 Charlotte V. Hobbs
Charlotte V. Hobbs Alka Khaitan
Alka Khaitan Brian M. Kirmse4
Brian M. Kirmse4
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pediatr. , 24 November 2020
Sec. Pediatric Infectious Diseases
Volume 8 - 2020 | https://doi.org/10.3389/fped.2020.593455
During the COVID-19 pandemic, children have had markedly different clinical presentations and outcomes compared to adults. In the acute phase of infection, younger children are relatively spared the severe consequences reported in adults. Yet, they are uniquely susceptible to the newly described Multisystem Inflammatory Syndrome in Children (MIS-C). This may result from the developmental “immunodeficiency” resulting from a Th2 polarization that starts in utero and is maintained for most of the first decade of life. MIS-C may be due to IgA complexes in a Th2 environment or a Th1-like response to COVID-19 antigens that developed slowly. Alternatively, MIS-C may occur in vulnerable hosts with genetic susceptibilities in other immune and non-immune pathways. Herein, we present a brief overview of the host immune response, virologic and genetic factors, and comparable inflammatory syndromes that may explain the pathophysiology leading to drastic differences in clinical presentation and outcomes of COVID-19 between children and adults.
SARS-CoV-2, a novel coronavirus, was first reported in December 2019 from Wuhan, China. The disease caused by SARS-CoV-2, COVID-19, subsequently spread in pandemic fashion over the following 10 months causing a wide spectrum of clinical symptoms from asymptomatic disease to death (1). Mortality correlated with preexisting inflammatory conditions including obesity, hypertension, sex (mortality rates in males 1.5 higher than in females), and most notably, age (2).
The natural history of COVID-19 in children has been different. In adults, initial reports of case fatality rates approached 15% compared to <1% in children (3). An initial report of a Chinese cohort of 36 children with documented COVID-19 infection revealed relatively mild symptoms (4). A larger cohort of 110 children documented mostly asymptomatic infection with a shorter duration of viral detection (11 vs. 17 days). These early reports indicated that symptomatic children were more likely to have fever, pneumonia, and lymphopenia (5).
Considerable information on COVID-19 is already available based on review of clinical and laboratory data, allowing for molecular, epidemiological, physiologic and clinical disease characterization. Given the information we have thus far, we construct hypotheses to explain some of the variation in infection and clinical outcomes of COVID-19 in children. This report attempts to apply our knowledge of virology, developmental immunology, comparable inflammatory syndromes, and host genetics to explain the drastic differences in clinical presentation and outcomes between children and adults.
Molecular sequencing identified SARS-CoV-2 as a relative of the SARS Coronavirus, most closely related to a bat coronavirus (6). Spread to humans may have occurred via an intermediate host, possibly the pangolin, and perhaps in the setting of exotic food markets (7). Experience from Wuhan suggested viral doubling time was every 2–3 days early in the epidemic (8), and the infection soon was recognized in Europe (9). It is now recognized that viral evolution has resulted in strains dominant in different parts of the world with amino acid mutations that impact infectivity (10).
Using surveillance and contact tracing, earlier studies suggested that children appear to acquire COVID-19 infection at a rate similar to that seen in adults (11). However, other studies document a risk 30% of that seen in exposed adults (12), and more recent mathematical modeling studies based on available epidemiologic data suggest that under age 20, susceptibility to infection is half that of those over age 20 (13). In contrast, a study indicated higher viral loads (lower threshold concentrations on RT PCR nasopharyngeal specimens) in children <5 years of age with mild to moderate COVID-19 (14). Children have been studied as index cases in households, and some studies show that children over age 10 transmit COVID-19 at rates similar to adults (15), but other studies with slightly different age breakdowns suggest this is not the case (16). Indeed, with few exceptions, while children may not suffer from significant disease with COVID-19, their role in transmission remains to be clarified (17), although recent meta-analyses suggest children have a lower risk of being infected with SARS CoV2 and “play a lesser role than adults in transmission at a population level (18).” These factors have significant implications for back-to-school programs for children and need to be evaluated carefully in the context of existing data and return-to-school programs ongoing in other countries, such as Denmark. Indeed, a recent study in Germany suggested that child to child transmission, in the context of reduced class size, face mask wearing (inside), exclusion of sick children, and with frequent ventilation of rooms, even with limited physical distancing among children, resulted in the maintenance of low COVID-19 transmission rates in children (19), and summer camps with stricter COVID-related measures (20). Nonetheless, transmission from children infected in daycare settings has been demonstrated (21). In addition, with heterogeneous approaches and persistently high levels of community transmission in the context of schools returning to in-person learning, such as is the current situation in states like Mississippi (Hobbs, C. pers. comm.), other locations will continue to suffer higher rates of pediatric COVID-19 infection (22).
The major host target for SARS-CoV-2 is the ACE type 2 receptor (ACEr2). This receptor target distinguishes SARS-CoV-2 from bat coronaviruses, SARS and MERS. Infection of pulmonary and other epithelial cell types is facilitated by a protease, particularly a membrane embedded cysteine cell protease, TMPRSS2 (23). The viral entry receptor, ACEr2 is expressed mainly in lung type 2 alveoli but also in some nasal epithelial cells, and there is limited expression on pulmonary alveolar cells (24). It is also present in blood vessels, kidneys, cardiac and neural tissue (25). It has been speculated that younger children suffer less severe acute COVID-19 due to reduced ACEr2 expression (26), although recent studies have shown younger children have lower ACEr2 expression but not viral load (27). Expression is a function of clinical states (e.g., hypertension) and the presence of TMPRSS2 (a serine protease encoded on Chromosome 21) which increases ACEr2 expression, allows the virus to enter the cell after cleavage. Recently, it has been shown that COVID-19 can also infect small intestinal enterocytes via TMPRSS2 and TMPRSS4, another serine protease which facilitates fusogenic function of the virus (28). Androgen levels increase the protease expression (29), perhaps explaining the increase in infection among males. Importantly, ACEr2 gene expression is lower in children (30).
Innate responses are variable in early life and generally below what is seen in adults, but likely sufficient to deal with low levels of virus seen early in infection. Early antiviral defense is mediated by the innate immune system with a variety of extracellular RNA sensors (TLRs) and intracellular sensors (IFIH1/MDA5, ZBP1, DDX58, RIG-1) to elicit a protective response. These innate defenses are genetically regulated and vary across different species (31). A variety of cytokines and chemokines, often under genetic control, would be expected to activate inflammatory responses (STAT1 or STAT3) that could eliminate low-level infection. This involves innate immune natural killer cells and polymorphonuclear cells. A recent study by transcriptomics highlights the importance of activated dendritic and neutrophils in the respiratory tract of subjects with COVID-19 (32). Viral antigens have been shown to subvert these responses. Innate immune responses (particularly IFNλ or IL28/29), play a significant role in determining the likelihood of developing bronchiolitis after RSV infection (33). One study showed that an interferon response element (IFTM3) is associated with excessive immune response to virus in an age dependent manner (34). Interferon lambda, a cytokine in the IL-10 family of cytokines (35) made by epithelial and dendritic cells, plays a significant role in defense against low levels of respiratory viruses. It binds to IFNLR1 and IL10RB, eliciting responses different from that of IFN1/2 (36). Different allelic variants of an IFN lambda SNP affect the outcome of Dengue infection in children, suggesting such genetic variation may be at play with other RNA viruses (37).
If early immune responses fail to eliminate virus, adaptive responses are initiated to interfere with pulmonary infection. Local mucosal responses precede pulmonary responses and might be capable of eliminating virus sufficiently to modify pulmonary disease. A recent publication suggests that T cell trafficking to different target organs is highly associated with sex and age, perhaps determining where inflammation will occur (38). Although lymphocytopenia is associated with disease activity, this may be a result of trafficking from blood to target organs. Autopsy material reveals an influx of mononuclear T cells, particularly Th1 CD4 T cells and Tc1 CD8 T cells. Also seen are microthrombi and fibrin, possibly trapping viral antigens (39, 40). An excessive release of proinflammatory IL-12, interferon gamma (IFNγ), tumor necrosis factor (TNF) and IL-6 produce a “cytokines storm” which can lead to tissue damage and death (39, 40). Subsequent “control” of viral infection may be impaired due to immune exhaustion of the T cells (41), perhaps prolonging the clinical state in the face of other inflammation. In this later stage, treatment with methylprednisolone and/or anti-IL6 monoclonal antibodies may reverse the storm and slow down the damage (42).
An important factor determining T cell tropism is antigen concentration (43). Like other coronaviruses, there are several proteins involved in viral replication, the S or spike protein, the M or matrix protein, the E or envelope protein, and the N or nucleoprotein. Viral titer in the throat, peaks in the 1st week and declines by day 14. Antibody is documented in some by day 7 and most by day 14. They consist of IgM, IgA and IgG to spike or nucleoprotein antigens. The appearance of antibody did not preclude the ability to detect virus in some as they may not be neutralizing antibodies (44). Moreover, some individuals with mild SARS-CoV-2 infection developed T cell responses in the absence of antibody seroconversion (45). Experience derived from SARS and MERS showed that neutralizing antibody and B cell responses fade after 1–2 years (46). However, HLA-restricted central memory CD4 and effector memory CD8 T cell responses to spike proteins persist for several years (47, 48).
In children, adaptive responses are more Th2 polarized (49). This holds true for responses to respiratory viruses. When there is pulmonary pathology seen, airway CD8 Tc1 response is seen (50). Th2 responses downmodulate Th1 responses which likely induces tissue damage of infected cells. This Th2 predominant response would ameliorate the cytokine “storm” seen in adults and resulting in significant clinical disease. After infection with the SARS virus in children, high levels of IL-1 were seen in the 1st week. The traditional Th1 cytokines, IL-6 and TNF, that were high in adults with COVID-19, were not overly expressed at first and declined thereafter in children (51).
Children commonly experience multiple viral infections, such as rhinovirus, RSV, and influenza, causing respiratory symptoms, including rhinorrhea in most, and often cough and gastrointestinal complaints (52). Previous infection or coinfection with these may modify the immune response to the COVID-19 virus. Children are frequently infected with multiple agents at the same time (53–55). However, there is a paucity of data related to coinfections due to shortage of viral transport media paired with lack of availability to make home-brew media based on CDC recommendations1, and so the true co-infection rates at this time require further study.
Seasonal human coronaviruses HCoV-229E, -NL63, -OC43, and -HKU1 contribute to a considerable share of upper and lower respiratory tract infection in children. The majority of children are seropositive for HCoV-229E and -NL63 by the age of 3.5 years (56, 57). Interestingly, there may be cross-reactive immunity between seasonal human coronaviruses and COVID-19. In one study of samples stored prior to the COVID-19 pandemic, nearly 50% of healthy adults were found to have SARS-CoV2-specific CD4 T cells (58). In this study, there were also SARS-CoV2-specific CD8 T cells in a smaller subset of COVID-19 unexposed adults. It is conceivable that these pre-existing SARS-CoV2-specific T cells contribute to an overly robust T cell response after COVID-19 infection leading to cytokine storm and severe disease. Young children may be naïve to seasonal human coronavirus strains or mount weaker T cell responses that cross-react with SARS-CoV2, ironically protecting them from cytokine storm and the inflammatory process associated with severe disease. Indeed, among children hospitalized in intensive care units, over half were above the age of 10 years (59), which parallels when we would expect higher seropositivity to seasonal human coronaviruses and immunologic maturity that has shifted from Th2 to Th1 predominance and stronger T cell responses.
Conversely, smaller studies have shown that children who develop multi-system inflammatory syndrome in children (MIS-C) were less likely to have antibodies to seasonal coronaviruses: this could be itself because MIS-C children represent an older age group, or because having these antibodies plays a role in controlling later hyperinflammatory COVID-19 associated complications (60). Interestingly, a study from Seattle Children's in the early pandemic showed a surprisingly low seroprevalence of SARS CoV2 antibody in children, although neutralizing antibody activity was detected in children in whom SARS CoV2 infection had not been suspected (61). Perhaps this is not surprising though given that we know in adults, detectable antibody is more likely to be seen those who are symptomatic (62). Moreover, some individuals with mild SARS-CoV-2 infection develop T cell responses in the absence of antibody seroconversion (45). In addition, a recent study examining immune responses in pediatric compared with adult patients with COVID-19 infection (and for pediatrics, acute COVID-19 and MIS-C patients were included) found that adults with COVID-19 were more likely to have neutralizing antibody titers compared with children, as well as lower IL-17A and interferon gamma responses, and children in this study did not have differences in anti-spike antibody titers when compared to adults (63). Of note, some of these studies so far have relatively small numbers and larger studies to clarify the immune response profile distinctions amongst children with acute COVID-19 compared with MIS-C compared with adults with acute COVID-19 (severe and less severe/asymptomatic) are needed, especially in the context of the COVID-19 vaccine development efforts.
In adults, coexisting health issues associated with inflammatory states such as obesity, type 2 diabetes, cardiac disease with impaired function, pulmonary insufficiency associated with exposure to respiratory toxins, liver disease facilitated by alcohol exposure, and declining renal function were associated with worse outcomes (64). Similarly, a recent report from pediatric ICUs in the United States revealed that among 48 COVID-19 infected children the mortality was 4% (59). Almost 90% of these children had preexisting comorbidities (59). Management of acute COVID-19 based on available evidence is reviewed in the Infectious Disease Society of America COVID-19 Guidelines (65).
However, the spectrum of disease in children continues to be described, and co-morbidities in children, as they are in adults, may not account for the entire picture. Basic knowledge of immunologic ontogeny, paired with what we have seen thus far with COVID-19, suggests that infected children develop varied manifestations of the COVID-19 infection: (1) asymptomatic infection, if innate immunity resolves upper respiratory infection; or (2) mild fever with upper respiratory symptoms; or (3) lower respiratory infection with some symptomatology which resolves after a few days. These would be expected with a Th2 preponderance, based on immunologic maturity. If the virus were allowed to disseminate to other tissue and the child developed adaptive responses to the virus over the following one or more weeks, the response to SARS-CoV-2 antigens might provoke a delayed inflammatory state. Therefore; (4) IgA immune complexes may result in systemic inflammatory responses that mimic Th1 responses and that also may produce a diverse set of syndromes that they are genetically at risk for.
There have been recent individual reports of hyperinflammatory disease with parallels to other multisystem inflammatory disease syndromes in children, experienced by relatively small numbers of children with evidence of COVID-19 infection by PCR or antibody testing (66). The first and largest case series were published from the Italian, British, and American (New York) experiences, prompting emergency announcements regarding what is being termed as “Multisystem Inflammatory Syndrome in Children” or MIS-C, and case definitions released by the U.S. and European Centers for Disease Control as well as the WHO on May 14 and 15, 2020 (67–69). Many of these cases cluster at least a month after the peak of community transmission, with the vast majority testing SARS-CoV-2 IgG positive. The definitions comprise the clinical symptoms and lab findings of inflammation as well as evidence of, or exposure to, COVID-19. Abnormal labs include overall increased WBC, low lymphocytes, increased CRP, lower albumin, increased ferritin, increased troponin, elevated d-dimers, increased LDH, and abnormal troponin and BNP (67).
Children are presenting with a spectrum of manifestations with prolonged fevers in the absence of alternative diagnosis, and in worst cases, frank cardiogenic shock with myocardial dysfunction (70). There is no known predictive factor as to which children will have worse disease, and there may be host genetics at play as these cases were described in Europe and have not yet been described in East Asian countries (for example, China) that have gone through COVID-19 epidemic peaks (71). In addition, a recent small study from Paris reported MIS-C was more commonly seen in children of African ancestry (72). The initial reports of this condition from the U.K. showed this diagnosis also was frequent in children of Afro-Caribbean descent (68), and data from the Centers for Disease Control and Prevention suggest this condition is more commonly described in racial and ethnic minority (Hispanic and black) children (73). Moreover, we know MIS-C is a relatively rare complication, suggesting potential host genetic factors at play. In addition, we do know that acute COVID-19 is more commonly diagnosed in racial and ethnic minority children, and the more frequent occurrence of MIS-C in racial and ethnic minority populations may reflect factors that contribute to acute COVID-19 being also more frequent in these populations.
While there is data for treatment guidelines continues to be gathered at this time, the current consensus or recurring themes of treatment include treatment with immunomodulatory treatment (e.g., IVIG, steroids), anticoagulant therapy, frequent echocardiograms, and extreme caution with fluid resuscitation and IVIG administration if it is used. Indeed the American Academy of Pediatrics has published interim guidance at this time, and the American College of Rheumatology has the first set of published guidelines for this (74). Optimum follow-up for cardiac complications also requires further study (75). Other more specific immunomodulators have also been employed, including anti-IL-6 and anti-IL-1 inhibitors, with a preference toward the latter due to its safety profile and short half-life. Parallels between MIS-C and the hyperinflammatory response in the acute phase of COVID-19 in adults have been drawn, but whether the mechanisms are similar or different remains to be defined. Implications for vaccine development are also a concern given the severe post-inflammatory syndrome seen with these children.
The clinical phenomena of MIS-C shares features of other genetic, inflammatory and post-infectious childhood diseases, and in fact, calls to streamline the case definition have been made to avoid diagnostic overlap (76). Some of these syndromes share features of Monogenic Autoinflammatory Disease which is marked by overexpression of IL-1 and in some who develop Macrophage Activating Syndrome, overexpression of IL-18. A review of clinical and laboratory features referenced below and peculiar to each disease is summarized in Table 1.

Table 1. Characteristics of MIS-C and other Hyperinflammatory Syndromes.
Kawaski Disease (KD) demonstrates some pathophysiologic features of COVID-19 related inflammation. KD-experts are very cautious to distinguish MIS-C from KD, though, and there remains the question of what exactly MIS-C is, as it is not KD itself. MIS-C occurs in older children (although there are possibly cases in adults) (77), and presenting symptoms include more prominent gastrointestinal symptoms and cardiac features, with parallel distinct laboratory features such as elevated ferritin, D-dimers, and triglycerides (67), which bear more resemblance to Macrophage Activation Syndrome (MAS) (71).
Clinical diagnostic criteria of KD include persistent fever, rash, cervical lymphadenopathy, bilateral conjunctival erythema, mucositis and peripheral swelling, which overlap with MIS-C. In KD, there are increased levels of IL-1, IL-6 and IL-8. Neutrophils predominate in the 1st week and may invade the wall of medium-sized arteries such as the coronaries. With time they are joined by macrophages, dendritic cells and CD8 T cells. Immune complexes are prevalent but there is little evidence for a major role in pathogenesis, and no target antigen has been identified reliably. Many different stimuli (e.g., commensal bacteria, mycoplasma, candida, different viral classes) have been suggested. Early treatment with high dose IVIG and/or steroid plus aspirin resolves fever and prevents coronary artery aneurysms.
Children who do not meet diagnostic criteria may have “Atypical” KD and are treated as if they are “classical,” with good resolution of the symptoms. The pathogenesis may share common mechanisms but also elements of macrophage activation, resulting in inflammatory cytokine release and thrombotic phenomena (78). However, atypical KD often affects older children. Their platelet counts, which continue to rise to over 500,000 in KD, are often low or normal in the atypical disease. In comparison, the ability of COVID-19 to produce a hypercoagulable state may allow for clots to develop without elevated platelet counts.
COVID-19 infection induces antibody production 2–3 weeks after infection, although these antibody levels are higher in symptomatic patients compared with asymptomatic, and fall within months of infection (62). Although most antigens typically produce IgM followed by IgG, this infection produces IgA at the time IgM is seen, and the response is stronger and more persistent than IgM (79). Patients with COVID-19 produce increased IgA-secreting cells following TLR9 stimulation (80). IgA complexes can promote proinflammatory cytokines (TNFa, IL-1b, IL-6, IL-23) through FCalphaR1-TLR (TLR 3,4,5) crosstalk via gene transcription of macrophages, monocytes and Kupffer cells (81). Such a process would result in Th1-like inflammation that occurs several weeks after COVID-19 infection. A mouse model of KD vasculitis demonstrated that intestinal permeability and IgA provoked an immune vasculitis linked to cardiovascular inflammation (82).
HLH, in its familiar form, is typically seen in children under 18 months and is associated with genetic abnormalities. It can occur sporadically at any age. Two-thirds of patients have genetic, autoimmune or cancer predispositions (83). Prolonged PTT with hypofibrinogenemia and increased fibrin-split products are associated with easy bruising. Evidence of hemophagocytosis in the bone marrow is pathognomonic. There are increased levels of soluble CD25 (IL-2) and CD163 (macrophage marker). This disorder may be a result of an infectious trigger such as COVID-19 re-exposure.
Some children have developed a late-onset purpuric skin lesion in the lower extremities. While this may represent easy bruising, it also resembles the vasculitis rash of Henoch-Schonlein Purpura (HSP) in which there are deposits of IgA and IgG complexes in the affected area. There are also mesenteric areas with the same complexes, causing pain and perhaps diarrhea. The COVID-19 individuals also have enteric symptomatology and renal issues such as hematuria and proteinuria. There are likely genetic factors that play a role in HSP, particularly MHC class 2 and polymorphisms in the renin-angiotensin system, which may parallel COVID-19 pathophysiology (84). In contrast to some other inflammatory conditions, HSP doesn't respond well to corticosteroids but does improve with colchicine, a macrophage stabilizer.
Some children with MIS-C have developed macrophage-activated phenotypes, seen in HLH and MAS. In a multicenter study of 362 patients which predates COVID-19 and which aimed to describe MAS, symptoms included: fever (95%), hepatomegaly (70%), splenomegaly (58%), cardiac involvement (26%), hemorrhagic manifestations (20%), renal involvement (15%) and CNS symptomatology. Increased levels of ferritin, D-dimer, ALT, triglycerides and LDH are seen (85). Platelet counts and albumin levels are depressed. Treatment consists of anti-inflammatory drugs such as corticosteroids, cyclosporin, anti-IL1 agents and etoposide. Plasma exchange is often needed. In a quarter of patients, infection with EBV or other Herpesviruses may be a trigger (85).
ARF set a precedent for the development of a systemic disease that occurred weeks after an initial infection with a pathogen, and comparisons to COVID-19 and its associated syndromes have been explored (86). ARF followed a pharyngeal infection with a common bacterial pathogen, the Group A Streptococcus. The initial infection was frequently asymptomatic. In a few individuals (~1/1,000) over age 2, fever developed about 2 weeks later. In one study, the majority presented with pain in the joints, manifested as arthritis in 81% and arthralgia in 15%. Four percent presented with congestive heart failure. Carditis developed in 42%, with a high incidence of pericarditis (6%) and congestive heart failure (15%) and a mortality of 2% (87). In another study, although antibodies to streptococci developed, and may have recognized myosin, the disease was due to T cell stimulation (88). The M protein of certain streptococci can produce superantigen-like stimulation of T cells, resulting in cytokine release (89–91). Treatment of ARF with anti-inflammatory doses of aspirin or corticosteroids resolved the inflammation. If treatment was initiated early enough, the arthritis and/or carditis resolved. If treatment occurred later, valvular damage was irrevocable. There were instances of recurrent inflammation after exposure to group A streptococcus, which was prevented by antibiotic prophylaxis. When recurrence did occur, there was a reprise of the original syndrome of carditis or arthritis. This suggests a likely genetic predisposition, which is supported by twin studies showing a high concordance of ARF in monozygotic twins (92). Genetic and genome-wide association studies have reproducibly found a link between ARF and the HLA locus on chromosome 6 as well as the immunoglobulin heavy chain locus which includes IGHV4-61 (93).
ARF, while not uncommon prior to 1970 in the USA, has become uncommon since then. Its rarity in children under 2 years was thought to be related to their relative inability to develop sufficiently high levels of Th1 responses. The delay from primary SARS-CoV-2 infection to disease is reminiscent of ARF in the past, which also occurred in a small fraction of at-risk individuals.
The first genetic association studies looking at host genetic factors in COVID-19 patients published as preprints have suggested a link between severe disease and certain ABO blood types including A and B (32) and Rh positivity (94). Furthermore, preliminary results of a genome-wide association study of Italian and Spanish patients found not only an association between the ABO locus (chromosome 9q34) and more severe disease, but also a gene cluster on chromosome 3p21 which includes a gene that encodes a proline transporter that interacts with ACE2 (SLC6A20) as well as two chemokine genes, CC-motif chemokine receptor 9 (CCR9) and the C-X-C motif chemokine receptor 6 (CXCR6) (95). It has yet to be determined if either of these loci will play a role in the development of MIS-C or severe disease in children. There are several ongoing international collaborative efforts (including the COVID Human Genetic Effort and the COVID-19 Host Genetics Initiative) which aim to uncover common and rare host genetic variation that contributes to either COVID-19 infection severity or the development of the rarer complications and clinical syndromes, including those that seem to affect only younger patients and children (96, 97). Indeed, there is now at least one convincing report describing 2 pairs of brothers from the Netherlands who all developed severe COVID-related disease and after whole exome sequencing were found to have loss-of-function variants in the X-linked gene TLR7 which encodes toll- like receptor 7, an inducer of the interferon I and II responses in the innate immune system (98).
As we try to establish the host genetic factors underlying the emerging pediatric COVID-19-related syndromes like MIS-C, it may be helpful to review what is known about the genetic etiologies behind HLH and KD (Table 2). Indeed, COVID19-related illnesses in children (in particular MIS-C) may share patho-etiological origins, or genetic susceptibilities, with other pediatric hyperinflammatory syndromes. HLH is usually categorized into a primary (or familial) form and a secondary (or acquired) form (99). The former is a classic Mendelian disorder, inherited in an autosomal recessive fashion2. There is, however, also evidence that heterozygotes (carriers) of pathogenic alleles in one of the known HLH genes might also develop HLH (100). The pathological basis of HLH (or the macrophage-activating syndrome as it is sometimes called when it occurs in a patient with an antecedent rheumatological disease) is the disordered loading, moving, priming and/or docking of the cytotoxic T-lymphocyte's toxic granules leading to target cell death and overproduction of cytokines, in particular IFNγ and TNFα (101). The products of the genes that are associated with HLH all seem to play a role in this process and therefore may be potential susceptibility loci in those with severe COVID-19-related syndromes, especially wherein there is immunological and clinical evidence of an HLH-like process.
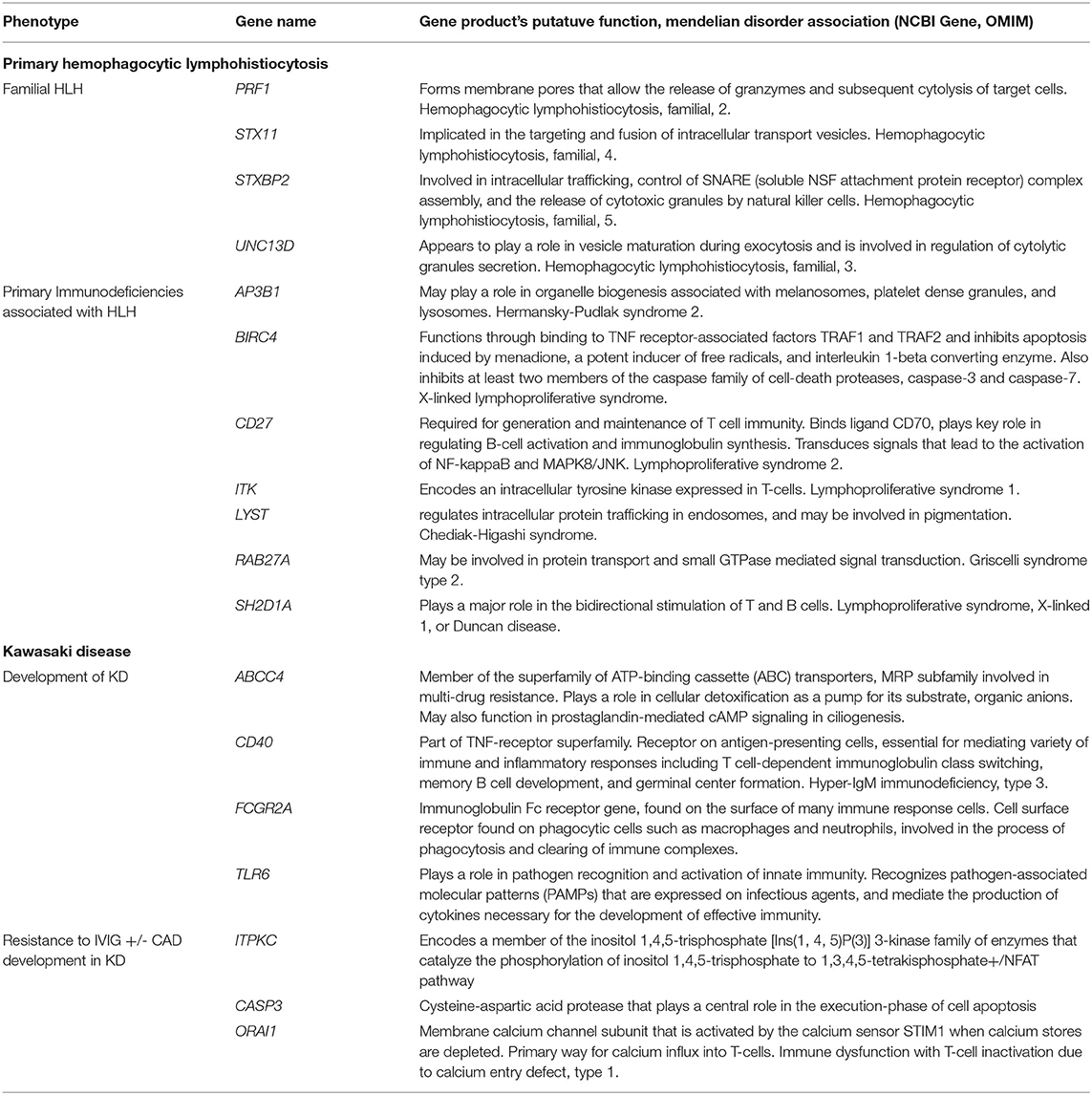
Table 2. Primary Hemophagocytic Lymphohistiocytosis and Kawasaki Disease-Associated Genes.
The host genetic factors contributing to the development of KD are less clear. The currently accepted hypothesis is that KD is a multifactorial disorder that develops in a genetically susceptible child after an environmental/infectious trigger. So far, no one has established any Mendelian forms of KD; KD does not have a Mendelian entry in OMIM. Twin studies in KD are conflicting but suggest some concordance in the development of KD-related vasculitis (102). There are also reports of familial clustering of KD in Japan (103). While the genes implicated in monogenic forms of HLH tend to converge on a single T-cell process, the genes so far associated with KD and its complications are more biologically diverse and roughly fall into four categories: T-cell activation, B-cell signaling, cellular apoptosis and dysfunctional transforming growth factor beta signaling (104). Furthermore, there is at least one report of a specific viral trigger (EBV) leading to KD in a patient with Familial Mediterranean Fever due to mutations in MEFV which encodes pyrin, a protein involved in the innate immune response (105). Interestingly, a rare complication of KD is HLH itself (106), suggesting that, at least in some, the pathological mechanisms of both disorders may overlap.
COVID-19 infection in children is markedly different from adults in a number of ways, including differences in transmission itself, as well as differences in severity and pathogenesis including viral genetic diversity, sex-related-impacted protease and viral susceptibility differences, and age-related and potentially genetic innate and adaptive immune response differences. Parallels to other hyperinflammatory syndromes and comparisons in the context of some of these factors may also continue to lead to understanding pediatric susceptibility to COVID-19 disease and its associated syndromes. Understanding transmission and pathogenesis in children is essential to optimizing the care of children, and also very practically, implementing strategies to allow children to attend school and adopt back to some degree of normalcy.
Children have relatively little symptomatology upon acute infection. This may be a consequence of developmental “immunodeficiency” resulting from a Th2 polarization that starts in utero and is maintained for most of the first decade of life. Th2 responses may suppress Th1 proinflammatory processes seen in adults and are responsible for some of the increasingly severe symptomatology in advancing age. Th2 responses also allow for improved antibody responses, although the contribution of antibody responses to pathology compared with control of infection remains to be clarified in adults and children. With maturation of this process in the second decade of life, increasing symptoms are sometimes seen with acute infection. In a very small subset of children, a diffuse set of inflammatory syndromes are seen several weeks after infection with COVID-19. These entities may develop in a genetically vulnerable population with susceptibilities that are similar to those already illuminated for other inflammatory syndromes (e.g. Kawasaki Disease, HLH/MAS and HSP). Another possibility is that these inflammatory syndromes occur as a consequence of IgA complexes resulting from the Th2 environment or as a result of a Th1-like response to COVID-19 antigens that were slow to develop after infection.
Fortunately, children respond to typical treatment for the syndromes. However, as children are certainly not just little adults, we need to learn and model through vaccines or immune-modulatory treatments how children handle this infection so much better than we do. We as adults can learn from our children–as we always should.
AK, CH, BK, and WB contributed wrote, reviewed, edited the manuscript, and tables. WB conceived the paper. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. ^CDC. Available online at: https://www.cdc.gov/coronavirus/2019-ncov/downloads/Viral-Transport-Medium.pdf (accessed May 20, 2020).
2. ^OMIM: Available online at: https://www.omim.org/entry/613101 (accessed May 16, 2020).
1. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. (2020) 395:497–506. doi: 10.1016/s0140-6736(20)30183-5
2. Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. (2020) 395:507–13. doi: 10.1016/s0140-6736(20)30211-7
3. Wu Z, McGoogan MJ. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72,314 cases from the Chinese center for disease control and prevention. JAMA. (2020) 323:1239–42. doi: 10.1001/jama.2020.2648
4. Qiu H, Wu J, Hong L, Luo Y, Song Q, Chen D. Clinical and epidemiological features of 36 children with coronavirus disease 2019 (COVID-19) in Zhejiang, China: an observational cohort study. Lancet Infect Dis. (2020) 20:689–96. doi: 10.1016/s1473-3099(20)30198-5
5. Lu Y, Li Y, Deng W, Liu M, He Y, Huang L, et al. Symptomatic infection is associated with prolonged duration of viral shedding in mild coronavirus disease 2019: a retrospective study of 110 children in Wuhan. Pediatr Infect Dis J. (2020) 39:e95–9. doi: 10.1097/inf.0000000000002729
6. Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. (2020) 395:565–74. doi: 10.1016/s0140-6736(20)30251-8
7. Xiao K, Zhai J, Feng Y, Zhou N, Zhang X, Zou JJ, et al. Isolation of SARS-CoV-2-related coronavirus from Malayan pangolins. Nature. (2020) 583:286–9. doi: 10.1038/s41586-020-2313-x
8. Sanche S, Lin YT, Xu C, Romero-Severson E, Hengartner N, Ke R. High contagiousness and rapid spread of severe acute respiratory syndrome coronavirus 2. Emerg Infect Dis. (2020) 26:1470–7. doi: 10.3201/eid2607.200282
9. Olsen SJ, Chen MY, Liu YL, Witschi M, Ardoin A, Calba C, et al. Early introduction of severe acute respiratory syndrome coronavirus 2 into Europe. Emerg Infect Dis. (2020) 26:1567–70. doi: 10.3201/eid2607.200359
10. Korber B, Fischer WM, Gnanakaran S, Yoon H, Theiler J, Abfalterer W, et al. tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell. (2020) 182:812–27.e19. doi: 10.1016/j.cell.2020.06.043
11. Bi Q, Wu Y, Mei S, Ye C, Zou X, Zhang Z, et al. Epidemiology and transmission of COVID-19 in 391 cases and 1286 of their close contacts in Shenzhen, China: a retrospective cohort study. Lancet Infect Dis. (2020) 20:911–9. doi: 10.1016/s1473-3099(20)30287-5
12. Zhang J, Litvinova M, Liang Y, Wang Y, Wang W, Zhao S, et al. Changes in contact patterns shape the dynamics of the COVID-19 outbreak in China. Science. (2020) 368:1481–6. doi: 10.1126/science.abb8001
13. Davies NG, Klepac P, Liu Y, Prem K, Jit M, Eggo MR. Age-dependent effects in the transmission and control of COVID-19 epidemics. Nat Med. (2020) 26:1205–11. doi: 10.1038/s41591-020-0962-9
14. Heald-Sargent T, Muller WJ, Zheng X, Rippe J, Patel AB, Kociolek KL. Age-related differences in nasopharyngeal severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) levels in patients with mild to moderate coronavirus disease 2019 (COVID-19). JAMA Pediatr. (2020) 174:902–3. doi: 10.1001/jamapediatrics.2020.3651
15. Park YJ, Choe YJ, Park O, Park SY, Kim YM, Kim J, et al. Contact tracing during coronavirus disease outbreak, South Korea, 2020. Emerg Infect Dis. (2020) 20:2465–8. doi: 10.3201/eid2610.201315
16. Posfay-Barbe KM, Wagner N, Gauthey M, Moussaoui D, Loevy N, Diana A, et al. COVID-19 in children and the dynamics of infection in families. Pediatrics. (2020) 146:e20201576. doi: 10.1542/peds.2020-1576
17. Kelvin AA, Halperin S. COVID-19 in children: the link in the transmission chain. Lancet Infect Dis. (2020) 20:633–4. doi: 10.1016/s1473-3099(20)30236-x
18. Viner RM, Mytton OT, Bonell C, Melendez-Torres GJ, Ward J, Hudson L, et al. Susceptibility to SARS-CoV-2 infection among children and adolescents compared with adults: a systematic review and meta-analysis. JAMA Pediatr. (2020) e204573. doi: 10.1001/jamapediatrics.2020.4573
19. Ehrhardt J, Ekinci A, Krehl H, Meincke M, Finci I, Klein J, et al. Transmission of SARS-CoV-2 in children aged 0 to 19 years in childcare facilities and schools after their reopening in May 2020, Baden-Württemberg, Germany. Euro Surveill. (2020) 25:2001587. doi: 10.2807/1560-7917.Es.2020.25.36.2001587
20. Blaisdell LL, Cohn W, Pavell JR, Rubin DS, Vergales EJ. Preventing and mitigating SARS-CoV-2 transmission - four overnight camps, Maine, June-August 2020. MMWR Morb Mortal Wkly Rep. (2020) 69:1216–20. doi: 10.15585/mmwr.mm6935e1
21. Lopez AS, Hill M, Antezano J, Vilven D, Rutner T, Bogdanow L, et al. Transmission dynamics of COVID-19 outbreaks associated with child care facilities - Salt Lake City, Utah, April-July 2020. MMWR Morb Mortal Wkly Rep. (2020) 69:1319–23. doi: 10.15585/mmwr.mm6937e3
22. American Academy of Pediatrics and the Children's Hospital Association: Children and COVID-19 State Data Report. Children and COVID-19 State Data Report (October 9, 2020).
23. Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. (2020) 181:271–80.e8. doi: 10.1016/j.cell.2020.02.052
24. Sungnak W, Huang N, Bécavin C, Berg M, Queen R, Litvinukova M, et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat Med. (2020) 26:681–7. doi: 10.1038/s41591-020-0868-6
25. Li MY, Li L, Zhang Y, Wang SX. Expression of the SARS-CoV-2 cell receptor gene ACE2 in a wide variety of human tissues. Infect Dis Poverty. (2020) 9:45. doi: 10.1186/s40249-020-00662-x
26. Buonsenso D, Sali M, Pata D, De Rose C, Sanguinetti M, Valentini P, et al. Children and COVID-19: microbiological and immunological insights. Pediatr Pulmonol. (2020). doi: 10.1002/ppul.24978. [Epub ahead of print].
27. Yonker LM, Neilan AM, Bartsch Y, Patel AB, Regan J, Arya P, et al. Pediatric severe acute respiratory syndrome coronavirus. 2 (SARS-CoV-2): clinical presentation, infectivity, immune responses. J Pediatr. (2020). doi: 10.1016/j.jpeds.2020.08.037. [Epub ahead of print].
28. Zang R, Gomez Castro MF, McCune BT, Zeng Q, Rothlauf PW, Sonnek NM, et al. TMPRSS2 and TMPRSS4 promote SARS-CoV-2 infection of human small intestinal enterocytes. Sci Immunol. (2020) 5:eabc3582. doi: 10.1126/sciimmunol.abc3582
29. Giagulli VA, Guastamacchia E, Magrone T, Jirillo E, Lisco G, De Pergola G, et al. Worse progression of COVID-19 in men: is testosterone a key factor? Andrology. (2020). doi: 10.1111/andr.12836. [Epub ahead of print].
30. Bunyavanich S, Do A, Vicencio A. Nasal gene expression of angiotensin-converting enzyme 2 in children and adults. JAMA. (2020) 323:2427–9. doi: 10.1001/jama.2020.8707
31. Fischer H, Tschachler E, Eckhart L. Pangolins lack IFIH1/MDA5, a cytoplasmic RNA sensor that initiates innate immune defense upon coronavirus infection. Front Immunol. (2020) 11:939. doi: 10.3389/fimmu.2020.00939
32. Zhao J, Yang Y, Huang H, Li D, Gu D, Lu X, et al. Relationship between the ABO blood group and the COVID-19 susceptibility. Clin Infect Dis. (2020). doi: 10.1093/cid/ciaa1150. [Epub ahead of print].
33. Hancock DG, Cavallaro EC, Doecke E, Reynolds M, Charles-Britton B, Dixon DL, et al. Immune biomarkers predicting bronchiolitis disease severity: a systematic review. Paediatr Respir Rev. (2019) 32:82–90. doi: 10.1016/j.prrv.2018.11.004
34. Zhang Y, Qin L, Zhao Y, Zhang P, Xu B, Li K, et al. Interferon-induced transmembrane protein-3 genetic variant rs12252-C is associated with disease severity in COVID-19. J Infect Dis. (2020) 222:34–7. doi: 10.1093/infdis/jiaa224
35. Selvaggi C, Pierangeli A, Fabiani M, Spano L, Nicolai A, Papoff P, et al. Interferon lambda 1-3 expression in infants hospitalized for RSV or HRV associated bronchiolitis. J Infect. (2014) 68:467–77. doi: 10.1016/j.jinf.2013.12.010
36. Kikkert M. Innate immune evasion by human respiratory RNA viruses. J Innate Immun. (2020) 12:4–20. doi: 10.1159/000503030
37. da Silva Cezar RD, da Silva Castanha PM, Matos Freire N, Mola C, Feliciano do Carmo R, Tenório Cordeiro M, et al. Association between interferon lambda 3 rs12979860 polymorphism and clinical outcome in dengue virus-infected children. Int J Immunogenet. (2020) 47:351–8. doi: 10.1111/iji.12477
38. Marderstein AR, Uppal M, Verma A, Bhinder B, Tayyebi Z, Mezey J, et al. Demographic and genetic factors influence the abundance of infiltrating immune cells in human tissues. Nat Commun. (2020) 11:2213. doi: 10.1038/s41467-020-16097-9
39. Liu J, Li S, Liu J, Liang B, Wang X, Wang H, et al. Longitudinal characteristics of lymphocyte responses and cytokine profiles in the peripheral blood of SARS-CoV-2 infected patients. EBioMedicine. (2020) 55:102763. doi: 10.1016/j.ebiom.2020.102763
40. Yao Z, Zheng Z, Wu K, Junhua Z. Immune environment modulation in pneumonia patients caused by coronavirus: SARS-CoV, MERS-CoV and SARS-CoV-2. Aging. (2020) 12:7639–51. doi: 10.18632/aging.103101
41. Diao B, Wang C, Tan Y, Chen X, Liu Y, Ning L, et al. Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID-19). Front Immunol. (2020) 11:827. doi: 10.3389/fimmu.2020.00827
42. Felsenstein S, Herbert JA, McNamara PS, Hedrich MC. COVID-19: immunology and treatment options. Clin Immunol. (2020) 215:108448. doi: 10.1016/j.clim.2020.108448
43. Szabo SJ, Sullivan BM, Peng SL, Glimcher HL. Molecular mechanisms regulating Th1 immune responses. Annu Rev Immunol. (2003) 21:713–58. doi: 10.1146/annurev.immunol.21.120601.140942
44. Wölfel R, Corman VM, Guggemos W, Seilmaier M, Zange S, Müller MA, et al. Virological assessment of hospitalized patients with COVID-2019. Nature. (2020) 581:465–9. doi: 10.1038/s41586-020-2196-x
45. Bozkus CC. SARS-CoV-2-specific T cells without antibodies. Nat Rev Immunol. (2020) 20:463–3. doi: 10.1038/s41577-020-0393-3
46. Channappanavar R, Zhao J, Perlman S. T cell-mediated immune response to respiratory coronaviruses. Immunol Res. (2014) 59:118–28. doi: 10.1007/s12026-014-8534-z
47. Fan YY, Huang ZT, Li L, Wu MH, Yu T, Koup RA, et al. Characterization of SARS-CoV-specific memory T cells from recovered individuals 4 years after infection. Arch Virol. (2009) 154:1093–9. doi: 10.1007/s00705-009-0409-6
48. Tang F, Quan Y, Xin ZT, Wrammert J, Ma MJ, Lv H, et al. Lack of peripheral memory B cell responses in recovered patients with severe acute respiratory syndrome: a six-year follow-up study. J Immunol. (2011) 186:7264–8. doi: 10.4049/jimmunol.0903490
49. Kollmann TR, Kampmann B, Mazmanian SK, Marchant A, Levy O. Protecting the newborn and young infant from infectious diseases: lessons from immune ontogeny. Immunity. (2017) 46:350–63. doi: 10.1016/j.immuni.2017.03.009
50. Connors TJ, Ravindranath TM, Bickham KL, Gordon CL, Zhang F, Levin B, et al. Airway CD8(+) T cells are associated with lung injury during infant viral respiratory tract infection. Am J Respir Cell Mol Biol. (2016) 54:822–30. doi: 10.1165/rcmb.2015-0297OC
51. Ng PC, Lam CW, Li AM, Wong CK, Cheng FW, Leung TF, et al. Inflammatory cytokine profile in children with severe acute respiratory syndrome. Pediatrics. (2004) 113:e7–14. doi: 10.1542/peds.113.1.e7
52. Krammer F, Smith GJD, Fouchier RAM, Peiris M, Kedzierska K, Doherty PC, et al. Influenza. Nat Rev Dis Primers. (2018) 4:3. doi: 10.1038/s41572-018-0002-y
53. Götzinger F, Santiago-García B, Noguera-Julián A, Lanaspa M, Lancella L, Calò Carducci FI, et al. COVID-19 in children and adolescents in Europe: a multinational, multicentre cohort study. Lancet Child Adolesc Health. (2020) 4:653–61. doi: 10.1016/s2352-4642(20)30177-2
54. Lu Y, Wang S, Zhang L, Xu C, Bian C, Wang Z, et al. Han: Epidemiology of human respiratory viruses in children with acute respiratory tract infections in Jinan, China. Clin Dev Immunol. (2013) 2013:210490. doi: 10.1155/2013/210490
55. Zimmermann P, Curtis N. COVID-19 in Children, pregnancy and neonates: a review of epidemiologic and clinical features. Pediatr Infect Dis J. (2020) 39:469–477. doi: 10.1097/inf.0000000000002700
56. Dijkman R, Jebbink MF, El Idrissi NB, Pyrc K, Muller MA, Kuijpers TW, et al. Human coronavirus NL63 and 229E seroconversion in children. J Clin Microbiol. (2008) 46:2368–73. doi: 10.1128/JCM.00533-08
57. Heimdal I, Moe N, Krokstad S, Christensen A, Skanke LH, Nordbo SA, Dollner H. Human coronavirus in hospitalized children with respiratory tract infections: a 9-year population-based study From Norway. J Infect Dis. (2019) 219:1198–206. doi: 10.1093/infdis/jiy646
58. Grifoni A, Weiskopf D, Ramirez SI, Mateus J, Dan JM, Moderbacher CR, et al. Targets of T cell responses to SARS-CoV-2 coronavirus in humans with COVID-19 disease and unexposed individuals. Cell. (2020) 181:1489–501.e15. doi: 10.1016/j.cell.2020.05.015
59. Shekerdemian LS, Mahmood NR, Wolfe KK, Riggs BJ, Ross CE, McKiernan CA, et al. Characteristics and outcomes of children with Coronavirus disease 2019 (COVID-19) infection admitted to us and canadian pediatric intensive care units. JAMA Pediatr. (2020) 174:868–73. doi: 10.1001/jamapediatrics.2020.1948
60. Consiglio CR, Cotugno N, Sardh F, Pou C, Amodio D, Rodriguez L, et al. The immunology of multisystem inflammatory syndrome in children with COVID-19. Cell. (2020). doi: 10.1016/j.cell.2020.09.016. [Epub ahead of print].
61. Dingens AS, Crawford KHD, Adler A, Steele SL, Lacombe K, Eguia R, et al. Serological identification of SARS-CoV-2 infections among children visiting a hospital during the initial Seattle outbreak. Nat Commun. (2020) 11:4378. doi: 10.1038/s41467-020-18178-1
62. Long Q-X, Tang X-J, Shi Q-L, Li Q, Deng H-J, Yuan J, et al. Clinical and immunological assessment of asymptomatic SARS-CoV-2 infections. Nat Med. (2020) 26:1200–4. doi: 10.1038/s41591-020-0965-6
63. Pierce CA, Preston-Hurlburt P, Dai Y, Aschner CB, Cheshenko N, Galen B, et al. Immune responses to SARS-CoV-2 infection in hospitalized pediatric and adult patients. Sci Transl Med. (2020) 12:eabd5487. doi: 10.1126/scitranslmed.abd5487
64. Ferrucci L, Corsi A, Lauretani F, Bandinelli S, Bartali B, Taub DD, et al. The origins of age-related proinflammatory state. Blood. (2005) 105:2294–9. doi: 10.1182/blood-2004-07-2599
65. IDSA: Infectious Disease Society of America Guidelines on the Treatment and Management of Patients with COVID-19. (2020). Available online at: https://www.idsociety.org/practice-guideline/covid-19-guideline-treatment-and-management/ (accessed August 5, 2020).
66. Jones VG, Mills M, Suarez D, Hogan CA, Yeh D, Segal JB, et al. COVID-19 and Kawasaki disease: novel virus and novel case. Hosp Pediatr. (2020) 10:537–40. doi: 10.1542/hpeds.2020-0123
67. Feldstein LR, Rose EB, Horwitz SM, Collins JP, Newhams MM, Son MBF, et al. Multisystem inflammatory syndrome in Children US, and adolescents. N Engl J Med. (2020) 383:334–6. doi: 10.1056/NEJMoa2021680
68. Riphagen S, Gomez X, Gonzalez-Martinez C, Wilkinson N, Theocharis P. Hyperinflammatory shock in children during COVID-19 pandemic. Lancet. (2020) 395:1607–8. doi: 10.1016/s0140-6736(20)31094-1
69. Verdoni L, Mazza A, Gervasoni A, Martelli L, Ruggeri M, Ciuffreda M, et al. An outbreak of severe Kawasaki-like disease at the Italian epicentre of the SARS-CoV-2 epidemic: an observational cohort study. Lancet. (2020) 395:1771–8. doi: 10.1016/s0140-6736(20)31103-x
70. Belhadjer Z, Méot M, Bajolle F, Khraiche D, Legendre A, Abakka S, et al. Acute heart failure in multisystem inflammatory syndrome in children (MIS-C) in the context of global SARS-CoV-2 pandemic. Circulation. (2020). doi: 10.1161/circulationaha.120.048360. [Epub ahead of print].
71. Shulman ST. Pediatric COVID-associated Multi-system Inflammatory Syndrome (PMIS). J Pediatric Infect Dis Soc. (2020) 9:285–6. doi: 10.1093/jpids/piaa062
72. Toubiana J, Poirault C, Corsia A, Bajolle F, Fourgeaud J, Angoulvant F, et al. Kawasaki-like multisystem inflammatory syndrome in children during the covid-19 pandemic in Paris, France: prospective observational study. BMJ. (2020) 369:m2094. doi: 10.1136/bmj.m2094
73. CDC: In: Health Department-Reported Cases of Multisystem Inflammatory Syndrome in Children (MIS-C) in the United States Centers for Disease Control. Prevention: Health Department Reported Cases of Multi-System Inflammatory Syndrome in Children (MIS-C) in the United States. Centers for Disease Control (2020). Available online at: https://www.cdc.gov/mis-c/cases/index.html (accessed October 6, 2020).
74. Henderson LA, Canna SW, Friedman KG, Gorelik M, Lapidus SK, Bassiri H, et al. American college of rheumatology clinical guidance for pediatric patients with multisystem inflammatory syndrome in children (MIS-C) associated with SARS-CoV-2 and hyperinflammation in COVID-19. Arthritis Rheumatol. (2020). doi: 10.1002/art.41454. [Epub ahead of print].
75. Simpson JM, Newburger WJ. Multi-system inflammatory syndrome in children in association with COVID-19. Circulation. (2020) 142:437–40. doi: 10.1161/circulationaha.120.048726
76. Rowley H. Understanding SARS-CoV-2-related multisystem inflammatory syndrome in children. Nat Rev Immunol, (2020) 20:453–54. (2020). doi: 10.1038/s41577-020-0367-5
77. Sokolovsky S, Soni P, Hoffman T, Kahn P, Scheers-Masters J. COVID-19 associated Kawasaki-like multisystem inflammatory disease in an adult. Am J Emerg Med. (2020). doi: 10.1016/j.ajem.2020.06.053. [Epub ahead of print].
78. Rowley AH, Shulman TS. Pathogenesis and management of Kawasaki disease. Expert Rev Anti Infect Ther. (2010) 8:197–203. doi: 10.1586/eri.09.109
79. Yu HQ, Sun BQ, Fang ZF, Zhao JC, Liu XY, Li YM, et al. Distinct features of SARS-CoV-2-specific IgA response in COVID-19 patients. Eur Respir J. (2020) 56:2001526. doi: 10.1183/13993003.01526-2020
80. Hicar MD. Antibodies and immunity during kawasaki disease. Front Cardiovasc Med. (2020) 7:94. doi: 10.3389/fcvm.2020.00094
81. Hansen IS, Hoepel W, Zaat SAJ, Baeten DLP, den Dunnen J. Serum IgA immune complexes promote proinflammatory cytokine production by human macrophages, monocytes, and kupffer cells through FcαRI-TLR Cross-Talk. J Immunol. (2017) 199:4124–31. doi: 10.4049/jimmunol.1700883
82. Noval M, Rivas Wakita D, Franklin MK, Carvalho TT, Abolhesn A, Gomez AC, et al. Intestinal permeability and IgA provoke immune vasculitis linked to cardiovascular inflammation. Immunity. (2019) 51:508–21.e6. doi: 10.1016/j.immuni.2019.05.021
83. Chinn IK, Eckstein OS, Peckham-Gregory EC, Goldberg BR, Forbes LR, Nicholas SK, et al. Genetic and mechanistic diversity in pediatric hemophagocytic lymphohistiocytosis. Blood. (2018) 132:89–100. doi: 10.1182/blood-2017-11-814244
84. He X, Yu C, Zhao P, Ding Y, Liang X, Zhao Y, et al. The genetics of Henoch-Schönlein purpura: a systematic review and meta-analysis. Rheumatol Int. (2013) 33:1387–95. doi: 10.1007/s00296-012-2661-4
85. Minoia F, Davì S, Horne A, Demirkaya E, Bovis F, Li C, et al. Clinical features, treatment, and outcome of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: a multinational, multicenter study of 362 patients. Arthritis Rheumatol. (2014) 66:3160–9. doi: 10.1002/art.38802
86. Buonsenso D, Riitano F, Valentini P. Pediatric inflammatory multisystem syndrome temporally related with SARS-CoV-2: immunological similarities with acute rheumatic fever and toxic shock syndrome. Front Pediatrics. (2020) 8:574. doi: 10.3389/fped.2020.00574
87. Majeed HA, Yousof AM, Shaltout A, Khuffash AF. Acute rheumatic fever below the age of five years: a prospective study of the clinical profile. Ann Trop Paediatr. (1984) 4:37–40. doi: 10.1080/02724936.1984.11748303
88. Read SE, Fischetti VA, Utermohlen V, Falk RE, Zabriskie BJ. Cellular reactivity studies to streptococcal antigens. migration inhibition studies in patients with streptococcal infections and rheumatic fever. J Clin Invest. (1974) 54:439–50. doi: 10.1172/jci107780
89. Quinn A, Kosanke S, Fischetti VA, Factor SM, Cunningham WM. Induction of autoimmune valvular heart disease by recombinant streptococcal m protein. Infect Immun. (2001) 69:4072–8. doi: 10.1128/iai.69.6.4072-4078.2001
90. Bhatnagar A, Grover A, Ganguly KN. Superantigen-induced T cell responses in acute rheumatic fever and chronic rheumatic heart disease patients. Clin Exp Immunol. (1999) 116:100–6. doi: 10.1046/j.1365-2249.1999.00853.x
91. Rasmussen SA, Smulian JC, Lednicky JA, Wen TS, Jamieson JD. Coronavirus disease 2019 (COVID-19) and pregnancy: what obstetricians need to know. Am J Obstet Gynecol. (2020) 222:415–26. doi: 10.1016/j.ajog.2020.02.017
92. Engel ME, Stander R, Vogel J, Adeyemo AA, Mayosi MB. Genetic susceptibility to acute rheumatic fever: a systematic review and meta-analysis of twin studies. PLoS ONE. (2011) 6:e25326. doi: 10.1371/journal.pone.0025326
93. Muhamed B, Parks T, Sliwa K. Genetics of rheumatic fever and rheumatic heart disease. Nat Rev Cardiol. (2020) 17:145–154. doi: 10.1038/s41569-019-0258-2
94. Latz CA, DeCarlo C, Boitano L, Png CYM, Patell R, Conrad MF, et al. Blood type and outcomes in patients with COVID-19. Ann Hematol. (2020) 99:2113–8. doi: 10.1007/s00277-020-04169-1
95. Ellinghaus D, Degenhardt F, Bujanda L, Buti M, Albillos A, Invernizzi P, et al. Genomewide association study of severe covid-19 with respiratory failure. N Engl J Med. (2020) 383:1522–34. doi: 10.1056/NEJMoa2020283
96. Casanova JL, Su HC, COVID Human Genetic Effort. A global effort to define the human genetics of protective immunity to SARS-CoV-2 infection. Cell. (2020) 181:1194–9. doi: 10.1016/j.cell.2020.05.016
97. C-HGI. The: The COVID-19 Host Genetics Initiative, a global initiative to elucidate the role of host genetic factors in susceptibility and severity of the SARS-CoV-2 virus pandemic. Euro J Human Genet. (2020) 28:715–8. doi: 10.1038/s41431-020-0636-6
98. van der Made Simons A, Schuurs-Hoeijmakers J, van den Heuvel G, Mantere T, Kersten S, van Deuren RC et al. Presence of genetic variants among young men with severe COVID-19. JAMA. (2020) 324:1–11. doi: 10.1001/jama.2020.13719
99. Long SS, Pickering LK, Prober GC. Principles and Practice of Pediatric Infectious Diseases. Philadelphia, PA: Elsevier (2018). p. 718.
100. Cetica V, Sieni E, Pende D, Danesino C, De Fusco C, Locatelli F, et al. Aricò: Genetic predisposition to hemophagocytic lymphohistiocytosis: Report on 500 patients from the Italian registry. J Allergy Clin Immunol. (2016) 137:188–96.e4. doi: 10.1016/j.jaci.2015.06.048
101. Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis. Annu Rev Med. (2012) 63:233–46. doi: 10.1146/annurev-med-041610-134208
102. Kottek A, Shimizu C, Burns CJ. Kawasaki disease in monozygotic twins. Pediatr Infect Dis J. (2011) 30:1114–6. doi: 10.1097/INF.0b013e31822ac4ff
103. Fujita Y, Nakamura Y, Sakata K, Hara N, Kobayashi M, Nagai M, et al. Kawasaki disease in families. Pediatrics. (1989) 84:666–9.
104. Kumrah R, Vignesh P, Rawat A, Singh S. Immunogenetics of Kawasaki disease. Clin Rev Allergy Immunol. (2020) 59:122–39. doi: 10.1007/s12016-020-08783-9
105. Maggio MC, Fabiano C, Corsello G. Kawasaki disease triggered by EBV virus in a child with familial mediterranean fever. Ital J Pediatr. (2019) 45:129. doi: 10.1186/s13052-019-0717-8
Keywords: COVID-19, SARS-CoV-2, children, immune response, genetics, multisystem inflammatory
Citation: Hobbs CV, Khaitan A, Kirmse BM and Borkowsky W (2020) COVID-19 in Children: A Review and Parallels to Other Hyperinflammatory Syndromes. Front. Pediatr. 8:593455. doi: 10.3389/fped.2020.593455
Received: 10 August 2020; Accepted: 15 October 2020;
Published: 24 November 2020.
Edited by:
Mark Fredric Cotton, Stellenbosch University, South AfricaReviewed by:
Steven Welch, University Hospitals Birmingham NHS Foundation Trust, United KingdomCopyright © 2020 Hobbs, Khaitan, Kirmse and Borkowsky. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: William Borkowsky, d2lsbGlhbS5ib3Jrb3dza3lAbnl1bGFuZ29uZS5vcmc=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.