDs (84).
In 2014, Kozlitina et al. reported that the TM6SF2 rs58542926 variant, a C-to-T substitution, encoding a glutamate to lysine change at codon 167 (E167K) and associated with high hepatic triglyceride content and elevated liver serum enzymes levels in adults enrolled in the Dallas Heart Study (85). TM6SF2 E167K variant carriers with MAFLD have impaired hepatic lipid synthesis from polyunsaturated fatty acids (86). Further studies reported the TM6SF2 rs58542926 variant also influences hepatic fibrosis and metabolic homeostasis (87, 88). In children, the association between TM6SF2 rs58542926 variant and MAFLD has been replicated in different ethnicities (45–47). Interestingly, the plasma levels of triglycerides are lower in TM6SF2 E167K variant carriers than in the non-carriers (89). The increase in hepatic steatosis for loss of function mutation in TM6SF2 is due to a double mechanism, namely a reduction in the lipidation of VLDL particles (84) and in the number of the secreted apolipoprotein B100 particles (90). In addition to its effects on the liver, it has been reported that TM6SF2 variants also affected the renal function in children (91) and adults (92) independently of MAFLD.
GCKR
Glucokinase regulator protein (GCKR) is an inhibitor of glucokinase which regulates glucose storage and disposal and controls de novo lipogenesis by regulating the flux of glucose into hepatocytes (93). In 2011, Speliotes et al. reported variants in or near GCKR are associated with liver fat content and histopathologic phenotypes at genome-wide significance levels (94). Subsequent meta-analysis provides evidence of significant association between GCKR rs780094 (an intronic variant) and risk of MAFLD (95). Silva et al. measured metabolites by mass spectrometry and found novel associations of the GCKR rs780094 variant with amino acids and their downstream metabolites, especially lipids (96). In addition, the progression of fibrosis in MAFLD could be influenced by the GCKR genotype (97). The effect of GCKR genotype on pediatric MAFLD have been reported. The variant GCKR rs780094 has been reported to confer susceptibility to MAFLD in obese school children and adolescents in Taiwan (48).
Another common variant in GCKR rs1260326, which is in linkage disequilibrium with rs780094, was associated with hepatic triglyceride content in the Dallas Heart Study (98). This variant encodes for a proline to leucine substitution at the 446 position (P446L), resulting in a loss of the affinity of the GCKR protein for the glucokinase. Consequently, more glucokinase is available in the cytoplasm to convert glucose into glucose-6-phosphate. The increased production of glucose-6-phosphate results in an increased rate of glycolysis, leading to increased production of malonyl-CoA, the precursor of de novo lipogenesis (99). The rs1260326 in GCKR gene is also linked to fatty liver in obese youths (49).
MBOAT7
The membrane-bound O-acyltransferase domain-containing protein 7 (MBOAT7) is a 6 transmembrane domain (100). Hepatocyte specific inactivation of this gene caused an increase in hepatic fat content due to a non-canonical triglyceride synthesis pathway related to a high turnover of phosphatidyl inositol (101). Down-regulation of MBOAT7 predisposes subjects to MAFLD (102).
In 2016, Mancina et al. first reported the rs641738 C>T variant in the MBOAT7 gene was associated with increased risk of MAFLD in adults of European descent (103). Subsequently, an association between MBOAT7 variant and liver fibrosis severity was confirmed by Krawczyk et al. (104). In an animal study, MBOAT7 over-expression was negatively correlated with obesity and insulin sensitive, driving the progression of MAFLD (105). A pediatric study showed the MBOAT7 rs641738 variant was associated with plasma concentrations of ALT in obese children (50, 51). Umano et al. reported that another variant (rs626283) in MBOAT7 gene was associated with MAFLD in Caucasian obese children (52). Notably, conflicting data has reported that MBOAT7 rs641738 polymorphism does not influence hepatic steatosis and liver injury as determined by serum levels of CK-18 fragment in obese Taiwanese children of Han Chinese ethnicity (47). The difference in the effect of MBOAT7 variant on MAFLD might be due to different ethnicities.
HSD17B13
Increasing data demonstrate hydroxysteroid 17-beta dehydrogenase 13 (HSD17B13) acts a pivotal part in hepatic lipid homeostasis and the pathogenesis of MAFLD (106). In 2018, Abul-Husn et al. reported that a loss-of function splice variant (rs72613567:TA) of HSD17B13 gene was associated with a reduced risk of chronic liver disease and of MAFLD progression (107). More recently, an exome-wide association study confirmed the HSD17B13 rs72613567 variant influenced the susceptibility and histological severity of MAFLD (108). A pediatric study in Italy, reported obese children carrying the HSD17B13 variant had lower hepatic steatosis and pediatric MAFLD fibrosis index than non-carriers (53). A recent work also showed that this genetic variation provided kidney protection for children (109).
The HSD17B13 protein is a liver-specific LD-associated protein and exhibits retinol dehydrogenase activity. The HSD17B13 variant is supposed to alter mRNA splicing, yielding a truncated protein with reduced enzymatic activity. However, the exact role and function of HSD17B13 in MAFLD pathophysiology remains largely uncharacterized. Over-expression of HSD17B13 in human hepatoma cell lines or C57BL/6 mice leads to excessive lipid accumulation. HSD17B13 gene encodes a LD-associated protein, which is involved in regulating lipogenesis (110). Recently, the Rotman group published interesting studies on the inactivation of Hsd17b13 in mice (111) and the identification of an enzymatic active site metabolizing retinol (112).
Hepatic Lipid Metabolism
Excessive fat accumulation damages hepatocytes and leads to inflammatory response, cytokine production, oxidative stress, abnormal cellular signaling, and activation of stellate cells. The intracellular storage and utilization of lipids play an important role in supporting cellular energy homeostasis. In addition to the major MAFLD susceptible genes aforementioned, several important genetic variants have been identified to affect hepatic lipid metabolism.
IRGM
In 2009, Singh et al. first reported that LDs can be degraded in hepatocytes by a specific autophagy-related process “lipophagy” (113). The immunity-related GTPase family M protein encoded by the IRGM gene controls autophagy activation (114). Our previous study found obese children with the variant IRGM rs10065172 TT genotype have a higher risk of MAFLD and elevated ALT levels compared with subjects with wild type (54). A downregulation of IRGM in HepG2 cells decreased autophagic flux accompanied by an increased lipid accumulation. In contrast, overexpression of IRGM decreased LD content in HepG2 cells. This genetic association has been replicated in a cohort of obese Italian children (55). A recent murine study revealed liver specific suppression of Ifgga2 (the mice ortholog of human IRGM) increases hepatic fat content in a backcross of obese C57BL/6J New Zealand mice (115).
MTTP
The human microsomal triglyceride transfer protein (MTTP) works to lipidate and assemble the apoB-containing lipoproteins in the liver. It is critical to remove lipid from liver through the assembly and secretion of VLDL particles. Hsiao et al. reported the MTTP polymorphisms can modulate lipid homeostasis and determine the serum lipids and risk of MAFLD (116). A pediatric study also showed the association of MTTP rs2306986 variant and MAFLD in obese children (56).
LIPIN1
Lipin-1 encoded by the LPIN1 gene, expressed mainly in adipose and the liver, has phosphatidic phosphatase activity (117). Valenti et al. reported children, but not adult, carrying the LIPIN1 rs13412852 TT genotype had a lower prevalence of MAFLD, less severe liver damage and a lower liver fibrosis prevalence (57). The mutation in LIPIN1 gene may result in decreasing the flux of free fatty acids (FFAs) to the liver.
Insulin Resistance
Insulin resistance is a feature of the MAFLD pathophysiology and occurs in its early phases (118, 119). It affects metabolic syndrome and MAFLD in obese children and adolescents (120), even in non-obese patients (121). Peripheral insulin resistance leads to excessive lipolysis in adipose tissue, releasing a lot of FFAs into the circulation (8). The liver then uptakes excessive FFAs and exceeds its capacity to transfer FFAs into neutral triglycerides, causing hepatic steatosis, lipotoxicity, and endoplasmic reticulum stress (122).
IRS1, ENPP1
The insulin receptor substrate 1 (IRS1) and ectonucleotide pyrophosphate phosphodiesterase (ENPP1) genes play crucial roles in controlling cell signaling in response to insulin. Once insulin binds to the insulin receptor, the IRS1 protein regulates hepatic gene expression that coordinates glucose homeostasis. ENPP1 protein negatively modulates insulin receptors and induces insulin resistance if overexpressed.
Hepatic IRS1 overexpression is associated with histological progression in patients with MAFLD (123). The ENPP1 rs104449 and IRS1 rs1801278 variants decrease hepatic insulin signaling and predispose adult patients with MAFLD to liver damage (58). In children, Hudert et al. reported ENPP1 rs1044498, not IRS1 rs1801278 variant was associated with pediatric MAFLD (59).
Oxidative Stress and Inflammation
The two-hit hypothesis is widely recognized as a model of MAFLD progression (124). The first hit causes hepatic fat accumulation and the second hit causes inflammation and fibrosis. The second hit usually results from excessive oxidative stress, such as mitochondrial stress and insulin resistance. In the other words, oxidative stress plays a critical role in the progression from simple steatosis to steatohepatitis (125, 126).
GPR120
G protein-coupled receptor 120 (GPR120) is a functional omega-3 fatty acid receptor that mediates anti-inflammatory and insulin sensitivity (127, 128). In 2014, Marzuillo et al. reported the association between the GPR120 rs116454156 variant (R270H) and liver injury in obese children and adolescents (60). By regulating GPR120, docosahexaenoic acid (DHA) can reduce the inflammatory response of MAFLD in children (129).
UGT1A1
Genetic modifiers belonging to oxidative stress are involved in either the generation of reactive oxygen species (ROS) or the modulation of cellular antioxidant defense. Genetic variant in uridine-5'-diphosphoglucuronosyltransferase 1A1 (UGT1A1) increases serum bilirubin, which has anti-oxidative properties. Our previous study found that the variant UGT1A1*6 genotype was associated with a lower risk of MAFLD in obese Taiwanese children (61). A subsequent study showed an inverse relation between serum bilirubin levels and the presence of MAFLD on Italian children, replicating our previous finding (130).
HO-1
Increasing heme oxygenase-1 (HO-1) activity can reverse complications related to obesity, metabolic syndrome, and MAFLD (131). HO-1 is a stress-responsive protein, defensing against the oxidative process. The (GT)n dinucleotide repeat within the HO-1 gene promotor region is highly polymorphic. Our previous study found that obese children with the long repeat of HO-1 (GT)n dinucleotide were more susceptible to MAFLD (63). In obese mice, liraglutide, a glucagon-like peptide 1 analog, ameliorated MAFLD severity through upregulating Sestrin2-mediated Nrf2/HO-1 pathway in obese mice (132), suggesting HO-1 could be a therapeutic target for MAFLD.
PPARGC1A
The peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PPARGC1A) gene encodes a PGC-1α protein that regulates mitochondrial functions, oxidative stress, and lipogenesis (133). Our previous study revealed the PPARGC1A rs8192678 risk A allele was associated with an increased risk of MAFLD in obese Taiwanese children (62). An animal study revealed loss of estrogen signaling contributes to oxidative damage caused by low levels of PPARGC1A in liver in mice (134).
CNR2
Innate immunity and inflammation are the hallmarks of progressive MAFLD (135). Inflammatory cytokines are involved in the progression from simple steatosis to steatohepatitis. The cannabinoid receptor type 2 (CB2), encoded by the CNR2 gene, is a seven-transmembrane domain G protein-coupled receptor. Activation of CB2 receptor inhibits nuclear translocation of NF-kB, decreasing production of inflammatory cytokines. An animal experiment demonstrated CB2 activation can reduce hepatic injury and promote liver regeneration (136). Recent research revealed the role of the CNR2 rs35761398 variant in modulating the hepatic inflammation state in obese children with MAFLD and in increasing susceptibility to liver damage (64).
KLB
β-Klotho gene (KLB) encodes a transmembrane protein mainly expressed in the liver. A recent study indicated KLB rs17618244 variant increased the risk of hepatocellular ballooning and lobular inflammation in children with MAFLD (65). The precise mechanism of KLB protein on MAFLD is not clear.
Fibrogenesis
The liver fibrosis stage is the strongest predictor for disease-specific mortality in MAFLD (137). Accurate assessment of the risk of having advanced liver fibrosis in children with MAFLD is important in the clinical practice. However, because the fibrosis phenotype requires a period of cumulative damage, it is difficult to accurately measure the impact of genetic variation on the liver fibrosis in children with MAFLD. This is why there are fewer studies on the association between liver fibrosis and MAFLD in children. The limited available data on genetic variants associated with liver fibrosis in children with MAFLD are discussed below:
KLF6
Nobili et al. reported the Krueppel-like factor 6 (KLF6) rs3750861 variant reduced the risk of liver fibrosis in children with MAFLD (66). KLF6 is up-regulated by activated HSCs following liver injury (138).
FDFT1
The farnesyl-diphosphate farnesyltransferase 1 (FDFT1) gene encodes for squalene synthase and modulates the cholesterol biosynthesis. This FDFT1 rs2645424 variant has been reported to be associated with the MAFLD activity score and moderate/severe fibrosis in a multiethnic cohort of obese youths (67). It is not currently clear whether the FDFT1 rs2645424 variant affects the enzyme activity because it is an intronic variant.
Influences of Genetic Variants on MAFLD Pathogenesis
A schematic representation of the pathways in the pathogenesis of MAFLD development and progression affected by genetic factors is summarized in Figure 1. It currently remains difficult to accurately predict the development and progression of MAFLD because of the complicated interactions between genetic pathways and variable environmental influences.
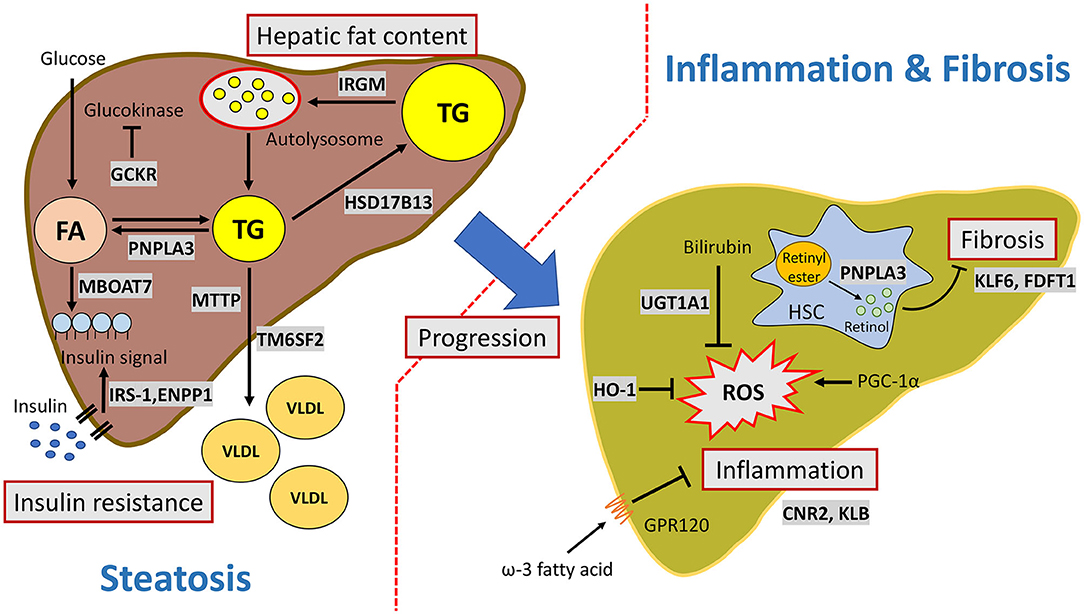
Figure 1. Major pathways involved in the genetic susceptibility toward MAFLD. PNPLA3 facilitates triglyceride hydrolysis in hepatocytes and mediates retinol release from retinyl ester in hepatic stellate cells. IRGM activates autophagosome formation and increases hepatic lipophagy. TM6SF2 transfers triglyceride from cytoplasmic to VLDL particles. MBOAT7 is involved in phospholipid remodeling. HSD17B13 is a lipid droplet-related protein and has retinol dehydrogenase activity. IRS-1 and ENPP1control cell signaling in response to insulin. GCKR regulates glucokinase activity and facilitates de novo lipogenesis. GPR120 is a functional omega-3 fatty acid (such as DHA) receptor which mediates anti-inflammatory effects. PGC-1α (encoded by PPARGC1A) is a master regulator of reactive oxygen species. HO-1 and UGT1A1are involved in the bilirubin metabolism and antioxidant activity. CB2 (encoded by CNR2) and KLB are involved in the inflammation process, but the mechanism is still unknown. KLF6 and squalene synthase (encode by FDFT1) are associated with fibrosis. KLF6 activates hepatic stellate cells. Squalene synthase modulates cholesterol biosynthesis.
Translational Implications of Genetic Variations for MAFLD
Genetic variations in PNPLA3, TM6SF2, GCKR, MBOAT7, and HSD17B13, etc. provide novel insights into the MAFLD pathophysiology and may be incorporated into predictive model for precision medicine in patients with MAFLD (139). Early recognition and treatment of MAFLD decreases long-term morbidity and mortality (140). Because MAFLD is a heterogeneous disease, the treatment option should be personal/ individualized. Personalized prediction is required to guide risk stratification and treatment. Ma et al. reported improved diet quality is more effective in individuals at a high genetic risk of MAFLD (141). DHA supplementation may not be as effective as non-carriers in reducing liver fat levels in PNPLA3 I148M carriers (142). Recently, Costanzo et al. reported a weighted-genetic risk score combining PNPLA3, GCKR, and TM6SF2 risk alleles was associated with an 8-fold higher risk of MAFLD in obese children (143). In this regard, polygenic risk scores may be applied in risk stratification and guide the treatment.
Modulation of the human genetics associated with MAFLD presents the opportunity to develop a precision medicine. High-throughput technologies, such as gene array and next generation sequencing, can facilitate the translation of genetic testing in the care of children with MAFLD (144). Considering this complexity, computational models may help design personalized treatment strategies which account for genetic and environmental factors (145).
Despite its promise, the utility of genetic testing in patients with MAFLD remains controversial due to the lack of established evidence related to clinical benefits. Societal guidelines from ESPGHAN (European Society for Pediatric Gastroenterology Hepatology and Nutrition) in 2012 and NASPGHAN (North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition) in 2017 recognize the genetic predisposition strongly affects the risk of MAFLD development in children, but they do not recommend routine genetic testing in children with NALFD (146, 147). In adults, 2016 EASL (European Association for the Study of the Liver) and 2018 AASLD (American Association for the Study of Liver Diseases) guidelines claimed that testing for genetic variants of MAFLD in routine clinical care is currently not advocated (148, 149). More trials are needed to be conducted to test the role of gene-based diagnosis and treatment for MAFLD before its clinical use.
Conclusion
This review article summarizes current knowledge and new advances related to the genetics of pediatric MAFLD. Overall, understanding human genetics and molecular mechanisms can give important information not only for prediction of risk but also on how to design drugs (150). To date, genetic studies have successfully advanced our understanding in the pathogenesis of MAFLD, but there are still gaps in translating these genetic studies into clinical applications in the real world. In the future, by analyzing more comprehensive personalized genetic and environmental factors, we will be able to accurately assess the risk of children with MAFLD and adopt personalized treatment.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was funded by Ministry of Science and Technology, Executive Yuan, Taiwan (MOST 109-2314-B-418-009-MY3), Far Eastern Memorial Hospital (FEMH 109-2314-B-418-009-MY3, FEMH-2020-C-011), and Far Eastern Memorial Hospital - National Taiwan University Hospital Joint Research Program (109-FTN07).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Perumpail BJ, Khan MA, Yoo ER, Cholankeril G, Kim D, Ahmed A. Clinical epidemiology and disease burden of nonalcoholic fatty liver disease. World J Gastroenterol. (2017) 23:8263–76. doi: 10.3748/wjg.v23.i47.8263
2. Li J, Zou B, Yeo YH, Feng Y, Xie X, Lee DH, et al. Prevalence, incidence, and outcome of non-alcoholic fatty liver disease in Asia, 1999–2019: a systematic review and meta-analysis. Lancet Gastroenterol Hepatol. (2019) 4:389–98. doi: 10.1016/S2468-1253(19)30039-1
3. Le MH, Devaki P, Ha NB, Jun DW, Te HS, Cheung RC, et al. Prevalence of non-alcoholic fatty liver disease and risk factors for advanced fibrosis and mortality in the United States. PLoS ONE. (2017) 12:e0173499. doi: 10.1371/journal.pone.0173499
4. Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. (2018) 15:11–20. doi: 10.1038/nrgastro.2017.109
5. Rafiq N, Bai C, Fang Y, Srishord M, McCullough A, Gramlich T, et al. Long-term follow-up of patients with nonalcoholic fatty liver. Clin Gastroenterol Hepatol. (2009) 7:234–8. doi: 10.1016/j.cgh.2008.11.005
6. Nobili V, Alisi A, Newton KP, Schwimmer JB. Comparison of the phenotype and approach to pediatric vs adult patients with nonalcoholic fatty liver disease. Gastroenterology. (2016) 150:1798–810. doi: 10.1053/j.gastro.2016.03.009
7. Zhang X, Ji X, Wang Q, Li JZ. New insight into inter-organ crosstalk contributing to the pathogenesis of non-alcoholic fatty liver disease (NAFLD). Protein Cell. (2018) 9:164–77. doi: 10.1007/s13238-017-0436-0
8. Sanyal AJ, Campbell-Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, et al. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology. (2001) 120:1183–92. doi: 10.1053/gast.2001.23256
9. Di Sessa A, Umano GR, Miraglia Del Giudice E, Santoro N. From the liver to the heart: cardiac dysfunction in obese children with non-alcoholic fatty liver disease. World J Hepatol. (2017) 9:69–73. doi: 10.4254/wjh.v9.i2.69
10. Byrne CD, Targher G. NAFLD as a driver of chronic kidney disease. J Hepatol. (2020) 72:785–801. doi: 10.1016/j.jhep.2020.01.013
11. Di Costanzo A, Pacifico L, D'Erasmo L, Polito L, Martino MD, Perla FM, et al. Nonalcoholic Fatty Liver Disease (NAFLD), but not its susceptibility gene variants, influences the decrease of kidney function in overweight/obese children. Int J Mol Sci. (2019) 20:4444. doi: 10.3390/ijms20184444
12. Eslam M, Sanyal AJ, George J, International Consensus P. MAFLD: a consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology. (2020) 158:1999–2014. doi: 10.1053/j.gastro.2019.11.312
13. Godoy-Matos AF, Silva Junior WS, Valerio CM. NAFLD as a continuum: from obesity to metabolic syndrome and diabetes. Diabetol Metab Syndr. (2020) 12:60. doi: 10.1186/s13098-020-00570-y
14. Prokopowicz Z, Malecka-Tendera E, Matusik P. Predictive value of adiposity level, metabolic syndrome, and insulin resistance for the risk of nonalcoholic fatty liver disease diagnosis in obese children. Can J Gastroenterol Hepatol. (2018) 2018:9465784. doi: 10.1155/2018/9465784
15. Harrison SA, Oliver D, Arnold HL, Gogia S, Neuschwander-Tetri BA. Development and validation of a simple NAFLD clinical scoring system for identifying patients without advanced disease. Gut. (2008) 57:1441–7. doi: 10.1136/gut.2007.146019
16. Anderson EL, Howe LD, Jones HE, Higgins JP, Lawlor DA, Fraser A. The prevalence of non-alcoholic fatty liver disease in children and adolescents: a systematic review and meta-analysis. PLoS ONE. (2015) 10:e0140908. doi: 10.1371/journal.pone.0140908
17. Kleiner DE, Makhlouf HR. Histology of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis in adults and children. Clin Liver Dis. (2016) 20:293–312. doi: 10.1016/j.cld.2015.10.011
18. Brunt EM. Pathology of nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol. (2010) 7:195–203. doi: 10.1038/nrgastro.2010.21
19. Angulo P, Kleiner DE, Dam-Larsen S, Adams LA, Bjornsson ES, Charatcharoenwitthaya P, et al. Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology. (2015) 149:389–97.e10. doi: 10.1053/j.gastro.2015.04.043
20. Crespo M, Lappe S, Feldstein AE, Alkhouri N. Similarities and differences between pediatric and adult nonalcoholic fatty liver disease. Metabolism. (2016) 65:1161–71. doi: 10.1016/j.metabol.2016.01.008
21. Fitzpatrick E, Dhawan A. Childhood and adolescent nonalcoholic fatty liver disease: is it different from adults? J Clin Exp Hepatol. (2019) 9:716–22. doi: 10.1016/j.jceh.2019.05.005
22. Schwimmer JB, Behling C, Newbury R, Deutsch R, Nievergelt C, Schork NJ, et al. Histopathology of pediatric nonalcoholic fatty liver disease. Hepatology. (2005) 42:641–9. doi: 10.1002/hep.20842
23. Goldner D, Lavine JE. Nonalcoholic fatty liver disease in children: unique considerations and challenges. Gastroenterology. (2020) 158:1967–83.e1. doi: 10.1053/j.gastro.2020.01.048
24. Sherif ZA, Saeed A, Ghavimi S, Nouraie SM, Laiyemo AO, Brim H, et al. Global epidemiology of nonalcoholic fatty liver disease and perspectives on US minority populations. Dig Dis Sci. (2016) 61:1214–25. doi: 10.1007/s10620-016-4143-0
25. Loomba R, Sirlin CB, Schwimmer JB, Lavine JE. Advances in pediatric nonalcoholic fatty liver disease. Hepatology. (2009) 50:1282–93. doi: 10.1002/hep.23119
26. Yu EL, Golshan S, Harlow KE, Angeles JE, Durelle J, Goyal NP, et al. Prevalence of nonalcoholic fatty liver disease in children with obesity. J Pediatr. (2019) 207:64–70. doi: 10.1016/j.jpeds.2018.11.021
27. Kim D, Kim WR. Nonobese fatty liver disease. Clin Gastroenterol Hepatol. (2017) 15:474–85. doi: 10.1016/j.cgh.2016.08.028
28. VanWagner LB, Armstrong MJ. Lean NAFLD: a not so benign condition? Hepatol Commun. (2018) 2:5–8. doi: 10.1002/hep4.1143
29. Weston SR, Leyden W, Murphy R, Bass NM, Bell BP, Manos MM, et al. Racial and ethnic distribution of nonalcoholic fatty liver in persons with newly diagnosed chronic liver disease. Hepatology. (2005) 41:372–9. doi: 10.1002/hep.20554
30. Marzuillo P, del Miraglia Giudice E, Santoro N. Pediatric fatty liver disease: role of ethnicity and genetics. World J Gastroenterol. (2014) 20:7347–55. doi: 10.3748/wjg.v20.i23.7347
31. Schwimmer JB, McGreal N, Deutsch R, Finegold MJ, Lavine JE. Influence of gender, race, and ethnicity on suspected fatty liver in obese adolescents. Pediatrics. (2005) 115:e561–5. doi: 10.1542/peds.2004-1832
32. Loomba R, Sanyal AJ. The global NAFLD epidemic. Nat Rev Gastroenterol Hepatol. (2013) 10:686–90. doi: 10.1038/nrgastro.2013.171
33. Fan JG, Kim SU, Wong VW. New trends on obesity and NAFLD in Asia. J Hepatol. (2017) 67:862–73. doi: 10.1016/j.jhep.2017.06.003
34. Szanto KB, Li J, Cordero P, Oben JA. Ethnic differences and heterogeneity in genetic and metabolic makeup contributing to nonalcoholic fatty liver disease. Diabetes Metab Syndr Obes. (2019) 12:357–67. doi: 10.2147/DMSO.S182331
35. Loomba R, Schork N, Chen CH, Bettencourt R, Bhatt A, Ang B, et al. Heritability of hepatic fibrosis and steatosis based on a prospective twin study. Gastroenterology. (2015) 149:1784–93. doi: 10.1053/j.gastro.2015.08.011
36. Dongiovanni P, Anstee QM, Valenti L. Genetic predisposition in NAFLD and NASH impact on severity of liver disease and response to treatment. Curr Pharm Design. (2013) 19:5219–38. doi: 10.2174/13816128113199990381
37. Anstee QM, Daly AK, Day CP. Genetic modifiers of non-alcoholic fatty liver disease progression. Biochim Biophys Acta. (2011) 1812:1557–66. doi: 10.1016/j.bbadis.2011.07.017
38. Barbara M, Scott A, Alkhouri N. New insights into genetic predisposition and novel therapeutic targets for nonalcoholic fatty liver disease. Hepatobiliary Surg Nutr. (2018) 7:372–81. doi: 10.21037/hbsn.2018.08.05
39. Day CP. Genetic and environmental susceptibility to non-alcoholic fatty liver disease. Dig Dis. (2010) 28:255–60. doi: 10.1159/000282098
40. Kotronen A, Peltonen M, Hakkarainen A, Sevastianova K, Bergholm R, Johansson LM, et al. Prediction of non-alcoholic fatty liver disease and liver fat using metabolic and genetic factors. Gastroenterology. (2009) 137:865–72. doi: 10.1053/j.gastro.2009.06.005
41. Larrieta-Carrasco E, Leon-Mimila P, Villarreal-Molina T, Villamil-Ramirez H, Romero-Hidalgo S, Jacobo-Albavera L, et al. Association of the I148M/PNPLA3 variant with elevated alanine transaminase levels in normal-weight and overweight/obese Mexican children. Gene. (2013) 520:185–8. doi: 10.1016/j.gene.2013.03.038
42. Lin YC, Chang PF, Hu FC, Yang WS, Chang MH, Ni YH. A common variant in the PNPLA3 gene is a risk factor for non-alcoholic fatty liver disease in obese Taiwanese children. J Pediatr. (2011) 158:740–4. doi: 10.1016/j.jpeds.2010.11.016
43. Goran MI, Walker R, Le KA, Mahurkar S, Vikman S, Davis JN, et al. Effects of PNPLA3 on liver fat and metabolic profile in Hispanic children and adolescents. Diabetes. (2010) 59:3127–30. doi: 10.2337/db10-0554
44. Valenti L, Alisi A, Galmozzi E, Bartuli A, Del Menico B, Alterio A, et al. I148M patatin-like phospholipase domain-containing 3 gene variant and severity of pediatric nonalcoholic fatty liver disease. Hepatology. (2010) 52:1274–80. doi: 10.1002/hep.23823
45. Grandone A, Cozzolino D, Marzuillo P, Cirillo G, Di Sessa A, Ruggiero L, et al. TM6SF2 Glu167Lys polymorphism is associated with low levels of LDL-cholesterol and increased liver injury in obese children. Pediatr Obes. (2016) 11:115–9. doi: 10.1111/ijpo.12032
46. Goffredo M, Caprio S, Feldstein AE, D'Adamo E, Shaw MM, Pierpont B, et al. Role of TM6SF2 rs58542926 in the pathogenesis of nonalcoholic pediatric fatty liver disease: a multiethnic study. Hepatology. (2016) 63:117–25. doi: 10.1002/hep.28283
47. Lin YC, Chang PF, Chang MH, Ni YH. Genetic determinants of hepatic steatosis and serum cytokeratin-18 fragment levels in Taiwanese children. Liver Int. (2018) 38:1300–7. doi: 10.1111/liv.13689
48. Lin YC, Chang PF, Chang MH, Ni YH. Genetic variants in GCKR and PNPLA3 confer susceptibility to nonalcoholic fatty liver disease in obese individuals. Am J Clin Nutr. (2014) 99:869–74. doi: 10.3945/ajcn.113.079749
49. Santoro N, Zhang CK, Zhao H, Pakstis AJ, Kim G, Kursawe R, et al. Variant in the glucokinase regulatory protein (GCKR) gene is associated with fatty liver in obese children and adolescents. Hepatology. (2012) 55:781–9. doi: 10.1002/hep.24806
50. Di Sessa A, Umano GR, Cirillo G, Del Prete A, Iacomino R, Marzuillo P, et al. The membrane-bound O-acyltransferase7 rs641738 variant in pediatric nonalcoholic fatty liver disease. J Pediatr Gastroenterol Nutr. (2018) 67:69–74. doi: 10.1097/MPG.0000000000001979
51. Viitasalo A, Eloranta AM, Atalay M, Romeo S, Pihlajamaki J, Lakka TA. Association of MBOAT7 gene variant with plasma ALT levels in children: the PANIC study. Pediatr Res. (2016) 80:651–5. doi: 10.1038/pr.2016.139
52. Umano GR, Caprio S, Di Sessa A, Chalasani N, Dykas DJ, Pierpont B, et al. The rs626283 variant in the MBOAT7 gene is associated with insulin resistance and fatty liver in caucasian obese youth. Am J Gastroenterol. (2018) 113:376–83. doi: 10.1038/ajg.2018.1
53. Di Sessa A, Umano GR, Cirillo G, Marzuillo P, Arienzo MR, Pedulla M, et al. The rs72613567: TA variant in the hydroxysteroid 17-beta dehydrogenase 13 gene reduces liver damage in obese children. J Pediatr Gastroenterol Nutr. (2020) 70:371–4. doi: 10.1097/MPG.0000000000002573
54. Lin YC, Chang PF, Lin HF, Liu K, Chang MH, Ni YH. Variants in the autophagy-related gene IRGM confer susceptibility to non-alcoholic fatty liver disease by modulating lipophagy. J Hepatol. (2016) 65:1209–16. doi: 10.1016/j.jhep.2016.06.029
55. Bellini G, Miraglia Del Giudice E, Nobili V, Rossi F. The IRGM rs10065172 variant increases the risk for steatosis but not for liver damage progression in Italian obese children. J Hepatol. (2017) 67:653–5. doi: 10.1016/j.jhep.2017.02.037
56. Dai D, Wen F, Zhou S, Su Z, Liu G, Wang M, et al. Association of MTTP gene variants with pediatric NAFLD: a candidate-gene-based analysis of single nucleotide variations in obese children. PLoS ONE. (2017) 12:e0185396. doi: 10.1371/journal.pone.0185396
57. Valenti L, Motta BM, Alisi A, Sartorelli R, Buonaiuto G, Dongiovanni P, et al. LPIN1 rs13412852 polymorphism in pediatric nonalcoholic fatty liver disease. J Pediatr Gastroenterol Nutr. (2012) 54:588–93. doi: 10.1097/MPG.0b013e3182442a55
58. Dongiovanni P, Valenti L, Rametta R, Daly AK, Nobili V, Mozzi E, et al. Genetic variants regulating insulin receptor signalling are associated with the severity of liver damage in patients with non-alcoholic fatty liver disease. Gut. (2010) 59:267–73. doi: 10.1136/gut.2009.190801
59. Hudert CA, Selinski S, Rudolph B, Blaker H, Loddenkemper C, Thielhorn R, et al. Genetic determinants of steatosis and fibrosis progression in paediatric non-alcoholic fatty liver disease. Liver Int. (2019) 39:540–56. doi: 10.1111/liv.14006
60. Marzuillo P, Grandone A, Conte M, Capuano F, Cirillo G, Di Sessa A, et al. Novel association between a nonsynonymous variant (R270H) of the G-protein-coupled receptor 120 and liver injury in children and adolescents with obesity. J Pediatr Gastroenterol Nutr. (2014) 59:472–5. doi: 10.1097/MPG.0000000000000463
61. Lin YC, Chang PF, Hu FC, Chang MH, Ni YH. Variants in the UGT1A1 gene and the risk of pediatric nonalcoholic fatty liver disease. Pediatrics. (2009) 124:e1221–7. doi: 10.1542/peds.2008-3087
62. Lin Y-C, Chang P-F, Chang M-H, Ni Y-H. A common variant in the peroxisome proliferator–activated receptor-γ coactivator-1α gene is associated with nonalcoholic fatty liver disease in obese children. Am J Clin Nutr. (2013) 97:326–31. doi: 10.3945/ajcn.112.046417
63. Chang PF, Lin YC, Liu K, Yeh SJ, Ni YH. Heme oxygenase-1 gene promoter polymorphism and the risk of pediatric nonalcoholic fatty liver disease. Int J Obes. (2015) 39:1236–40. doi: 10.1038/ijo.2015.46
64. Rossi F, Bellini G, Alisi A, Alterio A, Maione S, Perrone L, et al. Cannabinoid receptor type 2 functional variant influences liver damage in children with non-alcoholic fatty liver disease. PLoS ONE. (2012) 7:e42259. doi: 10.1371/journal.pone.0042259
65. Dongiovanni P, Crudele A, Panera N, Romito I, Meroni M, De Stefanis C, et al. beta-Klotho gene variation is associated with liver damage in children with NAFLD. J Hepatol. (2020) 72:411–19. doi: 10.1016/j.jhep.2019.10.011
66. Nobili V, Donati B, Panera N, Vongsakulyanon A, Alisi A, Dallapiccola B, et al. A 4-polymorphism risk score predicts steatohepatitis in children with nonalcoholic fatty liver disease. J Pediatr Gastroenterol Nutr. (2014) 58:632–6. doi: 10.1097/MPG.0000000000000279
67. Santoro N, Feldstein AE, Enoksson E, Pierpont B, Kursawe R, Kim G, et al. The association between hepatic fat content and liver injury in obese children and adolescents: effects of ethnicity, insulin resistance, and common gene variants. Diabetes Care. (2013) 36:1353–60. doi: 10.2337/dc12-1791
68. Huang Y, He S, Li JZ, Seo YK, Osborne TF, Cohen JC, et al. A feed-forward loop amplifies nutritional regulation of PNPLA3. Proc Natl Acad Sci USA. (2010) 107:7892–7. doi: 10.1073/pnas.1003585107
69. Wilson PA, Gardner SD, Lambie NM, Commans SA, Crowther DJ. Characterization of the human patatin-like phospholipase family. J Lipid Res. (2006) 47:1940–9. doi: 10.1194/jlr.M600185-JLR200
70. Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. (2008) 40:1461–5. doi: 10.1038/ng.257
71. Romeo S, Sentinelli F, Cambuli VM, Incani M, Congiu T, Matta V, et al. The 148M allele of the PNPLA3 gene is associated with indices of liver damage early in life. J Hepatol. (2010) 53:335–8. doi: 10.1016/j.jhep.2010.02.034
72. Grimaudo S, Pipitone RM, Pennisi G, Celsa C, Camma C, Di Marco V, et al. Association between PNPLA3 rs738409 C>G variant and liver-related outcomes in patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. (2020) 18:935–44.e3. doi: 10.1016/j.cgh.2019.08.011
73. Liu YM, Moldes M, Bastard JP, Bruckert E, Viguerie N, Hainque B, et al. Adiponutrin: a new gene regulated by energy balance in human adipose tissue. J Clin Endocrinol Metab. (2004) 89:2684–9. doi: 10.1210/jc.2003-031978
74. BasuRay S, Smagris E, Cohen JC, Hobbs HH. The PNPLA3 variant associated with fatty liver disease (I148M) accumulates on lipid droplets by evading ubiquitylation. Hepatology. (2017) 66:1111–24. doi: 10.1002/hep.29273
75. Pirazzi C, Adiels M, Burza MA, Mancina RM, Levin M, Stahlman M, et al. Patatin-like phospholipase domain-containing 3 (PNPLA3) I148M (rs738409) affects hepatic VLDL secretion in humans and in vitro. J Hepatol. (2012) 57:1276–82. doi: 10.1016/j.jhep.2012.07.030
76. Sookoian S, Castano GO, Burgueno AL, Gianotti TF, Rosselli MS, Pirola CJ. A nonsynonymous gene variant in the adiponutrin gene is associated with nonalcoholic fatty liver disease severity. J Lipid Res. (2009) 50:2111–6. doi: 10.1194/jlr.P900013-JLR200
77. Pirazzi C, Valenti L, Motta BM, Pingitore P, Hedfalk K, Mancina RM, et al. PNPLA3 has retinyl-palmitate lipase activity in human hepatic stellate cells. Hum Mol Genet. (2014) 23:4077–85. doi: 10.1093/hmg/ddu121
78. Pingitore P, Dongiovanni P, Motta BM, Meroni M, Lepore SM, Mancina RM, et al. PNPLA3 overexpression results in reduction of proteins predisposing to fibrosis. Hum Mol Genet. (2016) 25:5212–22. doi: 10.1093/hmg/ddw341
79. Bruschi FV, Claudel T, Tardelli M, Caligiuri A, Stulnig TM, Marra F, et al. The PNPLA3 I148M variant modulates the fibrogenic phenotype of human hepatic stellate cells. Hepatology. (2017) 65:1875–90. doi: 10.1002/hep.29041
80. Sookoian S, Pirola CJ. Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology. (2011) 53:1883–94. doi: 10.1002/hep.24283
81. Linden D, Ahnmark A, Pingitore P, Ciociola E, Ahlstedt I, Andreasson AC, et al. Pnpla3 silencing with antisense oligonucleotides ameliorates nonalcoholic steatohepatitis and fibrosis in Pnpla3 I148M knock-in mice. Mol Metab. (2019) 22:49–61. doi: 10.1016/j.molmet.2019.01.013
82. Marzuillo P, Di Sessa A, Guarino S, Capalbo D, Umano GR, Pedulla M, et al. Nonalcoholic fatty liver disease and eGFR levels could be linked by the PNPLA3 I148M polymorphism in children with obesity. Pediatr Obes. (2019) 14:e12539. doi: 10.1111/ijpo.12539
83. Targher G, Mantovani A, Alisi A, Mosca A, Panera N, Byrne CD, et al. Relationship between PNPLA3 rs738409 polymorphism and decreased kidney function in children with NAFLD. Hepatology. (2019) 70:142–53. doi: 10.1002/hep.30625
84. Smagris E, Gilyard S, BasuRay S, Cohen JC, Hobbs HH. Inactivation of Tm6sf2, a gene defective in fatty liver disease, impairs lipidation but not secretion of very low density lipoproteins. J Biol Chem. (2016) 291:10659–76. doi: 10.1074/jbc.M116.719955
85. Kozlitina J, Smagris E, Stender S, Nordestgaard BG, Zhou HH, Tybjaerg-Hansen A, et al. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. (2014) 46:352–6. doi: 10.1038/ng.2901
86. Luukkonen PK, Zhou Y, Nidhina Haridas PA, Dwivedi OP, Hyotylainen T, Ali A, et al. Impaired hepatic lipid synthesis from polyunsaturated fatty acids in TM6SF2 E167K variant carriers with NAFLD. J Hepatol. (2017) 67:128–36. doi: 10.1016/j.jhep.2017.02.014
87. Liu YL, Reeves HL, Burt AD, Tiniakos D, McPherson S, Leathart JB, et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat Commun. (2014) 5:4309. doi: 10.1038/ncomms5309
88. Ehrhardt N, Doche ME, Chen S, Mao HZ, Walsh MT, Bedoya C, et al. Hepatic Tm6sf2 overexpression affects cellular ApoB-trafficking, plasma lipid levels, hepatic steatosis and atherosclerosis. Hum Mol Genet. (2017) 26:2719–31. doi: 10.1093/hmg/ddx159
89. Viitasalo A, Pihlajamaki J, Paananen J, Atalay M, Lindi V, Lakka TA. Associations of TM6SF2 167K allele with liver enzymes and lipid profile in children: the PANIC study. Pediatr Res. (2016) 79:684–8. doi: 10.1038/pr.2016.3
90. Prill S, Caddeo A, Baselli G, Jamialahmadi O, Dongiovanni P, Rametta R, et al. The TM6SF2 E167K genetic variant induces lipid biosynthesis and reduces apolipoprotein B secretion in human hepatic 3D spheroids. Sci Rep. (2019) 9:11585. doi: 10.1038/s41598-019-47737-w
91. Marzuillo P, Di Sessa A, Cirillo G, Umano GR, Pedulla M, La Manna A, et al. Transmembrane 6 superfamily member 2 167K allele improves renal function in children with obesity. Pediatr Res. (2020) 88:300–4. doi: 10.1038/s41390-020-0753-5
92. Musso G, Cassader M, Gambino R. PNPLA3 rs738409 and TM6SF2 rs58542926 gene variants affect renal disease and function in nonalcoholic fatty liver disease. Hepatology. (2015) 62:658–9. doi: 10.1002/hep.27643
93. Rees MG, Wincovitch S, Schultz J, Waterstradt R, Beer NL, Baltrusch S, et al. Cellular characterisation of the GCKR P446L variant associated with type 2 diabetes risk. Diabetologia. (2012) 55:114–22. doi: 10.1007/s00125-011-2348-5
94. Speliotes EK, Yerges-Armstrong LM, Wu J, Hernaez R, Kim LJ, Palmer CD, et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. (2011) 7:e1001324. doi: 10.1371/journal.pgen.1001324
95. Zain SM, Mohamed Z, Mohamed R. Common variant in the glucokinase regulatory gene rs780094 and risk of nonalcoholic fatty liver disease: a meta-analysis. J Gastroenterol Hepatol. (2015) 30:21–7. doi: 10.1111/jgh.12714
96. Fernandes Silva L, Vangipurapu J, Kuulasmaa T, Laakso M. An intronic variant in the GCKR gene is associated with multiple lipids. Sci Rep. (2019) 9:10240. doi: 10.1038/s41598-019-46750-3
97. Petta S, Miele L, Bugianesi E, Camma C, Rosso C, Boccia S, et al. Glucokinase regulatory protein gene polymorphism affects liver fibrosis in non-alcoholic fatty liver disease. PLoS ONE. (2014) 9:e87523. doi: 10.1371/journal.pone.0087523
98. Stender S, Kozlitina J, Nordestgaard BG, Tybjaerg-Hansen A, Hobbs HH, Cohen JC. Adiposity amplifies the genetic risk of fatty liver disease conferred by multiple loci. Nat Genet. (2017) 49:842–7. doi: 10.1038/ng.3855
99. Beer NL, Tribble ND, McCulloch LJ, Roos C, Johnson PR, Orho-Melander M, et al. The P446L variant in GCKR associated with fasting plasma glucose and triglyceride levels exerts its effect through increased glucokinase activity in liver. Hum Mol Genet. (2009) 18:4081–8. doi: 10.1093/hmg/ddp357
100. Caddeo A, Jamialahmadi O, Solinas G, Pujia A, Mancina RM, Pingitore P, et al. MBOAT7 is anchored to endomembranes by six transmembrane domains. J Struct Biol. (2019) 206:349–60. doi: 10.1016/j.jsb.2019.04.006
101. Tanaka Y, Shimanaka Y, Caddeo A, Kubo T, Mao Y, Kubota T, et al. LPIAT1/MBOAT7 depletion increases triglyceride synthesis fueled by high phosphatidylinositol turnover. Gut. (2020). doi: 10.1136/gutjnl-2020-320646. [Epub ahead of print].
102. Meroni M, Longo M, Fracanzani AL, Dongiovanni P. MBOAT7 down-regulation by genetic and environmental factors predisposes to MAFLD. EBioMedicine. (2020) 57:102866. doi: 10.1016/j.ebiom.2020.102866
103. Mancina RM, Dongiovanni P, Petta S, Pingitore P, Meroni M, Rametta R, et al. The MBOAT7-TMC4 variant rs641738 increases risk of nonalcoholic fatty liver disease in individuals of European descent. Gastroenterology. (2016) 150:1219–30.e6. doi: 10.1053/j.gastro.2016.01.032
104. Krawczyk M, Rau M, Schattenberg JM, Bantel H, Pathil A, Demir M, et al. Combined effects of the PNPLA3 rs738409, TM6SF2 rs58542926, and MBOAT7 rs641738 variants on NAFLD severity: a multicenter biopsy-based study. J Lipid Res. (2017) 58:247–55. doi: 10.1194/jlr.P067454
105. Helsley RN, Varadharajan V, Brown AL, Gromovsky AD, Schugar RC, Ramachandiran I, et al. Obesity-linked suppression of membrane-bound O-acyltransferase 7 (MBOAT7) drives non-alcoholic fatty liver disease. Elife. (2019) 8:e49882. doi: 10.7554/eLife.49882
106. Su W, Mao Z, Liu Y, Zhang X, Zhang W, Gustafsson JA, et al. Role of HSD17B13 in the liver physiology and pathophysiology. Mol Cell Endocrinol. (2019) 489:119–25. doi: 10.1016/j.mce.2018.10.014
107. Abul-Husn NS, Cheng X, Li AH, Xin Y, Schurmann C, Stevis P, et al. A protein-truncating HSD17B13 variant and protection from chronic liver disease. N Engl J Med. (2018) 378:1096–106. doi: 10.1056/NEJMoa1712191
108. Sookoian S, Arrese M, Pirola CJ. Genetics meets therapy? Exome-wide association study reveals a loss-of-function variant in 17-beta-hydroxysteroid dehydrogenase 13 that protects patients from liver damage and nonalcoholic fatty liver disease progression. Hepatology. (2019) 69:907–10. doi: 10.1002/hep.30209
109. Di Sessa A, Umano GR, Cirillo G, Passaro AP, Verde V, Cozzolino D, et al. Pediatric non-alcoholic fatty liver disease and kidney function: effect of HSD17B13 variant. World J Gastroenterol. (2020) 26:5474–83. doi: 10.3748/wjg.v26.i36.5474
110. Su W, Wang Y, Jia X, Wu W, Li L, Tian X, et al. Comparative proteomic study reveals 17beta-HSD13 as a pathogenic protein in nonalcoholic fatty liver disease. Proc Natl Acad Sci USA. (2014) 111:11437–42. doi: 10.1073/pnas.1410741111
111. Ma Y, Brown PM, Lin DD, Ma J, Feng D, Belyaeva OV, et al. Hsd17b13 deficiency does not protect mice from obesogenic diet injury. Hepatology. (2020). doi: 10.1002/hep.31517. [Epub ahead of print].
112. Ma Y, Karki S, Brown PM, Lin DD, Podszun MC, Zhou W, et al. Characterization of essential domains in HSD17B13 for cellular localization and enzymatic activity. J Lipid Res. (2020) 61:1400–9. doi: 10.1194/jlr.RA120000907
113. Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature. (2009) 458:1131–5. doi: 10.1038/nature07976
114. Chauhan S, Mandell MA, Deretic V. IRGM governs the core autophagy machinery to conduct antimicrobial defense. Mol Cell. (2015) 58:507–21. doi: 10.1016/j.molcel.2015.03.020
115. Schwerbel K, Kamitz A, Krahmer N, Hallahan N, Jahnert M, Gottmann P, et al. Immunity-related GTPase induces lipophagy to prevent excess hepatic lipid accumulation. J Hepatol. (2020) 73:771–82. doi: 10.1016/j.jhep.2020.04.031
116. Hsiao PJ, Lee MY, Wang YT, Jiang HJ, Lin PC, Yang YH, et al. MTTP-297H polymorphism reduced serum cholesterol but increased risk of non-alcoholic fatty liver disease-a cross-sectional study. BMC Med Genet. (2015) 16:93. doi: 10.1186/s12881-015-0242-6
117. Reue K, Zhang P. The lipin protein family: dual roles in lipid biosynthesis and gene expression. FEBS Letters. (2008) 582:90–6. doi: 10.1016/j.febslet.2007.11.014
118. Musso G, Cassader M, De Michieli F, Rosina F, Orlandi F, Gambino R. Nonalcoholic steatohepatitis versus steatosis: adipose tissue insulin resistance and dysfunctional response to fat ingestion predict liver injury and altered glucose and lipoprotein metabolism. Hepatology. (2012) 56:933–42. doi: 10.1002/hep.25739
119. Honma M, Sawada S, Ueno Y, Murakami K, Yamada T, Gao J, et al. Selective insulin resistance with differential expressions of IRS-1 and IRS-2 in human NAFLD livers. Int J Obes. (2018) 42:1544–55. doi: 10.1038/s41366-018-0062-9
120. Yang HR, Chang EJ. Insulin resistance, body composition, and fat distribution in obese children with nonalcoholic fatty liver disease. Asia Pac J Clin Nutr. (2016) 25:126–33. doi: 10.6133/apjcn.2016.25.1.15
121. Bugianesi E, Gastaldelli A, Vanni E, Gambino R, Cassader M, Baldi S, et al. Insulin resistance in non-diabetic patients with non-alcoholic fatty liver disease: sites and mechanisms. Diabetologia. (2005) 48:634–42. doi: 10.1007/s00125-005-1682-x
122. Ibrahim SH, Kohli R, Gores GJ. Mechanisms of lipotoxicity in NAFLD and clinical implications. J Pediatr Gastroenterol Nutr. (2011) 53:131–40. doi: 10.1097/MPG.0b013e31822578db
123. Enooku K, Kondo M, Fujiwara N, Sasako T, Shibahara J, Kado A, et al. Hepatic IRS1 and ss-catenin expression is associated with histological progression and overt diabetes emergence in NAFLD patients. J Gastroenterol. (2018) 53:1261–75. doi: 10.1007/s00535-018-1472-0
124. Dowman JK, Tomlinson JW, Newsome PN. Pathogenesis of non-alcoholic fatty liver disease. QJM. (2010) 103:71–83. doi: 10.1093/qjmed/hcp158
125. Nobili V, Parola M, Alisi A, Marra F, Piemonte F, Mombello C, et al. Oxidative stress parameters in paediatric non-alcoholic fatty liver disease. Int J Mol Med. (2010) 26:471–6. doi: 10.3892/ijmm_00000487
126. Spahis S, Delvin E, Borys JM, Levy E. Oxidative stress as a critical factor in nonalcoholic fatty liver disease pathogenesis. Antioxid Redox Signal. (2017) 26:519–41. doi: 10.1089/ars.2016.6776
127. Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, et al. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell. (2010) 142:687–98. doi: 10.1016/j.cell.2010.07.041
128. Im DS. Functions of omega-3 fatty acids and FFA4 (GPR120) in macrophages. Eur J Pharmacol. (2016) 785:36–43. doi: 10.1016/j.ejphar.2015.03.094
129. Nobili V, Carpino G, Alisi A, De Vito R, Franchitto A, Alpini G, et al. Role of docosahexaenoic acid treatment in improving liver histology in pediatric nonalcoholic fatty liver disease. PLoS ONE. (2014) 9:e88005. doi: 10.1371/journal.pone.0088005
130. Puri K, Nobili V, Melville K, Corte CD, Sartorelli MR, Lopez R, et al. Serum bilirubin level is inversely associated with nonalcoholic steatohepatitis in children. J Pediatr Gastroenterol Nutr. (2013) 57:114–8. doi: 10.1097/MPG.0b013e318291fefe
131. Drummond GS, Baum J, Greenberg M, Lewis D, Abraham NG. HO-1 overexpression and underexpression: clinical implications. Arch Biochem Biophys. (2019) 673:108073. doi: 10.1016/j.abb.2019.108073
132. Han X, Ding C, Zhang G, Pan R, Liu Y, Huang N, et al. Liraglutide ameliorates obesity-related nonalcoholic fatty liver disease by regulating Sestrin2-mediated Nrf2/HO-1 pathway. Biochem Biophys Res Commun. (2020) 525:895–901. doi: 10.1016/j.bbrc.2020.03.032
133. Soyal S, Krempler F, Oberkofler H, Patsch W. PGC-1alpha: a potent transcriptional cofactor involved in the pathogenesis of type 2 diabetes. Diabetologia. (2006) 49:1477–88. doi: 10.1007/s00125-006-0268-6
134. Besse-Patin A, Leveille M, Oropeza D, Nguyen BN, Prat A, Estall JL. Estrogen signals through peroxisome proliferator-activated receptor-gamma coactivator 1alpha to reduce oxidative damage associated with diet-induced fatty liver disease. Gastroenterology. (2017) 152:243–56. doi: 10.1053/j.gastro.2016.09.017
135. Arrese M, Cabrera D, Kalergis AM, Feldstein AE. Innate immunity and inflammation in NAFLD/NASH. Dig Dis Sci. (2016) 61:1294–303. doi: 10.1007/s10620-016-4049-x
136. Teixeira-Clerc F, Belot MP, Manin S, Deveaux V, Cadoudal T, Chobert MN, et al. Beneficial paracrine effects of cannabinoid receptor 2 on liver injury and regeneration. Hepatology. (2010) 52:1046–59. doi: 10.1002/hep.23779
137. Ekstedt M, Hagstrom H, Nasr P, Fredrikson M, Stal P, Kechagias S, et al. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology. (2015) 61:1547–54. doi: 10.1002/hep.27368
138. Miele L, Beale G, Patman G, Nobili V, Leathart J, Grieco A, et al. The Kruppel-like factor 6 genotype is associated with fibrosis in nonalcoholic fatty liver disease. Gastroenterology. (2008) 135:282–91. doi: 10.1053/j.gastro.2008.04.004
139. Carlsson B, Linden D, Brolen G, Liljeblad M, Bjursell M, Romeo S, et al. Review article: the emerging role of genetics in precision medicine for patients with non-alcoholic steatohepatitis. Aliment Pharmacol Ther. (2020) 51:1305–20. doi: 10.1111/apt.15738
140. Giorgio V, Prono F, Graziano F, Nobili V. Pediatric non alcoholic fatty liver disease: old and new concepts on development, progression, metabolic insight and potential treatment targets. BMC Pediatr. (2013) 13:40. doi: 10.1186/1471-2431-13-40
141. Ma J, Hennein R, Liu C, Long MT, Hoffmann U, Jacques PF, et al. Improved diet quality associates with reduction in liver fat, particularly in individuals with high genetic risk scores for nonalcoholic fatty liver disease. Gastroenterology. (2018) 155:107–17. doi: 10.1053/j.gastro.2018.03.038
142. Nobili V, Bedogni G, Donati B, Alisi A, Valenti L. The I148M variant of PNPLA3 reduces the response to docosahexaenoic acid in children with non-alcoholic fatty liver disease. J Med Food. (2013) 16:957–60. doi: 10.1089/jmf.2013.0043
143. Di Costanzo A, Pacifico L, Chiesa C, Perla FM, Ceci F, Angeloni A, et al. Genetic and metabolic predictors of hepatic fat content in a cohort of Italian children with obesity. Pediatr Res. (2019) 85:671–7. doi: 10.1038/s41390-019-0303-1
144. Kovalic AJ, Banerjee P, Tran QT, Singal AK, Satapathy SK. Genetic and epigenetic culprits in the pathogenesis of nonalcoholic fatty liver disease. J Clin Exp Hepatol. (2018) 8:390–402. doi: 10.1016/j.jceh.2018.04.001
145. Skubic C, Drakulic Z, Rozman D. Personalized therapy when tackling nonalcoholic fatty liver disease: a focus on sex, genes, and drugs. Expert Opin Drug Metab Toxicol. (2018) 14:831–41. doi: 10.1080/17425255.2018.1492552
146. Vajro P, Lenta S, Socha P, Dhawan A, McKiernan P, Baumann U, et al. Diagnosis of nonalcoholic fatty liver disease in children and adolescents: position paper of the ESPGHAN Hepatology Committee. J Pediatr Gastroenterol Nutr. (2012) 54:700–13. doi: 10.1097/MPG.0b013e318252a13f
147. Vos MB, Abrams SH, Barlow SE, Caprio S, Daniels SR, Kohli R, et al. NASPGHAN clinical practice guideline for the diagnosis and treatment of nonalcoholic fatty liver disease in children: recommendations from the expert committee on NAFLD (ECON) and the North American Society of Pediatric Gastroenterology, Hepatology and Nutrition (NASPGHAN). J Pediatr Gastroenterol Nutr. (2017) 64:319–34. doi: 10.1097/MPG.0000000000001482
148. European Association for the Study of the L European Association for the Study of D European Association for the Study of O. EASL-EASD-EASO clinical practice guidelines for the management of non-alcoholic fatty liver disease. J Hepatol. (2016) 64:1388–1402.
149. Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, et al. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the study of liver diseases. Hepatology. (2018) 67:328–57. doi: 10.1002/hep.29367
Keywords: fatty liver, genetics, sequence variation, precision medicine, pediatric, children, obesity
Citation: Lin Y-C, Wu C-C and Ni Y-H (2020) New Perspectives on Genetic Prediction for Pediatric Metabolic Associated Fatty Liver Disease. Front. Pediatr. 8:603654. doi: 10.3389/fped.2020.603654
Received: 07 September 2020; Accepted: 17 November 2020;
Published: 09 December 2020.
Edited by:
André Hörning, University Hospital Erlangen, GermanyReviewed by:
Pierluigi Marzuillo, Second University of Naples, ItalyDamjana Rozman, University of Ljubljana, Slovenia
Stefano Romeo, University of Gothenburg, Sweden
Copyright © 2020 Lin, Wu and Ni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yu-Cheng Lin, ZHJsaW55dWNoZW5nQG50dS5lZHUudHc=