
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Pediatr. , 07 April 2020
Sec. Pediatric Endocrinology
Volume 8 - 2020 | https://doi.org/10.3389/fped.2020.00145
 Petra Loid1,2,3*
Petra Loid1,2,3* Minna Pekkinen1,2,3Monica Reyes4Taina Mustila5,6
Minna Pekkinen1,2,3Monica Reyes4Taina Mustila5,6 Heli Viljakainen2,7Harald Jüppner4
Heli Viljakainen2,7Harald Jüppner4 Outi Mäkitie1,2,3,8
Outi Mäkitie1,2,3,8Context: Pseudohypoparathyroidism type Ia (PHP1A) is caused by inactivating mutations involving GNAS exons 1–13, encoding the alpha-subunit of the stimulatory G protein (Gsα). Particularly PHP1A, but also other disorders involving the Gsα-cAMP-signaling pathway, have been associated with early-onset obesity. Thus, patients with mutations in the genes encoding PDE4D and PRKAR1A can also be obese. Furthermore, epigenetic GNAS changes, as in pseudohypoparathyroidism type Ib (PHP1B), can lead to excessive weight.
Objective: Search for genetic variants in GNAS, PDE4D, and PRKAR1A and for methylation alterations at the GNAS locus in Finnish subjects with isolated severe obesity before age 10 years.
Methods: Next generation sequencing to identify pathogenic variants in the coding exons of GNAS, PDE4D, and PRKAR1A; Multiplex Ligation-dependent Probe Amplification (MLPA) and methylation-sensitive MLPA (MS-MLPA) to search for deletions in GNAS and STX16, and for epigenetic changes at the four differentially methylated regions (DMR) within GNAS.
Results: Among the 88 subjects (median age 13.8 years, median body mass index Z-score +3.9), we identified one rare heterozygous missense variant of uncertain significance in the XL exon of GNAS in a single patient. We did not identify clearly pathogenic variants in PDE4D and PRKAR1A, and no GNAS methylation changes were detected by MS-MLPA.
Conclusions: Our results suggest that coding GNAS mutations or methylation changes at the GNAS DMRs, or coding mutations in PDE4D and PRKAR1A are not common causes of isolated childhood obesity in Finland.
Pseudohypoparathyroidism (PHP) and related disorders have been associated with early-onset obesity (1, 2). These diseases, which can present with highly variable clinical findings (3), are caused by a defect in the stimulatory G protein-cAMP-signaling pathway.
GNAS is a complex imprinted locus on chromosome 20q13.3. Exons 1–13 encode the alpha-subunit of the stimulatory G protein (Gsα), a signaling protein mediating the functions of several hormones that require the second messenger cAMP down-stream of their cognate receptors (3). GNAS also encodes several other transcripts with incompletely defined biological actions, including the mRNAs encoding the neuroendocrine secretory protein 55 (NESP55) and the extra-large form of Gsα (XLαs), as well as the A/B and the antisense (AS) transcript.
The Gsα transcript is biallelically expressed in most but not all tissues; the paternal expression is reduced in several tissues, e.g., proximal renal tubules, thyroid, gonads, brown adipose tissue, hypothalamus, and pituitary gland (3). Clinical and laboratory manifestations of GNAS mutations are therefore determined by the parental origin of the mutated allele. Mutations involving exons 1–13 of the maternal GNAS allele underlie pseudohypoparathyroidism type 1A (PHP1A) in which patients develop resistance to PTH in the proximal renal tubules leading to elevated plasma PTH levels, hypocalcemia and hyperphosphatemia, and often resistance to multiple other hormones. Patients with PHP1A show clinical features of Albright's hereditary osteodystrophy (AHO), including short stature, brachydactyly, early-onset obesity, neurodevelopmental defects and subcutaneous ossifications. In contrast, inactivating mutations located on the paternal GNAS allele cause pseudopseudohypoparathyroidism (PPHP) characterized in most patients by several AHO features, but no hormonal resistance, no obesity, and no intellectual challenges. Methylation defects at GNAS exon A/B alone, or at several GNAS DMRs, are observed in pseudohypoparathyroidism type 1B (PHP1B), characterized by resistance to PTH and frequently TSH, but infrequently by AHO features (3).
Other genes involved in the Gsα-cAMP-signaling pathway are PRKAR1A and PDE4D. PRKAR1A encodes the regulatory subunit of cAMP-dependent protein kinase A (PKA) and PDE4D encodes the phosphodiesterase 4D. Heterozygous mutations in PDE4D and PRKAR1A lead to acrodysostosis because cAMP is either degraded too rapidly or unable to dissociate the regulatory from the catalytic subunit of PKA. The disorders caused by mutations in the latter genes present with clinical manifestations overlapping partly those encountered in PHP (4).
Early-onset obesity can be the first and only evidence for pseudohypoparathyroidism (2, 5). Several mechanisms can be responsible for the excessive weight gain. In patients with PHP1A, reduced energy expenditure caused by the decreased Gsα-mediated signaling in brown or beige fat cells, as well as impaired signaling at the melanocortin 4 receptor leading to hyperphagia and thus increased caloric intake have been proposed as important mechanisms leading to obesity (6–8). Furthermore, the resistance to TSH and growth hormone-releasing-hormone may play a role in the development of obesity (2). Early-onset and childhood obesity has also been observed in PHP1B (1, 9). Based on these observations, it has been proposed that genetic and epigenetic defects at the GNAS locus should be considered in patients with unexplained childhood obesity (2). In fact, mutations in the GNAS exons encoding Gsα were recently identified in a large British cohort of obese patients (10). This prompted us to look for genetic variants in GNAS, PDE4D, and PRKAR1A, and for methylation alterations of GNAS locus in a cohort of Finnish patients who had developed severe obesity already in early childhood.
This study included 88 subjects recruited through pediatric endocrine clinics at Children's Hospital at Helsinki University Hospital and Seinäjoki Central Hospital in Finland. The study was approved by the Research Ethics Committees of the Hospital District of Helsinki and Uusimaa and the Pirkanmaa Hospital District. Informed written consent was obtained from all participants or their guardians (subjects aged <18 years).
The inclusion criterion was severe obesity as defined according to the Finnish growth standards (11) as height-adjusted weight >60% before 10 years of age. The patients had been followed up by a pediatrician and patients who were diagnosed with a known underlying endocrine or genetic disorder were excluded from the study. Clinical and growth data were collected from hospital records. Height, weight and waist circumference were measured during a study visit. Sex-and age-specific BMI Z-scores were derived based on the World Health Organization reference values (www.who.int/childgrowth/standards).
Genomic DNA was isolated from peripheral blood samples according to standard procedures. The probes for targeted exome sequencing were designed using SeqCap EZ Choice Library and NimbleDesign (Roche NimbleGen, United States). DNA capture and Next Generation Sequencing were performed at Oxford Genomics Centre. The reads were aligned to the reference genome hg19. The software Platypus version 0.8.1 was used for variant calling and Ensembl Variant Effector Predictor (VEP) was used for functional annotation of the variants. Filtering of variants was performed using VarAFT 2.13 (http://varaft.eu). Exonic and splice site variants were considered. We compared the allele frequencies of the identified variants to those obtained from the Exome Aggregation Consortium (http://exac.broadinstitute.org), the Genome Aggregation Database (http://gnomad.broadinstitute.org), 1,000 Genomes Project (http://www.internationalgenome.org) and the Sequencing Initiative Suomi project (SISu) (http://sisuproject.fi). The potential pathogenicity of the sequence variants was evaluated using different variant prediction databases (Polyphen-2, SIFT, Combined Annotation Dependent Depletion (CADD) and MutationTaster2). Sanger sequencing was performed to confirm findings. Primers were designed using Primer3 software and PCR was performed using DreamTaqTM DNA Polymerase (Thermo Fisher Scientific) according to standard protocol and chromatograms were analyzed with Sequencer v5.0 software. Primer sequences are available upon request.
Multiplex Ligation-dependent Probe Amplification (MLPA) to search for GNAS or STX16 deletions and analysis of the GNAS methylation status, as assessed by methylation specific multiplex ligation-dependent probe amplification (MS-MLPA), was performed using kit ME031 GNAS (MRC-Holland, Amsterdam, The Netherland) following the manufacturer's instructions (https://www.mlpa.com/), as described (12). PCR products were examined using the ABI3730xl Genetic Analyzer at the DNA Core Facility of the Massachusetts General Hospital and Peak Scanner 2.0 software (http://peak-scanner-software.software.informer.com/2.0/). The results obtained for two to five probes at each of the different DMRs were averaged for each patient and mean± standard deviation (SD) were subsequently calculated for each DMR for the entire cohort.
The median age for the 88 study subjects (54.5% males) was 13.8 years [interquartile range (IQR) 11.1–17.0 years]. The median BMI Z-score at the time of the study visit was +3.9 (IQR +3.4 to +4.9). The study subjects fulfilled our inclusion criteria of severe obesity (height-adjusted weight >60%) at the median age of 5.3 years (IQR 4.1–7.0 years). They had reached height-adjusted weight >40% (corresponding to obesity according to Finnish growth standards) at the median age of 4.0 years (IQR 2.0–5.0 years) (Table 1). Of the 68 school-aged subjects for whom information on learning difficulties was available, 10 (15%) had learning difficulties and required special education.

Table 1. Characteristics of study subjects, n = 88.
The variants detected in GNAS, PDE4D, and PRKAR1A are presented in Table 2. We identified one rare heterozygous missense GNAS variant in exon XL (c.897C>A, p.S299R) in a female with severe early-onset obesity. This variant is found with allele frequency 0.00036 in the Finnish population in gnomAD. Polyphen-2 predicted the variant as possibly damaging, SIFT as tolerated, MutationTaster2 as disease-causing; the CADD score was high 23. The patient presented with obesity already at 3.5 years of age with weight 24.5 kg and height 105.5 cm (BMI Z-score +3.8). Presently at the age of 21 years, she has severe obesity: weigh 121 kg, height 170.5 cm (BMI 42) and waist circumference 120 cm. The patient did not present with any hormone resistance or clinical characteristics of AHO apart from obesity. Both parents were obese, but their DNA samples were unavailable for genotyping.
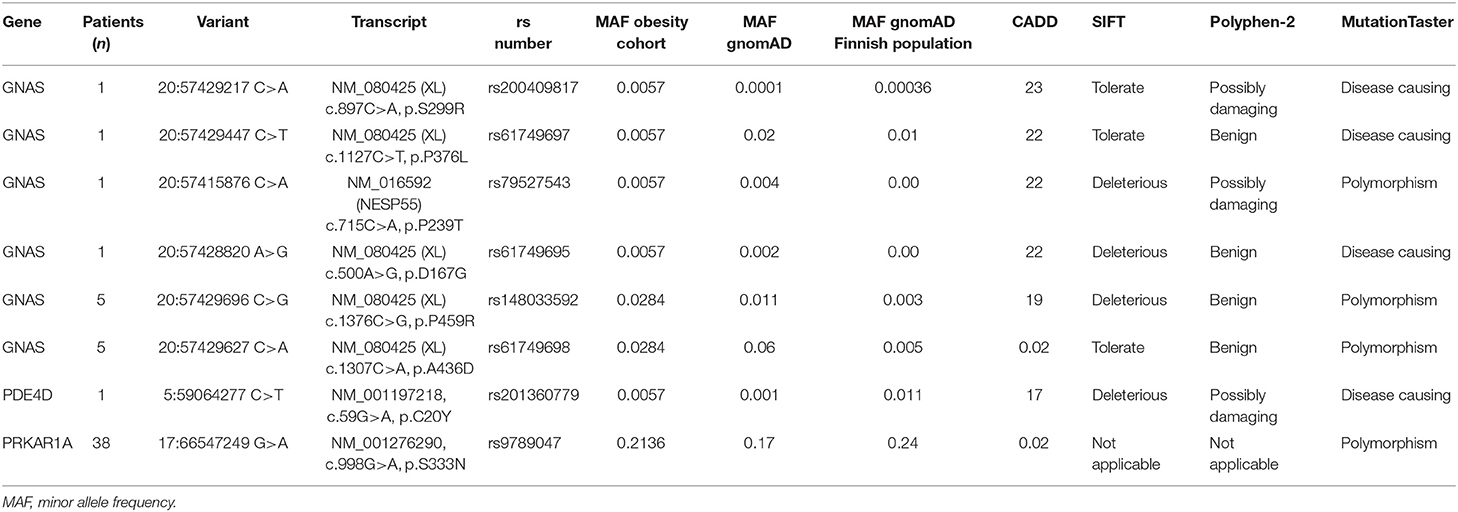
Table 2. Non-synonymous sequence variants found in GNAS, PRKAR1A, and PDE4D genes (reference genome hg19).
Furthermore, five heterozygous variants were identified that change single amino acids in GNAS exon XL and NESP, respectively. However, these variants have allele frequencies in gnomAD comparable to the allele frequencies observed in our cohort. No variants were identified in the GNAS exons encoding Gsα. In PDE4D or PRKAR1A, we did not detect any variants that have previously been shown to be disease-causing.
Analysis of GNAS methylation by MS-MPLA did not identify any epigenetic alterations at the GNAS locus (Supplemental Table 1); the DMRs showed no evidence for abnormal methylation at NESP (53.4 ± 2.5%), AS (53.9 ± 3.1%), XL (53.5 ± 2.9%), and A/B (52.4 ± 3.6%).
Pseudohypoparathyroidism and acrodysostosis are rare, heterogeneous disorders caused by different genetic/epigenetic defects (3). Differential diagnosis between these disorders can be challenging because of their overlapping clinical and/or laboratory phenotypes. The clinical features can be difficult to identify by physical examination and some patients show only minor clinical abnormalities that furthermore can vary with age (13). Furthermore, PTH-resistance in PHP1B may remain undetected until symptomatic hypocalcemia develops (2). Establishing the correct diagnosis can thus be challenging but would be of significant importance because of the multiple endocrine disturbances related to these disorders.
Previous studies have found that obesity is a common clinical finding in children with pseudohypoparathyroidism type 1A (2, 5). In a study investigating early features of PHP1A, the major clinical sign was obesity, present in 70% of children >2 years of age (5). Furthermore, it has been suggested that GNAS defects in patients with early-onset obesity may be underestimated, since Hendricks et al. (10) found several novel variants in GNAS in unselected patients with severe obesity, yet no obvious endocrine defects or short stature. However, many of the patients in the study by Hendricks et al. with GNAS variants had developmental delay (10).
We searched for pathogenic variants in GNAS, PDE4D, and PRKAR1A, and for GNAS methylation changes in 88 Finnish subjects with isolated severe early-onset obesity. We identified only one rare heterozygous missense variant in GNAS exon XL in a patient with isolated severe obesity. Unfortunately, parental DNA samples were not available. This variant does not affect the transcript encoding Gsα. The patient did not present with any skeletal, endocrine or developmental defect. The clinical significance of this missense variant thus remains uncertain. We furthermore identified four heterozygous non-synonymous amino acid changes in GNAS exon XL and one change in exon NESP, as well as single variants in PRKAR1A and in PDE4D. All these amino acid changes were present in gnomAD with comparable or higher allele frequencies and were not predicted to be disease-causing variants.
We did not identify any pathogenic defects in the investigated genes. Although we had carefully selected our patients presenting severe obesity with early onset, the relatively small cohort size of only 88 patients can be regarded as a limitation. Furthermore, patients with PHP and related disorders may have been identified earlier by pediatricians and could therefore not be included in our study.
Another limitation of our study was the lack of a control group of normal-weight subjects. However, we compared the allele frequencies of the identified variants with the Finnish population in gnomad and SISu project, which include genotype data of more than 10,000 individuals from the Finnish population.
In conclusion, our results suggest that mutations in GNAS, PDE4D, and PRKAR1A, and methylation changes in GNAS locus do not play a significant role in etiology of childhood-onset obesity in our Finnish cohort of patients, who lacked biochemical features of pseudohypoparathyroidism and related disorders.
Data cannot be shared publicly because the data consists of sensitive patient data. More specifically the data consists of individual clinical data and individual genotypes for young children. Data are available from the Helsinki University Hospital's Institutional Data Access/Ethics Committee for researchers who meet the criteria for access to confidential data. Data availability contact: Outi Mäkitie MD, PhD.
The study was approved by the Research Ethics Committees of the Hospital District of Helsinki and Uusimaa and the Pirkanmaa Hospital District. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Study design: PL, MP, TM, HV, HJ, and OM. Data collection: PL, TM, and HV. Data analysis and data interpretation: PL, MP, MR, HJ, and OM. Drafting of manuscript: PL, MP, HJ, and OM. All authors reviewed manuscript content and approved the final version.
This study was financially supported by the Academy of Finland, Sigrid Jusélius Foundation, Foundation for Pediatric Research, Folkhälsan Research Foundation, Päivikki and Sakari Sohlberg Foundation, Stiftelsen Dorothea Olivia, Karl Walter och Jarl Walter Perkléns minne, the Swedish Research Council, the Novo Nordisk Foundation, University of Helsinki through the Doctoral Program in Clinical Research, Helsinki University Hospital research funds and the National Institutes of Health, RO1-DK046718 (to HJ).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank all the patients and their family members for participating in the study. We thank RN Päivi Turunen for help with data collection.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2020.00145/full#supplementary-material
1. Hanna P, Grybek V, Perez de Nanclares G, Tran LC, de Sanctis L, Elli F, et al. Genetic and epigenetic defects at the GNAS locus lead to distinct patterns of skeletal growth but similar early-onset obesity. J Bone Miner Res. (2018) 33:1480–8. doi: 10.1002/jbmr.3450
2. Gruters-Kieslich A, Reyes M, Sharma A, Demirci C, DeClue TJ, Lankes E, et al. Early-onset obesity: unrecognized first evidence for GNAS mutations and methylation changes. J Clin Endocrinol Metab. (2017) 102:2670–7. doi: 10.1210/jc.2017-00395
3. Linglart A, Levine MA, Juppner H. Pseudohypoparathyroidism. Endocrinol Metab Clin North Am. (2018) 47:865–88. doi: 10.1016/j.ecl.2018.07.011
4. Mantovani G, Elli FM. Multiple hormone resistance and alterations of G-protein-coupled receptors signaling. Best Pract Res Clin Endocrinol Metab. (2018) 32:141–54. doi: 10.1016/j.beem.2018.01.002
5. Kayemba-Kay's S, Tripon C, Heron A, Hindmarsh P. Pseudohypoparathyroidism type 1A-subclinical hypothyroidism and rapid weight gain as early clinical signs: a clinical review of 10 cases. J Clin Res Pediatr Endocrinol. (2016) 8:432–8. doi: 10.4274/jcrpe.2743
6. Roizen JD, Danzig J, Groleau V, McCormack S, Casella A, Harrington J, et al. Resting energy expenditure is decreased in pseudohypoparathyroidism type 1A. J Clin Endocrinol Metab. (2016) 101:880–8. doi: 10.1210/jc.2015-3895
7. Chen M, Shrestha YB, Podyma B, Cui Z, Naglieri B, Sun H, et al. Gsalpha deficiency in the dorsomedial hypothalamus underlies obesity associated with Gsalpha mutations. J Clin Invest. (2017) 127:500–10. doi: 10.1172/JCI88622
8. Shoemaker AH, Juppner H. Nonclassic features of pseudohypoparathyroidism type 1A. Curr Opin Endocrinol Diabetes Obes. (2017) 24:33–8. doi: 10.1097/MED.0000000000000306
9. de Lange IM, Verrijn Stuart AA, van der Luijt RB, Ploos van Amstel HK, van Haelst MM. Macrosomia, obesity, and macrocephaly as first clinical presentation of PHP1b caused by STX16 deletion. Am J Med Genet A. (2016) 170:2431–5. doi: 10.1002/ajmg.a.37818
10. Hendricks AE, Bochukova EG, Marenne G, Keogh JM, Atanassova N, Bounds R, et al. Rare variant analysis of human and rodent obesity genes in individuals with severe childhood obesity. Sci Rep. (2017) 7:4394. doi: 10.1038/s41598-017-03054-8
11. Saari A, Sankilampi U, Hannila ML, Kiviniemi V, Kesseli K, Dunkel L. New Finnish growth references for children and adolescents aged 0 to 20 years: length/height-for-age, weight-for-length/height, and body mass index-for-age. Ann Med. (2011) 43:235–48. doi: 10.3109/07853890.2010.515603
12. Garin I, Mantovani G, Aguirre U, Barlier A, Brix B, Elli FM, et al. European guidance for the molecular diagnosis of pseudohypoparathyroidism not caused by point genetic variants at GNAS: an EQA study. Eur J Hum Genet. (2015) 23:438–44. doi: 10.1038/ejhg.2014.127
Keywords: GNAS, G protein-cAMP-signaling, childhood-onset obesity, pseudohypoparathyroidism, acrodysostosis
Citation: Loid P, Pekkinen M, Reyes M, Mustila T, Viljakainen H, Jüppner H and Mäkitie O (2020) GNAS, PDE4D, and PRKAR1A Mutations and GNAS Methylation Changes Are Not a Common Cause of Isolated Early-Onset Severe Obesity Among Finnish Children. Front. Pediatr. 8:145. doi: 10.3389/fped.2020.00145
Received: 14 January 2020; Accepted: 13 March 2020;
Published: 07 April 2020.
Edited by:
Tsutomu Ogata, Hamamatsu University School of Medicine, JapanReviewed by:
Keisuke Nagasaki, Niigata University, JapanCopyright © 2020 Loid, Pekkinen, Reyes, Mustila, Viljakainen, Jüppner and Mäkitie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Petra Loid, cGV0cmEubG9pZEBoZWxzaW5raS5maQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.