 Jason A. Tye-Din
Jason A. Tye-Din Heather J. Galipeau
Heather J. Galipeau Daniel Agardh
Daniel Agardh- 1Immunology Division, The Walter and Eliza Hall Institute, Parkville, VIC, Australia
- 2Department of Medical Biology, University of Melbourne, Parkville, VIC, Australia
- 3Department of Gastroenterology, The Royal Melbourne Hospital, Parkville, VIC, Australia
- 4Centre for Food & Allergy Research, Murdoch Children's Research Institute, Parkville, VIC, Australia
- 5Farncombe Family Digestive Health Research Institute, McMaster University, Hamilton, ON, Canada
- 6The Diabetes and Celiac Disease Unit, Department of Clinical Sciences, Lund University, Malmö, Sweden
- 7Unit of Endocrinology and Gastroenterology, Department of Pediatrics, Skåne University Hospital, Malmö, Sweden
Our understanding of celiac disease and how it develops has evolved significantly over the last half century. Although traditionally viewed as a pediatric illness characterized by malabsorption, it is now better seen as an immune illness with systemic manifestations affecting all ages. Population studies reveal this global disease is common and, in many countries, increasing in prevalence. These studies underscore the importance of specific HLA susceptibility genes and gluten consumption in disease development and suggest that other genetic and environmental factors could also play a role. The emerging data on viral and bacterial microbe-host interactions and their alterations in celiac disease provides a plausible mechanism linking environmental risk and disease development. Although the inflammatory lesion of celiac disease is complex, the strong HLA association highlights a central role for pathogenic T cells responding to select gluten peptides that have now been defined for the most common genetic form of celiac disease. What remains less understood is how loss of tolerance to gluten occurs. New insights into celiac disease are now providing opportunities to intervene in its development, course, diagnosis, and treatment.
Introduction
Celiac disease (CeD) is a chronicimmune-mediated enteropathyprecipitated by to dietary gluten in genetically predisposed individuals (1). Current diagnosis is based on demonstrating the enteropathy in small intestinal biopsies where histologic examination shows villous atrophy, crypt hyperplasia and intraepithelial lymphocytosis, and the presence of circulating CeD-specific antibodies to tissue transglutaminase (tTG), deamidated gliadin peptides (DGP), and endomysium (EMA). In children who have symptoms suggestive of CeD, a strongly positive tTG antibody (tTGA) titre and a CeD-associated HLA genotype, the diagnosis of CeD may be possible without the need for small intestinal biopsy (2). Since the 1950s when gluten was identified as the causative trigger of CeD, a strict and lifelong gluten-free diet (GFD) has been the mainstay of treatment.
While CeD is common around the globe and is rising in prevalence in many populations, it is frequently undetected in clinical practice (3). Symptomatic and untreated disease is associated with elevated morbidity and mortality and impaired quality of life (4–7). The clinical presentation is broad and includes gastrointestinal upset, chronic fatigue, nutrient deficiencies, poor growth, and failure to thrive. Extra-intestinal manifestations are common, and while they were once considered more frequent in adults than children with CeD, recent data suggests the frequency is similar in CeD children, although the types and rates of recovery differ (8). In children, extra-intestinal manifestations include short stature, anemia, delayed puberty, dental enamel hypoplasia, reduced bone density, oral ulcers, liver and biliary disease, and dermatitis herpetiformis. Poor growth and anemia tend to be the most common and there is a correlation with more severe histological damage at diagnosis compared to children with a gastrointestinal presentation or screen-detected cases (9). Insidious effects of undiagnosed CeD in children include behavioral disturbances and reduced educational performance (10).
CeD is also associated with an increased risk of autoimmune diseases including Hashimoto's thyroiditis, Graves' disease and type 1 diabetes (T1D) (11–14). A large Danish population study revealed the prevalence of autoimmune diseases was 16.4% among CeD patients compared with 5.3% in the general population in 2016 (12). Approximately 5% of patients with CeD have T1D and ~6% of T1D patients have CeD (15). In Northern Sardinia, a population with a high prevalence of CeD (16), patients with autoimmune thyroiditis had a 4-fold greater prevalence of CeD than the general population and while iron deficiency was present in almost half, none had gastrointestinal symptoms (17). The co-occurrence of autoimmune diseases supports the concept of shared genetic and immune pathways contributing to immune dysregulation and loss of self-tolerance, however it remains unclear whether CeD directly leads to other autoimmune disease and whether early diagnosis and treatment with a GFD alters this risk (18). Next generation sequencing of the HLA region shows that extended class II haplotypes differ between populations (19); this may partially explain regional differences in the degree of association between CeD and autoimmune disease. The strong association of CeD with autoimmunity, especially T1D and autoimmune thyroid disease, supports screening for these conditions in CeD patients.
The consumption of gluten containing foods is necessary for CeD to develop. Gluten is the viscoelastic protein that remains after washing dough and is composed of alcohol-soluble gliadins and alcohol-insoluble glutenins. The rheological properties of gluten allow it to impart a light and extensible texture to food making it highly favored in the food industry. Modern wheat gluten arises from a hexaploid genome, making it heterogeneous and more genetically complex than the human genome. Similar proteins rich in glutamine and proline (thus, the collective term prolamins) are found in barley and rye and termed hordeins and secalins, respectively, and are also toxic in CeD. Avenin, the prolamin component in oats, is phylogenetically distinct from wheat, barley and rye prolamin. Oats are considered safe for consumption by most people with CeD, although adverse immune and clinical effects have been reported (20–22) and some cultivars may be more immunogenic (23). Several expert reviews have concluded that further research into the toxicity of oats in CeD is needed (24, 25).
Why CeD develops in some people remains unanswered. Epidemiologic and prospective observational studies implicate a range of environmental factors that impact disease development. A role for the microbiome in the development of tolerance and disease pathogenesis is also emerging. Genetic and immunological studies have revealed the importance of key HLA and non-HLA susceptibility genes in disease development and a long-lived pathogenic population of gluten-specific T cells targeting certain gluten peptides (T cell epitopes). The emerging story of CeD development is one where environmental factors increase the risk of CeD in genetically predisposed individuals by shaping the immunologic context in which gluten is presented and shifting the balance from gluten tolerance to reactivity, and that this may be in part mediated through microbiome-host interactions. Contemporary clinical issues of importance include expediting the detection and diagnosis of CeD, improving and quality of life and health outcomes for those diagnosed, and developing treatments that are more effective and less burdensome than the current approach of a lifelong and strict GFD. This article will review current knowledge about CeD development and pathogenesis and how this knowledge is being applied to explore novel diagnostic, treatment and preventative approaches.
The Role of Environment in Disease Development
CeD is a global disease that has been reported in Western and Eastern Europe, North America, South America, Asia, Oceania, and Africa (3, 26). It appears to be relatively uncommon in southeast Asia and sub-Saharan Africa. Recent reports from China suggest the illness could be substantially unrecognized there however more biopsy-based studies are required (27). In a systematic review and meta-analysis, the global seroprevalence and biopsy-confirmed prevalence of CeD was estimated to be 1.4% and 0.7%, respectively (3). The sero-prevalence of CeD in the US from National Health and Nutrition Examination Surveys (NHANES) was 0.7% and, consistent with a range of population studies from around the globe, showed that most cases remain undiagnosed in the community (28). Since CeD is frequently undiagnosed an active case-finding approach is considered best practice.
The prevalence of CeD varies with sex, age, and geographic location with the frequency of predisposing HLA haplotypes in the general population and per-capita wheat consumption the two major determinants of prevalence. There is a modest gender bias in favor of females (29). Familial clustering in CeD is common with 10% of first-degree relatives of a CeD sufferer affected. The high concordance rate for monozygotic twins (~80%) compared to HLA-identical siblings (~30%) and dizygotic twins (~10%) underscores the importance of both genetic factors (HLA and non-HLA genes) and the environment in CeD risk (30).
The high prevalence of CeD observed among populations living in areas with a high consumption of wheat products are highly suggestive for involvement of dietary gluten in CeD development (26). Although intake of gluten is necessary for CeD to develop, it does not solely explain why not all genetically-predisposed individuals consuming gluten develop CeD and why the disease can develop later in life despite many decades of gluten intake. Significant differences in the prevalence of CeD between people of similar genetic background and wheat intake living in nearby regions (for example, Finland and Russian Karelia) is strong evidence that the risk of CeD is influenced by other factors apart from genetic susceptibility and wheat consumption (31). Indeed, population studies have implicated a range of environmental factors associated with CeD risk (summarized in Table 1). Heterogeneity of study design has yielded conflicting results in the search for triggers in CeD. We have limited our review to focus on the association between CeD and diet, infections, antibiotic use, and delivery mode.
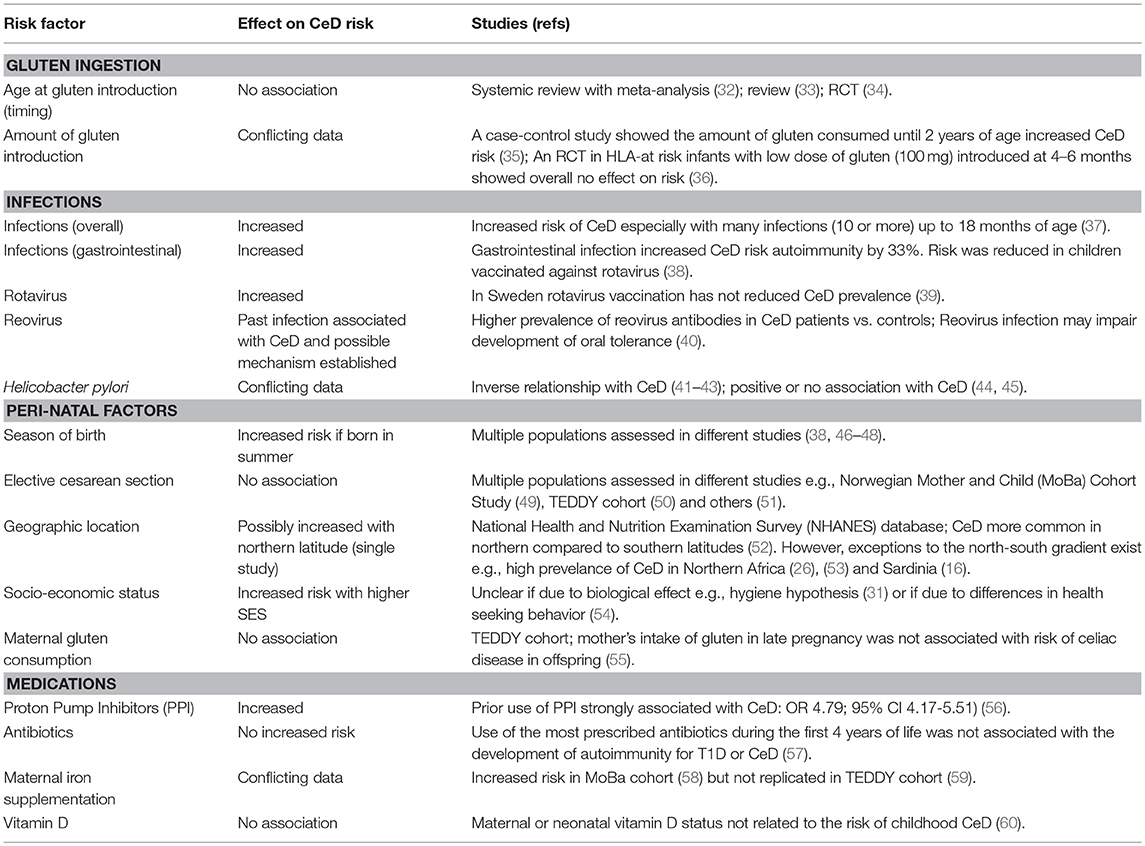
Table 1. Environmental factors potentially associated with CeD development.
Infant Feeding
The steep rise in CeD incidence in young children after changes in the Swedish national infant feeding recommendation in the mid-1980s that suggested postponing gluten introduction from 4 to 6 months of age hinted that timing of gluten intake influenced CeD risk (61). However, the epidemic of CeD that occurred in Sweden occurred simultaneously with companies raising the gluten content in commercial baby formulas and was confounded by an observed protective effect of long breastfeeding duration (62). This made it difficult to disentangle whether timing or amount of gluten intake in relation to weaning impacted on the risk of CeD. The hypothesis that timing of first gluten exposure was associated with CeD was further supported in a study that found infants exposed to gluten either early (< 4 months) or late (>7 months) were at an increased risk (63). Since this first prospective study was published several follow-up papers from larger longitudinal prospective birth cohorts summarized in two recent systemic reviews with meta-analysis (32, 33) have not been able to confirm the previous findings that either age of gluten introduction or breast-feeding influence CeD risk.
Although there are wide differences in gluten intake between countries (64), it is not entirely clear whether the quantity of gluten intake during early childhood affects the risk of CeD. A Swedish retrospective case-control study indicated that children that later developed CeD consumed larger amounts of gluten before the age of 2 years than healthy children (62). This finding was in line with another cross-sectional study from the same group that observed a lower prevalence of CeD in a birth cohort reporting a lower gluten consumption in children born after (65) as compared to children born during the years of the Swedish epidemic (66). In a nested case-control study, a high intake of gluten amount increased the risk for CeD in Swedish children (35). However, whether gluten intake contributes to CeD development is still controversial as another multicenter study consisting of five other European countries found no association with CeD and gluten amount except for children carrying the lower-risk HLA-DQ2.2/DQ7 haplotype (67). Larger prospective studies with a longer follow-up are underway and will shed light on whether gluten intake is an independent risk factor in CeD.
Infections
Several studies have shown that children that later develop CeD are more frequently affected by infections during early life (37, 68, 69). One limitation is that these studies are based on questionnaires filled in by parents and the type and site of infection is not specified. In a multicenter, prospective birth cohort study parents that reported a gastrointestinal infection 3 months prior to seroconversion of tTGA were at an increased risk of CeD autoimmunity later in life (38). There is also an effect of seasonality on the risk of developing CeD, hypothesized to be caused by viral infections occurring during a vulnerable period of immune development. This is supported by the association with frequent rotavirus infections and increased risk of CeD autoimmunity from longitudinal prospective studies (39) and a protective effect of rotavirus vaccination (38).
How infections trigger CeD development remains unexplained. Gastrointestinal infections may increase gastrointestinal permeability to increase the passage of gluten across the mucosa, or elevate tTG expression that can increase the generation of immunogenic gluten peptides. Molecular mimicry could possibly occur if the foreign antigen (such as a virus or bacteria) shares sequence or structural similarities with gluten itself and then initiates an anti-gluten response. Several studies have shown antibodies to adenovirus (70–72) and rotavirus peptides (73) circulating in CeD sera but further studies are required to determine the significance of these associations with disease pathogenesis. In recent work in mice, viral infection led to a break in oral tolerance to dietary proteins (40). Some reoviruses can promote a proinflammatory phenotype in mouse dendritic cells (DCs) which lose their capacity to promote tolerance toward food antigens and cause a pathogenic T cell response instead. Reovirus infection causes increased signaling by type 1 interferons and increased expression of the transcription factor interferon regulatory factor 1 (IRF1) which can block the conversion of T cells into regulatory T cells (Tregs) and promote a proinflammatory TH1 response to dietary antigens, respectively. Supporting relevance in humans, patients with CeD tended to have higher anti-reovirus antibody titers. Importantly, reovirus infections are often silent or asymptomatic in humans and a large proportion of the population is exposed to self-limiting gastrointestinal infections during childhood. The findings provide a mechanistic explanation that links an apparently innocuous virus with the loss of tolerance to a common food antigen. More research is required to unravel the significance of viral, bacterial or other microbial host interactions or infections in the development of CeD.
Antibiotics and Delivery Mode
Early case-control studies reported a link between prior antibiotic use and subsequent CeD development in both adults (74) and children (69). Similarly, children with CeD were more likely to have been born by cesarean section (c-section) (75). A large case-control study found that while emergency c-section was not associated with later CeD development, elective cesarean delivery was (76). However, conflicting data has been reported. For instance, no link between increased CeD risk and antibiotic use during the first 6 months of life (68) or antibiotic use during pregnancy (77) was found. The Environmental Determinants of Diabetes in the Young (TEDDY) study is a multicenter observational cohort study that aims to identify environmental factors associated with T1D and CeD in children at HLA risk followed from birth (78). It found no association between antibiotic use and CeD autoimmunity during the first 4 years of life (57) or between delivery by c-section and increased CeD risk (50). Similarly, large observational studies found no link between c-section and CeD development (49, 69, 79). Finally, a large register-based study, that included children from two independent cohorts, found that birth delivery mode was not associated with increased risk of diagnosed CeD (51). Although the data is conflicting, the potential links between early events that can alter the microbiota composition, such as antibiotic use or birth delivery mode, and later CeD implicate a role of the microbiome in disease development.
The Microbiome in Celiac Disease
Microbial colonization occurs at birth and shapes the development of the mucosal and systemic immune system and the intestinal barrier. These host-microbe interactions continue throughout life, and a disruption of these interactions, through altered bacterial composition or functions, have been hypothesized to increase the risk of a range of autoimmune or inflammatory diseases such as CeD. Altered microbiota composition in patients with CeD may represent an environmental modifier of CeD development.
An early study described the presence of rod-shaped bacteria in duodenal biopsies of Swedish children with CeD born during the epidemic, which weren't observed in biopsies of control children (80), or in children born following the epidemic (81). The bacteria were subsequently identified as Clostridium spp, Prevotella spp, and Actinomyces spp, and their presence was suggested to be a risk factor for CeD that contributed to the increase in disease incidence in Sweden from 1985-1995 (81). Subsequent clinical studies have described differences in both fecal and duodenal microbial composition in children and adults with active compared with treated CeD, or healthy controls (82). While no specific microbial signature has been described for CeD, many groups have described increases in the proportions of Bacteroides and members of Proteobacteria, and decreases in Lactobacillus and Bifidobacterium (83, 84). In addition, CeD patients suffering from persistent symptoms were shown to have increased abundance of Proteobacteria compared to those who were asymptomatic (85). While these studies suggest an association between altered microbial composition and development of CeD, studies exploring mechanisms and causality are lacking. Moreover, whether alterations in the microbial composition are a cause or consequence of small intestinal inflammation has not been fully elucidated.
Recent studies have suggested that the microbiota from CeD patients may harbor more pathogenic or pro-inflammatory bacteria. Reports of CeD diagnosis following Campylobacter jejuni infection (86) suggest that bacterial infections could precede CeD development. Escherichia coli clones isolated from CeD patients expressed a higher number of virulent genes compared to those isolated from healthy controls (87). Similarly, the presence of virulent genes were higher in Staphylococcus spp and in Bacteroides fragilis strains isolated from CeD patients compared to healthy controls (88, 89). Importantly, strains isolated from CeD patients were more pro-inflammatory in vitro and stimulated altered DC morphology, characteristic of DC maturation, increased pro-inflammatory cytokine production, and altered epithelial barrier integrity. Similarly, Neisseria flavescens, a member of Proteobacteria, was identified in the duodenum of active CeD patients but not from control subjects and induced an inflammatory phenotype in human and murine DCs (90).
In contrast to the above studies, bacterial infections may also protect against CeD development. Some studies indicate an inverse relationship between the presence of Helicobacter pylori and CeD in both adults and children (41–43) whereas other studies have shown a positive or no association (44, 45). Mechanisms underlying this association have not been elucidated and inconsistencies across studies may relate to differences in techniques used to determine H. pylori status or H. pylori virulence. Less virulent strains may exacerbate the mucosal response in CeD whereas more virulent strains may provide protection against CeD (45, 91).
Functional differences in the microbiota could also affect metabolic processes important in CeD pathogenesis. The gastrointestinal tract harbors diverse bacteria that participate in gluten metabolism in vitro and this may differ between healthy individuals and those with CeD (92–94). As most studies have measured microbial composition in active or treated CeD compared to healthy controls it is difficult to determine whether functional differences are present prior to disease onset.
To gain insight into the potential role of microbial factors in CeD development, previous studies have profiled the fecal microbial composition of genetically at-risk children. High-risk children were shown to harbor a different microbiota compared to children who were at low genetic risk for CeD (95, 96), suggesting that the high-risk genotype may influence early gut microbiota composition. Infants at the highest risk for CeD had a higher prevalence of enterotoxigenic E. coli compared to those at low or intermediate risk for CeD (97). In addition, in a cohort of 164 infants, those at risk for CeD had lower numbers of Bifidobacterium spp and B. longum and increased numbers of B. fragilis and Staphylococcus spp. The differences in Bacteroides and bifidobacteria were attenuated by breastfeeding (98). At-risk children that later developed CeD were recently shown to have an altered microbial trajectory that coincided with immune changes. These changes were suggestive of a “premature maturation” of the gut microbiota in children who went on to develop CeD (99). On the other hand, the fecal microbiota of at-risk infants who went on to develop CeD was similar at 9–12 months to those infants that remained healthy by the age of four (100). Whether the duodenal microbial composition or function is altered in at-risk individuals that go on to develop CeD needs to be investigated further in larger clinical trials.
Diet and environment also determine gut microbiota composition (101, 102), highlighting the complexity of delineating the influence of genotype and environment on shaping the microbiota. Larger clinical trials where both the composition and function of the microbiota is studied in at-risk individuals and followed over time are needed to help understand gene-microbe interactions in CeD development.
The Role of Genetics in Disease Development
While environmental factors are important for CeD development a notable feature of CeD is its high heritability and strong HLA association (103). This strong genetic association reflects the central role of CD4+ T cells as the HLA molecules associated with CeD bind specific gluten peptides that activate T cells (104). Ninety percent of Caucasian CeD patients possess the HLA-DQ2.5 haplotype (encoded by the DQA1*05:01 and DQB1*02:01 alleles) either in cis or trans positions, and the remaining carry either HLA-DQ8 (encoded by the DQA1*03:01 and DQB1*03:02 alleles), HLA-DQ2.2 alone (encoded by the DQB1*02:02 allele) or HLA-DQ7 alone (encoded by the DQA1*05:01 allele). Less than 1% of CeD patients lack these HLA haplotypes (105) and their absence can be exploited in the clinical setting to assist in excluding a diagnosis of CeD.
A “gene-dose effect” related to the number of copies of the DQB1*02 allele has been reported to affect CeD risk, clinical phenotype and patient responses to a T cell targeted therapy. The presumed basis for this effect is that gluten presented by APCs in HLA-DQ2.5 homozygous (i.e., two copies of DQB1*02) individuals can induce at least a 4-fold higher T-cell response compared with gluten presented by APCs in HLA-DQ2.5 heterozygous (i.e., one copy of DQB1*02) individuals (106). The CeD risk in HLA-DQ2.5 homozygous patients is ~2.5 and 5 times that conferred by HLA-DQ2.5 heterozygosity and lower risk HLA groups, respectively (107). A prospective Italian study (Celiac Disease and Age at Gluten Introduction study; CELIPREV) followed newborns with a family history of CeD and showed the risk of CeD autoimmunity (positive CeD-serology panel) at 10 years of age was far higher among children who were HLA-DQ2.5 homozygous (or who had two copies of DQB1*02 than among those who were HLA-DQ2.5 heterozygous or HLA-DQ8 (38 vs. 19%, P = 0.001), as was the risk of overt CeD (26 vs. 16%, P = 0.05) (34). In this cohort, 80% of those in whom CeD developed did so during the first 3 years of life. In the TEDDY study following 6403 US and European genetically at risk children at for CeD, the risks of CeD autoimmunity and confirmed CeD by age 5 were 11 and 3%, respectively in the heterozygous children and 26 and 11%, respectively, in those who were homozygous (108).
In addition, HLA-DQ2.5 homozygosity has been associated with a more severe CeD phenotype with earlier disease onset, greater villous atrophy, diarrhea, and lower hemoglobin at presentation, and a slower rate of villous healing on a GFD (109), plus a higher rate of refractory (non-responsive) CeD (110). In a recent clinical trial of an immunotherapy targeting gluten-specific CD4+ T cells, CeD subjects who were HLA-DQ2.5 homozygous were more likely to experience gastrointestinal symptoms following systemic administration compared to those who were heterozygous (111).
The contribution of non-HLA genes to CeD risk susceptibility is much less strong (OR < 1.5) than the HLA-associated haplotypes (OR >5) but collectively are significant. More than 70 candidate genes in over 40 non-HLA loci have been implicated in CeD heritability (112–117). These loci encode proteins involved in a range of immune pathways affecting T and B cell activation, chemokine receptor activity and cell migration, cytokine binding, thymic differentiation of CD4+ and CD8+ T cells, stress pathways and innate immunity. Only one gene has been shown to be gut specific (RGS1), underscoring the systemic nature of immune dysregulation in CeD (114). To date there is no evidence to implicate specific alleles encoding gastrointestinal proteases or tTG. Mirroring the frequent disease co-occurrence, there is substantial overlap between genetic risk factors for CeD and those of autoimmune diseases such as rheumatoid arthritis, multiple sclerosis and T1D (112, 115, 118, 119). Furthermore, despite a much weaker clinical association with CeD, there is overlap of genetic risk loci for inflammatory bowel disease such as Crohn's disease (18, 120). Intriguingly, 90% of the identified risk loci map to non-coding regions such as promoter regions, enhancers or non-coding RNA genes, suggesting that regulation of gene expression rather than changes at the protein-coding level are more important for CeD susceptibility and development (121).
A Key Role for T Cells In Pathogenesis
The role of CD4+ helper T cells in CeD was confirmed with the isolation of pro-inflammatory gluten-specific CD4+ T cells from intestinal tissue of CeD patients (122) (Figure 1). These pathogenic T cells have a Th1 phenotype characterized by production of IFN-γ and TNF-α (123) and almost all are HLA-DQ2- and/or DQ8-restricted (122, 124, 125). Gluten peptides that have been post-translationally modified by the enzyme tTG in a process called deamidation can effectively activate these T cells (126, 127). Deamidation converts specific glutamine residues to glutamate and this modification enhances the gluten peptide's binding affinity to disease-associated HLA dimers (128–130). Deamidation is crucial in converting poorly immunogenic wild-type gluten peptides to highly immunogenic antigens for CD4+ T cells. The structural requirements that generate effective binding of gluten peptides to HLA-DQ2 or DQ8 and T cells via the T cell receptor (TCR) have been further elucidated in structural studies (131, 132) and assessment of the biased use of TCR genes (133, 134).
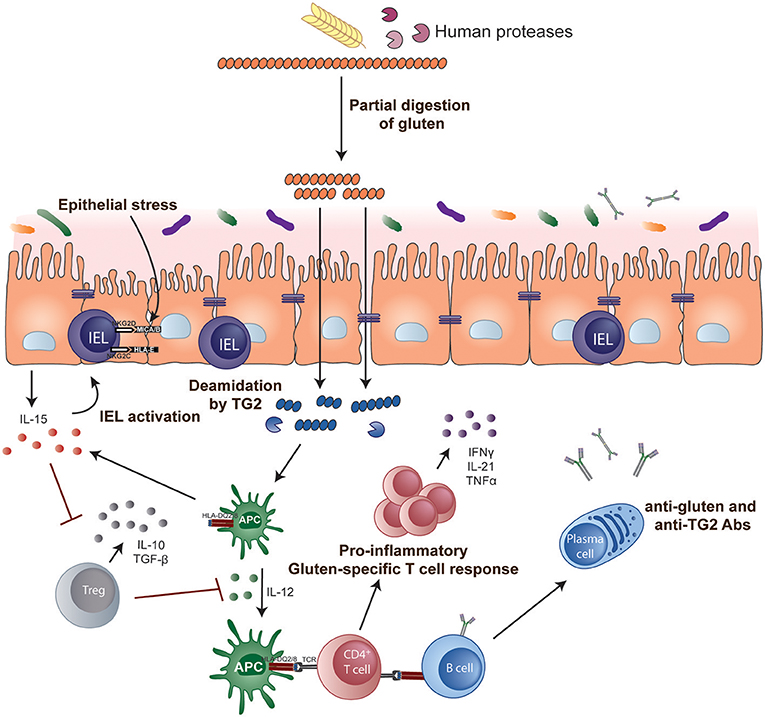
Figure 1. Key steps in CeD pathogenesis. Gluten peptides containing T-cell epitopes resist gastrointestinal degradation. tTG catalyses the deamidation of gluten peptides, which can then bind more efficiently to the disease-relevant HLA-DQ molecules on APCs. Activated gluten-specific CD4+ T cells secrete a variety of pro-inflammatory cytokines such as IFN-γ and IL-21 that contribute to the intestinal lesion and promote activation of IELs and stimulate B-cell responses. Activated IELs transform into cytolytic NK-like cells that mediate destruction of enterocytes expressing stress signals. IL-15 renders effector T cells resistant to the suppressive effects of Tregs and, in the lamina propria, endows mucosal DCs with inflammatory properties promoting pro-inflammatory responses and preventing Treg differentiation.
Studies of T cells isolated from the intestine of CeD patients, or from their blood after short-term oral gluten challenge, have been used to define the gluten peptides (specifically, the T cell epitopes) immunogenic in CeD. Most studies have focused on the 90% of CeD patients who are HLA-DQ2.5 and a range of immunodominant T cell epitopes have been defined (135). Less is known about the nature of the T cell response to gluten in CeD patients without HLA-DQ2.5. The most immunogenic gluten epitopes for CeD patients with HLA-DQ2.5 after wheat ingestion reside in α-gliadin and ω-gliadin (136). Much of the field has focused on the immunogenicity of T cell epitopes in α-gliadin, specifically those encompassed within a protease resistant 33mer (137). However, gluten challenge studies show that the most immunogenic peptides induced by gluten ingestion depends on whether wheat, rye or barley is ingested, and that a sequence from ω-gliadin (encompassing the T cell epitopes DQ2.5-glia-ω1 and DQ2.5-glia-ω2) is dominant irrespective of which grain is consumed (136). Despite the multitude of immunogenic peptides, just three peptides from wheat and barley appear to recapitulate most of the response to gluten in CeD patients with HLA-DQ2.5. Interestingly, after oat ingestion, about 8% of CeD patients have detectable T cells specific for avenin peptides that share close sequence homology with barley hordein, suggesting that cross-reactive T cells may mediate immune responses following oats ingestion in some CeD patients (21).
While early work suggested T cells from children with CeD displayed a different pattern of reactivity to gluten compared to adults (138), more recent studies show gluten-specific T cells in blood induced by oral wheat challenge (139), or expanded from the small intestine during active disease (140), share the same specificity for deamidated, immunodominant T cell epitopes across all ages. The same gluten-specific T cell clonotypes persist in patients' blood and intestinal tissue up to several decades and share the same TCR gene use motifs in CeD patients from Norway, Finland and Australia (134, 139, 141, 142). Their stability over such long periods of time may be maintained by ongoing gluten exposure as inadvertent gluten intake is common in CeD even when a strict gluten-free diet is attempted (143, 144).
Antibodies and B Cells In Celiac Disease
Measurement of tTGA is a useful screening test for CeD as the titer reflects disease activity caused by gluten, however a direct role in disease pathogenesis is less clear. As tTGA are anti-angiogenic they may contribute to some extra-intestinal manifestations of CeD (145). As tTGA are detectable in intestinal tissue prior to the typical enteropathy of CeD and predict future disease onset, antibody production is likely to occur early in disease development (146). However, approximately 50% of children with positive CeD serology normalize their levels despite ongoing gluten exposure, suggesting that gluten immunity that leads to a pathogenic response is not necessarily fixed once it has commenced (147). Supporting this concept is the observation that almost 20% of adults with CeD diagnosed during childhood who elect to resume gluten intake have no evidence of active disease (148).
Production of tTGA appears reliant on T cells, as antibody formation to tTG and DGP is strictly dependent on the presence of CeD-associated HLA types as well as gluten. This supports the idea that tTG-specific B cells internalize tTG in complex with gluten peptides and present gluten-derived peptides to gluten-specific T cells, effectively amplifying the T cell response. These T cells then provide the required “help” to the B cell, resulting in production of tTGA and DGP antibodies (149, 150).
In active CeD, a large number of plasma cells can be found in the intestinal lesion and tTG-specific plasma cells made up a large proportion of them (5–25%) (151). There is much that still needs to be understood about how these antibody producing cells are selected and mature. tTG can form covalently linked multimers with itself that readily bind gluten peptides and can be taken up by tTG-specific BCR transduced cells and activate gluten-specific T cells with increased capacity compared to tTG monomers (152). Immunoglobulin expressed on B cells could act as substrates for tTG, in particular IgD, resulting in BCR/tTG cross-linking (153). As B cell epitopes are in close proximity to immunodominant T cell epitopes (154) and react to a higher degree to deamidated peptides (155) future work needs to examine the B cell and T cell interaction during the gluten-specific immune response.
The Development of Intestinal Villous Atrophy
The events that culminate in the histological changes of CeD are incompletely understood. Gluten T cell epitopes cluster in regions of high proline making them resistant to the effect of gastrointestinal proteases (156). These peptides may pass across the epithelium via transcellular (157–159) and paracellular (receptor or antibody mediated) pathways (160). Increased epithelial permeability, a feature of active CeD, may be mediated by a direct effect of gliadin acting via the chemokine receptor CXCR3 in intestinal epithelial cells on tight junctions (160). DCs are presumed to play a major role in presentation of gluten peptides to CD4+ T cells (161), however little is known about their identity, where this presentation occurs, and the extent to which this role is undertaken by gluten-specific B cells. Activated CD4+ T cells produce large amounts of IFN-γ, that may induce cytotoxicity of intraepithelial lymphocytes (IELs), as well as IL-21, which plays a role in T-cell-dependent B cell responses (162). IL-17 producing CD4+ T cells have been reported in untreated CeD (163) but their role and that of IL-17A production in CeD pathogenesis is less clear (162, 164). IL-15 and IFN-α feature prominently in the inflamed tissue in CeD patients (165, 166). IELs are believed to play an important effector role in mediating destruction of enterocytes in CeD in a TCR-independent manner. When activated by stress signals on intestinal epithelial cells such as HLA-E and MIC-A (167), IELs express high levels of NK activating receptors such as NKG2D and CD94/NKG2C and adopt a cytolytic phenotype capable of destroying enterocytes (168). IL-15 plays a key role by upregulating the activating NKG2D receptor and acting as a co-stimulatory molecule. The effect is to ‘license' cytotoxic IELs with the ability to kill intestinal epithelial cells expressing the stress-induced MIC molecules. Adaptive immunity to gluten and epithelial stress where cytotoxic IELs have acquired an activated NK cell phenotype may both be required for villous atrophy to develop in CeD (169).
The causes of epithelial stress that trigger IEL activation and transformation in CeD are not known. Some hypothesize it is driven by gluten itself or other stimuli such as those resulting from a microbe-host interaction (Figure 2). The innate immune system is a pre-programmed form of host defense that responds rapidly to stimuli. Responses are triggered when pattern recognition receptors, for example, toll-like receptors on macrophages, bind molecules with conserved structures. An innate immune stimulatory effect of a gliadin sequence (A-gliadin p31-43) has been reported, but this work has not been replicated (170). No other gluten peptides activating innate immunity in humans have been defined. Gliadin may function as a stress signal for the activation of MICA expression only in the initial stages of disease and decline to baseline levels once the inflammatory lesion is established. In contrast to the critical role of HLA-restricted, gluten-specific CD4+ T cells in CeD pathogenesis, the relative contribution of innate immunity to disease has not been established in genetic or functional studies and further research in this area is required.
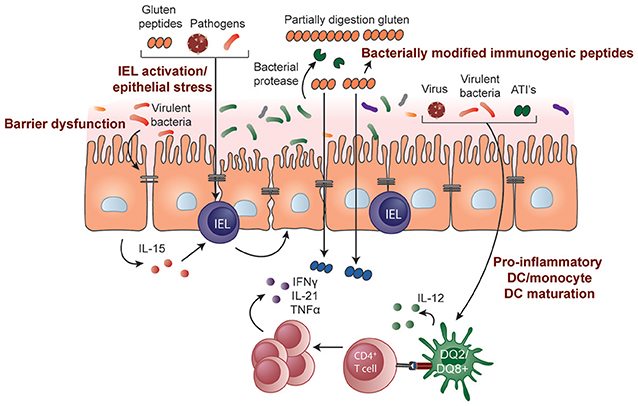
Figure 2. Potential role of microbes and environmental triggers in CeD pathogenesis. Microbes that include both commensals and opportunistic pathogens may contribute to the development of CeD by influencing gluten peptide digestion, intestinal barrier function, epithelial cell stress, or IEL activation/upregulation through IL-15 regulation. Pathogenic bacteria, viruses, and non-gluten components of wheat, such as amylase-trypsin inhibitors (ATIs), may also induce DC maturation and proinflammatory cytokine production, modulating the induction of CD4+ T-cell responses.
How Does Loss of Tolerance to Gluten Develop?
Although gluten consumption is common in the Western diet most individuals who possess HLA susceptibility for CeD develop immunologic tolerance to it as they do for other food proteins. Why a small percentage fail to develop or lose tolerance to gluten remains unresolved. Tregs are an important immune component contributing to intestinal homeostasis and when functioning normally inhibit pathogenic responses to dietary antigens and help maintain oral tolerance. This role suggests they may be important in CeD, however identifying and studying this cell population has been limited by the technical challenge of identifying a population of cells truly gluten-specific and functionally suppressive in vivo. These challenges have meant that studies of Treg function in CeD have been conflicting, with some indicating reduced suppressive function (171–173) and some showing it is retained (174, 175). Further studies that definitively isolate true gluten-specific Tregs and assess their function in CeD are needed.
Interestingly, the immunosuppressive cytokines IL-10 and TGF-β can be detected in high amounts in the CeD intestinal lesion but appear insufficient to counter active disease (176, 177). The high levels of pro-inflammatory cytokines such as IFN-γ and IL-15 may render this anti-inflammatory response inadequate (178, 179). IL-15 is a pleiotropic cytokine that may promote inflammation through several pathways, including driving the accumulation of cytotoxic IELs in the CeD lesion, interfering with the suppressive activity of Tregs (175), impairing TGF-β signaling (176), or allowing activation of disease-specific CD4+ T cells (180). IL-15 also drives the expansion of aberrant IEL clones that can lead to the development of enteropathy-associated T cell lymphoma. The multiple pro-inflammatory effects of IL-15 make it a rational target for therapeutic blockade, and clinical trials of anti-IL-15 biologic agents are underway in refractory CeD (181).
Additional insights into molecular pathways altered in loss of tolerance may be gained by study of gene expression profiles in CeD patients. Analysis of blood-derived non-gluten stimulated CD4+ T cells in CeD showed significant upregulation of the IFN-γ gene and reduced expression of a network of BACH2 regulated genes (182). BACH2 is a transcription factor that plays an important immunoregulatory role in inhibiting T effector cell development and promoting development of Tregs. Advances in techniques such as RNAseq now affords the opportunity to analyze multiple transcriptional and phenotypic features of antigen-specific effector T cells and Tregs to provide insights at the single cell level into these heterogeneous populations.
The findings on Treg numbers in the CeD lesion suggest that the defect in tolerance is not simply due to defects in numbers or recruitment of suppressive cells, but that the pro-inflammatory environment characteristic of CeD overcomes the tolerogenic milieu that normally maintains balance and inhibits abnormal immune responses. However, further studies on disease-relevant Tregs from the small intestine of CeD patients that assess antigen-specificity, function in vivo and the factors that impact their function are required.
From Pathogenesis to Clinical Care
Insights From Animal Models
Modeling CeD has been a challenge as no single animal model that encompasses all elements of disease exists. As a result, mouse models have played a limited role in the development or pre-clinical testing of new therapies (183) and have more often been used to investigate specific mechanisms related to disease pathogenesis (184). The transfer of gluten-specific T cells into immunodeficient mice have been used to study the role of CD4+ T cells in mediating tissue damage (185, 186). Transgenic mouse models have also been used to investigate specific cytokines or genetic components in CeD pathogenesis. For example, mice over expressing IL-15 in the lamina propria (165) or in the epithelium (180) have shed light on the role of innate mediators in the development of the intestinal lesion in CeD. Mice that express human HLA-DQ2 or -DQ8 develop gluten-specific T cells and some innate immune activation following sensitization to gliadin with an adjuvant. However, they do not progress to full blown gluten-induced enteropathy (183, 187–189), emphasizing the importance of additional genetic, immune, or environmental factors in triggering tissue destruction in CeD. This lack of spontaneous loss of tolerance to gluten in transgenic mouse models can be taken advantage of and utilized to better understand environmental factors that participate in the loss of tolerance to gluten. For example, the mechanisms through which microbes contribute to CeD development can be studied by manipulating the microbiota composition or exposing transgenic mouse models to certain bacteria.
In mice expressing human HLA-DQ8 the composition of the gut microbiota was found to influence the degree of gluten-induced immunopathology (190). Mice harboring a limited microbiota devoid of Proteobacteria and opportunistic pathogens were protected from gluten-induced pathology and immune responses. However, this protective effect was lost when these mice were supplemented with an enteroadherent strain of E. coli that was isolated from a CeD patient. Similarly, treatment of specific pathogen free mice with vancomycin increased Proteobacteria levels, including Escherichia, and led to more severe gluten-induced pathology. While the mechanisms remain elusive, the results provide a proof-of-concept that microbes could alter how a host responds to gluten and could therefore be targeted as a prophylactic approach.
Gnotobiotic mice, or mice colonized with known microbes, provide a model where the impact of specific bacteria on gluten-mediated responses in vivo can be studied in a controlled environment. Studies of mice colonized with bacteria isolated from the duodenum of CeD patients or from healthy controls have shown that bacteria participate in gluten metabolism in vivo (94). Interestingly, the immunogenicity of the end products generated by bacterially-mediated gluten digestion differed depending on the type of bacteria. Following human protease digestion, elastase from Pseudomonas aeruginosa generated highly immunogenic gluten peptides that could strongly activate gluten-specific T cells from human CeD patients. These peptides were better able to translocate the epithelial barrier, potentially facilitating immune cell-peptide interactions. Conversely, gluten peptides produced following digestion by human proteases or by elastase from P. aeruginosa were detoxified or degraded by Lactobacillus spp, a core member of a healthy microbiome. The continued use of gnotobiotic models will be critical for understanding how microbes or pathogens may interact with the host and/or gluten to contribute to CeD pathogenesis (Figure 2). Importantly, these models can also be used to test microbiota-targeted therapies for CeD.
Can Celiac Disease Be Prevented?
Population studies have provided important insights into environmental factors associated with CeD but are unable to establish true causality or mechanism. If factors that impact CeD risk can be identified and modified, then prevention of CeD may be possible. Randomized controlled trials (RCTs) do allow a controlled assessment of how a factor impacts CeD risk and several have now been undertaken or are underway in genetically at-risk infants or children (Table 2). The results have sometimes differed from assumptions made in population studies, underscoring the importance of running well designed and controlled intervention studies and undertaking research to examine mechanism.

Table 2. Prospective trials in infants/children looking at factors impacting celiac disease development.
Two independent RCTs assessed whether low amounts of gluten can prevent genetically at-risk children from developing CeD. The idea of a “window of opportunity” during which gluten could be introduced in small amounts to induce tolerance was based on previous experience from the Swedish epidemic and supported by a prospective study showing infants exposed to gluten either early (< 4 months) or late (>7 months) were at an increased CeD risk (63). The optimal window was proposed between 4 and 6 months, preferably during ongoing breast-feeding (191). In the multicenter PreventCD study, almost 1,000 genetically at-risk children with at least one first-degree relative with CeD were randomized to a double-blind, placebo-controlled dietary-intervention to receive 100 mg of gluten daily or placebo from 16 to 24 weeks of age (36). Neither breast-feeding nor the introduction of small quantities of gluten at 16–24 weeks of age reduced the risk of CeD by 3 years of age in this group of high-risk children (36). Published in the same journal, the CELIPREV group randomised over 800 newborns who had a first-degree relative with CeD to have dietary gluten introduced at either 6 or 12 months (34). The results from the CELIPREV study concorded with the results from the PreventCD study, showing that neither the delayed introduction of gluten nor breast-feeding modified the risk of CeD among at-risk infants (34). Although CeD was not prevented by delaying the introduction of gluten, it was associated with a delayed onset of disease (34). Since these two RCTs were published, other clinical trials have been performed or are currently being conducted.
Another potential approach to CeD prevention is through modification of the host-microbe interactions in at-risk individuals. Trials to assess the impact of probiotic supplementation in preventing CeD in genetically at-risk children are underway (Table 2). If successful, they would support the idea that altered function of the microbiome is a major event underpinning the development of CeD, and that preventing dysregulated host-microbe interactions may be of prophylactic benefit. Furthermore, a better understanding of the mechanisms through which microbes contribute to CeD development can provide further rationale and a more targeted approach for microbiota-modulating preventative strategies.
Improving the Diagnosis of Celiac Disease
While villous atrophy remains the cornerstone of CeD diagnosis there is the growing realization that this “gold-standard” has limitations. For example, results are affected by the number of samples collected and how the biopsies are oriented and reported (192–195). Ultra-short CeD where villous atrophy is present only in the duodenal bulb and “mild enteropathy CeD” where villous atrophy is absent in the setting of positive CeD serology both present diagnostic challenges and highlight potential shortcomings of histology (196, 197). Improvement in the quality of serological testing for CeD and the requirement for specific HLA genotypes for CeD to develop has meant that a serogenetic approach to CeD diagnosis is appealing and may be sufficient for diagnosis in the right clinical situations (2, 198). As expeditious treatment of CeD may avoid or reduce the risk of many CeD-associated complications such as impaired bone density and stunted growth in children, improving early diagnosis remains a clinical and research priority.
In recent years the high rate of community adoption of the GFD, including in children (199), has compounded the challenge of CeD diagnosis as the accuracy of current serological and histological approaches depend on active gluten intake. In order to make a CeD diagnosis reintroduction of dietary gluten, generally for several weeks to months, is recommended prior to testing but patients are often reluctant to undertake this and for those that do many fail to tolerate it. As the serologic and histologic response to gluten challenge is highly heterogeneous the optimal duration of gluten challenge required for definitive diagnosis of CeD remains uncertain (200–202). Immune diagnostics that measure the gluten-specific immune response target a fundamental component of CeD and may overcome the limitations of current diagnostics. The use of tetramers (203) or cytokine release assays (204) to identify gluten-specific T cells induced in blood after short-term oral gluten challenge is highly sensitive and specific for CeD (205). Diagnostics that are accurate with limited or even no gluten exposure such as tetramer-based detection of gluten-specfic T cells (206) are particularly appealing to clinicians and patients as they may avoid the need for prolonged gluten challenge prior to testing with serology and histology. Large multi-center validation studies to confirm the accuracy of assessing disease-specific T cells as a CeD diagnostic are required, and if successful, may force a re-think of how CeD should be classified. Arguably, CeD may be better defined by the HLA-linked, T cell mediated systemic response to gluten rather than histologic changes in the proximal small intestine or circulating antibodies that indirectly reflect disease activity.
Improving the Treatment of Celiac Disease
While adherence to a strict and lifelong GFD still remains the single proven and available treatment for CeD, it is for many patients complicated, onerous, and expensive. In adults with CeD, daily consumption of as little as 50 mg of gluten, equivalent to that contained in 1/100th of a slice of standard wheat bread, over three months can damage the small intestine (207). A safe gluten “dose” threshold relevant to children with CeD has not been assessed in a controlled trial. Several longitudinal studies in adults with CeD indicate that failure to achieve mucosal healing is common even in those appearing to maintain good dietary adherence over many years (208–213). While healing is considered to be more complete and faster in children with CeD treated with a GFD one study showed 19% had persistent disease activity after 12 months on a GFD (214). Assuming enough time has elapsed on the GFD, persistent mucosal activity may be driven by ongoing, potentially intermittent, gluten exposure (143), such as that inadvertently consumed in contaminated meals when eating out (144). The challenge in maintaining adequately strict gluten exclusion and persistent disease activity is a major driver for research into new therapeutic approaches. While several therapies are under development it is notable that none of them have yet been evaluated in children.
Clinical trials of novel therapies for CeD have increased substantially in recent years but compared to other illnesses such as inflammatory bowel disease the field is still in its infancy. No therapeutic approach for CeD has yet completed Phase 3 clinical trials. An understanding of the optimal goals of treatment and the methods to assess efficacy are an evolving area and have been shaped by the requirements of regulatory bodies such as the FDA. Symptom improvement is now regarded as a key outcome measure and this has driven interest in validating patient reported outcome measures (215) and understanding the basis for gluten-induced symptoms in CeD. A standardized approach to reporting small intestinal histology based on quantitative assessment of morphology (villus height, crypt depth and their ratio) and inflammation (density of intraepithelial lymphocytes) is now commonly employed in CeD clinical trials (192). Confirming adequate dietary gluten exclusion during studies is a major challenge as symptom records, serology, histology, and dietary history are indirect measures of GFD adherence (216). New technology based on the detection of urinary or fecal gluten immunogenic peptides (GIPs) derived from the 33mer peptide in wheat α-gliadin provides objective evidence of dietary gluten exposure (217, 218). In addition to a role in the clinic, it may be a promising tool for evaluating and selecting patients for CeD clinical trials where controlling for inadvertent gluten exposure is important, such as therapies designed to prevent symptoms due to inadvertent gluten exposure (215).
Insight into the molecular mechanisms underpinning CeD pathogenesis provide several opportunities for novel therapeutics development and a range of pharmaceuticals are currently being assessed in pre-clinical and clinical trials (Table 3). These can be broadly classified into luminal approaches that aim to quantitatively reduce the load of gluten available to trigger the immune response and qualitative approaches that aim to induce gluten tolerance. A third category, not discussed in this review, encompass immunomodulators (e.g., budesonide, azathioprine), biologics (e.g., anti-IL-15, anti-CD52), and chemotherapy (e.g., cladribine) used to treat refractory CeD (219).
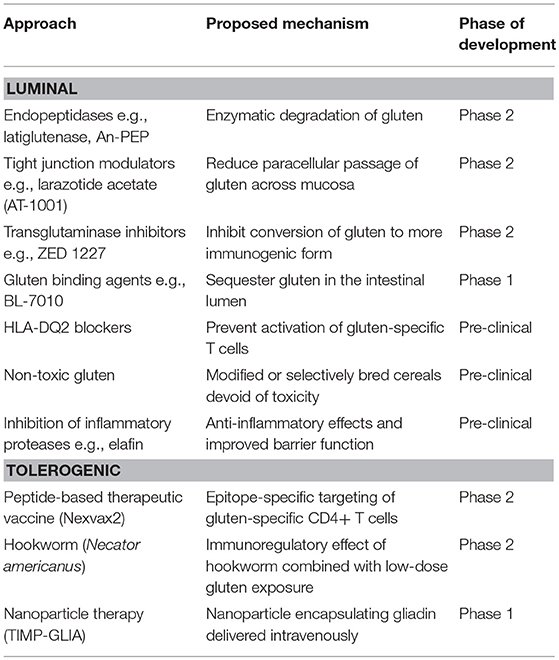
Table 3. Experimental therapies for celiac disease in pre-clinical or clinical development.
Quantitative approaches include the use of (i) endopeptidase enzymes (glutenases) derived from plants, bacteria or fungi that have a gluten degrading effect, such as latiglutenase (ALV003) (220, 221) and AN-PEP (222) (ii) agents to reduce paracellular passage of gluten i.e., larazotide acetate, an intestinal tight junction regulator that may enhance barrier function (223–225), and (iii) compounds that bind gluten in the gut lumen to reduce absorption, such as the polymer BL-7010 (226). Supplements to the GFD which render small amounts of dietary gluten harmless could substantially improve the quality of life of patients by allowing them to dine out with less fear of adverse effects resulting from contamination by small amounts of gluten. Enzymatic approaches could also be applied during the baking process to reduce gluten immunogenicity (227). Genetic modification of wheat with a variety of targeted techniques such as RNA interference (228) and CRISPR (229) can reduce gluten T cell epitope content and immunogenicity however clinical feeding trials are awaited. The recent publication of the first fully annotated reference wheat genome is an important advance that may help guide targeted approaches (230). Use of protease inhibitors, such as elafin, which is decreased in the mucosa of patients with active CeD has been proposed as it has barrier enhancing and anti-inflammatory effects in gluten-sensitive mice (231).
A phase 2 RCT of latiglutenase taken orally showed it could attenuate small intestinal mucosal injury in CeD patients induced by 2 g of ingested gluten (221). In symptomatic CeD patients following a GFD, latiglutenase reduced symptoms in the subgroup who were seropositive (232), suggesting that gluten exposure was necessary in order to demonstrate a positive effect of the enzyme. In a Phase 2 trial larazotide acetate was shown to reduce symptoms in CeD patients on a GFD better than a GFD alone, but only at the lowest dose of 0.5 mg (225). More studies are required to establish the efficacy of these approaches and how they can be safely used by patients. A controlled gluten challenge will be an important component of the study design in order to demonstrate efficacy and establish the amount of ingested gluten patients can be protected from.
Qualitative approaches aim to establish durable immune tolerance to gluten. One way this might be achieved is by targeting the long-lived population of gluten-specific T cells and deleting or rendering them functionally unresponsive (anergic) and inducing suppressive Tregs (233). As the target T cell population is stable in established CeD (139, 140, 142), it is anticipated these approaches will apply similarly to children as they do in adults with CeD. Phase 1 studies of Nexvax2, a therapeutic vaccine composed of three gluten peptides encompassing immunodominant HLA-DQ2.5-restricted T cell epitopes, initially caused gastrointestinal symptoms similar to those triggered by gluten, however after later administration of Nexvax2 symptoms were no different from those after placebo (111, 234). The recall immune response to gluten was modified in people with CeD receiving Nexvax2. A phase 2 clinical trial of infection with the hookworm Necator Americanus combined with a micro-gluten challenge in 12 CeD patients showed immune modifying effects and clinical protection against gluten (235) and a larger controlled study is underway.
Conclusion and Future Perspectives
Our view of CeD has evolved from a gastrointestinal illness to an immune disease characterized by the presence of specific HLA genes, CD4+ T cells responding to specific gluten peptides, circulating antibodies to tTG and systemic clinical manifestations. Aside from the fact it is driven by an exogenous, dietary antigen, the genetic and immunologic basis for CeD overlaps with that of traditional autoimmune diseases. While HLA susceptibility and wheat consumption and are major determinants of disease development it is apparent that non-HLA genes and a range of environmental factors are important for disease development. Prospective studies have established that the timing of gluten introduction and breastfeeding do not impact the development of CeD. More results from multicenter, prospective longitudinal studies are needed to understand the long-term effects of a high amount of gluten intake and to identify if other environmental exposures might trigger the disease.
Furthermore, although in vitro and in vivo studies suggests there are host-microbe or gluten-microbe interactions that promote gluten-specific immune responses, larger clinical trials where both the composition and function of the microbiota is studied in at-risk individuals and followed over time are needed to help understand gene-microbe interactions in CeD development. These kinds of studies may provide insight into the microbial events leading to CeD development that could be targeted as preventative or therapeutic strategies.
Finally, understanding how tolerance to gluten is lost in CeD is a fundamental question that needs more study. Insights into disease relevant pathways may come from analysis of the genome and gene expression in CeD, and epigenetic studies are needed to examine the impact of environment on gene expression and disease development. While novel therapies for CeD have not yet been tested in children, emerging studies on the role of environmental factors and the microbiome and how they might impact gluten immunity and tolerance may one day make disease prevention possible. For now, as primary prevention of CeD is a highly attractive, but as yet unrealized goal, the focus must be on driving expeditious diagnosis and treatment in symptomatic children and adults.
Author Contributions
JT-D wrote the first draft of the manuscript. DA and HG wrote sections of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Conflict of Interest Statement
JT-D has served as a consultant and scientific advisory board member for ImmusanT Inc., USA, and owns shares in Nexpep Pty Ltd and is a co-inventor of patents pertaining to the use of gluten peptides in CeD therapeutics, diagnostics and nontoxic gluten. Nexpep Pty. Ltd. and ImmusanT Inc. were formed to develop novel diagnostics and treatments for CeD. DA has served as principal investigator for probiotic studies in collaboration with Probi AB, Lund, Sweden, and is the co-inventor of patents pertaining to the use of L. plantarum (strain HEAL9) and L. paracasei (strain 8700:2) in CeD.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Ludvigsson JF, Leffler DA, Bai JC, Biagi F, Fasano A, Green PH, et al. The Oslo definitions for coeliac disease and related terms. Gut (2013) 62:43–52. doi: 10.1136/gutjnl-2011-301346
2. Husby S, Koletzko S, Korponay-Szabo IR, Mearin ML, Phillips A, Shamir R, et al. European society for pediatric gastroenterology, hepatology, nutrition guidelines for the diagnosis of coeliac disease. J Pediatr Gastroenterol Nutr. (2012) 54:136–60. doi: 10.1097/MPG.0b013e31821a23d0
3. Singh P, Arora A, Strand TA, Leffler DA, Catassi C, Green PH, et al. Global prevalence of celiac disease: systematic review and meta-analysis. Clin Gastroenterol Hepatol. (2018) 16:823–36.e2. doi: 10.1016/j.cgh.2017.06.037
4. Rubio-Tapia A, Kyle RA, Kaplan EL, Johnson DR, Page W, Erdtmann F, et al. Increased prevalence and mortality in undiagnosed celiac disease. Gastroenterology (2009) 137:88–93. doi: 10.1053/j.gastro.2009.03.059
5. Kaukinen K, Peraaho M, Lindfors K, Partanen J, Woolley N, Pikkarainen P, et al. Persistent small bowel mucosal villous atrophy without symptoms in coeliac disease. Aliment Pharmacol Therapeut. (2007) 25:1237–45. doi: 10.1111/j.1365-2036.2007.03311.x
6. Godfrey JD, Brantner TL, Brinjikji W, Christensen KN, Brogan DL, Van Dyke CT, et al. Morbidity and mortality among older individuals with undiagnosed celiac disease. Gastroenterology (2010) 139:763–9. doi: 10.1053/j.gastro.2010.05.041
7. Green PHR, Stavropoulos SN, Panagi SG, Goldstein SL, McMahon DJ, Absan H, et al. Characteristics of adult celiac disease in the USA: results of a national survey. Am J Gastroenterol. (2001) 96:126–31. doi: 10.1111/j.1572-0241.2001.03462.x
8. Jericho H, Guandalini S. Extra-intestinal manifestation of celiac disease in children. Nutrients (2018) 10:755. doi: 10.3390/nu10060755
9. Nurminen S, Kivela L, Huhtala H, Kaukinen K, Kurppa K. Extraintestinal manifestations were common in children with coeliac disease and were more prevalent in patients with more severe clinical and histological presentation. Acta Paediatr. (2018). 107:1–7. doi: 10.1111/apa.14324
10. Smith LB, Lynch KF, Kurppa K, Koletzko S, Krischer J, Liu E, et al. Psychological manifestations of celiac disease autoimmunity in young children. Pediatrics (2017) 139:e20162848. doi: 10.1542/peds.2016-2848
11. Hagopian W, Lee HS, Liu E, Rewers M, She JX, Ziegler AG, et al. Co-occurrence of type 1 diabetes and celiac disease autoimmunity. Pediatrics (2017) 140:e20171305. doi: 10.1542/peds.2017-1305
12. Canova C, Pitter G, Ludvigsson JF, Romor P, Zanier L, Zanotti R, et al. Celiac disease and risk of autoimmune disorders: a population-based matched birth cohort study. J Pediatr. (2016) 174:146–52 e1. doi: 10.1016/j.jpeds.2016.02.058
13. Assa A, Frenkel-Nir Y, Tzur D, Katz LH, Shamir R. Large population study shows that adolescents with celiac disease have an increased risk of multiple autoimmune and nonautoimmune comorbidities. Acta Paediatr. (2017) 106:967–72. doi: 10.1111/apa.13808
14. Stordal K, Bakken IJ, Suren P, Stene LC. Epidemiology of coeliac disease and comorbidity in Norwegian children. J Pediatr Gastroenterol Nutr. (2013) 57:467–71. doi: 10.1097/MPG.0b013e3182a455dd
15. Elfstrom P, Sundstrom J, Ludvigsson JF. Systematic review with meta-analysis: associations between coeliac disease and type 1 diabetes. Aliment Pharmacol Ther. (2014) 40:1123–32. doi: 10.1111/apt.12973
16. Meloni G, Dore A, Fanciulli G, Tanda F, Bottazzo GF. Subclinical coeliac disease in schoolchildren from northern Sardinia. Lancet (1999) 353:37. doi: 10.1016/S0140-6736(05)74871-6
17. Meloni GF, Tomasi PA, Bertoncelli A, Fanciulli G, Delitala G, Meloni T. Prevalence of silent celiac disease in patients with autoimmune thyroiditis from Northern Sardinia. J Endocrinol Invest. (2001) 24:298–302. doi: 10.1007/BF03343864
18. Lundin KE, Wijmenga C. Coeliac disease and autoimmune disease-genetic overlap and screening. Nat Rev Gastroenterol Hepatol. (2015) 12:507–15. doi: 10.1038/nrgastro.2015.136
19. Alshiekh S, Zhao LP, Lernmark A, Geraghty DE, Naluai AT, Agardh D. Different DRB1*03:01-DQB1*02:01 haplotypes confer different risk for celiac disease. HLA (2017) 90:95–101. doi: 10.1111/tan.13065
20. Lundin KE, Nilsen EM, Scott HG, Loberg EM, Gjoen A, Bratlie J, et al. Oats induced villous atrophy in coeliac disease. Gut (2003) 52:1649–52. doi: 10.1136/gut.52.11.1649
21. Hardy MY, Tye-Din JA, Stewart JA, Schmitz F, Dudek NL, Hanchapola I, et al. Ingestion of oats and barley in patients with celiac disease mobilizes cross-reactive T cells activated by avenin peptides and immuno-dominant hordein peptides. J Autoimmun. (2015) 56:56–65. doi: 10.1016/j.jaut.2014.10.003
22. Arentz-Hansen H, Fleckenstein B, Molberg O, Scott H, Koning F, Jung G, et al. The molecular basis for oat intolerance in patients with celiac disease. PLoS Med. (2004) 1:e1. doi: 10.1371/journal.pmed.0010001
23. Comino I, Real A, de Lorenzo L, Cornell H, Lopez-Casado MA, Barro F, et al. Diversity in oat potential immunogenicity: basis for the selection of oat varieties with no toxicity in coeliac disease. Gut (2011) 60:915–22. doi: 10.1136/gut.2010.225268
24. Pinto-Sanchez MI, Causada-Calo N, Bercik P, Ford AC, Murray JA, Armstrong D, et al. Safety of adding oats to a gluten-free diet for patients with celiac disease: systematic review and meta-analysis of clinical and observational studies. Gastroenterology (2017) 153:395–409 e3. doi: 10.1053/j.gastro.2017.04.009
25. Ciacci C, Ciclitira P, Hadjivassiliou M, Kaukinen K, Ludvigsson JF, McGough N, et al. The gluten-free diet and its current application in coeliac disease and dermatitis herpetiformis. U Eur Gastroenterol J. (2015) 3:121–35. doi: 10.1177/2050640614559263
26. Lionetti E, Gatti S, Pulvirenti A, Catassi C. Celiac disease from a global perspective. Best Pract Res Clin Gastroenterol. (2015) 29:365–79. doi: 10.1016/j.bpg.2015.05.004
27. Yuan J, Zhou C, Gao J, Li J, Yu F, Lu J, et al. Prevalence of celiac disease autoimmunity among adolescents and young adults in China. Clin Gastroenterol Hepatol. (2017) 15:1572–9 e1. doi: 10.1016/j.cgh.2017.04.025
28. Rubio-Tapia A, Ludvigsson JF, Brantner TL, Murray JA, Everhart JE. The prevalence of celiac disease in the United States. Am J Gastroenterol. (2012) 107:1538–44; quiz 1537:1545. doi: 10.1038/ajg.2012.219
29. Jansson-Knodell CL, King KS, Larson JJ, Van Dyke CT, Murray JA, Rubio-Tapia A. gender-based differences in a population-based cohort with celiac disease: more alike than unalike. Dig Dis Sci. (2018) 63:184–92. doi: 10.1007/s10620-017-4835-0
30. Greco L, Romino R, Coto I, Di Cosmo N, Percopo S, Maglio M, et al. The first large population based twin study of coeliac disease. Gut (2002) 50:624–8. doi: 10.1136/gut.50.5.624
31. Kondrashova A, Mustalahti K, Kaukinen K, Viskari H, Volodicheva V, Haapala AM, et al. Lower economic status and inferior hygienic environment may protect against celiac disease. Ann Med. (2008) 40:223–31. doi: 10.1080/07853890701678689
32. Szajewska H, Shamir R, Chmielewska A, Piescik-Lech M, Auricchio R, Ivarsson A, et al. Systematic review with meta-analysis: early infant feeding and coeliac disease–update 2015. Aliment Pharmacol Ther. (2015) 41:1038–54. doi: 10.1111/apt.13163
33. Chmielewska A, Piescik-Lech M, Szajewska H, Shamir R. Primary prevention of celiac disease: environmental factors with a focus on early nutrition. Ann Nutr Metabo. (2015) 67 (Suppl. 2):43–50. doi: 10.1159/000440992
34. Lionetti E, Castellaneta S, Francavilla R, Pulvirenti A, Tonutti E, Amarri S, et al. Weaning and Risk CD. introduction of gluten, HLA status, and the risk of celiac disease in children. N Engl J Med. (2014) 371:1295–303. doi: 10.1056/NEJMoa1400697
35. Andren Aronsson C, Lee HS, Koletzko S, Uusitalo U, Yang J, Virtanen SM, et al. Effects of gluten intake on risk of celiac disease: a case-control study on a swedish birth cohort. Clin Gastroenterol Hepatol. (2016) 14:403–9 e3. doi: 10.1016/j.cgh.2015.09.030
36. Vriezinga SL, Auricchio R, Bravi E, Castillejo G, Chmielewska A, Crespo Escobar P, et al. Randomized feeding intervention in infants at high risk for celiac disease. N Engl J Med. (2014) 371:1304–15. doi: 10.1056/NEJMoa1404172
37. Marild K, Kahrs CR, Tapia G, Stene LC, Stordal K. Infections and risk of celiac disease in childhood: a prospective nationwide cohort study. Am J Gastroenterol. (2015) 110:1475–84. doi: 10.1038/ajg.2015.287
38. Kemppainen KM, Lynch KF, Liu E, Lonnrot M, Simell V, Briese T, et al. Factors that increase risk of celiac disease autoimmunity after a gastrointestinal infection in early life. Clin Gastroenterol Hepatol. (2017) 15:694–702 e5. doi: 10.1016/j.cgh.2016.10.033
39. Stene LC, Honeyman MC, Hoffenberg EJ, Haas JE, Sokol RJ, Emery L, et al. Rotavirus infection frequency and risk of celiac disease autoimmunity in early childhood: a longitudinal study. Am J Gastroenterol. (2006) 101:2333–40. doi: 10.1111/j.1572-0241.2006.00741.x
40. Bouziat R, Hinterleitner R, Brown JJ, Stencel-Baerenwald JE, Ikizler M, Mayassi T, et al. Reovirus infection triggers inflammatory responses to dietary antigens and development of celiac disease. Science (2017) 356:44–50. doi: 10.1126/science.aah5298
41. Lebwohl B, Blaser MJ, Ludvigsson JF, Green PH, Rundle A, Sonnenberg A, et al. Decreased risk of celiac disease in patients with Helicobacter pylori colonization. Am J Epidemiol. (2013) 178:1721–30. doi: 10.1093/aje/kwt234
42. Ciacci C, Squillante A, Rendina D, Limauro S, Bencivenga C, Labanca F, et al. Helicobacter pylori infection and peptic disease in coeliac disease. Eur J Gastroenterol Hepatol. (2000) 12:1283–7. doi: 10.1097/00042737-200012120-00004
43. Narang M, Puri AS, Sachdeva S, Singh J, Kumar A, Saran RK. Celiac disease and Helicobacter pylori infection in children: is there any association? J Gastroenterol Hepatol. (2017) 32:1178–82. doi: 10.1111/jgh.13654
44. Dore MP, Salis R, Loria MF, Villanacci V, Bassotti G, Pes GM. Helicobacter pylori infection and occurrence of celiac disease in subjects HLA-DQ2/DQ8 positive: a prospective study. Helicobacter (2018) 23:e12465. doi: 10.1111/hel.12465
45. Konturek PC, Karczewska E, Dieterich W, Hahn EG, Schuppan D. Increased prevalence of Helicobacter pylori infection in patients with celiac disease. Am J Gastroenterol. (2000) 95:3682–3. doi: 10.1111/j.1572-0241.2000.03421.x
46. Lebwohl B, Green PH, Murray JA, Ludvigsson JF. Season of birth in a nationwide cohort of coeliac disease patients. Arch Dis Child. (2013) 98:48–51. doi: 10.1136/archdischild-2012-302360
47. Assa A, Waisbourd-Zinman O, Daher S, Shamir R. Birth month as a risk factor for the diagnosis of celiac disease later in life: a population-based study. J Pediatr Gastroenterol Nutr. (2018) 67:367–70. doi: 10.1097/MPG.0000000000002001
48. Ivarsson A, Hernell O, Nystrom L, Persson LA. Children born in the summer have increased risk for coeliac disease. J Epidemiol Community Health (2003) 57:36–9. doi: 10.1136/jech.57.1.36
49. Emilsson L, Magnus MC, Stordal K. Perinatal risk factors for development of celiac disease in children, based on the prospective Norwegian Mother and Child Cohort Study. Clin Gastroenterol Hepatol. (2015) 13:921–7. doi: 10.1016/j.cgh.2014.10.012
50. Koletzko S, Lee HS, Beyerlein A, Aronsson CA, Hummel M, Liu E, et al. Cesarean section on the risk of celiac disease in the offspring: the teddy study. J Pediatr Gastroenterol Nutri. (2018) 66:417–24. doi: 10.1097/MPG.0000000000001682
51. Dydensborg Sander S, Hansen AV, Stordal K A, -Andersen MN, Murray JA, Husby S. Mode of delivery is not associated with celiac disease. Clin Epidemiol. (2018) 10:323–32. doi: 10.2147/CLEP.S152168
52. Unalp-Arida A, Ruhl CE, Choung RS, Brantner TL, Murray JA. Lower prevalence of celiac disease and gluten-related disorders in persons living in Southern vs Northern Latitudes of the United States. Gastroenterology (2017) 152:1922–32.e2. doi: 10.1053/j.gastro.2017.02.012
53. Teresi S, Crapisi M, Vallejo MD, Castellaneta SP, Francavilla R, Iacono G, et al. Celiac disease seropositivity in Saharawi children: a follow-up and family study. J Pediatr Gastroenterol Nutr. (2010) 50:506–9. doi: 10.1097/MPG.0b013e3181bab30c
54. Roy A, Mehra S, Kelly CP, Tariq S, Pallav K, Dennis M, et al. The association between socioeconomic status and the symptoms at diagnosis of celiac disease: a retrospective cohort study. Therap Adv Gastroenterol. (2016) 9:495–502. doi: 10.1177/1756283X16637532
55. Uusitalo U, Lee HS, Aronsson CA, Yang J, Virtanen SM, Norris J, et al. Environmental determinants of the diabetes in the young study: gluten consumption during late pregnancy and risk of celiac disease in the offspring: the TEDDY birth cohort. Am J Clin Nutr. (2015) 102:1216–21. doi: 10.3945/ajcn.115.119370
56. Lebwohl B, Spechler SJ, Wang TC, Green PH, Ludvigsson JF. Use of proton pump inhibitors and subsequent risk of celiac disease. Dig Liver Dis. (2014) 46:36–40. doi: 10.1016/j.dld.2013.08.128
57. Kemppainen KM, Vehik K, Lynch KF, Larsson HE, Canepa RJ, Simell V, et al. Environmental determinants of diabetes in the young study: association between early-life antibiotic use and the risk of islet or celiac disease autoimmunity. JAMA Pediatr. (2017) 171:1217–25. doi: 10.1001/jamapediatrics.2017.2905
58. Stordal K, Haugen M, Brantsaeter AL, Lundin KE, Stene LC. Association between maternal iron supplementation during pregnancy and risk of celiac disease in children. Clin Gastroenterol Hepatol. (2014) 12:624–31 e1-2. doi: 10.1016/j.cgh.2013.09.061
59. Yang J, Tamura RN, Aronsson CA, Uusitalo UM, Lernmark A, Rewers M, et al. Environmental determinants of diabetes in the young study: maternal use of dietary supplements during pregnancy is not associated with coeliac disease in the offspring: the environmental determinants of diabetes in the young (TEDDY) study. Br J Nutr. (2017) 117:466–72. doi: 10.1017/S0007114517000332
60. Marild K, Tapia G, Haugen M, Dahl SR, Cohen AS, Lundqvist M, et al. Maternal and neonatal vitamin D status, genotype and childhood celiac disease. PLoS ONE (2017) 12:e0179080. doi: 10.1371/journal.pone.0179080
61. Ivarsson A, Persson LA, Nystrom L, Ascher H, Cavell B, Danielsson L, et al. Epidemic of coeliac disease in Swedish children. Acta Paediatr. (2000) 89:165–71. doi: 10.1111/j.1651-2227.2000.tb01210.x
62. Ivarsson A, Hernell O, Stenlund H, Persson LA. Breast-feeding protects against celiac disease. Am J Clin Nutr. (2002) 75:914–21. doi: 10.1093/ajcn/75.5.914
63. Norris JM, Barriga K, Hoffenberg EJ, Taki I, Miao D, Haas JE, et al. Risk of celiac disease autoimmunity and timing of gluten introduction in the diet of infants at increased risk of disease. JAMA (2005) 293:2343–51. doi: 10.1001/jama.293.19.2343
64. Aronsson CA, Lee HS, Liu E, Uusitalo U, Hummel S, Yang J, et al. Age at gluten introduction and risk of celiac disease. Pediatrics (2015) 135:239–45. doi: 10.1542/peds.2014-1787
65. Ivarsson A, Myleus A, Norstrom F, van der Pals M, Rosen A, Hogberg L, Danielsson L, et al. Prevalence of childhood celiac disease and changes in infant feeding. Pediatrics (2013) 131:e687–94. doi: 10.1542/peds.2012-1015
66. Myleus A, Ivarsson A, Webb C, Danielsson L, Hernell O, Hogberg L, et al. Celiac disease revealed in 3% of Swedish 12-year-olds born during an epidemic. J Pediatr Gastroenterol Nutr. (2009) 49:170–6. doi: 10.1097/MPG.0b013e31818c52cc
67. Crespo-Escobar P, Mearin ML, Hervas D, Auricchio R, Castillejo G, Gyimesi J, et al. The role of gluten consumption at an early age in celiac disease development: a further analysis of the prospective PreventCD cohort study. Am J Clin Nutr. (2017) 105:890–6. doi: 10.3945/ajcn.116.144352
68. Myleus A, Hernell O, Gothefors L, Hammarstrom ML, Persson LA, Stenlund H, et al. Early infections are associated with increased risk for celiac disease: an incident case-referent study. BMC Pediatr. (2012) 12:194. doi: 10.1186/1471-2431-12-194
69. Canova C, Zabeo V, Pitter G, Romor P, Baldovin T, Zanotti R, et al. Association of maternal education, early infections, antibiotic use with celiac disease: a population-based birth cohort study in northeastern Italy. Am J Epidemiol. (2014) 180:76–85. doi: 10.1093/aje/kwu101
70. Kagnoff MF, Paterson YJ, Kumar PJ, Kasarda DD, Carbone FR, Unsworth DJ, et al. Evidence for the role of a human intestinal adenovirus in the pathogenesis of coeliac disease. Gut (1987) 28:995–1001. doi: 10.1136/gut.28.8.995
71. Lahdeaho ML, Parkkonen P, Reunala T, Maki M, Lehtinen M. Antibodies to E1b protein-derived peptides of enteric adenovirus type 40 are associated with celiac disease and dermatitis herpetiformis. Clin Immunol Immunopathol. (1993) 69:300–5. doi: 10.1006/clin.1993.1184
72. Lahdeaho ML, Lehtinen M, Rissa HR, Hyoty H, Reunala T, Maki M. Antipeptide antibodies to adenovirus E1b protein indicate enhanced risk of celiac disease and dermatitis herpetiformis. Int Arch Allergy Immunol. (1993) 101:272–6. doi: 10.1159/000236457
73. Zanoni G, Navone R, Lunardi C, Tridente G, Bason C, Sivori S, et al. In celiac disease, a subset of autoantibodies against transglutaminase binds toll-like receptor 4 and induces activation of monocytes. PLoS Med (2006) 3:e358. doi: 10.1371/journal.pmed.0030358
74. Marild K, Ye W, Lebwohl B, Green PH, Blaser MJ, Card T, et al. Antibiotic exposure and the development of coeliac disease: a nationwide case-control study. BMC Gastroenterol. (2013) 13:109. doi: 10.1186/1471-230X-13-109
75. Decker E, Engelmann G, Findeisen A, Gerner P, Laass M, Ney D, et al. Cesarean delivery is associated with celiac disease but not inflammatory bowel disease in children. Pediatrics (2010) 125:e1433–40. doi: 10.1542/peds.2009-2260
76. Marild K, Stephansson O, Montgomery S, Murray JA, Ludvigsson JF. Pregnancy outcome and risk of celiac disease in offspring: a nationwide case-control study. Gastroenterology (2012) 142:39–45 e3. doi: 10.1053/j.gastro.2011.09.047
77. Marild K, Ludvigsson J, Sanz Y, Ludvigsson JF. Antibiotic exposure in pregnancy and risk of coeliac disease in offspring: a cohort study. BMC Gastroenterol. (2014) 14:75. doi: 10.1186/1471-230X-14-75
78. Agardh D, Lee HS, Kurppa K, Simell V, Aronsson CA, Jorneus O, et al. Clinical features of celiac disease: a prospective birth cohort. Pediatrics (2015) 135:627–34. doi: 10.1542/peds.2014-3675
79. Sevelsted A, Stokholm J, Bonnelykke K, Bisgaard H. Cesarean section and chronic immune disorders. Pediatrics (2015) 135:e92–8. doi: 10.1542/peds.2014-0596
80. Forsberg G, Fahlgren A, Horstedt P, Hammarstrom S, Hernell O, Hammarstrom ML. Presence of bacteria and innate immunity of intestinal epithelium in childhood celiac disease. Am J Gastroenterol. (2004) 99:894–904. doi: 10.1111/j.1572-0241.2004.04157.x
81. Ou G, Hedberg M, Hörstedt P, Baranov V, Forsberg G, Drobni M, et al. Proximal small intestinal microbiota and identification of rod-shaped bacteria associated with childhood celiac disease. Am J Gastroenterol. (2009) 104:3058–67. doi: 10.1038/ajg.2009.524
82. Verdu EF, Galipeau HJ, Jabri B. Novel players in coeliac disease pathogenesis: role of the gut microbiota. Nat Rev Gastroenterol Hepatol. (2015) 12:497–506. doi: 10.1038/nrgastro.2015.90
83. Sanchez E, Donat E, Ribes-Koninckx C, Fernandez-Murga ML, Sanz Y. Duodenal-mucosal bacteria associated with celiac disease in children. Appl Environ Microbiol. (2013) 79:5472–9. doi: 10.1128/AEM.00869-13
84. Pozo-Rubio T, Olivares M, Nova E, De Palma G, M2ujico JR, Ferrer MD, et al. Immune development and intestinal microbiota in celiac disease. Clin Dev Immunol. (2012) 2012:654143. doi: 10.1155/2012/654143
85. Wacklin P, Laurikka P, Lindfors K, Collin P, Salmi T, Lahdeaho ML, et al. Altered duodenal microbiota composition in celiac disease patients suffering from persistent symptoms on a long-term gluten-free diet. Am J Gastroenterol. (2014) 109:1933–41. doi: 10.1038/ajg.2014.355
86. Verdu EF, Mauro M, Bourgeois J, Armstrong D. Clinical onset of celiac disease after an episode of Campylobacter jejuni enteritis. Can J Gastroenterol. (2007) 21:453–5. doi: 10.1155/2007/169591
87. Sanchez E, Nadal I, Donat E, Ribes-Koninckx C, Calabuig M, Sanz Y. Reduced diversity and increased virulence-gene carriage in intestinal enterobacteria of coeliac children. BMC Gastroenterol. (2008) 8:50. doi: 10.1186/1471-230X-8-50
88. Sanchez E, Ribes-Koninckx C, Calabuig M, Sanz Y. Intestinal Staphylococcus spp. and virulent features associated with coeliac disease. J Clin Pathol. (2012) 65:830–4. doi: 10.1136/jclinpath-2012-200759
89. Sanchez E, Laparra JM, Sanz Y. Discerning the role of Bacteroides fragilis in celiac disease pathogenesis. Appl Environ Microbiol. (2012) 78:6507–15. doi: 10.1128/AEM.00563-12
90. D'Argenio V, Casaburi G, Precone V, Pagliuca C, Colicchio R, Sarnataro D, et al. Metagenomics reveals dysbiosis and a potentially pathogenic n. flavescens strain in duodenum of adult celiac patients. Am J Gastroenterol. (2016) 111:879–90. doi: 10.1038/ajg.2016.95
91. Aydogdu S, Cakir M, Yuksekkaya HA, Tumgor G, Baran M, Arikan C, et al. Helicobacter pylori infection in children with celiac disease. Scand J Gastroenterol. (2008) 43:1088–93. doi: 10.1080/00365520802101846
92. Caminero A, Herrán AR, Nistal E, Pérez-Andrés J, Vaquero L, Vivas S, et al. Diversity of the cultivable human gut microbiome involved in gluten metabolism: isolation of microorganisms with potential interest for coeliac disease. FEMS Microbiol Ecol. (2014) 88:309–19. doi: 10.1111/1574-6941.12295
93. Caminero A, Nistal E, Herrán AR, Pérez-Andrés J, Ferrero MA, Vaquero Ayala L, et al. Differences in gluten metabolism among healthy volunteers, coeliac disease patients and first-degree relatives. Br J Nutr. (2015) 114:1157–67. doi: 10.1017/S0007114515002767
94. Caminero A, Galipeau HJ, McCarville JL, Johnston CW, Bernier SP, Russell AK, et al. Duodenal bacteria from patients with celiac disease and healthy subjects distinctly affect gluten breakdown and immunogenicity. Gastroenterology (2016) 151:670–83. doi: 10.1053/j.gastro.2016.06.041
95. Olivares M, Neef A, Castillejo G, Palma GD, Varea V, Capilla A, et al. The HLA-DQ2 genotype selects for early intestinal microbiota composition in infants at high risk of developing coeliac disease. Gut (2015) 64:406–17. doi: 10.1136/gutjnl-2014-306931
96. Sellitto M, Bai G, Serena G, Fricke WF, Sturgeon C, Gajer P, et al. Proof of concept of microbiome-metabolome analysis and delayed gluten exposure on celiac disease autoimmunity in genetically at-risk infants. PLoS ONE (2012) 7:e33387. doi: 10.1371/journal.pone.0033387
97. Olivares M, Benitez-Paez A, de Palma G, Capilla A, Nova E, Castillejo G, et al. Increased prevalence of pathogenic bacteria in the gut microbiota of infants at risk of developing celiac disease: the proficel study. Gut Microbes (2018) 9:1–8. doi: 10.1080/19490976.2018.1451276
98. Palma GD, Capilla A, Nova E, Castillejo G, Varea V, Pozo T, et al. Influence of milk-feeding type and genetic risk of developing coeliac disease on intestinal microbiota of infants: the PROFICEL study. PLoS ONE (2012) 7:e30791. doi: 10.1371/journal.pone.0030791
99. Olivares M, Walker AW, Capilla A, Benítez-Páez A, Palau F, Parkhill J, et al. Gut microbiota trajectory in early life may predict development of celiac disease. Microbiome (2018) 6:36. doi: 10.1186/s40168-018-0415-6
100. Rintala A, Riikonen I, Toivonen A, Pietilä S, Munukka E, Pursiheimo JP, et al. Early fecal microbiota composition in children who later develop celiac disease and associated autoimmunity. Scand J Gastroenterol. (2018) 53:403–9. doi: 10.1080/00365521.2018.1444788
101. Rothschild D, Weissbrod O, Barkan E, Kurilshikov A, Korem T, Zeevi D, et al. Environment dominates over host genetics in shaping human gut microbiota. Nature (2018) 555:210–5. doi: 10.1038/nature25973
102. Carmody RN, Gerber GK, Luevano JM, Gatti DM, Somes L, Svenson KL, et al. Diet dominates host genotype in shaping the murine gut microbiota. Cell Host Microbe. (2015) 17:72–84. doi: 10.1016/j.chom.2014.11.010
103. Kuja-Halkola R, Lebwohl B, Halfvarson J, Wijmenga C, Magnusson PK, Ludvigsson JF. Heritability of non-HLA genetics in coeliac disease: a population-based study in 107 000 twins. Gut (2016) 65:1793–8. doi: 10.1136/gutjnl-2016-311713
104. Sollid LM, Markussen G, Ek J, Gjerde H, Vartdal F, Thorsby E. Evidence for a primary association of celiac disease to a particular HLA-DQ alpha/beta heterodimer. J Exp Med. (1989) 169:345–50. doi: 10.1084/jem.169.1.345
105. Karell K, Louka AS, Moodie SJ, Ascher H, Clot F, Greco L, et al. HLA types in celiac disease patients not carrying the DQA1*05-DQB1*02 (DQ2) heterodimer: results from the European genetics cluster on celiac disease. Human Immunol. (2003) 64:469–77. doi: 10.1016/S0198-8859(03)00027-2
106. Vader W, Stepniak D, Kooy Y, Mearin L, Thompson A, van Rood JJ, et al. The HLA-DQ2 gene dose effect in celiac disease is directly related to the magnitude and breadth of gluten-specific T cell responses. Proc Natl Acad Sci USA. (2003) 100:12390–5. doi: 10.1073/pnas.2135229100
107. Liu E, Lee HS, Aronsson CA, Hagopian WA, Koletzko S, Rewers MJ, et al. Risk of pediatric celiac disease according to HLA haplotype and country. N Engl J Med. (2014) 371:42–9. doi: 10.1056/NEJMoa1313977
108. Liu E, Lee HS, Agardh D. Risk of celiac disease according to HLA haplotype and country. N Engl J Med. (2014) 371:1074. doi: 10.1056/NEJMc1409252
109. Karinen H, Karkkainen P, Pihlajamaki J, Janatuinen E, Heikkinen M, Julkunen R, et al. Gene dose effect of the DQB1*0201 allele contributes to severity of coeliac disease. Scand J Gastroenterol. (2006) 41:191–9. doi: 10.1080/00365520500206277
110. Al-Toma A, Goerres MS, Meijer JW, Pena AS, Crusius JB, Mulder CJ. Human leukocyte antigen-DQ2 homozygosity and the development of refractory celiac disease and enteropathy-associated T-cell lymphoma. Clin Gastroenterol Hepatol. (2006) 4:315–9. doi: 10.1016/j.cgh.2005.12.011
111. Goel G, King T, Daveson AJ, Andrews JM, Krishnarajah J, Krause R, et al. Epitope-specific immunotherapy targeting CD4-positive T cells in coeliac disease: two randomised, double-blind, placebo-controlled phase 1 studies. Lancet Gastroenterol Hepatol. (2017) 2:479–93. doi: 10.1016/S2468-1253(17)30110-3
112. Dubois PC, Trynka G, Franke L, Hunt KA, Romanos J, Curtotti A, et al. Multiple common variants for celiac disease influencing immune gene expression. Nat Genet. (2010) 42:295–302. doi: 10.1038/ng.543
113. van Heel DA, Franke L, Hunt KA, Gwilliam R, Zhernakova A, Inouye M, et al. A genome-wide association study for celiac disease identifies risk variants in the region harboring IL2 and IL21. Nat Genet. (2007) 39:827–9. doi: 10.1038/ng2058
114. Hunt KA, Zhernakova A, Turner G, Heap GA, Franke L, Bruinenberg M, et al. Newly identified genetic risk variants for celiac disease related to the immune response. Nat Genet. (2008) 40:395–402. doi: 10.1038/ng.102
115. Trynka G, Hunt KA, Bockett NA, Romanos J, Mistry V, Szperl A, et al. Dense genotyping identifies and localizes multiple common and rare variant association signals in celiac disease. Nat Genet. (2011) 43:1193–201. doi: 10.1038/ng.998
116. Gutierrez-Achury J, Zhernakova A, Pulit SL, Trynka G, Hunt KA, Romanos J, et al. Fine mapping in the MHC region accounts for 18% additional genetic risk for celiac disease. Nat Genet. (2015) 47:577–8. doi: 10.1038/ng.3268
117. Sharma A, Liu X, Hadley D, Hagopian W, Liu E, Chen WM, et al. Identification of Non-HLA genes associated with celiac disease and country-specific differences in a large, international pediatric cohort. PLoS ONE (2016) 11:e0152476. doi: 10.1371/journal.pone.0152476
118. Smyth DJ, Plagnol V, Walker NM, Cooper JD, Downes K, Yang JH, et al. Shared and distinct genetic variants in type 1 diabetes and celiac disease. N Engl J Med. (2008) 359:2767–77. doi: 10.1056/NEJMoa0807917
119. Zhernakova A, Stahl EA, Trynka G, Raychaudhuri S, Festen EA, Franke L, et al. Meta-analysis of genome-wide association studies in celiac disease and rheumatoid arthritis identifies fourteen non-HLA shared loci. PLoS Genet. (2011) 7:e1002004. doi: 10.1371/journal.pgen.1002004
120. Festen EA, Goyette P, Green T, Boucher G, Beauchamp C, Trynka G, et al. A meta-analysis of genome-wide association scans identifies IL18RAP, PTPN2, TAGAP, and PUS10 as shared risk loci for Crohn's disease and celiac disease. PLoS Genet. (2011) 7:e1001283. doi: 10.1371/journal.pgen.1001283
121. Castellanos-Rubio A, Fernandez-Jimenez N, Kratchmarov R, Luo X, Bhagat G, Green PH, et al. A long noncoding RNA associated with susceptibility to celiac disease. Science (2016) 352:91–5. doi: 10.1126/science.aad0467
122. Lundin KE, Scott H, Hansen T, Paulsen G, Halstensen TS, Fausa O, et al. Gliadin-specific, HLA-DQ(alpha 1*0501,beta 1*0201) restricted T cells isolated from the small intestinal mucosa of celiac disease patients. J Exp Med. (1993) 178:187–96. doi: 10.1084/jem.178.1.187
123. Nilsen EM, Lundin KE, Krajci P, Scott H, Sollid LM, Brandtzaeg P. Gluten specific, HLA-DQ restricted T cells from coeliac mucosa produce cytokines with Th1 or Th0 profile dominated by interferon gamma. Gut (1995) 37:766–76. doi: 10.1136/gut.37.6.766
124. Molberg O, Kett K, Scott H, Thorsby E, Sollid LM, Lundin KE. Gliadin specific, HLA DQ2-restricted T cells are commonly found in small intestinal biopsies from coeliac disease patients, but not from controls. Scand J Immunol. (1997) 46:103–8.
125. Lundin KE, Scott H, Fausa O, Thorsby E, Sollid LM. T cells from the small intestinal mucosa of a DR4, DQ7/DR4, DQ8 celiac disease patient preferentially recognize gliadin when presented by DQ8. Human Immunol. (1994) 41:285–91. doi: 10.1016/0198-8859(94)90047-7
126. Fleckenstein B, Molberg O, Qiao SW, Schmid DG, von der Mulbe F, Elgstoen K, et al. Gliadin T cell epitope selection by tissue transglutaminase in celiac disease. role of enzyme specificity and pH influence on the transamidation versus deamidation process. J Biol Chem. (2002) 277:34109–16. doi: 10.1074/jbc.M204521200
127. Vader LW, de Ru AY, van der Wal Kooy YM, Benckhuijsen W, Mearin ML, Drijfhout JW, et al. Specificity of tissue transglutaminase explains cereal toxicity in celiac disease. J Exp Med. (2002) 195:643–9. doi: 10.1084/jem.20012028
128. Quarsten H, Molberg O, Fugger L, McAdam SN, Sollid LM. HLA binding and T cell recognition of a tissue transglutaminase-modified gliadin epitope. Eur J Immunol. (1999) 29:2506–14. doi: 10.1002/(SICI)1521-4141(199908)29:08<2506::AID-IMMU2506>3.0.CO;2-9
129. van de Wal Y, Kooy Y, van Veelen P, Pena S, Mearin L, Papadopoulos G, et al. Selective deamidation by tissue transglutaminase strongly enhances gliadin-specific T cell reactivity. J Immunol. (1998) 161:1585–8.
130. Molberg O, McAdam SN, Korner R, Quarsten H, Kristiansen C, Madsen L, et al. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat Med. (1998) 4:713–7. doi: 10.1038/nm0698-713
131. Kim CY, Quarsten H, Bergseng E, Khosla C, Sollid LM. Structural basis for HLA-DQ2-mediated presentation of gluten epitopes in celiac disease. Proc Nat Acad Sci USA. (2004) 101:4175–9. doi: 10.1073/pnas.0306885101
132. Henderson KN, Tye-Din JA, Reid HH, Chen Z, Borg NA, Beissbarth T, et al. A structural and immunological basis for the role of human leukocyte antigen DQ8 in celiac disease. Immunity (2007) 27:23–34. doi: 10.1016/j.immuni.2007.05.015
133. Broughton SE, Petersen J, Theodossis A, Scally SW, Loh KL, Thompson A, et al. Biased T cell receptor usage directed against human leukocyte antigen DQ8-restricted gliadin peptides is associated with celiac disease. Immunity (2012) 37:611–21. doi: 10.1016/j.immuni.2012.07.013
134. Qiao SW, Raki M, Gunnarsen KS, Loset GA, Lundin KE, Sandlie I, et al. Posttranslational modification of gluten shapes TCR usage in celiac disease. J Immunol. (2011) 187:3064–71. doi: 10.4049/jimmunol.1101526
135. Sollid LM, Qiao SW, Anderson RP, Gianfrani C, Koning F. Nomenclature and listing of celiac disease relevant gluten T-cell epitopes restricted by HLA-DQ molecules. Immunogenetics (2012) 64:455–60. doi: 10.1007/s00251-012-0599-z
136. Tye-Din JA, Stewart JA, Dromey JA, Beissbarth T, van Heel DA, Tatham A, et al. Comprehensive, quantitative mapping of T cell epitopes in gluten in celiac disease. Sci Trans Med. (2010) 2:1–14. doi: 10.1126/scitranslmed.3001012
137. Shan L, Molberg O, Parrot I, Hausch F, Filiz F, Gray GM, et al. Structural basis for gluten intolerance in celiac sprue. Science (2002) 297:2275–9. doi: 10.1126/science.1074129
138. Vader W, Kooy Y, Van Veelen P, De Ru A, Harris D, Benckhuijsen W, et al. The gluten response in children with celiac disease is directed toward multiple gliadin and glutenin peptides. Gastroenterology (2002) 122:1729–37. doi: 10.1053/gast.2002.33606
139. Hardy MY, Girardin A, Pizzey C, Cameron DJ, Watson KA, Picascia S, et al. Consistency in polyclonal T-cell responses to gluten between children and adults with celiac disease. Gastroenterology (2015) 149:1541–52 e2. doi: 10.1053/j.gastro.2015.07.013
140. Raki M, Dahal-Koirala S, Yu H, Korponay-Szabo IR, Gyimesi J, Castillejo G, et al. Similar responses of intestinal T cells from untreated children and adults with celiac disease to deamidated gluten epitopes. Gastroenterology (2017) 153:787–98 e4. doi: 10.1053/j.gastro.2017.05.016
141. Yohannes DA, Freitag TL, de Kauwe A, Kaukinen K, Kurppa K, Wacklin P, et al. Deep sequencing of blood and gut T-cell receptor beta-chains reveals gluten-induced immune signatures in celiac disease. Sci Rep. (2017) 7:17977. doi: 10.1038/s41598-017-18137-9
142. Risnes LF, Christophersen A, Dahal-Koirala S, Neumann RS, Sandve GK, Sarna VK, et al. Disease-driving CD4+ T cell clonotypes persist for decades in celiac disease. J Clin Invest. (2018) 128:2642–50. doi: 10.1172/JCI98819
143. Syage JA, Kelly CP, Dickason MA, Ramirez AC, Leon F, Dominguez R, et al. Determination of gluten consumption in celiac disease patients on a gluten-free diet. Am J Clin Nutr. (2018) 107:201–7. doi: 10.1093/ajcn/nqx049
144. Halmos EP, Di Bella CA, Webster R, Deng M, Tye-Din JA. Gluten in “gluten-free” food from food outlets in Melbourne: a cross-sectional study. Med J Aust. (2018) 209:42–3. doi: 10.5694/mja17.00883
145. Di Simone N, Silano M, Castellani R, Di Nicuolo F, D'Alessio MC, Franceschi F, et al. Anti-tissue transglutaminase antibodies from celiac patients are responsible for trophoblast damage via apoptosis in vitro. Am J Gastroenterol. (2010) 105:2254–61. doi: 10.1038/ajg.2010.233
146. Salmi TT, Collin P, Jarvinen O, Haimila K, Partanen J, Laurila K, et al. Immunoglobulin a autoantibodies against transglutaminase 2 in the small intestinal mucosa predict forthcoming coeliac disease. Aliment Pharmacol Ther. (2006) 24:541–52. doi: 10.1111/j.1365-2036.2006.02997.x
147. Simell S, Kupila A, Hoppu S, Hekkala A, Simell T, Stahlberg MR, et al. Natural history of transglutaminase autoantibodies and mucosal changes in children carrying HLA-conferred celiac disease susceptibility. Scand J Gastroenterol. (2005) 40:1182–91. doi: 10.1080/00365520510024034
148. Matysiak-Budnik T, Malamut G, de Serre NP, Grosdidier E, Seguier S, Brousse N, et al. Long-term follow-up of 61 coeliac patients diagnosed in childhood: evolution toward latency is possible on a normal diet. Gut (2007) 56:1379–86. doi: 10.1136/gut.2006.100511
149. Sollid LM, Molberg O, McAdam S, Lundin KE. Autoantibodies in coeliac disease: tissue transglutaminase–guilt by association? Gut (1997) 41:851–2. doi: 10.1136/gut.41.6.851
150. Koning F. Celiac disease: caught between a rock and a hard place. Gastroenterology (2005) 129:1294–301. doi: 10.1053/j.gastro.2005.07.030
151. Di Niro R, Snir O, Kaukinen K, Yaari G, Lundin KE, Gupta NT, et al. Responsive population dynamics and wide seeding into the duodenal lamina propria of transglutaminase-2-specific plasma cells in celiac disease. Mucosal Immunol. (2016) 9:254–64. doi: 10.1038/mi.2015.57
152. Stamnaes J, Iversen R, du Pre MF, Chen X, Sollid LM. Enhanced B-cell receptor recognition of the autoantigen transglutaminase 2 by efficient catalytic self-multimerization. PLoS ONE (2015) 10:e0134922. doi: 10.1371/journal.pone.0134922
153. Iversen RM, Fleur du PreM, Di Niro R, Sollid LM. Igs as substrates for transglutaminase 2: implications for autoantibody production in celiac disease. J Immunol. (2015) 195:5159–68. doi: 10.4049/jimmunol.1501363
154. Stamnaes J, Sollid LM. Celiac disease: autoimmunity in response to food antigen. Semin Immunol. (2015) 27:343–52. doi: 10.1016/j.smim.2015.11.001
155. Steinsbo O, Henry Dunand CJ, Huang M, Mesin L, Salgado-Ferrer M, Lundin KE, et al. Restricted VH/VL usage and limited mutations in gluten-specific IgA of coeliac disease lesion plasma cells. Nat Commun. (2014) 5:4041. doi: 10.1038/ncomms5041
156. Arentz-Hansen H, McAdam SN, Molberg O, Fleckenstein B, Lundin KE, Jorgensen TJ, et al. Celiac lesion T cells recognize epitopes that cluster in regions of gliadins rich in proline residues. Gastroenterology (2002) 123:803–9. doi: 10.1053/gast.2002.35381
157. Matysiak-Budnik T, Moura IC, Arcos-Fajardo M, Lebreton C, Menard S, Candalh C, et al. Secretory IgA mediates retrotranscytosis of intact gliadin peptides via the transferrin receptor in celiac disease. J Exp Med. (2008) 205:143–54. doi: 10.1084/jem.20071204
158. Lebreton C, Menard S, Abed J, Moura IC, Coppo R, Dugave C, et al. Interactions among secretory immunoglobulin A, CD71, and transglutaminase-2 affect permeability of intestinal epithelial cells to gliadin peptides. Gastroenterology (2012) 143:698–707.e4. doi: 10.1053/j.gastro.2012.05.051
159. Rauhavirta T, Qiao SW, Jiang Z, Myrsky E, Loponen J, Korponay-Szabo IR, et al. Epithelial transport and deamidation of gliadin peptides: a role for coeliac disease patient immunoglobulin A. Clin Exp Immunol. (2011) 164:127–36. doi: 10.1111/j.1365-2249.2010.04317.x
160. Lammers KM, Lu R, Brownley J, Lu B, Gerard C, Thomas K, et al. Gliadin induces an increase in intestinal permeability and zonulin release by binding to the chemokine receptor CXCR3. Gastroenterology (2008) 135:194–204 e3. doi: 10.1053/j.gastro.2008.03.023
161. Beitnes AC, Raki M, Brottveit M, Lundin KE, Jahnsen FL, Sollid LM. Rapid accumulation of CD14+CD11c+ dendritic cells in gut mucosa of celiac disease after in vivo gluten challenge. PLoS ONE (2012) 7:e33556. doi: 10.1371/journal.pone.0033556
162. Bodd M, Raki M, Tollefsen S, Fallang LE, Bergseng E, Lundin KE, et al. HLA-DQ2-restricted gluten-reactive T cells produce IL-21 but not IL-17 or IL-22. Mucosal Immunol. (2010) 3:594–601. doi: 10.1038/mi.2010.36
163. Monteleone I, Sarra MG, Del Vecchio Blanco G, Paoluzi OA, Franze E, Fina D, et al. Characterization of IL-17A-producing cells in celiac disease mucosa. J Immunol. (2010) 184:2211–8. doi: 10.4049/jimmunol.0901919
164. Fernandez S, Molina IJ, Romero P, Gonzalez R, Pena J, Sanchez F, et al. Characterization of gliadin-specific Th17 cells from the mucosa of celiac disease patients. Am J Gastroenterol. (2011) 106:528–38. doi: 10.1038/ajg.2010.465
165. DePaolo RW, Abadie V, Tang F, Fehlner-Peach H, Hall JA, Wang W, et al. Co-adjuvant effects of retinoic acid and IL-15 induce inflammatory immunity to dietary antigens. Nature (2011) 471:220–4. doi: 10.1038/nature09849
166. Monteleone G, Pender SL, Alstead E, Hauer AC, Lionetti P, McKenzie C, et al. Role of interferon alpha in promoting T helper cell type 1 responses in the small intestine in coeliac disease. Gut (2001) 48:425–9. doi: 10.1136/gut.48.3.425
167. Malamut G, El Machhour R, Montcuquet N, Martin-Lanneree S, Dusanter-Fourt I, Verkarre V, et al. IL-15 triggers an antiapoptotic pathway in human intraepithelial lymphocytes that is a potential new target in celiac disease-associated inflammation and lymphomagenesis. J Clin Invest. (2010) 120:2131–43. doi: 10.1172/JCI41344
168. Meresse B, Chen Z, Ciszewski C, Tretiakova M, Bhagat G, Krausz TN, et al. Coordinated induction by IL15 of a TCR-independent NKG2D signaling pathway converts CTL into lymphokine-activated killer cells in celiac disease. Immunity (2004) 21:357–66. doi: 10.1016/j.immuni.2004.06.020
169. Setty M, Discepolo V, Abadie V, Kamhawi S, Mayassi T, Kent A, et al. Distinct and synergistic contributions of epithelial stress and adaptive immunity to functions of intraepithelial killer cells and active celiac disease. Gastroenterology (2015) 149:681–91 e10. doi: 10.1053/j.gastro.2015.05.013
170. Maiuri L, Ciacci C, Ricciardelli I, Vacca L, Raia V, Auricchio S, et al. Association between innate response to gliadin and activation of pathogenic T cells in coeliac disease. Lancet (2003) 362:30–7. doi: 10.1016/S0140-6736(03)13803-2
171. Hmida NB, Ben Ahmed M, Moussa A, Rejeb MB, Said Y, Kourda N, et al. Impaired control of effector T cells by regulatory T cells: a clue to loss of oral tolerance and autoimmunity in celiac disease? Am J Gastroenterol. (2012) 107:604–11. doi: 10.1038/ajg.2011.397
172. Granzotto M, dal Bo S, Quaglia S, Tommasini A, Piscianz E, Valencic E, et al. Regulatory T-cell function is impaired in celiac disease. Dig Dis Sci. (2009) 54:1513–9. doi: 10.1007/s10620-008-0501-x
173. Cook L, Munier CML, Seddiki N, van Bockel D, Ontiveros N, Hardy MY, et al. Circulating gluten-specific FOXP3(+)CD39(+) regulatory T cells have impaired suppressive function in patients with celiac disease. J Allergy Clin Immunol. (2017) 140:1592–603 e8. doi: 10.1016/j.jaci.2017.02.015
174. Frisullo G, Nociti V, Iorio R, Patanella AK, Marti A, Assunta B, et al. Increased CD4+CD25+Foxp3+ T cells in peripheral blood of celiac disease patients: correlation with dietary treatment. Hum Immunol. (2009) 70:430–5. doi: 10.1016/j.humimm.2009.04.006
175. Zanzi D, Stefanile R, Santagata S, Iaffaldano L, Iaquinto G, Giardullo N, et al. IL-15 interferes with suppressive activity of intestinal regulatory T cells expanded in Celiac disease. Am J Gastroenterol. (2011) 106:1308–17. doi: 10.1038/ajg.2011.80
176. Benahmed M, Meresse B, Arnulf B, Barbe U, Mention JJ, Verkarre V, et al. Inhibition of TGF-beta signaling by IL-15: a new role for IL-15 in the loss of immune homeostasis in celiac disease. Gastroenterology (2007) 132:994–1008. doi: 10.1053/j.gastro.2006.12.025
177. Forsberg G, Hernell O, Melgar S, Israelsson A, Hammarstrom S, Hammarstrom ML. Paradoxical coexpression of proinflammatory and down-regulatory cytokines in intestinal T cells in childhood celiac disease. Gastroenterology (2002) 123:667–78. doi: 10.1053/gast.2002.35355
178. Di Sabatino A, Ciccocioppo R, Cupelli F, Cinque B, Millimaggi D, Clarkson MM, et al. Epithelium derived interleukin 15 regulates intraepithelial lymphocyte Th1 cytokine production, cytotoxicity, and survival in coeliac disease. Gut (2006) 55:469–77. doi: 10.1136/gut.2005.068684
179. Maiuri L, Ciacci C, Vacca L, Ricciardelli I, Auricchio S, Quaratino S, et al. IL-15 drives the specific migration of CD94+ and TCR-gammadelta+ intraepithelial lymphocytes in organ cultures of treated celiac patients. Am J Gastroenterol. (2001) 96:150–6. doi: 10.1111/j.1572-0241.2001.03437.x
180. Korneychuk N, Ramiro-Puig E, Ettersperger J, Schulthess J, Montcuquet N, Kiyono H, et al. Interleukin 15 and CD4(+) T cells cooperate to promote small intestinal enteropathy in response to dietary antigen. Gastroenterology (2014) 146:1017–27. doi: 10.1053/j.gastro.2013.12.023
181. Vicari AP, Schoepfer AM, Meresse B, Goffin L, Leger O, Josserand S, et al. Discovery and characterization of a novel humanized anti-IL-15 antibody and its relevance for the treatment of refractory celiac disease and eosinophilic esophagitis. MAbs (2017) 9:927–44. doi: 10.1080/19420862.2017.1332553
182. Quinn EM, Coleman C, Molloy B, Dominguez Castro P, Cormican P, Trimble V, et al. Transcriptome Analysis of CD4+ T Cells in coeliac disease reveals imprint of BACH2 and IFNgamma regulation. PLoS ONE (2015) 10:e0140049. doi: 10.1371/journal.pone.0140049
183. de Kauwe AL, Chen Z, Anderson RP, Keech CL, Price JD, Wijburg O, et al. Resistance to celiac disease in humanized HLA-DR3-DQ2-transgenic mice expressing specific anti-gliadin CD4+ T cells. J Immunol. (2009) 182:7440–50. doi: 10.4049/jimmunol.0900233
184. Korneychuk N, Meresse B, Cerf-Bensussan N. Lessons from rodent models in celiac disease. Mucosal Immunol. (2015) 8:18–28. doi: 10.1038/mi.2014.102
185. Freitag TL, Loponen J, Messing M, Zevallos V, Andersson LC, Sontag-Strohm T, et al. Testing safety of germinated rye sourdough in a celiac disease model based on the adoptive transfer of prolamin-primed memory T cells into lymphopenic mice. Am J Physiol. (2014) 306:G526–34. doi: 10.1152/ajpgi.00136.2013
186. Freitag TL, Rietdijk S, Junker Y, Popov Y, Bhan AK, Kelly CP, et al. Gliadin-primed CD4+CD45RBlowCD25- T cells drive gluten-dependent small intestinal damage after adoptive transfer into lymphopenic mice. Gut (2009) 58:1597–605. doi: 10.1136/gut.2009.186361
187. Black KE, Murray JA, David CS. HLA-DQ determines the response to exogenous wheat proteins: a model of gluten sensitivity in transgenic knockout mice. J Immunol. (2002) 169:5595–600. doi: 10.4049/jimmunol.169.10.5595
188. Galipeau HJ, Rulli NE, Jury J, Huang X, Araya R, Murray JA, et al. Sensitization to gliadin induces moderate enteropathy and insulitis in nonobese diabetic-DQ8 mice. J Immunol. (2011) 187:4338–46. doi: 10.4049/jimmunol.1100854
189. Verdu EF, Huang X, Natividad J, Lu J, Blennerhassett PA, David CS, et al. Gliadin-dependent neuromuscular and epithelial secretory responses in gluten-sensitive HLA-DQ8 transgenic mice. Am J Physiol. (2008) 294:G217–25. doi: 10.1152/ajpgi.00225.2007
190. Galipeau HJ, McCarville JL, Huebener S, Litwin O, Meisel M, Jabri B, et al. Intestinal microbiota modulates gluten-induced immunopathology in humanized mice. Am J Pathol. (2015) 185:2969–82. doi: 10.1016/j.ajpath.2015.07.018
191. Silano M, Agostoni C, Sanz Y, Guandalini S. Infant feeding and risk of developing celiac disease: a systematic review. BMJ Open (2016) 6:e009163. doi: 10.1136/bmjopen-2015-009163
192. Taavela J, Koskinen O, Huhtala H, Lahdeaho ML, Popp A, Laurila K, et al. Validation of morphometric analyses of small-intestinal biopsy readouts in celiac disease. PLoS ONE (2013) 8:e76163. doi: 10.1371/journal.pone.0076163
193. Arguelles-Grande C, Tennyson CA, Lewis SK, Green PH, Bhagat G. Variability in small bowel histopathology reporting between different pathology practice settings: impact on the diagnosis of coeliac disease. J Clin Pathol. (2012) 65:242–7. doi: 10.1136/jclinpath-2011-200372
194. Corazza GR, Villanacci V, Zambelli C, Milione M, Luinetti O, Vindigni C, et al. Comparison of the interobserver reproducibility with different histologic criteria used in celiac disease. Clin Gastroenterol Hepatol. (2007) 5:838–43. doi: 10.1016/j.cgh.2007.03.019
195. Villanacci V, Ceppa P, Tavani E, Vindigni C, Volta U. Coeliac disease: the histology report. Dig Liver Dis. (2011) 43 (Suppl. 4):S385–95. doi: 10.1016/S1590-8658(11)60594-X
196. Kurppa K, Collin P, Viljamaa M, Haimila K, Saavalainen P, Partanen J, et al. Diagnosing mild enteropathy celiac disease: a randomized, controlled clinical study. Gastroenterology (2009) 136:816–23. doi: 10.1053/j.gastro.2008.11.040
197. Mooney PD, Kurien M, Evans KE, Rosario E, Cross SS, Vergani P, et al. Clinical and immunologic features of ultra-short celiac disease. Gastroenterology (2016) 150:1125–34. doi: 10.1053/j.gastro.2016.01.029
198. Anderson RP, Henry MJ, Taylor R, Duncan EL, Danoy P, Costa MJ, et al. A novel serogenetic approach determines the community prevalence of celiac disease and informs improved diagnostic pathways. BMC Med. (2013) 11:1–13. doi: 10.1186/1741-7015-11-188
199. Tanpowpong P, Ingham TR, Lampshire PK, Kirchberg FF, Epton MJ, Crane J, et al. Coeliac disease and gluten avoidance in New Zealand children. Arch Dis Child. (2012) 97:12–6. doi: 10.1136/archdischild-2011-300248
200. Leffler D, Schuppan D, Pallav K, Najarian R, Goldsmith JD, Hansen J, et al. Kinetics of the histological, serological and symptomatic responses to gluten challenge in adults with coeliac disease. Gut (2013) 62:996–1004. doi: 10.1136/gutjnl-2012-302196
201. Sarna VK, Skodje GI, Reims HM, Risnes LF, Dahal-Koirala S, Sollid LM, et al. HLA-DQ:gluten tetramer test in blood gives better detection of coeliac patients than biopsy after 14-day gluten challenge. Gut (2018) 67:1606–13. doi: 10.1136/gutjnl-2017-314461
202. Bruins MJ. The clinical response to gluten challenge: a review of the literature. Nutrients (2013) 5:4614–41. doi: 10.3390/nu5114614
203. Raki M, Fallang LE, Brottveit M, Bergseng E, Quarsten H, Lundin KE, et al. Tetramer visualization of gut-homing gluten-specific T cells in the peripheral blood of celiac disease patients. Proc Natl Acad Sci USA. (2007) 104:2831–6. doi: 10.1073/pnas.0608610104
204. Ontiveros N, Tye-Din JA, Hardy MY, Anderson RP. Ex-vivo whole blood secretion of interferon (IFN)-gamma and IFN-gamma-inducible protein-10 measured by enzyme-linked immunosorbent assay are as sensitive as IFN-gamma enzyme-linked immunospot for the detection of gluten-reactive T cells in human leucocyte antigen (HLA)-DQ2.5(+) -associated coeliac disease. Clin Exp Immunol. (2014) 175:305–15. doi: 10.1111/cei.12232
205. Picascia S, Mandile R, Auricchio R, Troncone R, Gianfrani C. Gliadin-Specific T-cells mobilized in the peripheral blood of coeliac patients by short oral gluten challenge: clinical applications. Nutrients (2015) 7:10020–31. doi: 10.3390/nu7125515
206. Sarna VK, Lundin KE A, Morkrid L, Qiao SW, Sollid LM, Christophersen A. HLA-DQ-gluten tetramer blood test accurately identifies patients with and without celiac disease in absence of gluten consumption. Gastroenterology (2018) 154:886–96 e6. doi: 10.1053/j.gastro.2017.11.006
207. Catassi C, Fabiani E, Iacono G, D'Agate C, Francavilla R, Biagi F, et al. A prospective, double-blind, placebo-controlled trial to establish a safe gluten threshold for patients with celiac disease. Am J Clin Nutr. (2007) 85:160–6. doi: 10.1093/ajcn/85.1.160
208. Bardella MT, Velio P, Cesana BM, Prampolini L, Casella G, Di Bella C, et al. Coeliac disease: a histological follow-up study. Histopathology (2007) 50:465–71. doi: 10.1111/j.1365-2559.2007.02621.x
209. Newnham ED, Shepherd SJ, Strauss BJ, Hosking P, Gibson PR. Adherence to the gluten-free diet can achieve the therapeutic goals in almost all patients with coeliac disease: a 5-year longitudinal study from diagnosis. J Gastroenterol Hepatol. (2016) 31:342–9. doi: 10.1111/jgh.13060
210. Rubio-Tapia A, Rahim MW, See JA, Lahr BD, Wu TT, Murray JA. Mucosal recovery and mortality in adults with celiac disease after treatment with a gluten-free diet. Am J Gastroenterol. (2010) 105:1412–20. doi: 10.1038/ajg.2010.10
211. Wahab PJ, Meijer JW, Mulder CJ. Histologic follow-up of people with celiac disease on a gluten-free diet: slow and incomplete recovery. Am J Clin Pathol. (2002) 118:459–63. doi: 10.1309/EVXT-851X-WHLC-RLX9
212. Lanzini A, Lanzarotto F, Villanacci V, Mora A, Bertolazzi S, Turini D, et al. Complete recovery of intestinal mucosa occurs very rarely in adult coeliac patients despite adherence to gluten-free diet. Aliment Pharmacol Ther. (2009) 29:1299–308. doi: 10.1111/j.1365-2036.2009.03992.x
213. Lebwohl B, Murray JA, Rubio-Tapia A, Green PH, Ludvigsson JF. Predictors of persistent villous atrophy in coeliac disease: a population-based study. Aliment Pharmacol Ther. (2014) 39:488–95. doi: 10.1111/apt.12621
214. Leonard MM, Weir DC, DeGroote M, Mitchell PD, Singh P, Silvester JA, et al. Value of IgA tTG in predicting mucosal recovery in children with celiac disease on a gluten-free diet. J Pediatr Gastroenterol Nutr. (2017) 64:286–291. doi: 10.1097/MPG.0000000000001460
215. Ludvigsson JF, Ciacci C, Green PH, Kaukinen K, Korponay-Szabo IR, Kurppa K, et al. Outcome measures in coeliac disease trials: the tampere recommendations. Gut (2018) 67:1410–24. doi: 10.1136/gutjnl-2017-314853
216. Moreno ML, Rodriguez-Herrera A, Sousa C, Comino I. Biomarkers to monitor gluten-free diet compliance in celiac patients. Nutrients (2017) 9:46. doi: 10.3390/nu9010046
217. Comino I, Fernandez-Banares F, Esteve M, Ortigosa L, Castillejo G, Fambuena B, et al. Fecal gluten peptides reveal limitations of serological tests and food questionnaires for monitoring gluten-free diet in celiac disease patients. Am J Gastroenterol. (2016) 111:1456–65. doi: 10.1038/ajg.2016.439
218. Moreno ML, Cebolla A, Munoz-Suano A, Carrillo-Carrion C, Comino I, Pizarro A, et al. Detection of gluten immunogenic peptides in the urine of patients with coeliac disease reveals transgressions in the gluten-free diet and incomplete mucosal healing. Gut (2017) 66:250–7. doi: 10.1136/gutjnl-2015-310148
219. Kurada S, Yadav A, Leffler DA. Current and novel therapeutic strategies in celiac disease. Expert Rev Clin Pharmacol. (2016) 9:1211–23. doi: 10.1080/17512433.2016.1200463
220. Tye-Din JA, Anderson RP, Ffrench RA, Brown GJ, Hodsman P, Siegel M, et al. The effects of ALV003 pre-digestion of gluten on immune response and symptoms in celiac disease in vivo. Clin Immunol. (2010) 134:289–95. doi: 10.1016/j.clim.2009.11.001
221. Lahdeaho ML, Kaukinen K, Laurila K, Vuotikka P, Koivurova OP, Karja-Lahdensuu T, et al. Glutenase ALV003 attenuates gluten-induced mucosal injury in patients with celiac disease. Gastroenterology (2014) 146:1649–58. doi: 10.1053/j.gastro.2014.02.031
222. Tack GJ, van de Water JM, Bruins MJ, Kooy-Winkelaar EM, van Bergen J, Bonnet P, Vreugdenhil AC, et al. Consumption of gluten with gluten-degrading enzyme by celiac patients: a pilot-study. World J Gastroenterol. (2013) 19:5837–47. doi: 10.3748/wjg.v19.i35.5837
223. Kelly CP, Green PH, Murray JA, Dimarino A, Colatrella A, Leffler DA, et al. Larazotide acetate celiac disease study: larazotide acetate in patients with coeliac disease undergoing a gluten challenge: a randomised placebo-controlled study. Aliment Pharmacol Ther. (2013) 37:252–62. doi: 10.1111/apt.12147
224. Leffler DA, Kelly CP, Abdallah HZ, Colatrella AM, Harris LA, Leon F, et al. A randomized, double-blind study of larazotide acetate to prevent the activation of celiac disease during gluten challenge. Am J Gastroenterol. (2012) 107:1554–62. doi: 10.1038/ajg.2012.211
225. Leffler DA, Kelly CP, Green PH, Fedorak RN, DiMarino A, Perrow W, et al. Larazotide acetate for persistent symptoms of celiac disease despite a gluten-free diet: a randomized controlled trial. Gastroenterology (2015) 148:1311–9 e6. doi: 10.1053/j.gastro.2015.02.008
226. McCarville JL, Nisemblat Y, Galipeau HJ, Jury J, Tabakman R, Cohen A, et al. BL-7010 demonstrates specific binding to gliadin and reduces gluten-associated pathology in a chronic mouse model of gliadin sensitivity. PLoS ONE (2014) 9:e109972. doi: 10.1371/journal.pone.0109972
227. Gianfrani C, Siciliano RA, Facchiano AM, Camarca A, Mazzeo MF, Costantini S, et al. Transamidation of wheat flour inhibits the response to gliadin of intestinal T cells in celiac disease. Gastroenterology (2007) 133:780–9. doi: 10.1053/j.gastro.2007.06.023
228. Gil-Humanes J, Piston F, Tollefsen S, Sollid LM, Barro F. Effective shutdown in the expression of celiac disease-related wheat gliadin T-cell epitopes by RNA interference. Proc Natl Acad Sci USA. (2010) 107:17023–8. doi: 10.1073/pnas.1007773107
229. Sanchez-Leon S, Gil-Humanes J, Ozuna CV, Gimenez MJ, Sousa C, Voytas DF, et al. Low-gluten, nontransgenic wheat engineered with CRISPR/Cas9. Plant Biotechnol J. (2018) 16:902–10. doi: 10.1111/pbi.12837
230. Juhász A, Belova T, Florides C, Maulis C, Fischer I, Gell G, et al. Genome mapping of seed borne allergens and immune-responsive proteins in wheat. Sci Adv. (2018) 4:eaar8602. doi: 10.1126/sciadv.aar8602
231. Galipeau HJ, Wiepjes M, Motta JP, Schulz JD, Jury J, Natividad JM, et al. Novel role of the serine protease inhibitor elafin in gluten-related disorders. Am J Gastroenterol. (2014) 109:748–56. doi: 10.1038/ajg.2014.48
232. Syage JA, Murray JA, Green PH R, Khosla C. Latiglutenase improves symptoms in seropositive celiac disease patients while on a gluten-free diet. Dig Dis Sci. (2017) 62:2428–32. doi: 10.1007/s10620-017-4687-7
233. Anderson RP, Jabri B. Vaccine against autoimmune disease: antigen-specific immunotherapy. Curr Opin Immunol. (2013) 25:410–7. doi: 10.1016/j.coi.2013.02.004
234. Daveson AJM, Ee HC, Andrews JM, King T, Goldstein KE, Dzuris JL, et al. Epitope-specific immunotherapy targeting CD4-Positive T Cells in celiac disease: safety, pharmacokinetics, and effects on intestinal histology and plasma cytokines with escalating dose regimens of nexvax2 in a randomized, double-blind, placebo-controlled phase 1 study. EBioMedicine (2017) 26:78–90. doi: 10.1016/j.ebiom.2017.11.018
Keywords: celiac disease, gluten, T cells, microbiome, pathogenesis
Citation: Tye-Din JA, Galipeau HJ and Agardh D (2018) Celiac Disease: A Review of Current Concepts in Pathogenesis, Prevention, and Novel Therapies. Front. Pediatr. 6:350. doi: 10.3389/fped.2018.00350
Received: 22 August 2018; Accepted: 29 October 2018;
Published: 21 November 2018.
Edited by:
Ron Shaoul, Rambam Health Care Campus, IsraelReviewed by:
Gianfranco Meloni, University of Sassari, ItalyTudor Lucian Pop, Iuliu Haţieganu University of Medicine and Pharmacy, Romania
Copyright © 2018 Tye-Din, Galipeau and Agardh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jason A. Tye-Din, dHllZGluQHdlaGkuZWR1LmF1