
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr., 15 October 2019
Sec. Pediatric Immunology
Volume 7 - 2019 | https://doi.org/10.3389/fped.2019.00418
This article is part of the Research TopicMeaningful Cases of Primary Immunodeficiencies: Volume 2View all 21 articles
 Lucia Leonardi1*
Lucia Leonardi1* Giulia Lorenzetti1
Giulia Lorenzetti1 Rita Carsetti2
Rita Carsetti2 Simona Ferrari3
Simona Ferrari3 Alessia Di Felice1Bianca Cinicola1
Alessia Di Felice1Bianca Cinicola1 Marzia Duse1
Marzia Duse1Common variable immunodeficiency (CVID) is the most common and clinically relevant primary immunodeficiency (PID). Genetic basis of CVID remains largely unknown. However, in a minority of CVID patients, a number of distinct genetic defects affecting the normal processes of B cell maturation and differentiation into memory B cells have now been identified, resulting in markedly reduced serum levels of immunoglobulin G (IgG) and low immunoglobulin A (IgA) or immunoglobulin M (IgM), with impaired antibody responses, despite the presence of normal levels of B cells. Patients with CVID develop recurrent and chronic infections of respiratory and gastrointestinal tracts, autoimmune diseases, lymphoproliferative complications, malignancies, and granulomatous disease. We report the case of a boy admitted to our unit for the first time at the age of three for reduced gamma globulin levels and a clinical history positive for two episodes of pneumonia. Our patient incompletely met ESID diagnostic criteria for CVID, but molecular genetic analysis, a NGS panel including 47 PID-associated genes was performed in the proband and in his parents, revealing the presence of a heterozygous nucleotide substitution in exon 4 (c.579C>A) of TNFRSF13B encoding TACI. This mutation has been described only in two CVID adult patients and in a child with selective IgA deficiency (sIgAD). We highlighted the same mutation in the asymptomatic mother and detected two extra heterozygous mutations of RIG1 and LIG1. We promptly started intravenous immunoglobulin (IVIG) therapy with good tolerance. Despite the diagnosis of CVID remains clinical, in this case report we underline the importance of considering and planning genetic workup in all subjects with unclear diagnosis and of reporting new molecular diagnosis especially in case of rare mutations.
Hypogammaglobulinemia in children is sustained by a large spectrum of clinical conditions. Transient hypogammaglobulinemia of infancy (THI) should be considered during the first years of life, due to age dependent maturation of adaptive immunity. THI is characterized by immunoglobulin G (IgG) below age-related normal value in absence of defined causes of hypogammaglobulinemia, detected in the first 3 years of life (measured at least twice), with spontaneous resolution approximately after the 4th birthday. In children in the first 3 years of life with low IgG levels the definition of unclassified antibody deficiency (UNPAD) should be considered, reserving THI for those children who recover by the age of four. To rule out THI, a definite diagnosis of common variable immunodeficiency (CVID) should not be given before the age of four (1). However, alternative diagnosis for unclassified antibody deficiency includes early-onset CVID.
CVID is the most common primary immunodeficiency (PID) affecting about 20% of all PIDs patients in Europe and 25% of CVID patients are diagnosed in childhood (2, 3). Despite the majority of CVID cases seems to occur sporadically, about 5 to 25% of patients have a positive family history and a monogenic defect has been identified in 2–10% of patients with CVID phenotype (2). However, the current opinion is that, apart from these rare monogenic forms, CVID is more likely a complex rather than a Mendelian disease and modifier genes may play a crucial role in the development of disease (1). The spectrum of TACI mutations is becoming broader as more patients are screened worldwide. TNFRSF13B/TACI defects represent, indeed, the most common DNA sequence variations in individuals affected by CVID, being found in about 10–20% of subjects (4) and associated with CVID specific phenotypes, including the presence of benign lymphoproliferation and autoimmunity. After the initial description of an association with both CVID and selective immunoglobulin A deficiency (sIgAD) (5, 6), further analyses have shown the existence of some TNFRSF13B variants also in healthy individuals (7) and unaffected family members of patients with CVID (8–11). In addition, the functional effects of TNFRSF13B mutations in relation to the development of the disease have not been completely established yet and seem to include both causal and modifier mutations.
We report the case of a boy admitted to our unit for the first time at the age of three for reduced gamma globulin levels and a clinical history positive for two episodes of pneumonia.
The patient was born by cesarean section after a full-term uncomplicated gestation. He was admitted at kindergarten at 1 year of age and completed a regular immunization schedule. His parents were non-consanguineous and family history was negative for PIDs, but positive for coeliac disease (sister), allergic rhinitis (paternal grandmother), atopic asthma (mother/father). The child had a clinical history of bronchitis with wheezing since the age of 4 months. He had no known allergies. At the age of 29 months he was diagnosed with bronchopneumonia involving the right lower lobe. Seven months later he was admitted to our pediatric unit for persistent fever unresponsive to antibiotic therapy and he was diagnosed with bronchopneumonia involving the right middle lobe. Routine blood tests showed low serum immunoglobulin for his age: IgG 330 mg/dl (n.v. > 462); IgA 20 mg/dl (n.v. >27) and IgM 69 mg/dl (n.v >62). Cell blood count and lymphocyte subsets assay as well as IgG subclass were in the normal range, while anti-tetanus IgG (0.1 UI/ml) and anti-pneumococcal IgG (0.9 mg/l) were not protective despite previous vaccination in the first year of life. Booster doses of tetanus and pneumococcal vaccines were administered and, after 21 days, anti-tetanus toxoid IgG and anti-pneumococcal capsular polysaccharide IgG achieved a borderline protective level (anti-tetanus IgG 0.44 UI/ml, anti-Pneumococcal IgG 36 mg/l) suggesting a potential THI. Screening for autoimmunity, including coeliac disease, was negative. In addition, it was found a positivity of EBV VCA IgM and IgG with negative EBV DNA (RT-PCR) in absence of specific clinical manifestation. An accurate follow-up was planned, including periodical evaluation of clinical conditions and laboratory findings. Three months later, clinical conditions remained good (the patient did not present severe infections or autoimmune manifestations), but immunoglobulin levels were persistently lower than the normal range and low switched memory B cells for age were detected (Figure 1) as well as persistent positivity of EBV VCA IgM and IgG with negative RT-PCR. We therefore performed an NGS PID panel covering 47 classical genes associated with humoral and combined immunodeficiencies and immune dysregulation diseases (ADA, BTK, CD19, CD79A, IGLL1, CD79B, PIK3R1, TCF3, DCLRE1C, TNFRSF13B, CD81, MS4A1, CR2, NFKB1, NFKB2, PIK3CD, CTLA4, LRBA, CASP10, IL2RA, PTEN, RAG1, RAG2, CECR1, AICDA, CD40LG, CD40, ATM, UNG, TTC7A, IKZF1, LIG1, OAS1, DKC1, STAT1, STAT3, XIAP, SH2D1A, FAS, FASLG, CASP8, NFKBIA, PLCG2, PRKCD, PRF1, NRAS, KRAS). The genes on the panel have been carefully selected based on scientific literature, mutations databases, and our experience.
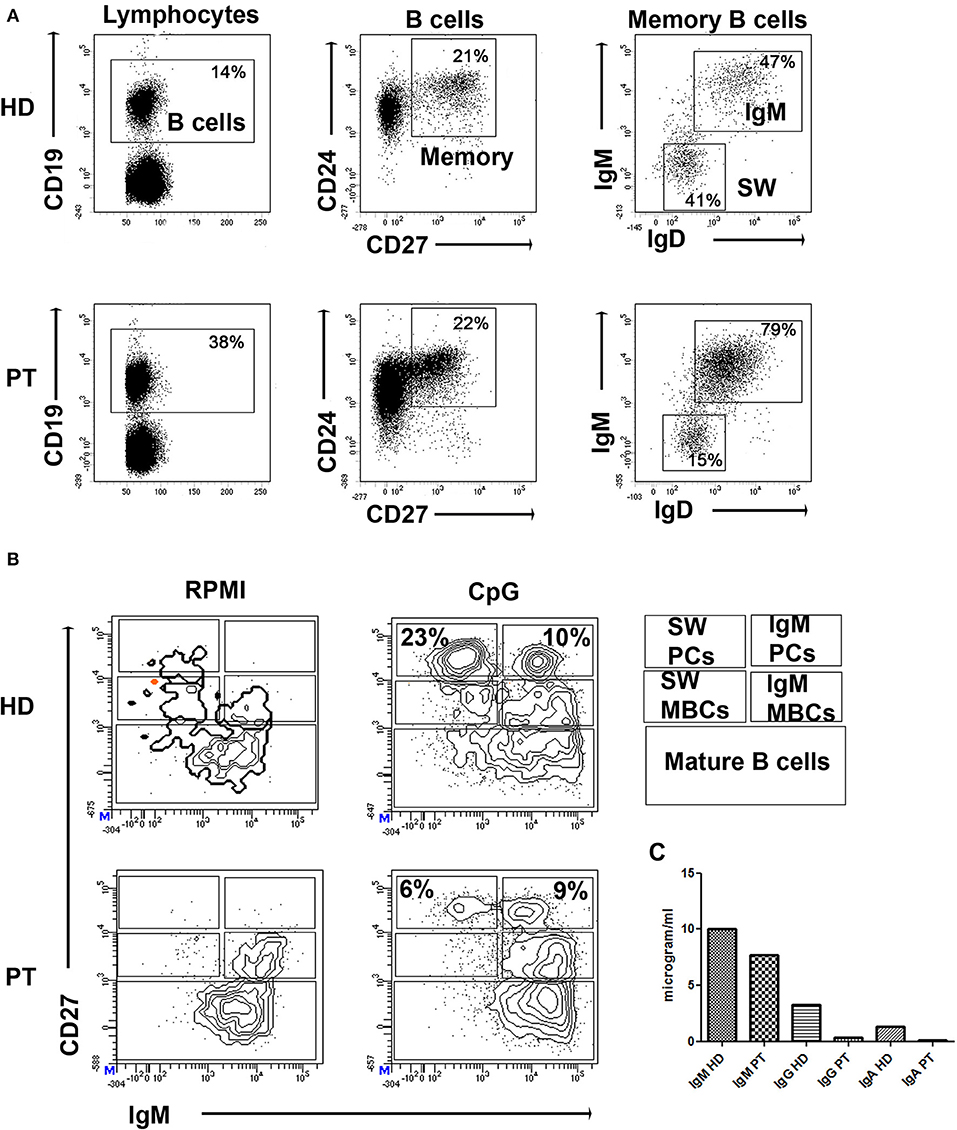
Figure 1. (A) Dot plots show the gates used to identify B cells (CD19pos), CD24posCD27pos memory B cells (MBCs), and inside MBCs the IgMpos and IgMneg (SW). (B) PBMCs were kept in culture (RPMI) or stimulated in vitro with CpG for 7 days (CpG). Staining with CD27 and IgM identifies IgM MBCs (IgMposCD27pos, indicated as IgM MBCs) and switched MBCs (IgMnegCD27pos, indicated as SW MBCs). Plasma cells have higher levels of CD27 and express either IgM (IgM PCs) or switched isotypes (SW PCs). CD27negIgMpos cells are mature B cells. (C) Levels of secretory immunoglobulin in cell medium of HD and patient (PT) after stimulation with CpG.
Molecular analysis revealed a heterozygous nucleotide substitution in the exon 4 (c.579C>A) of TNFRSF13B (ClinVar: NA; ExAC: 0.00005), being inherited from the asymptomatic mother. This determines the substitution of a cysteine (C) with a stop codon (X) causing premature termination of the protein translation (p.C193X). Moreover, two extra heterozygous mutation were detected: LIG1 p.Val753Met (ClinVar:NA;ExAC:0.007) inherited from the mother, and RAG1 p.Pro1028Leu (ClinVar:NA; ExAC: 0.000008) inherited from the father who was asymptomatic as well.
Following molecular results and in consideration of the progressive decreasing IgG levels to 290 mg/dl, the proband started intravenous immunoglobulin therapy (IVIG) once a month, without any adverse effects. Nowadays the patient is in good general condition, no further major infectious episode nor autoimmunity manifestations and lymphoproliferation occurred.
Written informed consent was obtained from the patient's parents prior to the publication of this case report.
Our case was firstly diagnosed as unclassified antibody deficiency as alternative diagnosis of potential THI because of age of onset, low serum IgG, slightly low serum IgA, and borderline protective antibody levels after a booster injection of tetanus and pneumococcal vaccines. At the age of 39 months clinical conditions and serological values were unchanged. Our extended NGS analysis (47 PID genes) revealed the presence of a heterozygous nucleotide substitution in the exon 4 (c.579C>A) of TNFRSF13B. This determines the substitution of a cysteine (C) with a stop codon (X) causing the premature termination of protein translation (p.C193X). C193X variant is described in the ExAc population with a frequency of 0.00005. The analysis showed that our patient inherited this mutation from his mother, who was completely asymptomatic.
Moreover, two extra heterozygous mutation were detected: LIG1 p.Val753Met (ClinVar:NA; ExAC:0.007) inherited from the mother, and RAG1 p.Pro1028Leu (ClinVar:NA; ExAC:0.000008) inherited from the father who was asymptomatic as well. Both parents showed normal serum immunoglobulin, lymphocyte subsets and B lymphocytes phenotype.
Literature search for TNFRSF13B mutation (C193X) identified two adult patients diagnosed with CVID and a child with sIgAD. Salzer et al. (11) evaluated the genetic, immunologic, and clinical condition of 50 individuals with TNFRSF13B alterations: the same mutation of our patient was described in heterozygosis in a 50-year-old man with CVID and autoimmune haemolytic anemia, coeliac disease and splenomegaly. This mutation was also described by Pulvirenti et al. (12) in homozygosis in a 49-year-old CVID patient with severe B-cell lymphopenia (<1%), autoimmune haemolytic anemia, lymph nodes enlargement and granulomas. In the same article, the same mutation is described in heterozygosis in a 6-year-old child diagnosed with sIgAD (with no clinical manifestations of autoimmunity). In the Italian cohort of 189 CVID patients described by Pulvirenti et al. the C193X allelic frequency is in accordance with the Hardy-Weinberg equilibrium (12). However, the frequency of whole TNFRSF13B monoallelic mutations is significantly higher in CVID patients compared to healthy controls (p = 0.0034, as calculated using two-tailed Fisher's exact test). ClinVar did not report on the pathogenicity of this variant. Nevertheless, there are evidence in literature that TACI haploinsufficiency is involved in B cell dysfunction (13, 14). On the other hand, the existence of healthy controls with heterozygous mutations in this gene and the lack of a clear Mendelian inheritance in families have led to consider some of TNFRSF13B mutations as risk factors (9, 11), which could be determinant to cause disease only in homozygous individuals (6). Thus, TNFRSF13B could be considered a modifier gene rather than a causal gene in monoallelic cases (2).
The monoallelic LIG1 and RAG1 mutations detected have not been associated with CVID phenotype while TNFRSF13B variants are described in heterozygosis in CVID patients. Since our patient mutation is inherited from the asymptomatic mother, we speculate that this TACI mutation could be a modifier in a RAG1-LIG1 digenic disease. However, it remains necessary to understand the functional role of these genes' mutations.
Molecular findings were crucial for the diagnosis of CVID in our patient, as ESID diagnostic criteria for CVID were not completely met (15). More specifically, our patient presented marked decrease of IgG, slightly decreased IgA with normal IgM, low switched memory B cells for his age (Figure 1), and borderline protective antibody response to vaccines after the second booster. He did not present autoimmune manifestations, granulomatous disease, unexplained polyclonal lymphoproliferation, affected family members with antibody deficiency.
A strict clinical follow up has been planned in order to promptly detect any possible evolution of the clinical picture toward autoimmune, lymphoproliferative and granulomatous disease, while IVIG was started. Clinical follow up will clarify if early introduction of IVIG therapy can slow down the evolution of the immunodeficiency. It would also be interesting to know more about the clinical history evolution of the described pediatric case carrying the same mutation and diagnosed with sIgAD.
In conclusion, current opinion is that in case of hypogammaglobulinemia in children younger than 4 years molecular investigation for CVID is not required (1). However, last version of ESID criteria may be unable to identify all CVID cases, so that many patients under 4 years of age remain undiagnosed. Therefore, considering that the spectrum of mutations causing CVID is becoming broader as more patients are screened worldwide, an extended NGS panel is useful in all suspected cases to obtain information leading to a diagnosis that anticipate complete clinical presentation and allow a prompt therapeutic intervention. In addition, we highlight the importance of reporting new molecular diagnosis in order to better understand the pathophysiology of CVID, clarifying the clinical significance of rare mutations, differentiating disease-causing alterations and disease-modifying ones.
All datasets generated for this study are included in the manuscript/supplementary files.
LL, GL, AD, BC, and MD wrote the first draft of the manuscript. SF performed the molecular analysis. RC performed the immunological functional study. LL and GL wrote the final version of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Bonilla FA, Barlan I, Chapel H, Costa-Carvalho BT, Cunningham-Rundles C, de la Morena MT, et al. International Consensus Document (ICON): common variable immunodeficiency disorders. J Allergy Clin Immunol Pract. (2016) 4:38–59. doi: 10.1016/j.jaip.2015.07.025
2. Bogaert DJA, Dullaers M, Lambrecht BN, Vermaelen K, De Baere E, Haerynck F. Genes associated with common variable immunodeficiency: one diagnosis to rule them all? J Med Genet. (2016) 53:575–90. doi: 10.1136/jmedgenet-2015-103690
3. Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood. (2012) 119:1650–7. doi: 10.1182/blood-2011-09-377945
4. Park MA, Li JT, Hagan JB, Maddox DE, Abraham RS. Common variable immunodeficiency: a new look at an old disease. Lancet. (2008) 372:489–502. doi: 10.1016/S0140-6736(08)61199-X
5. Castigli E, Wilson SA, Garibyan L, Rachid R, Bonilla F, Schneider L, et al. TACI is mutant in common variable immunodeficiency and IgA deficiency. Nat Genet. (2005) 37:829–34. doi: 10.1038/ng1601
6. Salzer U, Chapel HM, Webster AD, Pan-Hammarstrom Q, Schmitt-Graeff A, Schlesier M, et al. Mutations in TNFRSF13B encoding TACI are associated with common variable immunodeficiency in humans. Nat Genet. (2005) 37:820–8. doi: 10.1038/ng1600
7. Zhang L, Radigan L, Salzer U, Behrens TW, Grimbacher B, Diaz G, et al. Transmembrane activator and calcium-modulating cyclophilin ligand interactor mutations in common variable immunodeficiency: clinical and immunologic outcomes in heterozygotes. J Allergy Clin Immunol. (2007) 120:1178–85. doi: 10.1016/j.jaci.2007.10.001
8. Castigli E, Wilson SA, Elkhal A, Ozcan E, Garibyan L, Geha RS. Transmembrane activator and calcium modulator and cyclophilin ligand interactor (TACI) enhances CD40 driven plasma cell differentiation. J Allergy Clin Immunol. (2007) 120:885–91. doi: 10.1016/j.jaci.2007.06.012
9. Pan-Hammarstrom Q, Salzer U, Du L, Björkander J, Cunningham-Rundles C, Nelson DL, et al. Reexamining the role of TACI coding variants in common variable immunodeficiency and selective IgA deficiency. Nat Genet. (2007) 39:429–30. doi: 10.1038/ng0407-429
10. Lee JJ, Ozcan E, Rauter I, Geha RS. Transmembrane activator and calcium–modulator and cyclophilin ligand interactor mutations in common variable immunodeficiency. Curr Opin Allergy Clin Immunol. (2008) 8:520–6. doi: 10.1097/ACI.0b013e3283141200
11. Salzer U, Bacchelli C, Buckridge S, Pan-Hammarström Q, Jennings S, Lougaris V, et al. Relevance of biallelic versus monoallelic TNFRSF13B mutations in distinguishing disease-causing from risk-increasing TNFRSF13B variants in antibody deficiency syndromes. Blood. (2009) 113:1967–76. doi: 10.1182/blood-2008-02-141937
12. Pulvirenti F, Zuntini R, Milito C, Specchia F, Spadaro G, Danieli MG, et al. Clinical Associations of Biallelic andMonoallelic TNFRSF13B Variants in Italian Primary Antibody Deficiency Syndromes. J Immunol Res. (2016) 2016:8390356. doi: 10.1155/2016/8390356
13. Chinen J, Shearer WT. Advances in basic and clinical immunology in 2011. J Allergy Clin Immunol. (2012) 129:342–8. doi: 10.1016/j.jaci.2011.11.047
14. Jabara HH, Lee JJ, Janssen E, Ullas S, Liadaki K, Garibyan L, et al. Heterozygosity for transmembrane activator and calcium modulator ligand interactor A144E causes haploinsufficiency and pneumococcal susceptibility in mice. J Allergy Clin Immunol. (2017) 139:1293–301.e4. doi: 10.1016/j.jaci.2016.07.028
15. European Society for Immunodeficiencies. Registry Working Party Diagnosis Criteria. Available online at: https://esid.org/Working-Parties/RegistryWorking-Party/Diagnosis-criteria (accessed January 22, 2019).
Keywords: CVID phenotype, TNFRSF13B, C193X, RAG1, LIG1
Citation: Leonardi L, Lorenzetti G, Carsetti R, Ferrari S, Di Felice A, Cinicola B and Duse M (2019) Rare TACI Mutation in a 3-Year-Old Boy With CVID Phenotype. Front. Pediatr. 7:418. doi: 10.3389/fped.2019.00418
Received: 20 September 2018; Accepted: 30 September 2019;
Published: 15 October 2019.
Edited by:
Henner Morbach, University Hospital Würzburg, GermanyReviewed by:
Antonio Condino-Neto, University of São Paulo, BrazilCopyright © 2019 Leonardi, Lorenzetti, Carsetti, Ferrari, Di Felice, Cinicola and Duse. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lucia Leonardi, bHVjaWFsZW9uYXJkaUB5YWhvby5pdA==; bHVjaWEubGVvbmFyZGlAdW5pcm9tYTEuaXQ=; bHVjaWFsZW9uYXJkaUBwZWMuaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.