 Neha Gupta
Neha Gupta Robert Richter1
Robert Richter1 Stephen Robert
Stephen Robert Michele Kong
Michele Kong
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pediatr., 18 September 2018
Sec. Pediatric Critical Care
Volume 6 - 2018 | https://doi.org/10.3389/fped.2018.00252
This article is part of the Research TopicSepsis in Neonates and ChildrenView all 23 articles
Sepsis in children is typically presumed to be bacterial in origin until proven otherwise, but frequently bacterial cultures ultimately return negative. Although viruses may be important causative agents of culture-negative sepsis worldwide, the incidence, disease burden and mortality of viral-induced sepsis is poorly elucidated. Consideration of viral sepsis is critical as its recognition carries implications on appropriate use of antibacterial agents, infection control measures, and, in some cases, specific, time-sensitive antiviral therapies. This review outlines our current understanding of viral sepsis in children and addresses its epidemiology and pathophysiology, including pathogen-host interaction during active infection. Clinical manifestation, diagnostic testing, and management options unique to viral infections will be outlined.
Sepsis is a leading cause of pediatric mortality (1). Defined as systemic inflammatory response syndrome in the presence of a suspected or confirmed infection, it is a clinical syndrome principally characterized by dysregulation of the host innate immune response and may result in an immune phenotype of coexistent systemic inflammation and immunosuppression (2). Pathological cross-talk between inflammatory and coagulation cascades, complement activation, and neuroendocrine signals wreak havoc on homeostatic controls. This hyperinflammatory response has untoward effects on the cardiopulmonary system, vascular endothelium, and gut, precipitating progressive organ dysfunction until the host succumbs (3). The morbidity, mortality, and costs associated with pediatric sepsis impose a significant burden on the healthcare community and global economy (4, 5). Watson et al reported a mean hospital length of stay of 31 days, with approximately $2 billion spent annually in healthcare cost associated with severe pediatric sepsis (1). International guidelines for management of sepsis and septic shock stress the importance of rapid resuscitation, prompt antimicrobial administration, and supportive care of organ dysfunction as the mainstays of pediatric sepsis treatment (6).
Viral sepsis can be defined as a severe inflammatory response to a suspected or confirmed viral infection. However, making the definitive diagnosis of viral sepsis in a child is particularly challenging for clinicians. The astute clinician must incorporate the patient's history of present illness, physical exam, laboratory and radiographic data to determine the likelihood of a viral etiology for sepsis. Even with a positive viral test, limitations of the testing result should be considered. Despite these challenges, timely diagnosis of viral sepsis has significant implications on clinical management, including guiding the use of appropriate antiviral therapy and informing isolation and containment strategies. Moreover, timely diagnosis of viral sepsis may prevent unnecessarily prolonged antibacterial treatment exposure and thus could help prevent consequent antibacterial resistance and deleterious effects on the host microbiome. This review outlines our current understanding of viral sepsis in children, including its epidemiology and the pathophysiology of the viral-host response during active infection. The clinical manifestations, appropriate diagnostic testing, and management unique to viral infections are outlined.
The true incidence of viral sepsis, particularly in the pediatric population, remains unknown. Since bacterial sepsis is amenable to treatment and is presumably more common, viral testing is frequently foregone in the acute presentation of sepsis. However, a recent study of adult patients with sepsis showed that viral respiratory pathogens, namely influenza A virus, human metapneumovirus, coronavirus, and respiratory syncytial virus (RSV), were overlooked in 70% of patients (7). In a multi-national epidemiological study of children with severe sepsis, an infectious etiology was only proven in 65% of patients and out of these, approximately one-third had a viral infection (8). The most frequent sites of infection were the respiratory tract (40%) and bloodstream (20%), with rhinovirus, RSV, and adenovirus most commonly isolated. In contrast, the Australia and New Zealand sepsis study group identified a pathogen in approximately 50% of patients with sepsis and septic shock (9). Of these patients, only one-fifth had a viral etiology, with RSV, cytomegalovirus (CMV), Epstein-Barr virus (EBV), herpes simplex virus (HSV), varicella zoster virus (VZV) and influenza being the most common viruses identified in this study. Recently, Ames et al. reported that 16% of pediatric patients who presented with septic shock had a primary viral disease (10). In another study of neonates with sepsis, bacterial etiology was found in only approximately 15% of cases, making viral infection more likely as a plausible cause of sepsis in these patients (11).
In the pediatric intensive care unit (PICU), influenza virus is a leading cause of viral sepsis and caries an especially high mortality rate (12). RSV has also been found to cause severe bronchiolitis and may present with sepsis, especially in children with history of premature birth, chronic lung disease, congenital heart disease or primary immunodeficiency (13, 14). Sepsis has also been observed in neonates with HSV, human parechovirus (HPeV) and enteroviral infection (15–18). Patients with immunodeficiency due to human immunodeficiency virus (HIV) infection are highly susceptible to viral sepsis depending on the stage of disease and access and response to the treatment (19). In these patients, common viral infections observed to cause sepsis include RSV, influenza, parainfluenza, adenovirus, CMV, EBV, and VZV (19). Diarrheal diseases secondary to viral infections can also lead to sepsis, especially in developing countries (20). Although rare, rotavirus has been associated with sepsis due to bacterial coinfection (21). Despite several large studies on viral sepsis in general (ref as above), as well as on specific viruses, in the absence of routine viral testing during the diagnostic evaluation of sepsis, the true incidence of viral infection as the cause of sepsis remains unclear.
The host response to infection consists of a multitude of simultaneous processes designed to neutralize the infectious threat and initiate repair of injured tissue. Sepsis is characterized by systemic and dysregulated inflammation, which can lead to a vicious cycle of vascular endotheliopathy, microcirculatory hypoperfusion, intestinal barrier dysfunction, circulatory shock, mitochondrial failure, and death (22–24). Moreover, the concomitant compensatory anti-inflammatory response syndrome that is characterized by lymphocyte apoptosis and immune paralysis predisposes the host to secondary nosocomial infection and latent viral activation (25, 26). The type of mechanisms employed vary by virus but generally result in some combination of (1) cytokine release, (2) endotheliopathy, and (3) host cytotoxicity (27). While an in-depth review of the pathogenesis of all human disease-causing viruses is beyond the scope of this manuscript, we have outlined the general pathophysiology below, highlighting major illustrative viral examples where possible.
Pathogen-recognition receptors (PRRs) are cellular sensors that recognize specific molecular structure of a pathogen (28). Toll-like receptors (TLRs) and retinoic acid-inducible gene-I (RIG-I)-like receptors (RLRs) are two types of PRRs that are involved in viral sensing (28). TLRs, which are found on the cell surface or within endosomes of monocytes, macrophages, dendritic, epithelial and endothelial cells, encounter pathogen-associated molecular patterns (PAMPs) (29, 30). Intracellular TLR-7 and TLR-8 recognize single-stranded Ribonucleic Acid (RNA) of viruses like HPeV, the enteroviruses, human metapneumovirus, and influenza; intracellular TLR-9 recognizes double-stranded (ds) DNA of viruses like the herpes viruses (e.g., HSV-1 and -2, EBV), adenovirus, and CMV; and TLR-3 recognizes dsRNA produced during intracellular viral replication (31). TLR activation culminates in myeloid differentiation primary response 88 (MyD88, through TLR-7,-8, and -9) or Toll/Interleukin (IL)-1 receptor domain-containing adapter protein inducing IFN-β (TRIF, through TLR-3) activation (32). These proteins, in turn, activate nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and IFN-regulatory factor (IFR)-mediated cytokine transcription (33–35). RLRs are cytosolic innate immunity sensors for viral RNA. Three members of RLRs have been identified: RIG-I, melanoma differentiation associated factor 5 (MDA5], and laboratory of genetics and physiology 2 (LGP2) (36). RIG-I and MDA5 recognize dsRNAs in response to different RNA viruses and signal the production of pro-inflammatory cytokines and type-1 IFNs (37). Cytokine proliferation instigates a pro-inflammatory cascade that results in complement activation, neutrophil chemotaxis, cytotoxic cluster of differentiation (CD) 8+ T-cell recruitment, and protease release from leukocytes and endothelial cells, particularly trypsin (38) and heparanase (39). Trypsin is upregulated and released by the vascular endothelium (38) and has been shown to cleave circulating pro-matrix metalloproteinase (pro-MMP) released from macrophages to form activated MMPs (40). MMPs, in conjunction with heparanase, degrade the endothelial glycocalyx (41, 42). Moreover, viral particles induce reactive oxygen species generation by circulating neutrophils, eosinophils, and macrophages (43, 44) that further injure the endothelial glycocalyx (45) and activate NF-κB cell-signaling (46), propagating a positive-feedback loop that results in endotheliopathy and end-organ damage.
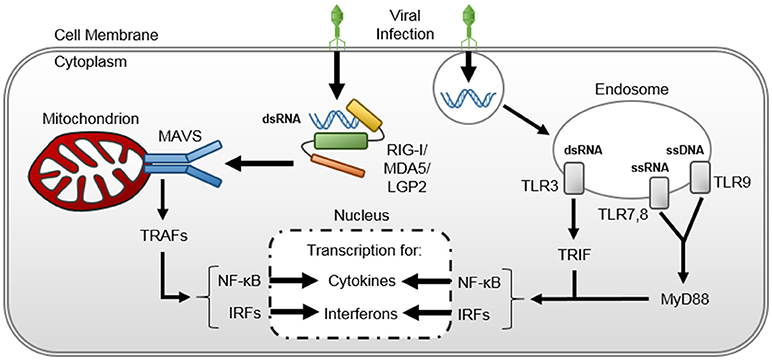
Figure 1. Viral-induced cytokine upregulation and release. Double stranded viral ribonucleic acids are (dsRNA) recognized within the host cellular cytosol by retinoic acid-inducible gene-I (RIG-I)-like receptors (RLRs)- RIG-I, MDA5, and LGP2. The RLRs bound to viral dsRNA undergo conformational change and complex with mitochondrial antiviral signaling (MAVS) protein on the mitochondrion surface. The RLR-MAVS interaction instigates an assembly of host proteins to activate TNF- receptor-associated factors (TRAFs), thereby inducing nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and interferon regulator factor (IRF)-mediated cytokine transcription in the host cell nucleus. Viral nucleic acids are also recognized within Toll-like receptors (TLRs) within host cell endosomes, triggering myeloid differentiation primary response 88 (MyD88) and Toll/interleukin-1 receptor-domain-containing adaptor-inducing interferon-β (TRIF) pathways that also activate NF-κB and IRF-mediated cytokine transcription in the host cell nucleus.
Systemic viral dissemination appears to be the etiology of viral sepsis. The exact mechanisms by which viruses that are normally isolated to the respiratory or integumentary epithelium reach the bloodstream are not known. However, it is plausible that viremia occurs through direct invasion of epithelial cells (or neurons as in case of HSV or varicella disease) to reach the surrounding vasculature (47). Once in the blood, the virus may induce endothelial glycocalyx degradation by activating leukocytes, platelets, and endothelial cells to secrete MMPs and heparanase that target glycocalyx components (39, 48). Endothelial glycocalyx disruption exposes selectins and intracellular adhesion molecules, making them available for leukocyte adhesion and activation (49). Glycocalyx degradation also releases heparan sulfate that may bind and activate antithrombin III and exposes membrane-bound glycoprotein Ib/IX/V complexes (50) that can bind circulating von Willebrand factor (51) and P-selectins on platelets (52, 53), precipitating coagulopathy. Moreover, loss of integrity of the protein-rich glycocalyx alters the microvascular fluid equilibrium between the vascular lumen and subglycocalyx, increasing fluid and macromolecule filtration through the endothelium to the surrounding interstitium (54). The end result of endothelial glycocalyx damage is pro-inflammatory propagation and vascular leak that can compromise organ function.
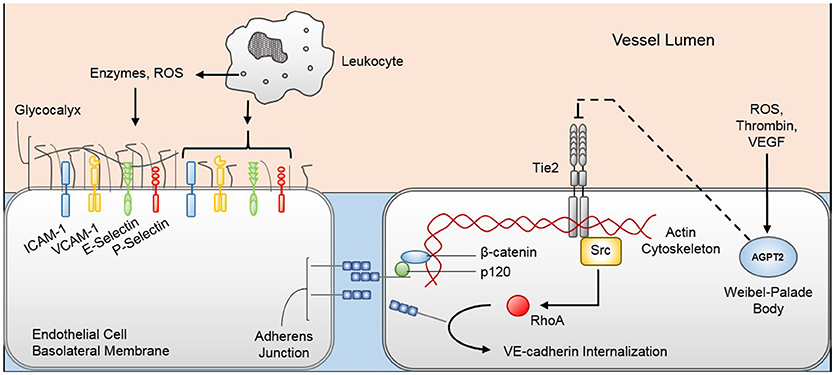
Figure 2. Pathophysiologic mechanisms of viral-induced endotheliopathy. Innate immune system activation during viral sepsis precipitates leukocyte degranulation and release of enzymes and reactive oxygen species (ROS) that degrade the endothelial glycocalyx. Denuded endothelial glycocalyx exposes cellular adhesion molecules (e.g., ICAM-1, VCAM-1, E-selectin, P-selectin) that increase the margination and activation of leukocytes, further promoting the inflammatory response. Additionally, inflammatory mediators, namely thrombin, ROS, and vascular endothelial growth factor (VEGF), promote Weibel-Palade body exocytosis, releasing angiopoietin-2 (Agpt-2) into the circulation. Agpt-2 antagonizes the endothelial cell Tie2 receptor, allowing the Src-mediated RhoA enzyme to reconfigure the endothelial cell cytoskeleton and promote VE-cadherin internalization from the adherens junction. Loss of glycocalyx and adherens junction integrity permits increased trans-cellular protein and fluid movement from the vascular lumen to the interstitium. ICAM-1, intercellular adhesion molecule 1; VCAM-1, vascular cell adhesion molecule 1.
Circulating viral particles may also induce endothelial cell structural changes that lead to barrier disruption and further vascular leak. Endothelial cells are anchored to each other through adherens junctions comprised predominantly of vascular endothelial (VE)-cadherin, which is attached to the endothelial cytoskeleton through beta-catenin and p120-catenin (55, 56). In human endothelial cells, pathogenic strains of hantavirus appear to bind to cellular surface β3-integrins, thereby promoting VE-cadherin internalization and adherens junction destabilization (57). VE-cadherin destabilization may also be mediated through the cellular membrane Tie2 receptor. Tie2 receptor is activated by angiopoietin-1 (Agpt-1) that is derived by periendothelial cells (58, 59). When activated, the Tie2 receptor activates PI3K/Akt cell-survival signaling and Rac1-mediated cytoskeletal stabilization (60). Inflammatory mediators, such as thrombin (61), reactive oxygen species (62), and VEGF (63), stimulate endothelial cell Weibel-Palade body exocytosis, releasing the Tie2 antagonist Agpt-2 (64). Agpt-2 acts in an autocrine fashion to inhibit Tie2 signaling, thereby promoting RhoA kinase activity and VE-cadherin destabilization (60). Mice infected with a pathogenic strain of H3N2 influenza virus develop acute lung injury that is rescued by the Tie2 agonist vasculotide (65), suggesting that the Tie2 receptor is integral in the development of endotheliopathy during viral sepsis. The exact mechanisms each virus employs to induce endothelial cell dysfunction are not clear; however, the typical presentation of capillary leak with viral sepsis suggests a common pathway by which endothelial integrity is compromised.
Viral-induced host cytotoxicity is mediated by cytopathic effects, cellular reprogramming, and/or initiation of host immune cytotoxic responses. Viruses take over and utilize host intracellular machinery to replicate, depleting host cells of energy stores and transcription potential. Furthermore, viral-infected cells may be activated for caspase-dependent apoptosis (66). Viruses may also indirectly promote apoptosis through macrophage reprogramming. Macrophages infected with H5N1 avian influenza have been shown to upregulate production and release of tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) that promote T-cell apoptosis (67). Though the effect of TRAIL on other cell lineages was not determined, it is plausible that the effect seen in T-cells may be more diffuse. Systemic viral dissemination from sites of primary infection (e.g., human metapneumovirus in the respiratory tract or HPeV in the gastrointestinal tract) may occur through these apoptotic mechanisms, whereby new virions are released, infect local endothelial cells, and cause further cellular apoptosis and systemic viral spread. The invasiveness and pervasiveness of the viral infection is likely dictated by cell tropism and genetically determined virulence as viruses with greater cytopathogenicity, such as H5N1 avian influenza (66) and HPeV-3 (68), are more likely to cause sepsis in immunocompetent hosts than viruses with typically minimal associated cytopathology (e.g., RSV or parainfluenza) (69). Lastly, effectors of the host immune system, such as natural killer (NK), cytotoxic CD8+ T cells and complement, attack, and destroy virally infected host cells.
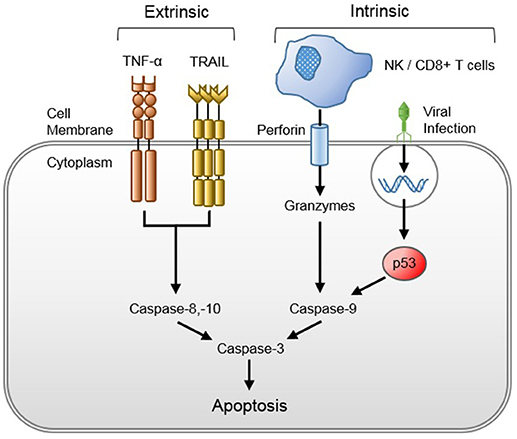
Figure 3. Viral-induced host cytotoxicity through the extrinsic and intrinsic apoptotic pathways. Tumor necrosis factor-alpha (TNF-α) is released after viral recognition by the innate immune system, and TNF-related apoptosis-inducing ligand (TRAIL) is released from viral-reprogrammed macrophages, both of which bind their respective host cell surface death receptors to activate the extrinsic apoptotic pathway. Natural killer (NK) and cluster of differentiation 8 (CD8+) T cells inject perforin into the membranes of infected host cells, through which they secrete granzymes that elicit the intrinsic apoptotic pathway. The intrinsic pathway may be activated after viral protein recognition by the host cell or suicide gene insertion by cytotoxic viruses.
Viral pathogenesis in children also varies according to the degree of host immunocompetence. Generally, young infants have significantly reduced TLR expression, antigen-presenting cell activity, NK cell responsiveness, T-cell functionality, B-cell maturity, and complement concentration (70). This immaturity of the developing immune system places young infants at significantly higher risk for severe disease from viruses that would typically cause minimal harm to older children and adults (e.g., HSV, HPeV, enteroviruses, CMV). Similarly, children with congenital or acquired immunodeficiencies are more susceptible to viral pathogens. Specific immunodeficiencies that place children at higher risk for viral sepsis include NK cell deficiency, interferon (IFN)-γ receptor deficiency, TLR-3 deficiency, nuclear factor-kappa B essential modulator deficiency, severe combined immunodeficiency, severe T-cell lymphopenia in DiGeorge syndrome, agammaglobulinemia, and hyperIgM syndrome (71). Severe RNA viral infections have also been observed in patients with loss-of-function mutation of the IFN induced with helicase C domain 1 (IFIH1) gene that encodes the RLR MDA5 (36, 37, 72). Moreover, children receiving immunomodulatory or immunosuppressive therapies due to malignancy, transplantation, or autoimmune disease are more susceptible to viral infection or reactivation.
The constitutional symptoms and clinical features of viral sepsis are frequently indistinguishable from bacterial or fungal sepsis. Presenting symptoms and signs include fever, chills, rash, respiratory distress, nausea, vomiting, diarrhea, dysuria, confusion, and altered mental status. None of these symptoms is pathognomonic of sepsis, let alone viral induced sepsis. Moreover, classic features of systemic inflammation might not be seen in every individual, especially in immunocompromised children. Fever is one of the most common symptoms seen in septic children, attributable to the pyrogenic activity of IL-1, IL-6, IFNs, and TNF-α. It has been observed that these substances increase prostaglandin E2 synthesis in the hypothalamus (73, 74), resulting in the elevation in the host central nervous system core temperature set-point regulated by the pre-optic and dorsomedial hypothalamic nuclei (75). Hypothermia, on the other hand, is a less frequent but more specific indicator of sepsis that may be predictive of illness severity and death, especially in younger children and chronically debilitated patients (74). Injury to the vascular endothelium may result in broad array of failing organs that manifests as confusion, nausea, vomiting, diarrhea, oliguria, and coagulopathy. A myriad of cardiopulmonary manifestations ranging from mild tachypnea and tachycardia to acute respiratory distress syndrome and shock can be seen (76). The presenting symptoms usually depend on the type of virus. Clinical presentation in patients with respiratory viral infections can range from completely asymptomatic to severe respiratory distress due to pneumonia. Diarrheal illness has been observed in patients infected with rotavirus, norovirus, enterovirus, and adenovirus. VZV and HSV infection may present with vesicular rash. Children with HSV or arbovirus infection may have confusion, altered mental status or seizures from encephalitis. Elevated transaminases are common with HSV and enteroviral infections which may be complicated by hepatitis, coagulopathy and encephalitis. Neonatal HPeV infection can mimic other enteroviral infections in the initial presentation. Often these patients present with fever, rash, irritability, feeding intolerance, and seizures (17). They can develop sepsis like illness and encephalitis. Patients with acute HIV infection often have flu-like symptoms such as fever, headache and rash, which usually resolve spontaneously. These patients soon enter a phase of clinical latency until they develop acquired immunodeficiency syndrome, usually heralded by acquisition of an opportunistic infection.
As with other types of sepsis, virus-induced sepsis requires a high index of suspicion, especially in very young children and those with chronic medical conditions. Neonates and young infants are at higher risk of sepsis from HSV, HPeVs, and enterovirus. HSV is usually acquired perinatally from mothers with genital herpes. Mothers with primary herpes are more likely to transmit the infection when compared to those with recurrent and non-primary herpes (77). Nielsen et al reported that second born children are at higher risk of HPeV-3 infection than the firstborn (78). Seizures, drowsiness and lethargy, and absence of oral lesions are associated with severe enteroviral infection in children (79). In RSV infection, comorbid conditions reported to increase the risk for severe infection include the history of prematurity, congenital heart disease, chronic lung disease, and immunodeficiency (13). In a recent study, Eggleston et al found that patients with metapneumovirus infection were more likely to be older and have congenital heart disease compared to RSV infected patients (80). In contrast, asthmatics and premature infants were at higher risk for rhinovirus infection (81). Finally, predisposing conditions for severe pediatric influenza infection include age less than 2 years; asthma; cardiac, renal, hepatic, hematologic, neurologic or neuromuscular conditions; long-term aspirin therapy; immunosuppressive therapy and residence in a chronic care facility (82). Risk of mother-to-child perinatal HIV transmission is higher in mothers with CD4 count < 200 cells/μL and lower in infants receiving antiretroviral prophylaxis (83, 84). If patients with any of these conditions present with sepsis, diagnostic viral testing and appropriate empiric antiviral treatment should be strongly considered according to the individual's risk factors.
Secondary bacterial infections are commonly associated with respiratory viral infections (85). In the winter of 1995–96, an outbreak of Streptococcus pneumoniae pneumonia developed in otherwise healthy children who had a preceding influenza A viral illness (86). During the 2009–10 influenza A pandemic, one third of critically ill children afflicted with influenza were diagnosed with concurrent bacterial infections (87). In this study, the leading three bacterial coinfections were Staphylococcus aureus, Pseudomonas spp., and Haemophilus influenza (87). In children hospitalized for RSV, Haemophilus influenzae and Streptococcus pneumoniae were the most common organisms isolated in those who developed bacteremia (88). These secondary bacterial infections may exacerbate innate immune dysfunction (89) and convey substantially increased risk of worse outcomes (90, 91). However, to date, the mechanisms underlying bacterial synergism and increased susceptibility to secondary bacterial infection in the setting of a preceding respiratory viral infection remain unclear. In general, this phenomenon appears to involve impairment of respiratory epithelial and innate immune system defenses. Viral destruction of the airway epithelium affects mucociliary clearance, allowing bacterial attachment to mucins and eventual colonization of the respiratory tract (92, 93). Additionally, viral-induced upregulation of IFN-γ and TNF-α may lead to a dysregulated host T-cell response, decreased neutrophil chemotaxis, and impaired macrophage phagocytosis that increases the host susceptibility to secondary bacterial pathogens (94). Upregulation of the surface platelet-activating factor receptor on epithelial cells and leukocytes by pro-inflammatory cytokines may also increase adhesion and invasion of certain virulent pneumococcal strains (95).
Rotavirus infection has also been associated with secondary bacterial infections (21). Although, the exact mechanisms leading to sepsis and organ dysfunction are unknown, a leading hypothesis entails translocation of bacteria and endotoxin through damaged intestinal epithelium into the splanchnic circulation, systemically increasing production of nitric oxide and circulating pro-inflammatory cytokines like TNF and IL-1β, and high mobility group box 1 protein, resulting in sequential organ failure (96). HIV infection can lead to apoptosis of CD4 T-lymphocytes, defective T and B lymphocyte function, decreased production of IFN-γ, IL-2 and immunoglobulins, and decreased NK cell activity (97–99). This leads to not only increased risk of secondary bacterial infections but also increased susceptibility to other viruses and intracellular organisms such as mycobacteria and Pneumocystis jiroveci.
Similar to bacterial coinfections, critically ill children can be simultaneously infected by multiple viruses. The course of illness in patients with viral coinfections depend on virus-virus interaction. Various mechanisms for disease virulence in viral coinfections have been proposed, including viral gene interactions, immunologic interactions and alteration in host environment (100). Even though the clinical significance such interactions is unknown, a study by Rhedin et al. reported increased risk of severe respiratory disease in patients with viral coinfections compared to those with single viral infections (101). Approximately 20% of the patients had viral coinfection and RSV, bocavirus and adenovirus were the most common viruses associated with coinfections (101). In another study performed in Canada on patients with respiratory viral infections, approximately 17% of the patients had viral coinfections (102). There was no difference in the risk of hospitalization or the severity of illness in patients with single viral infections and those with viral coinfections (102). Another study done in Greece revealed a much higher viral coinfection rate (42%) with most common coinfections with RSV, influenza, rhinovirus and parainfluenza viruses (103). Increased risk of hospitalization has been observed in patients with viral coinfections (103). However, systematic reviews and meta-analyses of children with viral coinfections have not shown any association with increased clinical severity (104, 105). Patients infected with HIV are at high risk of secondary viral infections such as CMV, HSV and respiratory viruses like RSV, influenza and metapneumovirus.
The diagnosis of viral sepsis is typically one of exclusion. Bacterial sepsis, whether primary or secondary, is usually of higher initial concern because failure to recognize this diagnosis and promptly administer systemic antibiotics has lethal consequences. Unfortunately, in our current state of limited antiviral therapies, even the prompt recognition and treatment of viral sepsis may not quickly improve a patient's clinical course. Nonetheless, early, definitive diagnosis of a primary viral septic process may inform treatment decision-making and help limit unnecessary systemic antibiotic administration. In symptomatic critically ill children, identification of a viral etiology can play an important role in the management and impact the outcome of these patients.
There are currently no standard approaches to viral diagnostic testing. Point-of-care (POC) antigen-based testing is relatively inexpensive and provides rapid detection of common respiratory viruses from a nasopharyngeal swab, such as RSV or common strains of human influenza. However, POC testing may lack the sensitivity needed to determine the etiology of life-threatening sepsis (106). Direct fluorescent antibody (DFA) testing may provide better specificity and a broader range of viral strain detection than POC testing, but the test depends on the collection of sufficient numbers of epithelial cells for adequate viral detection (107). Cell culture is the traditional gold-standard for viral diagnoses, including for HSV, however the long turn-around time for results significantly limits its utility for expedient diagnosis (108). Commercial or laboratory-developed nucleic acid amplification tests (NAATs) (e.g., polymerase chain reaction, PCR, or reverse transcription-loop-mediated isothermal amplification) may provide greater sensitivity and specificity than POC or DFA testing but requires sophisticated equipment and specially trained laboratory staff to complete (109, 110). NAATs have the added benefit of being highly multiplexed with new commercially available technology like Biofire® FilmArray® multiplex PCR (111). Unfortunately, the use of NAATs is limited by the high cost, delay in results and the inability to distinguish between viral nucleic acids from live viruses (112). Ultimately, the methods available for timely viral detection are limited by technique of sample collection and institutional resource availability.
Several limitations to the current diagnostic testing for causative viruses are worth noting, and results of viral testing always need to be interpreted with caution. For instance, although a type of enterovirus, HPeV cannot be detected on routine enterovirus PCR assay. HPeV-specific PCR is required to detect this virus in respiratory, CSF and stool samples of infected children and should be considered as a part of workup for neonates and young children presenting with sepsis (113). The clinical utility of viral respiratory PCR panels is also limited by their high rates of positive detections without clinical correlates. Detection of a virus in a patient with sepsis does not necessarily indicate causation. Some studies have shown that a respiratory virus can be detected in about one third of asymptomatic children (114, 115). Viral PCR testing is particularly difficult to interpret due to its high sensitivity for viral nucleic acids, making it challenging for the clinician to distinguish between active viral disease and viral nucleic acid or live viral carriage (112, 116, 117). A positive test could be a result of asymptomatic colonization, prolonged viral shedding or viral coinfection. In a study by Rhedin et al. comparing PCR results between symptomatic and asymptomatic patients, RSV, metapneumovirus and parainfluenza viruses had a significantly higher detection rate in children with acute respiratory infection, suggesting causation; however, other viruses (enterovirus, coronavirus, bocavirus, rhinovirus, and adenovirus) had an equally high detection rates in asymptomatic children (101). Because of these positive viral detections in asymptomatic children, it is important that clinicians consider the big picture, and factor in other pertinent information that may indicate an active ongoing bacterial infection before discontinuing antibacterial agents based on a viral assay. The use of serum biomarkers (see section below) and whole blood gene expression analysis (118) may serve a crucial role in this setting to discern viral from bacterial sepsis. When faced with a septic child who has an unusual sepsis presentation, does not respond to usual therapies, or has persistently negative diagnostic evaluations, a viral etiology must be considered and consultation with an infectious disease expert is recommended.
Several serum biomarkers, including lactate, C-reactive protein (CRP), and procalcitonin (PCT), are used to guide management in sepsis and are an important part of early goal directed therapy (119). PCT is more commonly used in the ICU setting for early determination of the likelihood of a bacterial etiology for sepsis and to guide antimicrobial duration (120, 121). Serum PCT has been shown to be elevated in bacterial infections and is superior to CRP in assessing the severity and course of the disease (122). However, the mean sensitivity of PCT as a biomarker of sepsis remains low at 77%, with a specificity of 79% (123, 124). Unfortunately, in general, these biomarkers are non-specific in distinguishing bacterial vs. viral infection (125).
In recent years, several viral-specific biomarkers have been identified. Many transcriptional signatures have been designed to distinguish viral infections from bacterial infections as well as non-infectious conditions (126, 127). Zaas et al. identified a 30-gene signature to discriminate symptomatic influenza A-infected subjects from both healthy and bacterially-infected subjects (128). In a recent study by Herberg et al., a 2-transcript RNA signature [FAM89A and IFI44L) showed promising results in its ability to distinguish between bacterial and viral infections, demonstrating that the expression of IFI44L was increased in patients with viral infection, whereas expression of FAM89A was increased in patients with bacterial infection (118). Another recent study identified a four-gene expression signature in whole blood to distinguish viral infections from other etiologies (129). Human myxovirus resistance protein 1 (MxA) is an important intermediate product in the IFN-mediated antiviral response against a variety of viruses. Serum MxA levels are significantly higher in patients with viral infections compared to bacterial infections in pediatric population and thus may be an additionally useful biomarker to discriminate viral from bacterial illness (130).
There is a paucity of data regarding treatment and management of viral infection. Supportive care is the current mainstay of therapy for most viral infections, particularly for respiratory viruses. Though broad-spectrum antibiotic therapy may be prudent until a bacterial source for sepsis has been definitively ruled-out, sustained antibiotic treatment has no role in the management of viral sepsis except in the case of bacterial coinfections. Many viral infections can be prevented with the use of hand hygiene, environmental decontamination, use of personal protective equipment, elimination of second-hand smoke, and isolation of infected children (131). Additional protection can be conferred by administering vaccines for common communicable viruses. These preventive strategies are of particular importance in high-risk patients. As the scope of available vaccines and anti-viral therapies remains rather limited, development of novel vaccines and treatment is critical (131).
For RSV infection, management is currently limited to passive immunization for at-risk infants. Palivizumab, an RSV-specific monoclonal antibody, is Food and Drug Administration (FDA) approved for the prevention of infection in high-risk infants during RSV season. The American Academy of Pediatrics has issued more clear recommendations for palivizumab use, stating that it should be administered as a monthly injection during RSV season in children born less than 29 weeks, 0 days gestation and are less than 12 months of age or in children with congenital heart disease, chronic lung disease (132). Studies have shown variable efficacy of palivizumab, with reduction in RSV hospitalization rate by approximately 60% (133). Currently, aerosolized ribavirin is the only FDA-approved treatment available for the management of RSV infection, though its use remains controversial (134). To date, RSV vaccines and antiviral therapies remain an active area of investigation (135). A randomized, controlled trial performed in adult patients with RSV infection compared the RSV entry inhibitor GS-5806 to placebo and demonstrated a decrease in both viral load and the clinical severity of infection in patients treated with GS-5806 (136). Similar fusion inhibitors such as ALX-0171 (137), JNJ-2408068 (138), MDT-637 (139), and VP14637 (138) demonstrate efficacy in vitro, and ALX-0171 is undergoing a phase II clinical trial in infants hospitalized for RSV (clinicaltrials.gov registration no. NCT02979431). The use of ALS-008176, an RSV polymerase inhibitor, has similarly been shown to reduce viral load, rapidly clear RSV, and improve the severity of disease in adults with RSV infection (140). ALN-RSV01 is a lipid-based nanoparticulate system, containing small-interfering RNA (siRNA] that demonstrates promising antiviral effects against RSV in lung transplant patients (141) by targeting the mRNA of the RSV nucleocapsid protein, thereby limiting viral replication (142). However, until these novel treatments have undergone appropriate clinical trials, the pediatric medical community must continue to wait for effective RSV antiviral therapy.
Unlike RSV, seasonal vaccines and several antiviral therapies are available to treat influenza viral infections. The seasonal influenza vaccine has demonstrated reasonable efficacy at attenuating influenza A and B viral disease (143). Currently, two forms of the influenza vaccine are available for use in children: a live attenuated vaccine in the form of a nasal spray and an inactivated vaccine in an injectable form. Antiviral agents used in the treatment and post-exposure prophylaxis of influenza infections include neuraminidase inhibitors (oseltamivir and zanamivir) and the adamantanes (amantadine and rimantadine). Oseltamivir is the most commonly used medication due to high prevalence of adamantane resistance. Oseltamivir has shown to be beneficial and tolerable in children with influenza if received within first 48 h of illness (144, 145). However, in cases of severe infection, initiation of oseltamivir beyond 48 h of symptom onset may still provide benefit (146). Nanotechnology-based vaccines are also being developed for influenza virus. InflexalR V and InfluvacR are two virosomal vaccines that have been shown to be efficacious against influenza infection (147, 148). STP702, another nanotherapeutic agent, is an siRNA under development designed to inhibit conserved regions in H1N1 and H5N1 strains of the influenza virus and prevent viral replication (116). Nanotraps such as sialylneolacto-N-tetraose c (LSTc)-bearing liposomal decoys bind to hemagglutinins on the influenza virus and prevent viral spread in vitro, demonstrating the potential have shown to be effective against influenza virus (117). The influenza polymerase inhibitor T-705 (favipiravir) has been demonstrated significant attenuation of influenza virus activity (149). Interestingly, at higher concentration, it has also shown to be effective against poliovirus, rhinovirus and RSV (149). Other agents under investigation include CS-8958, a long-acting neuraminidase inhibitor, and DAS181, an attachment inhibitor (150). Animal studies have also shown promising results with combination therapy (150). Various immunomodulatory agents have also been posited to temper the dysregulated host inflammatory response in severe influenza (151), including cyclooxygenase-2 inhibitors (152), doxycycline (153), glucocorticoids (154), macrolides (155, 156), peroxisome proliferator-activated receptor agonists such as gemfibrozil (157), sphingosine-1-phosphate (158), and the Tie2 receptor activator vasculotide (65). Further studies are needed to determine the efficacy of these treatments in human influenza infection.
Pleconaril, an orally administered viral capsid inhibitor, has shown to be effective against picornaviruses, especially enteroviruses and rhinoviruses (159). Abzug et al. reported greater survival in patients with neonatal enteroviral sepsis who received pleconaril (159). Similarly, patient with rhinovirus infection treated with pleconaril have shorter duration of symptoms, depending on susceptibility of the virus to the medication (160). No antiviral activity has been observed from pleconaril against HPeV (18). Intravenous immunoglobulin has shown potential benefit in management of enteroviral infections (161).
Prevention of neonatal HSV infection is more elusive as neonatal HSV disease often occurs after transmission from asymptomatic women with primary HSV infection (162). In cases of active maternal genital herpes, cesarean sections can decrease the incidence of neonatal HSV infection, especially when performed within 4 h of rupture of membranes (163). A subunit HSV vaccine has shown promising results in prevention of genital herpes and is currently under Phase III trial (164). Although not routinely recommended, antiviral prophylaxis with acyclovir in late pregnancy has been demonstrated to decrease viral shedding, leading to reduction in cesarean rates and recurrent herpes (165, 166). Patients with severe neonatal HSV infection (those with disseminated disease and CNS infection) should be treated with intravenous acyclovir for 21 days (167).
Viral sepsis may occur in HIV-infected children due to opportunistic or other secondary viral infections (19). Increasing use of highly active antiretroviral therapy (HAART) has significantly improved survival of HIV infected children by decreasing the progression to acquired immunodeficiency syndrome (AIDS), thereby maintaining host immunocompetence that protects against the development of viral sepsis (168–171). However, HAART is associated with potentially deleterious sequelae, making timing of the therapy very controversial in patients with active sepsis (19).
Extensive studies have not been done to characterize the effect of viral sepsis on outcomes. In a recent study, Hon et al. found no difference in mortality between patients with and without viral infections who were admitted to PICU (172). Shi et al performed a systematic review of RSV infections in 2015 and estimated case fatality rates in children with RSV infection to be around 2.2% (<6 months of age) and 2.4% (6–11 months of age) in developing countries. Case fatality rates in higher income countries were significantly lower (0.2 for <6 months and 0.9 for 6–11 months) (173). In another study, the highest mortality from RSV infection was seen at mean age of 6.2–7.5 months with three quarters of these cases associated with comorbid conditions (174).
Seasonal influenza epidemics and various pandemics have historically led to significant morbidity and mortality in the past, either due to exacerbation of an underlying condition or due to secondary bacterial infections. Mortality with influenza varies not only with season, but with predominant influenza strain and effectiveness of influenza vaccine each season. During the first year of the pandemic 2009 H1N1, global mortality in children aged 0–17 years was estimated to be as high as ~ 45,000 cases, with majority of deaths occurring in Southeast Asia and Africa (175). Both pediatric and adult patients during this pandemic had a very rapid progression to respiratory failure and required prolonged mechanical ventilation and vasopressor support (176, 177). Various extrapulmonary complications secondary to influenza sepsis have been reported in the literature. These include, but are not limited to renal failure, rhabdomyolysis, encephalopathy, myocarditis, and multiorgan failure. These complications also lead to poorer outcomes (178).
Sepsis from HPeV can lead to significant morbidity in neonates and young children. Although, most infections are self-limited, long-term neurological deficits such as learning disability, developmental delay, paralysis and epilepsy have been observed in these patients (179, 180). HPeV infections have also been associated with encephalitis, hepatitis and coagulopathy (18). In addition, rare complications have also been observed in these patients including necrotizing enterocolitis, myocarditis, myositis, hemolytic uremic syndrome, and Reye's syndrome (18). Other enteroviral infections can lead to similar complications and long-term neurological deficits. Hepatic and cardiac dysfunction can also be observed in these patients (181–183). In HSV infection, neurological complications such as developmental delay and seizures have been observed in infected neonates (15). Mortality from systemic HSV infection is usually due to severe coagulopathy, hepatitis and pneumonitis (15). In a multicenter study, Spaeder et al. observed a mortality rate of 9% in patients with severe metapneumovirus infection (184). Increased mortality from metapneumovirus infection has been observed in children with chronic medical conditions, female gender and patients who acquired the infection in the hospital (184). Similarly, RSV, parainfluenza and influenza infections, when acquired in hospital, have been associated with increased mortality (185). Rotavirus infection can lead to extraintestinal complications like seizures and meningoencephalitis (186, 187). In HIV patients, likelihood of progression to AIDS and of mortality are impacted by time of acquisition of HIV, viral load, CD4 count and timing of HAART initiation. Approximately 80% mortality has been observed in developing countries with limited access to HAART (188). Complications that lead to increased morbidity and mortality in these patients include severe CMV infection, encephalopathy, recurrent life-threatening bacterial infections, tuberculosis, and pneumocystis infection (189).
End-organ failure is a major contributor to mortality in sepsis and septic shock, including virus-induced sepsis. Complications such as acute respiratory distress syndrome, disseminated intravascular coagulation, and acute renal injury often leads to a worse prognosis. Developing countries often have disproportionately higher mortality in patients with viral infections (190), likely due to delayed diagnosis and treatment. Risk of severe sepsis is also related to the site of infection, with endocarditis and CNS infections being associated with mortality as high as 20% (1). Besides the site of infection, the type of virus also determines the risk of mortality. For example, meningitis from HPeVs is a common cause of sepsis in neonates and young children but consequent mortality is low in these patients (179). HPeV3 is associated with more severe disease than HPeV1 (113). Moreover, the extent of systemic involvement can predict the development of multiple organ failure and thus mortality (191). Sepsis related mortality has been reported in other viral infections including dengue fever (192). However, further studies are necessary in order to estimate the burden of viral sepsis on outcomes including morbidity, mortality, and health care related costs.
Although the incidence of viral-induced sepsis is not precisely known, it is suspected to be common and may represent an important subset of children with “culture-negative sepsis.” It is therefore critical for clinicians to suspect and test for viral infection in children with culture-negative sepsis if appropriate infection containment measures are to be instituted in a timely fashion and in the interest of early identification of children with viral infections amenable to treatment. These considerations are especially urgent for high-risk children, such as those born prematurely or those having congenital heart disease, chronic lung disease, or immunodeficiency. Appropriate diagnosis of viral sepsis may provide the clinician added confidence to limit the duration of empiric antibacterial exposure in children with sepsis, and therefore may be helpful in the fight against antibiotic-resistant bacteria. Further studies are needed to identify novel viral-specific biomarkers and therapeutics.
NG, RR, SR, and MK contributed to the conception, writing, and final edits of this manuscript.
This work was funded by the National Institutes of Health (NHLBI 5K08HL119359-02) to MK.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Watson RS, Carcillo JA, Linde-Zwirble WT, Clermont G, Lidicker J, Angus DC. The epidemiology of severe sepsis in children in the United States. Am J Respir Crit Care Med. (2003) 167:695–701. doi: 10.1164/rccm.200207-682OC
2. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA (2016) 315:801–10. doi: 10.1001/jama.2016.0287
3. Bone RC. The sepsis syndrome. Definition and general approach to management. Clin Chest Med. (1996) 17:175–81.
4. Marshall JC. Understanding the global burden of pediatric sepsis. Am J Respir Crit Care Med. (2015) 191:1096–8. doi: 10.1164/rccm.201503-0594ED
5. Fleischmann-Struzek C, Goldfarb DM, Schlattmann P, Schlapbach LJ, Reinhart K, Kissoon N. The global burden of paediatric and neonatal sepsis: a systematic review. Lancet Respir Med. (2018) 6:223–30. doi: 10.1016/S2213-2600(18)30063-8
6. Rhodes A, Evans LE, Alhazzani W, Levy MM, Antonelli M, Ferrer R, et al. Surviving sepsis campaign: international guidelines for management of sepsis and septic shock: 2016. Intensive Care Med. (2017) 43:304–77. doi: 10.1007/s00134-017-4683-6
7. Ljungstrom LR, Jacobsson G, Claesson BEB, Andersson R, Enroth H. Respiratory viral infections are underdiagnosed in patients with suspected sepsis. Eur J Clin Microbiol Infect Dis. (2017) 36:1767–76. doi: 10.1007/s10096-017-2990-z
8. Weiss SL, Fitzgerald JC, Pappachan J, Wheeler D, Jaramillo-Bustamante JC, Salloo A, et al. Global epidemiology of pediatric severe sepsis: the sepsis prevalence, outcomes, and therapies study. Am J Respir Crit Care Med. (2015) 191:1147–57. doi: 10.1164/rccm.201412-2323OC
9. Schlapbach LJ, Straney L, Alexander J, MacLaren G, Festa M, Schibler A, et al. Mortality related to invasive infections, sepsis, and septic shock in critically ill children in Australia and New Zealand, 2002–13: a multicentre retrospective cohort study. Lancet Infect Dis. (2015) 15:46–54. doi: 10.1016/S1473-3099(14)71003-5
10. Ames SG, Workman JK, Olson JA, Korgenski EK, Masotti S, Knackstedt ED, et al. Infectious etiologies and patient outcomes in pediatric septic shock. J Pediatric Infect Dis Soc. (2017) 6:80–6. doi: 10.1093/jpids/piv108
11. Byington CL, Enriquez FR, Hoff C, Tuohy R, Taggart EW, Hillyard DR, et al. Serious bacterial infections in febrile infants 1 to 90 days old with and without viral infections. Pediatrics (2004) 113:1662–6.
12. Bhat N, Wright JG, Broder KR, Murray EL, Greenberg ME, Glover MJ, et al. Influenza-associated deaths among children in the United States, 2003-2004. N Engl J Med. (2005) 353:2559–67. doi: 10.1056/NEJMoa051721
13. Caballero MT, Polack FP. Respiratory syncytial virus is an “opportunistic” killer. Pediatr Pulmonol. (2018) 53:664–7. doi: 10.1002/ppul.23963
14. Law BJ, Carbonell-Estrany X, Simoes EA. An update on respiratory syncytial virus epidemiology: a developed country perspective. Respir Med. (2002) 96 (Suppl. B):S1–7. doi: 10.1016/S0954-6111(02)90064-8
15. Kimberlin DW. Herpes simplex virus infections in neonates and early childhood. Semin Pediatr Infect Dis. (2005) 16:271–81. doi: 10.1053/j.spid.2005.06.007
16. Sharp J, Harrison CJ, Puckett K, Selvaraju SB, Penaranda S, Nix WA, et al. Characteristics of young infants in whom human parechovirus, enterovirus or neither were detected in cerebrospinal fluid during sepsis evaluations. Pediatr Infect Dis J. (2013) 32:213–6. doi: 10.1097/INF.0b013e318276b328
17. Verboon-Maciolek MA, Krediet TG, Gerards LJ, de Vries LS, Groenendaal F, van Loon AM. Severe neonatal parechovirus infection and similarity with enterovirus infection. Pediatr Infect Dis J. (2008) 27:241–5. doi: 10.1097/INF.0b013e31815c1b07
18. Harvala H, Wolthers KC, Simmonds P. Parechoviruses in children: understanding a new infection. Curr Opin Infect Dis. (2010) 23:224–30. doi: 10.1097/QCO.0b013e32833890ca
19. Hatherill M. Sepsis predisposition in children with human immunodeficiency virus. Pediatr Crit Care Med. (2005) 6(Suppl. 3):S92–8. doi: 10.1097/01.PCC.0000161579.39050.6B
20. Randolph AG, McCulloh RJ. Pediatric sepsis: important considerations for diagnosing and managing severe infections in infants, children, and adolescents. Virulence (2014) 5:179–89. doi: 10.4161/viru.27045
21. Scheier E, Aviner S. Septicemia following rotavirus gastroenteritis. Isr Med Assoc J. (2013) 15:166–9.
22. Klingensmith NJ, Coopersmith CM. The gut as the motor of multiple organ dysfunction in critical illness. Crit Care Clin. (2016) 32:203–12. doi: 10.1016/j.ccc.2015.11.004
23. Singer M. The role of mitochondrial dysfunction in sepsis-induced multi-organ failure. Virulence (2014) 5:66–72. doi: 10.4161/viru.26907
24. Lewis AJ, Billiar TR, Rosengart MR. Biology and metabolism of sepsis: innate immunity, bioenergetics, and autophagy. Surg Infect. (2016) 17:286–93. doi: 10.1089/sur.2015.262
25. Florescu DF, Kalil AC. Cytomegalovirus infections in non-immunocompromised and immunocompromised patients in the intensive care unit. Infect Disord Drug Targets (2011) 11:354–64.
26. Carcillo JA, Podd B, Aneja R, Weiss SL, Hall MW, Cornell TT, et al. Pathophysiology of pediatric multiple organ dysfunction syndrome. Pediatr Crit Care Med. (2017) 18(Suppl. 1):S32–45. doi: 10.1097/PCC.0000000000001052
27. Steinberg BE, Goldenberg NM, Lee WL. Do viral infections mimic bacterial sepsis? The role of microvascular permeability: a review of mechanisms and methods. Antiviral Res. (2012) 93:2–15. doi: 10.1016/j.antiviral.2011.10.019
28. Ng CS, Kato H, Fujita T. Recognition of viruses in the cytoplasm by RLRs and other helicases–how conformational changes, mitochondrial dynamics and ubiquitination control innate immune responses. Int Immunol. (2012) 24:739–49. doi: 10.1093/intimm/dxs099
29. Jensen S, Thomsen AR. Sensing of RNA viruses: a review of innate immune receptors involved in recognizing RNA virus invasion. J Virol. (2012) 86:2900–10. doi: 10.1128/JVI.05738-11
30. Iwasaki A, Pillai PS. Innate immunity to influenza virus infection. Nat Rev Immunol. (2014) 14:315–28. doi: 10.1038/nri3665
31. Chaturvedi A, Pierce SK. How location governs toll-like receptor signaling. Traffic (2009) 10:621–8. doi: 10.1111/j.1600-0854.2009.00899.x
32. Kaisho T, Akira S. Toll-like receptor function and signaling. J Allergy Clin Immunol. (2006) 117:979–87; quiz 88. doi: 10.1016/j.jaci.2006.02.023
33. Wang JQ, Jeelall YS, Ferguson LL, Horikawa K. Toll-like receptors and cancer: myd88 mutation and inflammation. Front Immunol. (2014) 5:367. doi: 10.3389/fimmu.2014.00367
34. Nimmerjahn F, Dudziak D, Dirmeier U, Hobom G, Riedel A, Schlee M, et al. Active NF-kappaB signalling is a prerequisite for influenza virus infection. J Gen Virol. (2004) 85:2347–56. doi: 10.1099/vir.0.79958-0
35. Sprague AH, Khalil RA. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem Pharmacol. (2009) 78:539–52. doi: 10.1016/j.bcp.2009.04.029
36. Loo YM, Gale M, Jr. Immune signaling by RIG-I-like receptors. Immunity (2011) 34:680–92. doi: 10.1016/j.immuni.2011.05.003
37. Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature (2006) 441:101–5. doi: 10.1038/nature04734
38. Wang S, Le TQ, Kurihara N, Chida J, Cisse Y, Yano M, et al. Influenza virus-cytokine-protease cycle in the pathogenesis of vascular hyperpermeability in severe influenza. J Infect Dis. (2010) 202:991–1001. doi: 10.1086/656044
39. Puerta-Guardo H, Glasner DR, Harris E. Dengue virus NS1 disrupts the endothelial glycocalyx, leading to hyperpermeability. PLoS Pathog. (2016) 12:e1005738. doi: 10.1371/journal.ppat.1005738
40. Duncan ME, Richardson JP, Murray GI, Melvin WT, Fothergill JE. Human matrix metalloproteinase-9: activation by limited trypsin treatment and generation of monoclonal antibodies specific for the activated form. Eur J Biochem. (1998) 258:37–43. doi: 10.1046/j.1432-1327.1998.2580037.x
41. Ramnath R, Foster RR, Qiu Y, Cope G, Butler MJ, Salmon AH, et al. Matrix metalloproteinase 9-mediated shedding of syndecan 4 in response to tumor necrosis factor alpha: a contributor to endothelial cell glycocalyx dysfunction. FASEB J. (2014) 28:4686–99. doi: 10.1096/fj.14-252221
42. Chappell D, Jacob M, Rehm M, Stoeckelhuber M, Welsch U, Conzen P, et al. Heparinase selectively sheds heparan sulphate from the endothelial glycocalyx. Biol Chem. (2008) 389:79–82. doi: 10.1515/BC.2008.005
43. Schwarz KB. Oxidative stress during viral infection: a review. Free Radic Biol Med. (1996) 21:641–9.
44. Perrone LA, Belser JA, Wadford DA, Katz JM, Tumpey TM. Inducible nitric oxide contributes to viral pathogenesis following highly pathogenic influenza virus infection in mice. J Infect Dis. (2013) 207:1576–84. doi: 10.1093/infdis/jit062
45. Rubio-Gayosso I, Platts SH, Duling BR. Reactive oxygen species mediate modification of glycocalyx during ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. (2006) 290:H2247–56. doi: 10.1152/ajpheart.00796.2005
46. Garofalo RP, Kolli D, Casola A. Respiratory syncytial virus infection: mechanisms of redox control and novel therapeutic opportunities. Antioxid Redox Signal. (2013) 18:186–217. doi: 10.1089/ars.2011.4307
47. Armstrong SM, Mubareka S, Lee WL. The lung microvascular endothelium as a therapeutic target in severe influenza. Antiviral Res. (2013) 99:113–8. doi: 10.1016/j.antiviral.2013.05.003
48. Chen HR, Chao CH, Liu CC, Ho TS, Tsai HP, Perng GC, et al. Macrophage migration inhibitory factor is critical for dengue NS1-induced endothelial glycocalyx degradation and hyperpermeability. PLoS Pathog. (2018) 14:e1007033. doi: 10.1371/journal.ppat.1007033
49. Constantinescu AA, Vink H, Spaan JA. Endothelial cell glycocalyx modulates immobilization of leukocytes at the endothelial surface. Arterioscler Thromb Vasc Biol. (2003) 23:1541–7. doi: 10.1161/01.ATV.0000085630.24353.3D
50. Wu G, Essex DW, Meloni FJ, Takafuta T, Fujimura K, Konkle BA, et al. Human endothelial cells in culture and in vivo express on their surface all four components of the glycoprotein Ib/IX/V complex. Blood (1997) 90:2660–9.
51. Ruggeri ZM. Von Willebrand factor, platelets and endothelial cell interactions. J Thromb Haemost. (2003) 1:1335–42.
52. Koedam JA, Cramer EM, Briend E, Furie B, Furie BC, Wagner DD. P-selectin, a granule membrane protein of platelets and endothelial cells, follows the regulated secretory pathway in AtT-20 cells. J Cell Biol. (1992) 116:617–25.
53. Berndt MC, Shen Y, Dopheide SM, Gardiner EE, Andrews RK. The vascular biology of the glycoprotein Ib-IX-V complex. Thromb Haemost. (2001) 86:178–88.
54. Levick JR, Michel CC. Microvascular fluid exchange and the revised Starling principle. Cardiovasc Res. (2010) 87:198–210. doi: 10.1093/cvr/cvq062
55. Xiao K, Allison DF, Kottke MD, Summers S, Sorescu GP, Faundez V, et al. Mechanisms of VE-cadherin processing and degradation in microvascular endothelial cells. J Biol Chem. (2003) 278:19199–208. doi: 10.1074/jbc.M211746200
56. Potter MD, Barbero S, Cheresh DA. Tyrosine phosphorylation of VE-cadherin prevents binding of p120- and beta-catenin and maintains the cellular mesenchymal state. J Biol Chem. (2005) 280:31906–12. doi: 10.1074/jbc.M505568200
57. Gorbunova E, Gavrilovskaya IN, Mackow ER. Pathogenic hantaviruses Andes virus and Hantaan virus induce adherens junction disassembly by directing vascular endothelial cadherin internalization in human endothelial cells. J Virol. (2010) 84:7405–11. doi: 10.1128/JVI.00576-10
58. Davis S, Aldrich TH, Jones PF, Acheson A, Compton DL, Jain V, et al. Isolation of angiopoietin-1, a ligand for the TIE2 receptor, by secretion-trap expression cloning. Cell (1996) 87:1161–9.
59. Brindle NP, Saharinen P, Alitalo K. Signaling and functions of angiopoietin-1 in vascular protection. Circ Res. (2006) 98:1014–23. doi: 10.1161/01.RES.0000218275.54089.12
60. Milam KE, Parikh SM. The angiopoietin-Tie2 signaling axis in the vascular leakage of systemic inflammation. Tissue Barriers (2015) 3:e957508. doi: 10.4161/21688362.2014.957508
61. Levine JD, Harlan JM, Harker LA, Joseph ML, Counts RB. Thrombin-mediated release of factor VIII antigen from human umbilical vein endothelial cells in culture. Blood (1982) 60:531–4.
62. Vischer UM, Jornot L, Wollheim CB, Theler JM. Reactive oxygen intermediates induce regulated secretion of von Willebrand factor from cultured human vascular endothelial cells. Blood (1995) 85:3164–72.
63. Matsushita K, Yamakuchi M, Morrell CN, Ozaki M, O'Rourke B, Irani K, et al. Vascular endothelial growth factor regulation of Weibel-Palade-body exocytosis. Blood (2005) 105:207–14. doi: 10.1182/blood-2004-04-1519
64. Fiedler U, Scharpfenecker M, Koidl S, Hegen A, Grunow V, Schmidt JM, et al. The Tie-2 ligand angiopoietin-2 is stored in and rapidly released upon stimulation from endothelial cell Weibel-Palade bodies. Blood (2004) 103:4150–6. doi: 10.1182/blood-2003-10-3685
65. Sugiyama MG, Armstrong SM, Wang C, Hwang D, Leong-Poi H, Advani A, et al. The Tie2-agonist vasculotide rescues mice from influenza virus infection. Sci Rep. (2015) 5:11030. doi: 10.1038/srep11030
66. Daidoji T, Koma T, Du A, Yang CS, Ueda M, Ikuta K, et al. H5N1 avian influenza virus induces apoptotic cell death in mammalian airway epithelial cells. J Virol. (2008) 82:11294–307. doi: 10.1128/JVI.01192-08
67. Zhou J, Law HK, Cheung CY, Ng IH, Peiris JS, Lau YL. Functional tumor necrosis factor-related apoptosis-inducing ligand production by avian influenza virus-infected macrophages. J Infect Dis. (2006) 193:945–53. doi: 10.1086/500954
68. Olijve L, Jennings L, Walls T. Human parechovirus: an increasingly recognized cause of sepsis-like illness in young infants. Clin Microbiol Rev. (2018) 31:e00047–17. doi: 10.1128/CMR.00047-17
69. Zhang L, Peeples ME, Boucher RC, Collins PL, Pickles RJ. Respiratory syncytial virus infection of human airway epithelial cells is polarized, specific to ciliated cells, and without obvious cytopathology. J Virol. (2002) 76:5654–66. doi: 10.1128/JVI.76.11.5654-5666.2002
70. Simon AK, Hollander GA, McMichael A. Evolution of the immune system in humans from infancy to old age. Proc Biol Sci. (2015) 282:20143085. doi: 10.1098/rspb.2014.3085
71. Orange JS. Congenital immunodeficiencies and sepsis. Pediatr Crit Care Med. (2005) 6(Suppl−3):S99–107. doi: 10.1097/01.PCC.0000164488.19810.DB
72. Asgari S, Schlapbach LJ, Anchisi S, Hammer C, Bartha I, Junier T, et al. Severe viral respiratory infections in children with IFIH1 loss-of-function mutations. Proc Natl Acad Sci USA. (2017) 114:8342–7. doi: 10.1073/pnas.1704259114
73. Dinarello CA, Kent EF, Jr. Chemical characterization of an interleukin-1-inducing substance derived from human mixed leukocyte reactions: IL-1-inducing substance is not gamma interferon. Yale J Biol Med. (1985) 58:101–13.
74. Harris RL, Musher DM, Bloom K, Gathe J, Rice L, Sugarman B, et al. Manifestations of sepsis. Arch Intern Med. (1987) 147:1895–906.
75. Zhao ZD, Yang WZ, Gao C, Fu X, Zhang W, Zhou Q, et al. A hypothalamic circuit that controls body temperature. Proc Natl Acad Sci USA. (2017) 114:2042–7. doi: 10.1073/pnas.1616255114
76. Krausz MM, Perel A, Eimerl D, Cotev S. Cardiopulmonary effects of volume loading in patients in septic shock. Ann Surg. (1977) 185:429–34.
77. Brown ZA, Wald A, Morrow RA, Selke S, Zeh J, Corey L. Effect of serologic status and cesarean delivery on transmission rates of herpes simplex virus from mother to infant. JAMA (2003) 289:203–9.
78. Nielsen NM, Midgley SE, Nielsen AC, Christiansen CB, Fischer TK. Severe human parechovirus infections in infants and the role of older siblings.Am J Epidemiol. (2016) 183:664–70. doi: 10.1093/aje/kwv206
79. Owatanapanich S, Wutthanarungsan R, Jaksupa W, Thisyakorn U. Risk factors for severe enteroviral infections in children. J Med Assoc Thai. (2016) 99:322–30.
80. Eggleston HA, Gunville CF, Miller JI, Sontag MK, Mourani PM. A comparison of characteristics and outcomes in severe human metapneumovirus and respiratory syncytial virus infections in children treated in an intensive care unit. Pediatr Infect Dis J. (2013) 32:1330–4. doi: 10.1097/INF.0b013e3182a2261b
81. Drysdale SB, Mejias A, Ramilo O. Rhinovirus - not just the common cold. J Infect. (2017) 74(Suppl 1):S41–6. doi: 10.1016/S0163-4453(17)30190-1
82. Committee on Infectious Diseases. Recommendations for prevention and control of influenza in children, 2017 - 2018. Pediatrics (2017) 140:e20172550. doi: 10.1542/peds.2017-2550
83. Ngwende S, Gombe NT, Midzi S, Tshimanga M, Shambira G, Chadambuka A. Factors associated with HIV infection among children born to mothers on the prevention of mother to child transmission programme at Chitungwiza Hospital, Zimbabwe, 2008. BMC Public Health (2013) 13:1181. doi: 10.1186/1471-2458-13-1181
84. de Lemos LM, Lippi J, Rutherford GW, Duarte GS, Martins NG, Santos VS, et al. Maternal risk factors for HIV infection in infants in northeastern Brazil. Int J Infect Dis. (2013) 17:e913–8. doi: 10.1016/j.ijid.2013.04.015
85. Hendaus MA, Jomha FA, Alhammadi AH. Virus-induced secondary bacterial infection: a concise review. Ther Clin Risk Manag. (2015) 11:1265–71. doi: 10.2147/TCRM.S87789
86. O'Brien KL, Walters MI, Sellman J, Quinlisk P, Regnery H, Schwartz B, et al. Severe pneumococcal pneumonia in previously healthy children: the role of preceding influenza infection. Clin Infect Dis. (2000) 30:784–9. doi: 10.1086/313772
87. Randolph AG, Vaughn F, Sullivan R, Rubinson L, Thompson BT, Yoon G, et al. Critically ill children during the 2009-2010 influenza pandemic in the United States. Pediatrics (2011) 128:e1450–8. doi: 10.1542/peds.2011-0774
88. Cebey-Lopez M, Pardo-Seco J, Gomez-Carballa A, Martinon-Torres N, Martinon-Sanchez JM, Justicia-Grande A, et al. Bacteremia in children hospitalized with respiratory syncytial virus infection. PLoS ONE (2016) 11:e0146599. doi: 10.1371/journal.pone.0146599
89. Hall MW, Geyer SM, Guo CY, Panoskaltsis-Mortari A, Jouvet P, Ferdinands J, et al. Innate immune function and mortality in critically ill children with influenza: a multicenter study. Crit Care Med. (2013) 41:224–36. doi: 10.1097/CCM.0b013e318267633c
90. Morens DM, Taubenberger JK, Fauci AS. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J Infect Dis. (2008) 198:962–70. doi: 10.1086/591708
91. Finelli L, Fiore A, Dhara R, Brammer L, Shay DK, Kamimoto L, et al. Influenza-associated pediatric mortality in the United States: increase of Staphylococcus aureus coinfection. Pediatrics (2008) 122:805–11. doi: 10.1542/peds.2008-1336
92. Wilson R, Dowling RB, Jackson AD. The biology of bacterial colonization and invasion of the respiratory mucosa. Eur Respir J. (1996) 9:1523–30.
93. Avadhanula V, Rodriguez CA, Devincenzo JP, Wang Y, Webby RJ, Ulett GC, et al. Respiratory viruses augment the adhesion of bacterial pathogens to respiratory epithelium in a viral species- and cell type-dependent manner. J Virol. (2006) 80:1629–36. doi: 10.1128/JVI.80.4.1629-1636.2006
94. Rynda-Apple A, Robinson KM, Alcorn JF. Influenza and bacterial superinfection: illuminating the immunologic mechanisms of disease. Infect Immun. (2015) 83:3764–70. doi: 10.1128/IAI.00298-15
95. Cundell DR, Gerard NP, Gerard C, Idanpaan-Heikkila I, Tuomanen EI. Streptococcus pneumoniae anchor to activated human cells by the receptor for platelet-activating factor. Nature (1995) 377:435–8. doi: 10.1038/377435a0
96. Lowenthal A, Livni G, Amir J, Samra Z, Ashkenazi S. Secondary bacteremia after rotavirus gastroenteritis in infancy. Pediatrics (2006) 117:224–6. doi: 10.1542/peds.2005-0177
97. Goulder PJ, Jeena P, Tudor-Williams G, Burchett S. Paediatric HIV infection: correlates of protective immunity and global perspectives in prevention and management. Br Med Bull. (2001) 58:89–108.
98. Tudor-Williams G, Pizzo PA. Pediatric human immunodeficiency virus infection. In: Stiehm ER, editor. Immunologic Disorders in Infants and Children. 4th edn. Philadelphia, PA: WB Saunders Company (1996). p. 510–52.
99. Hogan CM, Hammer SM. Host determinants in HIV infection and disease. Part 1: cellular and humoral immune responses. Ann Intern Med. (2001) 134:761–76.
100. DaPalma T, Doonan BP, Trager NM, Kasman LM. A systematic approach to virus-virus interactions. Virus Res. (2010) 149:1–9. doi: 10.1016/j.virusres.2010.01.002
101. Rhedin S, Lindstrand A, Rotzen-Ostlund M, Tolfvenstam T, Ohrmalm L, Rinder MR, et al. Clinical utility of PCR for common viruses in acute respiratory illness. Pediatrics (2014) 133:e538–45. doi: 10.1542/peds.2013-3042
102. Asner SA, Rose W, Petrich A, Richardson S, Tran DJ. Is virus coinfection a predictor of severity in children with viral respiratory infections? Clin Microbiol Infect. (2015) 21:264.e1–6. doi: 10.1016/j.cmi.2014.08.024
103. Kouni S, Karakitsos P, Chranioti A, Theodoridou M, Chrousos G, Michos A. Evaluation of viral co-infections in hospitalized and non-hospitalized children with respiratory infections using microarrays. Clin Microbiol Infect. (2013) 19:772–7. doi: 10.1111/1469-0691.12015
104. Lim FJ, de Klerk N, Blyth CC, Fathima P, Moore HC. Systematic review and meta-analysis of respiratory viral coinfections in children. Respirology (2016) 21:648–55. doi: 10.1111/resp.12741
105. Scotta MC, Chakr VC, de Moura A, Becker RG, de Souza AP, Jones MH, et al. Respiratory viral coinfection and disease severity in children: a systematic review and meta-analysis. J Clin Virol. (2016) 80:45–56. doi: 10.1016/j.jcv.2016.04.019
106. Aslanzadeh J, Zheng X, Li H, Tetreault J, Ratkiewicz I, Meng S, et al. Prospective evaluation of rapid antigen tests for diagnosis of respiratory syncytial virus and human metapneumovirus infections. J Clin Microbiol. (2008) 46:1682–5. doi: 10.1128/JCM.00008-08
107. Griffiths C, Drews SJ, Marchant DJ. Respiratory syncytial virus: infection, detection, and new options for prevention and treatment. Clin Microbiol Rev. (2017) 30:277–319. doi: 10.1128/CMR.00010-16
108. Leland DS, Ginocchio CC. Role of cell culture for virus detection in the age of technology. Clin Microbiol Rev. (2007) 20:49–78. doi: 10.1128/CMR.00002-06
109. Shirato K, Nishimura H, Saijo M, Okamoto M, Noda M, Tashiro M, et al. Diagnosis of human respiratory syncytial virus infection using reverse transcription loop-mediated isothermal amplification. J Virol Methods (2007) 139:78–84. doi: 10.1016/j.jviromet.2006.09.014
110. de-Paris F, Beck C, Machado AB, Paiva RM, da Silva Menezes D, de Souza Nunes L, et al. Optimization of one-step duplex real-time RT-PCR for detection of influenza and respiratory syncytial virus in nasopharyngeal aspirates. J Virol Methods (2012) 186:189–92. doi: 10.1016/j.jviromet.2012.07.008
111. Couturier MR, Barney T, Alger G, Hymas WC, Stevenson JB, Hillyard D, et al. Evaluation of the filmarray(R) respiratory panel for clinical use in a large children's hospital. J Clin Lab Anal. (2013) 27:148–54. doi: 10.1002/jcla.21576
112. Gill PJ, Richardson SE, Ostrow O, Friedman JN. Testing for respiratory viruses in children: to swab or not to swab. JAMA Pediatr. (2017) 171:798–804. doi: 10.1001/jamapediatrics.2017.0786
113. Wolthers KC, Benschop KS, Schinkel J, Molenkamp R, Bergevoet RM, Spijkerman IJ, et al. Human parechoviruses as an important viral cause of sepsislike illness and meningitis in young children. Clin Infect Dis. (2008) 47:358–63. doi: 10.1086/589752
114. Moe N, Pedersen B, Nordbo SA, Skanke LH, Krokstad S, Smyrnaios A, et al. Respiratory virus detection and clinical diagnosis in children attending day care. PLoS ONE (2016) 11:e0159196. doi: 10.1371/journal.pone.0159196
115. Martin ET, Fairchok MP, Kuypers J, Magaret A, Zerr DM, Wald A, et al. Frequent and prolonged shedding of bocavirus in young children attending daycare. J Infect Dis. (2010) 201:1625–32. doi: 10.1086/652405
117. Hendricks GL, Weirich KL, Viswanathan K, Li J, Shriver ZH, Ashour J, et al. Sialylneolacto-N-tetraose c (LSTc)-bearing liposomal decoys capture influenza A virus. J Biol Chem. (2013) 288:8061–73. doi: 10.1074/jbc.M112.437202
118. Herberg JA, Kaforou M, Wright VJ, Shailes H, Eleftherohorinou H, Hoggart CJ, et al. Diagnostic test accuracy of a 2-transcript host RNA signature for discriminating bacterial vs viral infection in febrile children. JAMA (2016) 316:835–45. doi: 10.1001/jama.2016.11236
119. Rivers E, Nguyen B, Havstad S, Ressler J, Muzzin A, Knoblich B, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med. (2001) 345:1368–77. doi: 10.1056/NEJMoa010307
120. Prkno A, Wacker C, Brunkhorst FM, Schlattmann P. Procalcitonin-guided therapy in intensive care unit patients with severe sepsis and septic shock–a systematic review and meta-analysis. Crit Care (2013) 17:R291. doi: 10.1186/cc13157
121. Downes KJ, Weiss SL, Gerber JS, Klieger SB, Fitzgerald JC, Balamuth F, et al. A pragmatic biomarker-driven algorithm to guide antibiotic use in the pediatric intensive care unit: The Optimizing Antibiotic Strategies in Sepsis (OASIS) study. J Pediatric Infect Dis Soc. (2017) 6:134–41. doi: 10.1093/jpids/piw023
122. Meisner M, Tschaikowsky K, Palmaers T, Schmidt J. Comparison of procalcitonin (PCT) and C-reactive protein (CRP) plasma concentrations at different SOFA scores during the course of sepsis and MODS. Crit Care (1999) 3:45–50. doi: 10.1186/cc306
123. Afshari A, Harbarth S. Procalcitonin as diagnostic biomarker of sepsis. Lancet Infect Dis. (2013) 13:382–4. doi: 10.1016/S1473-3099(13)70026-4
124. Wacker C, Prkno A, Brunkhorst FM, Schlattmann P. Procalcitonin as a diagnostic marker for sepsis: a systematic review and meta-analysis. Lancet Infect Dis. (2013) 13:426–35. doi: 10.1016/S1473-3099(12)70323-7
125. Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med. (1999) 340:448–54. doi: 10.1056/NEJM199902113400607
126. Hu X, Yu J, Crosby SD, Storch GA. Gene expression profiles in febrile children with defined viral and bacterial infection. Proc Natl Acad Sci USA. (2013) 110:12792–7. doi: 10.1073/pnas.1302968110
127. Woods CW, McClain MT, Chen M, Zaas AK, Nicholson BP, Varkey J, et al. A host transcriptional signature for presymptomatic detection of infection in humans exposed to influenza H1N1 or H3N2. PLoS ONE (2013) 8:e52198. doi: 10.1371/journal.pone.0052198
128. Zaas AK, Chen M, Varkey J, Veldman T, Hero AO III, Lucas J, et al. Gene expression signatures diagnose influenza and other symptomatic respiratory viral infections in humans. Cell Host Microbe (2009) 6:207–17. doi: 10.1016/j.chom.2009.07.006
129. Sampson DL, Fox BA, Yager TD, Bhide S, Cermelli S, McHugh LC, et al. A four-biomarker blood signature discriminates systemic inflammation due to viral infection versus other etiologies. Sci Rep. (2017) 7:2914. doi: 10.1038/s41598-017-02325-8
130. Engelmann I, Dubos F, Lobert PE, Houssin C, Degas V, Sardet A, et al. Diagnosis of viral infections using myxovirus resistance protein A (MxA). Pediatrics (2015) 135:e985–93. doi: 10.1542/peds.2014-1946
131. Alter SJ, Bennett JS, Koranyi K, Kreppel A, Simon R. Common childhood viral infections. Curr Probl Pediatr Adolesc Health Care (2015) 45:21–53. doi: 10.1016/j.cppeds.2014.12.001
132. American Academy of Pediatrics Committee on Infectious D, American Academy of Pediatrics Bronchiolitis Guidelines C. Updated guidance for palivizumab prophylaxis among infants and young children at increased risk of hospitalization for respiratory syncytial virus infection. Pediatrics (2014) 134:415–20. doi: 10.1542/peds.2014-1665
133. Committee on Infectious D. From the american academy of pediatrics: policy statements–modified recommendations for use of palivizumab for prevention of respiratory syncytial virus infections. Pediatrics (2009) 124:1694–701. doi: 10.1542/peds.2009-2345
134. Conrad DA, Christenson JC, Waner JL, Marks MI. Aerosolized ribavirin treatment of respiratory syncytial virus infection in infants hospitalized during an epidemic. Pediatr Infect Dis J. (1987) 6:152–8.
135. Mazur NI, Martinon-Torres F, Baraldi E, Fauroux B, Greenough A, Heikkinen T, et al. Lower respiratory tract infection caused by respiratory syncytial virus: current management and new therapeutics. Lancet Respir Med. (2015) 3:888–900. doi: 10.1016/S2213-2600(15)00255-6
136. DeVincenzo JP, Whitley RJ, Mackman RL, Scaglioni-Weinlich C, Harrison L, Farrell E, et al. Oral GS-5806 activity in a respiratory syncytial virus challenge study. N Engl J Med. (2014) 371:711–22. doi: 10.1056/NEJMoa1401184
137. Detalle L, Stohr T, Palomo C, Piedra PA, Gilbert BE, Mas V, et al. Generation and Characterization of ALX-0171, a potent novel therapeutic nanobody for the treatment of respiratory syncytial virus infection. Antimicrob Agents Chemother. (2016) 60:6–13. doi: 10.1128/AAC.01802-15
138. Douglas JL, Panis ML, Ho E, Lin KY, Krawczyk SH, Grant DM, et al. Small molecules VP-14637 and JNJ-2408068 inhibit respiratory syncytial virus fusion by similar mechanisms. Antimicrob Agents Chemother. (2005) 49:2460–6. doi: 10.1128/AAC.49.6.2460-2466.2005
139. Kim YI, Pareek R, Murphy R, Harrison L, Farrell E, Cook R, et al. The antiviral effects of RSV fusion inhibitor, MDT-637, on clinical isolates, vs its achievable concentrations in the human respiratory tract and comparison to ribavirin. Influenza Other Respir Viruses (2017) 11:525–30. doi: 10.1111/irv.12503
140. DeVincenzo JP, McClure MW, Symons JA, Fathi H, Westland C, Chanda S, et al. Activity of oral ALS-008176 in a respiratory syncytial virus challenge study. N Engl J Med. (2015) 373:2048–58. doi: 10.1056/NEJMoa1413275
141. Zamora MR, Budev M, Rolfe M, Gottlieb J, Humar A, Devincenzo J, et al. RNA interference therapy in lung transplant patients infected with respiratory syncytial virus. Am J Respir Crit Care Med. (2011) 183:531–8. doi: 10.1164/rccm.201003-0422OC
142. Burnett JC, Rossi JJ, Tiemann K. Current progress of siRNA/shRNA therapeutics in clinical trials. Biotechnol J. (2011) 6:1130–46. doi: 10.1002/biot.201100054
143. Flannery B, Reynolds SB, Blanton L, Santibanez TA, O'Halloran A, Lu PJ, et al. Influenza vaccine effectiveness against pediatric deaths: 2010-2014. Pediatrics (2017) 139:977–981. doi: 10.1542/peds.2016-4244
144. Malosh RE, Martin ET, Heikkinen T, Brooks WA, Whitley RJ, Monto AS. Efficacy and Safety of oseltamivir in children: systematic review and individual patient data meta-analysis of randomized controlled trials. Clin Infect Dis. (2018) 66:1492–500. doi: 10.1093/cid/cix1040
145. Whitley RJ, Hayden FG, Reisinger KS, Young N, Dutkowski R, Ipe D, et al. Oral oseltamivir treatment of influenza in children. Pediatr Infect Dis J. (2001) 20:127–33.
146. Pickering LKBC, Kimberlin DW, Long SS. Influenza. Red Book: 2012 Report of the Committee on Infectious Diseases, American Academy of Pediatrics (2012) pp. 439–53.
147. Zuccotti GV, Fabiano V. Influvac, a trivalent inactivated subunit influenza vaccine. Expert Opin Biol Ther. (2011) 11:89–98. doi: 10.1517/14712598.2011.541436
148. Herzog C, Hartmann K, Kunzi V, Kursteiner O, Mischler R, Lazar H, et al. Eleven years of Inflexal V-a virosomal adjuvanted influenza vaccine. Vaccine (2009) 27:4381–7. doi: 10.1016/j.vaccine.2009.05.029
149. Furuta Y, Takahashi K, Fukuda Y, Kuno M, Kamiyama T, Kozaki K, et al. In vitro and in vivo activities of anti-influenza virus compound T-705. Antimicrob Agents Chemother. (2002) 46:977–81. doi: 10.1128/AAC.46.4.977-981.2002
150. Hayden F. Developing new antiviral agents for influenza treatment: what does the future hold? Clin Infect Dis. (2009) 48 (Suppl. 1):S3–13. doi: 10.1086/591851
151. Darwish I, Mubareka S, Liles WC. Immunomodulatory therapy for severe influenza. Expert Rev Anti Infect Ther. (2011) 9:807–22. doi: 10.1586/eri.11.56
152. Zheng BJ, Chan KW, Lin YP, Zhao GY, Chan C, Zhang HJ, et al. Delayed antiviral plus immunomodulator treatment still reduces mortality in mice infected by high inoculum of influenza A/H5N1 virus. Proc Natl Acad Sci USA. (2008) 105:8091–6. doi: 10.1073/pnas.0711942105
153. Ng HH, Narasaraju T, Phoon MC, Sim MK, Seet JE, Chow VT. Doxycycline treatment attenuates acute lung injury in mice infected with virulent influenza H3N2 virus: involvement of matrix metalloproteinases. Exp Mol Pathol. (2012) 92:287–95. doi: 10.1016/j.yexmp.2012.03.003
154. Quispe-Laime AM, Bracco JD, Barberio PA, Campagne CG, Rolfo VE, Umberger R, et al. H1N1 influenza a virus-associated acute lung injury: response to combination oseltamivir and prolonged corticosteroid treatment. Intensive Care Med. (2010) 36:33–41. doi: 10.1007/s00134-009-1727-6
155. Sato K, Suga M, Akaike T, Fujii S, Muranaka H, Doi T, et al. Therapeutic effect of erythromycin on influenza virus-induced lung injury in mice. Am J Respir Crit Care Med. (1998) 157(3 Pt 1):853–7. doi: 10.1164/ajrccm.157.3.9703098
156. Yamaya M, Shinya K, Hatachi Y, Kubo H, Asada M, Yasuda H, et al. Clarithromycin inhibits type a seasonal influenza virus infection in human airway epithelial cells. J Pharmacol Exp Ther. (2010) 333:81–90. doi: 10.1124/jpet.109.162149
157. Budd A, Alleva L, Alsharifi M, Koskinen A, Smythe V, Mullbacher A, et al. Increased survival after gemfibrozil treatment of severe mouse influenza. Antimicrob Agents Chemother. (2007) 51:2965–8. doi: 10.1128/AAC.00219-07
158. Walsh KB, Teijaro JR, Rosen H, Oldstone MB. Quelling the storm: utilization of sphingosine-1-phosphate receptor signaling to ameliorate influenza virus-induced cytokine storm. Immunol Res. (2011) 51:15–25. doi: 10.1007/s12026-011-8240-z
159. Abzug MJ, Michaels MG, Wald E, Jacobs RF, Romero JR, Sanchez PJ, et al. A Randomized, double-blind, placebo-controlled trial of pleconaril for the treatment of neonates with enterovirus sepsis. J Pediatric Infect Dis Soc. (2016) 5:53–62. doi: 10.1093/jpids/piv015
160. Pevear DC, Hayden FG, Demenczuk TM, Barone LR, McKinlay MA, Collett MS. Relationship of pleconaril susceptibility and clinical outcomes in treatment of common colds caused by rhinoviruses. Antimicrob Agents Chemother. (2005) 49:4492–9. doi: 10.1128/AAC.49.11.4492-4499.2005
161. Abzug MJ, Keyserling HL, Lee ML, Levin MJ, Rotbart HA. Neonatal enterovirus infection: virology, serology, and effects of intravenous immune globulin. Clin Infect Dis. (1995) 20:1201–6.
162. Whitley RJ, Corey L, Arvin A, Lakeman FD, Sumaya CV, Wright PF, et al. Changing presentation of herpes simplex virus infection in neonates. J Infect Dis. (1988) 158:109–16.
163. Nahmias A, Josey WE, Naib ZM, Freeman MG, Fernandez RJ, Wheeler JH. Perinatal risk associated with maternal genital herpes simplex virus infection. Am J Obstetr Gynecol. (1971) 110:825–37. doi: 10.1016/0002-9378(71)90580-1
164. Stanberry LR, Spruance SL, Cunningham AL, Bernstein DI, Mindel A, Sacks S, et al. Glycoprotein-D-adjuvant vaccine to prevent genital herpes. N Engl J Med. (2002) 347:1652–61. doi: 10.1056/NEJMoa011915
165. Braig S, Luton D, Sibony O, Edlinger C, Boissinot C, Blot P, et al. Acyclovir prophylaxis in late pregnancy prevents recurrent genital herpes and viral shedding. Eur J Obstet Gynecol Reprod Biol. (2001) 96:55–8. doi: 10.1016/S0301-2115(00)00406-1
166. Scott LL, Sanchez PJ, Jackson GL, Zeray F, Wendel GD, Jr. Acyclovir suppression to prevent cesarean delivery after first-episode genital herpes. Obstet Gynecol. (1996) 87:69–73.
167. Pickering LKBC, Kimberlin DW, Long SS (editors). Herpes simplex. In: Red Book: Report of the Committee on Infectious Diseases. 25th ed. Elk Grove Village, IL: American Academy of Pediatrics (2000).
168. Gibb DM, Duong T, Tookey PA, Sharland M, Tudor-Williams G, Novelli V, et al. Decline in mortality, AIDS, and hospital admissions in perinatally HIV-1 infected children in the United Kingdom and Ireland. BMJ (2003) 327:1019. doi: 10.1136/bmj.327.7422.1019
169. Sánchez JM, Amador JTR, De Miguel SF, Tomé MIG, Conejo PR, Vivas PF, et al. Impact of highly active antiretroviral therapy on the morbidity and mortality in Spanish human immunodeficiency virus-infected children. Pediatr Infect Disease J. (2003) 22:863–8. doi: 10.1097/01.inf.0000091282.70253.5f
170. de Martino M, Tovo PA, Balducci M, Galli L, Gabiano C, Rezza G, et al. Reduction in mortality with availability of antiretroviral therapy for children with perinatal HIV-1 infection. Italian register for HIV infection in children and the Italian national AIDS registry. JAMA (2000) 284:190–7.
171. Gortmaker SL, Hughes M, Cervia J, Brady M, Johnson GM, Seage GR III, et al. Effect of combination therapy including protease inhibitors on mortality among children and adolescents infected with HIV-1. N Engl J Med. (2001) 345:1522–8. doi: 10.1056/NEJMoa011157
172. Hon KL, Luk MP, Fung WM, Li CY, Yeung HL, Liu PK, et al. Mortality, length of stay, bloodstream and respiratory viral infections in a pediatric intensive care unit. J Crit Care (2017) 38:57–61. doi: 10.1016/j.jcrc.2016.09.019
173. Shi T, McAllister DA, O'Brien KL, Simoes EAF, Madhi SA, Gessner BD, et al. Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in young children in 2015: a systematic review and modelling study. Lancet (2017) 390:946–58. doi: 10.1016/S0140-6736(17)30938-8
174. Byington CL, Wilkes J, Korgenski K, Sheng X. Respiratory syncytial virus-associated mortality in hospitalized infants and young children. Pediatrics (2015) 135:e24–31. doi: 10.1542/peds.2014-2151
175. Dawood FS, Iuliano AD, Reed C, Meltzer MI, Shay DK, Cheng PY, et al. Estimated global mortality associated with the first 12 months of 2009 pandemic influenza A H1N1 virus circulation: a modelling study. Lancet Infect Dis. (2012) 12:687–95. doi: 10.1016/S1473-3099(12)70121-4
176. Kumar A, Zarychanski R, Pinto R, Cook DJ, Marshall J, Lacroix J, et al. Critically ill patients with 2009 influenza A(H1N1) infection in Canada. JAMA (2009) 302:1872–9. doi: 10.1001/jama.2009.1496
177. Dominguez-Cherit G, Lapinsky SE, Macias AE, Pinto R, Espinosa-Perez L, de la Torre A, et al. Critically Ill patients with 2009 influenza A(H1N1) in Mexico. JAMA (2009) 302:1880–7. doi: 10.1001/jama.2009.1536
178. Sellers SA, Hagan RS, Hayden FG, Fischer WA II. The hidden burden of influenza: a review of the extra-pulmonary complications of influenza infection. Influenza Other Respir Viruses (2017) 11:372–93. doi: 10.1111/irv.12470
179. Khatami A, McMullan BJ, Webber M, Stewart P, Francis S, Timmers KJ, et al. Sepsis-like disease in infants due to human parechovirus type 3 during an outbreak in Australia. Clin Infect Dis. (2015) 60:228–36. doi: 10.1093/cid/ciu784
180. Verboon-Maciolek MA, Groenendaal F, Hahn CD, Hellmann J, van Loon AM, Boivin G, et al. Human parechovirus causes encephalitis with white matter injury in neonates. Ann Neurol. (2008) 64:266–73. doi: 10.1002/ana.21445
181. Abzug MJ. Prognosis for neonates with enterovirus hepatitis and coagulopathy. Pediatr Infect Dis J. (2001) 20:758–63.
182. Freund MW, Kleinveld G, Krediet TG, van Loon AM, Verboon-Maciolek MA. Prognosis for neonates with enterovirus myocarditis. Arch Dis Child Fetal Neonatal Ed. (2010) 95:F206–12. doi: 10.1136/adc.2009.165183
183. Tebruegge M, Curtis N. Enterovirus infections in neonates. Semin Fetal Neonatal Med. (2009) 14:222–7. doi: 10.1016/j.siny.2009.02.002
184. Spaeder MC, Custer JW, Bembea MM, Aganga DO, Song X, Scafidi S. A multicenter outcomes analysis of children with severe viral respiratory infection due to human metapneumovirus. Pediatr Crit Care Med. (2013) 14:268–72. doi: 10.1097/PCC.0b013e3182720fc7
185. Spaeder MC, Fackler JC. Hospital-acquired viral infection increases mortality in children with severe viral respiratory infection. Pediatr Crit Care Med. (2011) 12:e317–21. doi: 10.1097/PCC.0b013e3182230f6e
186. Sugata K, Taniguchi K, Yui A, Miyake F, Suga S, Asano Y, et al. Analysis of rotavirus antigenemia and extraintestinal manifestations in children with rotavirus gastroenteritis. Pediatrics (2008) 122:392–7. doi: 10.1542/peds.2007-2290
187. Dickey M, Jamison L, Michaud L, Care M, Bernstein DI, Staat MA. Rotavirus meningoencephalitis in a previously healthy child and a review of the literature. Pediatr Infect Dis J. (2009) 28:318–21. doi: 10.1097/INF.0b013e31818ddbe9
188. Taha TE, Graham SM, Kumwenda NI, Broadhead RL, Hoover DR, Markakis D, et al. Morbidity among human immunodeficiency virus-1-infected and -uninfected African children. Pediatrics (2000) 106:E77. doi: 10.1097/INF.0b013e31822cca05
189. Centers for Disease Control and Prevention. 1994 Revised classification system for human immunodeficiency virus infection in children less than 13 years of age; Official authorized addenda: human immunodeficiency virus infection codes and official guidelines for coding and reporting ICD-9-CM. Morbid. Mortal. Week. Rep. (1994) 43:1–10.
190. Zanone SM, Krause LK, Madhi SA, Bassat Q, Jha P, Simoes EA, et al. Challenges in estimating RSV-associated mortality rates. Lancet Respir Med. (2016) 4:345–7. doi: 10.1016/S2213-2600(16)30042-X
191. Leclerc F, Leteurtre S, Duhamel A, Grandbastien B, Proulx F, Martinot A, et al. Cumulative influence of organ dysfunctions and septic state on mortality of critically ill children. Am J Respir Crit Care Med. (2005) 171:348–53. doi: 10.1164/rccm.200405-630OC
Keywords: viral sepsis, sepsis, viral infections, viral coinfections, secondary bacterial infections, pediatric, children
Citation: Gupta N, Richter R, Robert S and Kong M (2018) Viral Sepsis in Children. Front. Pediatr. 6:252. doi: 10.3389/fped.2018.00252
Received: 11 June 2018; Accepted: 28 August 2018;
Published: 18 September 2018.
Edited by:
Luregn J. Schlapbach, The University of Queensland, AustraliaReviewed by:
Katie Maree Moynihan, Harvard University, United StatesCopyright © 2018 Gupta, Richter, Robert and Kong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michele Kong, bWtvbmdAcGVkcy51YWIuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.