 Emily Y. Tan1
Emily Y. Tan1 Jaap Jan Boelens2
Jaap Jan Boelens2 Simon A. Jones3
Simon A. Jones3 Robert F. Wynn3* on behalf of the Inborn Errors Working Party of the EBMT
Robert F. Wynn3* on behalf of the Inborn Errors Working Party of the EBMT- 1Faculty of Health and Medical Sciences, University of Western Australia, Perth, WA, Australia
- 2Stem Cell Transplant and Cellular Therapies, Memorial Sloan Kettering Cancer Center, New York, NY, United States
- 3Metabolic and Blood and Marrow Transplant Units, Royal Manchester Children's Hospital, Manchester, United Kingdom
Hematopoietic stem cell transplantation (HSCT) has been established as an effective therapy for selected inborn errors of metabolism. The success of HSCT in metabolic disease is best exemplified through the treatment of Hurler's syndrome, a lysosomal storage disease. Through the collaborative effort of several international centers, factors that predict successful patient and transplant outcomes have been identified. In this review, we discuss the principles that underlie the use of HSCT in metabolic diseases. We consider the clinical indications, conditioning regimens, and disease-specific follow-up for HSCT in different metabolic diseases. We highlight persisting challenges in HSCT to delay progression of certain organ systems that remain refractory to HSCT and the relatively high rates of aplastic graft failure. Finally, we evaluate the variable applicability of these principles to other inherited metabolic disorders including peroxisomal, mitochondrial, and other lysosomal storage diseases.
Key Messages
1. Hematopoietic Stem Cell Transplantation (HSCT) for metabolic diseases is specialized medicine. It requires multidisciplinary management and continuous collaboration with other specialties and allied health professionals before, during and beyond the period of transplantation.
2. HSCT is better at preventing disease progression than in reversing already established disease manifestations.
3. Early intervention with HSCT is associated with better outcomes.
4. Transplant outcomes should be distinguished from disease outcomes in metabolic disease, and much progress has been made in recent years from careful, multi-center and collaborative analysis of the factors that separately influence each.
5. Engraftment rates are improved with pharmacokinetic analysis-guided, myeloablative busulfan without ex-vivo T-cell depletion.
6. Autologous HSCT using gene therapy may provide an improved treatment option for inherited metabolic diseases in the future, both by reducing allogeneic treatment-related toxicities, and by improving efficacy through augmented graft enzyme delivery.
Introduction
Inherited metabolic disorders comprise a large, diverse, and complex group of diseases caused by defects in genes that code for proteins involved in metabolic pathways. HSCT is an option and even standard of care for specific metabolic diseases, where other available therapies are less effective and where the benefit of HSCT outweighs the risk of a transplant. This chapter will serially discuss the use of HSCT in certain lysosomal storage and peroxisomal diseases where HSCT is standard of care. Furthermore, it will discuss its conditional role in other metabolic disease including mitochondrial disease (Table 1).
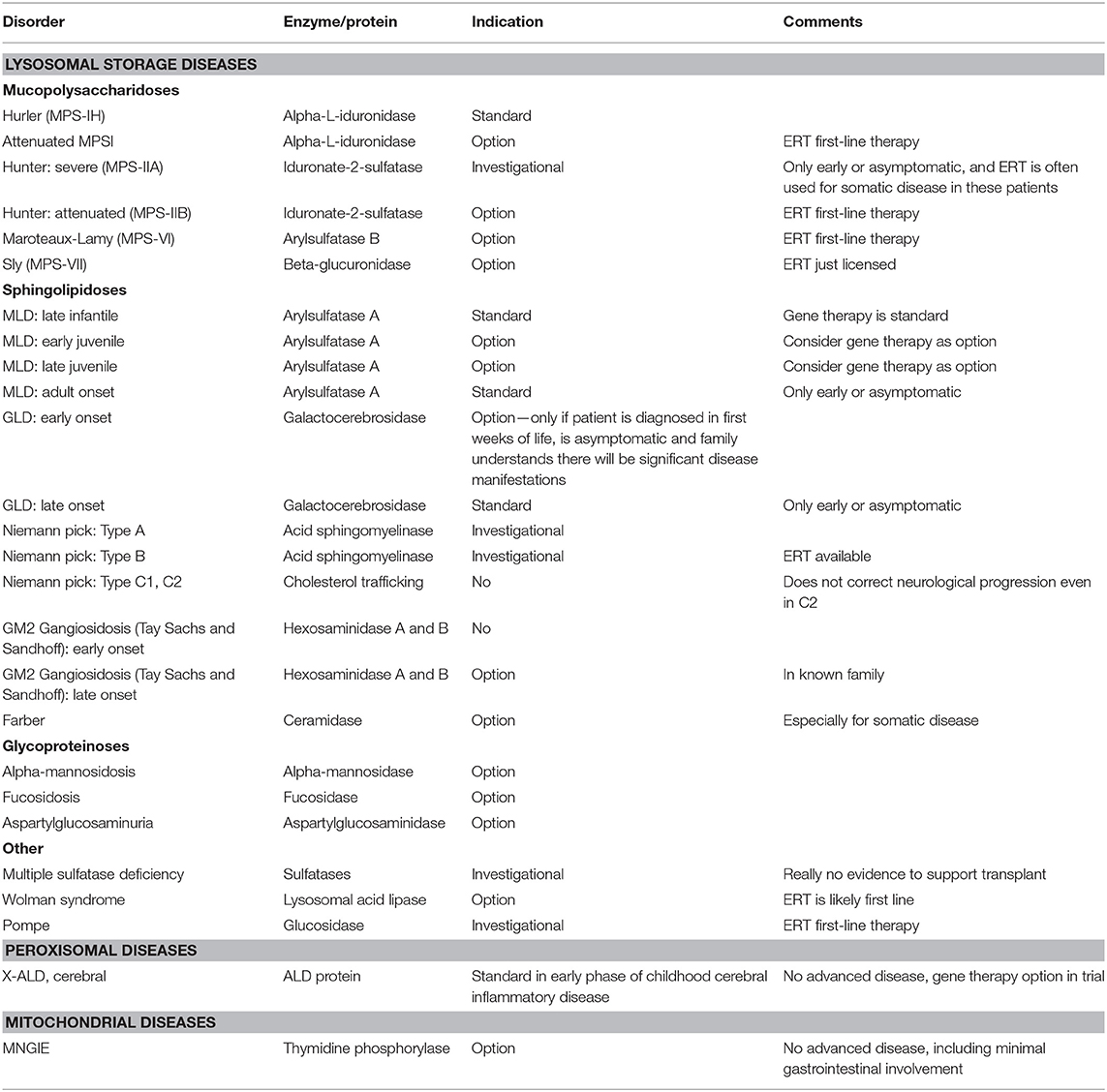
Table 1. Inherited metabolic disorders where HSCT may be indicated.
Lysosomal Storage Diseases
Pathophysiology
The Lysosomal Storage Diseases (LSDs) encompass over 70 diseases, which comprise genetic defects in specific lysosomal proteins. In the past decade, the function of lysosomes has extended beyond their involvement in degradation and recycling of extracellular and intracellular material. They play a crucial role in plasma membrane repair, lipid and metabolite exchange between organelles and have recently been found to regulate energy metabolism via calcium signaling (1–3). Understandably, genetic defects in lysosomal proteins involved in any of these microdomains can have broad functional consequences. The pathogenesis of cellular injury is not fully understood but stems from the primary accumulation of undigested substrates within lysosomes and subsequent downstream pathology (4).
Mucopolysaccharidosis Type 1 (MPS I) is an autosomal recessive LSD characterized by lysosomal accumulation of mucopolysaccharides or glycosaminoglycans (GAGs) (5). In MPS I, patients have a defective mutation in the IDUA gene which codes for alpha-L-iduronidase, resulting in ineffective catabolism of heparan and dermatan sulfate (5). Accumulation and subsequent deposition of these GAGs in vital organs causes significant multiorgan dysfunction. This can manifest as progressive mental retardation, skeletal deformities, gastrointestinal pathology, and visual and auditory impairment (6). The clinical severity of MPS I is observed across a vast spectrum. MPS IH, or Hurler's Syndrome, is the more severe phenotype of MPS I where patients have an early-onset, rapidly progressive disease with neurological involvement. In untreated children with MPS IH, death is usual in the first decade of life, often from cardiac or respiratory complications (7, 8).
Indication for HSCT
LSDs require early intervention and multi-disciplinary management to optimize treatment response, quality of life and prevent premature mortality. The principle of HSCT in LSDs is in cross-correction. HSCT provides the recipient with a continuous source of enzyme produced by donor-derived myeloid cells, which are then taken up by enzyme-deficient host cells (9). Furthermore, the superiority of HSCT to enzyme replacement therapy (ERT) lies in its exploitation of donor-derived cells to migrate across the blood brain barrier and differentiate into tissue macrophages, known as microglia, which secrete the deficient enzyme to the central nervous system, improving neurocognitive outcomes (10). MPS IH is the paradigm of successful HSCT in metabolic disease. HSCT is the gold-standard treatment option for MPS IH patients who are younger than 2 years of age who have no or minimal cognitive impairment (11). Currently available ERT is ineffective in preventing cognitive decline as it is unable to cross the blood brain barrier in sufficient doses and long-term therapy with ERT is limited by the induction of anti-enzyme antibodies, diminishing substrate reduction (10, 12–14).
Approach to HSCT, Outcomes and Disease-Specific Follow-Up
Conditioning
Full intensity myeloablative conditioning with fludarabine and pharmacokinetic-guided busulfan dosing is the current recommendation for LSDs (15). Parenteral busulfan with therapeutic drug monitoring has facilitated more precise dose delivery (16, 17). This has mitigated previously high incidences of hepatic veno-occlusive disease (VOD) associated with increased busulfan exposure, while ensuring adequate therapeutic levels are achieved to avoid graft rejection (18). Furthermore, although cyclophosphamide (CY) was originally used instead of fludarabine, the readily described CY-associated cardiac toxicity and reduced duration of neutropenia with fludarabine, as well as reduced rates of VOD, has limited the use of CY in pre-transplant conditioning (19) (see the review “Conditioning Perspectives for Primary Immunodeficiencies”).
Transplant Outcomes
In the past two decades, the proportion of MPS IH patients with graft failure has declined by more than 3-fold (20). Preferential use of umbilical cord blood (UCB) has shown superiority in achieving full-donor chimerism, where an increased number of patients have more than 95% of donor-derived haematopoiesis, compared to other cell sources (20, 21). The interval to transplant is reduced in UCB transplant, and there is better tolerance of HLA-mismatch. In addition, UCB is associated with greater delivery of normal enzyme levels, improving disease-related outcomes (17, 22). Some optimization of the UCB transplant procedure is required since there remain relatively high rates of aplastic graft failure requiring re-transplant, and of immune-mediated cytopenia (IMC) (20, 23). Assessment and optimization of contributing factors to graft failure and IMC is an active area of investigation.
Patient Outcomes
HSCT undoubtedly improves the clinical course of patients with MPS IH. Following HSCT, early disease-related mortality is reduced and prolonged survival possible and even expected (24, 25). However, there remain disease manifestations that require long term follow up. Earlier age at HSCT and graft enzyme output are the twin predictors of superior clinical outcomes in MPS IH, including preservation of neurocognition (24). Somatic outcomes including cardiac disease and corneal clouding stabilizes and has shown improvement in the majority of MPS IH patients post-transplant (24). In contrast, orthopedic complications continue to progress despite transplant, with the majority of patients requiring surgical intervention (24). Thus, the capacity for HSCT to ameliorate disease progression varies widely between organ systems, and certain organs remain relatively refractory to HSCT. Enzyme delivery after transplant may be augmented using an ex vivo stem cell gene therapy procedure and this may improve skeletal outcomes.
Follow Up
Multidisciplinary management beyond the period of transplantation is fundamental in the care of patients with LSD. HSCT improves the clinical course of disease but it is not curative. Regular, long-term follow up with several specialties is mandatory including a bone marrow transplant clinician, metabolic disease specialist, endocrinologist, orthopedic specialist, and spinal surgeon. The psychosocial impact of the disease should not be neglected and psychological support should be offered.
The Role of HSCT in Other LSDs
MPS IH has exemplified core underlying principles of HSCT in LSDs. It has demonstrated the concept of efficacious enzyme delivery via cross-correction, facilitated optimization of a cell source hierarchy that includes UCB, and optimization of a fludarabine and pharmacokinetic-guided busulfan conditioning regimen. It has highlighted the importance of performing HSCT early and pre-symptomatically. While some of these principles are applicable to a few LSDs, it is not indicated in others as it fails to improve patient outcomes.
In Alpha Mannosidosis (AM), HSCT is considered standard of care based on limited published evidence (26). Deficiency of alpha-mannosidase causes a clinically heterogenous disease of neurocognitive impairment and musculoskeletal abnormalities (27, 28). In AM, earlier age at HSCT predicts superior clinical outcomes (29). Similarly, it was thought that early, pre-symptomatic HSCT was crucial for altering the course of rapid neurological disease in infantile Globoid Cell Leukodystrophy (GLD) (30). However, the introduction of newborn screening (NBS) in New York yielded controversial patient outcomes, showing significant HSCT-associated morbidity and mortality (31). Long term collaborative multi-center data may provide better insight into the utility of HSCT in GLD.
There is no role for HSCT in Sanfilippo syndrome (MPS III) or infantile Metachromatic Leukodystrophy (MLD). MLD is caused by deficiency of arylsulfatase A and subsequent accumulation of sulfatides in the central and peripheral nervous system, resulting in widespread demyelination (32, 33). What has been learnt from MPS IH has not reliably translated to infantile MLD or MPS III. Despite pre-symptomatic HSCT and utilization of UCB, HSCT does not halt disease progression in infantile MLD (brain seems modified, but peripheral nerve system not) or MPS III (34–36). Patient outcomes for infantile MLD following HSCT is complicated by peripheral neuropathy and significant HSCT-associated morbidity (36, 37). Even with full engraftment in MPS III patients, there is no biochemical correction of the disease in the CSF (35). In MLD, transplant failure may be largely attributable to the slow and gradual replacement of resident tissue macrophages and microglia populations by donor-derived progeny compared with the rapid progression of disease. Furthermore, donor-derived microglial cells may secrete insufficient amounts of enzyme to correct neuronal tissue in these LSDs. Ex-vivo stem cell gene therapy of autologous HSC improves graft enzyme delivery and has been shown to be dramatically beneficial in modifying disease progression in infantile MLD (38) (see the review “Autologous stem cell-based gene therapy for inherited disorders: state-of-the-art and future prospects”).
Peroxisomal Diseases: X-ALD
Pathophysiology
Adrenoleukodystrophy (X-ALD) is caused by a genetic mutation in ABCD1, resulting in a deficiency of ALD, a membrane transporter which transports substrates from the cytosol into the peroxisome (39). Subsequently, very long-chain fatty acids (VLCFA) are insufficiently degraded via peroxisomal beta-oxidation, accumulate in the cytosol, and incorporate into different complex lipids in varying degrees across cell types, although the nervous system, and adrenal glands are particularly vulnerable (40, 41). The vast phenotypic variation of X-ALD can be simplified into four categories; asymptomatic, adrenal failure, adrenomyeloneuropathy, and inflammatory cerebral disease (42). Childhood cerebral ALD (CCALD) is the most severe manifestation, associated with rapid neurological decline and mortality if untreated (42). The initial episode of cerebral demyelination that occurs with CCALD could be attributed to selective accumulation of VLCFA in myelin, leading to progressive destabilization of myelin sheaths and subsequent demyelination (43). Complex lipids containing VLCFA may cause microglial activation, apoptosis, and diminish their capacity to oxidize VLCFA or provide neuroprotective factors for neighboring cells, leading to rapidly progressive inflammatory demyelination (44, 45).
Indication for HSCT
HSCT is only indicated in CCALD and is the single treatment modality available in this phenotype to demonstrate amelioration of disease (46). The efficacy of HSCT in X-ALD is unlikely to be in cross-correction. HSCT may arrest the neuroinflammatory demyelinating process by replacing dysfunctional microglia with bone-marrow derived macrophages but this mechanism has not been fully elucidated (47).
Approach to HSCT, Outcomes and Disease-Specific Follow-Up
Myeloablative conditioning with busulfan and CY with or without total body irradiation (TBI) has originally been used for HSCT in X-ALD (48). Recently, a reduced intensity conditioning regimen with fludarabine, melphalan, and low-dose TBI has been trialed, demonstrating promising survival rates and modest neurological stabilization, both clinically and on imaging (49, 50). Optimal patient outcomes are achieved in patients with limited clinical evidence of cerebral disease at HSCT (48, 51). To reduce time to transplant, NBS may facilitate early detection of HSCT-eligible individuals and identification of transplant donors which can be recruited once the need for HSCT manifests (52).
Regular evaluation with multi-disciplinary input is necessary to provide appropriate assessment and management for the diverse complications that may arise with X-ALD. This includes follow-up by an endocrinologist, neurologist, and ongoing radiological monitoring by MRI to detect early cerebral disease in genetically affected boys, or evaluate neurological response to treatment.
Mitochondrial Diseases: MNGIE
Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) is an autosomal recessive disease characterized by deficiency of thymidine phosphorylase (TP) (53). Nucleosides accumulate and cause mitochondrial DNA instability (54). This manifests as a constellation of clinical features that includes peripheral neuropathy, external ophthalmoplegia, gastrointestinal dysmotility, and leukoencephalopathy (53). HSCT may pose as a viable treatment option where donor-derived normalization of TP enzymatic activity can eliminate accumulated nucleosides. While there is limited published evidence for HSCT, the results are promising and suggest that HSCT should only be considered in younger patients before severe gastrointestinal dysmotility develop and only if a fully matched donor is available (55, 56).
Summary and Future Perspectives
• The collaborative effort of several international studies has led to the success of HSCT in treating metabolic disease, particularly MPS IH.
• The principle of cross-correction does not reliably translate to all LSDs or other metabolic diseases, and persistent progression of organs that are refractory to HSCT cause significant morbidity following transplant.
• Autologous HSCT using gene therapy may facilitate supranormal enzyme production by transducing a patient's HSCs with a viral vector ex vivo, and infusing them back into the patient following appropriate conditioning.
• These gene-corrected cells can integrate into the host genome, where certain viral vectors such as lentiviruses can do so with a lower risk of insertional mutagenesis.
• This may help treat refractory diseases such as MLD and MPSIIIA, and better correct refractory organs in a responding disease, such as the skeleton in MPSIH, while avoiding significant HSCT-associated morbidity. Pre-clinical experiments in animal models are promising and clinical trials are ongoing.
Author Contributions
ET and RW were the main authors with contributions by JB and SJ. All authors reviewed the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Medina DL, Ballabio A. Lysosomal calcium regulates autophagy. Autophagy. (2015) 11:970–1. doi: 10.1080/15548627.2015.1047130
2. Reddy A, Caler EV, Andrews NW. Plasma membrane repair is mediated by Ca(2+)-regulated exocytosis of lysosomes. Cell. (2001) 106:157–69. doi: 10.1016/S0092-8674(01)00421-4
3. Todkar K, Ilamathi HS, Germain M. Mitochondria and lysosomes: discovering bonds. Front Cell Dev Biol. (2017) 5:106. doi: 10.3389/fcell.2017.00106
4. Campos D, Monaga M. Mucopolysaccharidosis type I: current knowledge on its pathophysiological mechanisms. Metab Brain Dis. (2012) 27:121–9. doi: 10.1007/s11011-012-9302-1
5. Muenzer J. Overview of the mucopolysaccharidoses. Rheumatology. (2011) 50(Suppl. 5):v4–12. doi: 10.1093/rheumatology/ker394
6. Cleary M, Wraith J. The presenting features of mucopolysaccharidosis type IH (Hurler syndrome). Acta Paediatr. (1995) 84:337–9. doi: 10.1111/j.1651-2227.1995.tb13640.x
7. Mohan UR, Hay AA, Cleary MA, Wraith JE, Patel RG. Cardiovascular changes in children with mucopolysaccharide disorders. Acta Paediatr. (2002) 91:799–804. doi: 10.1080/08035250213211
8. Leal GN, de Paula AC, Leone C, Kim CA. Echocardiographic study of paediatric patients with mucopolysaccharidosis. Cardiol Young. (2010) 20:254–61. doi: 10.1017/S104795110999062X
9. Di Ferrante N, Nichols BL, Donnelly PV, Neri G, Hrgovcic R, Berglund RK. Induced degradation of glycosaminoglycans in Hurler's and Hunter's syndromes by plasma infusion. Proc Natl Acad Sci USA. (1971) 68:303–7. doi: 10.1073/pnas.68.2.303
10. Fratantoni JC, Hall CW, Neufeld EF. Hurler and Hunter syndromes: mutual correction of the defect in cultured fibroblasts. Science. (1968) 162:570–2. doi: 10.1126/science.162.3853.570
11. Muenzer J, Wraith JE, Clarke LA. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics. (2009) 123:19–29. doi: 10.1542/peds.2008-0416
12. Fratantoni JC, Hall CW, Neufeld EF. The defect in Hurler and Hunter syndromes. II. Deficiency of specific factors involved in mucopolysaccharide degradation. Proc Natl Acad Sci USA. (1969) 64:360–6. doi: 10.1073/pnas.64.1.360
13. Dickson P, Peinovich M, McEntee M, Lester T, Le S, Krieger A, et al. Immune tolerance improves the efficacy of enzyme replacement therapy in canine mucopolysaccharidosis I. J Clin Invest. (2008) 118:2868–76. doi: 10.1172/JCI34676
14. Kakkis ED, Muenzer J, Tiller GE, Waber L, Belmont J, Passage M, et al. Enzyme-replacement therapy in mucopolysaccharidosis I. N Engl J Med. (2001) 344:182–8. doi: 10.1056/NEJM200101183440304
15. Bartelink IH, van Reij EML, Gerhardt CE, van Maarseveen EM, de Wildt A, Versluys B, et al. Fludarabine and exposure-targeted busulfan compares favorably with busulfan/cyclophosphamide-based regimens in pediatric hematopoietic cell transplantation: maintaining efficacy with less toxicity. Biol Blood Marrow Transplant. (2014) 20:345–53. doi: 10.1016/j.bbmt.2013.11.027
16. Andersson BS, Madden T, Tran HT, Hu WW, Blume KG, Chow DS, et al. Acute safety and pharmacokinetics of intravenous busulfan when used with oral busulfan and cyclophosphamide as pretransplantation conditioning therapy: a phase I study. Biology Blood Marrow Transplant. (2000) 6:548–54. doi: 10.1016/S1083-8791(00)70064-4
17. Boelens JJ, Aldenhoven M, Purtill D, Ruggeri A, Defor T, Wynn R, et al. Outcomes of transplantation using various hematopoietic cell sources in children with Hurler syndrome after myeloablative conditioning. Blood. (2013) 121:3981–7. doi: 10.1182/blood-2012-09-455238
18. Slattery JT, Sanders JE, Buckner CD, Schaffer RL, Lambert KW, Langer FP, et al. Graft-rejection and toxicity following bone marrow transplantation in relation to busulfan pharmacokinetics. Bone Marrow Transplant. (1995) 16:31–42.
19. Ben-Barouch S, Cohen O, Vidal L, Avivi I, Ram R. Busulfan fludarabine vs busulfan cyclophosphamide as a preparative regimen before allogeneic hematopoietic cell transplantation: systematic review and meta-analysis. Bone Marrow Transplant. (2016) 51:232–40. doi: 10.1038/bmt.2015.238
20. Lum SH, Miller WP, Jones S, Poulton K, Ogden W, Lee H, et al. Changes in the incidence, patterns and outcomes of graft failure following hematopoietic stem cell transplantation for Hurler syndrome. Bone Marrow Transplant. (2017) 52:846–53. doi: 10.1038/bmt.2017.5
21. Boelens JJ, Wynn RF, O'Meara A, Veys P, Bertrand Y, Souillet G, et al. Outcomes of hematopoietic stem cell transplantation for Hurler's syndrome in Europe: a risk factor analysis for graft failure. Bone Marrow Transplant. (2007) 40:225. doi: 10.1038/sj.bmt.1705718
22. Church H, Tylee K, Cooper A, Thornley M, Mercer J, Wraith E, et al. Biochemical monitoring after haemopoietic stem cell transplant for Hurler syndrome (MPSIH): implications for functional outcome after transplant in metabolic disease. Bone Marrow Transplant. (2007) 39:207. doi: 10.1038/sj.bmt.1705569
23. Deambrosis D, Lum SH, Hum RM, Poulton K, Ogden W, Jones S, et al. Immune cytopenia post-cord transplant in Hurler syndrome is a forme fruste of graft rejection. Blood Adv. (2019) 3:570–4. doi: 10.1182/bloodadvances.2018026963
24. Aldenhoven M, Wynn RF, Orchard PJ, O'Meara A, Veys P, Fischer A, et al. Long-term outcome of Hurler syndrome patients after hematopoietic cell transplantation: an international multicenter study. Blood. (2015) 125:2164–72. doi: 10.1182/blood-2014-11-608075
25. Lum SH, Stepien KM, Ghosh A, Broomfield A, Church H, Mercer J, et al. Long term survival and cardiopulmonary outcome in children with Hurler syndrome after haematopoietic stem cell transplantation. J Inherit Metab Dis. (2017) 40:455–60. doi: 10.1007/s10545-017-0034-6
26. Mynarek M, Tolar J, Albert MH, Escolar ML, Boelens JJ, Cowan MJ, et al. Allogeneic hematopoietic SCT for alpha-mannosidosis: an analysis of 17 patients. Bone Marrow Transplant. (2011) 47:352. doi: 10.1038/bmt.2011.99
27. Beck M, Olsen KJ, Wraith JE, Zeman J, Michalski JC, Saftig P, et al. Natural history of alpha mannosidosis a longitudinal study. Orphanet J Rare Dis. (2013) 8:88. doi: 10.1186/1750-1172-8-88
28. Riise Stensland HM, Klenow HB, Van Nguyen L, Hansen GM, Malm D, Nilssen O. Identification of 83 novel alpha-mannosidosis-associated sequence variants: functional analysis of MAN2B1 missense mutations. Hum Mutat. (2012) 33:511–20. doi: 10.1002/humu.22005
29. Broomfield AA, Chakrapani A, Wraith JE. The effects of early and late bone marrow transplantation in siblings with alpha-mannosidosis. Is early haematopoietic cell transplantation the preferred treatment option? J Inherit Metab Dis. (2010) 33(Suppl. 3):S123–7. doi: 10.1007/s10545-009-9035-4
30. Escolar ML, Poe MD, Provenzale JM, Richards KC, Allison J, Wood S, et al. Transplantation of umbilical-cord blood in babies with infantile Krabbe's disease. N Engl J Med. (2005) 352:2069–81. doi: 10.1056/NEJMoa042604
31. Wasserstein MP, Andriola M, Arnold G, Aron A, Duffner P, Erbe RW, et al. Clinical outcomes of children with abnormal newborn screening results for Krabbe disease in New York State. Genet Med. (2016) 18:1235. doi: 10.1038/gim.2016.35
32. Jeon SB, Yoon HJ, Park SH, Kim IH, Park EJ. Sulfatide, a major lipid component of myelin sheath, activates inflammatory responses as an endogenous stimulator in brain-resident immune cells. J Immunol. (2008) 181:8077–87. doi: 10.4049/jimmunol.181.11.8077
33. MacFaul R, Cavanagh N, Lake BD, Stephens R, Whitfield AE. Metachromatic leucodystrophy: review of 38 cases. Arch Dis Child. (1982) 57:168–75. doi: 10.1136/adc.57.3.168
34. Bredius RG, Laan LA, Lankester AC, Poorthuis BJ, van Tol MJ, Egeler RM, et al. Early marrow transplantation in a pre-symptomatic neonate with late infantile metachromatic leukodystrophy does not halt disease progression. Bone Marrow Transplant. (2007) 39:309–10. doi: 10.1038/sj.bmt.1705581
35. Welling L, Marchal JP, van Hasselt P, van der Ploeg AT, Wijburg FA, Boelens JJ. Early umbilical cord blood-derived stem cell transplantation does not prevent neurological deterioration in mucopolysaccharidosis type III. JIMD Rep. (2014) 18:63–8. doi: 10.1007/8904_2014_350
36. Boucher AA, Miller W, Shanley R, Ziegler R, Lund T, Raymond G, et al. Long-term outcomes after allogeneic hematopoietic stem cell transplantation for metachromatic leukodystrophy: the largest single-institution cohort report. Orphanet J Rare Dis. (2015) 10:94. doi: 10.1186/s13023-015-0313-y
37. Martin HR, Poe MD, Provenzale JM, Kurtzberg J, Mendizabal A, Escolar ML. Neurodevelopmental outcomes of umbilical cord blood transplantation in metachromatic leukodystrophy. Biol Blood Marrow Transplant. (2013) 19:616–24. doi: 10.1016/j.bbmt.2013.01.010
38. Biffi A, Montini E, Lorioli L, Cesani M, Fumagalli F, Plati T, et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. (2013) 341:1233158. doi: 10.1126/science.1233158
39. Mosser J, Douar AM, Sarde CO, Kioschis P, Feil R, Moser H, et al. Putative X-linked adrenoleukodystrophy gene shares unexpected homology with ABC transporters. Nature. (1993) 361:726–30. doi: 10.1038/361726a0
40. Singh I, Moser AE, Moser HW, Kishimoto Y. Adrenoleukodystrophy: impaired oxidation of very long chain fatty acids in white blood cells, cultured skin fibroblasts, and amniocytes. Pediatr Res. (1984) 18:286–90. doi: 10.1203/00006450-198403000-00016
41. Hubbard WC, Moser AB, Tortorelli S, Liu A, Jones D, Moser H. Combined liquid chromatography-tandem mass spectrometry as an analytical method for high throughput screening for X-linked adrenoleukodystrophy and other peroxisomal disorders: preliminary findings. Mol Genet Metab. (2006) 89:185–7. doi: 10.1016/j.ymgme.2006.05.001
42. Moser HW, Moser AB, Smith KD, Bergin A, Borel J, Shankroff J, et al. Adrenoleukodystrophy: phenotypic variability and implications for therapy. J Inherit Metab Dis. (1992) 15:645–64. doi: 10.1007/BF01799621
43. Ho JK, Moser H, Kishimoto Y, Hamilton JA. Interactions of a very long chain fatty acid with model membranes and serum albumin. Implications for the pathogenesis of adrenoleukodystrophy. J Clin Invest. (1995) 96:1455–63. doi: 10.1172/JCI118182
44. Lund TC, Stadem PS, Panoskaltsis-Mortari A, Raymond G, Miller WP, Tolar J, et al. Elevated cerebral spinal fluid cytokine levels in boys with cerebral adrenoleukodystrophy correlates with MRI severity. PLoS ONE. (2012) 7:e32218. doi: 10.1371/journal.pone.0032218
45. Eichler FS, Ren J-Q, Cossoy M, Rietsch AM, Nagpal S, Moser AB, et al. Is microglial apoptosis an early pathogenic change in cerebral X-linked adrenoleukodystrophy? Ann Neurol. (2008) 63:729–42. doi: 10.1002/ana.21391
46. Mahmood A, Raymond GV, Dubey P, Peters C, Moser HW. Survival analysis of haematopoietic cell transplantation for childhood cerebral X-linked adrenoleukodystrophy: a comparison study. Lancet Neurol. (2007) 6:687–92. doi: 10.1016/S1474-4422(07)70177-1
47. Ferrer I, Aubourg P, Pujol A. General aspects and neuropathology of x-linked adrenoleukodystrophy. Brain Pathol. (2010) 20:817–30. doi: 10.1111/j.1750-3639.2010.00390.x
48. Peters C, Charnas LR, Tan Y, Ziegler RS, Shapiro EG, DeFor T, et al. Cerebral X-linked adrenoleukodystrophy: the international hematopoietic cell transplantation experience from 1982 to 1999. Blood. (2004) 104:881–8. doi: 10.1182/blood-2003-10-3402
49. Kato K, Maemura R, Wakamatsu M, Yamamori A, Hamada M, Kataoka S, et al. Allogeneic stem cell transplantation with reduced intensity conditioning for patients with adrenoleukodystrophy. Mol Genet Metab. (2018) 18:1–6. doi: 10.1016/j.ymgmr.2018.11.001
50. Niizuma H, Uematsu M, Sakamoto O, Uchiyama T, Horino S, Onuma M, et al. Successful cord blood transplantation with reduced-intensity conditioning for childhood cerebral X-linked adrenoleukodystrophy at advanced and early stages. Pediatr Transplant. (2012) 16:E63–70. doi: 10.1111/j.1399-3046.2011.01539.x
51. Miller WP, Rothman SM, Nascene D, Kivisto T, DeFor TE, Ziegler RS, et al. Outcomes after allogeneic hematopoietic cell transplantation for childhood cerebral adrenoleukodystrophy: the largest single-institution cohort report. Blood. (2011) 118:1971–8. doi: 10.1182/blood-2011-01-329235
52. Kemper AR, Brosco J, Comeau AM, Green NS, Grosse SD, Jones E, et al. Newborn screening for X-linked adrenoleukodystrophy: evidence summary and advisory committee recommendation. Genet Med. (2016) 19:121. doi: 10.1038/gim.2016.68
53. Hirano M, Silvestri G, Blake DM, Lombes A, Minetti C, Bonilla E, et al. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE). Clinical, biochemical, and genetic features of an autosomal recessive mitochondrial disorder. Neurology. (1994) 44:721–7. doi: 10.1212/WNL.44.4.721
54. Hirano M, Lagier-Tourenne C, Valentino ML, Marti R, Nishigaki Y. Thymidine phosphorylase mutations cause instability of mitochondrial DNA. Gene. (2005) 354:152–6. doi: 10.1016/j.gene.2005.04.041
55. Halter JP, Michael W, Schüpbach M, Mandel H, Casali C, Orchard K, et al. Allogeneic haematopoietic stem cell transplantation for mitochondrial neurogastrointestinal encephalomyopathy. Brain. (2015) 138(Pt 10):2847–58. doi: 10.1093/brain/awv226
56. Halter J, Schüpbach WMM, Casali C, Elhasid R, Fay K, Hammans S, et al. Allogeneic hematopoietic SCT as treatment option for patients with mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): a consensus conference proposal for a standardized approach. Bone Marrow Transplant. (2010) 46:330. doi: 10.1038/bmt.2010.100
Keywords: hematopoietic stem cell transplantation, bone marrow transplant, inborn errors of metabolism, lysosomal storage disease, peroxisomal disease, mitochondrial disease
Citation: Tan EY, Boelens JJ, Jones SA and Wynn RF (2019) Hematopoietic Stem Cell Transplantation in Inborn Errors of Metabolism. Front. Pediatr. 7:433. doi: 10.3389/fped.2019.00433
Received: 30 July 2019; Accepted: 07 October 2019;
Published: 25 October 2019.
Edited by:
Andrew R. Gennery, Newcastle University, United KingdomReviewed by:
Eduardo Lopez-Granados, University Hospital La Paz, SpainSatiro De Oliveira, University of California, Los Angeles, United States
Copyright © 2019 Tan, Boelens, Jones and Wynn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert F. Wynn, cm9iZXJ0Lnd5bm5AbWZ0Lm5ocy51aw==