 Akihiro Nakamura
Akihiro Nakamura Kazuyuki Ikeda
Kazuyuki Ikeda Kenji Hamaoka
Kenji Hamaoka
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Pediatr. , 28 June 2019
Sec. Pediatric Immunology
Volume 7 - 2019 | https://doi.org/10.3389/fped.2019.00244
This article is part of the Research Topic Infection-Related Immune-Mediated Diseases and Microbiota View all 10 articles
Kawasaki disease (KD) is a pediatric vasculitis syndrome that is often involves coronary artery lesions (e. g., coronary artery aneurysms). Although its causal factors and entire pathogenesis remain elusive, the available evidence indicates that the pathogenesis of KD is closely associated with dysregulation of immune responses to various viruses or microbes. In this short review, we address several essential aspects of the etiology of KD with respect to the immune response to infectious stimuli: 1) the role of viral infections, 2) the role of bacterial infections and the superantigen hypothesis, 3) involvement of innate immune response including pathogens/microbe-associated molecular patterns and complement pathways, and 4) the influence of genetic background on the response to infectious stimuli. Based on the clinical and experimental evidence, we discuss the possibility that a wide range of microbes and viruses could cause KD through common and distinct immune processes.
Kawasaki disease (KD), named after its discoverer Dr. Tomisaku Kawasaki, is a pediatric vasculitis syndrome which is characterized by clinical manifestations including fever persisting for 5 days or more, swelling of the cervical lymph nodes, conjunctival infection, changes in oral mucosa and the tongue, skin rash, and redness of the palms and soles of the feet (1, 2). Although KD shows a systemic vascular inflammation, the coronary arteries are one of the worst affected sites. Without adequate treatment in the acute phase, approximately 30% of patients exhibit coronary artery lesions (CALs) including coronary arterial dilation, stenosis, and aneurysms (3).
Treatment of KD typically features intravenous immunoglobulin (IVIG) therapy (4). In fact, IVIG has markedly decreased the mortality rate in patients with acute KD. However, a persisting concern is that the disease may impair cardiovascular health in adults with a history of KD. Furthermore, approximately 20% of acute KD patients show a low response to IVIG (5). The therapeutic resistance also results in an increased risk of CALs and future cardiovascular events.
The aetiological mechanism of KD remains unclear and the causal factors are also unknown (6). Although there is no definitive evidence that KD is an infectious disease, recent studies support the view that a dysregulated immune response to a variety of infectious stimuli is likely to contribute to KD pathogenesis (6, 7). Based on these studies, this short review explores the possible relationship between KD and the immune response to various infectious agents (Figure 1).

Figure 1. Possible causal microorganisms of Kawasaki disease. Most of the listed microorganisms were identified on the basis of PCR or serological examinations of clinical specimens. The asterisks indicate experimental evidence from animal models, not clinical specimens.
The incidence of KD exhibits seasonality and outbreaks occurred in Japan in 1979, 1982, and 1986 (8, 9). This has led to the speculation that viral infection may underlie KD pathogenesis. Based on serological and polymerase chain reaction (PCR) based-analyses of clinical specimens, at least 14 species of the virus have been reported to be relevant to KD (10). We consider here three possible candidates: Epstein-Barr virus (EBV), human adenovirus, and a putative KD-associated RNA virus.
EBV is a type of human herpes virus. Kikuta et al. (11, 12) reported that the EBV DNA sequence was detected in 83% of KD patients and in 18% of control subjects. Chronic active EBV infection sometimes involves CALs, including coronary artery aneurysms (13, 14). Although the pathogenesis of EBV-infection-associated CALs is unclear, it has been demonstrated in vitro that deoxyuridine 5'-triphosphate nucleotidohydrolase (dUTPase), an EBV-encoded protein, stimulates monocyte-derived macrophages through Toll-like receptor 2 (TLR2)-dependent signaling (15). This up-regulates the production of interleukin-6 (IL-6) (15), which activates endothelial cells (ECs) and platelets. Contrary evidence has also been presented on the involvement of EBV in KD (16, 17).
It has been known that adenoviral infection exhibits seasonal pattern and some symptoms similar to that of KD (18, 19). However, semi-quantitative PCR-based investigations found no association between adenovirus and KD (20). Gene microarray of the blood samples also showed the distinct pattern between KD and adenovirus-infected patients (21). Further study is needed to clarify the involvement of adenoviral infection in KD (22).
Immunoglobulin A (IgA)-producing plasma cells are observed in the affected arterial tissue of KD patients (23, 24). Rowley et al. (25, 26) detected RNA virus-like inclusion bodies in the cytoplasm of bronchoepithelial cells of KD patients. It was detected using synthetic antibodies generated from the alpha and kappa chain-encoding DNA sequences, which were cloned from the affected arterial tissue of KD patients (27). However, the putative KD-associated RNA virus has not been identified yet.
Previous studies suggest that the involvement of viruses in KD is still very controversial. However, a recent transcriptomic study reported the significant up-regulation of a set of type I interferon (INF)-induced genes closely related to cellular antiviral processes in the coronary arteries of KD patients (28). Furthermore, the increased plasma level of C-X-C motif chemokine ligand 10 (CXCL10/IP-10), a representative INF-alpha2a/gamma-inducible protein was recently reported as a promising biomarker for the early acute phase of KD (29). These two studies raise the possibility that KD pathogenesis might be associated with a common immune response to viral infections.
Superantigens (SAgs) are a group of proteins, which can activate approximately 20% of the T cells in the peripheral blood. SAgs stimulate these cells by forming a bridge between the T-cell receptor and the major histocompatibility complex class II (MHC-II) of antigen presenting cells in the absence of any antigenic peptide presentation (30, 31). This results in the overproduction of pro-inflammatory cytokines, including tumor necrosis factor-alpha (TNF-α), by the activated T cells (32). Human MHC-class II and co-stimulators are also expressed in the endothelial cells (33). In fact, SAgs can directly injure these cells in conjunction with T cells (34, 35) in vitro.
SAgs are produced by a wide range of organisms, including bacteria, viruses, fungi, and plants (36–38). The pathological role of SAgs has been well-studied with respect to toxic shock syndrome and scarlet fever, which are caused by Staphylococcus aureus and Streptococcus pyogenes, respectively (39, 40). KD displays some clinical similarities to these two diseases (41).
Staphylococcus aureus produces a SAg designated TTS toxin-1 (TSST-1), which induces expansion of Vβ2 T cell receptor (TCR)-positive T cells. Some early studies reported the frequent detection of TSST-1-producing S. aureus or the specific antibody to the SAg in KD-patients (42, 43). As ECs express MHC-II and co-stimulators, TSST-1 can activate human umbilical vein ECs in vitro in the presence of T cells (34). Considering that specific types of vessels (e.g., coronary arteries), are preferentially affected in KD., it might be worth investigating the pathophysiological significance of the role of ECs as a semi-professional antigen-presenting cells in KD (44, 45).
Streptococcal pyrogenic exotoxin C (SPEC) selectively activates Vβ2-bearing T-cell subsets and Vβ6.5-bearing ones (46). The number of Vβ2-/Vβ6.5-bearing T-cell subsets and anti-SPEC antibody levels are increased in the peripheral blood of acute KD patients (47). DNA fragments encoding these SAgs were also significantly more prevalent in the stool of KD patients than in that of non-KD febrile subjects (48), indicating that these bacteria-derived SAgs are involved in KD pathogenesis. Streptococcus pyogenes infection occasionally triggers autoimmune heart diseases, such as rheumatic fever. The serum level of IgM-type autoantibody to the cardiac myosin heavy chain, which is highly homologous to group A streptococcal M5 protein (49), was increased in KD patients (50). A variety of autoantibodies has also been detected in KD patients (51–53). Besides molecular mimicry, SAgs could be implicated in the activation of potentially autoreactive peripheral T and B cells (54).
Although the physiological role of circulating follicular T helper cells (cTfh) remains elusive, the cells could be stimulated by SAgs and/or other pathogen-derived molecules. Xu et al. (55) reported that IL-4 and the cTfh2 subpopulation of total cTfh cells significantly increased during the acute phase of KD. Increases in cTfh2 and IL-4 are observed in IgA-vasculitis and IgG4-related disease (IgG4-RD) (56–58). Although the general clinical features of IgG4-RD are apparently dissimilar to those of KD, IgG4-RD occasionally involves abnormalities of large- and intermediate-sized vessels, including coronary arteritis (59).
Despite the foregoing evidence, the involvement of SAgs in KD has not been definitively confirmed. Some studies have independently found no significant elevation of antibodies against S. aureus or S. pyogenes-derived SAgs in KD patients (60, 61). Although these bacterial-derived SAgs may provide a rationale to understand the pathogenesis of KD, its aetiological significance is still very controversial.
Based on the similarity in clinical manifestations and the data from some serological studies, it has been argued that Y. pseudotuberculosis (YP) infections might be involved in the pathogenesis of KD (62). Some species of YP produce YP-derived mitogens (YPM), which are SAg-like virulence factors (63). Consistent with the increased prevalence of KD in Far East countries, YPM-positive pathogenic YP are also predominantly distributed in these countries and are less frequent in western countries (64). However, in a recent clinical study involving 108 Japanese KD patients, it was found that 90% of patients were negative for antibodies to YP and YPM (65). Vβ2-bearing T-cell subsets be preferentially activated in KD, whereas Vβ3, Vβ9, Vβ13.1, or Vβ13.2-bearing T-cell subsets are activated in YP infection (66).
Reactivation of Bacillus Calmette-Guérin (BCG) scar is a well-established clinical manifestation in acute KD, indicating that Mycobacteria or immunologically related pathogens might be involved in KD. Although Mycobacteria have not been isolated from KD patients as a causal pathogen, antibody and CD4+/CD8+ T-cell clones specific to Mycobacterium heat shock protein 65 have been detected in KD patients (67, 68).
Besides M. tuberculosis, M. leprae, and M. lepromatosis, non-tuberculous mycobacteria (NTM) can cause self-limited infections in humans. M. avium complex (MAC) is a representative NTM. The immune response to MAC infections involves peculiar macrophages, which are characterized by the co-expression of anti-inflammatory M2 macrophage markers (e.g., CD163, IL10) and markers of pro-inflammatory M1 macrophages (e.g., CCR7, IL1β) (69). These cells promote Th17 cell-differentiation (69). Intriguingly, the coronary arteries affected in acute KD also often feature marked infiltration of macrophages, which are negative for CD80 (an M1 marker), and positive for CD163 (an M2 marker) (70). Plasma level of Th17-related cytokine sets are also increased (71). Considering these recent findings and BCG scar reactivation in KD, re-visiting the possible implication of mycobacteria in KD could be warranted.
Although there is no definitive clinical evidence suggesting that fungi are a causal factor for KD, it has been well-established that Candida albicans extracts develop KD-like experimental model of vasculitis in mice. Based on the meteorological and enviromentological study, Rodo et al. (72) recently reported that KD is associated with tropospheric winds containing Candida species as a predominant fungus. Similar investigation in Canada also suggested the implication of westerly wind-associated fungi in KD (73).
It might be possible to validate phlogogenic activity of the wind-blown microbes / molecules with established animal models for KD.
Except Y. pseudotuberculosis infection (65, 74, 75), few cases satisfy the six diagnostic criteria for Kawasaki disease (KD) among the infectious diseases caused by the aforementioned agents. Considering that genetic background is closely associated with the susceptibility to KD (see section Influence of Genetic Background Affecting Response to Infectious Stimuli), polymorphisms in some specific genes of infected children might affect the clinical futures of the infectious diseases (76). Moreover, it is not exclusive that unidentified agents could be involved in the onset of KD. Caution should be exercised when considering the possible causal agents on the basis of the symptom similarity.
While innate immunity is the first line of self-defense against infectious agents, it is also accompanied by inflammatory reaction (77). Thus, its inadequate regulation gives rise to inflammatory tissue damage. Accumulating evidence indicates that KD could be associated with dysregulated innate immune response.
Apart from SAgs, a variety of microbes or virus-derived biomolecules (e.g., lipopolysaccharides, glucans, and nucleotides) can stimulate immune cells or other cell types, including ECs. These biomolecules are designated pathogen-associated molecular patterns (PAMPs) and microbe-associated molecular patterns (MAMPs). PAMPs and MAMPs are recognized by a type of cellular or soluble receptors, which are designated pattern recognition receptors (PRRs) (e.g., TLRs) to trigger innate immune responses, including the production of inflammatory cytokines through intracellular signaling pathways. A possible implication of these PAMPs or MAMPs in KD has been demonstrated in mouse models and more recently in clinical specimens from KD patients (78, 79).
KD-like vasculitis can be induced in rabbit, swine, and mouse models by various methods (80–83). The known genetic background and ease of genetic manipulation have made mouse models the preferred model to investigate molecular pathogenesis of KD and to discover its therapeutic targets (84–88).
In a murine model of vasculitis, Candida albicans water-soluble fraction (CAWS) is used as the inducer. Marked inflammatory change is observed in the aortic root, including the coronary arteries in CAWS-treated mice (89, 90). While CCR2 and GM-CSF play an indispensable role in its pathogenesis (89, 90), T and B cells are also involved in the vasculitis (89). Sensitivity and severity of CAWS-induced vasculitis apparently depend on mouse strain, suggesting that genetic background affects its pathogenesis (also see section Influence of Genetic Background Affecting Response to Infectious Stimuli).
Another KD-like murine coronary arteritis model involves induction by Lactobacillus casei cell wall extract (LCWE). TLR2-dependent signaling, ILβ-dependent signaling, and CD8+ T cells (cytotoxic T cells) play a crucial role in its pathogenesis (91–93).
KD-like coronary arteritis can also be induced in mice by NOD1 ligand, FK565 (94, 95), a synthetic derivative of acylpeptide produced by Streptomyces olivaceogriseus (96). The experimental model of vasculitis reportedly underlies the interaction between the CCR2 expressed in monocytes and the CCL2 induced by the NOD1-dependent signaling in EC (78).
It is important to consider whether these models accurately represent the actual molecular mechanisms of KD (97). However, some evidences indicate that the experimental murine vasculitis is relevant to KD pathogenesis. The antibodies to β-glucan, another major CAWS component, was increased in KD patients (98). Regarding findings in CAWS-induced or FK565-induced mouse model, it was also reported that genetic polymorphisms in the CCR3-CCR2-CCR5 gene cluster are associated with KD susceptibility (99). Furthermore, it has been found that lipophilic substances almost identical to MAMPs derived from Bacillus cereus, Bacillus subtilis, Y. pseudotuberculosis, and S. aureus, have been detected in the serum of KD patients (79). This finding supports the possibility that KD underlies the molecular pathogenesis similar to the aforementioned mouse models. The possible involvement of Y. pseudotuberculosis and S. aureus in KD has been argued based on other clinical evidence (also see sections Staphylococcus aureus and Streptococcus pyogenes and Yersinia pseudotuberculosis). The collective findings indicate the possible relationship between PAMPs/MAMPs and the aetiological mechanism of KD vasculitis.
Regarding the implication of PRRs, Huang et al. (100) reported that the CpG sites of TLR genes were reversibly hypomethylated in the peripheral whole blood cells of acute KD patients, resulting in upregulated expression of TLRs. This transient epigenetic change supposedly potentiates the TLR-dependent innate immune response. DNA demethylation in immune cells is also observed in patients infected with mycobacteria and certain viruses (101, 102), possibly implicating these intracellular-living pathogens in KD.
While killer immunoglobulin-like receptor (KIR) expressed in natural killer cells interacts with HLA-I to suppress activation of NK cells, KIR3DL1/2, a subtype of KIR, also recognizes and KIR3DL2 engulfs pathogen-derived CpG-oligodeoxynucleotide (CpG-ODN) to activate TLR9-dependent signaling (103). A recent case-control study of HLA-A and -B genotypes in Caucasian KD patients proposed the hypothesis that high abundance of HLA ligands for KIR3DL1/2 in KD patients interferes with KIR-dependent cellular CpG-ODN sensing to impair effective clearance of pathogens during infection (104). Consequently this impaired clearance might allow expansion of PAMPs/MAMPs. Although accumulating evidence suggests that PAMPs/MAMPs are involved in KD pathogenesis, their pathophysiological significance may be more complex than is currently appreciated.
Three complement pathways (classical, lectin, and alternative pathway) are major components of the innate immune system against infectious agents. However, their excess activation causes inflammatory tissue injury. Indeed, accumulating evidence suggests that their dysregulated activation underlies the pathogenesis of vascular inflammation and aortic aneurysms (105, 106). Nevertheless, a limited number of studies had been undertaken regarding the involvement of complement pathways in KD (107, 108).
Recently Okuzaki et al. (109) found that ficolin-1, a circulating soluble PRR that is responsible for activating the lectin pathway, was increased in acute KD. They also demonstrated that KD-like murine vasculitis is ameliorated by infusion of an inhibitory antibody to ficolin-1 (110), suggesting that the lectin pathway could participate in KD pathogenesis. The lectin pathway is triggered by activation of mannose-binding lectin-associated serine proteinases (MASPs). In addition to their role in the complement system, MASPs are involved in coagulation and EC activation (111), both of which are closely connected to KD.
Complement systems are rigorously regulated by more than ten protein species to prevent their undesirable excess activation. Genetic polymorphisms of these proteins might affect susceptibility of KD under infectious condition (106, 112).
While a variety of microorganisms could be causative agents of KD, the prevalence of KD in children is potentially limited, suggesting that the genetic background of an individual likely affects the disease susceptibility. A series of clinical genetic investigations have revealed KD-related gene polymorphisms in more than 20 genes (113), although its aetiological significance is elucidated only in a limited number of these polymorphisms.
Statistical genetic studies identified an SNP associated with KD susceptibility and resistance to IVIG therapy in the gene encoding inositol-triphosphate 3-kinase C (ITPKC), which suppresses T-cell activation and regulates inflammasome activity in macrophages (114, 115). This SNP destabilizes ITPKC mRNA and reduces the cellular level of ITPKC protein (115). KD-associated gene polymorphisms have been discovered in the CASP3 gene, which might also influence the down-regulation of activated immune cells (116). In addition, Onouchi et al. (114) reported KD-associated polymorphisms in the gene encoding ORAI1, a calcium release-activated calcium channel protein 1. Notably, these proteins are also involved in the calcium-dependent nuclear factor-activated T cell (NFAT) pathway (117, 118).which regulates the function of T and B cells. EBV-encoded latent membrane protein 1 (LMP-1) up-regulates ORAI1 (119). Considering that EBV is a possible trigger for KD (see section Epstein-Barr Virus), it is intriguing whether the identified genetic polymorphisms could affect immune response to EBV infection.
The collective molecular genetic data indicate that most genes with KD-associated polymorphisms are responsible for the modulation of inflammatory responses, including T-cell activation, which is compatible with the postulated roles of SAg and PAMPs/MAMPs. This strengthens the possibility that the dysregulated immune response to infectious stimuli underlies the pathogenesis of KD.
Induced pluripotent stem cell technique has been successfully used to establish some human EC lines harboring genetic backgrounds of KD patients (120). Studies with these cells enable the verification of the aetiological significance of individual genetic backgrounds in KD in vitro.
Although the etiology of KD is far from being resolved, the evidence collected so far drives the following hypothesis (Figure 2): Diverse pathogens could be potential causative agents of KD. However, such different infectious stimuli converge on a similar/common immune process associated with the activation of T cells, innate immune cells, and ECs. The genetic background of infected children affects the magnitude of the immune responses to develop KD in a limited number of children.
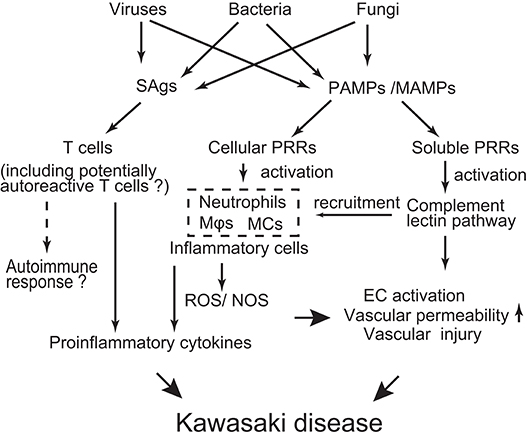
Figure 2. Schematic depicting possible mechanisms of KD pathogenesis. Various infectious agents produce superantigens (SAgs) and pathogen/microbe-associated molecular patterns (PAMPs/MAMPs). SAgs non-specifically activate T cells, probably including potentially autoreactive T cells. PAMPs/MAMPs also stimulate immune cells [e.g., macrophages (Mϕ), dendritic cells (DCs), monocytes (MCs)] and endothelial cells (ECs) through cellular pattern recognition receptors (PRRs) (e.g., TLRs, NOD1, Dectin-1/-2). This stimulation up-regulates production of pro-inflammatory cytokines/chemokines and reactive oxygen/nitrogen species (ROS/NOS), leading to a systemic inflammatory reaction. On the other hand, PAMPs/MAMPs also activate the complement lectin pathway through soluble PRRs (e.g., ficolin-1, mannose binding lectin-2). Activated complement pathways can elicit inflammatory vascular damage through recruitment of innate inflammatory cells and direct injury to ECs. The extent of the inflammatory reaction is influenced by the genetic backgrounds of the individuals, resulting in a limited number of children developing KD in response to infectious stimuli.
The manuscript was written by AN, and it was edited by KI and KH.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Kawasaki T. Acute febrile mucocutaneous syndrome with lymphoid involvement with specific desquamation of the fingers and toes in children. Arerugi. (1967) 16:178–222.
2. AHA Scientific Statement. Diagnostic guidelines for Kawasaki disease. Circulation. (2001) 103:335–6. doi: 10.1161/01.CIR.103.2.335
3. Kuo H. Preventing coronary artery lesions in Kawasaki disease. Biomed J. (2017) 40:141–6. doi: 10.1016/j.bj.2017.04.002
4. Lo MS, Newburger JW. Role of intravenous immunoglobulin in the treatment of Kawasaki disease. Int J Rheum Dis. (2018) 21:64–9. doi: 10.1111/1756-185X.13220
5. Uehara R, Belay ED, Maddox RA, Holman RC, Nakamura Y, Yashiro M, et al. Analysis of potential risk factors associated with nonresponse to initial intravenous immunoglobulin treatment among Kawasaki disease patients in Japan. Pediatr Infect Dis J. (2008) 27:155–60. doi: 10.1097/INF.0b013e31815922b5
6. Shulman ST, Rowley AH. Kawasaki disease: insights into pathogenesis and approaches to treatment. Nat Rev Rheumatol. (2015) 11:475–82. doi: 10.1038/nrrheum.2015.54
7. Chen KYH, Messina N, Germano S, Bonnici R, Freyne B, Cheung M, et al. Innate immune responses following Kawasaki disease and toxic shock syndrome. PLoS ONE. (2018) 13:e0191830. doi: 10.1371/journal.pone.0191830
8. Burns JC, Herzog L, Fabri O, Tremoulet AH, Rodó X, Uehara R, et al. Kawasaki disease global climate consortium. Seasonality of Kawasaki disease: a global perspective. PLoS ONE. (2013) 8:e74529. doi: 10.1371/journal.pone.0074529
9. Nakamura Y, Yashiro M, Uehara R, Sadakane A, Tsuboi S, Aoyama Y, et al. Epidemiologic features of Kawasaki disease in Japan: results of the 2009–2010 nationwide survey. J Epidemiol. (2012) 22:216–21. doi: 10.2188/jea.JE20110126
10. Principi N, Rigante D, Esposito S. The role of infection in Kawasaki syndrome. J Infect. (2013) 67:1–10. doi: 10.1016/j.jinf.2013.04.004
11. Kikuta H, Taguchi Y, Tomizawa K, Kojima K, Kawamura N, Ishizaka A, et al. Epstein-Barr virus genome-positive T lymphocytes in a boy with chronic active EBV infection associated with Kawasaki-like disease. Nature. (1988) 333:455–7. doi: 10.1038/333455a0
12. Kikuta H, Matsumoto S, Yanase Y, Kawasaki T, Mizuno F, Osato T. Recurrence of Kawasaki disease and Epstein-Barr virus infection. J Infect Dis. (1990) 162:1215. doi: 10.1093/infdis/162.5.1215
13. Kikuta H, Sakiyama Y, Matsumoto S, Hamada I, Yazaki M, Iwaki T, et al. Detection of Epstein-Barr virus DNA in cardiac and aortic tissues from chronic, active Epstein-Barr virus infection associated with Kawasaki disease-like coronary artery aneurysms. J Pediatr. (1993) 123:90–2. doi: 10.1016/S0022-3476(05)81546-X
14. Muneuchi J, Ohga S, Ishimura M, Ikeda K, Yamaguchi K, Nomura A, et al. Cardiovascular complications associated with chronic active Epstein-Barr virus infection. Pediatr Cardiol. (2009) 30:274–81. doi: 10.1007/s00246-008-9343-8
15. Ariza ME, Glaser R, Kaumaya PT, Jones C, Williams MV. The EBV-encoded dUTPase activates NF-kappa B through the TLR2 and MyD88-dependent signaling pathway. J Immunol. (2009) 182:851–9. doi: 10.4049/jimmunol.182.2.851
16. Fuse S, Fujinaga E, Mori T, Hotsubo T, Kuroiwa Y, Morii M. Children with Kawasaki disease are not infected with Epstein-Barr virus. Pediatr Infect Dis J. (2010) 29:286–7. doi: 10.1097/INF.0b013e3181c3f111
17. Marchette NJ, Melish ME, Hicks R, Kihara S, Sam E, Ching D. Epstein-Barr virus and other herpesvirus infections in Kawasaki syndrome. J Infect Dis. (1990) 161:680–4. doi: 10.1093/infdis/161.4.680
18. Shike H, Shimizu C, Kanegaye JT, Foley JL, Schnurr DP, Wold LJ, et al. Adenovirus, adeno-associated virus and Kawasaki disease. Pediatr Infect Dis J. (2005) 24:1011–4. doi: 10.1097/01.inf.0000183769.31951.1e
19. Okano M, Thiele GM, Sakiyama Y, Matsumoto S, Purtilo DT. Adenovirus infection in patients with Kawasaki disease. J Med Viol. (1990) 32:53–7. doi: 10.1002/jmv.1890320109
20. Jaggi P, Kajon AE, Mejias A, Ramilo O, Leber A. Human adenovirus infection in Kawasaki disease: a confounding bystander? Clin Infect Dis. (2013) 56:58–64. doi: 10.1093/cid/cis807
21. Popper SJ, Watson VE, Shimizu C, Kanegaye JT, Burns JC, Relman DA. Gene transcript abundance profiles distinguish Kawasaki disease from adenovirus infection. J Infect Dis. (2009) 200:657–66. doi: 10.1086/603538
22. Song E, Kajon AE, Wang H, Salamon D, Texter K, Ramilo O, et al. Clinical and virologic characteristics may aid distinction of acute adenovirus disease from Kawasaki disease with incidental adenovirus detection. J Pediatr. (2016) 170:325–30. doi: 10.1016/j.jpeds.2015.11.021
23. Rowley AH, Shulman ST, Spike BT, Mask CA, Baker SC. Oligoclonal IgA response in the vascular wall in acute Kawasaki disease. J Immunol. (2001) 166:1334–43. doi: 10.4049/jimmunol.166.2.1334
24. Rowley AH, Shulman ST, Mask CA, Finn LS, Terai M, Baker SC, et al. IgA plasma cell infiltration of proximal respiratory tract, pancreas, kidney, and coronary artery in acute Kawasaki disease. J Infect Dis. (2000) 182:1183–91. doi: 10.1086/315832
25. Rowley AH, Baker SC, Orenstein JM, Shulman ST. Searching for the cause of Kawasaki disease–cytoplasmic inclusion bodies provide new insight. Nat Rev Microbiol. (2008) 6:394–401. doi: 10.1038/nrmicro1853
26. Rowley AH, Baker SC, Shulman ST, Garcia FL, Fox LM, Kos IM, et al. RNA-containing cytoplasmic inclusion bodies in ciliated bronchial epithelium months to years after acute Kawasaki disease. PLoS ONE. (2008) 3:e1582. doi: 10.1371/journal.pone.0001582
27. Rowley AH, Shulman ST, Garcia FL, Guzman-Cottrill JA, Miura M, Lee HL, et al. Cloning the arterial IgA antibody response during acute Kawasaki disease. J Immunol. (2005) 175:8386–91. doi: 10.4049/jimmunol.175.12.8386
28. Rowley AH, Wylie KM, Kim KY, Pink AJ, Yang A, Reindel R, et al. The transcriptional profile of coronary arteritis in Kawasaki disease. BMC Genomics. (2015) 16:1076. doi: 10.1186/s12864-015-2323-5
29. Ko TM, Kuo HC, Chang JS, Chen SP, Liu YM, Chen HW, et al. CXCL10/IP-10 is a biomarker and mediator for Kawasaki disease. Circ Res. (2015) 116:876–83. doi: 10.1161/CIRCRESAHA.116.305834
30. Sriskandan S, Faulkner L, Hopkins P. Streptococcus pyogenes: insight into the function of the Streptococcal superantigens. Int J Biochem Cell Biol. (2007) 39:12–9. doi: 10.1016/j.biocel.2006.08.009
31. Spaulding AR, Salgado-Pabón W, Kohler PL, Horswill AR, Leung DY, Schlievert PM. Staphylococcal and Streptococcal superantigen exotoxins. Clin Microbiol Rev. (2013) 26:422–47. doi: 10.1128/CMR.00104-12
32. Faulkner L, Cooper A, Fantino C, Altmann DM, Sriskandan S. The mechanism of superantigen-mediated toxic shock: not a simple Th1 cytokine storm. J Immunol. (2005) 175:6870–7. doi: 10.4049/jimmunol.175.10.6870
33. Turesson C. Endothelial expression of MHC class II molecules in autoimmune disease. Curr Pharm. (2004) 10:129–43. doi: 10.2174/1381612043453414
34. Brogan PA, Shah V, Klein N, Dillon MJ. Vbeta-restricted T cell adherence to endothelial cells: a mechanism for superantigen-dependent vascular injury. Arthritis Rheum. (2004) 50:589–97. doi: 10.1002/art.20021
35. Kulhankova K, Kinney KJ, Stach JM, Gourronc FA, Grumbach IM, Klingelhutz AJ, et al. The superantigen toxic shock syndrome toxin-1 alters human aortic endothelial cell function. Infect Immun. (2017) 11:IAI.00848–17. doi: 10.1128/IAI.00848-17
37. Proft T, Fraser JD. Bacterial superantigens. Clin Exp Immunol. (2003) 133:299–306. doi: 10.1046/j.1365-2249.2003.02203.x
38. Devore-Carter D, Kar S, Vellucci V, Bhattacherjee V, Domanski P, Hostetter MK. Superantigen-like effects of a Candida albicans polypeptide. J Infect Dis. (2008) 197:981–9. doi: 10.1086/529203
39. Bohach GA, Fast DJ, Nelson RD, Schlievert PM. Staphylococcal and streptococcal pyrogenic toxins involved in toxic shock syndrome and related illnesses. Crit Rev Microbiol. (1990) 17:251–72. doi: 10.3109/10408419009105728
40. O'Connell J, Sloand E. Kawasaki disease and Streptococcal scarlet fever. J Nurse Pract. (2013) 9:259–64. doi: 10.1016/j.nurpra.2012.12.013
41. Hall M, Hoyt L, Ferrieri P, Schlievert PM, Jenson HB. Kawasaki syndrome-like illness associated with infection caused by enterotoxin B-secreting Staphylococcus aureus. Clin Infect Dis. (1999) 29:586–9. doi: 10.1086/598638
42. Leung DY, Meissner HC, Fulton DR, Murray DL, Kotzin BL, Schlievert PM. Toxic shock syndrome toxin-secreting Staphylococcus aureus in Kawasaki syndrome. Lancet. (1993) 342:1385–8. doi: 10.1016/0140-6736(93)92752-F
43. Matsubara K, Fukaya T, Miwa K, Shibayama N, Nigami H, Harigaya H, et al. Development of serum IgM antibodies against superantigens of Staphylococcus aureus and Streptococcus pyogenes in Kawasaki disease. Clin Exp Immunol. (2006) 143:427–34. doi: 10.1111/j.1365-2249.2006.03015.x
44. Waisman A, Johann L. Antigen-presenting cell diversity for T cell reactivation in central nervous system autoimmunity. J Mol Med. (2018) 96:1279–92. doi: 10.1007/s00109-018-1709-7
45. Razakandrainibe R, Pelleau S, Grau GE, Jambou R. Antigen presentation by endothelial cells: what role in the pathophysiology of malaria? Trends Parasitol. (2012) 28:151–60. doi: 10.1016/j.pt.2012.01.004
46. Toyosaki T, Yoshioka T, Tsuruta Y, Yutsudo T, Iwasaki M, Suzuki R. Definition of the mitogenic factor (MF) as a novel streptococcal superantigen that is different from streptococcal pyrogenic exotoxins A, B, and C. Eur J Immunol. (1996) 26:2693–701. doi: 10.1002/eji.1830261122
47. Yoshioka T, Matsutani T, Iwagami S, Toyosaki-Maeda T, Yutsudo T, Tsuruta Y, Suzuki H, et al. Polyclonal expansion of TCRBV2- and TCRBV6-bearing T-cells in patients with Kawasaki disease. Immunology. (1999) 96:465–72. doi: 10.1046/j.1365-2567.1999.00695.x
48. Suenaga T, Suzuki H, Shibuta S, Takeuchi T, Yoshikawa N. Detection of multiple superantigen genes in stools of patients with Kawasaki disease. J Pediatr. (2009) 155:266–70. doi: 10.1016/j.jpeds.2009.03.013
49. Cunningham MW, Antone SM, Smart M, Liu R, Kosanke S. Molecular analysis of human cardiac myosin-cross-reactive B- and T-cell epitopes of the group A streptococcal M5 protein. Infect Immun. (1997) 65:3913–23.
50. Cunningham MW, Meissner HC, Heuser JS, Pietra BA, Kurahara DK, Leung DY. Anti-human cardiac myosin autoantibodies in Kawasaki syndrome. J Immunol. (1999) 163:1060–5.
51. Nussinovitch U, Shoenfeld Y. The clinical and diagnostic significance of anti-myosin autoantibodies in cardiac disease. Clin Rev Allergy Immunol. (2013) 44:98–108. doi: 10.1007/s12016-010-8229-8
52. Matsunaga A, Harita Y, Shibagaki Y, Shimizu N, Shibuya K, Ono H, et al. Identification of 4-trimethylaminobutyraldehyde dehydrogenase (TMABA-DH) as a candidate serum autoantibody target for Kawasaki disease. PLoS ONE. (2015) 10:e0128189. doi: 10.1371/journal.pone.0128189
53. Fujieda M, Karasawa R, Takasugi H, Yamamoto M, Kataoka K, Yudoh K, et al. A novel anti-peroxiredoxin autoantibody in patients with Kawasaki disease. Microbiol Immunol. (2012) 56:56–61. doi: 10.1111/j.1348-0421.2011.00393.x
54. Hurst JR, Kasper KJ, Sule AN, McCormick JK. Streptococcal pharyngitis and rheumatic heart disease: the superantigen hypothesis revisited. Infect Genet Evol. (2018) 61:160–75. doi: 10.1016/j.meegid.2018.03.006
55. Xu M, Jiang Y, Wang J, Liu D, Wang S, Yi H, et al. Distribution of distinct subsets of circulating T follicular helper cells in Kawasaki disease. BMC Pediatr. (2019) 19:43. doi: 10.1186/s12887-019-1412-z
56. Liu D, Liu J, Wang J, Guo L, Liu C, Jiang Y, et al. Distribution of circulating T follicular helper cell subsets is altered in immunoglobulin A vasculitis in children. PLoS ONE. (2017) 12:e0189133. doi: 10.1371/journal.pone.0189133
57. Rigante D, Castellazzi L, Bosco A, Esposito S. Is there a crossroad between infections, genetics and Henoch-Schönlein purpura? Autoimmunity Rev. (2013) 12:1016–21. doi: 10.1016/j.autrev.2013.04.003
58. Akiyama M, Yasuoka H, Yoshimoto K, Takeuchi T. Interleukin-4 contributes to the shift of balance of IgG subclasses toward IgG4 in IgG4-related disease. Cytokine. (2018) 110:416–9. doi: 10.1016/j.cyto.2018.05.009
59. Ishizaka N. IgG4-related disease underlying the pathogenesis of coronary artery disease. Clin Chim Acta. (2013) 415:20–5. doi: 10.1016/j.cca.2012.11.003
60. Morita A, Imada Y, Igarashi H, Yutsudo T. Serologic evidence that streptococcal superantigens are not involved in the pathogenesis of Kawasaki disease. Microbiol Immunol. (1997) 41:895–900. doi: 10.1111/j.1348-0421.1997.tb01947.x
61. Gupta-Malhotra M, Viteri-Jackson A, Thomas W, Zabriskie JB. Antibodies to highly conserved peptide sequence of staphylococcal and streptococcal superantigens in Kawasaki disease. Exp Mol Pathol. (2004) 76:117–21. doi: 10.1016/j.yexmp.2003.12.003
62. Vincent P, Salo E, Skurnik M, Fukushima H, Simonet M. Similarities of Kawasaki disease and Yersinia pseudotuberculosis infection epidemiology. Pediatric Infect Dis J. (2007) 26:629–31. doi: 10.1097/INF.0b013e3180616d3c
63. Donadini R, Fields BA. Yersinia pseudotuberculosis superantigens. Chem Immunol Allergy. (2007) 93:77–91. doi: 10.1159/000100859
64. Fukushima H, Matsuda Y, Seki R, Tsubokura M, Takeda N, Shubin FN, et al. Geographical heterogeneity between Far Eastern and Western countries in prevalence of the virulence plasmid, the superantigen Yersinia pseudotuberculosis-derived mitogen, and the high-pathogenicity island among Yersinia pseudotuberculosis strains. J Clin Microbiol. (2001) 39:3541–7. doi: 10.1128/JCM.39.10.3541-3547.2001
65. Horinouchi T, Nozu K, Hamahira K, Inaguma Y, Abe J, Nakajima H, et al. Yersinia pseudotuberculosis infection in Kawasaki disease and its clinical characteristics. BMC Pediatr. (2015) 15:177. doi: 10.1186/s12887-015-0497-2
66. Goubard A, Loïez C, Abe J, Fichel C, Herwegh S, Faveeuw C, et al. Superantigenic Yersinia pseudotuberculosis induces the expression of granzymes and perforin by CD4+ T cells. Infect Immun. (2015) 83:2053–64. doi: 10.1128/IAI.02339-14
67. Yokota S, Tsubaki K, Kuriyama T, Shimizu H, Ibe M, Mitsuda T, et al. Presence in Kawasaki disease of antibodies to mycobacterial heat-shock protein HSP65 and autoantibodies to epitopes of human HSP65 cognate antigen. Clin Immunol Immunopathol. (1993) 67:163–70. doi: 10.1006/clin.1993.1060
68. Sireci G, Dieli F, Salerno A. T cells recognize an immunodominant epitope of heat shock protein 65 in Kawasaki disease. Mol Med. (2000) 6:581–90. doi: 10.1007/BF03401796
69. Tatano Y, Shimizu T, Tomioka H. Unique macrophages different from M1/M2 macrophages inhibit T cell mitogenesis while upregulating Th17 polarization. Sci Rep. (2014) 4:4146. doi: 10.1038/srep04146
70. Takahashi K, Oharaseki T, Yokouchi Y. Histopathological aspects of cardiovascular lesions in Kawasaki disease. Int J Rheumatic Dis. (2018) 21:31–5. doi: 10.1111/1756-185X.13207
71. Guo MM, Tseng WN, Ko CH, Pan HM, Hsieh KS, Kuo HC. Th17- and Treg-related cytokine and mRNA expression are associated with acute and resolving Kawasaki disease. Allergy. (2015) 70:310–8. doi: 10.1111/all.12558
72. Rodó X, Curcoll R, Robinson M, Ballester J, Burns JC, Cayan DR, et al. Tropospheric winds from northeastern China carry the etiologic agent of Kawasaki disease from its source to Japan. Proc Natl Acad Sci USA. (2014) 111:7952–7. doi: 10.1073/pnas.1400380111
73. Manlhiot C, Mueller B, O'Shea S, Majeed H, Bernknopf B, Labelle M, et al. Environmental epidemiology of Kawasaki disease: Linking disease etiology, pathogenesis and global distribution. PLoS ONE. (2018) 13:e0191087. doi: 10.1371/journal.pone.0191087
74. Baba K, Takeda N, Tanaka M. Cases of Yersinia pseudotuberculosis infection having diagnostic criteria of Kawasaki disease. Contrib Microbiol Immunol. (1991) 12:292–6.
75. Sato K. Yersinia pseudotuberculosis infection in children. Contrib Microbiol Immunol. (1987) 9:111–6.
76. Vollmer-Conna U, Piraino BF, Cameron B, Davenport T, Hickie I, Wakefield D, et al. Cytokine polymorphisms have a synergistic effect on severity of the acute sickness response to infection. Clin Infect Dis. (2008) 47:1418–25. doi: 10.1086/592967
77. Xiao TS. Innate immunity and inflammation. Cell Mol Immunol. (2017) 14:1–3. doi: 10.1038/cmi.2016.45
78. Hara T, Nakashima Y, Sakai Y, Nishio H, Motomura Y, Yamasaki S. Kawasaki disease: a matter of innate immunity. Clin Exp Immunol. (2016) 186:134–43. doi: 10.1111/cei.12832
79. Kusuda T, Nakashima Y, Murata K, Kanno S, Nishio H, Saito M, et al. Kawasaki disease-specific molecules in the sera are linked to microbe-associated molecular patterns in the biofilms. PLoS ONE. (2014) 9:e113054. doi: 10.1371/journal.pone.0113054
80. Hamaoka-Okamoto A, Suzuki C, Yahata T, Ikeda K, Nagi-Miura N, Ohno N, et al. The involvement of the vasa vasorum in the development of vasculitis in animal model of Kawasaki disease. Pediatr Rheumatol Online J. (2014) 12:12. doi: 10.1186/1546-0096-12-12
81. Ohno N. Chemistry and biology of angiitis inducer, Candida albicans water-soluble mannoprotein-beta-glucan complex (CAWS). Microbiol Immunol. (2003) 47:479–90. doi: 10.1111/j.1348-0421.2003.tb03409.x
82. Fujii M, Tanaka H, Nakamura A, Suzuki C, Harada Y, Takamatsu T, et al. Histopathological characteristics of post-inflamed coronary arteries in Kawasaki disease-like vasculitis of rabbits. Acta Histochem Cytochem. (2016) 49:29–36. doi: 10.1267/ahc.15028
83. Philip S, Lee WC, Wu MH, Mammen CK, Lue HC. Histopathological evaluation of horse serum-induced immune complex vasculitis in swine: implication to coronary artery lesions in Kawasaki disease. Pediatr Neonatol. (2014) 55:297–305. doi: 10.1016/j.pedneo.2013.10.012
84. Yoshikane Y, Koga M, Imanaka-Yoshida K, Cho T, Yamamoto Y, Yoshida T, et al. JNK is critical for the development of Candida albicans-induced vascular lesions in a mouse model of Kawasaki disease. Cardiovasc Pathol. (2015) 24:33–40. doi: 10.1016/j.carpath.2014.08.005
85. Miyabe C, Miyabe Y, Miura NN, Takahashi K, Terashima Y, Toda E, et al. Am80, a retinoic acid receptor agonist, ameliorates murine vasculitis through the suppression of neutrophil migration and activation. Arthritis Rheum. (2013) 65:503–12. doi: 10.1002/art.37784
86. Suzuki C, Nakamura A, Miura N, Fukai K, Ohno N, Yahata T, et al. Non-receptor type, proline-rich protein tyrosine kinase 2 (Pyk2) is a possible therapeutic target for Kawasaki disease. Clin Immunol. (2017) 179:17–24. doi: 10.1016/j.clim.2017.01.013
87. Hirata N, Ishibashi K, Sato W, Nagi-Miura N, Adachi Y, Ohta S, et al. β-mannosyl linkages inhibit CAWS arteritis by negatively regulating dectin-2-dependent signaling in spleen and dendritic cells. Immunopharmacol Immunotoxicol. (2013) 35:594–604. doi: 10.3109/08923973.2013.830124
88. Matundan HH, Sin J, Rivas MN, Fishbein MC, Lehman TJ, Chen S, et al. Myocardial fibrosis after adrenergic stimulation as a long-term sequela in a mouse model of Kawasaki disease vasculitis. JCI Insight. (2019) 4:126279. doi: 10.1172/jci.insight.126279
89. Martinez HG, Quinones MP, Jimenez F, Estrada C, Clark KM, Suzuki K, et al. (2012) Important role of CCR2 in a murine model of coronary vasculitis. BMC Immunol. 13:56. doi: 10.1186/1471-2172-13-56
90. Stock AT, Hansen JA, Sleeman MA, McKenzie BS, Wicks IP. GM-CSF primes cardiac inflammation in a mouse model of Kawasaki disease. J Exp Med. (2016) 213:1983–98. doi: 10.1084/jem.20151853
91. Lee Y, Schulte DJ, Shimada K, Chen S, Crother TR, Chiba N, et al. Interleukin-1β is crucial for the induction of coronary artery inflammation in a mouse model of Kawasaki disease. Circulation. (2012) 125:1542–50. doi: 10.1161/CIRCULATIONAHA.111.072769
92. Rosenkranz ME, Schulte DJ, Agle LM, Wong MH, Zhang W, Ivashkiv L, et al. TLR2 and MyD88 contribute to Lactobacillus casei extract-induced focal coronary arteritis in a mouse model of Kawasaki disease. Circulation. (2005) 112:2966–73. doi: 10.1161/CIRCULATIONAHA.105.537530
93. Noval Rivas M, Lee Y, Wakita D, Chiba N, Dagvadorj J, Shimada K, et al. CD8+ T cells contribute to the development of coronary arteritis in the Lactobacillus casei cell wall extract-induced murine model of Kawasaki disease. Arthritis Rheumatol. (2017) 69:410–21. doi: 10.1002/art.39939
94. Nishio H, Kanno S, Onoyama S, Ikeda K, Tanaka T, Kusuhara K, et al. Nod1 ligands induce site-specific vascular inflammation. Arterioscler Thromb Vasc Biol. (2011) 31:1093–9. doi: 10.1161/ATVBAHA.110.216325
95. Motomura Y, Kanno S, Asano K, Tanaka M, Hasegawa Y, Katagiri H, et al. Identification of pathogenic cardiac CD11c+ macrophages in Nod1-mediated acute coronary arteritis. Arterioscler Thromb Vasc Biol. (2015) 35:1423–33. doi: 10.1161/ATVBAHA.114.304846
96. Kitaura Y, Nakaguchi O, Takeno H, Okada S, Yonishi S, Hemmi K, et al. N2-(gamma-D-Glutamyl)-meso-2(L),2'(D)-diaminopimelic acid as the minimal prerequisite structure of FK-156: its acyl derivatives with potent immunostimulating activity. J Med Chem. (1982) 25:335–7. doi: 10.1021/jm00346a001
97. Osterburg AR, Hexley P, Supp DM, Robinson CT, Noel G, Ogle C, et al. Concerns over interspecies transcriptional comparisons in mice and humans after trauma. Proc Natl Acad Sci USA. (2013) 110:E3370. doi: 10.1073/pnas.1306033110
98. Ishibashi K, Fukazawa R, Miura NN, Adachi Y, Ogawa S, Ohno N. Diagnostic potential of antibody titres against Candida cell wall β-glucan in Kawasaki disease. Clin Exp Immunol. (2014) 177:161–7. doi: 10.1111/cei.12328
99. Breunis WB, Biezeveld MH, Geissler J, Kuipers IM, Lam J, Ottenkamp J, et al. Polymorphisms in chemokine receptor genes and susceptibility to Kawasaki disease. Clin Exp Immunol. (2007) 150:83–90. doi: 10.1111/j.1365-2249.2007.03457.x
100. Huang YH, Li SC, Huang LH, Chen PC, Lin YY, Lin CC, et al. Identifying genetic hypomethylation and upregulation of Toll-like receptors in Kawasaki disease. Oncotarget. (2017) 8:11249–58. doi: 10.18632/oncotarget.14497
101. Pacis A, Tailleux L, Morin AM, Lambourne J, MacIsaac JL, Yotova V, et al. Bacterial infection remodels the DNA methylation landscape of human dendritic cells. Genome Res. (2015) 25:1801–11. doi: 10.1101/gr.192005.115
102. Hernando H, Shannon-Lowe C, Islam AB, Al-Shahrour F, Rodríguez-Ubreva J, Rodríguez-Cortez VC, et al. The B cell transcription program mediates hypomethylation and overexpression of key genes in Epstein-Barr virus-associated proliferative conversion. Genome Biol. (2013) 14:R3. doi: 10.1186/gb-2013-14-1-r3
103. Sivori S, Falco M, Carlomagno S, Romeo E, Soldani C, Bensussan A, et al. A novel KIR-associated function: evidence that CpG DNA uptake and shuttling to early endosomes is mediated by KIR3DL2. Blood. (2010) 116:1637–47. doi: 10.1182/blood-2009-12-256586
104. Capittini C, Emmi G, Mannarino S, Bossi G, Dellepiane RM, Salice P, et al. An immune-molecular hypothesis supporting infectious aetiopathogenesis of Kawasaki disease in children. Eur J Immunol. (2018) 48:543–5. doi: 10.1002/eji.201747226
105. Chimenti MS, Ballanti E, Triggianese P, Perricone R. Vasculitides and the complement system. Clin Rev Allergy Immunol. (2015) 49:333–46. doi: 10.1007/s12016-014-8453-8
106. Zhou HF, Yan H, Bertram P, Hu Y, Springer LE, Thompson RW, et al. Fibrinogen-specific antibody induces abdominal aortic aneurysm in mice through complement lectin pathway activation. Proc Natl Acad Sci USA. (2013) 110:E4335–44. doi: 10.1073/pnas.1315512110
107. Kohsaka T, Abe J, Asahina T, Kobayashi N. Classical pathway complement activation in Kawasaki syndrome. J Allergy Clin Immunol. (1994) 93:520–5. doi: 10.1016/0091-6749(94)90362-X
108. Biezeveld MH, Geissler J, Weverling GJ, Kuipers IM, Lam J, Ottenkamp J, et al. Polymorphisms in the mannose-binding lectin gene as determinants of age-defined risk of coronary artery lesions in Kawasaki disease. Arthritis Rheum. (2006) 54:369–76. doi: 10.1002/art.21529
109. Okuzaki D, Ota K, Takatsuki SI, Akiyoshi Y, Naoi K, Yabuta N, et al. FCN1 (M-ficolin), which directly associates with immunoglobulin G, is a molecular target of intravenous immunoglobulin therapy for Kawasaki disease. Sci Rep. (2017) 7:11334. doi: 10.1038/s41598-017-11108-0
110. Katayama M, Ota K, Nagi-Miura N, Ohno N, Yabuta N, Nojima H, et al. Ficolin-1 is a promising therapeutic target for autoimmune diseases. Int Immunol. (2019) 6:23–32. doi: 10.1093/intimm/dxy056
111. Dobó J, Pál G, Cervenak L, Gál P. The emerging roles of mannose-binding lectin-associated serine proteases (MASPs) in the lectin pathway of complement and beyond. Immunol Rev. (2016) 274:98–111. doi: 10.1111/imr.12460
112. Sato S, Kawashima H, Kashiwagi Y, Fujioka T, Takekuma K, Hoshika A. Association of mannose-binding lectin gene polymorphisms with Kawasaki disease in the Japanese. Association of mannose-binding lectin gene polymorphisms with Kawasaki disease in the Japanese. Int J Rheumatol. (2009) 12:307–10. doi: 10.1111/j.1756-185X.2009.01428.x
113. Onouchi Y. Molecular genetics of Kawasaki disease. Pediatr Res. (2009) 65 (5 Pt 2):46R−54R. doi: 10.1203/PDR.0b013e31819dba60
114. Onouchi Y, Gunji T, Burns JC, Shimizu C, Newburger JW, Yashiro M, et al. ITPKC functional polymorphism associated with Kawasaki disease susceptibility and formation of coronary artery aneurysms. Nat Genet. (2008) 40:35–42. doi: 10.1038/ng.2007.59
115. Alphonse MP, Duong TT, Shumitzu C, Hoang TL, McCrindle BW, Franco A, et al. Inositol-triphosphate 3-kinase C mediates inflammasome activation and treatment response in Kawasaki disease. J Immunol. (2016) 197:3481–9. doi: 10.4049/jimmunol.1600388
116. Onouchi Y, Ozaki K, Buns JC, Shimizu C, Hamada H, Honda T, et al. Common variants in CASP3 confer susceptibility to Kawasaki disease. Hum Mol Genet. (2010) 19:2898–906. doi: 10.1093/hmg/ddq176
117. Onouchi Y, Fukazawa R, Yamamura K, Suzuki H, Kakimoto N, Suenaga T, et al. Variations in ORAI1 gene associated with Kawasaki disease. PLoS ONE. (2016) 11:e0145486. doi: 10.1371/journal.pone.0145486
118. Srikanth S, Gwack Y. Orai1-NFAT signalling pathway triggered by T cell receptor stimulation. Mol Cells. (2013) 35:182–94. doi: 10.1007/s10059-013-0073-2
119. Dellis O, Arbabian A, Papp B, Rowe M, Joab I, Chomienne C. Epstein-Barr virus latent membrane protein 1 increases calcium influx through store-operated channels in B lymphoid cells. J Biol Chem. (2011) 286:18583–92. doi: 10.1074/jbc.M111.222257
120. Ikeda K, Mizoro Y, Ameku T, Nomiya Y, Mae SI, Matsui S, et al. Transcriptional analysis of intravenous immunoglobulin resistance in Kawasaki disease using an induced pluripotent stem cell disease model. Circ J. (2016) 81:110–8. doi: 10.1253/circj.CJ-16-0541
121. Dominguez SR, Anderson MS, Glodé MP, Robinson CC, Holmes KV. Blinded case-control study of the relationship between human coronavirus NL63 and Kawasaki syndrome. J Infect Dis. (2006) 194:1697–701. doi: 10.1086/509509
122. Chang LY, Chiang BL, Kao CL, Wu MH, Chen PJ, Berkhout B, et al. Kawasaki Disease Research Group. Lack of association between infection with a novel human coronavirus (HCoV), HCoV-NH, and Kawasaki disease in Taiwan. J Infect Dis. (2006) 193:283–6. doi: 10.1086/498875
123. Williams MV, Cox B, Ariza ME. Herpesviruses dUTPases: a new family of pathogen-associated molecular pattern (PAMP) proteins with implications for human disease. Pathogens. (2016) 6:E2. doi: 10.3390/pathogens6010002
124. Johnson RM, Bergmann KR, Manaloor JJ, Yu X, Slaven JE, Kharbanda AB. Pediatric Kawasaki disease and adult human immunodeficiency virus kawasaki-like syndrome are likely the same malady. Open Forum Infect Dis. (2016) 3:ofw160. doi: 10.1093/ofid/ofw160
125. Bajolle F, Meritet JF, Rozenberg F, Chalumeau M, Bonnet D, Gendrel D, et al. Markers of a recent bocavirus infection in children with Kawasaki disease: “a year prospective study”. Pathol Biol. (2014) 62:356–8. doi: 10.1016/j.patbio.2014.06.002
126. Thissen JB, Isshiki M, Jaing C, Nagao Y, Lebron Aldea D, Allen JE, et al. A novel variant of torque teno virus 7 identified in patients with Kawasaki disease. PLoS ONE. (2018) 13:e0209683. doi: 10.1371/journal.pone.0209683
127. Nigro G, Zerbini M, Krzysztofiak A, Gentilomi G, Porcaro MA, Mango T, et al. Active or recent parvovirus B19 infection in children with Kawasaki disease. Lancet. (1994) 343:1260–1. doi: 10.1016/S0140-6736(94)92154-7
128. Lin IC, Suen JL, Huang SK, Huang SC, Huang HC, Kuo HC, et al. Dectin-1/Syk signaling is involved in Lactobacillus casei cell wall extract-induced mouse model of Kawasaki disease. Immunobiology. (2013) 218:201–12. doi: 10.1016/j.imbio.2012.04.004
129. Vitale EA, La Torre F, Calcagno G, Infricciori G, Fede C, Conti G, et al. Mycoplasma pneumoniae: a possible trigger of kawasaki disease or a mere coincidental association? Report of the first four Italian cases. Minerva Pediatr. (2010) 62:605–7.
130. Lehman TJ, Mahnovski V. Animal models of vasculitis. Lessons we can learn to improve our understanding of Kawasaki disease. Rheum Dis Clin North Am. (1998) 14:479–87.
131. Yonekawa A, Saijo S, Hoshino Y, Miyake Y, Ishikawa E, Suzukawa M, et al. Dectin-2 is a direct receptor for mannose-capped lipoarabinomannan of mycobacteria. Immunity. (2014) 41:402–13. doi: 10.1016/j.immuni.2014.08.005
132. Oharaseki T, Yokouchi Y, Enomoto Y, Sato W, Ishibashi K, Miura N, et al. Recognition of alpha-mannan by dectin 2 is essential for onset of Kawasaki disease-like murine vasculitis induced by Candida albicans cell-wall polysaccharide. Mod Rheumatol. (2019) 29:1–8. doi: 10.1080/14397595.2019.1601852
Keywords: Kawasaki disease, vasculitis, animal models, infection, superantigens, pattern recognition receptors
Citation: Nakamura A, Ikeda K and Hamaoka K (2019) Aetiological Significance of Infectious Stimuli in Kawasaki Disease. Front. Pediatr. 7:244. doi: 10.3389/fped.2019.00244
Received: 31 December 2018; Accepted: 29 May 2019;
Published: 28 June 2019.
Edited by:
Hiromichi Hamada, Tokyo Women's Medical University Yachiyo Medical Center, JapanReviewed by:
Ying-Hsien Huang, Kaohsiung Chang Gung Memorial Hospital, TaiwanCopyright © 2019 Nakamura, Ikeda and Hamaoka. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Akihiro Nakamura, bmFrYW05OTNAa290by5rcHUtbS5hYy5qcA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.