 Xiao Li1,2,3,4,5
Xiao Li1,2,3,4,5 Ling-Ling Xie1,2,3,4,5
Ling-Ling Xie1,2,3,4,5 Wei Han1,2,3,4,5Si-Qi Hong1,2,3,4,5
Wei Han1,2,3,4,5Si-Qi Hong1,2,3,4,5 Jian-Nan Ma1,2,3,4,5Juan Wang1,2,3,4,5Li Jiang1,2,3,4,5*
Jian-Nan Ma1,2,3,4,5Juan Wang1,2,3,4,5Li Jiang1,2,3,4,5*- 1Department of Neurology, Children's Hospital of Chongqing Medical University, Chongqing, China
- 2Ministry of Education Key Laboratory of Child Development and Disorders, Chongqing, China
- 3National Clinical Research Center for Child Health and Disorders, Chongqing, China
- 4China International Science and Technology Cooperation base of Child Development and Critical Disorders, Chongqing, China
- 5Chongqing Key Laboratory of Pediatrics, Chongqing, China
Objective: This study aims to analyze the electroclinical characteristics and gene test results of children on the severe end of the epilepsy aphasia spectrum (EAS) and also the correlation of EAS-related GRIN2A genes to explore the genotype-phenotype relationships, as well as potential pathogenic mechanism of EAS.
Methods: A retrospective study was conducted on the participants diagnosed with Landau-Kleffner syndrome (LKS), epileptic encephalopathy with continuous spike-and-wave during sleep (CSWS), and atypical benign partial epilepsy (ABPE) at the Children's Hospital of Chongqing Medical University from January 2013 to June 2019. Whole-exome sequencing was performed in six patients, and epileptic panel was carried out in two. In addition, we reviewed all the published literatures reporting EAS patients with pathogenic variants until June 2019 and conducted Gene Ontology (GO) analysis, as well as protein-protein interaction (PPI) network.
Results: The mean age at seizure onset was 55.4 ± 27.0 months. The baseline severity of the spike-wave index (SWI) was not significantly correlated with intellectual disability (ID) level. Two pathogenic de novo GRIN2A null variants were identified in patients with ABPE who had less severe ID, despite the electrical status epilepticus during slow-wave sleep (ESES). By literature reviewing, 18 GRIN2A missense mutations and 11 GRIN2A truncating mutations which lead to N-methyl-D-aspartate receptors' loss of function has been reported. Of these mutations, 9 (31.0%) are situated in amino (N)-terminal domain, 6 (20.7%) in linger-binding domain S1, and 10 (34.5%) in linger-binding domain S2. EAS-related genes were enriched in the biological process of chemical synaptic transmission and vocalization (FDR, <0.01). The hub protein in PPI network is GluN2A, which might affect language function via foxp2-srpx2/uPAR signal network.
Conclusion: Our data suggested that when children suspected with benign epilepsy of children with centrotemporal spikes (BECTs) have early-onset age, changed seizure semiology, and deterioration of behavior/cognition/motor function, neurologists should be alert of the appearance of ESES. The neuropsychological deterioration in children with EAS might not only be completely affected by electric discharge severity but also genetic etiology. Our finding also enforced the current genotype-phenotype relationship theory about EAS. For EAS children, GRIN2A-FOXP2-SRPX2/uPAR signal network might contribute to the mechanism of their language deficit.
Introduction
Benign epilepsy of children with centrotemporal spikes (BECTs) is the most common idiopathic focal epileptic syndrome in childhood, accounting for 15–25% of childhood-onset epilepsy. It is characterized by focal sensorimotor attack and typical EEG signal showing high amplitude spike wave in central temporal area, followed by a slow wave. The prognosis of BECTs was considered to be good, mostly relieved before puberty. However, with the broader utilization of neuropsychological evaluations, some cognitive and linguistic deficits have been found in up to 50% of the patients with BECTs (1, 2). Also, a deteriorate evolution of clinical manifestation and EEG has frequently been found in patients with atypical BECTs (3, 4).
The atypical evolution of BECTs may lead to atypical benign childhood partial epilepsy (ABPE), status epilepticus of BECTS (SEBECTS), Landau-Kleffner syndrome (LKS), and epileptic encephalopathy with continuous spike-and-wave during sleep (CSWS). These diseases are now considered to be different entities but part of a wide single spectrum of disorders named epileptic-aphasia spectrum (EAS), with BECTs situated at the mildest end having a relatively good prognosis while CSWS located at the most severe end has a mostly poor prognosis. EAS, a spectrum of epileptic, cognitive, and language disorders, is associated with (and presumably influenced by) the presence of electrical status epilepticus during slow-wave sleep (ESES) and imposes a catastrophic effect on the family and the growth of children (5–10).
Recently, some studies showed that children with EAS share some common epilepsy genes, including GRIN2A (glutamate receptor, ionotropic, N-methyl D-aspartate 2A), which is the most advocated one (7–15). Genetic identification of EAS offers the potential for understanding pathogenic mechanisms and targeted treatment options. For now, Gene Ontology (GO) analysis and protein-protein interaction (PPI) network related to EAS genes have never been studied and the study of genotype and phenotype relationship of EAS patients carrying pathogenic GRIN2A variants are currently a hot research area which has important value for early diagnosis and prognosis.
Furthermore, since ESES has been taken as the main object to explore the influence it has on cognition and prognosis in many previous studies, the potential pathogenic genetic factors attributing to cognitive function, treatment, and long-term prognosis have been studied poorly.
Therefore, in our research, we first summarized the electroclinical characteristics of children diagnosed with ABPE, LKS, or CSWS in our cohort. Secondly, we applied whole-exome sequencing (WES) test to six EAS children and epileptic panel to two EAS individuals whose parents were willing to cooperate and analyzed their genotype-phenotype relationships. Next, we summarized the manifestation features, treatments, and variant distribution of all the published literature reporting patients with GRIN2A missense mutations and truncation variants which led to NMDAR loss-of-function (LOF) change. In the end, the GO and PPI network of genes linked to EAS were further explored.
Study Design
Case Source
Patients diagnosed with ABPE, LKS, and CSWS were recruited from the neurological outpatient and inpatient units of the Chongqing Medical University's Affiliated Children's Hospital during January 2013 to June 2019. We retrospectively reviewed the clinical records and the EEG recording (including at least one entire sleep cycle) of all subjects.
Inclusion and Exclusion Criteria
All subjects who possessed complete longitudinal clinical follow-up record, brain MRI results, EEG record, and at least one neuropsychological evaluation were included. Children with abnormal birth history and/or abnormal development history and/or significant abnormalities in brain MRI and/or abnormal hematuria metabolism screen results were excluded.
This study was approved by the Ethics Committee of the Chongqing Medical University's Affiliated Children's Hospital in China. Written informed consent was obtained from all the participants or their parents.
Clinical Data Collection
Clinical information including gender, perinatal and family history, physical examination, age at the onset of seizure or ESES, seizure types, psychomotor evaluation before and/or after ESES, treatment, prognosis, metabolism screening, and neuroimaging results were collected through a review of clinical records combined with follow up telephone calls. Neuropsychological assessment: Wechsler Intelligence Scale for Children (WISC) and/or Test of Everyday Attention for Children (TEA-Ch) and/or Peabody Picture Vocabulary Test (PPVT) and/or Autism Behavior Check list (ABC) + Modified Checklist for Autism in Toddlers (M-CHAT) Scale were repeatedly performed during the admission and/or follow-up period.
Definition (16)
ABPE
(1) The early course of ABPE is similar to BECTs, but patients exhibit more severe seizures, including epileptic negative myoclonus, atonic, atypical absence, myoclonic seizures, opercular syndrome, and frequent nocturnal focal seizures; (2) patients also suffer from mild neurocognitive and linguistic deficits; and (3) their EEG shows centro-temporal spikes (CTSs) which may develop into the ESES pattern. Classification: type I—Children with epileptic negative myoclonus and/or atypical absence and/or myoclonic seizures and/or frequent nocturnal focal seizures, manifesting as torpor, fine/gross motor clumsiness, drooping of the upper limbs head nodding, unexpected sudden falling, and mild cognitive impairment (17). Type II—Children with speech disorder/oropharynx manifesting as dysarthria, even complete aphasia, salivating, swallowing and tongue movement disorders when eating, and even opercular syndrome, such as dysphagia and bucking, without deficit in understanding speech and instruction (17). The intelligence of those children is usually normal.
LKS
(1) There is an abrupt or gradual onset of acquired language regression due to verbal auditory agnosia, with or without focal seizures, secondarily generalized tonic-clonic seizures, absences, or atonic seizures and (2) the EEG of LKS exhibits mainly bilateral synchronous or asynchronous temporal spikes which may develop into ESES.
CSWS
(1) Multiple types of seizures; (2) with moderate to severe global or selective regression affecting behavior, language, and intelligence; and (3) the EEG mostly shows ESES with a Spike-Wave Index (SWI) ≥50% during non-rapid eye movement (NREM) sleep.
ESES
EEG shows diffuse or bilaterally synchronous or non-synchronous spikes and waves at 1–3.5 Hz during NREM sleep, and the range of SWI is over 50% (The number of seconds containing epileptiform discharges was divided by the total number of seconds in the epoch, which was 600 s). EEG was recorded by scalp electrodes placed over the scalp according to the 10–20 International system. Children were divided into three groups, based on the SWI: group A (SWI ≥85%), group B (SWI 50–84%), and group C (SWI <50%).
Developmental Delay/Intellectual Disability (DD/ID)
DD/ID was evaluated according to the Diagnostic and Statistical Manual of Mental Disorders (DSM-5). DD/ID of patients were classified as follows: normal DD/ID = 0 points, borderline DD/ID = 1 point, mild DD/ID = 2 points, moderate DD/ID = 3 points, and severe DD/ID = 4 points. The terms DD and ID are used interchangeably here.
The Effects of AEDs
➀ seizure control: the seizure disappeared completely; ➁ seizure reduction: the frequency of seizures decreased ≥50%; and ➂ no effect: the frequency of seizures decreased ≤50%.
The Effects of Methylpyridines Pulse Therapy
➀ sensitive: SWI complete disappearance and response (SWI decreased to <50 or >20% reduction but still >50% or epileptic seizures completely disappeared); ➁ resistant: SWI did not change significantly (decreased <20%) or the reduction of seizures was <50%; ➂ dependent: SWI increased again or seizures reappeared/increased during or after the oral steroid withdrawal.
Sequencing
A total of 535 genes known to be related to epileptic encephalopathy were included in our gene panel; the genes are listed in Supplementary Material. Two EAS individuals were conducted with epilepsy panel sequencing, while the rest six EAS patients with the WES. The processes for analyzing the sequencing data were as follows:
DNA Extraction
Genomic DNA was obtained from the peripheral blood of eight patients using standard methods and fragmented using Covaris sonicator (Covaris S2, USA).
Library Construction
Fragments were end repaired and adaptors were ligated using a standard Illumina paired-end (PE) adapter. Then, the ligated products were amplified through polymerase chain reaction (PCR). Subsequently, quality testing of the DNA library was performed through Nanodrop2000 and agarose gel electrophoresis, and the unqualified products were excluded.
Target Region Capture
Sequence capture, enrichment, and elution were performed following the manufacturer's instructions.
Sequencing
The final captured DNA libraries were analyzed by an Illumina HiSeq 2000 Sequencer following the manufacturer's sequencing protocols. The mean coverage and depth of all genes were 98% and 10 × average, respectively, which fulfilled the quality test.
Functional Annotation of Genetic Variants
The variants were annotated using public databases (Annotation Dependent Depletion (CADD) scores, HapMap database, dbSNP 144, ESP6500, and EXAC, 1,000 genome variants database) against the GRCh37 human reference genome and classified as missense, nonsense, splice-site, insertion, deletion, synonymous, or non-coding mutations.
All the novel variants were considered to be pathogenic if they met either: (1) stop/frameshift variants; (2) missense mutations situated in the amino acid conservative region across species; (3) splice-site variations fulfilling the GT-AT rules; or (4) predicted to be possibly damaging or disease causing by more than two bioinformatic programs, as follows: Sorting Intolerant from Tolerant (SIFT), PolyPhen-2, Mutation Taster, or Protein Variation Effect Analyzer (PROVEAN); {http://provean.jcvi.org/index.php}.
Sanger Sequencing
Sanger sequencing was performed to determine the variant validation and segregation analyses following standard protocols.
Statistical Analysis
Continuous variables are reported as mean ± standard deviation. The Kruskal-Wallis test was used to analyze the correlation between the EEG-SWI and IQ level. Categorical variables are presented as frequencies and percentages. All analyses were conducted in SPSS 19.0 (SPSS Inc., Chicago, IL). The t-test and two-tailed p-values were used for all the analyses in this study, with p < 0.05 considered statistically significant.
Results
From January 2013 to June 2019, 120 children were diagnosed with EAS in the Neurology Department of Chongqing Medical University's Affiliated Children's Hospital. Only 18 of them met the inclusion and exclusion criteria after our strict screening, and 33.3% (6/18) had LKS, 27.8% (5/18) had CSWS, and 38.9% (7/18) had ABPE. According to the ABPE classification method mentioned above, seven children with ABPE were divided into three type-I ABPE, three type-II ABPE, and one mixed-type ABPE. Gender ratio: (male:female = 5:4).
Clinical Findings
Epilepsy
Sixteen out of eighteen children had seizure attacks, and their mean age at seizure onset was 55.4 ± 27.0 months (ranging from 27 to 107 months). Among that, 13 children's EEG showed ESES signal [ABPE (5), LKS (3), CSWS (5)]. The average time from seizure onset to ESES signal onset was 22.7 ± 15.5 months (ranging from 1 to 53 months). Seizure types are shown in Table 1. Eight patients had new epileptic attack forms within 3 months before or after the onset of ESES, in which five children had new types of seizure before ESES; three had it concurrently with ESES. The average time from seizure onset to appearance of new forms of attacks was 24.3 ± 15.2 months.
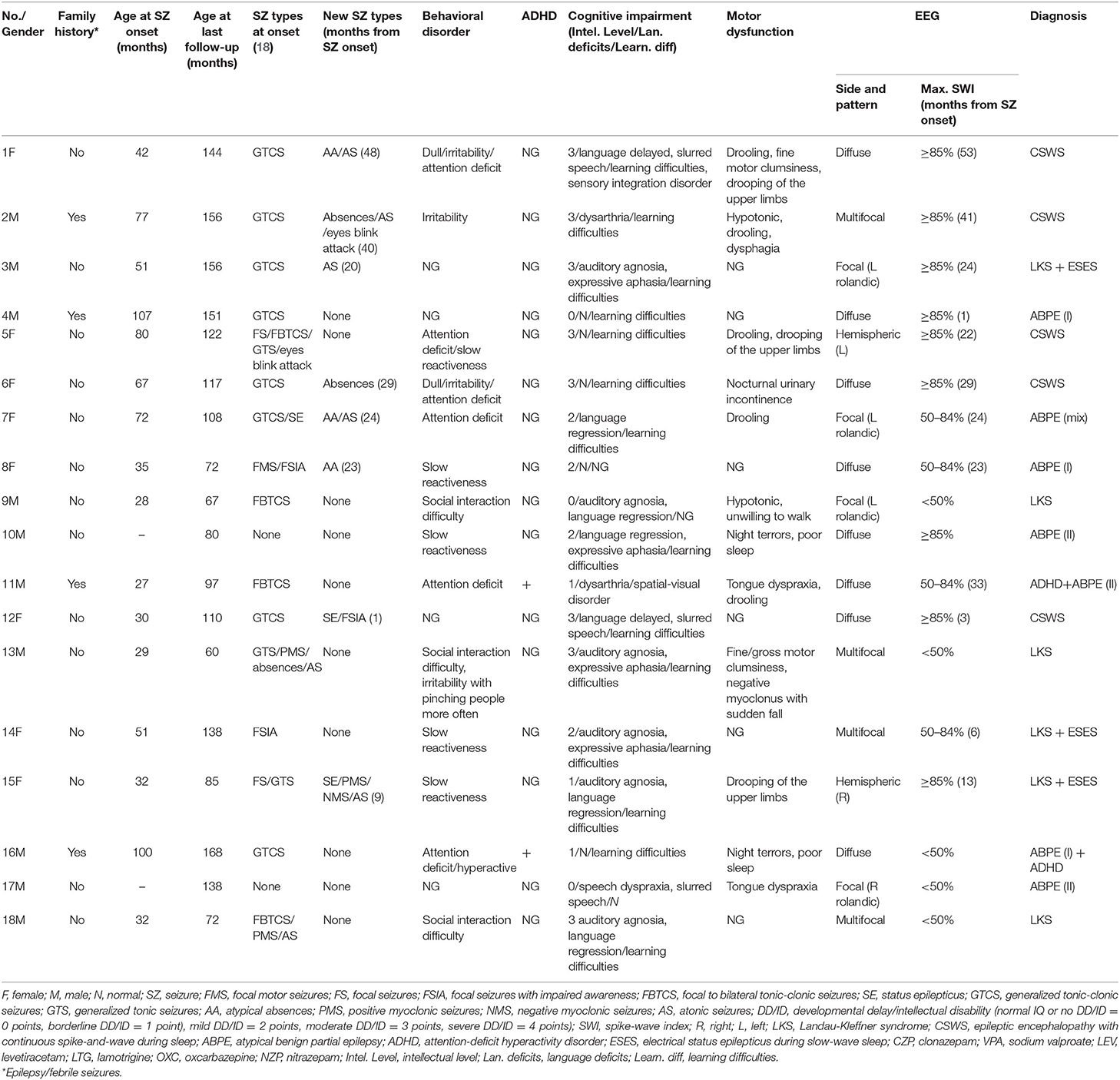
Table 1. Electroclinical features and diagnosis of 18 patients with EAS.
Two children had no seizure (patients No. 10 and No. 17). According to their clinical manifestation, with aphasia and oropharyngeal dyskinesia with normal auditory tests, they were diagnosed as ABPE type II. The EEG signal of patient No. 10 showed a diffuse spike-slow wave in the bilateral center and posterior temporal area (SWI ≥ 85%), while the EEG of patient No. 17 showed a focal spike-slow wave in the rolandic area on the right hemisphere (SWI <50%).
Cognitive/Behavioral/Motor Symptoms
The cognitive, behavioral, and motor function impairment of 18 children are shown in Table 2. Among the 13 patients with ESES, 10 (76.9%) presented one or more behavioral abnormalities, such as dullness, irritability, aggressive behavior, attention deficit, and slow reactiveness, and one was diagnosed as having attention-deficit and hyperactivity disorder (ADHD). All patients (100%) with ESES showed cognitive impairment, which presented as intellectual impairment [1/13 (7.7%) normal, 2/13 (15.3%) borderline, 4/13 (30.8%) mild, 6/13 (46.2%) moderate to severe], learning difficulties, visual motor/visual spatial impairment, sensory integration disorder, and language impairment. Eight of 13 patients (61.5%) had motor dysfunction, such as decreased motor coordination, fine/gross motor clumsiness, orofacial dyspraxia with drooling, negative myoclonus with sudden falls, and negative myoclonus with drooping of the upper limbs.
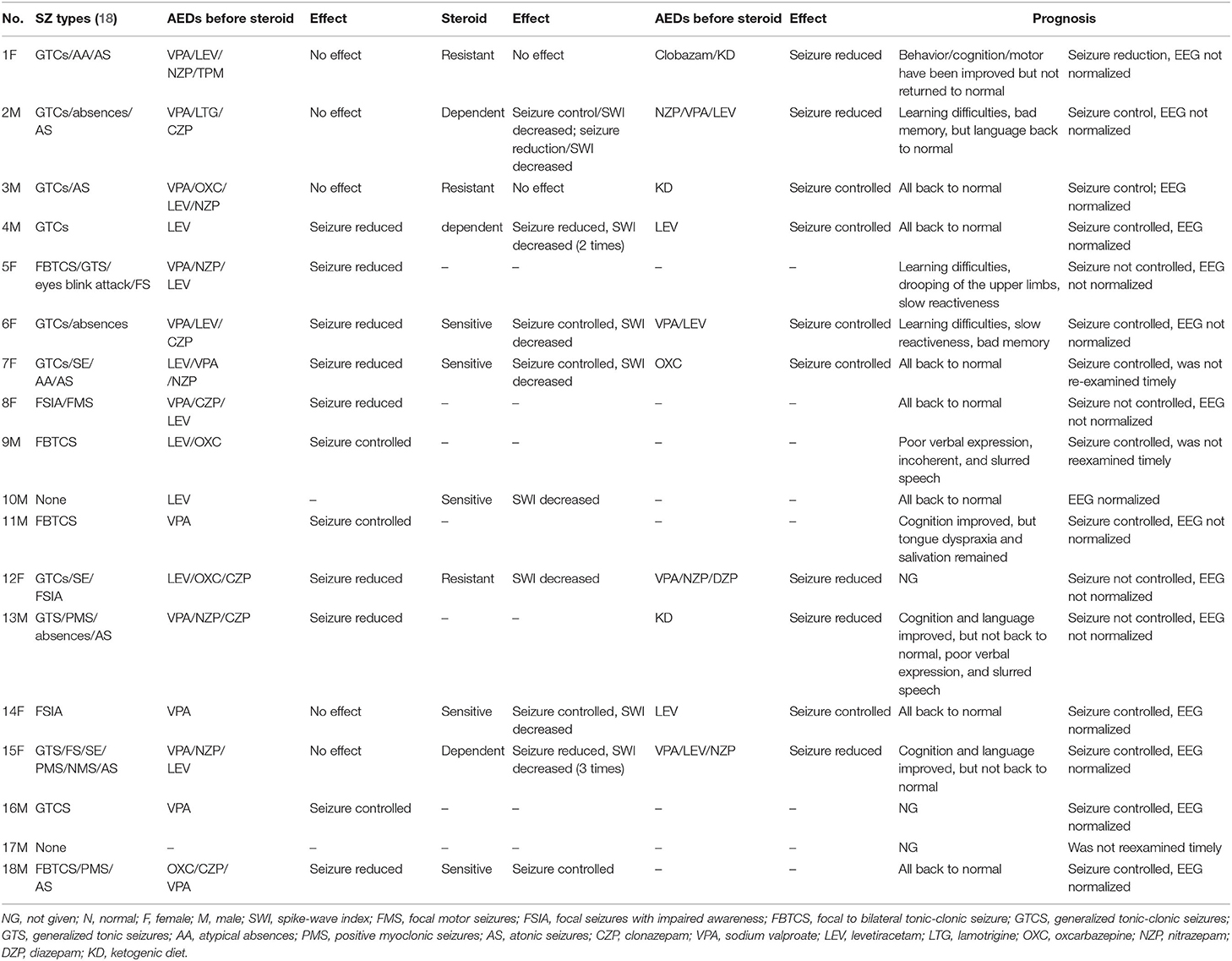
Table 2. The cognitive, behavioral, and motor function impairment of 18 children.
Among the five patients without ESES, four (80%) manifested behavioral abnormalities, such as attention deficit, hyperactivity, irritability, and impaired social interactions, and one was also diagnosed with ADHD. All patients without ESES (100%) had cognitive impairment, which presented the same as what was mentioned above in the group of patients with ESES, but a different degree (intellectual impairment: 2/5 (40%) normal, 1/5 (20%) borderline, 2 (40%) moderate to severe). Four of five patients (80%) had motor dysfunctions, such as fine/gross motor clumsiness, dysarthria, tongue dyspraxia, hypotonia, negative myoclonus with sudden fall, and night terrors.
A total of 13/18 (72.2%) children had language impairment. Global intelligence quotient (IQ) ranged from 41 to 96 in the entire sample, with an average score of 66.7 ± 17.0. Neuropsychiatric comorbidity was found in two individuals with ADHD (10%).
Notably, in our study, ID of children diagnosed with LKS ranged from normal to moderate-severe (Table 3), and two who had moderate-severe ID had EEG-SWI <50% while one with borderline IQ had EEG-SWI ≥85%.
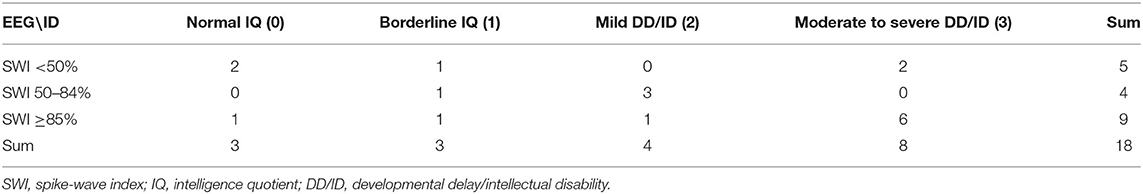
Table 3. SWI and IQ or DD/ID characteristics of 18 patients.
Accessory Examination
Brain MRI
All of the 18 patients went through MRI. Four patients had subtle demyelination anomalies (No. 3, No. 4, No. 11, and No. 18), and the brain MRIs of the remaining individuals were normal. However, after the reassessment of two neurologists, the mild abnormality in the four patients' MRI was found to be of no clinical significance.
Twenty-Four-Hours EEG
In our study, 50% (9/18) of the individuals had an EEG-SWI ≥85%, 22.2% (4/18) had an EEG-SWI between 50 and 84%, and 27.8% (5/18) had an EEG-SWI <50%. The baseline severity of the SWI in groups A, B, and C was not significantly correlated with their DD/ID severity (rho = 0.364, p = 0.138, n = 18) (Table 4). Notably, the deterioration of clinical manifestation (including the onset of new seizures or appearance of behavioral/cognitive/motor impairment) in children with ESES all appeared before the onset or at the onset of their ESES.

Table 4. Intelligence distribution of children diagnosed as ABPE/LKS/CSWS.
Pathogenic Variants
2 of the 8 patients (25.0%) who were sequenced were found to carry de novo pathogenic GRIN2A mutations (BioSample accessions: SRR12681660, SRR12681661). These two GRIN2A mutations were detected in two male patients with ABPE (II) patient No. 10 (Proband 1): cDNA c.2094T>G (p.Y698*) and patient No. 11 (Proband 2): cDNA c.2041C>T (p.Y681*) and were found to be pathogenic according to the guidelines of the American College of Medical Genetics and Genomics (ACMG) (the evidence levels of the two variants were both PVS + PS). Patient No. 9 was found to carry an inherited GRIN2A mutation: cDNA c.4126C>T (p.R1376C) which was ruled out later because of no segregation found in the proband's family (even Mutation Taster and PolyPhen-2 outcomes showed “disease causing”). The mutation of gene SLC2A1: cDNA c.668G>A (p.R223Q) was identified in patient No. 1 who was diagnosed with CSWS and had cerebrospinal fluid (CSF)/periphery blood glucose ratio of 0.56 (borderline CSF glucose for children, 2.7 mmol/l). However, this mutation was ruled out later as well, because the functional experiment of R233Q had been performed in 2012 with an outcome as “no functional change.”
Proband 1 (Tyr698*) is a male aged 6.8 years old with a negative family history and a normal perinatal period. He could sing children's songs and had normal neuromotor development before becoming ill. His language regression initially started at 3 years and 11 months as he gradually lost the ability to speak or only unconsciously mumbled “BABA” in 1 month. However, he did not have any difficulty in following auditory instructions. His Wechsler Intelligence Scale score was 70 points. He did not have any seizures, but with ESES affecting centrotemporal area on the left side and parietal, occipital, and temporal lobes on the right side. Brain MRI was normal. Levetiracetam combined with a high dose of intravenous methylprednisolone (two episodes) was given when he was 4 years old, and subsequently, the linguistic impairment and intellectual deficit gradually improved.
Proband 2 (Arg681*) is a male aged 8.1 years old with normal perinatal period and a positive family history, as his father had febrile seizure once during childhood. The proband suffered recurrent partial seizures (including focal to bilateral tonic-clonic seizures) which occasionally happened after febris or afebris, six times in total and lasting 10–30 min per time since he was 2 years and 3 months old. His score on the Wechsler Intelligence Scale was 85 points, whereas neuropsychological evaluation presented spatial-visual disorder, expressive language impairment, and ADHD. The EEG showed a spike-slow wave on both sides of the front and the middle temporal lobe areas during the wake-up and sleeping period, especially during the sleeping period. MRI result presented abnormal signal on the right-side temporal lobe at the age of 2.5 but was normal after being rescanned at 4 and 7 years old, respectively. He was treated with valproic acid around the age of 3 years old, and after that, his seizures relieved in 60 months while language impairment remained.
One of the identified pathogenic GRIN2A mutations-de novo c.2094T>G (p.Y698*) has not been reported before, whereas the GRIN2A mutation-de novo c.2041C>T (p.Y681*) has been reported in a family with LKS in 2013 (inherited). Both of the GRIN2A variants led to the end of the protein transcription/translation and caused NMDAR loss-of-function change (Table 5; Figure 1). Three-dimensional protein chain structures of these two variants were constructed with SWISS-MODEL, which showed both of the variants affecting the S2-LBD area (Figure 2).
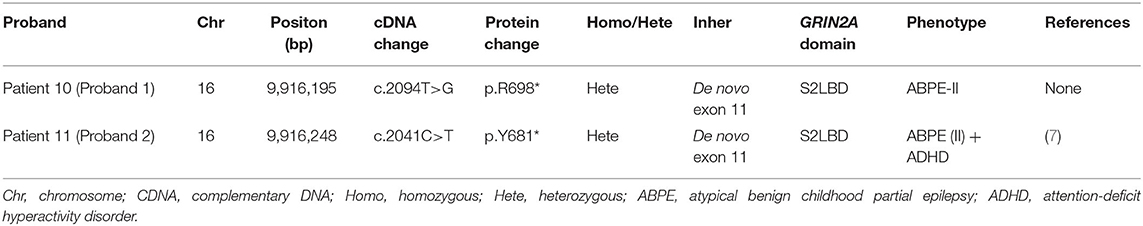
Table 5. Genetic and protein change data of patient 10 and patient 11.
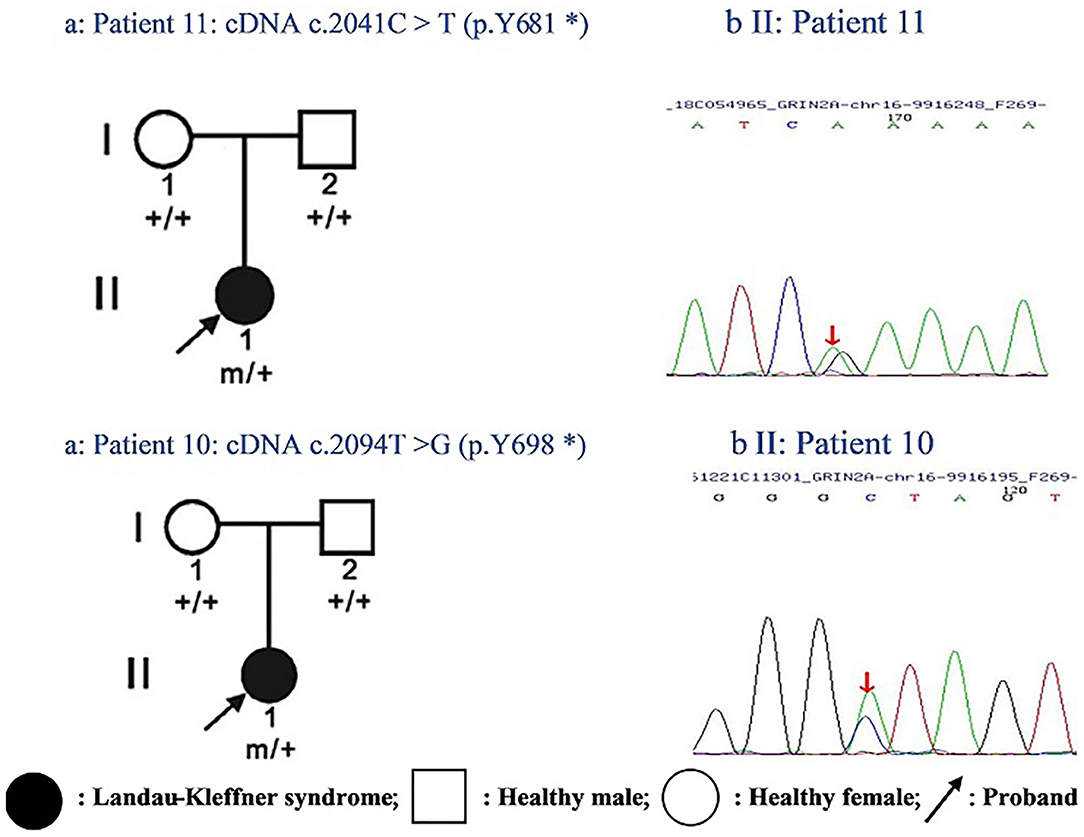
Figure 1. Pedigrees of two cases with epilepsy-aphasia disorders and identified GRIN2A mutations: (a) Filled-in circles indicate individuals with Landau-Kleffner syndrome; empty circles/squares indicate unaffected women/men. Arrows indicate the probands. Individuals with the GRIN2A mutation are indicated by m/+, and mutation-negative individuals are indicated by +/+. (b) Chromatograms of GRIN2A mutation detected in two probands. Arrows show the position of the mutation.
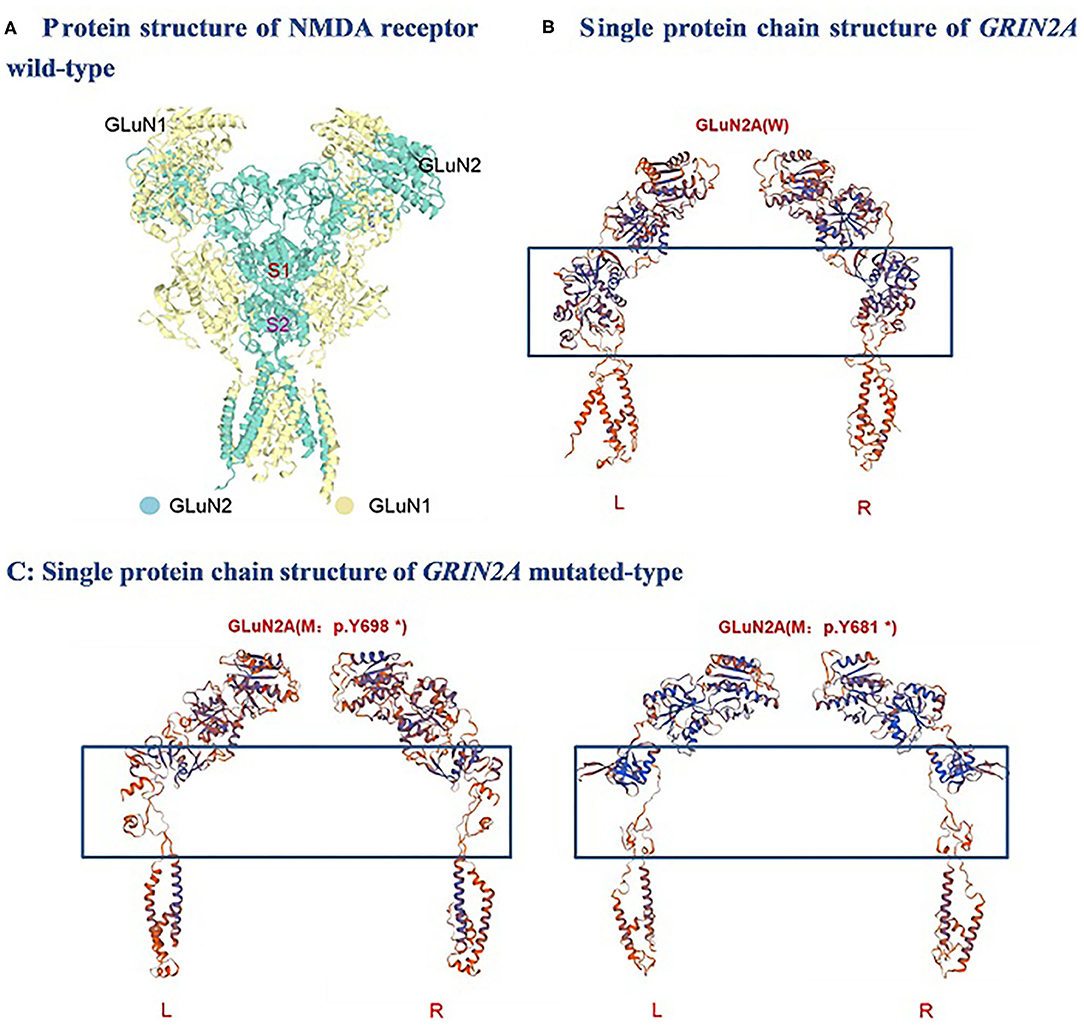
Figure 2. Molecular analysis of GRIN2A variants: 3-D protein chain structural changes in GRIN2A mutated-type compared with the wild-type were predicted with SWISS-MODEL. S1, S1 ligand-binding domains; S2, S2 ligand-binding domains; NMDA, N-methyl-D-aspartate; W, Wild-type; M, mutated-type; L, Left; R, Right.
Treatment
AEDs
In this cohort, 16 patients had seizures and 17 patients were treated with 1–5 (average 2.8) different types of AEDs. Among them, only 2 (12.5%, 2/16) patients had their seizures controlled with monotherapy, and only one had EEG normalization. Three (18.8%, 3/16) patients received therapy with multiple AEDs, and only one of which reached controlled seizures while none of which had EEG normalization. Eleven (68.7%, 11/16) patients were treated with multiple AEDs combined with steroids, and six had their seizures controlled while four had EEG normalization. In addition, three patients who were resistant to AEDs or steroid therapy were treated with a ketogenic diet (KD) subsequently, and one of them had their seizures controlled, EEG normalized, and neuropsychological function recovery (2 years after the ketogenic diet was introduced) eventually. Moreover, the other two children treated with KD also had their seizures and EEG significantly improved 4–8 months later.
In this study, nine different kinds of AEDs were added, including valproate, levetiracetam, nitrazepam, topiramate, lamotrigine, clonazepam, oxcarbazepine, clobazam, and diazepam. The anticonvulsants initially used in this cohort were mostly valproic acid (64.7% of patients), levetiracetam (29.4%), and oxcarbazepine (5.9%). The most effective (seizure control + seizure reduction) AEDs were valproate (4/17, 23.5%), valproate + nitrazepam/clonazepam (5/17, 29.4%), and levetiracetam (2/17, 11.8%). It should be noted that there were two children who suffered from absence after the addition of levetiracetam.
Steroids
Of the 11 patients who received steroid treatment because their SWI continued to increase despite taking AEDs, 45.4% (5/11) were sensitive to steroids, 27.3% (3/11) were resistant to steroids, and 27.3% (3/11) were steroid-dependent.
Prognosis
Until August 2019, the average time of follow-up was 61.7 ± 26.4 months (31–114 months). For 16 patients who had seizures, 11 were seizure free, and seven had EEG normalization. The mean time from the onset of attack to being seizure free was 27.5 ± 23.3 months (n = 11), and the mean time for the EEG to return to normal was 37 ± 25.4 months (n = 7). The cognitive/behavioral/motor functions were all back to normal in seven patients. Among the patients who were seizure free at the last follow-up, five still suffered from different kinds of deficits. However, in five cases, their cognitive/behavioral/motor functions almost returned to a normal level after EEG normalization (Table 2).
Literature Review of Pathogenic GRIN2A-Lof Variants
We reviewed all the published literature reporting patients with pathogenic GRIN2A variants, and missense mutations which were proved to result in NMDAR-LOF change were selected from those literatures along with GRIN2A truncation variants (Table 6). There were 18 GRIN2A missense mutations and 11 GRIN2A truncating mutations in total which lead to NMDAR-LOF change. Of these mutations, 9 (31.0%) were situated in NTD, 6 (20.7%) in LBD (S1), 10 (34.5%) in LBD (S2), 2 (6.9%) in CTD, and only 2 (6.9%) in TMD and linker domain, respectively. Among those literatures we reviewed, the language situations of 22 cases were descripted in detail; 95.5% of them manifested language deficit, such as speech/language developmental delay, language regression, acquired aphasia, auditory agnosia, verbal dyspraxia, slurred speech, and dysarthria. Among the 14 cases which had clear intellectual evaluation in the literatures, 28.6% had normal IQ, 21.4% had mild ID/DD, 28.6% had moderate ID/DD, 14.3% had mild-moderate ID/DD, and only 7.1% had severe ID/DD. To the best of our knowledge, the treatment process of only two patients has been described in detail in all of the published literature in Table 6 (11, 22). One of the two patients diagnosed with LKS were treated with prednisone and immunoglobin after being unresponsive to multiple AEDs, and the cognition of this patient improved gradually 6 months after prednisone and immunoglobin treatment. Another patient diagnosed with atypical rolandic epilepsy was treated with four AEDs but responded poorly. Both of their EEGs showed ESES.
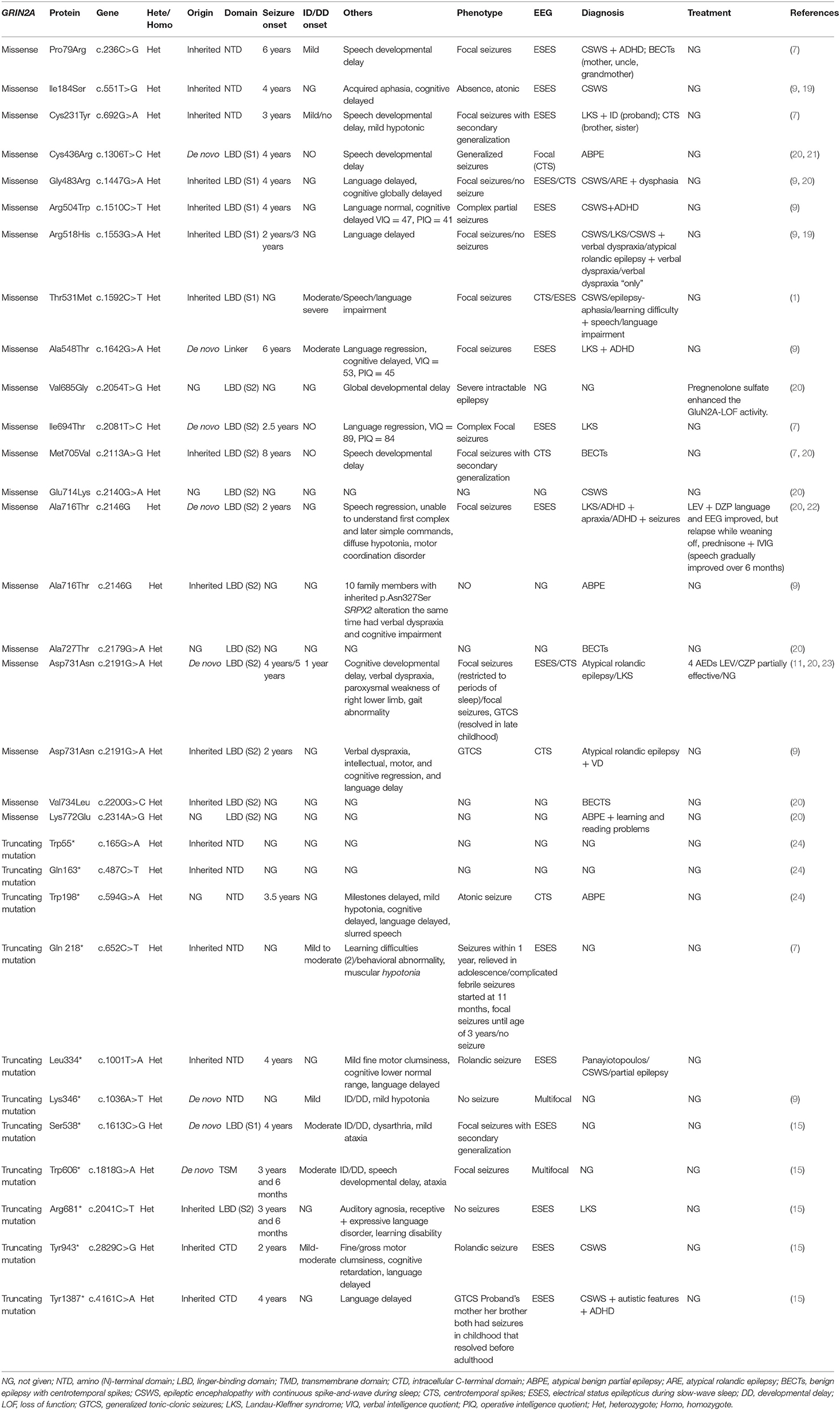
Table 6. Characteristics of patients with GRIN2A-LOF variants (missense + truncation).
Correlation Analysis of EAS-Related Genes
We conducted systematic literature review to identify all of the EAS-related genes in our previous work (25). In this study, we applied string software to study the PPI of those EAS-related genes, and the results are shown in Figure 3.
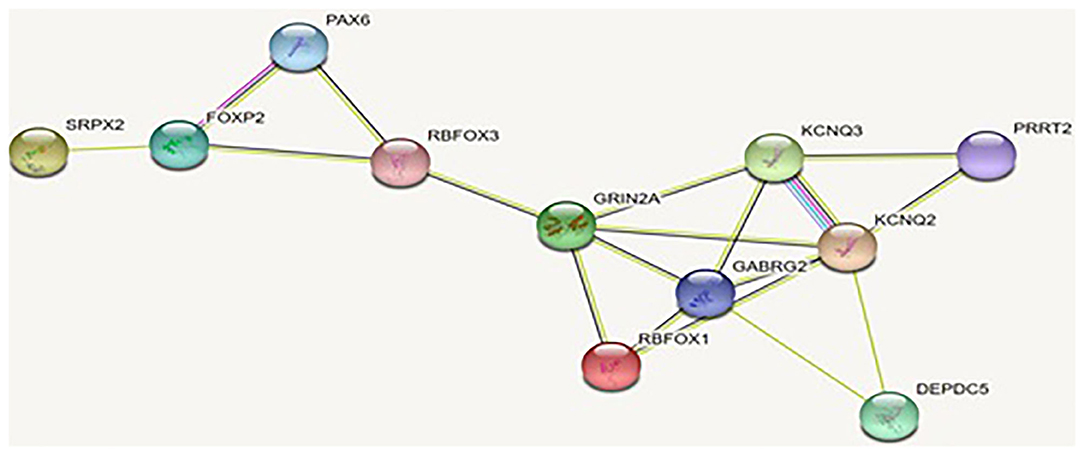
Figure 3. Interaction of the proteins encoded by the genes related to EAS: the interaction of these proteins is stronger than a group of similar size proteins randomly selected from the genome. Such enrichment suggests that these proteins are biologically related and that the GRIN2A gene appears to be the hub gene of PPI.
Further, we analyzed the gene ontology to understand the biological process, molecular function, and cellular components [false-discovery rate (FDR), <0.01] in EAS-related genes. The results are shown in Table 7.
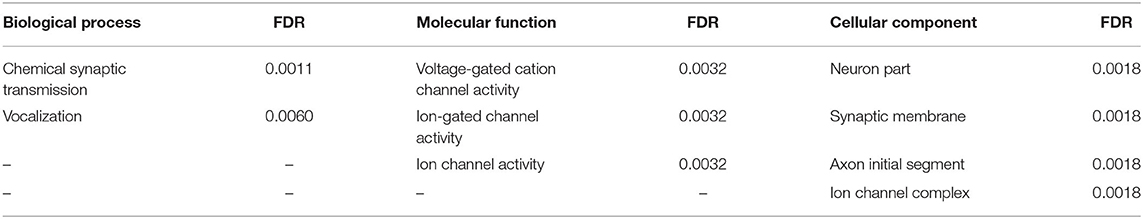
Table 7. Gene ontology of genes related to EAS.
Discussion
Electroclinical Characteristics
Epilepsy is the most typical feature in our group, and the average age of onset was 4.5 ± 2.2 years, which is significantly earlier than that of a previous large-scale study of Chinese classic BECTS (6.85 ± 2.45 years, 1,817 children) (26).
In our group, the onset of new seizures or the appearance of behavioral/cognitive/motor impairment in children with ESES all appeared earlier or at the same time as the onset of their ESES. To the best of our knowledge, there are few research studies on the time relationship between ESES and clinical deterioration.
Therefore, this data might suggest that when children with suspected BECTs have an early onset, changed seizure semiology, and deterioration of behavior/cognition/motor function, neurologists should be alert on the appearance of ESES.
Now, there are inconsistent agreements on the relationship between the EEG SWI severity and the degree of cognitive impairment in children with EAS. Previous studies have reported that the severity of cognitive impairment in EAS patients seem to be related to the EEG SWI, but in recent years, some studies have disagreed with that. Wolff et al. reported that cognitive deficits were associated with spike location but not with the SWI in 20 children with IFE (27), whereas Nicolai et al. reported that the correlation between cognitive deficits and EEG location was not consistent (28). Overvliet et al. found that there was a relationship between the cognitive deficits and SWI in nocturne, which indicated that cognitive and behavioral functional deficits were concordant with the magnitude of the EEG discharge index (29). In our study, however, the baseline severity of the SWI was not significantly correlated with the DD/ID severity. The possible explanation might be as follows: first, the neuropsychological evaluation did not always parallel with the EEG evaluation due to the poor compliance of some patients. Furthermore, inflammation may be one of the mechanisms. In 2016, Munckhof et al. found that the levels of IL-1A, IL-6, and CXCL8/IL-8 of patients with ESES were significantly higher than those of the control group. After immunotherapy (steroid or immunoglobulin) was given, the level of IL-6 decreased significantly. The change of IL-6 was accompanied by the improvement of EEG SWI and neuropsychological evaluation (30). Third, sleep structure disruption may also contribute to the cognitive deficit. Sleep has been demonstrated to play an important role in neurophysiological and neurochemical processes underlying the memory and learning consolidation (31). Epilepsy and pathognomonic sleep-related EEG pattern as ESES in EAS may impair patients' cognition by affecting normal sleep-awareness cycles, resulting in decreased total sleep time, lower sleep efficiency, and higher arousal index. Moreover, sleep fragmentation disorders could further aggravate the seizure situation as a vicious circle (32). However, the sleep quality evaluation of patients in this study has not been performed due to limited recording instrument and poor compliance. Finally, genetic factors might contribute to the intellectual impairment in some EAS children.
In conclusion, ESES is limited in estimating the degree of cognitive deficits because the deterioration of clinical manifestations was ahead of or synchronized with the ESES occurrence in our study and the severity of the SWI did not parallel with the DD/ID severity either. Therefore, we hypothesize that the neuropsychological deterioration in children with EAS is not only completely affected by electric discharge but also may be affected by autoimmune response, sleep disorder, and genetic etiology. However, the small number of subjects included in this study is one of our limitations.
Genetic Characteristics
In our series, the identification of EAS-related pathogenic variants further confirmed its genetic predisposition. So far, EAS was believed to be a complex polygenic hereditary disorder, while multiple genes were found to be related to EAS, such as GRIN2A, KCNQ2, KCNQ3, DEPDC5, PAX6, GABRG2, RbFOX1/RbFOX3, SRPX2, FOXP2, PRRT2, and other genes (5–16, 33). Recently, several studies have confirmed that the GRIN2A gene is the most likely candidate for EAS. The GRIN2A gene is located in chromosome 16p13.2 and encodes the GluN2A subunit of the N-methyl-D-aspartate receptor (NMDAR). NMDARs are hetero-tetramers usually composed of two GluN1 and two GluN2 (A-D) subunits and involved in neurodevelopment and brain plasticity, as well as learning, memory, and cognitive processes. NMDARs are activated following the binding of glycine and glutamate to the GluN1 and GluN2 subunits, respectively. GluN2 (A-D) subunit mainly consist of four domains: the extracellular amino (N)-terminal domain (NTD) which is involved in subunit assembly and allosteric modulation; the agonist-binding domain (ABD) which is made up of two discontinuous segments (S1 + S2); the transmembrane domain (TMD) which is composed of three and a half transmembrane helices (TM1–4) and forms the channel pore; and the intracellular C-terminal domain (CTD) involved in receptor trafficking (1, 15).
So far, more than 80 GRIN2A gene variants have been found in epilepsies, especially in EAS (1, 34). The more severe the phenotype is, the higher the positive rate of GRIN2A mutation is. It has been found that the positive rate of GRIN2A gene in the classical BECTS is 4.9%, in CSWs 17.6%, and in LKS 20% (7).
A recent study shows null variants of GRIN2A were associated with less severe EAS phenotypes and NMDAR-LOF (15). This finding is consistent with our outcomes: two ABPE (II) patients carrying GRIN2A null variants presented less severe DD/ID, despite the ESES-EEG existence. This finding further enforced that the severity of DD/ID might not parallel the severity of SWI, but genotype variants. However, due to the expensive testing fees, another limitation of this study is that only for eight cases whole-exome sequencing was available.
De novo GRIN2A variant c.2041C>T (p.Arg 681*) identified in our study was found to cause LKS (inherited) in 2013 (7). However, the symptoms of the patient in the previous study (manifesting no seizures, but with learning disability, auditory agnosia and receptive/expressive language disorder) were partially different from ours with linguistic impairments in common. The explanation could be the different inheritance pattern and phenotypic heterogeneity, which is worth further investigation.
As shown in Figure 3 and Table 7, EAS-related genes are enriched in the biological process of chemical synaptic transmission and vocalization (FDR, <0.01). As the key protein in PPI network, GluN2a protein coded by GRIN2A is linked to Foxp2-SRPX2 and the Pax6 protein through Rbfox3. The signal pathway of foxp2-srpx2/uPAR network is proved to be related to language disorders (35). The foxp2 protein is also regulated by the Pax6 protein. Therefore, we speculated that the GRIN2A gene might contribute to the language disorder of EAS through the foxp2-srpx2/uPAR pathway which is worth studying further.
Treatment
Among the 17 patients treated with antiepileptic drugs, 12.5% were treated with monotherapy, 18.8% with multiple AED therapy, and 68.7% with multiple AEDs combined with steroids. Compared with the study of classic BECT children in Liu's group, where single drug treatment accounted for 62.9%, and multiple therapies accounted for 10.6% (26). Among the 11 children who received AEDs combined with steroid therapy, 72.7% of them had a good response (seizures reduction + seizure control), including one with the c.2094T>G (p.Y698*) variant. Our data suggested that the effect of monotherapy was not sufficient for severe subtypes of EAS or atypical BECTs, and most patients needed further methylpyridine pulse therapy after adding multiple AEDs. This was in concordance with current studies (36, 37). In 2015, a meta-analysis of 575 patients with ESES showed that corticosteroids and surgical treatment were the most effective way to reduce seizures and epileptic discharge and improve cognition, with an effective rate of 81 and 90%, respectively (36). A large-scale study of 44 patients with ESES also indicated that steroid therapy had a significant effect on the improvement of EEG and clinical neuropsychology of patients with ESES (38).
Notably, one patient with EEG-ESES carrying GRIN2A truncation variants which led to NMADR-LOF change our study reached cognitive recovery and SWI normalization ultimately by steroid treatment after given unresponsive AEDs. The mechanism could be as follows: (1) Autoimmune response was proved to contribute partially to the etiology of EAS. GRIN2A mutation itself could increase the antigenicity of extracellular components of NMDA receptor (22, 24, 39), and the seizure process might also increase the self-antigenicity of NMDA receptors (22). (2) Most likely, recent research presented that steroids can enhance the function of NMDA receptors and regulate glutamate activity. In 2012, Cynthia et al. found that hormones can increase the NMDA receptor numbers in 5-hydroxytryptamine neurons and enhance the activity of glutamate (40). In 2016, Swanger also found that pregnenolone sulfate can enhance NMDA function in LOF mutation of GRIN2A in vitro (20). Similar to our study, the previously reported patient with LKS who were treated with prednisone and immunoglobin after given unresponsive multiple AEDs, also noted significant improvement (22).
Therefore, for children carrying the GRIN2A mutation resulting in LOF of NMDAR, researchers may need to study whether neurologists should consider providing steroid treatment in time after given unresponsive AEDs, despite the absence of ESES signal on EEG to ensure a better prognosis.
For patients with pathogenic GRIN2A variants leading to NMDAR-GOF change, there are studies which argued that they mostly result in epileptic encephalopathy with more severe ID/DD and have poor prognosis (15). And the situation of these patients could be significant improved by memantine, a non-competitive antagonist of the NMDAR (41, 42). Those case reports guarantee the importance of precise treatment (19, 21, 23, 41, 42).
For now, the principle of treatment for EAS is not only to control the seizures but also to effectively decrease the SWI and improve the cognitive and linguistic outcomes (36). Earlier and appropriate intervention is always the priority to guarantee a good prognosis. Therefore, it is necessary to combine the epileptic syndromes, seizure types, neuropsychological status, SWI, and genetic characteristics of patients all together to conduct a comprehensive plan for the selection of AEDs. The effects of AEDs alone on cognitive improvement and seizure control are always insufficient for patients with EAS. So, methylpyridines pulse therapy should be given in a timely manner and monitored carefully due to the high relapse rate. However, under what certain circumstance should steroid be added still remains unclear in EAS. Especially for patients carrying GRIN2A-LOF variants present no ESES signal.
Conclusions
In this study, we retrospectively reviewed the electroclinical characteristics and gene test results of children diagnosed with ABPE, LKS, and CSWS. Then, we further explored the GO of EAS-related genes and the PPI of those genes expressed. Our data suggested that when children suspected with BECTs have early-onset age, changed seizure semiology, and deterioration of behavior/cognition/motor function, neurologists should be alert on the appearance of ESES. The neuropsychological deterioration in children with EAS might not only be completely affected by electric discharge severity but also genetic etiology. Our finding also enforced the current genotype-phenotype relationship theory about EAS. For EAS children, GRIN2A-FOXP2-SRPX2/uPAR signal network might contribute to the mechanism of their language deficit.
Data Availability Statement
The authors acknowledge that the data presented in this study must be deposited and made publicly available in an acceptable repository, prior to publication. Frontiers cannot accept a article that does not adhere to our open data policies.
Ethics Statement
The studies involving human participants were reviewed and approved by the ethics committee of Children's Hospital of Chongqing Medical University. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
XL and LJ contributed to coming up with the ideas and data collection. XL and L-LX participated in co-writing. WH, S-QH, J-NM, and JW participated in the critical review of the manuscript. All authors have approved the manuscript for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2020.574803/full#supplementary-material
References
1. Carvill GL, Regan BM, Yendle SC, O'Roak BJ, Lozovaya N, Bruneau N, et al. GRIN2A mutations cause epilepsy-aphasia spectrum disorders. Nature genetics. (2013) 45:1073–6. doi: 10.1038/ng.2727
2. Deonna T, Zesiger P, Davidoff V, Maeder M, Mayor C, Roulet E. Benign partial epilepsy of childhood: a longitudinal neuropsychological and EEG study of cognitive function. Dev Med Child Neurol. (2000) 42:595–603. doi: 10.1017/s0012162200001122
3. Fejerman N, Caraballo R, Tenembaum SN. Atypical evolutions of benign localization-related epilepsies in children: are they predictable? Epilepsia. (2000) 41:380–90. doi: 10.1111/j.1528-1157.2000.tb00177.x
4. Aicardi J, Chevrie JJ. Atypical benign partial epilepsy of childhood. Dev Med Child Neurol. (1982) 24:281–92. doi: 10.1111/j.1469-8749.1982.tb13620.x
5. Lee YJ, Hwang SK, Kwon S. The clinical spectrum of benign epilepsy with centro-temporal spikes: a challenge in categorization and predictability. J Epilepsy Res. (2017) 7:1–6. doi: 10.14581/jer.17001
6. Baumer FM, Cardon AL, Porter BE. Language dysfunction in pediatric epilepsy. J Pediatr. (2018) 194:13–21. doi: 10.1016/j.jpeds.2017.10.031
7. Lemke JR, Lal D, Reinthaler EM, Steiner I, Nothnagel M, Alber M, et al. Mutations in GRIN2A cause idiopathic focal epilepsy with Rolandic spikes. Nat Genet. (2013) 45:1067–72. doi: 10.1038/ng.2728
8. Rudolf G, Valenti MP, Hirsch E, Szepetowski P. From Rolandic epilepsy to continuous spike-and-waves during sleep and Landau-Kleffner syndromes: insights into possible genetic factors. Epilepsia. (2009) 50:25–8. doi: 10.1111/j.1528-1167.2009.02214.x
9. Lesca G, Rudolf G, Bruneau N, Lozovaya N, Labalme A, Boutry-Kryza N, et al. GRIN2A mutations in acquired epileptic aphasia and related childhood focal epilepsies and encephalopathies with speech and language dysfunction. Nat Genet. (2013) 45:1061–6. doi: 10.1038/ng.2726
10. Chen J, Yang Z, Liu X, Ji T, Fu N, Wu Y, et al. Efficacy of methylprednisolone therapy for electrical status epilepticus during sleep in children. Zhonghua Er Ke Za Zhi. (2014) 52:678–82. doi: 10.3760/cma.j.issn.0578-1310.2014.09.008
11. Gao K, Tankovic A, Zhang Y, Kusumoto H, Zhang J, Chen W, et al. A de novo loss-of-function GRIN2A mutation associated with childhood focal epilepsy and acquired epileptic aphasia. PLoS ONE. (2017) 12:e0170818. doi: 10.1371/journal.pone.0170818
12. Tsai MH, Vears DF, Turner SJ, Smith RL, Berkovic SF, Sadleir LG, et al. Clinical genetic study of the epilepsy-aphasia spectrum. Epilepsia. (2013) 54:280–287. doi: 10.1111/epi.12065
13. Turner SJ, Morgan AT, Perez ER, Scheffer IE. New genes for focal epilepsies with speech and language disorders. Curr Neurol Neurosci Rep. (2015) 15:35. doi: 10.1007/s11910-015-0554-0
14. Bobbili DR, Lal D, May P, Reinthaler EM, Jabbari K, Thiele H, et al. Exome-wide analysis of mutational burden in patients with typical and atypical Rolandic epilepsy. Eur J Hum Genet. (2018) 26:258–264. doi: 10.1038/s41431-017-0034-x
15. Strehlow V, Heyne HO, Vlaskamp DRM, Marwick KFM, Rudolf G, de Bellescize J, et al. GRIN2A-related disorders: genotype and functional consequence predict phenotype. Brain. (2019) 142:80–92. doi: 10.1093/brain/awy304
16. International League Against Epilepsy (Epilepsy Syndromes). Available online at: https://www.epilepsydiagnosis.org/syndrome/eecsws-overview.html
17. Zhang YH, Liu XY, Yang ZX, Bao XH, Xiong H, Wu Y, et al. Electroclinical features of the variants of benign childhood epilepsy with central temporal spikes. Chinese J Prac Pediatr Dec. (2010) 25:12.
18. Fisher RS, Cross JH, French JA, Higurashi N, Hirsch E, Jansen FE, et al. Operational classification of seizure types by the international league against epilepsy: position paper of the Ilae Commission for Classification and Terminology. Epilepsia. (2017) 58:522–30. doi: 10.1111/epi.13670
19. Sibarov DA, Bruneau N, Antonov SM, Szepetowski P, Burnashev N, Giniatullin R. Functional properties of human NMDA receptors associated with epilepsy-related mutations of glun2a subunit. Front Cell Neurosci. (2017) 11:155. doi: 10.3389/fncel.2017.00155
20. Swanger SA, Chen W, Wells G, Burger PB, Tankovic A, Bhattacharya S, et al. Mechanistic insight into NMDA receptor dysregulation by rare variants in the GluN2A and GluN2B agonist binding domains. Am J Hum Genet. (2016) 99:1261–80. doi: 10.1016/j.ajhg.2016.10.002
21. Addis L, Virdee JK, Vidler LR, Collier DA, Pal DK, Ursu D. Epilepsy-associated GRIN2A mutations reduce NMDA receptor trafficking and agonist potency–molecular profiling and functional rescue. Sci Rep. (2017) 7:66. doi: 10.1038/s41598-017-00115-w
22. Fainberg N, Harper A, Tchapyjnikov D, Mikati MA. Response to immunotherapy in a patient with Landau-Kleffner syndrome and GRIN2A mutation. Epileptic Disord. (2016) 18:97–100. doi: 10.1684/epd.2016.0791
23. Dyment DA, Tétreault M, Beaulieu CL, Hartley T, Ferreira P, Chardon JW, et al. Whole-exome sequencing broadens the phenotypic spectrum of rare pediatric epilepsy: a retrospective study. Clin Genet. (2015) 88:34–40. doi: 10.1111/cge.12464
24. Endele S, Rosenberger G, Geider K, Popp B, Tamer C, Stefanova I, et al. Mutations in GRIN2A and GRIN2B encoding regulatory subunits of NMDA receptors cause variable neurodevelopmental phenotypes. Nat Genet. (2010) 42:1021–6. doi: 10.1038/ng.677
25. Xiao L, Li J. Research progress in genetic studies of Rolandic epilepsy. Chin J Appl Clin Pediatr. (2020) 35:93–6. doi: 10.3760/cma.j.cn101070-20190306-00167
26. Liu MJ, Su XJ, Shi XY, Wu GF, Zhang YQ, Gao L, et al. Clinical features of benign epilepsy of childhood with centrotemporal spikes in Chinese children. Medicine. (2017) 96:e5623. doi: 10.1097/MD.000000000000562
27. Wolff M, Weiskopf N, Serra E, Preissl H, Birbaumer N, Kraegeloh-Mann I. Benign partial epilepsy in childhood selective cognitive deficits are related to the location of focal spikes determined by combined EEG. Epilepsia. (2005) 46:1661–667. doi: 10.1111/j.1528-1167.2005.00255.x
28. Nicolai J, van der Linden I, Arends JB, van Mil SG, Weber JW, Vles JS, et al. EEG characteristics related to educational impairments in children with benign childhood epilepsy with centrotemporal spikes. Epilepsia. (2007) 48:2093–100. doi: 10.1111/j.1528-1167.2007.01203.x
29. Overvliet GM, Besseling RM, Vles JS, Hofman PA, Backes WH, van Hall MH, et al. Nocturnal epileptiform EEG discharges, nocturnal epileptic seizures, and language impairments in children: review of the literature. Epilepsy Behav. (2010) 19:550–8. doi: 10.1016/j.yebeh.2010.09.015
30. van den Munckhof B, de Vries EE, Braun KP, Boss HM, Willemsen MA, van Royen-Kerkhof A, et al. Serum inflammatory mediators correlate with disease activity in electrical status epilepticus in sleep (ESES) syndrome. Epilepsia. (2016) 57:e45–50. doi: 10.1111/epi.13274
31. Parisi P, Bruni O, Pia Villa M, Verrotti A, Miano S, Luchetti A, et al. The relationship between sleep and epilepsy: the effect on cognitive functioning in children. Dev Med Child Neurol. (2010) 52:805–10. doi: 10.1111/j.1469-8749.2010.03662.x
32. Carotenuto M, Parisi P, Esposito M, Cortese S, Elia M. Sleep alterations in children with refractory epileptic encephalopathies: a polysomnographic study. Epilepsy Behav. (2014) 35:50–3. doi: 10.1016/j.yebeh.2014.03.009
33. Dimassi S, Labalme A, Lesca G, Rudolf G, Bruneau N, Hirsch E, et al. A subset of genomic alterations detected in Rolandic epilepsies contains candidate or known epilepsy genes including GRIN2A and PRRT2. Epilepsia. (2014) 55:370–378. doi: 10.1111/epi.12502
34. Xu XX, Liu XR, Fan CY, Lai JX, Shi YW, Yang W, et al. Functional investigation of a GRIN2A variant associated with Rolandic epilepsy. Neurosci Bull. (2018) 34:237–46. doi: 10.1007/s12264-017-0182-6
35. Lai CS, Fisher SE, Hurst JA, Vargha-Khadem F, Monaco AP. A forkhead-domain gene is mutated in a severe speech and language disorder. Nature. (2001) 413:519–23. doi: 10.1038/35097076
36. van den Munckhof B, van Dee V, Sagi L, Caraballo RH, Veggiotti P, Liukkonen E, et al. Treatment of electrical status epilepticus in sleep: a pooled analysis of 575 cases. Epilepsia. (2015) 56:1738–46. doi: 10.1111/epi.13128
37. Kramer U, Sagi L, Goldberg-Stern H, Zelnik N, Nissenkorn A, Ben-Zeev B. Clinical spectrum and medical treatment of children with electrical status epilepticus in sleep (ESES). Epilepsia. (2009) 50:1517–24. doi: 10.1111/j.1528-1167.2008.01891.x
38. Buzatu M, Bulteau C, Altuzarra C, Dulac O, Van Bogaert P. Corticosteroids as treatment of epileptic syndromes with continuous spike-waves during slow-wave sleep. Epilepsia. (2009) 50:68–72. doi: 10.1111/j.1528-1167.2009.02224.x
39. Lu Y, Sareddy GR, Wang J, Wang R, Li Y, Dong Y, et al. Neuron-derived estrogen regulates synaptic plasticity and memory. J Neurosci. (2019) 39:2792–809. doi: 10.1523/JNEUROSCI.1970-18.2019
40. Bethea CL, Reddy AP. Ovarian steroids increase glutamatergic related gene expression in serotonin neurons of macaques. Mol Cell Neurosci. (2012) 49:251–62. doi: 10.1016/j.mcn.2011.11.005
41. Pierson TM, Yuan H, Marsh ED, Fuentes-Fajardo K, Adams DR, Markello T, et al. GRIN2A mutation and early-onset epileptic encephalopathy: personalized therapy with memantine. Ann Clin Transl Neurol. (2014) 1:190–8. doi: 10.1002/acn3.39
Keywords: Landau-Kleffner syndrome (LKS), epileptic encephalopathy with continuous spike-and-wave during sleep (CSWS), atypical benign partial epilepsy (ABPE), genotype-phenotype relationship, epileptic-aphasia spectrum disorder
Citation: Li X, Xie L-L, Han W, Hong S-Q, Ma J-N, Wang J and Jiang L (2020) Clinical Forms and GRIN2A Genotype of Severe End of Epileptic-Aphasia Spectrum Disorder. Front. Pediatr. 8:574803. doi: 10.3389/fped.2020.574803
Received: 21 June 2020; Accepted: 28 September 2020;
Published: 06 November 2020.
Edited by:
Pasquale Parisi, Sapienza University of Rome, ItalyReviewed by:
Alice Bonuccelli, Pisana University Hospital, ItalyMaurizio Elia, Oasi Research Institute (IRCCS), Italy
Copyright © 2020 Li, Xie, Han, Hong, Ma, Wang and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Jiang, bGppYW5nQGhvc3BpdGFsLmNxbXUuZWR1LmNu