 Cristina Gug1†
Cristina Gug1† Dorina Stoicanescu1†
Dorina Stoicanescu1† Ioana Mozos2,3*
Ioana Mozos2,3* Laura Nussbaum4
Laura Nussbaum4 Mariana Cevei5Danae Stambouli6Anca Gabriela Pavel6Gabriela Doros7
Mariana Cevei5Danae Stambouli6Anca Gabriela Pavel6Gabriela Doros7- 1Department of Microscopic Morphology, Victor Babes University of Medicine and Pharmacy, Timisoara, Romania
- 2Department of Functional Sciences, Victor Babes University of Medicine and Pharmacy, Timisoara, Romania
- 3Center for Translational Research and Systems Medicine, Victor Babes University of Medicine and Pharmacy, Timisoara, Romania
- 4Department of Neurosciences, Victor Babes University of Medicine and Pharmacy, Timisoara, Romania
- 5Department of Psychoneuro Sciences and Rehabilitation, Faculty of Medicine and Pharmacy, University of Oradea, Oradea, Romania
- 6Department of Molecular Genetics and Cytogenetics, Cytogenomic Medical Laboratory, Bucharest, Romania
- 7Department of Pediatrics, Victor Babes University of Medicine and Pharmacy, Timisoara, Romania
Duplications of chromosome 8p lead to rare genetic conditions characterized by variable phenotypes. 8p21 and 8p23 duplications were associated with mental retardation but only 8p23 duplication was associated with heart defects. 8p22→ p21.3 duplications were associated with an autism spectrum disorder in several cases. We present a rare case with a de novo duplication of the entire 8p21.3→ p23.3 region, documented by karyotype, FISH, and array CGH, with t(4;8)(q35;p21.3) translocation in a 7 years-old girl. She was referred for genetic counseling at the age of 20 months due to mild dysmorphic facial features, psychomotor retardation, and a noncyanotic heart defect. Another examination carried out at the age of 5 years, enabled the diagnosis of autism spectrum disorder and attention deficit hyperactivity disorder. Upon re-examination after two years she was diagnosed with autism spectrum disorder, attention deficit hyperactivity disorder, liminal intellect with cognitive disharmony, delay in psychomotor acquisitions, developmental language delay, an instrumental disorder, and motor coordination disorder. Cytogenetic analysis using GTG technique revealed the following karyotype: 46,XX,der(4),t(4;8)(q35;p21.3). The translocation of the duplicated 8pter region to the telomeric region 4q was confirmed by FISH analysis (DJ580L5 probe). Array CGH showed: arr[GRCh37]8p23.3p21.3(125733_22400607) × 3. It identified a terminal duplication, a 22.3 Mb copy number gain of chromosome 8p23.3–p21.3, between 125,733 and 22,400,607. In this case, there is a de novo duplication of a large chromosomal segment, which was translocated to chromosome 4q. Our report provides additional data regarding neuropsychiatric features in chromosome 8p duplication. The phenotypic consequences in our patient allow clinical-cytogenetic correlations and may also reveal candidate genes for the phenotypic features.
Introduction
Significance of segmental duplications leading to abnormal gene dosage and consequently to different disorders has been intensively studied in recent years (1).
Duplications of the short arm of chromosome 8 are structural chromosomal abnormalities that lead to rare genetic conditions characterized by a variable phenotype. Depending on the size and part of the chromosome that is duplicated, some individuals may be apparently normal, while others may have a range of clinical features from mild to very severe (2, 3). The duplicated material from 8p may remain on the same chromosome or, rarer, can be translocated to a different chromosome. It seems that the phenotypic effects are not influenced by the position of the duplicated part (4, 5). Some chromosome 8p duplications may be familial, but an appreciable number of cases result from de novo mutations (6–9). Although inverted duplications adjacent to terminal deletions of chromosome 8p are the most common, cases with direct duplications have been also described (10, 11).
Known and Novel Scientific Information
• 8p21→ 8p23 duplications were associated with mental retardation (MR), interstitial duplication 8p23 was associated with congenital heart defects (CHDs).
• 8p23.1 duplication syndrome defined as a 3.75 Mb duplication of most band 8p23.1 is characterized by mild to moderate developmental delay, mental retardation (MR), mild facial dysmorphism, and CHDs.
• Duplications in chromosome 8p21.3→ p22 were previously associated with autism spectrum disorder (ASD).
We present a de novo duplication of the entire region 8p21.3→ p23.3, documented by karyotype, FISH, and array CGH, with translocation t(4;8)(q35;p21.3) in a 7 years-old girl with liminal intellect with cognitive disharmony, ASD, attention deficit hyperactivity disorder (ADHD) and cardiac defects.
• In this case, there is a de novo duplication of a large chromosomal segment, the duplicate part being translocated to chromosome 4q, without deletion of the 4q35.2 telomere, which has not been reported before.
• The phenotypic consequences in our patient allow clinical-cytogenetic correlations and may reveal candidate genes for the phenotypic features. Our case has a large duplication of the entire 8p21.3→ p23.1 region and displays a combination of clinical features: MR, ASD, ADHD, CHD, previously described separately for different duplicated chromosomal segments.
• The 8p23.1 region was associated with facial dysmorphism, especially prominent forehead, an important sign, noticed during infancy, that could guide investigations toward the identification of 8p duplication.
• We hypothesized that our patient's phenotype was entirely determined by the large duplication of 8p21.3→ p23.1 region.
Clinical Presentation and Family History
We report the case of a 7-year-old female patient, referred for genetic counseling due to dysmorphic features, psychomotor retardation, and noncyanotic CHD. She was the first child of an apparently healthy, non-consanguineous couple. Both parents were 28 years old when the patient was born. Pregnancy was uneventful; the mother denied any exposure to alcohol, radiation, or infectious agents. Family history was negative for mental retardation and congenital defects and the karyotype of the parents was normal. However, the mother had two miscarriages before the proband was born. The patient has two healthy younger sisters, the prenatal diagnosis was performed during both pregnancies.
Medical History
Clinical examination at birth revealed systolic murmur gr 3/6, heard over second, third, and fourth intercostal spaces at the left sternal border. Echocardiography performed 1 month later revealed noncyanotic CHD with ventricular septal defect (VSD) and patent foramen ovale. Developmental milestones acquisition delay was noticed at 20 months. There was also a delayed acquisition of language milestones; the patient had not verbally spoken a word. Nonetheless, nonverbal communication was relatively good, she smiled, but could not concentrate, did not establish visual contact, and did not respond when her name was called. She was referred for genetic counseling at this age due to mild dysmorphic facial features (prominent forehead, flat nasal bridge, low-set ears), psychomotor retardation, and noncyanotic CHD. aCGH was performed and showed a 22.3Mb duplication of the 8p21.3→ p22 region. The karyotype revealed the translocation of the duplicate fragment on chromosome 4q, terminal. The FISH analysis confirmed the duplication and translocation and showed that the 4q telomeres were preserved. At 2 years and 10 months, the first psychological assessment was performed. Low or average levels of all skills (cognitive, emotional, social, personal autonomy) were recorded. At 3 years and 9 months Early Childhood Inventory-4 screening used to assess common symptoms of psychiatric disorders in children aged 3–7 years, indicated ADHD characterized by hyperactive and impulsive inattentive behavior, in which attention deficit with medium-high severity prevailed. Other findings were physical and symbolic aggression, with specific phobia, selective mutism, and elimination disorders. The psychological evaluation using the Portage-Scale indicated that at the chronological age of 4 years and 8 months she had a mental age of 3 years and 9 months and DQ = 81 (developmental quotient). The pediatric neuropsychiatrist monitored the evolution, establishing mild mental retardation at the age of 5 years (IQ = 75 BS; Binet-Simon Intelligence scale), ASD, and ADHD.
Methodology
Clinical Evaluation
The patient underwent a multidisciplinary evaluation, involving health professionals from the following specialities: genetics, cardiology, pediatrics, pediatric neuropsychiatry, speech pathology, and psychology.
The patient's parents gave written informed consent (including for publication of images) considering the Declaration of Helsinki. This study was approved by the Ethics Committee for Scientific Research of the Emergency Hospital for Children “Louis Turcanu,” Timisoara.
Neuropsychiatric Evaluation
The neuropsychiatric diagnoses were put through clinical examination, assessment of the diagnostic criteria after the Diagnostic and Statistical Manual of Mental Disorders (DSM5) and confirmed through Kiddie Schedule for Affective Disorders and Schizophrenia (K-SADS). The patient was also evaluated considering the Childhood Autism Rating Scale (CARS), Autism Diagnostic Interview-Revised (ADI-R), Autism Diagnostic Observation Schedule (ADOS), the Portage-Scale and ADHD-Rating Scale.
Peripheral Blood Karyotype
Peripheral blood lymphocytes were cultured in a growth medium (PB-MAX™ Karyotyping Medium, Gibco). Metaphase chromosomes were harvested, and slides were made for analysis. GTG banding was used for staining (at the 550-band level). Chromosomal analysis was performed using LUCIA Karyo-G software, and the aberrations and karyotypes were classified according to the ISCN 2016 system.
Fluorescence in situ Hybridization (FISH)
Aquarius ®Cytocell FISH probes were applied to metaphases. The probes were: (a) Subtelomere Specific Probes 8p (DJ580L5-red) and (d) 8q (489D14-green), (b) Wolf-Hirschhorn Syndrome Critical Region (WHSCR) Probe with Subtelomere Specific Probe red, which corresponds to 4p16.3, and (DJ963k6-green), which corresponds to 4q35.2, (c) alpha-satellite 8 probe (D8Z2-green) and Whole Chromosome Painting Probe (wcp4-red).
Array Comparative Genomic Hybridization (aCGH) analysis was performed using CytoChip Focus Constitutional BAC array (Illumina Inc., U.S.) and the used reference was the normal human male genomic DNA (Promega, WI). The DNA probes were derived from BAC (Bacterial Artificial Chromosomes) DNA clones from human genome collection. The selected BACs were replicated to a high degree, 3 × and 4 ×, providing a more robust signal for each data point. The target solution was hybridized to the 2 × 180K array and data were acquired using the InnoScan 710 microarray scanner. A composite image was obtained and imported into the Blue Fuse Multi Software (v.3.1) microarray software, Genome Assembly NCBI Build 37hg19, for data analysis. The average whole-genome resolution was approximately 1Mb. The DGV (Database Genomic Variation), Decipher (Database of Chromosomal Imbalance and Phenotype in Human using Ensembl Resources), ISCA (International Standards for Cytogenomic Arrays), OMIM (Online Mendelian Inherited in Man) international databases were used to interpret the results.
Results
Clinical evaluation at the ages of 1 and 5 years showed mild dysmorphic facial features: prominent forehead, flat nasal bridge, and diastema (Figure 1). In the differential diagnosis of the prominent forehead, the most striking dysmorphic feature, Crouzon syndrome, Hurler syndrome, Pfeiffer syndrome, Rubinstein–Taybi syndrome, and Russell–Silver syndrome have been considered. They were ruled out due to the absence of other specific clinical features.
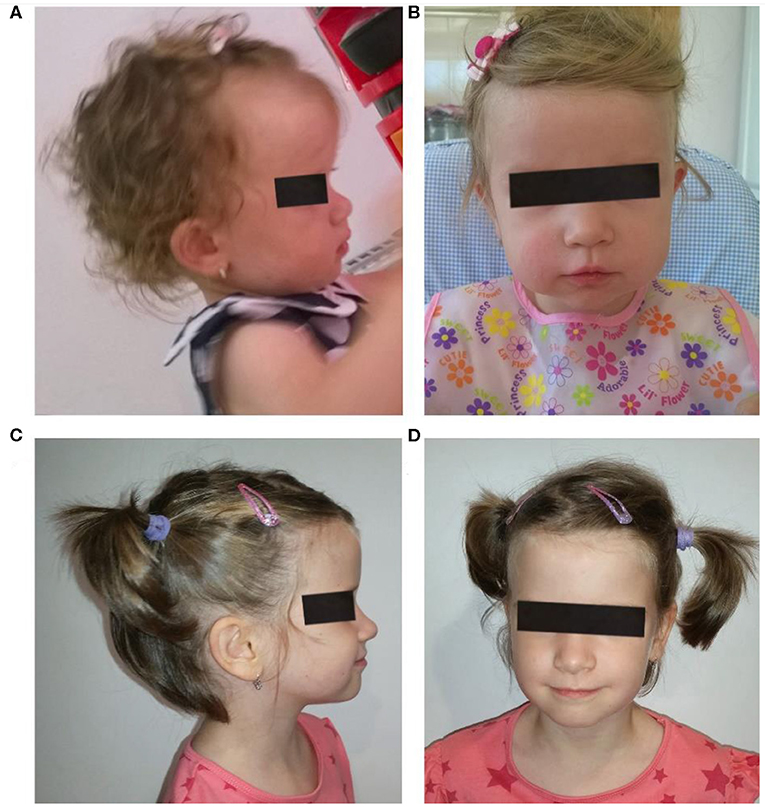
Figure 1. Patient face and profile at the age of 1 year (A,B) and 6 years (C,D). Mild facial dysmorphism with prominent forehead, flattened nose base, and low set ears can be noticed.
At the age of 6 years and 11 months, she was evaluated by the pediatric neuropsychiatrist, her diagnosis being ASD, ADHD, delay in psychomotor acquisitions with mild delay in the expressive language acquisition with polymorphic dyslalia, motor coordination disorder and instrumental disorders, liminal intellect with cognitive disharmony (IQ = 83 WISC; Wechsler Intelligence Scale for Children), minimal brain lesions indices, organic brain background. Related to the diagnosis of ASD, the patient showed qualitative deterioration of social interaction and cognition, communication, lack of emotional reciprocity and empathy, no make-believe play, lack of imaginative play, and also stereotyped, repetitive behavioral patterns and mannerisms and also resistance to change.
In the first stage of the differential diagnosis for ASD, we excluded organic disorders, which could show an “autistic-like” behavior, through the neurological examination and paraclinical investigations. In the second stage, we made an accurate differential diagnosis with all the psychiatric disorders, revealing an “autistic-like” behavior–mental retardation, language development disorders, ADHD, sensory disorders, reactive attachment disorders, early-onset schizophrenia. The clinical evaluation, the psychiatric exam, the psychological evaluations, and the applied scales guided us.
We also made a differential diagnosis of ADHD, ruling out other medical and psychiatric disorders with cognitive, attentional and executive functioning deficits and hyperkinetic impulsive behavior or agitation, restlessness–anxiety disorders, attachment disorders, posttraumatic stress disorder, and mood disorders, including depressive or bipolar disorders. In children, ADHD and bipolar disorders can have overlapping symptoms. Both can present with distractibility, increased energy, and mood lability, and, therefore, a thorough history is essential for the diagnosis.
The prognosis is guarded and symptomatic psychopharmacologic treatment, psychological therapies, psycho-sensorial stimulation, preschool special education, occupational, physical, speech, developmental, and behavioral therapies were helpful for the management of this complex case. Antipsychotic, mood-stabilizing, and neurotrophic medication but also behavioral therapies, like ABA (Applied Behavioral Analysis), PECS (Picture Exchange Communication Systems), or TEACCH (Treatment and Education for Autistic and Communication Handicapped Children) were recommended and might improve the prognosis and the clinical evolution of the patient.
Echocardiography revealed noncyanotic CHD with restrictive perimembranous DSV with a left to right shunt, covered by excess tissue from the tricuspid valve, patent foramen ovale, and bicuspid aortic valve with gr I aortic insufficiency. Cardiological monitoring was recommended and performance sports were contraindicated.
Cytogenetic analysis of the proband revealed a female karyotype with derivative chromosome 4. The following karyotype was revealed: 46,XX,der(4),t(4;8)(q35;p21.3). Hence, a partial trisomy of chromosome 8p, from band p21.3 to p23.3, resulted (Figures 2A,B). The duplicated fragment was translocated to chromosome 4q, terminal. The abnormality was found in all metaphases. The karyotype of the parents was normal; therefore, the abnormality is de novo.
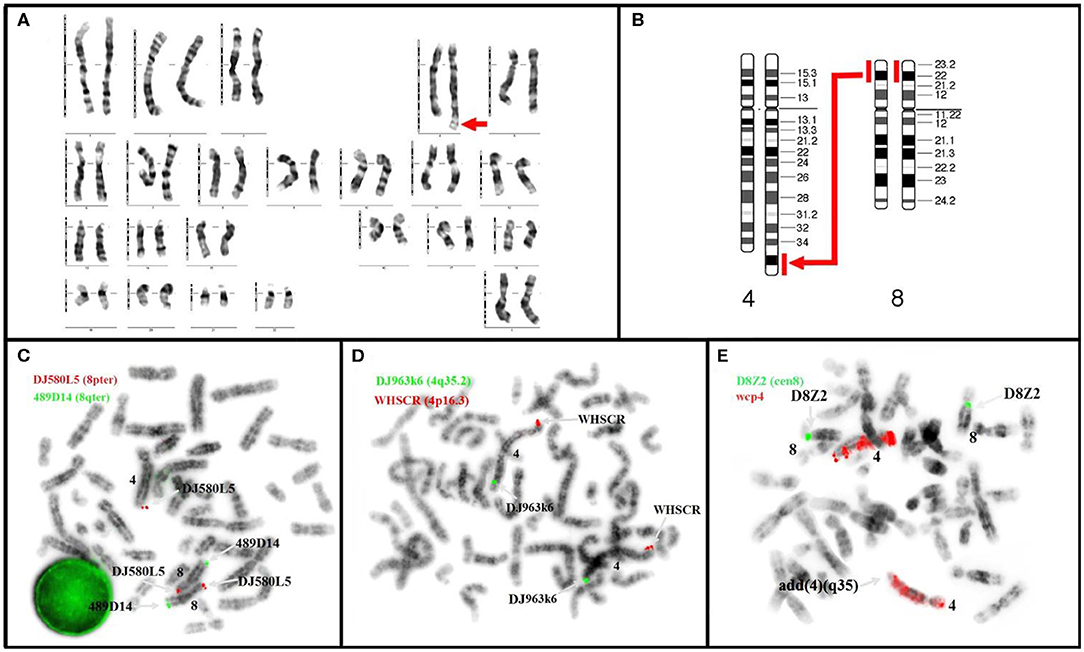
Figure 2. Karyotype (A,B) and metaphase FISH (C,D) of our patient revealed: (A) de novo duplication (p21.3→ p23.3) with translocation t(4;8)(q35;p21.3) (arrow); (B) diagram of partial trisomy 8p with the large arrow indicating the location of the duplicate segment; (C) Kit Aquarius® Specific Probes Red (DJ580L5) mark subtelomeric 8p and Green (489D14) mark subtelomeric 8q; 3 copies are noticed for 8p of which 2 correctly positioned and the third translocated to the telomeres of chromosome 4q, ish der(4)dup(4)t(4;8)(489D14+,DJ580L5++) (D) Kit Aquarius® Specific Probes Red (WHSCR) mark subtelomeric 4p16.3 and Green (DJ963k6) mark subtelomeric 4q35.2; the image prove s the presence of 4q telomeres: ish der(4)t(4;8)(WHSCR+,DJ963k6+). (E) Kit Aquarius® Whole Chromosome Painting Probes wcp 4 Red and Kit Aquarius® Satellite Enumeration Probes α-satellite 8 (D8Z2).
FISH result was 46,XX,der(4),t(4;8)(q35;p21.3).ish der(4),t(4;8)(q35;p21.3)(wcp4+,WHSCR+,DJ963k6+,DJ580L5+). The duplication of the 8pter region and translocation to the telomeric region 4q were confirmed by FISH analysis (DJ580L5 probe) (Figures 2C,D).
Array CGH showed: arr[GRCh37] 8p23.3p21.3(125733_22400607) × 3. It identified a terminal duplication, a 22.3 Mb copy number gain of chromosome 8p23.3–p21.3, between 125,733 and 22,400,607. Duplication limits using array platforms are illustrated in Figure 3. In the cytogenetic location delimited by the genomic coordinates (GRCh37-Genome Reference Consortium Human Build 37), there are 134 genes, located as follows: 1 gene in band 8p21; 30 genes in band 8p21.3; 39 genes in band 8p22; 57 genes in band 8p23.1; and 6 genes in band 8p23.3 (12).
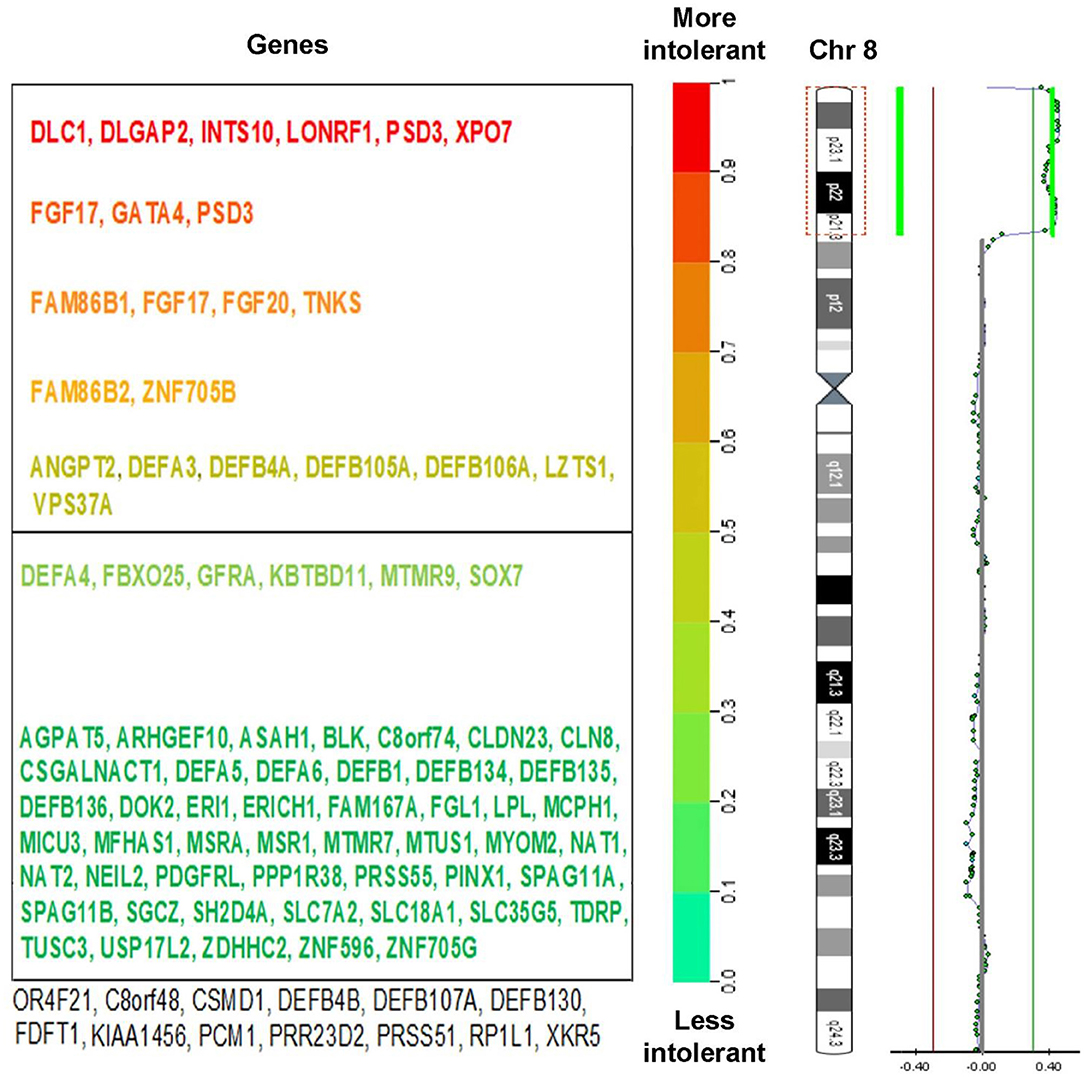
Figure 3. (Right) Chromosome view of a 22.3 Mb duplication of the 8p23.3-21.3 bands, arr[GRCh37] 8p23.3p21 (125733_22400607) × 3 detected by aCGH analysis and ideogram of chromosome 8. (Left) The genes within this region were marked with different colors, depending on their intolerance to mutations. Known pathogenic genes are marked in red (according to DECIPHER v9.31 database).
Discussion
Chromosome 8p duplication is a rare chromosomal aberration with unknown prevalence. Phenotypic features may be nonspecific and a combination of complementary tests that include karyotype, FISH analysis, and microarray are required for diagnosis. Table 1 summarizes the main information about 8p21.3→ p23.3 microduplication syndrome. The present study shows a rare case with a de novo duplication of the entire region 8p21.3–p23.3, which has not been reported before.
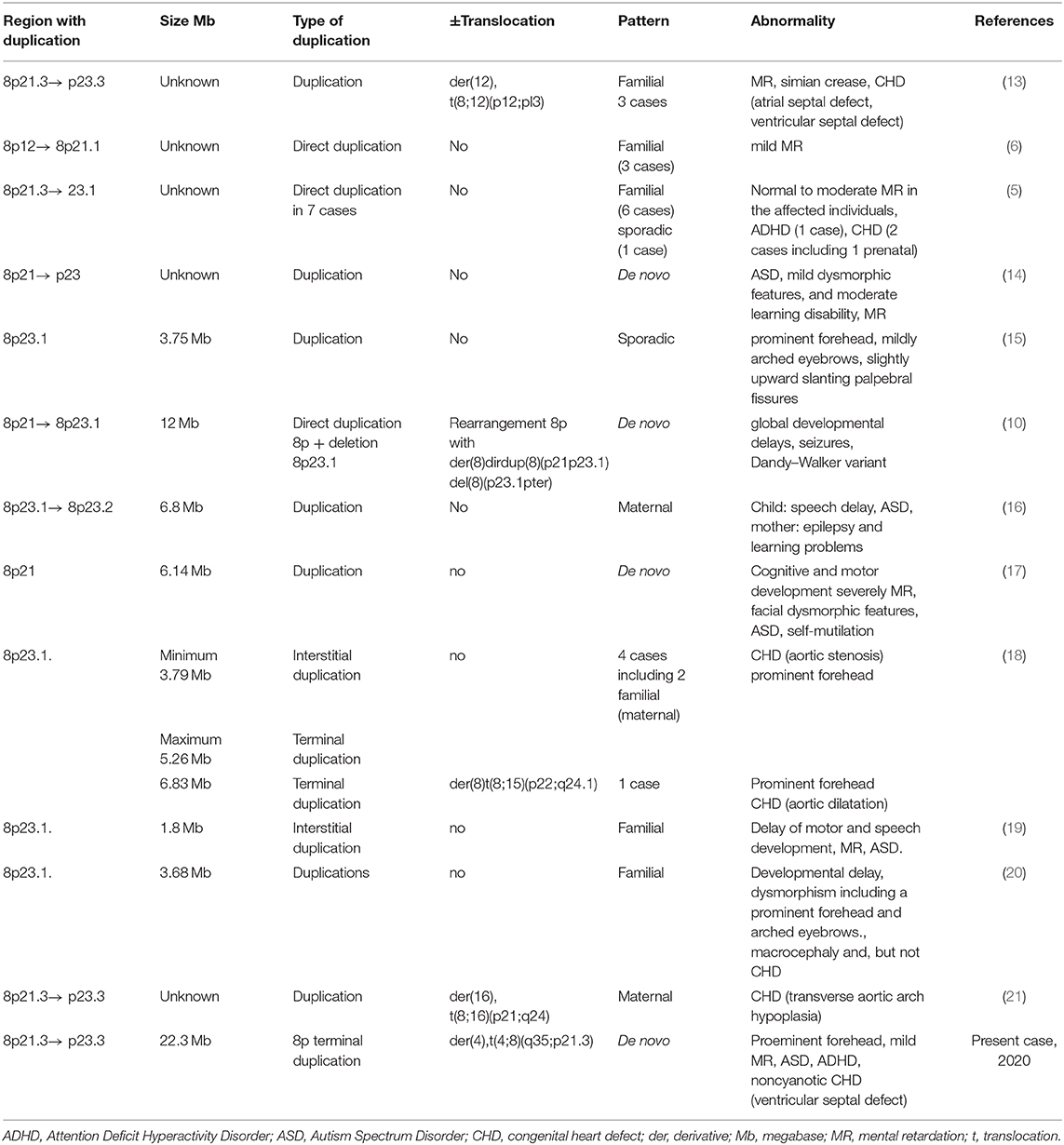
Table 1. 8p21.3→ p23.3 Duplication syndrome (in chronological order of the reports).
More than 50 cases with 8p inverted duplication/deletion syndrome have been described but, direct duplications of chromosome 8p are less common (5, 6, 11).
8p23.1 duplication syndrome defined as a 3.75 Mb duplication, most of band 8p23.1, has a prevalence of 1–9/100,000 (15, 22). The core 3.68 Mb duplication contains 32 genes of which five were considered candidates for the phenotypic features. Barber et al. (20) suggested that 8p23.1 duplication syndrome is an oligogenic condition, largely caused by the duplication and interactions of the SOX7 and GATA4 transcription factors. They noticed that eight genes led to developmental delay and dysmorphism including macrocephaly, prominent forehead, and arched eyebrows. Centromeric 8p23.1 microduplications including the GATA4 gene could enable the development of non-syndromic congenital heart defects (15, 20). An evaluation of 1645 pediatric patients with different developmental disorders by high-resolution microarray based CGH found four cases with a ≈4.0 Mb interstitial duplication of 8p23.1 (18). A patient with delayed motor and speech development and intellectual disability had a 1.80 Mb duplication in 8p23.1. SOX7 and TNKS1 genes and possibly MIR124-1 and MIR598, located within this interval, were probably responsible for the pathognomonic features of the syndrome (19).
Direct 8p21.3→ p23.3 duplication has been reported in one case with the following karyotype: 46,XX,der(16)t(8;16)(p21;q24)mat. Unlike our patient who had a de novo duplication, the other case resulted from a balanced maternal translocation (21).
The duplicated region in our case included 134 genes (12). The identified copy number variation was associated with 8p21.3→ p23.3 microduplication syndrome, being a genomic imbalance, described in international databases as having pathological significance. The duplicated region has an important gene content associated with the development of pathological conditions, particularly with growth retardation, dysmorphic features, skeletal abnormalities, which may predispose to restrictive lung disease and congenital heart defects (23, 24). Other phenotypic consequences are developmental delay, mental retardation, and behavioral problems. Our patient had facial dysmorphism, mild mental retardation/in dynamics liminal intellect with cognitive disharmony, delay in psychomotor acquisitions, developmental language delay, cognitive disharmony, instrumental and motor coordination disorder, ASD, and ADHD. The chromosomal segment 8p21.1→ p21.3 seems to be the critical region for 8p duplication syndrome (5). A mild clinical outcome for trisomy 8p22→ 8pter was reported in a study, in contrast to the severe findings when the duplication involved a longer, more proximal segment (25).
An 8p23.1→ 8p23.2 duplication spread over 6.8 Mb has been reported in a child with speech delay and autism and his mother, with epilepsy and learning problems. The interval included 41 known genes and 32 new genes among which the MCPH1 gene was thought to be the only plausible candidate gene for autism (16).
Papanikolaou et al. (14) described a patient with partial trisomy 8p(21-23) associated with autism, mild dysmorphic features, and moderate learning disability. Pinto et al. (26) suggested that the DLGAP2 gene, located on chromosome 8p23.3, could be a novel candidate gene for ASD. Further evidence for the DLGAP2 gene as a strong candidate gene has been provided by Poquet et al. (27) based on several cases with de novo duplications involving the DLGAP2 gene and presenting with ASD. Our patient also had a de novo duplication that included 8p23.3, DLGAP2 gene, respectively. Different chromosomal interstitial 8p rearrangements, including duplications, have been associated with ASD (17, 28, 29).
The size of the duplicated region correlated with the degree of cognitive deficiency in a patient with severe developmental and intellectual disability, severe impairment of expressive speech, and language (30).
CHDs were found only in a few patients with 8p23.1 duplication containing the GATA4 gene (18). These defects are different, even within the same family there were cases with aortic stenosis or aortic dilatation (18). Transverse aortic arch hypoplasia has been reported in two fetuses prenatally diagnosed with 8p23.3–p21.3 trisomy and with direct duplication of 8p21.3–p23.1, respectively (5, 21). Cardiac defects can manifest in unexpected forms in different syndromes, but they are always an important element in assessing complex cases (31).
Only extremely rarely, the 8p duplicate material is translocated to another chromosome. In our patient the cytogenetic analysis identified the translocation t(4;8)(q35;p21.3), therefore karyotyping remains a useful method to detect chromosomal rearrangements (32–34). The trisomic fragment 8p has been translocated to the end of 4q with the complete preservation of 4q telomeres. FISH analysis indicated preservation of telomeres; therefore, we hypothesize that our patient's phenotype is determined entirely by the large duplication of 8p21.3→ p23 region.
A search of PubMed using the keywords: “chromosome 8p duplication” and “translocation” revealed several reports referring to various chromosomal rearrangements involving chromosome 8. We excluded cases that had combined chromosome anomalies, and we found three reports of 8p duplication in offspring of carriers of balanced translocations involving chromosome 8. A report presented a family with t(8;12)(p12;pl3) carriers and affected descendants with 8p21.3→ p23.3 duplication, associated with MR and CHD (13). Translocation t(8;15)(p22;q24.1) and duplication 8p associated with CHD and prominent forehead in the offspring have been found in another family, as well. In the third family, a pregnant woman was a carrier of a balanced translocation t(8;16)(p21;q24), while the fetus had an unbalanced translocation 46,XX,der(16)t(8;16)(p21;q24)mat, associated with CHD (21).
Clinico-cytogenetic correlations revealed associations between phenotypic features and certain duplicate regions. Thus, 8p12 region was associated with MR (6), 8p23.1 region was associated with MR, ASD (16, 19), facial dysmorphism, especially prominent forehead (15, 18, 20), and CHD (18). 8p21.3→ p23.1 region was associated with MR (5, 13, 14, 17), ADHD (5, 17), ASD (27) and CHD (5, 13, 21). This study shows only one case that has a large duplication involving the entire 8p21.3→ p23.1 region, associated with mild facial dysmorphism with a prominent forehead, MR, ADHD, ASD, and CHD. This is preliminary evidence that indicates that all patient's symptoms are caused by this de novo duplication.
The origin of the duplicated segment has not been determined in our study. A report of 52 cases of de novo unbalanced translocations indicated that the primary driver for their occurrence was a maternal meiotic non-disjunction, followed by partial trisomy rescue (35).
A FISH analysis study found a direct tandem 8p duplication and, unlike inv dup del(8p), this was not derived from parental submicroscopic inversion (10). FISH method alone or in combination with other tests is highly informative (36). The phenotype of inverted duplications 8p is distinct from and much more severe than the clinical effect of partial trisomy 8p due to direct duplications known so far (6). Genetic counseling must consider that gonadal mosaicism cannot be excluded (37, 38). For this reason, prenatal diagnosis was performed in subsequent pregnancies (39–41).
Conclusions
We present a mildly affected phenotype correlated with a de novo 22.3Mb copy number gain of chromosome 8p21.3–p23.3, in a patient with liminal intellect with cognitive disharmony, autism spectrum disorder, attention deficit hyperactivity disorder, delay in psychomotor acquisitions and a noncyanotic congenital heart defect. Our results suggest that our patient's phenotype can be explained by the large duplication of 8p21.3→ p23.1 region. Our report emphasizes the diagnostic value of molecular cytogenetics in children with an autism spectrum disorder. It also provides additional data regarding neuropsychiatric features in chromosome 8p duplication.
Data Availability Statement
All datasets generated for this study are included in the article/supplementary material.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee for Scientific Research of Emergency Hospital for Children ‘Louis Turcanu,' Timisoara, Romania. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author Contributions
CG and DSto are the co-first authors. CG performed the cytogenetic analysis, the genetic counseling, and wrote the first draft of the manuscript. DSto coordinated and supervised data collection and interpretation, and critically reviewed the manuscript for important intellectual content. GD is the cardiologist who followed the child throughout the evaluation and made essential contributions to the manuscript writing. LN, MC, DSta, and AP collected the data, analyzed, and interpreted the findings, critically revised, and reviewed the manuscript for important intellectual content. IM revised and improved the first draft of the manuscript and is the corresponding author. All authors have read and approved the final manuscript for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We gratefully acknowledge the participation of the parents and for allowing us to learn from their stories. We would like to thank Vasilica Plaiasu MD, PhD, and biologist Ozunu Diana for excellent technical support in the cytogenetic laboratory for the FISH method.
Abbreviations
ABA, Applied Behavioral Analysis; aCGH, Array Comparative genomic hybridization; ADHD, Attention deficit hyperactivity disorder; ADI-R, Autism Diagnostic Interview-Revised; ADOS, Autism Diagnostic Observation Schedule; ASD, Autism spectrum disorder; BAC, Bacterial Artificial Chromosomes; BS, Binet-Simon Intelligence scale; CARS, Childhood Autism Rating Scale; CHD, Congenital heart defect; DGV, Database Genomic Variation; DSM-5, Diagnostic and Statistical Manual of Mental Disorders; GTG banding, Giemsa Trypsin G-band; IQ, Intelligence quotient; ISCA, International Standards for Cytogenomic Arrays; K-SADS, Kiddie Schedule for Affective Disorders and Schizophrenia; Mb, megabase; MR, mental retardation; PECS, Picture Exchange Communication Systems; t, translocation; TEACCH, Treatment and Education for Autistic and other Communication Handicapped Children; VSD, Ventricular septal defect; WAIS, Wechsler intelligence scales; WISC, Wechsler Intelligence Scale for Children.
References
1. Emanuel BS, Shaikh TH. Segmental duplications: an ‘expanding' role in genomic instability and disease. Nat Rev Genet. (2001) 2:791–800. doi: 10.1038/35093500
2. Liehr T, Weise A. Frequency of small supernumerary marker chromosomes in prenatal, newborn, developmentally retarded and infertility diagnostics. Int J Mol Med. (2007) 19:719–31. doi: 10.3892/ijmm.19.5.719
3. Liehr T. Small supernumerary marker chromosomes known to be correlated with specific syndromes. In Liehr T, editor. Small Supernumerary Marker Chromosomes (sSMC). Berlin; Heidelberg: Springer-Verlag (2012). p. 47–68.
4. Available, online at: https://www.rarechromo.org/media/information/Chromosome%20%208/8p%20duplications%20FTNW.pdf.
5. Fan YS, Siu VM, Jung JH, Farrell SA, Côté GB. Direct duplication of 8p21.3–>p23.1: a cytogenetic anomaly associated with developmental delay without consistent clinical features. Am J Med Genet. (2001) 103:231–4. doi: 10.1002/ajmg.1534
6. Moog U, Engelen JJ, Albrechts JC, Baars LG, de Die-Smulders CE. Familial dup(8)(p12p21.1): mild phenotypic effect and review of partial 8p duplications. Am J Med Genet. (2000) 94:306–10. doi: 10.1002/1096-8628(20001002)94:4<306::AID-AJMG8>3.0.CO;2-V
7. Basinko A, Douet-Guilbert N, Le Bris MJ, Parent P, Ansquer H, Morel F, et al. Molecular cytogenetic characterization of an 8p22-8p23.2 duplication derived from a maternal intrachromosomal insertion in a child with congenital heart malformation, delayed puberty, and learning disabilities. Am J Med Genet Part A. (2008) 146A:2950–54. doi: 10.1002/ajmg.a.32522
8. Puvabanditsin S, Gengel N, Botti C, Jacob M, Jalil M, Cabrera K, et al. 8p11 microduplication is associated with neonatal stridor. Mol Syndromol. (2018) 9:324–7. doi: 10.1159/000494796
9. Marafie MJ, Abu-Henedi MM, Abulhasan SJ, Al-Wadaani A. Partial duplication of chromosome 8p: report of 5 patients and review of literature. Egypt J Hum Genet. (2007) 8:199–208.
10. Cooke SL, Northup JK, Champaige NL, Zinser W, Edwards PAW, Lockhart LH, et al. Molecular cytogenetic characterization of a unique and complex de novo 8p rearrangement. Am J Med Genet Part A. (2008) 146A:1166–72. doi: 10.1002/ajmg.a.32248
11. Sireteanu A, Braha E, Popescu R, Gramescu M, Gorduza EV, Rusu C, et al. Inverted duplication deletion of 8P: characterization by standard cytogenetic and SNP array analyses. Rev Med Chir Soc Med Nat Iasi. (2013) 117:731–4.
12. DECIPHER, v9,.31. Available online at: https://decipher.sanger.ac.uk/browser#q/8:125733-22400607/location/8:159194-22684074 (Accesed February 17, 2020).
13. Moreno Fuenmayor HM, Meilinger KL, Rucknagel DL, Mohrenweiser HL, Chu EH. Duplication 8p syndrome: studies in a family with a reciprocal translocation between chromosomes 8 and 12. Am J Med Genet. (1980) 7:361–8. doi: 10.1002/ajmg.1320070318
14. Papanikolaou K, Paliokosta E, Gyftodimou J, Kolaitis G, Vgenopoulou S, Sarri C, et al. A case of partial trisomy of chromosome 8p associated with autism. J Autism Dev Disord. (2006) 36:705–9. doi: 10.1007/s10803-006-0104-3
15. Barber J, Maloney V, Huang S, Bunyan D, Cresswell L, Kinning E, et al. 8p23.1 duplication syndrome; a novel genomic condition with unexpected complexity revealed by array CGH. Eur J Hum Genet. (2008) 16:18–27. doi: 10.1038/sj.ejhg.5201932
16. Glancy M, Barnicoat A, Vijeratnam R, de Souza S, Gilmore J, Huang S, et al. Transmitted duplication of 8p23.1-8p23.2 associated with speech delay, autism and learning difficulties. Eur J Hum Genet. (2009) 17:37–43. doi: 10.1038/ejhg.2008.133
17. Ozgen HM, Staal WG, Barber JC, de Jonge MV, Eleveld MJ, Beemer FA, et al. A novel 6.14 Mb duplication of chromosome 8p21 in a patient with autism and self mutilation. J Autism Dev Disord. (2009) 39:322–9. doi: 10.1007/s10803-008-0627-x
18. Yu S, Zhou XG, Fiedler SD, Brawner SJ, Joyce JM, Liu HY, et al. Cardiac defects are infrequent findings in individuals with 8p23.1 genomic duplications containing GATA4. Circ Cardiovasc Genet. (2011) 4:620–25. doi: 10.1161/CIRCGENETICS.111.960302
19. Weber A, Köhler A, Hahn A, Müller U. 8p23.1 duplication syndrome: narrowing of critical interval to 1.80 Mbp. Mol Cytogenet. (2014) 7:94. doi: 10.1186/s13039-014-0094-3
20. Barber JCK, Rosenfeld JA, Graham JM, Kramer N, Lachlan KL, Bateman MS, et al. Inside the 8p23.1 duplication syndrome; eight microduplications of likely or uncertainclinical significance. Am J Med Genet A. (2015) 167A:2052–64. doi: 10.1002/ajmg.a.37120
21. Guo C, Wang J, Zhao L, Liu J, Wang J, Xiao J, et al. Prenatal diagnosis of a fetus with partial trisomy 8p resulting from a balanced maternal translocation by array-based comparative genomic hybridization. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. (2015) 32:375–7. doi: 10.3760/cma.j.issn.1003-9406.2015.03.016
22. Available, online at: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=EN&Expert=251076
23. Barber JCK. Directly transmitted unbalanced chromosome abnormalities and euchromatic variants. J Med Genet. (2005) 42:609–29. doi: 10.1136/jmg.2004.026955
24. Amaricai E, Suciu O, Onofrei RR, Miclaus RS, Iacob RE, Catan L, et al. Respiratory function, functional capacity, and physical activity behaviours in children and adolescents with scoliosis. J Int Med Res. (2019). doi: 10.1177/0300060519895093. [Epub ahead of print].
25. Sklower Brooks SS, Genovese M, Gu H, Duncan CJ, Shanske A, Jenkins EC, et al. Normal adaptive function with learning disability in duplication 8p including p22. Am J Med Genet. (1998) 78:114–7. doi: 10.1002/(SICI)1096-8628(19980630)78:2<114::AID-AJMG3>3.0.CO;2-O
26. Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. (2010) 466:368–72. doi: 10.1038/nature09146
27. Poquet H, Faivre L, El Chehadeh S, Morton J, McMullan D, Goel H. Further evidence for DLGAP2 as strong autism spectrum disorders/intellectual disability candidate gene. Autism Open Access. (2017) 6:197. doi: 10.4172/2165-7890.1000197
28. Tabares-Seisdedos R, Rubenstein JLR. Chromosome 8p as a potential hub for developmental neuropsychiatric disorders: implications for schizophrenia, autism and cancer. Mol Psychiatry. (2009) 14:563–89. doi: 10.1038/mp.2009.2
29. Dong P, Xu Q, An Y, Zhou B-R, Lu P, Liu R-C, et al. Novel 1.0 Mb duplication of chromosome 8p22-21.3 in a patient with autism spectrum disorder. Child Neurol Open. (2015) 2:1–6. doi: 10.1177/2329048X15580673
30. Fisch GS, Davis R, Youngblom J, Gregg J. Genotype-phenotype association studies of chromosome 8p inverted duplication deletion syndrome. Behav Genet. (2011) 41:373–80. doi: 10.1007/s10519-011-9447-4
31. Gug C, Gorduza EV, Lăcătuşu A, Vaida MA, Bîrsăşteanu F, Puiu M, et al. CHARGE syndrome associated with de novo (I1460Rfs*15) frameshift mutation of CHD7 gene in a patient with arteria lusoria and horseshoe kidney. Exp Ther Med. (2020) 20:479–85. doi: 10.3892/etm.2020.8683
32. Caba L, Rusu C, Plăiaşu V, Gug C, Grămescu M, Bujoran C, et al. Ring autosomes: some unexpected findings. Balkan J Med Genet. (2012) 15:35–46. doi: 10.2478/bjmg-2013-0005
33. Gug C, Cioata I, Cretu A, Tuduce I. Familial reciprocal translocation t(1;5)(q23;p12), three cases in two generations. Chromosome Res. (2007) 15(Suppl 1):69.
34. Gug C. Cytogenetic analysis in prenatal diagnosis – a report of 550 pregnancy. Chromosome Res. (2009) 17:241.
35. Bonaglia MC, Kurtas NE, Errichiello E, Bertuzzo S, Beri S, Mehrjouy MM. De novo unbalanced translocations have a complex history/aetiology. Hum Genet. (2018) 137:817–29. doi: 10.1007/s00439-018-1941-9
36. Belengeanu A, Muresan A, Stoicanescu D, Lazar E. Amplification of HER-2 gene in breast cancer: immunohistochemical and FISH assessment. Rom J Morphol Embryol. (2010) 51:321–6.
37. Gug C, Huţanu D, Vaida M, Doroş G, Popa C, Stroescu R, et al. De novo unbalanced translocation t(15;22)(q26.2;q12) with velo-cardio-facial syndrome: a case report and review of the literature. Exp Ther Med. (2018) 16:3589–95. doi: 10.3892/etm.2018.6609
38. Gug C, Caba L, Mozos I, Stoian D, Atasie D, Gug M, et al. Rare splicing mutation in COL1A1 gene identified by whole exomes sequencing in a patient with osteogenesis imperfecta type I followed by prenatal diagnosis: a case report and review of the literature. Gene. (2020) 741:144565. doi: 10.1016/j.gene.2020.144565
39. Gug C, Raţiu A, Navolan D, Drăgan I, Groza IM, Păpurică M, et al. Incidence and spectrum of chromosome abnormalities in miscarriage samples: a retrospective study of 330 cases. Cytogenet Genome Res. (2019) 158:171–83. doi: 10.1159/000502304
40. Gug C, Burada F, Ioana M, Riza AL, Moldovan M, Mozoţ I, et al. Polyploidy in first and second trimester pregnancies in Romania. Clin Lab. (2020) 66:517–27. doi: 10.7754/Clin.Lab.2019.190649
Keywords: 8p(21.3–p23.3) duplication, translocation(4;8), de novo, array CGH, FISH, mental retardation, autism spectrum disorder, congenital heart defects
Citation: Gug C, Stoicanescu D, Mozos I, Nussbaum L, Cevei M, Stambouli D, Pavel AG and Doros G (2020) De novo 8p21.3→ p23.3 Duplication With t(4;8)(q35;p21.3) Translocation Associated With Mental Retardation, Autism Spectrum Disorder, and Congenital Heart Defects: Case Report With Literature Review. Front. Pediatr. 8:375. doi: 10.3389/fped.2020.00375
Received: 14 March 2020; Accepted: 03 June 2020;
Published: 08 July 2020.
Edited by:
Enrico Baruffini, University of Parma, ItalyReviewed by:
Xiu Xu, Fudan University, ChinaAmal Mahmoud Mohamed, National Research Centre (Egypt), Egypt
Copyright © 2020 Gug, Stoicanescu, Mozos, Nussbaum, Cevei, Stambouli, Pavel and Doros. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ioana Mozos, aW9hbmFfbW96b3MmI3gwMDA0MDt5YWhvby5jb20=
†These authors share first authorship