 Kai Lehmberg
Kai Lehmberg Despina Moshous
Despina Moshous Claire Booth3,4
Claire Booth3,4- 1Division of Paediatric Stem Cell Transplantation and Immunology, University Medical Centre Hamburg Eppendorf, Hamburg, Germany
- 2Department of Immunohematology, Necker-Enfants Malades Hospital, APHP, and Imagine Institute, Inserm U 1163, Descartes University, Paris Sorbonne Cité, Paris, France
- 3Department of Paediatric Immunology, Great Ormond Street Hospital, London, United Kingdom
- 4Molecular and Cellular Immunology Section, UCL Great Ormond Street Institute of Child Health, London, United Kingdom
Haematopoietic stem cell transplantation currently remains the only curative treatment of primary forms of haemophagocytic lymphohistiocytosis (HLH). Rapid diagnosis, efficient primary treatment of hyperinflammation, and conditioning regimens tailored to this demanding condition have substantially improved prognosis in the past 40 years. However, refractory hyperinflammation, central nervous system (CNS) involvement, unavailability of matched donors, susceptibility to conditioning-related toxicities, and a high frequency of mixed chimaerism remain a challenge in a substantial proportion of patients. Gene therapeutic approaches for several genetic defects of primary HLH are being developed at pre-clinical and translational levels.
Key Messages
• Remission at HSCT is a key factor of survival in HLH patients. To avoid reactivations, time to transplant should be kept at a minimum, which may require alternative donors.
• The high prevalence of fatal veno-occlusive disease after full myeloablative busulfan-based conditioning in HLH patients has been substantially curtailed with conditioning regimens of reduced toxicity.
• Achieving high levels of sustainable donor chimerism in the absence of GvHD remains a challenge in HLH patients when using RIC regimens.
Introduction
Haemophagocytic lymphohistiocytosis (HLH) is a hyperinflammatory condition that may occur in a primary or secondary form. The clinical picture is characterized by the triad of fever, splenomegaly, and symptoms of cytopenia. In combination with laboratory and cytological findings, they form a set of defining criteria (HLH-2004). Untreated full scale primary HLH is usually fatal (1).
Genetic defects predisposing to primary HLH are found in autosomal recessive familial HLH (FHL) 2-5, Griscelli syndrome type 2 (GS2), Chediak-Higashi syndrome (CHS), X-linked lymphoproliferative disease type 1 (XLP1), and X-linked inhibitor of apoptosis (XIAP) deficiency. In primary HLH, defective cytotoxic function of CD8+ T cells and NK cells lead to excessive secretion of numerous inflammatory cytokines that can be made responsible for different clinical features of HLH (2). However, other immunodeficiencies (e.g., chronic granulomatous disease) display a predisposition to HLH as well (3). Pathogenesis of secondary HLH, associated with infectious, malignant, autoimmune, autoinflammatory, or metabolic diseases, is less well-elucidated.
Taking into account the rarity of primary HLH and the retrospective nature of the majority of published studies on HSCT in this condition, data summarized in this review must be interpreted with caution.
Pre-HSCT Treatment
Treatment of HLH aims at decreasing inflammation and treating rigorously the underlying trigger, if any. The principal goal of induction therapy is to suppress the life-threatening inflammatory process. Once remission of HLH is achieved, patients with primary HLH require allogeneic haematopoietic stem cell transplantation (HSCT), the only curative therapy to date preventing relapses and central nervous system (CNS) disease progression. Isolated CNS-HLH remains a challenge for diagnosis and treatment. The quality of remission prior to HSCT is an important survival factor, especially in patients who lack HLA identical donors (4, 5). Despite significant treatment progress, mortality remains high. The etoposide-based treatment induction in the HLH-94 (dexamethasone, etoposide, intrathecal methotrexate) and HLH-2004 studies (addition of cyclosporine A upfront) resulted in 20–30% mortality before HSCT and 5-year overall survival (including HSCT) of ~60% (6, 7). Etoposide-based regimens may be associated with toxicities contributing to pre HSCT mortality, including bone marrow suppression and infection, as well as secondary malignancy.
A better understanding of the pathophysiology of primary HLH has opened new avenues for targeted immunotherapy. Based on previous observations showing the efficacy of immunotherapy directed selectively against T cells by using anti-thymocyte globulin (ATG) in FHL patients (8), the anti-CD52 antibody alemtuzumab has been used efficaciously in refractory HLH (9). A prospective trial with alemtuzumab as first line treatment in primary HLH is currently ongoing (NCT02472054).
New treatment options are emerging in cytokine-driven immune disorders. In fact, many of the elevated cytokines in HLH signal via Janus Kinase (JAK) and Signal Transducer and Activator of Transcription (STAT)-associated receptors, therefore JAK1/2 have become targets for therapeutic intervention. Ruxolitinib, a JAK1/2, inhibitor has shown efficacy in mouse models of HLH (10, 11); it has also been used in humans in isolated cases of refractory primary and secondary HLH (12–14). The role of interferon (IFN)-γ in the development of HLH has led to the development of the anti-IFN-γ monoclonal antibody emapalumab which proved efficient in mice (15, 16), clinical studies in HLH patients are ongoing (NCT03312751).
Conditioning Regimens and Adverse Events
HSCT with myeloablative conditioning regimens containing busulfan, cyclophosphamide, and VP16 resulted in substantial transplantation related mortality (overall survival 43–64%), in particular related to veno-occlusive disease (VOD) (4, 5, 17, 18). The advent of conditioning regimens with reduced toxicity based on melphalan as alkylating agent in combination with fludarabine led to a substantial improvement of survival with a favorable toxicity profile and a very low rate of lethal VOD (18–20). Treosulfan-based schemes with thiotepa rendered similar results (21). Mostly retrospectively determined survival rates in patients with HLH have been reported at 44–100% (21–25) for treosulfan-based protocols and 51–92% (18, 23, 26–28) for melphalan-based regimens (Table 1). Whether sub-myeloablative busulfan will constitute an alternative approach remains to be demonstrated.
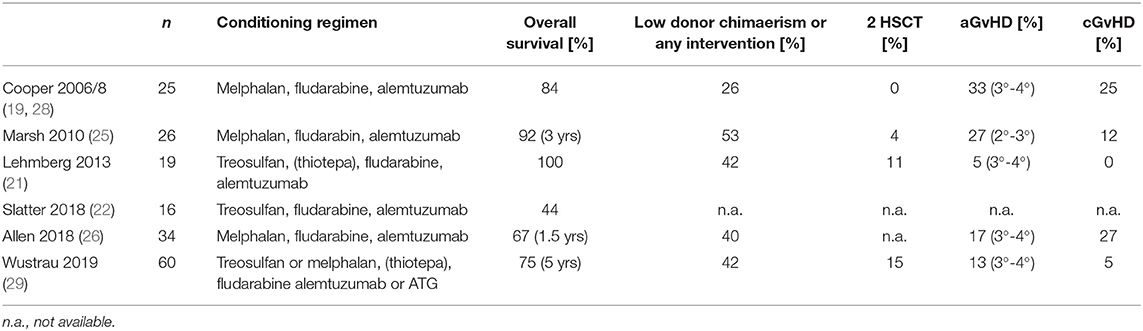
Table 1. Compilation of selected publications on HSCT for HLH after conditioning regimens with reduced toxicity.
It is challenging to tailor conditioning and serotherapy achieving high levels of donor chimerism in the absence of graft-versus-host disease (GvHD), particularly in mismatched donors (21). A donor chimerism of ≥20–30% has been shown to protect against disease reactivation; and even lower rates do not inevitably result in recurrence of HLH (30). However, the dynamics of incipient appearance of recipient cells is poorly predictable. Studies and case series using treosulfan or melphalan in HLH patients report 30–100% of patients with mixed chimerism and frequent necessity for secondary cellular cell therapy, including donor lymphocyte infusions and secondary HSCT (18, 22, 26, 27).
A prospective study reported a chronic GvHD rate of 27% (26). Serotherapy has a dual role in HLH patients, as it not only prevents GvHD and rejection but equally dampens residual disease activity. ATG and alemtuzumab have been reported as lymphodepleting agents in larger series. In a retrospective analysis of 60 patients, no significant difference regarding the occurrence of mixed chimerism between both agents could be detected (29). A “distal” administration of serotherapy reduces the risk of mixed chimerism, however leads to an elevated rate of graft-versus-host disease (GvHD) (31).
Alternative Donors
In primary HLH, allogeneic hematopoietic stem cell transplantation is the only curative treatment option and should ideally be offered as soon as remission from the activation syndrome is achieved. Especially in patients lacking HLA identical donors, the quality of remission is essential to improve survival rates (4, 5). Alternative donors should therefore be considered early. Unrelated umbilical cord blood transplantation may provide a readily available alternative donor source, and 6-year overall survival of 55% has been reported in a recent analysis of 118 patients receiving mostly busulfan-based conditioning (32).
Haploidentical parental donors provide the opportunity for virtually all patients to benefit from HSCT. However, T-lymphocyte depleted haplo-identical transplantation is associated with high risk of graft rejection when omitting cyclophosphamide and using busulfan/fludarabine or treosulfan/fludarabine in the conditioning regimen as currently recommended in the EBMT/ESID guidelines (33). T-lymphocyte replete HSCT with administration of cyclophosphamide post transplantation in order to reduce the incidence of GvHD while sparing other T-lymphocytes seems to be a valid option in primary HLH patients who lack a conventional donor. In a small series of 10 patients with primary HLH, treatment related mortality was 20% with one additional death in a patient who died from central line sepsis on parenteral nutrition 9 months post HSCT, 6 out these 10 patients have been already reported (34). The possibility of further reducing the conditioning intensity to decrease toxicity needs to be assessed.
Special Considerations
Although HSCT is a recognized curative treatment for primary HLH, certain conditions can prove more challenging, particularly when deciding if, or when, to proceed with transplant. It is well-established that patients entering HSCT with active disease have a worse outcome, and pre-emptive transplant is generally advocated in patients with proven genetic lesions and HLH associated with immune deficiency (e.g., XLP). Overall survival of second affected children in families with primary HLH was reported to be 93%, as compared to 64% in their index siblings (35).
HLH conditions associated with pigmentary disorders have variable risks of HLH and often significant neurological involvement. Patients with Griscelli syndrome (GS2) are at high risk of developing HLH but results of HSCT are favorable (36–38). Most series of GS2 patients report 40–50% significant neurological sequelae mostly related to CNS HLH (36) and families must be counseled appropriately. The risks of developing HLH are lower in Chediak-Higashi syndrome where decision to transplant should be based on genetic and cytotoxic function (39) and lower still in Hermansky-Pudlak syndrome type 2 where pre-emptive HSCT is not justified (40).
XIAP deficient patients can be particularly difficult to treat. HSCT related mortality in this patient population is worse than patients undergoing HSCT for other forms of familial HLH, even when using a reduced intensity conditioning regime (57% probability of survival at 1 year and 43% probability of long-term survival) (41). These patients may develop significant GvHD that can be resistant to therapy, contributing to morbidity and mortality (42). Interestingly a case series from Japan reported a 90% survival but 50% of patients developed inflammatory episodes post-HSCT. A high proportion of patients will manifest inflammatory enteropathy as part of their disease, and this can be slow to resolve post-HSCT.
Some patients with FHL5 due to mutations in STXBP2, may present severe, osmotic diarrhea, and renal proximal tubular dysfunction. These clinical manifestations are related to defective membrane trafficking in the gut and kidney and persist also after successful HSCT (43–45).
Although CNS involvement is present in around 30% of cases of systemic HLH (7), isolated CNS HLH is becoming increasingly recognized. Diagnosis and management can be challenging, and specialist advice must be sought early (46). Early intervention with CNS directed therapy appears key. HSCT is curative in these cases, but cannot revert manifest CNS damage (47).
Gene Therapy for HLH
Autologous HSC gene therapy (GT) has been developed for several primary immunodeficiencies and offers an efficacious alternative treatment option for patients lacking a suitable donor for HSCT (see review on “Autologous stem cell-based gene therapy for inherited disorders: state-of-the-art and future prospects”). Preclinical and translational studies of HSC GT are now underway for several forms of primary HLH including FHL2, FHL3, XLP, and XIAP deficiency. In addition, proof of concept for adoptive transfer of autologous gene corrected T-lymphocytes has now been published in three of these conditions (48); a therapeutic strategy, which may act as a bridge to transplant or perhaps offer longer-term clinical benefit.
Lentiviral transfer of the perforin gene into haematopoietic progenitors in a Prf −/− murine model led to the recovery of NK cell function, CD8 T-lymphocyte cytotoxicity and reduced IFN-ɤ secretion in vitro alongside improvement in cytopaenias and cytokine hypersecretion when gene corrected mice were challenged with lymphocytic choriomeningitis virus (LCMV) to induce an HLH phenotype (49). Subsequently, this group also demonstrated that gene correction of T-lymphocytes reduced disease activity and prevented HLH in vivo alongside restoring cytotoxicity in patient T-lymphocytes (48). Soheili et al. demonstrated that both an HSC and T-lymphocyte approach can ameliorate defects in FHL3 with again, reduction of HLH symptoms and biomarkers in an LCMV challenged Jinx mouse model following transfer of gene modified HSCs (50) and rescue of cytotoxicity in patient T-lymphocytes in a tumor model and degranulation assay (51). Dettmer et al. showed it was possible to efficiently transduce hyperactivated patients T-lymphocytes with a retroviral vector containing the UNC13D gene generating functionally corrected cells (52).
Preclinical studies have demonstrated proof of concept for HSC and T-lymphocyte gene therapy strategies for XLP with correction of immune abnormalities using both approaches. Sap−/y mice reconstituted with lentiviral mediated HSC gene correction showed recovery of NK cell cytotoxicity and humoral defects including immunoglobulin levels and T-lymphocyte dependent antigen responses (53). Transfer of gene corrected T-lymphocytes in Sap−/y mice restored T-lymphocyte dependent antigen responses and germinal center formation whilst gene transfer in patient T-lymphocytes led to functional recovery of T follicular helper lymphocytes and cytotoxicity (both in vitro and in vivo using a xenograft lymphoma model) (54). Preclinical studies are underway investigating HSC gene therapy as a possible therapeutic option for XIAP deficiency (unpublished data).
Conclusions and Outlook
Despite significant progress, front-line treatment and HSCT procedures still require improvement to further reduce mortality and long-term sequelae of this potentially devastating condition. New therapeutic agents may complement current standards of care, optimization of conditioning regimens may overcome remaining challenges, and gene therapy approaches will probably be available in the future for the most frequent HLH defects.
Author Contributions
All authors wrote the review and approved of the final version.
Funding
KL received a grant from Deutsche Kinderkrebsstiftung (DKS 2016.04, DKS 2018.04) for the HLH Registry of Histiocyte Society and European Society of Immunodeficiencies.
Conflict of Interest
KL is member of an advisory board of SOBI. CB has received consulting fees from SOBI and Novimmune.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor AG declared a current collaboration with the author CB.
Abbreviations
ATG, anti-thymocyte globulin; CHS, Chediak Higashi syndrome; CNS, central nervous system; EBMT, European Society of Bone Marrow Transplantation; ESID, European Society of Immunodeficiencies; FHL, familial HLH; GS2, Griscelli syndrome type 2; GT, gene therapy; GvHD, graft-versus-host disease; HLH, haemophagocytic lymphohistiocytosis; HSCT, haematopoietic stem cell transplantation; IFN, interferon; JAK, janus kinase; LCMV, lymphocytic choriomeningitis virus; STAT, Signal Transducer and Activator of Transcription; VOD, veno-occlusive disease; XIAP, x-linked inhibitor of apoptosis; XLP, x-linked lymphoproliferative disease.
References
1. Janka GE, Lehmberg K. Hemophagocytic syndromes–an update. Blood Rev. (2014) 28:135–42. doi: 10.1016/j.blre.2014.03.002
2. Humblet-Baron S, Franckaert D, Dooley J, Ailal F, Bousfiha A, Deswarte C, et al. IFN-gamma and CD25 drive distinct pathologic features during hemophagocytic lymphohistiocytosis. J Allergy Clin Immunol. (2019) 143:2215–26.e7. doi: 10.1016/j.jaci.2018.10.068
3. Bode SF, Ammann S, Al-Herz W, Bataneant M, Dvorak CC, Gehring S, et al. The syndrome of hemophagocytic lymphohistiocytosis in primary immunodeficiencies: implications for differential diagnosis and pathogenesis. Haematologica. (2015) 100:978–88. doi: 10.3324/haematol.2014.121608
4. Ouachee-Chardin M, Elie C, de Saint Basile G, Le Deist F, Mahlaoui N, Picard C, et al. Hematopoietic stem cell transplantation in hemophagocytic lymphohistiocytosis: a single-center report of 48 patients. Pediatrics. (2006) 117:e743–50. doi: 10.1542/peds.2005-1789
5. Horne A, Janka G, Maarten Egeler R, Gadner H, Imashuku S, Ladisch S, et al. Haematopoietic stem cell transplantation in haemophagocytic lymphohistiocytosis. Br J Haematol. (2005) 129:622–30. doi: 10.1111/j.1365-2141.2005.05501.x
6. Trottestam H, Horne A, Arico M, Egeler RM, Filipovich AH, Gadner H, et al. Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: long-term results of the HLH-94 treatment protocol. Blood. (2011) 118:4577–84. doi: 10.1182/blood-2011-06-356261
7. Bergsten E, Horne A, Arico M, Astigarraga I, Egeler RM, Filipovich AH, et al. Confirmed efficacy of etoposide and dexamethasone in HLH treatment: long-term results of the cooperative HLH-2004 study. Blood. (2017) 130:2728–38. doi: 10.1182/blood-2017-06-788349
8. Mahlaoui N, Ouachee-Chardin M, de Saint Basile G, Neven B, Picard C, Blanche S, et al. Immunotherapy of familial hemophagocytic lymphohistiocytosis with antithymocyte globulins: a single-center retrospective report of 38 patients. Pediatrics. (2007) 120:e622–8. doi: 10.1542/peds.2006-3164
9. Marsh RA, Allen CE, McClain KL, Weinstein JL, Kanter J, Skiles J, et al. Salvage therapy of refractory hemophagocytic lymphohistiocytosis with alemtuzumab. Pediatr Blood Cancer. (2013) 60:101–9. doi: 10.1002/pbc.24188
10. Das R, Guan P, Sprague L, Verbist K, Tedrick P, An QA, et al. Janus kinase inhibition lessens inflammation and ameliorates disease in murine models of hemophagocytic lymphohistiocytosis. Blood. (2016) 127:1666–75. doi: 10.1182/blood-2015-12-684399
11. Maschalidi S, Sepulveda FE, Garrigue A, Fischer A, de Saint Basile G. Therapeutic effect of JAK1/2 blockade on the manifestations of hemophagocytic lymphohistiocytosis in mice. Blood. (2016) 128:60–71. doi: 10.1182/blood-2016-02-700013
12. Broglie L, Pommert L, Rao S, Thakar M, Phelan R, Margolis D, et al. Ruxolitinib for treatment of refractory hemophagocytic lymphohistiocytosis. Blood Adv. (2017) 1:1533–6. doi: 10.1182/bloodadvances.2017007526
13. Sin JH, Zangardi ML. Ruxolitinib for secondary hemophagocytic lymphohistiocytosis: First case report. Hematol Oncol Stem Cell Ther. (2017) 12:166–70. doi: 10.1016/j.hemonc.2017.07.002
14. Slostad J, Hoversten P, Haddox CL, Cisak K, Paludo J, Tefferi A. Ruxolitinib as first-line treatment in secondary hemophagocytic lymphohistiocytosis: a single patient experience. Am J Hematol. (2018) 93:E47–9. doi: 10.1002/ajh.24971
15. Pachlopnik Schmid J, Ho CH, Chretien F, Lefebvre JM, Pivert G, Kosco-Vilbois M, et al. Neutralization of IFNgamma defeats haemophagocytosis in LCMV-infected perforin- and Rab27a-deficient mice. EMBO Mol Med. (2009) 1:112–24. doi: 10.1002/emmm.200900009
16. Jordan MB, Hildeman D, Kappler J, Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood. (2004) 104:735–43. doi: 10.1182/blood-2003-10-3413
17. Baker KS, Filipovich AH, Gross TG, Grossman WJ, Hale GA, Hayashi RJ, et al. Unrelated donor hematopoietic cell transplantation for hemophagocytic lymphohistiocytosis. Bone Marrow Transplant. (2008) 42:175–80. doi: 10.1038/bmt.2008.133
18. Marsh RA, Madden L, Kitchen BJ, Mody R, McClimon B, Jordan MB, et al. XIAP deficiency: a unique primary immunodeficiency best classified as X-linked familial hemophagocytic lymphohistiocytosis and not as X-linked lymphoproliferative disease. Blood. (2010) 116:1079–82. doi: 10.1182/blood-2010-01-256099
19. Cooper N, Rao K, Gilmour K, Hadad L, Adams S, Cale C, et al. Stem cell transplantation with reduced-intensity conditioning for hemophagocytic lymphohistiocytosis. Blood. (2006) 107:1233–6. doi: 10.1182/blood-2005-05-1819
20. Rao K, Adams S, Qasim W, Allwood Z, Worth A, Silva J, et al. Effect of stem cell source on long-term chimerism and event-free survival in children with primary immunodeficiency disorders after fludarabine and melphalan conditioning regimen. J Allergy Clin Immunol. (2016) 138:1152–60. doi: 10.1016/j.jaci.2016.01.053
21. Lehmberg K, Albert MH, Beier R, Beutel K, Gruhn B, Kroger N, et al. Treosulfan-based conditioning regimen for children and adolescents with hemophagocytic lymphohistiocytosis. Haematologica. (2014) 99:180–4. doi: 10.3324/haematol.2013.094730
22. Slatter MA, Rao K, Abd Hamid IJ, Nademi Z, Chiesa R, Elfeky R, et al. Treosulfan and fludarabine conditioning for hematopoietic stem cell transplantation in children with primary immunodeficiency: UK experience. Biol Blood Marrow Transplant. (2018) 24:529–36. doi: 10.1016/j.bbmt.2017.11.009
23. Messina C, Zecca M, Fagioli F, Rovelli A, Giardino S, Merli P, et al. Outcomes of children with hemophagocytic lymphohistiocytosis given allogeneic hematopoietic stem cell transplantation in Italy. Biol Blood Marrow Transplant. (2018) 24:1223–31. doi: 10.1016/j.bbmt.2018.01.022
24. Slatter MA, Boztug H, Potschger U, Sykora KW, Lankester A, Yaniv I, et al. Treosulfan-based conditioning regimens for allogeneic haematopoietic stem cell transplantation in children with non-malignant diseases. Bone Marrow Transplant. (2015) 50:1536–41. doi: 10.1038/bmt.2015.171
25. Marsh RA, Vaughn G, Kim MO, Li D, Jodele S, Joshi S, et al. Reduced-intensity conditioning significantly improves survival of patients with hemophagocytic lymphohistiocytosis undergoing allogeneic hematopoietic cell transplantation. Blood. (2010) 116:5824–31. doi: 10.1182/blood-2010-04-282392
26. Allen CE, Marsh R, Dawson P, Bollard CM, Shenoy S, Roehrs P, et al. Reduced-intensity conditioning for hematopoietic cell transplant for HLH and primary immune deficiencies. Blood. (2018) 132:1438–51. doi: 10.1182/blood-2018-01-828277
27. Oshrine BR, Olson TS, Bunin N. Mixed chimerism and graft loss in pediatric recipients of an alemtuzumab-based reduced-intensity conditioning regimen for non-malignant disease. Pediatr Blood Cancer. (2014) 61:1852–9. doi: 10.1002/pbc.25113
28. Cooper N, Rao K, Goulden N, Webb D, Amrolia P, Veys P. The use of reduced-intensity stem cell transplantation in haemophagocytic lymphohistiocytosis and Langerhans cell histiocytosis. Bone Marrow Transplant. (2008) 42(Suppl. 2):S47–50. doi: 10.1038/bmt.2008.283
29. Wustrau K. Risk factors for mixed chimerism after treosulfan or melphalan based stem cell transplantation in children with primary hemophagocytic lymphohistiocytosis. In: 35 Annual Meeting of the Histiocyte Society. Memphis, TN (2019).
30. Hartz B, Marsh R, Rao K, Henter JI, Jordan M, Filipovich L, et al. The minimum required level of donor chimerism in hereditary hemophagocytic lymphohistiocytosis. Blood. (2016) 127:3281–90. doi: 10.1182/blood-2015-12-684498
31. Marsh RA, Rao MB, Gefen A, Bellman D, Mehta PA, Khandelwal P, et al. Experience with alemtuzumab, fludarabine, and melphalan reduced-intensity conditioning hematopoietic cell transplantation in patients with nonmalignant diseases reveals good outcomes and that the risk of mixed chimerism depends on underlying disease, stem cell source, and alemtuzumab regimen. Biol Blood Marrow Transplant. (2015) 21:1460–70. doi: 10.1016/j.bbmt.2015.04.009
32. Furtado-Silva JM, Paviglianiti A, Ruggeri A, Boelens JJ, Veys P, Ahmari AA, et al. Risk factors affecting outcome of unrelated cord blood transplantation for children with familial haemophagocytic lymphohistiocytosis. Br J Haematol. (2019) 184:397–404. doi: 10.1111/bjh.15642
33. EBMT/ESID. Guidelines for Hematopoietic Stem Cell Transplantation for Primary Immunodeficiencies. (2017). Available online at: https://esid.org/layout/set/print/content/download/15402/422689/file/ESID%20EBMT%20HSCT%20Guidelines%202017.pdf
34. Neven B, Diana JS, Castelle M, Magnani A, Rosain J, Touzot F, et al. Haploidentical hematopoietic stem cell transplantation with post-transplant cyclophosphamide for primary immunodeficiencies and inherited disorders in children. Biol Blood Marrow Transplant. (2019) 25:1363–73. doi: 10.1016/j.bbmt.2019.03.009
35. Lucchini G, Marsh R, Gilmour K, Worth A, Nademi Z, Rao A, et al. Treatment dilemmas in asymptomatic children with primary hemophagocytic lymphohistiocytosis. Blood. (2018) 132:2088–96. doi: 10.1182/blood-2018-01-827485
36. Pachlopnik Schmid J, Moshous D, Boddaert N, Neven B, Dal Cortivo L, Tardieu M, et al. Hematopoietic stem cell transplantation in Griscelli syndrome type 2: a single-center report on 10 patients. Blood. (2009) 114:211–8. doi: 10.1182/blood-2009-02-207845
37. Al-Ahmari A, Al-Ghonaium A, Al-Mansoori M, Hawwari A, Eldali A, Ayas M, et al. Hematopoietic SCT in children with Griscelli syndrome: a single-center experience. Bone Marrow Transplant. (2010) 45:1294–9. doi: 10.1038/bmt.2009.358
38. Kuskonmaz B, Ayvaz D, Gokce M, Ozgur TT, Okur FV, Cetin M, et al. Hematopoietic stem cell transplantation in children with Griscelli syndrome: a single-center experience. Pediatr Transplant. (2017) 21:e13040. doi: 10.1111/petr.13040
39. Nagai K, Ochi F, Terui K, Maeda M, Ohga S, Kanegane H, et al. Clinical characteristics and outcomes of chediak-Higashi syndrome: a nationwide survey of Japan. Pediatr Blood Cancer. (2013) 60:1582–6. doi: 10.1002/pbc.24637
40. Jessen B, Bode SF, Ammann S, Chakravorty S, Davies G, Diestelhorst J, et al. The risk of hemophagocytic lymphohistiocytosis in Hermansky-Pudlak syndrome type 2. Blood. (2013) 121:2943–51. doi: 10.1182/blood-2012-10-463166
41. Marsh RA, Rao K, Satwani P, Lehmberg K, Muller I, Li D, et al. Allogeneic hematopoietic cell transplantation for XIAP deficiency: an international survey reveals poor outcomes. Blood. (2013) 121:877–83. doi: 10.1182/blood-2012-06-432500
42. Muller N, Fischer JC, Yabal M, Haas T, Poeck H, Jost PJ. XIAP deficiency in hematopoietic recipient cells drives donor T-cell activation and GvHD in mice. Eur J Immunol. (2019) 49:504–7. doi: 10.1002/eji.201847818
43. Mosa MH, Nicolle O, Maschalidi S, Sepulveda FE, Bidaud-Meynard A, Menche C, et al. Dynamic formation of microvillus inclusions during enterocyte differentiation in Munc18-2-deficient intestinal organoids. Cell Mol Gastroenterol Hepatol. (2018) 6:477–93.e1. doi: 10.1016/j.jcmgh.2018.08.001
44. Vogel GF, van Rijn JM, Krainer IM, Janecke AR, Posovszky C, Cohen M, et al. Disrupted apical exocytosis of cargo vesicles causes enteropathy in FHL5 patients with Munc18-2 mutations. JCI Insight. (2017) 2:e94564. doi: 10.1172/jci.insight.94564
45. Stepensky P, Bartram J, Barth TF, Lehmberg K, Walther P, Amann K, et al. Persistent defective membrane trafficking in epithelial cells of patients with familial hemophagocytic lymphohistiocytosis type 5 due to STXBP2/MUNC18-2 mutations. Pediatr Blood Cancer. (2013) 60:1215–22. doi: 10.1002/pbc.24475
46. Horne A, Wickstrom R, Jordan MB, Yeh EA, Naqvi A, Henter JI, et al. How to treat involvement of the central nervous system in hemophagocytic lymphohistiocytosis? Curr Treat Options Neurol. (2017) 19:3. doi: 10.1007/s11940-017-0439-4
47. Benson LA, Li H, Henderson LA, Solomon IH, Soldatos A, Murphy J, et al. Pediatric CNS-isolated hemophagocytic lymphohistiocytosis. Neurol Neuroimmunol Neuroinflamm. (2019) 6:e560. doi: 10.1212/NXI.0000000000000560
48. Ghosh S, Carmo M, Calero-Garcia M, Ricciardelli I, Bustamante Ogando JC, Blundell MP, et al. T-cell gene therapy for perforin deficiency corrects cytotoxicity defects and prevents hemophagocytic lymphohistiocytosis manifestations. J Allergy Clin Immunol. (2018) 142:904–13.e3. doi: 10.1016/j.jaci.2017.11.050
49. Carmo M, Risma KA, Arumugam P, Tiwari S, Hontz AE, Montiel CA, et al. Perforin gene transfer into hematopoietic stem cells improves immune dysregulation in murine models of perforin deficiency. Mol Ther. (2015) 23:737–45. doi: 10.1038/mt.2014.242
50. Soheili T, Durand A, Sepulveda FE, Riviere J, Lagresle-Peyrou C, Sadek H, et al. Gene transfer into hematopoietic stem cells reduces HLH manifestations in a murine model of Munc13-4 deficiency. Blood Adv. (2017) 1:2781–9. doi: 10.1182/bloodadvances.2017012088
51. Soheili T, Riviere J, Ricciardelli I, Durand A, Verhoeyen E, Derrien AC, et al. Gene-corrected human Munc13-4-deficient CD8+ T cells can efficiently restrict EBV-driven lymphoproliferation in immunodeficient mice. Blood. (2016) 128:2859–62. doi: 10.1182/blood-2016-07-729871
52. Dettmer V, Bloom K, Gross M, Weissert K, Aichele P, Ehl S, et al. Retroviral UNC13D gene transfer restores cytotoxic activity of T cells derived from familial hemophagocytic lymphohistiocytosis Type 3 patients in vitro. Hum Gene Ther. (2019) 30:975–84. doi: 10.1089/hum.2019.025
53. Rivat C, Booth C, Alonso-Ferrero M, Blundell M, Sebire NJ, Thrasher AJ, et al. SAP gene transfer restores cellular and humoral immune function in a murine model of X-linked lymphoproliferative disease. Blood. (2013) 121:1073–6. doi: 10.1182/blood-2012-07-445858
Keywords: haemophagocytic lymhohistiocytosis, macrophage activation syndrome, haematopoietic stem cell transplantation, gene therapy, veno-occlusive disease, mixed chimerism, reduced toxicity conditioning
Citation: Lehmberg K, Moshous D and Booth C (2019) Haematopoietic Stem Cell Transplantation for Primary Haemophagocytic Lymphohistiocytosis. Front. Pediatr. 7:435. doi: 10.3389/fped.2019.00435
Received: 10 July 2019; Accepted: 07 October 2019;
Published: 25 October 2019.
Edited by:
Andrew R. Gennery, Newcastle University, United KingdomReviewed by:
Mario Abinun, Newcastle upon Tyne Hospitals NHS Foundation Trust, United KingdomPietro Merli, Bambino Gesù Children Hospital (IRCCS), Italy
Copyright © 2019 Lehmberg, Moshous and Booth. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kai Lehmberg, ay5sZWhtYmVyZ0B1a2UuZGU=