 Xiaoxue Wang
Xiaoxue Wang Hui Pang2
Hui Pang2 Birju A. Shah
Birju A. Shah
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr. , 10 March 2020
Sec. Genetics of Common and Rare Diseases
Volume 8 - 2020 | https://doi.org/10.3389/fped.2020.00088
Kagami–Ogata syndrome (KOS) is a rare imprinting disorder characterized by skeletal abnormalities, dysmorphic facial features, growth retardation and developmental delay. The genetic etiology of KOS includes paternal uniparental disomy 14 [upd(14)pat], epimutations and microdeletions affecting the maternally derived imprinted region of chromosome 14q32.2. More than seventy KOS cases have been reported thus far; however, only 10, including two familial, are associated with upd(14)pat harboring Robertsonian translocation (ROB). Here, we reported a male infant with clinical manifestations of facial dysmorphism, bell-shaped small thorax, and omphalocele. Karyotype analyses identify a balanced ROB involving the long arms of chromosomes 13 and 14 both in the patient and his father. We further confirm the pattern of upd(14)pat utilizing DNA polymorphic markers. In conclusion, our case report provides a new male KOS case caused by upd(14)pat with paternally inherited Robertsonian translocation, which represents the second male case officially reported. Notably, a KOS case due to upd(14)pat and ROB is rare. An accurate diagnosis requires not only the identification of the characteristic clinical features but also systemic cytogenetic and molecular studies.
Genomic imprinting is an epigenetic phenomenon that restricts the expression of genes in an imprinted region to a single parental allele (1). Genomic or epigenetic changes of imprinted regions have been linked to certain genetic disorders (2). So far, six imprinted regions associated with nine clinically recognized imprinting disorders have been reported in human (3, 4). The common causes of imprinting disorders included genomic and epigenetic alterations such as mutations/copy number changes of imprinted genes, uniparental disomy (UPD) and increases or decreases of DNA methylation levels (5).
Kagami–Ogata syndrome (KOS; MIM 608149) is an imprinting disorder involving genes in chromosome 14q32.2 region and paternal uniparental disomy 14 [upd(14)pat] accounts for approximately two-thirds of KOS patients (6). The main clinical features of KOS include craniofacial dysmorphism, thoracic dysplasia (bell-shaped small thorax with coat-hanger appearance), abdominal wall defect, growth retardation, and developmental delay (7). UPD(14)pat has been reported as a de novo event as well as a result of a paternally inherited Robertsonian translocation (ROB) or due to a monosomy rescue which leads to paternal isodisomy (8). ROBs are whole-arm exchanges of acrocentric chromosomes which may cause unproperly segregation of homologous chromosomes in meiosis leading to aneuploidy conceptions. Many cases of UPD have been identified in carriers of ROBs and the risk of UPD in carriers of non-homologous ROB is 0.6–0.8% (9, 10). Only one of the ten reported KOS cases caused by ROB related UPD was from a male patient. Parental karyotype was performed on seven patients, and the results showed that five were de novo, and the other two were inherited from the father. Here, we presented another male patient with characteristic KOS features caused by upd(14)pat and balanced ROB inherited from the father.
This male infant was born at 36 weeks and 6 days of gestation by a cesarean section due to prenatal findings of congenital anomalies and polyhydramnios. The proband was conceived naturally to a 34-year-old father and a 31-year-old mother both of whom are Caucasian origin. The couple had a history of a previous miscarriage. There was no family history of genetic disorders, and parental consanguinity was denied. Ultrasound immediately prior to delivery was significant for omphalocele and amniotic fluid index of 38 (Normal value 5–25). Apgar scores were six and eight at 1 and 5 min of life, respectively. The admission exam was consistent with omphalocele, with a birth weight of 3,870 grams (Z score 2.08, >97th percentile), head circumference of 37 cm (Z score 2.55, >97th percentile), and length of 45 cm (Z score −1.29, 10th percentile). On physical examination at birth, dysmorphic facial features were noted including narrow forehead, frontal bossing, short palpebral fissures, depressed nasal bridge, anteverted nares, elongated philtrum, low-set posteriorly rotated ears, and micrognathia (Figures 1A,B). Additionally, short neck, bell-shaped chest, elongated fifth finger on each hand with flexion contractures, deep sacral dimple, bilateral undescended testes and right hydrocele were identified. There was no cardiac murmur or organomegaly. Echocardiography showed a patent foramen ovale. Abdominal ultrasound showed left-sided mild hydronephrosis, and skeletal survey displayed 11 pairs of ribs and bell-shaped thoracic cavity with coat-hanger looking ribs (Figures 1C,D). Primary repair of omphalocele on day 4 of life was performed under general endotracheal anesthesia. Later, gastrostomy tube was placed due to significant aspiration with penetration of thin liquids on dysphagiogram. Brain and spinal cord MRI showed echogenic scattered foci in periventricular white matter without restricted diffusion. Screening electroencephalogram was unremarkable for seizures. The placental pathology was unremarkable. The infant was discharged home on low-flow nasal cannula with supplemental oxygen, monitor and suction equipment on day 36 of life. Due to his multiple congenital anomalies, routine chromosome analysis was performed on peripheral blood and the results revealed an abnormal male karyotype with a balanced ROB:45,XY,der(13;14)(q10;q10) (Figure 2A). Further parental karyotyping indicated that this ROB chromosome was inherited from his father. The presence of paternally inherited der(13;14) combined with the patient's phenotype raised the concern for KOS caused by upd(14)pat. Therefore, microsatellite analysis was subsequently performed by utilizing multiple polymorphic DNA markers on subchromosomal regions 14q24.2(D14S77), 14q31.3(D14S68), and 14q32.2(D14S985). All three markers demonstrated that the patient inherited two homologous chromosome 14 from his father, confirming a paternal uniparental heterodisomy of 14q (Figures 2B,C). Then, the whole genome SNP array analysis were used to detect if there were additional small deletion or duplication. The test identified two homozygous regions with varied molecular sizes but did not detect any clinically relevant copy number change (Figure 2D). In the constellation of whole clinical picture, the results are consistent with the diagnosis of KOS caused by upd(14)pat. The follow-up investigation showed that the patient was transitioned from utilizing supplemental oxygen to room air. He's following pulmonology for surveillance of respiratory function secondary to thoracic dysplasia, urology for undescended testes and hydronephrosis, and genetics. Furthermore, the GI team is transitioning him to bolus feeding using a gastrostomy tube. He is 20-month-old and seems to be reassuring so far.
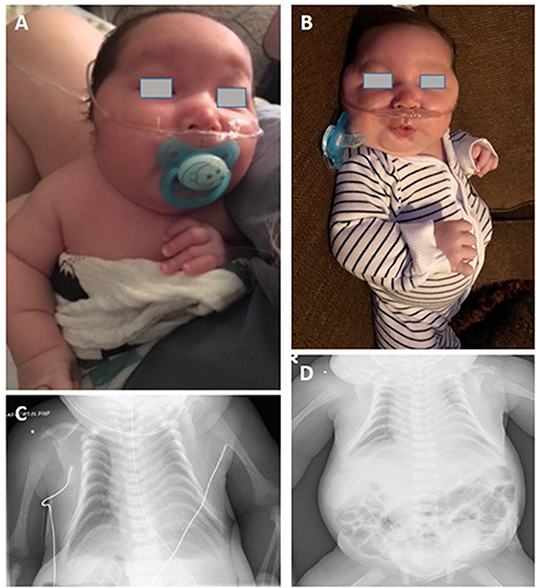
Figure 1. (A,B) Photograph of the patient. (C) Chest radiography of the patient. Thoracic dysplasia (bell-shaped small thorax with coat-hanger appearance). (D) Anterior abdominal wall defect with bowel herniation consistent with history of omphalocele. (The written informed consent was obtained from the parents of the patient for the publication of this image).
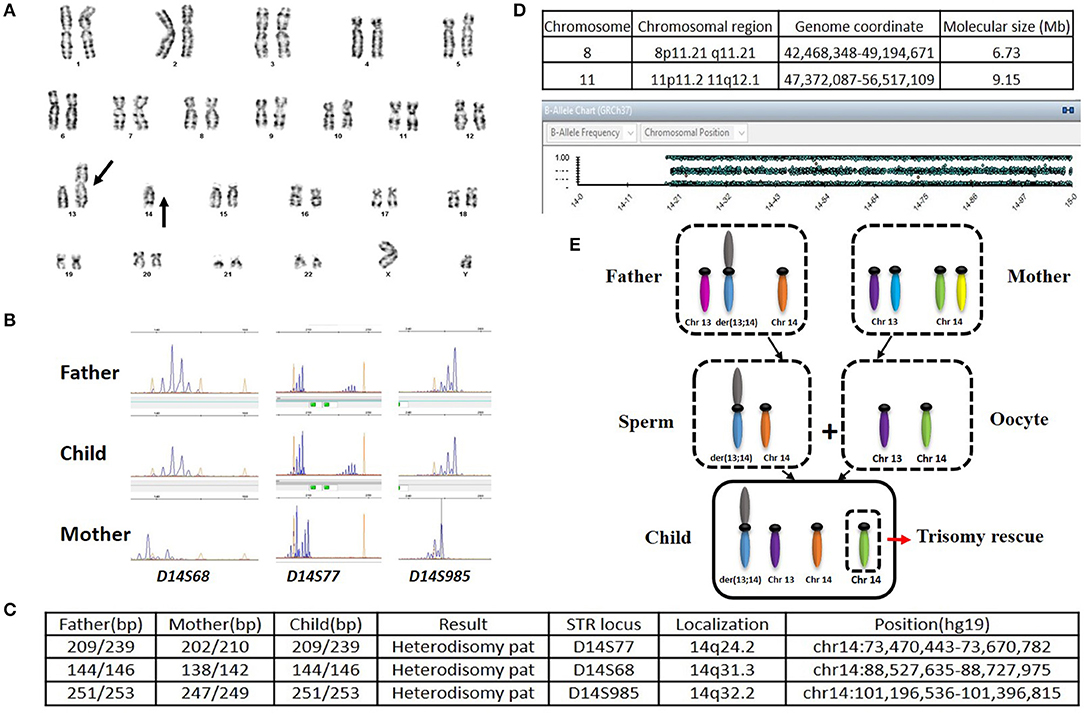
Figure 2. (A) Karyotype of the patient. (B) Microsatellite analysis. Maternally and paternally derived peaks are showed, respectively. (C) The short tandem repeats (STRs) on the long arm of chromosome 14 analyzed along with their position (hg19 map). (D) Result of SNP-Array. Homozygous regions in chromosome 8 and 11 were identified (Top); No homozygous region in chromosome 14 was detected (Bottom). (E) Schematic representation of the trisomy rescue (TR) mechanism leading to the heterodisomy upd(14)pat. A nondisjunction event between two chromosomes 14 occurred when a male ROB carrier produce a sperm. The segregation of the chromatids determined the formation of sperm with a ROB and a free chromosome 14. Oocyte has one of each normal chromosome 13 and 14. Then, after fertilization, there would be three chromosome 14 in the zygote. In order to avoid cell death of the three chromosome 14, the normal chromosome 14 from the mother will be excluded through the process of post-zygotic TR, which could result in paternal heterodisomy in the early embryo.
Among all the published KOS cases, more than 60% were caused by upd(14)pat, nearly 25% due to by microdeletions of chromosome 14q32 imprinted region, about 10% derived from epimutations (6, 11). The imprinted region on chromosome 14q32 harbor paternally expressed genes such as DLK1 and RTL1, and maternally expressed genes including MEG3, RTL1as (RTL1 antisense) and MEG8. Intergenic differentially methylated region (IG-DMR) between DLK1 and MEG3 as well as the post fertilization-derived secondary MEG3-DMR are of great importance for controlling imprinting (12, 13). The abnormalities of KOS caused by imprinting defects are due to abnormally expressed dosages of imprinted genes (overexpression of DLK1 and RTL1 as well as underexpression of MEGs). In our and not in this case, the abnormalities are probably originated from the overexpression of genes on the paternally inherited allele. Although phenotypes are comparable among patients with different genetic etiologies, placentomegaly or polyhydramnios may not present among KOS patients with microdeletion (13). Altogether, about 20% of the patients harboring UPD along with chromosome structural anomalies (acrocentric isochromosomes, ROB, marker chromosomes, derivative chromosomes, reciprocal translocations, etc.) displayed non-homologous ROB (14). The potential mechanisms leading to UPD include trisomy rescue (TR), gamete complementation (GC), monosomy rescue (MR), and post-fertilization mitotic error (PE) (15). The underlying mechanism of the strong correlation between ROB and hetero-upd(14)pat in our case is considered to be TR (Figure 2E). Interestingly, the numbers of the particular cases with upd(14)pat harboring ROB are relatively small in the current collection of surveys. Only eleven cases, including this one, have been reported; and most of the patients were female (14, 16–24). To provide a better understanding of KOS cases with upd(14)pat with ROB in particular, we review the published articles and the results are summary in Table 1. Furthermore, among the eight cases with detailed clinical information and karyotypes from both the patient and the parents, three cases present paternally derived ROB (16, 18) and our case is the only male case. The other male case was reported by Walter et al. (22) in 1996. The patient had similar symptoms to our case. It is not clear why the proportion of male patients is low. It is possible that it is due to a bias because of the low numbers. We need to accumulate more cases' information and improve the relevant examinations in order to better understand such disease.
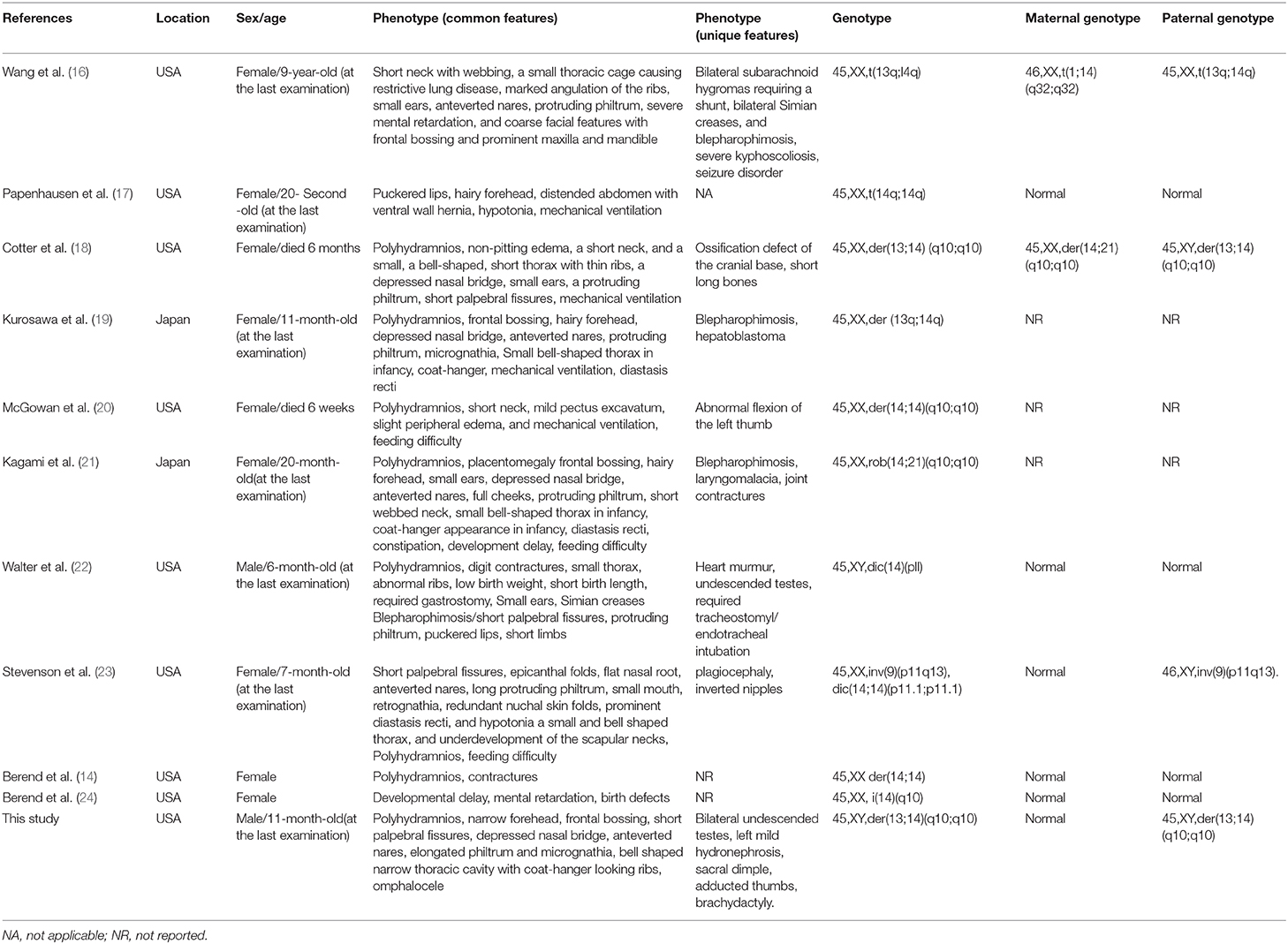
Table 1. Published case reports of upd(14)pat with Robertsonian translocation.
This study provides a new male case of KOS caused by upd(14)pat with paternally inherited ROB. For neonates with characteristic clinical features and ROB, further parental study and molecular studies for upd(14)pat should be considered. KOS due to upd(14)pat and ROB is rare, and cytogenetic and molecular studies can help for the diagnosis.
All datasets generated for this study are included in the article/supplementary material.
The studies involving human participants were reviewed and approved by Oklahoma University Health Science Center. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Written informed consent was obtained from the patient's parents for publication of this case report and the accompanying images.
XW: wrote the manuscript. HP and HG: responsible for the data analysis. BS: cared for the patient and provided clinical history. LZ: responsible for the revisions. HW: provided genetics consultation service and follow up of the patient, clinical data collection, and responsible for the revisions.
This study received financial support from the National Natural Science Foundation of China (NSFC, 81900153).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the patient's family for their cooperation in this study. We thank Shibo Li for scientific support and language modification. We thank Xianglan Lu for helping with karyotyping. We thank Jeremy K. Jinkins for technical assistance in UPD test. We thank Xianfu Wang for technical assistance in whole genome SNP-array analysis.
1. Eggermann T, Zerres K. Uniparental disomy and Robertsonian translocations: risk estimation and prenatal testing. Mol Diagn. (2003) 7:113–7. doi: 10.2165/00066982-200307020-00004
2. Kalish JM, Jiang C, Bartolomei MS. Epigenetics and imprinting in human disease. Int J Dev Biol. (2014) 58:291–8. doi: 10.1387/ijdb.140077mb
3. Peters J. The role of genomic imprinting in biology and disease: an expanding view. Nat Rev Genet. (2014) 15:517–30. doi: 10.1038/nrg3766
4. Mackay DJG, Temple IK. Human imprinting disorders: principles, practice, problems and progress. Eur J Med Genet. (2017) 60:618–26. doi: 10.1016/j.ejmg.2017.08.014
5. Giabicani E, Brioude F, Le Bouc Y, Netchine I. Imprinted disorders and growth. Ann Endocrinol. (2017) 78:112–3. doi: 10.1016/j.ando.2017.04.010
6. Jung HS, Vallee SE, Dinulos MB, Tsongalis GJ, Lefferts JA. Maternally inherited 133kb deletion of 14q32 causing Kagami-Ogata syndrome. J Hum Genet. (2018) 63:1231–9. doi: 10.1038/s10038-018-0506-z
7. Luk HM. Familial Kagami-Ogata syndrome in Chinese. Clin Dysmorphol. (2017) 26:124–7. doi: 10.1097/MCD.0000000000000158
8. Yip MY. Uniparental disomy in Robertsonian translocations: strategies for uniparental disomy testing. Transl Pediatr. (2014) 3:98–107. doi: 10.3978/j.issn.2224-4336.2014.03.03
9. Shaffer LG. Risk estimates for uniparental disomy following prenatal detection of a nonhomologous Robertsonian translocation. Prenat Diagn. (2006) 26:303–7. doi: 10.1002/pd.1384
10. Pilip L, Spinenko LA, Zukin VD, Bil'ko NM. [Meiotic segregation of chromosomes 13 and 14 of heterozygous Robertsonian translocation der(13;14) (q10; q10) carriers]. Tsitol Genet. (2014) 48:48–53.
11. van der Werf IM, Buiting K, Czeschik C, Reyniers E, Vandeweyer G, Vanhaesebrouck P, et al. Novel microdeletions on chromosome 14q32.2 suggest a potential role for non-coding RNAs in Kagami-Ogata syndrome. Eur J Hum Genet. (2016) 24:1724–29. doi: 10.1038/ejhg.2016.82
12. Kagami M, Matsubara K, Nakabayashi K, Nakamura A, Sano S, Okamura K, et al. Genome-wide multilocus imprinting disturbance analysis in Temple syndrome and Kagami-Ogata syndrome. Genet Med. (2017) 19:476–82. doi: 10.1038/gim.2016.123
13. Ogata T, Kagami M. Kagami-Ogata syndrome: a clinically recognizable upd(14)pat and related disorder affecting the chromosome 14q32.2 imprinted region. J Hum Genet. (2016) 61:87–94. doi: 10.1038/jhg.2015.113
14. Berend SA, Horwitz J, McCaskill C, Shaffer LG. Identification of uniparental disomy following prenatal detection of Robertsonian translocations and isochromosomes. Am J Hum Genet. (2000) 66:1787–93. doi: 10.1086/302916
15. Sensi A, Cavani S, Villa N, Pomponi MG, Fogli A, Gualandi F, et al. Nonhomologous Robertsonian translocations (NHRTs) and uniparental disomy (UPD) risk: an Italian multicentric prenatal survey. Prenat Diagn. (2004) 24:647–52. doi: 10.1002/pd.962
16. Wang JC, Passage MB, Yen PH, Shapiro LJ, Mohandas TK. Uniparental heterodisomy for chromosome 14 in a phenotypically abnormal familial balanced 13/14 Robertsonian translocation carrier. Am J Hum Genet. (1991) 48:1069–74.
17. Papenhausen PR, Mueller OT, Johnson VP, Sutcliffe M, Diamond TM, Kousseff BG. Uniparental isodisomy of chromosome 14 in two cases: an abnormal child and a normal adult. Am J Med Genet. (1995) 59:271–5. doi: 10.1002/ajmg.1320590302
18. Cotter PD, Kaffe S, McCurdy LD, Jhaveri M, Willner JP, Hirschhorn K. Paternal uniparental disomy for chromosome 14: a case report and review. Am J Med Genet. (1997) 70:74–9. doi: 10.1002/(SICI)1096-8628(19970502)70:1<74::AID-AJMG14>3.0.CO;2-U
19. Kurosawa K, Sasaki H, Sato Y, Yamanaka M, Shimizu M, Ito Y, et al. Paternal UPD14 is responsible for a distinctive malformation complex. Am J Med Genet. (2002) 110:268–72. doi: 10.1002/ajmg.10404
20. McGowan KD, Weiser JJ, Horwitz J, Berend SA, McCaskill C, Sutton VR, et al. The importance of investigating for uniparental disomy in prenatally identified balanced acrocentric rearrangements. Prenat Diagn. (2002) 22:141–3. doi: 10.1002/pd.279
21. Kagami M, Kurosawa K, Miyazaki O, Ishino F, Matsuoka K, Ogata T. Comprehensive clinical studies in 34 patients with molecularly defined UPD(14)pat and related conditions (Kagami-Ogata syndrome). Eur J Hum Genet. (2015) 23:1488–98. doi: 10.1038/ejhg.2015.13
22. Walter CA, Shaffer LG, Kaye CI, Huff RW, Ghidoni PD, McCaskill C, et al. Short-limb dwarfism and hypertrophic cardiomyopathy in a patient with paternal isodisomy 14:45,XY,idic(14)(p11). Am J Med Genet. (1996) 65:259–65. doi: 10.1002/(SICI)1096-8628(19961111)65:4<259::AID-AJMG2>3.0.CO;2-K
23. Stevenson DA, Brothman AR, Chen Z, Bayrak-Toydemir P, Longo N. Paternal uniparental disomy of chromosome 14: confirmation of a clinically-recognizable phenotype. Am J Med Genet A. (2004) 130A:88–91. doi: 10.1002/ajmg.a.30200
Keywords: uniparental disomy, Kagami–Ogata syndrome, imprinting disorders, Robertsonian translocation, heterodisomy
Citation: Wang X, Pang H, Shah BA, Gu H, Zhang L and Wang H (2020) A Male Case of Kagami-Ogata Syndrome Caused by Paternal Unipaternal Disomy 14 as a Result of a Robertsonian Translocation. Front. Pediatr. 8:88. doi: 10.3389/fped.2020.00088
Received: 25 November 2019; Accepted: 21 February 2020;
Published: 10 March 2020.
Edited by:
Enrico Baruffini, University of Parma, ItalyReviewed by:
Arturo Hernandez, Maine Medical Center, United StatesCopyright © 2020 Wang, Pang, Shah, Gu, Zhang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lijun Zhang, bHpoYW5nMjAyMDAzQDE2My5jb20=; Hua Wang, SHVhd2FuZ0BsbHUuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.