 Monique Radman
Monique Radman John McGuire
John McGuire Jerry Zimmerman
Jerry Zimmerman- Seattle Children's Hospital, Pediatric Critical Care, University of Washington, Seattle, WA, United States
Pediatric obesity is increasing in prevalence and is frequently an antecedent to adult obesity and adult obesity-associated morbidities such as atherosclerosis, type II diabetes, and chronic metabolic syndrome. Endothelial cell activation, one aspect of inflammation, is present in the early stages of atherosclerosis, often prior to the onset of symptoms. Endothelial activation is a pathological condition in which vasoconstricting, pro-thrombotic, and proliferative mediators predominate protective vasodilating, anti-thrombogenic, and anti-mitogenic mediators. Many studies report poor outcomes among obese children with systemic endothelial activation. Likewise, the link between childhood obesity and poor outcomes in critical illness is well-established. However, the link between obesity and severity of endothelial activation specifically in the setting of critical illness is largely unstudied. Although endothelial cell activation is believed to worsen disease in critically ill children, the nature and extent of this response is poorly understood due to the difficulty in measuring endothelial cell dysfunction and destruction. Based on the data available for the obese, asymptomatic population and the obese, critically ill population, the authors posit that obesity, and obesity-associated chronic inflammation, including oxidative stress and insulin resistance, may contribute to endothelial activation and associated worse outcomes among critically ill children. A research agenda to examine this hypothesis is suggested.
Introduction
Childhood obesity is a worldwide epidemic, resulting in a significant predisposition to adult obesity and increased cardiovascular morbidity and mortality (1–3). Endothelial activation (EA) consists of endothelial cell (EC) dysfunction, destruction and impaired repair and represents, in part, a pathological imbalance between endothelium-derived contracting and relaxing factors (4, 5). More importantly, it is an early and often asymptomatic sign of atherosclerotic disease in obese patients (6). Early EA detection may identify children at risk of developing cardiovascular disease so that interventions can be implemented to prevent and/or reduce disease progression and to mitigate exaggerated acute EA in acute or critical illness.
Normal Endothelial Function
Healthy endothelium responds to mechanical, neurogenic, and chemical signals with factors that regulate vascular tone, platelet aggregation, leukocyte adhesion and migration, mediator production, and smooth muscle cell proliferation (7). Endothelial vasoactive molecules dilate or constrict the microvasculature to balance tissue oxygen supply and metabolic demand, particularly in times of illness or injury.
Nitric oxide (NO), produced from the amino acid L-arginine through the action of constitutive endothelial NO synthase (eNOS) and cofactors such as tetrahydrobiopterin, is of central importance in all vascular beds (8). NO diffuses across the EC membrane to activate vessel wall smooth muscle cell guanylate cyclase to increase cyclic guanosine monophosphate concentrations with resultant smooth muscle relaxation. In healthy endothelium, eNOS is activated by adenosine, bradykinin, serotonin (produced during platelet aggregation), and vascular endothelial growth factor (VEGF) (stimulated by hypoxia) (9). Endothelium produced prostacyclin also mediates vasodilation (10).
Normal endothelium also generates vasoconstrictors including endothelin-1 (ET-1) and the conversion of EC-secreted angiotensin I to angiotensin II (ATII) by angiotensin-converting enzyme (ACE).
NO maintains endothelial quiescence by inhibiting inflammation, cellular proliferation, and thrombosis (11). Mechanically, laminar blood flow appears to enhance EC survival by suppressing apoptosis. Conversely, turbulent flow can trigger EC division (12–14). The endothelial glycocalyx is a network of membrane-bound proteoglycans and glycoproteins covering the endothelium lumen that integrates endothelium- and plasma-derived soluble molecules. Over the past decade, the role of the glycocalyx in vascular mechanotransduction, hemostasis, signaling, and blood cell–vessel wall interactions has been elucidated (15). Neurogenically, endothelial function is modulated by the “gateway reflex,” or neural circuits regulating entry of immune cells to the CNS by modulating the vascular EC barrier. The gateway reflex phenomenon also contributes to recruitment of immune cells to other tissues (16).
Endothelial Activation
While EA is often a normal immune response to insult or injury, it may become pathologic. This switch is characterized by three major components: EC dysfunction, destruction, and impaired repair. Based on the data summarized below from studies in obese, asymptomatic and obese, critically ill populations, it is plausible that obesity and obesity-associated chronic inflammation, oxidative stress, and insulin resistance (IR) may intensify EA and, in turn, contribute to worse outcomes among obese, critically ill children (17). Extreme nutrient excess may cause obese adipocyte cell death resulting in cytokine and fatty acid release. These may be sensed by inflammatory kinases, or Toll-like receptors (18). Kinases downstream of these receptors [c-jun N-terminal kinase (JNK), protein kinase R (PKR)] can, in turn, become activated and inhibit insulin signaling via phosphorylation of insulin receptor substrate 1 (IRS-1), thereby blocking insulin action and further inhibiting energy metabolism (19–21).
Insulin modulates vascular tone by regulating expression of eNOS gene in ECs, mediated by the activation of phosphatidylinositol-3 kinase (PI-3K). Activation of protein kinases such as PKR, as in IR, may inhibit PI-3K activity and eNOS expression (22). Superimposition of critical illness such as sepsis, acute respiratory distress syndrome (ARDS), multiple-organ dysfunction syndrome (MODS), trauma, and cardiopulmonary bypass (CPB) for cardiac surgery may compound the effects of obesity by further overwhelming these pathways (17).
Endothelial Cell Dysfunction
EC dysfunction constitutes a shift to a predominant generation of vasoconstrictors, including ET-1 and ATII and enhanced reduction-oxygenation (redox) signaling, resulting in reactive oxygen species (ROS), or free radicals that cause oxidative stress (5, 18). Superoxide anion () is generated and converted to hydrogen peroxide (H2O2) by superoxide dismutase (23, 24). H2O2 stimulates gene transcription and protease activation (25). Normally a key enzyme in NO-associated processes, eNOS may generate ROS like H2O2 and in the absence of L-arginine and tetrahydrobiopterin, respectively. This shift is referred to as “eNOS uncoupling,” a hallmark of pathologic EA (8). Obese patients live in a persistent state of relatively high ROS production and, consequently, a perpetual state of EA (26). In critical illness, decreased substrate delivery, and hypoxemia interrupt oxidative phosphorylation and more ROS may be produced, leading to further EA and inflammation (27–30).
Endothelial Cell Destruction
In the setting of cardiovascular risk factors, EC dysfunction may progress to destruction and loss of vessel endothelial integrity. For example, after ischemia/reperfusion, ECs swell and detach from the basement membrane (31). These cells suffer oxidative stress, leukocytes adhere/transmigrate, and vascular permeability increases (32–34). In animal models, ischemia/reperfusion leads to shedding of glycosaminoglycan chains and reduced glycocalyx thickness (35).
EC apoptosis results in circulation of both whole ECs and EC microparticles in the periphery as well as the coronary arteries. They are indicative of atherosclerosis or other inflammation-associated endothelial damage and have been independently quantified in both obese and critically ill individuals (36–40). The nature and extent of EC damage in obese, critically ill patients needs focused study.
Impaired Endothelial Cell Repair
Endothelial repair in response to injury or inflammation is crucial to vascular health (41). The key player in endothelial repair is the circulating endothelial progenitor cell (EPC). After release from the bone marrow, EPCs can differentiate into mature ECs. Paradoxically, EPCs are released from the bone marrow in response to NO. Among patients with impaired NO production (e.g., obesity), EA is further exacerbated (42). Interventions aimed at ameliorating cardiovascular risk factors such as exercise and statins increase EPC release and differentiation (39, 43, 44). Obesity interferes with both EPC mobilization and EPC function, and the number of risk factors for coronary artery disease is inversely related to the number and migratory activity of EPCs (39). Circulating EPCs, in an inflammatory milieu, can also differentiate into myeloid cells, such as macrophages, a key component of EA and atherosclerosis (45). It follows that a combination of obesity (chronic nutrient excess, metabolic pathway overload, and inflammation) and critical illness (acute inflammation) may exaggerate macrophage differentiation and pathologic EA.
Measuring Endothelial Function
The most commonly used non-invasive research method for assessing endothelial function is brachial artery diameter measurement using ultrasound before and after several minutes of blood flow occlusion. This change in arterial diameter is referred to as flow-mediated vasodilation (FMD), and the increase in blood flow as “reactive hyperemia.” When performed properly, this method correlates strongly with coronary artery endothelial function (46, 47).
Direct products of EA can be measured but currently have limited clinical application due to lack of specificity, assay availability, and performance variability. These include measures of NO bioavailability, adhesion molecules, inflammatory cytokines, mediators of thrombosis, and markers of endothelial damage and repair (EPCs). Despite the lack of mechanistic specificity, these serum markers remain the most frequently used research measures of endothelial function in inflammatory states including critical illness and obesity (48–50).
Childhood Obesity and Inflammation
Childhood obesity is particularly problematic as it independently contributes to adult morbidity (2, 51–53). Unlike obese adults, EA in obese children often silently affects the microcirculation rather than manifesting in the macrocirculation. Obese children rarely have atherosclerotic lesions and many have not yet developed type II diabetes or hypertension (46). Additionally, puberty-related pro-oxidative and pro-inflammatory changes and relative IR may impact the natural history of EA in obese children (54–56).
Obesity results in “metaflammation,” a chronic, low-grade inflammatory response to excess energy substrate by metabolic cells including adipocytes, hepatocytes, myocytes, pancreatic islets of Langerhans, and astrocytes, and neurons (57). Immune cells activated by metabolic cell inflammatory signaling exacerbate tissue inflammation.
Obesity-Associated Endothelial Activation
EA is an early and often asymptomatic sign of atherosclerotic disease in obese children and adults (46). Many of the pro-inflammatory and pro-atherogenic markers associated with vascular disease in adults have also been demonstrated in obese, otherwise healthy children.
Obese, Asymptomatic Children
Although atherosclerotic lesions are rare in children, obesity has a major impact on the development of EA and atherosclerosis. Schlager et al. measured microvascular function in obese, normotensive children with FMD. Compared to non-obese controls, obese children had higher peak perfusion during hyperemia and longer recovery time, indicative of impaired endothelium-dependent microcirculation vasodilation following ischemia and can be interpreted as an early sign of EA in obese children (58). In contrast, a previous study performed in lean, overweight and obese, hypertensive adolescents found no significant differences in endothelium-dependent microvascular reactivity among the three groups, suggesting that a blood pressure rise likely precedes endothelium-dependent microvascular function deterioration in juvenile essential hypertension pathogenesis (59). Thus, while obesity is strongly associated with hypertension, it is unclear whether EA precedes or is a consequence of other processes.
Chronic, low-grade inflammation, and resultant oxidative stress trigger early vascular damage in obese children. Associating markers of inflammation and oxidative stress with functional assessments of the vasculature provides an opportunity to understand mechanisms that may be targeted to mitigate cardiovascular disease. Studies have shown an inverse correlation between FMD and inflammatory markers [C-reactive protein (CRP), Interleukin (IL)-6, and Intercellular adhesion molecule (ICAM)-1 levels] in obese, otherwise healthy children (60–62).
Critical Illness
Several studies reported associations between poor outcomes and EA in critically ill children (63–67). Likewise, the link between childhood obesity and poor outcomes in critical illness is well-established (68–71). However, the link between obesity and EA severity specifically in the setting of critical illness is largely unstudied. Although EA may worsen disease in critically ill children, the nature and extent of this response is poorly understood due to the difficulty in measuring EC dysfunction and destruction. Based on the data available in the obese, asymptomatic population and the obese, critically ill population, it follows that worse EA-related outcomes in the PICU may, at least in part, be associated with obesity and associated chronic inflammation, oxidative stress and IR (Figure 1).
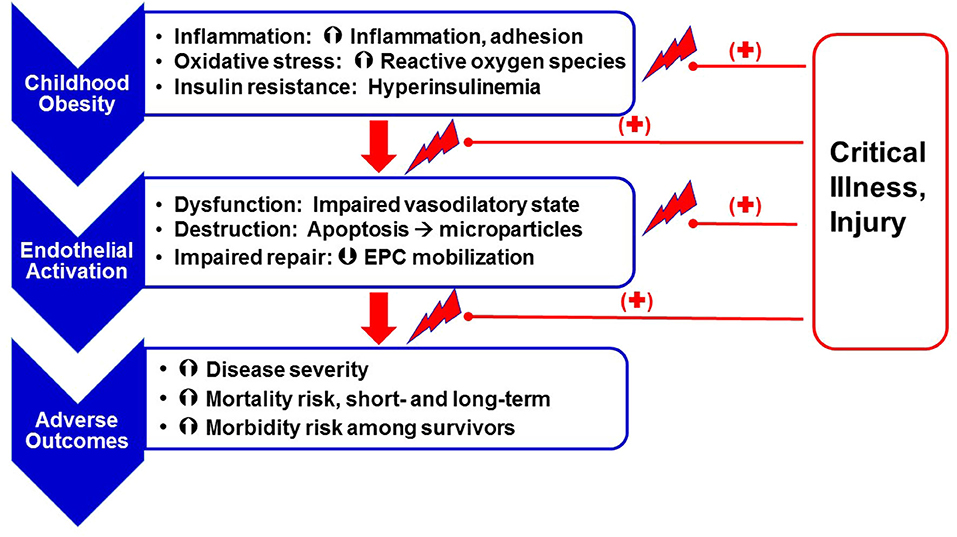
Figure 1. Obesity as a risk factor for chronic endothelial (microvascular) stress and adverse outcomes following critical illness. Obesity, secondary to excess nutrient intake, leads to a state of chronic inflammation [increased expression of pro-inflammatory mediators like Interleukin (IL)-6 and IL-8 and adhesion molecules like Intercellular adhesion molecule (ICAM)-1, Vascular cell adhesion molecule (VCAM) and E-selectin, enhanced oxidative stress (superoxide anion and hydrogen peroxide production), and insulin resistance]. These obesity-related effects lead to endothelial activation, including endothelial cell dysfunction (decreased production of vasodilatory mediators such as nitric oxide and prostaglandins E and I, and increased release of endothelin-1), cell destruction (resulting in release of endothelial microparticles), and impaired repair, in part, as a result of modulated endothelial progenitor cell mobilization. Obesity-related endothelial activation is associated with worse clinical outcomes. Superimposition of critical illness on a chronic state of endothelial activation further facilitates risk for adverse outcomes.
ECs act as targets and amplifiers of the cytokine signaling that characterizes critical illness including sepsis, ARDS, MODS, and CPB. The etiology of progression to MODS in the ICU is often unknown. However, the presence of ECs within every organ system suggests a pathophysiologic role (72). Adhesion molecules such as E-selectin are significantly higher in septic children with >3 failed organ systems compared to those with ≤ 3 failed organ systems, suggesting a role in EA (73). Pro-inflammatory markers, specifically IL-1, IL-6, and tumor necrosis factor, activate ECs to produce and express adhesion molecules (ICAM-1, E-selectin) to promote leukocyte motility and adhesion in the systemic inflammatory response (17). Derangements in endothelial and inflammatory biomarkers (higher VEGF, thrombomodulin, CRP, IL-6, IL-8) were shown in children upon presentation to the ICU with extrapulmonary sepsis with and without ARDS (74). Lastly, obesity-related EA among critically ill children with malignancies is largely unstudied. When not acutely ill, children with malignancies demonstrate worse FMD compared to controls. Lower FMD scores correlated with serum biomarkers of EA and increased waist circumference, pointing to adiposity as a potential exacerbating factor in critical illness (75).
Congenital Heart Disease and Cardiopulmonary Bypass
The impact of obesity on endothelial function and long-term cardiovascular health may be particularly important to children with preexisting acquired or congenital heart disease (CHD). Systemic and pulmonary vascular EA are well-documented in CHD and contribute to a lifetime of increased risk of mortality and morbidity as poor exercise capacity, ventricular dysfunction, and development of thromboembolic disease (76–78). A recent study estimated an overweight/obesity prevalence of 30% among single ventricular patients 5 years after undergoing a Fontan (79). Importantly, increased adiposity is independently associated with worse endothelial function and worse functional outcomes after the Fontan operation (80, 81). Understanding the relationship between vascular function and functional outcomes and the specific role of obesity is an important next step.
Low cardiac output syndrome (LCOS) following CPB in repair of CHD is characterized by a transient decrease in systemic perfusion secondary to myocardial dysfunction and is a manifestation of EA. LCOS contributes to postoperative morbidity including prolonged mechanical ventilation and hospitalization, increased risk of infection, and long-term adverse neurologic sequelae (82). Factors involved in LCOS include activation of inflammatory and complement cascades, altered blood flow, and ischemia-reperfusion injury (83).
CPB increases vasoconstrictors that contribute to vascular reactivity and decreased microcirculatory flow and result in myocardial injury. Additionally, CPB-induced EA shifts the anticoagulant phenotype to procoagulant. EC surface tissue factor expression increases, leading to deposition of fibrin in the microcirculation. Procoagulant properties are further propagated by the simultaneous downregulation of thrombomodulin (84). Lastly, CPB-associated EA stimulates surface protein expression that facilitates leukocyte-EC interactions. P-selectin release facilitates the strong bond between neutrophil surface integrins and adhesion molecules on the EA surface. This cascade precedes neutrophil infiltration into the perivascular tissue and, ultimately, the production and release of ROS and proteases that mediate cellular and end-organ damage (85).
Therapies
Determining nutritional goals for obese, critically ill children remains challenging, as commonly used equations to estimate caloric needs frequently under- or overestimate energy requirements (86). Overfeeding consequences include prolonged mechanical ventilation and hospitalization and hyperglycemia (87). The American Society for Parenteral and Enteral Nutrition recommends direct measurement of resting energy requirements utilizing indirect calorimetry. However, patients must be at “steady state” (no ongoing titration of oxygen and/or inotropes) to achieve accurate measurements, making this recommendation difficult to implement (88). Treatment options specifically for the obese child with presumed chronic inflammation and EA are outlined below. These treatments have the potential to reverse chronic EA and/or dampen acute EA in the setting of critical illness.
Exercise
Physical activity is promising as a therapeutic tool in obese children. Multiple studies have shown an improvement in FMD accompanied by a decrease in BMI with exercise interventions (89–91). Moreover, FMD may improve even in the absence of fat loss or decrease in BMI (92). These findings are likely explained by increased NO bioavailability due to exercise-induced shear stress (93). Importantly, exercise interventions that improved FMD without weight loss failed to decrease markers of inflammation and/or oxidative stress (94), underscoring the central role of fat mass in the inflammatory cascade. Watts et al. highlighted the importance of long-term, uninterrupted exercise in obese children by demonstrating a reversal of exercise-associated improvement in FMD after just 6 weeks of inactivity (95). In summary, an exercise program, even without dietary modifications, can improve baseline endothelial function, and may decrease vulnerability in the event of critical illness among overweight/obese children (89).
Statins
Two large, adult trials reported improvement in endothelial function with the use of cholesterol-lowering therapy (96, 97). HMG-CoA reductase inhibitors (statins) exhibit antioxidant, anti-inflammatory, and NO restorative properties (98), and beneficial effect on endothelial function has been shown in a broad range of patients (99–101). Statin therapy is now a first-line pharmacologic intervention for children with severe dyslipidemias failing treatment with diet and exercise alone (102). Statins also improve endothelial function in hypertension and hypercholesterolemia and may be useful alone or in combination with other agents.
L-Citrulline
L-citrulline is a naturally occurring amino acid and first intermediate in the urea cycle. Once produced, citrulline is transported from the mitochondria to the cytoplasm and converted to arginine, the precursor for NO. In multiple observational and clinical studies, plasma levels of citrulline, and arginine drop precipitously in CPB and do not recover for up to 48 h. In a recent study, CPB significantly decreased several urea cycle intermediates and NO metabolites after repair of unrestrictive ventricular septal defect and atrioventricular septal defect (103). In a phase IIb trial, patients receiving intravenous L-citrulline showed reduced duration of mechanical ventilation, inotropic needs, and ICU stay (104). Further study of associations between L-citrulline administration and endothelial function is needed in this population.
Preservation of The Glycocalyx
The endothelial glycocalyx is responsible for maintaining homeostasis of intravascular flow and dynamics. It is affected in inflammation and hyperglycemia and has a central role in capillary leak syndrome, or “endothelial failure syndrome,” particularly in sepsis (105). Sulodexide, a mix of glycosaminoglycan precursors, inhibits matrix metalloproteinases, and IL-6 while stimulating lipoprotein lipase activity and modulating the coagulation-fibrinolysis balance. In a recent animal sepsis study, sulodexide accelerated glycocalyx regeneration with decreased vascular permeability, and improved survival (106). Additionally, maintaining adequate levels of plasma proteins, particularly albumin, may promote glycocalyx repair (107, 108). Lastly, evidence suggests that resuscitation fluid volume and composition may impact glycocalyx stability and the extent of end-organ injury (109, 110). Further studies are needed to determine which therapies have direct effects on glycocalyx integrity and its relationship to ICU outcomes.
Other Treatments That May Regulate Vascular Function
Endothelin receptor antagonists (ERAs) have a high potential in the treatment of hypertension and renal diseases such as diabetic nephropathy by blunting endothelium-dependent vasoconstriction (111). The evidence for the potential benefits of ERAs is limited in children, and more data is needed.
Phosphodiesterase inhibitors (PDEIs) have promise as a treatment in specific pediatric populations. A recent phase III clinical trial demonstrated improved exercise performance after treatment with a PDEI among children who had undergone a Fontan procedure (112). Further investigation is needed to determine the effects of chronic treatment.
Other novel therapies with limited, but promising data that are outside of the scope of this review include metabolic therapies such as thiamine, vitamin C, tight glycemic control with insulin, lipid/triglyceride modulation, and periodic whole body acceleration.
Conclusions
The endothelium is crucial in maintaining vascular homeostasis in health and allostasis in critical illness. Under stress, ECs become activated, initiating inflammation, followed by new cell surface protein gene expression. While this response represents an attempt to neutralize infection and injury, it can be pathologic. Patients with baseline chronic inflammation, such as obese children, may be at risk for exaggerated EA and associated end-organ injury, morbidity, and mortality. However, this paradigm has not been adequately investigated among obese, critically ill children, and represents a current gap in pediatric research.
The relationship between obesity, EA, and outcomes of critically ill children is ripe with research potential. Further understanding of the relationships between weight loss and inflammation may inform specific diet and lifestyle modifications to prevent and/or reverse obesity-related EA. While EA is associated with higher illness severity and adverse outcomes, the severity and mechanisms involved are difficult to assess directly in critically ill children. Microcirculation markers such as capillary refill time frequently lack correlation with macrocirculation measures such as blood pressure or peripheral pulses (113). Additionally, studies on biomarkers of EA such as proteins or damaged ECs or EC particles continue to be an area of much needed future study.
Author Contributions
MR, JM, and JZ contributed to the focus and design of the review. MR wrote the first draft of the manuscript. All authors contributed to manuscript revisions, read, and approved the submitted version.
Funding
Open access publication fees provided by the Seattle Children's Hospital Department of Critical Care Medicine and Seattle Children's Research Institute Indirect Cost Return Allocation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Karnik S, Kanekar A. Childhood obesity: a global public health crisis. Int J Prev Med. (2012) 3:1–7. doi: 10.1201/b18227-3
2. Juonala M, Magnussen CG, Berenson GS, Venn A, Burns TL, Sabin MA, et al. Childhood adiposity, adult adiposity, and cardiovascular risk factors. N Engl J Med. (2011) 365:1876–85. doi: 10.1056/NEJMoa1010112
3. Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of obesity and trends in body mass index among US children and adolescents, 1999-2010. JAMA. (2012) 307:483–90. doi: 10.1001/jama.2012.40
4. Flammer AJ, Lüscher TF. Three decades of endothelium research: from the detection of nitric oxide to the everyday implementation of endothelial function measurements in cardiovascular diseases. Swiss Med Wkly. (2010) 140:w13122. doi: 10.4414/smw.2010.13122
5. Virdis A, Ghiadoni L, Taddei S. Human endothelial dysfunction: EDCFs. Pflugers Arch. (2010) 459:1015–23. doi: 10.1007/s00424-009-0783-7
6. Schächinger V, Zeiher AM. Atherosclerosis-associated endothelial dysfunction. Z Kardiol. (2000) 89(Suppl. 9):IX−70–4. doi: 10.1007/s003920070033
7. Flammer AJ, Anderson T, Celermajer DS, Creager MA, Deanfield J, Ganz P, et al. The assessment of endothelial function: from research into clinical practice. Circulation. (2012) 126:753–67. doi: 10.1161/CIRCULATIONAHA.112.093245
8. Förstermann U, Münzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation. (2006) 113:1708–14. doi: 10.1161/CIRCULATIONAHA.105.602532
9. Govers R, Rabelink TJ. Cellular regulation of endothelial nitric oxide synthase. Am J Physiol Renal Physiol. (2001) 280:F193–206. doi: 10.1152/ajprenal.2001.280.2.F193
10. Moncada S, Higgs EA, Vane JR. Human arterial and venous tissues generate prostacyclin (prostaglandin x), a potent inhibitor of platelet aggregation. Lancet. (1977) 1:18–20. doi: 10.1016/S0140-67367791655-5
11. Deanfield JE, Halcox JP, Rabelink TJ. Endothelial function and dysfunction: testing and clinical relevance. Circulation. (2007) 115:1285–95. doi: 10.1161/CIRCULATIONAHA.106.652859
12. García-Cardeña G, Anderson KR, Mauri L, Gimbrone MA. Distinct mechanical stimuli differentially regulate the PI3K/Akt survival pathway in endothelial cells. Ann N Y Acad Sci. (2000) 902:294–7. doi: 10.1111/j.1749-6632.2000.tb06325.x
13. Dimmeler S, Haendeler J, Rippmann V, Nehls M, Zeiher AM. Shear stress inhibits apoptosis of human endothelial cells. FEBS Lett. (1996) 399:71–74. doi: 10.1016/S0014-5793(96)01289-6
14. Gimbrone MA. Vascular endothelium, hemodynamic forces, and atherogenesis. Am J Pathol. (1999) 155:1–5. doi: 10.1016/S0002-9440(10)65090-0
15. Reitsma S, Slaaf DW, Vink H, van Zandvoort MAMJ, oude Egbrink MGA. The endothelial glycocalyx: composition, functions, and visualization. Pflugers Arch. (2007) 454:345–59. doi: 10.1007/s00424-007-0212-8
16. Stofkova A, Murakami M. Neural activity regulates autoimmune diseases through the gateway reflex. Bioelectron Med. (2019) 5:14–15. doi: 10.1186/s42234-019-0030-2
17. Pierce RW, Giuliano JS, Pober JS. Endothelial cell function and dysfunction in critically ill children. Pediatrics. (2017) 140:e20170355. doi: 10.1542/peds.2017-0355
18. Saghizadeh M, Ong JM, Garvey WT, Henry RR, Kern PA. The expression of TNF alpha by human muscle. relationship to insulin resistance. J Clin Invest. (1996) 97:1111–16. doi: 10.1172/JCI118504
19. Solinas G, Karin M. JNK1 and IKKbeta: molecular links between obesity and metabolic dysfunction. FASEB J. (2010) 24:2596–611. doi: 10.1096/fj.09-151340
20. Nakamura T, Furuhashi M, Li P, Cao H, Tuncman G, Sonenberg N, et al. Double-stranded RNA-dependent protein kinase links pathogen sensing with stress and metabolic homeostasis. Cell. (2010) 140:338–48. doi: 10.1016/j.cell.2010.01.001
21. Boura-Halfon S, Zick Y. Phosphorylation of IRS proteins, insulin action, and insulin resistance. Am J Physiol Endocrinol Metab. (2009) 296:E581–91. doi: 10.1152/ajpendo.90437.2008
22. Kuboki K, Jiang ZY, Takahara N, Ha SW, Igarashi M, Yamauchi T, et al. Regulation of endothelial constitutive nitric oxide synthase gene expression in endothelial cells and in vivo : a specific vascular action of insulin. Circulation. (2000) 101:676–81. doi: 10.1161/01.CIR.101.6.676
23. Spiekermann S, Landmesser U, Dikalov S, Bredt M, Gamez G, Tatge H, et al. Electron spin resonance characterization of vascular xanthine and NAD(P)H oxidase activity in patients with coronary artery disease: relation to endothelium-dependent vasodilation. Circulation. (2003) 107:1383–9. doi: 10.1161/01.CIR.0000056762.69302.46
24. Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res. (2000) 86:494–501. doi: 10.1161/01.RES.86.5.494
25. Rhee SG. Cell signaling. H2O2, a necessary evil for cell signaling. Science. (2006) 312:1882–3. doi: 10.1126/science.1130481
26. Montero D, Walther G, Perez-Martin A, Roche E, Vinet A. Endothelial dysfunction, inflammation, and oxidative stress in obese children and adolescents: markers and effect of lifestyle intervention. Obes Rev. (2012) 13:441–55. doi: 10.1111/j.1467-789X.2011.00956.x
27. Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes. Endocr Rev. (2002) 23:599–622. doi: 10.1210/er.2001-0039
28. Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. (2001) 414:813–20. doi: 10.1038/414813a
29. Moncada S, Erusalimsky JD. Does nitric oxide modulate mitochondrial energy generation and apoptosis? Nat Rev Mol Cell Biol. (2002) 3:214–20. doi: 10.1038/nrm762
30. Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. (2002) 109:S81–96. doi: 10.1016/S0092-8674(02)00703-1
31. Oliver MG, Specian RD, Perry MA, Granger DN. Morphologic assessment of leukocyte-endothelial cell interactions in mesenteric venules subjected to ischemia and reperfusion. Inflammation. (1991) 15:331–46. doi: 10.1007/BF00917350
32. Kupinski AM, Shah DM, Bell DR. Transvascular albumin flux in rabbit hindlimb after tourniquet ischemia. Am J Physiol. (1993) 264:H901–8. doi: 10.1152/ajpheart.1993.264.3.H901
33. Kurose I, Argenbright LW, Wolf R, Lianxi L, Granger DN. Ischemia/reperfusion-induced microvascular dysfunction: role of oxidants and lipid mediators. Am J Physiol. (1997) 272:H2976–82. doi: 10.1152/ajpheart.1997.272.6.H2976
34. Beuk RJ, Heineman E, Tangelder GJ, Quaedackers JS, Marks WH, Lieberman JM, et al. Total warm ischemia and reperfusion impairs flow in all rat gut layers but increases leukocyte-vessel wall interactions in the submucosa only. Ann Surg. (2000) 231:96–104. doi: 10.1097/00000658-200001000-00014
35. Mulivor AW, Lipowsky HH. Inflammation- and ischemia-induced shedding of venular glycocalyx. Am J Physiol Heart Circ Physiol. (2004) 286:H1672–80. doi: 10.1152/ajpheart.00832.2003
36. Woywodt A, Bahlmann FH, De Groot K, Haller H, Haubitz M. Circulating endothelial cells: life, death, detachment and repair of the endothelial cell layer. Nephrol Dial Transplant. (2002) 17:1728–30. doi: 10.1093/ndt/17.10.1728
37. Mallat Z, Benamer H, Hugel B, Benessiano J, Steg PG, Freyssinet JM, et al. Elevated levels of shed membrane microparticles with procoagulant potential in the peripheral circulating blood of patients with acute coronary syndromes. Circulation. (2000) 101:841–3. doi: 10.1161/01.CIR.101.8.841
38. Adawi M, Pastukh N, Saaida G, Sirchan R, Watad A, Blum A. Inhibition of endothelial progenitor cells may explain the high cardiovascular event rate in patients with rheumatoid arthritis. QJM. (2019) 112:161. doi: 10.1093/qjmed/hcy204
39. Vasa M, Fichtlscherer S, Aicher A, Adler K, Urbich C, Martin H, et al. Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Circ Res. (2001) 89:E1–7. doi: 10.1161/hh1301.093953
40. van Ierssel SH, Jorens PG, Van Craenenbroeck EM, Conraads VM. The endothelium, a protagonist in the pathophysiology of critical illness: focus on cellular markers. Biomed Res Int. (2014) 2014:985813–10. doi: 10.1155/2014/985813
41. Buijs den JO, Musters M, Verrips T, Post JA, Braam B, van Riel N. Mathematical modeling of vascular endothelial layer maintenance: the role of endothelial cell division, progenitor cell homing, and telomere shortening. Am J Physiol Heart Circ Physiol. (2004) 287:H2651–8. doi: 10.1152/ajpheart.00332.2004
42. Aicher A, Heeschen C, Mildner-Rihm C, Urbich C, Ihling C, Technau-Ihling K, et al. Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells. Nat Med. (2003) 9:1370–76. doi: 10.1038/nm948
43. Laufs U, Werner N, Link A, Endres M, Wassmann S, Jürgens K, et al. Physical training increases endothelial progenitor cells, inhibits neointima formation, and enhances angiogenesis. Circulation. (2004) 109:220–26. doi: 10.1161/01.CIR.0000109141.48980.37
44. Walter DH, Fichtlscherer S, Britten MB, Auch-Schwelk W, Schächinger V, Zeiher AM. Benefits of immediate initiation of statin therapy following successful coronary stent implantation in patients with stable and unstable angina pectoris and Q-wave acute myocardial infarction. Am J Cardiol. (2002) 89:1–6. doi: 10.1016/S0002-9149(01)02153-1
45. López M, San JR, Estrada V, Vispo E, Blanco F, Soriano V, et al. Endothelial dysfunction in HIV infection–the role of circulating endothelial cells, microparticles, endothelial progenitor cells and macrophages. AIDS Rev. (2012) 14:223–30.
46. Celermajer DS, Sorensen KE, Gooch VM, Spiegelhalter DJ, Miller OI, Sullivan ID, et al. Non-invasive detection of endothelial dysfunction in children and adults at risk of atherosclerosis. Lancet. (1992) 340:1111–15. doi: 10.1016/0140-6736(92)93147-F
47. Goldstein BH, Golbus JR, Sandelin AM, Warnke N, Gooding L, King KK, et al. Usefulness of peripheral vascular function to predict functional health status in patients with fontan circulation. Am J Cardiol. (2011) 108:428–34. doi: 10.1016/j.amjcard.2011.03.064
48. Pickkers P, Sprong T, Eijk LV, Hoeven HVD, Smits P, Deuren MV. Vascular endothelial growth factor is increased during the first 48 hours of human septic shock and correlates with vascular permeability. Shock. (2005) 24:508–12. doi: 10.1097/01.shk.0000190827.36406.6e
49. Oragui EE, Nadel S, Kyd P, Levin M. Increased excretion of urinary glycosaminoglycans in meningococcal septicemia and their relationship to proteinuria. Crit Care Med. (2000) 28:3002–8. doi: 10.1097/00003246-200008000-00054
50. Hazelzet JA, de Groot R, van Mierlo G, Joosten KF, van der Voort E, Eerenberg A, et al. Complement activation in relation to capillary leakage in children with septic shock and purpura. Infect Immun. (1998) 66:5350–56. doi: 10.1128/IAI.66.11.5350-5356.1998
51. Ford ES. Risks for all-cause mortality, cardiovascular disease, and diabetes associated with the metabolic syndrome: a summary of the evidence. Diabetes Care. (2005) 28:1769–78. doi: 10.2337/diacare.28.7.1769
52. Malik S, Wong ND, Franklin SS, Kamath TV, L'Italien GJ, Pio JR, et al. Impact of the metabolic syndrome on mortality from coronary heart disease, cardiovascular disease, and all causes in United States adults. Circulation. (2004) 110:1245–50. doi: 10.1161/01.CIR.0000140677.20606.0E
53. Wilson PWF, D'Agostino RB, Parise H, Sullivan L, Meigs JB. Metabolic syndrome as a precursor of cardiovascular disease and type 2 diabetes mellitus. Circulation. (2005) 112:3066–72. doi: 10.1161/CIRCULATIONAHA.105.539528
54. Hannon TS, Janosky J, Arslanian SA. Longitudinal study of physiologic insulin resistance and metabolic changes of puberty. Pediatr Res. (2006) 60:759–63. doi: 10.1203/01.pdr.0000246097.73031.27
55. Mauras N, Delgiorno C, Kollman C, Bird K, Morgan M, Sweeten S, et al. Obesity without established comorbidities of the metabolic syndrome is associated with a proinflammatory and prothrombotic state, even before the onset of puberty in children. J Clin Endocrinol Metab. (2010) 95:1060–68. doi: 10.1210/jc.2009-1887
56. Pérez-Navero JL, Benítez-Sillero JD, Gil-Campos M, Guillén-del Castillo M, Tasset I, Túnez I. [Changes in oxidative stress biomarkers induced by puberty]. An Pediatr. (2009) 70:424–8. doi: 10.1016/j.anpedi.2009.01.019
57. Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol. (2011) 29:415–45. doi: 10.1146/annurev-immunol-031210-101322
58. Schlager O, Willfort-Ehringer A, Hammer A, Steiner S, Fritsch M, Giurgea A, et al. Microvascular function is impaired in children with morbid obesity. Vasc Med. (2011) 16:97–102. doi: 10.1177/1358863X11400780
59. Monostori P, Baráth A, Fazekas I, Hódi E, Máté A, Farkas I, et al. Microvascular reactivity in lean, overweight, and obese hypertensive adolescents. Eur J Pediatr. (2010) 169:1369–74. doi: 10.1007/s00431-010-1234-3
60. Järvisalo MJ, Harmoinen A, Hakanen M, Paakkunainen U, Viikari J, Hartiala J, et al. Elevated serum C-reactive protein levels and early arterial changes in healthy children. Arterioscler Thromb Vasc Biol. (2002) 22:1323–8. doi: 10.1161/01.ATV.0000024222.06463.21
61. Kapiotis S, Holzer G, Schaller G, Haumer M, Widhalm H, Weghuber D, et al. A proinflammatory state is detectable in obese children and is accompanied by functional and morphological vascular changes. Arterioscler Thromb Vasc Biol. (2006) 26:2541–6. doi: 10.1161/01.ATV.0000245795.08139.70
62. Głowinska-Olszewska B, Tołwinska J, Urban M. Relationship between endothelial dysfunction, carotid artery intima media thickness and circulating markers of vascular inflammation in obese hypertensive children and adolescents. J Pediatr Endocrinol Metab. (2007) 20:1125–36. doi: 10.1515/JPEM.2007.20.10.1125
63. Kuiper JW, Tibboel D, Ince C. The vulnerable microcirculation in the critically ill pediatric patient. Crit Care. (2016) 20:352–12. doi: 10.1186/s13054-016-1496-x
64. Paize F, Sarginson R, Makwana N, Baines PB, Thomson APJ, Sinha I, et al. Changes in the sublingual microcirculation and endothelial adhesion molecules during the course of severe meningococcal disease treated in the paediatric intensive care unit. Intensive Care Med. (2012) 38:863–71. doi: 10.1007/s00134-012-2476-5
65. Whalen MJ, Doughty LA, Carlos TM, Wisniewski SR, Kochanek PM, Carcillo JA. Intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 are increased in the plasma of children with sepsis-induced multiple organ failure. Crit Care Med. (2000) 28:2600–7. doi: 10.1097/00003246-200007000-00070
66. Nadel S, Goldstein B, Williams MD, Dalton H, Peters M, Macias WL, et al. Drotrecogin alfa (activated) in children with severe sepsis: a multicentre phase III randomised controlled trial. Lancet. (2007) 369:836–43. doi: 10.1016/S0140-6736(07)60411-5
67. Caixeta DMDL, Fialho FMD, Azevedo ZMA, Collett-Solberg PF, Villela NR, Bouskela E. Evaluation of sublingual microcirculation in children with dengue shock. Clinics. (2013) 68:1061–4. doi: 10.6061/clinics/2013(07)26
68. Srinivasan V, Nadkarni VM, Helfaer MA, Carey SM, Berg RA. American heart association national registry of cardiopulmonary resuscitation investigators. childhood obesity and survival after in-hospital pediatric cardiopulmonary resuscitation. Pediatrics. (2010) 125:e481–8. doi: 10.1542/peds.2009-1324
69. Nafiu OO, Reynolds PI, Bamgbade OA, Tremper KK, Welch K, Kasa-Vubu JZ. Childhood body mass index and perioperative complications. Paediatr Anaesth. (2007) 17:426–30. doi: 10.1111/j.1460-9592.2006.02140.x
70. Rossano JW, Grenier MA, Dreyer WJ, Kim JJ, Price JF, Jefferies JL, et al. Effect of body mass index on outcome in pediatric heart transplant patients. J Heart Lung Transplant. (2007) 26:718–23. doi: 10.1016/j.healun.2007.05.001
71. Carroll CL, Bhandari A, Zucker AR, Schramm CM. Childhood obesity increases duration of therapy during severe asthma exacerbations. Pediatr Crit Care Med. (2006) 7:527–31. doi: 10.1097/01.PCC.0000243749.14555.E8
72. Aird WC. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood. (2003) 101:3765–77. doi: 10.1182/blood-2002-06-1887
73. Krueger M, Heinzmann A, Nauck M. Adhesion molecules in pediatric intensive care patients with organ dysfunction syndrome. Intensive Care Med. (2007) 33:359–63. doi: 10.1007/s00134-006-0453-6
74. Whitney JE, Zhang B, Koterba N, Chen F, Bush J, Graham K, et al. Systemic endothelial activation is associated with early acute respiratory distress syndrome in children with extrapulmonary sepsis. Crit Care Med. (2020) 48:344–52. doi: 10.1097/CCM.0000000000004091
75. Giordano P, Muggeo P, Delvecchio M, Carbonara S, Romano A, Altomare M, et al. Endothelial dysfunction and cardiovascular risk factors in childhood acute lymphoblastic leukemia survivors. Int J Cardiol. (2017) 228:621–7. doi: 10.1016/j.ijcard.2016.11.025
76. Uzark K, Jones K, Slusher J, Limbers CA, Burwinkle TM, Varni JW. Quality of life in children with heart disease as perceived by children and parents. Pediatrics. (2008) 121:e1060–7. doi: 10.1542/peds.2006-3778
77. Goldstein BH, Connor CE, Gooding L, Rocchini AP. Relation of systemic venous return, pulmonary vascular resistance, and diastolic dysfunction to exercise capacity in patients with single ventricle receiving fontan palliation. Am J Cardiol. (2010) 105:1169–75. doi: 10.1016/j.amjcard.2009.12.020
78. McCrindle BW, Williams RV, Mital S, Clark BJ, Russell JL, Klein G, et al. Physical activity levels in children and adolescents are reduced after the fontan procedure, independent of exercise capacity, and are associated with lower perceived general health. Arch Dis Child. (2007) 92:509–14. doi: 10.1136/adc.2006.105239
79. Wellnitz K, Harris IS, Sapru A, Fineman JR, Radman M. Longitudinal development of obesity in the post-fontan population. Eur J Clin Nutr. (2015) 69:1105–8. doi: 10.1038/ejcn.2015.68
80. Cohen MS, Zak V, Atz AM, Printz BF, Pinto N, Lambert L, et al. Anthropometric measures after fontan procedure: implications for suboptimal functional outcome. Am Heart J. (2010) 160:1092–98.e1. doi: 10.1016/j.ahj.2010.07.039
81. Goldstein BH, Urbina EM, Khoury PR, Gao Z, Amos MA, Mays WA, et al. Endothelial function and arterial stiffness relate to functional outcomes in adolescent and young adult fontan survivors. J Am Heart Assoc. (2016) 5:e004258. doi: 10.1161/JAHA.116.004258
82. Hoffman TM, Wernovsky G, Atz AM, Kulik TJ, Nelson DP, Chang AC, et al. Efficacy and safety of milrinone in preventing low cardiac output syndrome in infants and children after corrective surgery for congenital heart disease. Circulation. (2003) 107:996–1002. doi: 10.1161/01.CIR.0000051365.81920.28
83. Verrier ED, Morgan EN. Endothelial response to cardiopulmonary bypass surgery. Ann Thorac Surg. (1998) 66:S17–9. doi: 10.1016/S0003-4975(98)00965-5
84. Boyle EM, Pohlman TH, Cornejo CJ, Verrier ED. Endothelial cell injury in cardiovascular surgery: ischemia-reperfusion. Ann Thorac Surg. (1996) 62:1868–75. doi: 10.1016/S0003-4975(96)00950-2
85. Boyle EM, Verrier ED, Spiess BD. Endothelial cell injury in cardiovascular surgery: the procoagulant response. Ann Thorac Surg. (1996) 62:1549–57. doi: 10.1016/0003-4975(96)00836-3
86. Skillman HE, Wischmeyer PE. Nutrition therapy in critically ill infants and children. JPEN J Parenter Enteral Nutr. (2008) 32:520–34. doi: 10.1177/0148607108322398
87. Alaedeen DI, Walsh MC, Chwals WJ. Total parenteral nutrition-associated hyperglycemia correlates with prolonged mechanical ventilation and hospital stay in septic infants. J Pediatr Surg. (2006) 41:239–44. doi: 10.1016/j.jpedsurg.2005.10.045
88. Kyle UG, Arriaza A, Esposito M, Coss-Bu JA. Is indirect calorimetry a necessity or a luxury in the pediatric intensive care unit? JPEN J Parenter Enteral Nutr. (2012) 36:177–82. doi: 10.1177/0148607111415108
89. da Silva MR, Waclawovsky G, Perin L, Camboim I, Eibel B, Lehnen AM. Effects of high-intensity interval training on endothelial function, lipid profile, body composition and physical fitness in normal-weight and overweight-obese adolescents: a clinical trial. Physiol Behav. (2020) 213:112728. doi: 10.1016/j.physbeh.2019.112728
90. Tjønna AE, Stølen TO, Bye A, Volden M, Slørdahl SA, Odegård R, et al. Aerobic interval training reduces cardiovascular risk factors more than a multitreatment approach in overweight adolescents. Clin Sci. (2009) 116:317–26. doi: 10.1042/CS20080249
91. Meyer AA, Kundt G, Lenschow U, Schuff-Werner P, Kienast W. Improvement of early vascular changes and cardiovascular risk factors in obese children after a six-month exercise program. J Am Coll Cardiol. (2006) 48:1865–70. doi: 10.1016/j.jacc.2006.07.035
92. Murphy EC-S, Carson L, Neal W, Baylis C, Donley D, Yeater R. Effects of an exercise intervention using dance dance revolution on endothelial function and other risk factors in overweight children. Int J Pediatr Obes. (2009) 4:205–14. doi: 10.3109/17477160902846187
93. Tinken TM, Thijssen DHJ, Hopkins N, Dawson EA, Cable NT, Green DJ. Shear stress mediates endothelial adaptations to exercise training in humans. Hypertension. (2010) 55:312–18. doi: 10.1161/HYPERTENSIONAHA.109.146282
94. Kelly AS, Steinberger J, Olson TP, Dengel DR. In the absence of weight loss, exercise training does not improve adipokines or oxidative stress in overweight children. Metab Clin Exp. (2007) 56:1005–9. doi: 10.1016/j.metabol.2007.03.009
95. Watts K, Beye P, Siafarikas A, O'Driscoll G, Jones TW, Davis EA, et al. Effects of exercise training on vascular function in obese children. J Pediatr. (2004) 144:620–25. doi: 10.1016/j.jpeds.2004.02.027
96. Treasure CB, Klein JL, Weintraub WS, Talley JD, Stillabower ME, Kosinski AS, et al. Beneficial effects of cholesterol-lowering therapy on the coronary endothelium in patients with coronary artery disease. N Engl J Med. (1995) 332:481–7. doi: 10.1056/NEJM199502233320801
97. Anderson TJ, Meredith IT, Yeung AC, Frei B, Selwyn AP, Ganz P. The effect of cholesterol-lowering and antioxidant therapy on endothelium-dependent coronary vasomotion. N Engl J Med. (1995) 332:488–93. doi: 10.1056/NEJM199502233320802
98. Bonetti PO, Lerman LO, Napoli C, Lerman A. Statin effects beyond lipid lowering–are they clinically relevant? Eur Heart J. (2003) 24:225–48. doi: 10.1016/S0195-668X(02)00419-0
99. John S, Schlaich M, Langenfeld M, Weihprecht H, Schmitz G, Weidinger G, et al. Increased bioavailability of nitric oxide after lipid-lowering therapy in hypercholesterolemic patients: a randomized, placebo-controlled, double-blind study. Circulation. (1998) 98:211–16. doi: 10.1161/01.CIR.98.3.211
100. Dupuis J, Tardif JC, Cernacek P, Théroux P. Cholesterol reduction rapidly improves endothelial function after acute coronary syndromes. the RECIFE (reduction of cholesterol in ischemia and function of the endothelium) trial. Circulation. (1999) 99:3227–33. doi: 10.1161/01.CIR.99.25.3227
101. Wassmann S, Laufs U, Bäumer AT, Müller K, Ahlbory K, Linz W, et al. HMG-CoA reductase inhibitors improve endothelial dysfunction in normocholesterolemic hypertension via reduced production of reactive oxygen species. Hypertension. (2001) 37:1450–57. doi: 10.1161/01.HYP.37.6.1450
102. Expert panel on integrated guidelines for cardiovascular health and risk reduction in children and adolescents national heart lung and blood institute. Expert panel on integrated guidelines for cardiovascular health and risk reduction in children and adolescents: summary report. Pediatrics. (2011) 128(Suppl. 5):S213–56. doi: 10.1542/peds.2009-2107C
103. Barr FE, Beverley H, VanHook K, Cermak E, Christian K, Drinkwater D, et al. Effect of cardiopulmonary bypass on urea cycle intermediates and nitric oxide levels after congenital heart surgery. J Pediatr. (2003) 142:26–30. doi: 10.1067/mpd.2003.mpd0311
104. Barr FE, Tirona RG, Taylor MB, Rice G, Arnold J, Cunningham G, et al. Pharmacokinetics and safety of intravenously administered citrulline in children undergoing congenital heart surgery: potential therapy for postoperative pulmonary hypertension. J Thorac Cardiovasc Surg. (2007) 134:319–26. doi: 10.1016/j.jtcvs.2007.02.043
105. Fernández-Sarmiento J, Salazar-Peláez LM, Carcillo JA. The endothelial glycocalyx: a fundamental determinant of vascular permeability in sepsis. Pediatr Crit Care Med. (2020) 21:e291–e300. doi: 10.1097/PCC.0000000000002266
106. Song JW, Zullo JA, Liveris D, Dragovich M, Zhang XF, Goligorsky MS. Therapeutic restoration of endothelial glycocalyx in sepsis. J Pharmacol Exp Ther. (2017) 361:115–21. doi: 10.1124/jpet.116.239509
107. Zeng Y, Adamson RH, Curry F-RE, Tarbell JM. Sphingosine-1-phosphate protects endothelial glycocalyx by inhibiting syndecan-1 shedding. Am J Physiol Heart Circ Physiol. (2014) 306:H363–72. doi: 10.1152/ajpheart.00687.2013
108. Bark BP, Persson J, Grände P-O. Importance of the infusion rate for the plasma expanding effect of 5% albumin, 6% HES 130/0.4, 4% gelatin, and 0.9% NaCl in the septic rat. Crit Care Med. (2013) 41:857–66. doi: 10.1097/CCM.0b013e318274157e
109. Job KM, O'Callaghan R, Hlady V, Barabanova A, Dull RO. The biomechanical effects of resuscitation colloids on the compromised lung endothelial glycocalyx. Anesth Analg. (2016) 123:382–93. doi: 10.1213/ANE.0000000000001284
110. Long E, Babl FE, Oakley E, Sheridan B, Duke T. Pediatric research in emergency departments international collaborative (PREDICT). Cardiac index changes with fluid bolus therapy in children with sepsis-an observational study. Pediatr Crit Care Med. (2018) 19:513–18. doi: 10.1097/PCC.0000000000001534
111. Burnier M. Update on endothelin receptor antagonists in hypertension. Curr Hypertens Rep. (2018) 20:51–7. doi: 10.1007/s11906-018-0848-0
112. Goldberg DJ, Zak V, Goldstein BH, Schumacher KR, Rhodes J, Penny DJ, et al. Results of the FUEL trial. Circulation. (2020) 141:641–51. doi: 10.1161/CIRCULATIONAHA.119.044352
Keywords: obesity, endothelial, activation, dysfunction, critical illness, inflammation, pediatric
Citation: Radman M, McGuire J and Zimmerman J (2020) Childhood Obesity, Endothelial Cell Activation, and Critical Illness. Front. Pediatr. 8:441. doi: 10.3389/fped.2020.00441
Received: 11 May 2020; Accepted: 25 June 2020;
Published: 05 August 2020.
Edited by:
Andreas Schwingshackl, University of California, Los Angeles, United StatesReviewed by:
Masaaki Murakami, Institute for Genetic Medicine, Hokkaido University, JapanVijay Srinivasan, Children's Hospital of Philadelphia, United States
Copyright © 2020 Radman, McGuire and Zimmerman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Monique Radman, bW9uaXF1ZS5yYWRtYW4mI3gwMDA0MDtzZWF0dGxlY2hpbGRyZW5zLm9yZw==