
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pediatr. , 30 October 2018
Sec. Pediatric Gastroenterology, Hepatology and Nutrition
Volume 6 - 2018 | https://doi.org/10.3389/fped.2018.00317
This article is part of the Research Topic Pediatric Inflammatory Bowel Diseases: Looking to the Future View all 13 articles
 Deenaz Zaidi1,2
Deenaz Zaidi1,2 Eytan Wine1,2,3*
Eytan Wine1,2,3*Inflammatory bowel diseases (IBD), encompassing both Crohn Disease (CD) and ulcerative colitis (UC) are globally prevalent diseases, impacting children of all ages. The hallmark of IBD is a perturbed immune system that leads to continuous inflammation in the gut and challenges optimal treatment. Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κβ), a nuclear transcription factor, plays a major role in gut homeostasis and contributes significantly toward a balanced, homeostatic immune system. Dysregulation in the NF-κβ pathway and factors that regulate it lead to a state of uncontrolled inflammation and altered immunity, as typically observed in IBD. Levels of proinflammatory cytokines that are regulated through NF-κβ are increased in both CD and UC. Genes known to activate NF-κβ, such as, Nucleotide-binding oligomerization domain-containing protein 2 (NOD2) and Interleukin 23 (IL-23), are associated with IBD. Factors involved in inhibition of NF-κβ, such as A20 and TOLLIP, are also affected in IBD, resulting in failed inflammation suppression/regulation. NOD-2 and A20 have specifically been found to be strongly associated with pediatric IBD. Gut commensals are known to exert anti-inflammatory activities toward NF-κβ and can have a potential role in attenuating inflammation that likely occurs due to microbial dysbiosis in IBD. Failure to terminate/downregulate NF-κβ signaling results in chronic inflammation in IBD. Well-regulated control of inflammation in children with IBD can help better control the disease and suppress immune responses. Better understanding of factors that control NF-κβ can potentially lead toward discovering targeted therapeutic interventions for IBD. Suppression of NF-κβ can be achieved through many modalities including anti-sense oligonucleotides (ASOs), siRNA (small interfering RNA), factors regulating NF-κβ, and microbes. This review focuses on the role of NF-κβ, especially in pediatric IBD, and potential therapeutic venues for attenuating NF-κβ-induced inflammation.
The immune system of the gastrointestinal tract is normally well-tuned with the gut microenvironment, which enables the existence of a steady homeostatic state. The gut environment is continuously exposed to various exogenous materials, including food, xenobiotics, and microbial pathogens. Eradication of pathogens with simultaneous survival of gut commensals that are beneficial for maintaining homeostasis is a major challenge faced by the intestinal immune system. Nature has many protective mechanisms in place that help maintain this stable environment in the gut. This stability is disrupted in disease conditions affecting the gastrointestinal system, such as inflammatory bowel diseases (IBD). IBD, including both Crohn disease (CD) and ulcerative colitis (UC) have a debilitating impact on the lives of children and adults alike (1, 2). Some of the major features distinguishing between adult and pediatric IBD are nutritional challenges, poor bone health, delayed puberty, and growth failure, all of which are linked to inflammation (3, 4). Given the complex complications of IBD in children, it is critical to understand the basic factors that trigger inflammation and modulate treatment regimens accordingly.
Although many factors have been associated with IBD, the etiology is still not clearly understood, but seems to involve integrated mechanisms of uncontrolled immune response to various environmental/microbial stimuli in genetically susceptible hosts. Recent research has highlighted the importance of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κβ) in regulating immune responses in the gut. This review will focus on the role of NF-κβ in IBD and potential therapeutic mechanisms that can control NF-κβ-mediated inflammation in IBD, highlighting aspects especially relevant to children (5).
NF-κβ, a nuclear transcription factor, is a central player of sustaining a stable state of innate immunity in the gut. Disruptions and imbalances in the NF-κβ pathway lead to chronic inflammation, dysregulation of natural immune responses (6, 7), and altered immunity in IBD (8). Pathogenesis in both CD and UC is heavily marked by expression of multiple proinflammatory cytokines (9), many of which are regulated through NF-κβ. In fact, several of the key genes associated with IBD, such as Nucleotide-binding oligomerization domain-containing protein 2 (NOD2) and Interleukin 23 (IL-23), drive NF-κβ activation. On the flip side, dysregulation of NF-κβ inhibitory pathways, such as reduced expression of A20 (tumor necrosis factor α-induced protein 3; TNFAIP3) or TOLLIP (10), could also contribute to unremitting inflammation, as seen in NF-κβ essential modulator (NEMO) epithelial cell-specific knockout mice (8, 11). Thus, better understanding of factors that drive and control NF-κβ could lead to targeted therapeutic interventions for inflammatory conditions, including IBD.
As we hypothesize that dysregulation, or the inability to terminate NF-κβ signaling is critical for the persistence of chronic inflammation in IBD, we would argue that this is especially important in children as microbe-driven persistent inflammation cannot be turned off in this setting and is likely to drive chronic inflammation at its early stages. We have recently shown that A20, a negative regulator of NF-κβ, is specifically disrupted in pediatric IBD; despite an observed increase in A20 gene expression, protein levels and associated signaling are reduced, suggesting a pediatric-specific dysregulation of A20 and NF-κβ (12). In contrast, adult studies have shown variable expression of A20 (13).
Early life factors can define the immune milieu and microbial interactions and predispose for immune-mediated conditions, such as IBD; regulation of NF-κβ is likely to be critical for this process. For all these reasons, better defining how NF-κβ is regulated in children is likely to provide important insight into pathogenesis and guide future therapies. It is quite likely that as more therapies will target NF-κβ regulation, physicians and scientist caring for children with IBD would benefit from deeper understanding of this complex pathway.
NF-κβ structure consists of multiple protein subunits: p52, RelA (p65), p50, c-Rel, and RelB, which are coalesced in the cytoplasm bound to Iκβ proteins (Figure 1). N-terminal Rel homology domain (RHD) is shared by all subunits and is essential for dimer formation. The subunits are conjoined in the cytoplasm in the resting state and remain inactive while attached to Iκβ proteins. Gene transcription can be regulated by RelA, RelB, and c-Rel as they have a transcriptional activation domain (TAD). The transcriptional activity of the p50 and p52 subunits depends upon binding with proteins that have TAD, for example, RelA, RelB, and c-Rel (14).
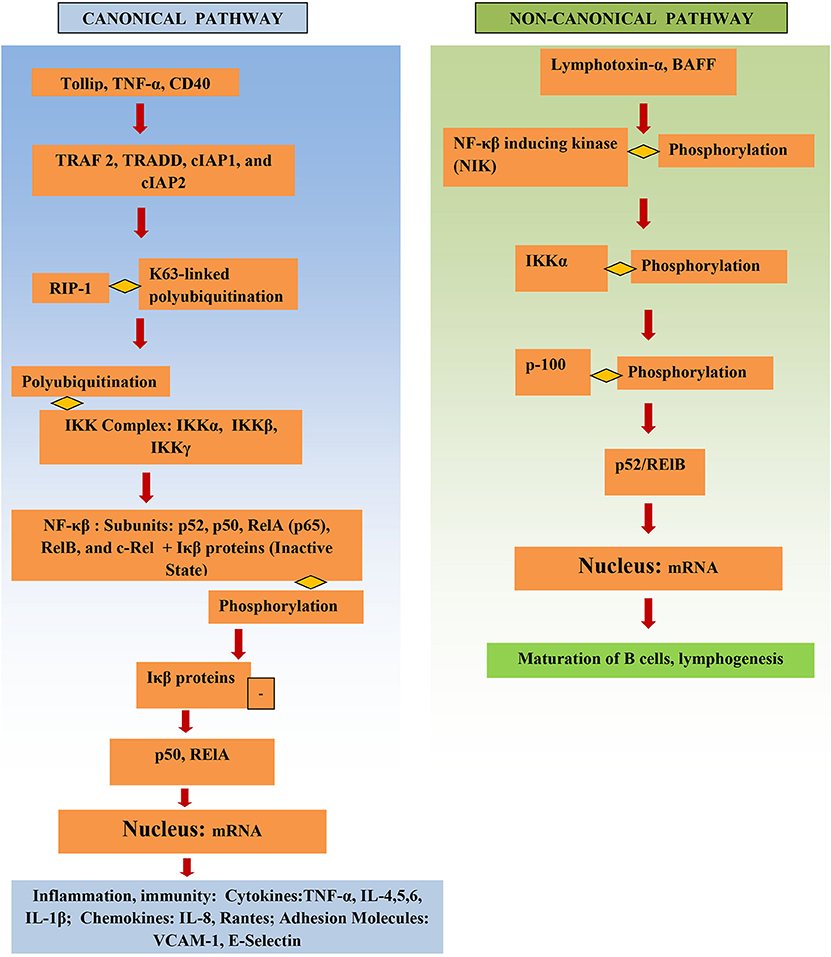
Figure 1. Structure and activation of NF-κβ.
The IKK complex is the major factor that activates the NF-κβ pathway. It consists of IKKγ (a non-catalytic protein) and the kinases IKKα and IKKβ. Phosphorylation of Iκβ by IKK results in its proteasomal degradation and thus activates NF-κβ, releasing NF-κβ, and resulting in NF-κβ subunits being translocated into the nucleus and leading to proinflammatory gene transcription.
The NF-κβ pathway is activated either through the canonical or the non-canonical pathway (Figure 1). Initiation of the canonical pathway occurs through a process of receptor-ligand binding. Binding and stimulation of TLRs with antigens leads adaptors, such as TRADD, TRAF 2, cIAP1, and cIAP2 and RIP1, to bind to NF-κβ receptor's cytoplasmic domain. K63-linked polyubiquitination of RIP1 aids the IKK complex in recruitment of NF-κβ to the activated receptor. This activates the IKK complex, which in turn phosphorylates Iκβ and causes it to degrade (15). Activation of NF-κβ via the canonical pathway triggers production of cytokines in IBD, such as TNF-α and IL-6 (16).
The non-canonical pathway depends upon activation of NF-κβ inducing kinase (NIK); this involves phosphorylation and subsequent activation of the IKKα complex by NIK. Activation leads to phosphorylation of p100 by IKKα, which in turn results in the formation of the p52/RelB active heterodimer. The p52/RelB active heterodimer is then translocated to the nucleus. Induction of the non-canonical pathway occurs through multiple factors, such as, IL-1β lymphotoxin-α, and BAFF. IκBα mediates the turn-off inhibitory signal for NF-κβ by binding with nuclear NF-κβ complexes and transferring them back to the cytoplasm (6). Several genes involved in the non-canonical pathway were significantly higher in diseased tissue of IBD patients vs. adjacent healthy areas and healthy controls (17).
Microbe-associated molecular patterns (MAMPs), damage-associated molecular pattern molecules (DAMPs), cytokines, oxidative stress, bacteria, viruses, and ischemia stimulate and activate the NF-κβ pathway (6).
There is a pathological shift in gut homeostasis in IBD that activates NF-κβ, which in turn further propagates inflammation (8). IBD patients had high levels of NF-κβ, and biopsies of inflamed regions showed a significantly higher number of NF-κβ positive cells compared to normal regions (18). NF-κβ activation has been linked to disease phenotype in CD patients, with high NF-κβ levels correlating with increased ileocolonic and less perianal involvement (19). While NF-κβ is closely linked to IBD, much remains unknown about the specific mechanisms of involvement in disease pathogenesis. Below are a few examples of what is known and how this could impact IBD.
Expression and activation of NF-κβ is greatly increased in the gut of IBD patients and is largely cell-specific. NF-κβ subunit p65 levels were found to be higher in the lamina propria of biopsy specimens from CD patients as compared to UC patients and controls (20). NF-κβ is involved in the induction and regulation of many cytokines, including IL-6, TNF-α, IL-1β (21), and IL-12 (9). IL-12 plays an important role in augmenting the differentiation of Th-1 helper cells, and other cytokines, such as TNF-α and IL-23 are also involved in this phenomenon that is critical for inflammation propagation (22, 23). Thus, the effect of NF-κβ on IBD is paramount, as TNF-α is involved in exerting extensive damage to the mucosa and extracellular matrix by being involved in the regulation of, and increasing levels of matrix metalloproteinase (24). In addition, it was found that NF-κβ was induced by IL-6 in colonic epithelial cells and caused an increase in the expression of intercellular adhesion molecule-1 in epithelial cells, which is essential for recruiting neutrophil granulocytes to places of inflammation (25). CD40L induced NF-κβ activation in fibroblasts of colonic epithelial cells, which in turn augmented the expression of IL-6 and IL-8 (26). The IL-6-STAT3 pathway is activated in pediatric IBD (27).
Several cytokines that are increased in IBD and contribute toward inflammation are especially relevant to children. For example, IL-6 is increased in intestinal lamina propria biopsies and serum of pediatric IBD patients (28). In children with IBD, low bone mineral density is attributed to high levels of IL-6 (29). Similar to adults there is an increase in TNF-α levels in the terminal ileum of pediatric CD patients (12). IL-7, IL-1β, IL-5, IL-16, interferon (IFN)-γ-inducible protein-10, leukemia inhibitory factor, monokine induced by IFN-γ, IFN-α2, and IFN-γ were also found to be increased in serum of pediatric IBD patients as compared to healthy control patients, whereas, IL-17, macrophage inhibitory protein-1β, and IL-2 were decreased; many of these cytokines are regulated by NF-κβ-related pathways (30). This imbalance in cytokine regulation indicates the need for further exploration of NF-κβ-related inflammatory pathway in pediatric IBD, as their role in propagating inflammation remains unclear.
Along with its association with inflammation, evidence suggests that NF-κβ has an anti-inflammatory role as well, as seen by increased intestinal inflammation, apoptosis, and reduced antimicrobial peptides in a NEMO-deficient epithelial cell mouse model (31). Similarly, NF-κβ RelA intestinal cell conditional knock-out mice were susceptible to develop DSS-induced colitis (32). These examples highlight the multifaceted, complex nature of innate immune control in the gut.
A20 is a cytoplasmic protein that acts as a significant inhibitor/regulator of NF-κβ-induced inflammation (33). A20 plays an important role in counter-regulating inflammation in the gut, as shown by the presence of damaged intestinal epithelium and increased apoptosis after intestinal epithelial cells-specific A20 knock-out mice were treated with TNF-α (34). A20 is an important inhibitor of TNF-α-induced NF-κβ inflammation (35). A20 also suppresses CD40 and IL-1, and pattern recognition receptors induced NF-κβ-mediated inflammation (36). A20 expression is increased in pediatric IBD patients with a simultaneous reduction in A20 protein levels, possibly due to destabilization of A20-chaperone factors in IBD (12). Genome-wide association studies (GWAS) have shown linkage between A20 and IBD (11). In adult IBD patients, A20 profiling has shown varying correlations with disease phenotype and severity. A20 expression was low in the colonic and terminal ileum (TI) mucosa (13) and was found to be high in colonic biopsies of adult UC, but not CD patients (37).
Gut commensals are integral to homeostasis and many regulatory functions, interacting with the mucosal immune system. As such, commensals are heavily involved in anti-inflammatory activities targeted toward NF-κβ in the gut, such as inhibition of NF-κβ activity through peroxisome proliferator activated receptor-γ (PPAR-γ) by Bacteroides thetaiotaomicron, which in turn suppresses transportation of the NF-κβ subunit RelA into the nucleus (38). Bifidobacterium infantis downregulates NF-κβ activity induced by LPS (39). Lactobacillus Casei counteracts inflammation induced by Shigella flexneri infection that causes increased transcription of inflammatory cytokines by acting on pathways that stabilize Iκβ and hence prevents translocation of NF-κβ to the nucleus (40). Microbial dysbiosis is a prominent factor involved in IBD and contributes toward inflammation.
IBD in children has been subclassified into different categories by age; above or below 10 years, very early onset IBD (VEOIBD) in children < 6 years, and infantile IBD in children < 2 years (41). Pediatric IBD differs from adult IBD in many aspects. Positive family history of IBD is more often the case in pediatric cases vs. in adults. Most often, at the initial stages, IBD occurs in the colon in young children, whereas in adults, small bowel is usually involved. Young children with Crohn disease have more colonic involvement than adults do. Pediatric IBD is more often refractory to medical and surgical treatments commonly used for management of IBD in older patients (42). The proportion of monogenetic causes of IBD-like presentation is highest in the VEOIBD and infantile groups and genetic defects that control NF-κβ, such as variations in TRIMM22, appear to be especially relevant in children (5). Defects and variations in IL-10 and IL-10 receptor are also significant in children with VEOIBD (43, 44). Alterations in other genes, such as, LRBA (45), XIAP (46), and TTC7A (47) is associated with a high risk of developing IBD mostly in childhood (48), but also in adults.
Changes in expression of NF-κβ and associated factors have been observed with several treatments for IBD. In IBD patients treated with corticosteroids, colonic epithelial, mononuclear, and endothelial cells had significantly less nuclear NF-κβ-p65 levels than cells from untreated patients. Corticosteroids increase the expression of IκBα, which retains NF-κβ in the cytoplasm and interacts physically with p65, thus preventing the activation of NF-κβ (49). However, prolonged use of corticosteroids affects linear growth and has the potential to cause hypertension, osteopenia, and increased susceptibility to infection (50).
Sulfasalazine was found to suppress IKKα and IKKβ, which in turn inhibit NF-κβ (51)
In vitro experiments revealed that when NF-κβ activation induced by TNF was suppressed by methotrexate, it appeared to be through prevention of phosphorylation and degradation of IκBα, which retains NF-κβ in the cytoplasm and interact physically with p65, thus preventing the activation of NF-κβ (52).
Infliximab treatment caused an increase in IκBα and IκBγ in colonic biopsies; this then inhibits NF-κβ activation and helps in maintaining remission in pediatric patients (53).
Exclusive enteral nutrition (EEN) has been proven to induce clinical remission and lead to mucosal healing in pediatric CD with matching or even superior efficacy to that of corticosteroids (54, 55). Although the use of EEN has been adopted across the globe (56), the mechanism of action remains unclear (54). Increased attenuation of inflammation was also observed in murine models of DSS-colitis along with suppression of TNF-α, IL-6, and IL-8 in colonic biopsies with administration of a novel nutritional polymeric formula. In vitro experiments also showed suppression of genes associated with the NF-κβ pathway including, TNF, TNFSF10, NF-κβ1, and RELB with polymeric formula (57). Arginine and glutamine present in polymeric formula suppress phosphorylation involved in the NF-κβ and P38 pathways preventing NF-κβ activation (58). Curcumin, glutamine, and arginine together suppressed IL-8, raising the option of addition of curcumin to polymeric formula to suppress inflammation in IBD (55).
Thus, although current standard therapies for IBD do exert an effect on NF-κβ to some extent, through its associated factors, additional therapies are required for better control of NF-κβ-associated inflammation, Furthermore, much of the published work was done in in vitro settings, which is a controlled environment and is starkly different than the actual gut microenvironment where multiple factors are simultaneously at play. Therefore, it is important to focus on the development of a translational approach to develop additional therapies that impact NF-κβ, as described in the following section.
Given the pathogenic role of NF-κβ in IBD pathogenesis, it would be attractive to reduce NF-κβ activity by manipulating its regulation. This could be achieved by either directly suppressing NF-κβ or indirectly, by enhancing factors that regulate it. The function of proteasomes differs amongst CD patients and healthy individuals as evident by an increased conversion of the precursor p105 toward its active form, p50 in CD patients (59). Proteasome inhibitors designed for proteasomes that convert the p105 precursor toward active p50 and enhance NF-κβ activation can be beneficial in breaking this pathway. Animal studies have reported successful treatment of TNBS-induced colitis with p65 antisense oligonucleotides that directly target NF-κβ proteins and block them, thus inhibiting the activation of NF-κβ (60). Similar studies have not yet been done in humans, highlighting the importance of early stages of developing drugs focused on enhancing immune regulation, in contrast to current mostly immunosuppressive approaches.
Alterations in gut microbial composition in IBD has been reported by many studies (61) and focus on future targeted microbe-altering therapies is under consideration (62). Fecal microbial transplant (FMT) has been used as a treatment for IBD, but results reported by different studies are variable (63, 64). While studies have shown that gut microbes do exert an inhibitory action on NF-κβ, experimental approaches for studying the mechanism of action of microbes toward NF-κβ in models of IBD and gut environment, such as, organoids derived from IBD patients, need to be developed. As “designer microbes” have been developed to induce immune regulation (through secretion of IL-10, for example) (65), it would be attractive to use microbes to directly regulate the NF-κβ pathway.
Treatments to prevent A20 degradation and administration of factors that stabilize it might be beneficial. A striking correlation was found between A20 and anti-TNF therapy within a Danish cohort of IBD patients, where functional polymorphisms in A20 were predictive of response to anti-TNF therapy (66). Probing another cohort revealed a correlation between A20 SNPs and response to anti-TNF therapy (67). These findings suggest the possibility of using polymorphisms in A20 as a genetic biomarker, a venue that should be explored further for practical application. Post-translation modification highly impacts the stability of A20. IKKβ, a kinase required to activate the NF-κβ pathway is also involved in the phosphorylation of A20 at the serine 381 (S381) site, and thus helps A20 to stabilize and stop NF-κβ signaling. As phosphorylation of A20 by IKKβ occurs in response to LPS and TNF (68), presence of IKKβ in inflammation could be a counteractive action to combat NF-κβ induced inflammation too, in addition to stimulating it. Perhaps the key is in studying the conditions that help IKKβ stimulate NF-κβ and altering them.
Paracaspase MALT-1 regulates T cell receptor signaling to NF-κβ and is essential for T cell activation. A20 is directed to a complex of MALT-1 and Bcl-10 upon T-cell receptor stimulation, resulting in its cleavage and rendering it unable to stop the NF-κβ signal (69). Thus, drugs that target MALT-1 can also have an indirect affect on A20, by removing MALT-1 from the cellular environment.
Interaction of A20 with other proteins, such as Tax1 binding protein 1 (TAX1BP-1) and A20-binding inhibitor of NF-κB activation 1 (ABIN-1) helps in attenuating inflammation. The ABIN family of proteins negatively regulate NF-κβ; they are ubiquitin binders and attach to NEMO (NF-κβ essential modulator complex; the IKK complex) (70). ABIN-1 aids A20 to attach to the IKK/NEMO complex and exert its deubiquitinating process (71). The expression of ABIN-1 is dependent upon NF-κβ (72). TAX1BP-1 is also involved in inhibition of NF-κβ-induced inflammation (73) and recruits A20 to the polyubiquitin chains, where A20 breaks and interrupts the IKK complex (74). Ensuring constant presence and stability of TAX1BP-1 and ABIN-1 therefore can play a very important role in suppressing NF-κβ induced inflammation.
As it was reported that CD patients with high NF-κβ levels had more ileocolonic disease and less perianal involvement than those with normal NF-κβ activity (19), it is important to confirm in larger cohorts whether NF-κβ activity is indeed site-specific and correlates with disease status. This could especially be important for pediatric patients, as IBD can possibly be better controlled in early stages of diagnosis and at an earlier age, without the presence of other co-morbidities.
Targeting controllers of NF-κβ to attenuate its activity directly has tremendous potential to suppress inflammation. Stability of tollip, protein that inhibit inflammation by preventing IL-1 interaction with IL-1Rs, inhibit IRAK phosphorylation and inhibit TLR-2 and TLR-4 mediated inflammation could be of benefit as well (75). In animal models, TNBS-induced colitis and DSS-induced colitis were attenuated through targeting NF-κβ 65 (60, 76), and NF-κβ 65 antisense oligonuceotides (ASO) are undergoing clinical trials currently. Better control of TNF-α (6), which activates the canonical pathway through Toll-like receptors (TLRs), through ASOs and siRNA (small interfering RNA) can be of significance important in precision therapy. Using phosphorothioate ASOs for CD40 also has anti-NF-κβ potential (77, 78). Resrevatrol, an immunomodulator and anti-cancer agent has been found to suppress p65 and IKKβ (79). Figure 2 illustrates the connection between factors affecting NF-κβ and potential therapeutic models that could be relevant to pediatric IBD.
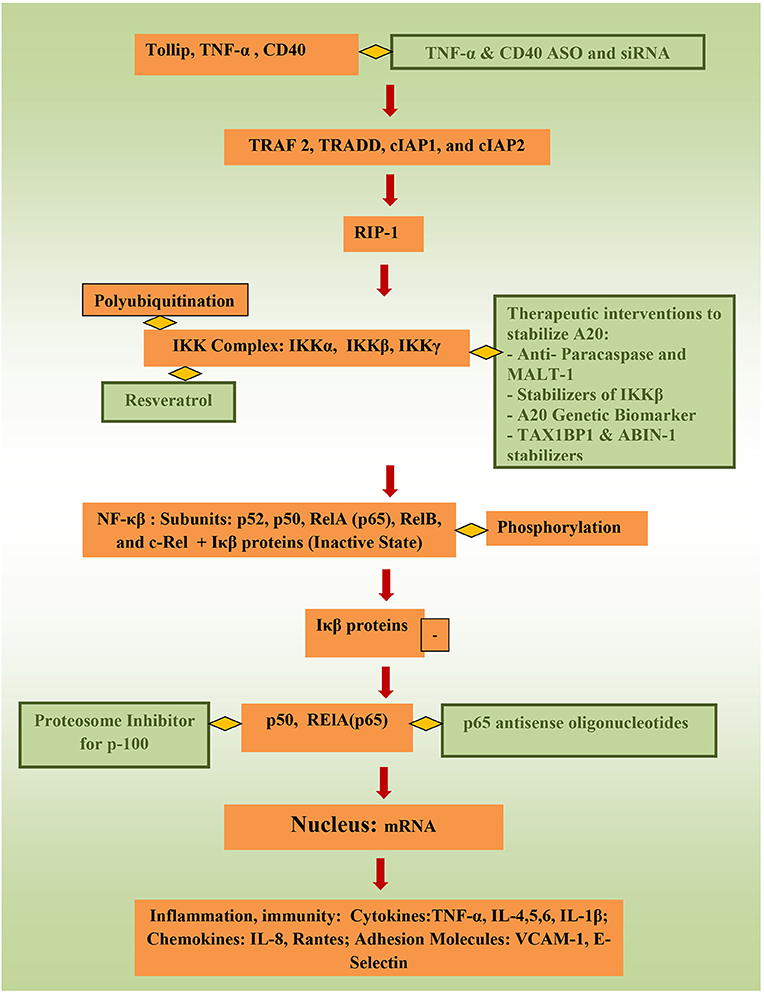
Figure 2. Potential therapeutic pathways for NF-κβ attentation (therapeutic interventions are shown in green).
Current IBD therapy is mostly aimed at sustaining immunosuppression. A definite cure is yet to be found. The aim of IBD treatment is induction and maintenance of remission, and prevention of flares and complications. Balance between drug safety and efficacy is a therapeutic challenge, as current medications have serious side effects, emphasizing the need of development of precision therapy for IBD patients. Current IBD treatments affecting NF-κβ are described in Table 1. Potential future treatments for IBD related to NF-κβ are described in Table 2.
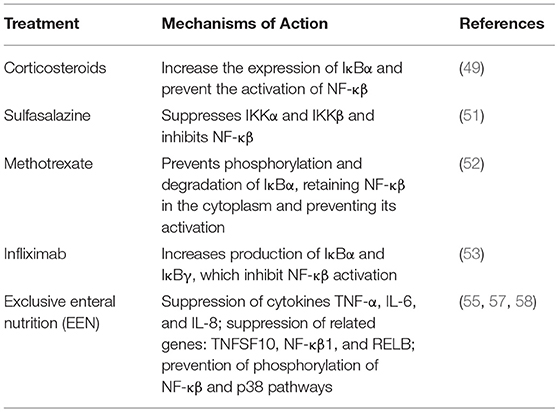
Table 1. Current IBD Treatments Affecting NF-κβ.

Table 2. Potential future treatments for IBD, related to NF-κβ.
Treatments directed toward suppressing NF-κβ have a huge therapeutic potential against diseases such as IBD, cancer, and other inflammatory conditions. This is especially relevant in children as controlling the inflammatory cascade at early stages can prevent the devastating long term effects of uncontrolled inflammation as seen in IBD. Systematic in vitro and in vivo studies in animal models need to be conducted to analyze factors that antagonize/attenuate NF-κβ, and results need to be interpreted with caution; however, in contrast to almost all IBD treatments used today, which suppress the immune response, modulating pathways, for example by enhancing NF-κβ regulating molecules, such as A20, could allow the gut environment to return to homeostasis, possibly without an increase in infection risk. Obviously, before translating potential treatments to treat humans, safety and efficacy of potential therapeutic regimens needs to be established. Development of medications specifically targeting NF-κβ to control its inflammatory activity can be of tremendous benefit to children with IBD, such as early control of symptoms and low risk of immunosuppression that is often associated with current IBD medications.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Eszter Muller K, Laszlo Lakatos P, Papp M, Veres G. (2014). Incidence and paris classification of pediatric inflammatory bowel disease. Gastroenterol Res Pract. (2014) 2014:904307. doi: 10.1155/2014/904307
2. Benchimol EI, Manuel DG, Guttmann A, Nguyen GC, Mojaverian N, Quach P, et al. Changing age demographics of inflammatory bowel disease in Ontario, Canada: a population-based cohort study of epidemiology trends. Inflamm Bowel Dis. (2014) 20:1761–9. doi: 10.1097/MIB.0000000000000103
3. Brain CE, Savage MO. Growth and puberty in chronic inflammatory bowel disease. Baillieres Clin Gastroenterol. (1994) 8:83–100.
4. Ezri J, Marques-Vidal P, Nydegger A. Impact of disease and treatments on growth and puberty of pediatric patients with inflammatory bowel disease. Digestion (2012) 85:308–19. doi: 10.1159/000336766
5. Li Q, Lee CH, Peters LA, Mastropaolo LA, Thoeni C, Elkadri A, et al. Variants in TRIM22 that affect NOD2 signaling are associated with very-early-onset inflammatory Bowel disease. Gastroenterology (2016) 150:1196–207. doi: 10.1053/j.gastro.2016.01.031
6. Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-κB signaling pathways. Nat Immunol. (2011) 12:695–708. doi: 10.1038/ni.2065
7. Hayden MS, Ghosh S. Shared principles in NF-κB signaling. Cell (2008) 132:344–62. doi: 10.1016/j.cell.2008.01.020
8. Atreya I, Atreya R, Neurath MF. NF-κB in inflammatory bowel disease. J Intern Med. (2008) 263:591–6. doi: 10.1111/j.1365-2796.2008.01953.x
9. Neurath MF. Cytokines in inflammatory bowel disease. Nat Rev Immunol. (2014) 14:329–42. doi: 10.1038/nri3661
10. Byun EB, Kim WS, Sung NY, Byun EH. Epigallocatechin-3-Gallate Regulates Anti-Inflammatory Action Through 67-kDa Laminin Receptor-Mediated Tollip Signaling Induction in Lipopolysaccharide-Stimulated Human Intestinal Epithelial Cells. Cell Physiol Biochem. (2018) 46:2072–81. doi: 10.1159/000489447
11. Ma A, Malynn BA. A20: linking a complex regulator of ubiquitylation to immunity and human disease. Nat Rev Immunol. (2012) 12:774–85. doi: 10.1038/nri3313
12. Zaidi D, Huynh HQ, Carroll MW, Baksh S, Wine E. Tumor necrosis factor α-induced protein 3 (A20) is dysregulated in pediatric Crohn disease. Clin Exp Gastroenterol. (2018) 11:217–31. doi: 10.2147/CEG.S148217
13. Arsenescu R, Bruno ME, Rogier EW, Stefka AT, McMahan AE, Wright TB, et al. Signature biomarkers in Crohn's disease: toward a molecular classification. Mucosal Immunol. (2008) 1:399–411. doi: 10.1038/mi.2008.32
14. Mechtler P, Ruchi S, Julia VK, Jonathan EB, Michael JB, Eugene SK. MicroRNA analysis suggests an additional level of feedback regulation in the NF-κB signaling cascade. Oncotarget (2015) 6:17097–106. doi: 10.18632/oncotarget.4005
15. Ruland J. Return to homeostasis: downregulation of NF-κB responses. Nat Immunol. (2011) 12:709–14. doi: 10.1038/ni.2055
16. Dylan KM, Kristin E, Veronica MR, Irving CA. Emerging roles for Non-canonical NF-κB signaling in the modulation of inflammatory bowel disease pathobiology. Inflamm Bowel Dis. (2016) 22:2265–79. doi: 10.1097/MIB.0000000000000858
17. Irving CA, Kristin E, Vu N, Kristin K, Dario S. Noncanonical NF-κB signaling is elevated in inflammatory bowel disease patients and may be associated with therapeutic response. J Immunol. (2017) 198(1 Suppl.) 197.5. Available online at: http://www.jimmunol.org/content/198/1_Supplement/197.5
18. Ellis RD, Goodlad JR, Limb GA, Powell JJ, Thompson RP, Punchard NA. Activation of nuclear factor kappa B in Crohn's disease. Inflamm Res Off J Eur Histamine Res Soc. Al (1998) 47:440–5.
19. Han YM, Koh J, Kim JW, Lee C, Koh SJ, Kim B, et al. NF-κB activation correlates with disease phenotype in Crohn's disease. PLoS ONE (2017) 12:e0182071. doi: 10.1371/journal.pone.0182071
20. Schreiber S, Nikolaus S, Hampe J. Activation of nuclear factor κB in inflammatory bowel disease. Gut (1998) 42:477–484.
21. Neurath MF, Pettersson S, Meyer Zum Buschenfelde KH, Strober W. Local administration of antisense phosphorothioate oligonucleotides to the p65 subunit of NF-κB abrogates established experimental colitis in mice. Nat Med. (1996) 2:998–1004.
22. Becker C, Wirtz S, Blessing M, Pirhonen J, Strand D, Bechthold O, et al. Constitutive p40 promoter activation and IL-23 production in the terminal ileum mediated by dendritic cells. J Clin Invest. (2003) 112:693–706. doi: 10.1172/JCI200317464
23. Pallone F, Monteleone G. Mechanisms of tissue damage in inflammatory bowel disease. Curr Opin Gastroenterol. (2001) 17:307–12.
24. Wang Y-D, Mao J-W. Expression of matrix metalloproteinase-1 and tumor necrosis factor-alpha in ulcerative colitis. World J. Gastroenterol. (2007) 13:5926–32. doi: 10.3748/wjg.v13.i44.5926
25. Wang L, Walia B, Evans J, Gewirtz AT, Merlin D, Sitaraman SV. IL-6 Induces NF-kB Activation in the Intestinal Epithelia. J Immunol. (2003) 171:3194–201. doi: 10.4049/jimmunol.171.6.3194
26. Gelbmann CM, Leeb SN, Vogl D, Maendel M, Herfarth H, Schölmerich J, et al. Inducible CD40 expression mediates NFkappaB activation and cytokine secretion in human colonic fibroblasts. Gut (2003) 52:1448–56. doi: 10.1136/gut.52.10.1448
27. Carey R, Jurickova I, Ballard E, Bonkowski E, Han X, Xu H, et al. Activation of an IL-6:STAT3-dependent transcriptome in pediatric-onset inflammatory bowel disease. Inflamm. Bowel Dis. (2008) 14:446–57. doi: 10.1002/ibd.20342
28. Brown KA, Back SJ, Ruchelli ED, Markowitz J, Mascarenhas M, Verma R, et al. Lamina propria and circulating interleukin-6 in newly diagnosed pediatric inflammatory bowel disease patients. Am. J. Gastroenterol. (2002) 97:2603–8. doi: 10.1111/j.1572-0241.2002.06030.x
29. Paganelli M, Albanese C, Borrelli O, Civitelli F, Canitano N, Viola F, et al. Inflammation is the main determinant of low bone mineral density in pediatric inflammatory bowel disease. Inflamm. Bowel Dis. (2007) 13:416–23. doi: 10.1002/ibd.20039
30. Kleiner G, Valentina Z, Lorenzo M, Sergio C, Lorenzo C, Daniela M, et al. Pediatric patients with inflammatory bowel disease exhibit increased serum levels of proinflammatory cytokines and chemokines, but decreased circulating levels of macrophage inhibitory protein-1β, interleukin-2 and interleukin-17. Exp Ther Med. (2015) 9:2047–52. doi: 10.3892/etm.2015.2370
31. Nenci A, Becker C, Wullaert A, Gareus R, van Loo G, Danese S, et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature (2007) 446:557–61. doi: 10.1038/nature05698
32. Steinbrecher KA, Harmel-Laws E, Sitcheran R, Baldwin AS. Loss of epithelial RelA results in deregulated intestinal proliferative/apoptotic homeostasis and susceptibility to inflammation. J Immunol. (2008) 180:2588–99. doi: 10.4049/jimmunol.180.4.2588
33. Coornaert B, Carpentier I, Beyaert R. A20: central gatekeeper in inflammation and immunity. J Biol Chem. (2009) 284:8217–21. doi: 10.1074/jbc.R800032200
34. Vereecke L, Sze M, Mc Guire C, Rogiers B, Chu Y, Schmidt-Supprian M, et al. Enterocyte-specific A20 deficiency sensitizes to tumor necrosis factor-induced toxicity and experimental colitis. J Exp Med. (2010) 207:1513–23. doi: 10.1084/jem.20092474
35. Catrysse L, Vereecke L, Beyaert R, van Loo G. A20 in inflammation and autoimmunity. Trends Immunol. (2014) 35:22–31. doi: 10.1016/j.it.2013.10.005
36. Beyaert R, Heyninck K, Van Huffel S. A20 and A20-binding proteins as cellular inhibitors of nuclear factor- kappa-B-dependent gene expression and apoptosis. Biochem Pharmacol. (2000) 60:1143–51. doi: 10.1016/S0006-2952(00)00404-4
37. Fernandes P, MacSharry J, Darby T, Fanning A, Shanahan F, Houston A, et al. Differential expression of key regulators of Toll-like receptors in ulcerative colitis and Crohn's disease: a role for Tollip and peroxisome proliferator-activated receptor gamma? Clin Exp Immunol. (2016)183:358–68. doi: 10.1111/cei.12732
38. Kelly D, Campbell JI, King TP, Grant G, Jansson EA, Coutts AG, et al. Commensal anaerobic gut bacteria attenuate inflammation by regulating nuclear-cytoplasmic shuttling of PPAR-gamma and RelA. Nat Immunol. (2004) 5:104–12. doi: 10.1038/ni1018
39. O'Mahony C, Scully P, O'Mahony D, Murphy S, O'Brien F, Lyons A, et al. Commensal-induced regulatory T cells mediate protection against pathogen-stimulated NF-kappaB activation. PLoS Pathog. (2008) 4:e1000112. doi: 10.1371/journal.ppat.1000112
40. Tien M-T, Girardin SE, Regnault B, Le Bourhis L, Dillies MA, Coppée JY, et al. Anti-inflammatory effect of Lactobacillus casei on Shigella-infected human intestinal epithelial cells. J Immunol Baltim. Md 1950 (2006) 176:1228–37. doi: 10.4049/jimmunol.176.2.1228
41. Benchimol EI, Bernstein CN, Bitton A, Carroll MW, Singh H, Otley AR, et al. Trends in Epidemiology of Pediatric Inflammatory Bowel Disease in Canada: Distributed Network Analysis of Multiple Population-Based Provincial Health Administrative Databases. Am J Gastroenterol (2017) 112:1120–34. doi: 10.1038/ajg.2017.97
42. Snapper SB. Very–early-onset inflammatory bowel disease. Gastroenterol Hepatol. (2015) 11:554–6.
43. Kotlarz D, Beier R, Murugan D, Diestelhorst J, Jensen O, Boztug K, et al. Loss of interleukin-10 signaling and infantile inflammatory bowel disease: implications for diagnosis and therapy. Gastroenterology (2012) 143:347–55. doi: 10.1053/j.gastro.2012.04.045
44. Glocker E-O, Kotlarz D, Boztug K, Gertz EM, Schäffer AA, Noyan F, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med. (2009) 361:2033–45. doi: 10.1056/NEJMoa0907206
45. Alangari A, Alsultan A, Adly N, Massaad MJ, Kiani IS, Aljebreen A, et al. LPS-responsive beige-like anchor (LRBA) gene mutation in a family with inflammatory bowel disease and combined immunodeficiency. J Allergy Clin Immunol. (2012) 130:481–488.e2. doi: 10.1016/j.jaci.2012.05.043
46. Amininejad L, Charloteaux B, Theatre E, Liefferinckx C, Dmitrieva J, Hayard P, et al. Analysis of genes associated with monogenic primary immunodeficiency identifies rare variants in XIAP in patients with Crohn's disease. Gastroenterology (2018) 1543:2165–77. doi: 10.1053/j.gastro.2018.02.028
47. Avitzur Y, Guo C, Mastropaolo LA, Bahrami E, Chen H, Zhao Z, et al. Mutations in tetratricopeptide repeat domain 7A result in a severe form of very early onset inflammatory bowel disease. Gastroenterology (2014) 146:1028–39. doi: 10.1053/j.gastro.2014.01.015
48. Miller TL, Lee D, Giefer M, Wahbeh G, Suskind DL. Nutritional therapy in very early-onset inflammatory bowel disease: a case report. Dig Dis Sci. (2017) 62:2196–200. doi: 10.1007/s10620-017-4616-9
49. Thiele K, Bierhaus A, Autschbach F, Hofmann M, Stremmel W, Thiele H, et al. Cell specific effects of glucocorticoid treatment on the NF-kappaBp65/IkappaBalpha system in patients with Crohn's disease. Gut (1999) 45:693–704.
50. Heuschkel RB, Menache CC, Megerian JT, Baird AE. Enteral nutrition and corticosteroids in the treatment of acute Crohn's disease in children. J Pediatr Gastroenterol Nutr. (2000) 31:8–15. doi: 10.1097/00005176-200007000-00005
51. Weber CK, Liptay S, Wirth T, Adler G, Schmid RM. Suppression of NF-kappaB activity by sulfasalazine is mediated by direct inhibition of IkappaB kinases alpha and beta. Gastroenterology (2000) 119:1209–1218. doi: 10.1053/gast.2000.19458
52. Majumdar S, Aggarwal BB. Methotrexate suppresses NF-κB activation through inhibition of IκBα phosphorylation and degradation. J Immunol Baltim. Md 1950 (2001) 167:2911–20. doi: 10.4049/jimmunol.167.5.2911
53. Guidi L, Costanzo M, Ciarniello M, De Vitis I, Pioli C, Gatta L, et al. Increased levels of NF-kappaB inhibitors (IκBα and IκBγ) in the intestinal mucosa of Crohn's disease patients during infliximab treatment. Int J Immunopathol Pharmacol. (2005) 18:155–64. doi: 10.1177/039463200501800116
54. Levine A, Wine E. Effects of enteral nutrition on Crohn's disease: clues to the impact of diet on disease pathogenesis. Inflamm Bowel Dis. (2013) 19:1322–9. doi: 10.1097/MIB.0b013e3182802acc
55. Alhagamhmad MH, Lemberg DA, Day AS, Tan LZ, Ooi CY, Krishnan U, et al. Advancing nutritional therapy: a novel polymeric formulation attenuates intestinal inflammation in a murine colitis model and suppresses pro-inflammatory cytokine production in ex-vivo cultured inflamed colonic biopsies. Clin Nutr (2017) 36:497–505. doi: 10.1016/j.clnu.2016.01.010
56. Lawley M, Wu JW, Navas-López VM, Huynh HQ, Carroll MW, Chen M, et al. Global Variation in Use of Enteral Nutrition for Pediatric Crohn Disease. J Pediatr Gastroenterol Nutr. (2018) 67:e22–9. doi: 10.1097/MPG.0000000000001946
57. Nahidi L, Corley SM, Wilkins MR, Wei J, Alhagamhmad M, Day AS, et al. The major pathway by which polymeric formula reduces inflammation in intestinal epithelial cells: a microarray-based analysis. Genes Nutr. (2015) 10:479. doi: 10.1007/s12263-015-0479-x
58. Alhagamhmad MH, Day AS, Lemberg DA, Leach ST. Exploring and Enhancing the Anti-Inflammatory Properties of Polymeric Formula. J Parenter Enter Nutr. (2017) 41:436–45. doi: 10.1177/0148607115625627
59. Visekruna A, Joeris T, Seidel D, Kroesen A, Loddenkemper C, Zeitz M, et al. Proteasome-mediated degradation of IkappaBalpha and processing of p105 in Crohn disease and ulcerative colitis. J Clin Invest. (2006) 116:3195–203. doi: 10.1172/JCI28804
60. Murano M, Maemura K, Hirata I, Toshina K, Nishikawa T, Hamamoto N, et al. Therapeutic effect of intracolonically administered nuclear factor kappa B (p65) antisense oligonucleotide on mouse dextran sulphate sodium (DSS)-induced colitis. Clin Exp Immunol. (2000) 120:51–58. doi: 10.1046/j.1365-2249.2000.01183.x
61. Dicksved J, Halfvarson J, Rosenquist M, Järnerot G, Tysk C, Apajalahti J, et al. Molecular analysis of the gut microbiota of identical twins with Crohn's disease. ISME J. (2008) 2:716–27. doi: 10.1038/ismej.2008.37
62. Wine E. Should we be treating the bugs instead of cytokines and T cells? Dig Dis. (2014) 32:403–9. doi: 10.1159/000358146
63. Colman RJ, Rubin DT. Fecal microbiota transplantation as therapy for inflammatory bowel disease: a systematic review and meta-analysis. J Crohns Colitis (2014) 8:1569–81. doi: 10.1016/j.crohns.2014.08.006
64. Moayyedi P, Surette MG, Kim PT, Libertucci J, Wolfe M, Onischi C, et al. Fecal microbiota transplantation induces remission in patients with active Ulcerative Colitis in a randomized controlled trial. Gastroenterology (2015) 149:102–9.e6. doi: 10.1053/j.gastro.2015.04.001
65. Quin MB, Schmidt-Dannert C. Designer microbes for biosynthesis. Curr Opin Biotechnol (2014) 29:55–61. doi: 10.1016/j.copbio.2014.02.014
66. Bank S, Andersen PS, Burisch J, Pedersen N, Roug S, Galsgaard J, et al. Associations between functional polymorphisms in the NFkappaB signaling pathway and response to anti-TNF treatment in Danish patients with inflammatory bowel disease. Pharmacogenomics J. (2014) 14:526–34. doi: 10.1038/tpj.2014.19
67. Vereecke L, Vieira-Silva S, Billiet T, van Es JH, Mc Guire C, Slowicka K, et al. A20 controls intestinal homeostasis through cell-specific activities. Nat Commun. (2014) 5:5103. doi: 10.1038/ncomms6103
68. Hutti JE, Turk BE, Asara JM, Ma A, Cantley LC, Abbott DW. I kappa B Kinase Phosphorylates the K63 Deubiquitinase A20 To Cause Feedback Inhibition of the NF-kB Pathway. Mol Cell Biol. (2007) 27:7451–61. doi: 10.1128/MCB.01101-07
69. Coornaert B, Baens M, Heyninck K, Bekaert T, Haegman M, Staal J, et al. T cell antigen receptor stimulation induces MALT1 paracaspase-mediated cleavage of the NF-kappaB inhibitor A20. Nat Immunol. (2008) 9:263–71. doi: 10.1038/ni1561
70. Wagner S, Carpentier I, Rogov V, Kreike M, Ikeda F, Löhr F, et al. Ubiquitin binding mediates the NF-kappaB inhibitory potential of ABIN proteins. Oncogene (2008) 27:3739–45. doi: 10.1038/sj.onc.1211042
71. Mauro C, Pacifico F, Lavorgna A, Mellone S, Iannetti A, Acquaviva R, et al. ABIN-1 binds to NEMO/IKK and co-operates with A20 in inhibiting NF-kB. J Biol Chem. (2006) 281:18482–8. doi: 10.1074/jbc.M601502200
72. Verstrepen L, Carpentier I, Verhelst K, Beyaert R. ABINs: A20 binding inhibitors of NF-kB and apoptosis signaling. Biochem Pharmacol. (2009) 78:105–14. doi: 10.1016/j.bcp.2009.02.009
73. Shembade N, Pujari R, Harhaj NS, Abbott DW, Harhaj EW. The kinase IKKα inhibits activation of the transcription factor NF-κB by phosphorylating the regulatory molecule TAX1BP1. Nat Immunol. (2011) 12:834–43. doi: 10.1038/ni.2066
74. Verstrepen L, Verhelst K, Carpentier I, Beyaert R. TAX1BP1, a ubiquitin-binding adaptor protein in innate immunity and beyond. Trends Biochem. Sci. (2011) 36:347–54. doi: 10.1016/j.tibs.2011.03.0
75. Zhang G, Ghosh S. Negative regulation of toll-like receptor-mediated signaling by Tollip. J Biol Chem. (2002) 277:7059–65. doi: 10.1074/jbc.M109537200
76. Lawrance IC, Wu F, Leite AZ, Willis J, West GA, Fiocchi C, et al. A murine model of chronic inflammation-induced intestinal fibrosis down-regulated by antisense NF-kappa B. Gastroenterology (2003) 125:1750–61. doi: 10.1053/j.gastro.2003.08.027
77. Takedatsu H. Nanomedicine and drug delivery strategies for treatment of inflammatory bowel disease. World J Gastroenterol. (2015) 21:11343. doi: 10.3748/wjg.v21.i40.11343
78. Ford AC, Sandborn WJ, Khan KJ, Hanauer SB, Talley NJ, Moayyedi P. Efficacy of biological therapies in inflammatory bowel disease: systematic review and meta-analysis. Am J Gastroenterol. (2011) 106:644–59. doi: 10.1038/ajg.2011.73
Keywords: nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κβ), inflammatory bowel diseases (IBD), Crohn Disease (CD), ulcerative colitis (UC), immunity, microbes, homeostasis
Citation: Zaidi D and Wine E (2018) Regulation of Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells (NF-κβ) in Inflammatory Bowel Diseases. Front. Pediatr. 6:317. doi: 10.3389/fped.2018.00317
Received: 23 July 2018; Accepted: 05 October 2018;
Published: 30 October 2018.
Edited by:
André Hörning, Universitätsklinikum Erlangen, GermanyReviewed by:
Moftah Hussin Alhagamhmad, Al-Arab Medical University, LibyaCopyright © 2018 Zaidi and Wine. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eytan Wine, d2luZUB1YWxiZXJ0YS5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.