 Lindsey M. Ludwig
Lindsey M. Ludwig Michele L. Nassin
Michele L. Nassin Abbas Hadji
Abbas Hadji James L. LaBelle
James L. LaBelle
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pediatr., 21 December 2016
Sec. Pediatric Hematology and Hematological Malignancies
Volume 4 - 2016 | https://doi.org/10.3389/fped.2016.00135
A crucial component of regulating organismal homeostasis is maintaining proper cell number and eliminating damaged or potentially malignant cells. Apoptosis, or programed cell death, is the mechanism responsible for this equilibrium. The intrinsic apoptotic pathway is also especially important in the development and maintenance of the immune system. Apoptosis is essential for proper positive and negative selection during B- and T-cell development and for efficient contraction of expanded lymphocytes following an immune response. Tight regulation of the apoptotic pathway is critical, as excessive cell death can lead to immunodeficiency while apoptotic resistance can lead to aberrant lymphoproliferation and autoimmune disease. Dysregulation of cell death is implicated in a wide range of hematological malignancies, and targeting various components of the apoptotic machinery in these cases is an attractive chemotherapeutic strategy. A wide array of compounds has been developed with the purpose of reactivating the intrinsic apoptotic pathway. These compounds, termed BH3 mimetics are garnering considerable attention as they gain greater clinical oncologic significance. As their use expands, it will be imperative to understand the effects these compounds have on immune homeostasis. Uncovering their potential immunomodulatory activity may allow for administration of BH3 mimetics for direct tumor cell killing as well as novel therapies for a wide range of immune-based directives. This review will summarize the major proteins involved in the intrinsic apoptotic pathway and define their roles in normal immune development and disease. Clinical and preclinical BH3 mimetics are described within the context of what is currently known about their ability to affect immune function. Prospects for future antitumor immune amplification and immune modulation are then proposed.
The apoptotic cascade can be divided into two main pathways, both of which culminate in the activation of effector caspases that cleave essential substrates and in turn mediate the ultimate destruction of the cell (1, 2). The extrinsic pathway is initiated through external signals propagated via death receptors on the cell surface such as FAS (CD95) or other members of the tumor necrosis factor receptor (TNFR) family. Ligand-induced receptor trimerization initiates cellular demise through adaptor protein association and initiator caspase-8 activation (3, 4). In contrast, the intrinsic pathway is activated in response to a variety of internal cellular stresses and is mediated primarily by the BCL-2 family of proteins. BCL-2 was first discovered as a part of a chromosomal translocation in B-cell lymphoma and was the first known oncogene to inhibit cell death as opposed to actively promoting proliferation (5–7). The BCL-2 proteins share one to four highly conserved regions in both sequence and structure, termed BCL-2 homology (BH) domains. Based on these domains, and in conjunction with their activity profile, the BCL-2 family is divided into three functional subgroups: the multidomain antiapoptotics (BCL-2, BCL-XL, BCL-W, MCL-1, BFL-1), the multidomain proapoptotics (BAK, BAX, BOK), and the BH3-only proteins (BIM, BID, BAD, NOXA, PUMA, BMF, BIK, HRK) (Figure 1). The BH3-only proteins, named so because they share only the third BH domain with the other BCL-2 family proteins, act as cellular sentinels that in times of stress bind discrete multidomain BCL-2 proteins and initiate the apoptotic cascade (8). This process can occur through two known mechanisms. BH3-only proteins can bind antiapoptotic BCL-2 members causing release of sequestered BAX and BAK (9). These are indirectly activating BH3-only proteins (e.g., BAD and NOXA). In addition, other BH3-only proteins, such as BIM, BID, and PUMA, can not only bind antiapoptotics but are also able to directly bind and activate BAK and BAX oligomerization (10). Once oligomerized, BAK and BAX form pores in the outer mitochondrial membrane causing mitochondrial outer membrane permeabilization (MOMP), which leads to the release of cytochrome c and other proapoptotic factors such as SMAC/DIABLO from the inner mitochondrial membrane space (11, 12). Cytochrome c associates with APAF and caspase-9 to form the apoptosome, which initiates the cleavage of effector caspases 3 and 7 leading to eventual cellular destruction (13). The contact interfaces between antiapoptotic and BH3-only proteins have been elucidated through crystal structure analyses. This has led to increasing interest and ability to design drugs that recapitulate these interactions in an effort to overcome apoptotic resistance. While these efforts have mainly focused on inducing cell death in the context of cancer therapy, there is potential to use these compounds as immunomodulators based upon the differential BCL-2 family member dependencies of immune cells (14).
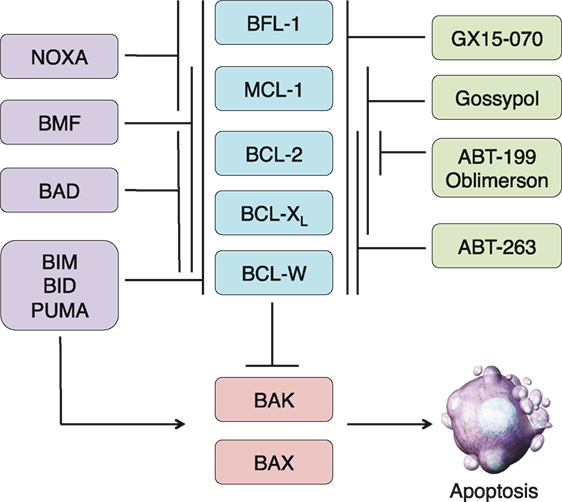
Figure 1. Overview of the BCL-2 family and BH3 mimetics in clinical trials. The BCL-2 family is divided into three subgroups: the multidomain antiapoptotics (blue), the multidomain proapoptotics (red), and the BH3-only proteins (purple). The antiapoptotic proteins sequester the proapoptotic proteins BAX and BAK. In times of cellular stress, BH3-only proteins can either bind to the antiapoptotic proteins and release the proapoptotics from their sequestration or directly bind and activate BAX and BAK. Once activated, BAX and BAK oligomerize and induce mitochondrial outer membrane permeabilization, leading to the release of other proapoptotic factors and eventual cellular destruction. The actions of the BH3-only proteins can be imitated by BH3 mimetics, some of which have reached clinical trials (green). Like the BH3-only proteins, these compounds have varying specificities for the antiapoptotic proteins.
The proapoptotic effector proteins BAK and BAX are considered to play redundant functional roles in the initiation of MOMP, as the deletion of either Bak or Bax alone leads to a minimal level of apoptotic defects (15). However, deletion of both proteins leads to a high incidence of embryonic lethality with surviving mice having a host of developmental and neuronal defects. Not surprisingly, BAX/BAK-deficient mice have a significant increase in both myeloid and lymphoid cells, leading to enlarged primary lymphoid organs and lymphocyte infiltration into peripheral organs (15). Lymphocytes from these animals are resistant to known activators of the intrinsic apoptotic pathway, including cytokine deprivation, etoposide, and irradiation (15). Mice with conditional T cell-specific Bax/Bak knockout have abnormal thymocyte development and increased accumulation of double-negative cells in the thymus (16). Thymocytes are resistant to apoptosis following treatment with γ-irradiation and animals develop T cell lymphoma with a median survival of only 10 months (16).
Because BAX and BAK activation is typically considered “the point of no return” in apoptosis induction, therapeutics that can directly activate their oligomerization would be potent initiators of apoptosis. However, there would be a lack of specificity in targeting these proteins directly and off-target effects may limit their clinical use. Direct BAX/BAK activators may find greater efficacy in combination with other, more specific BCL-2 family targeting agents. BH3 mimetics specific for discrete antiapoptotic proteins could potentially lower the apoptotic threshold in a targeted subset of lymphocytes, allowing for lower doses of BAX/BAK activators to exclusively induce apoptosis in these cells. The greatest limitation to this, however, is the inability at the present time to target BH3 mimetics to specific cell populations.
BCL-2 was the first member of the antiapoptotic subgroup to be extensively characterized. Overexpression of BCL-2 in Eμ-myc transgenic mice found that BCL-2 in conjunction with dysregulated c-myc promoted immature B cell proliferation and tumorigenesis. However, expression of BCL-2 alone allows for cell survival without any change in proliferation (7). Constitutive BCL-2 expression ultimately leads to increased numbers of pre-B cells, plasma cells, and T cells, all of which demonstrate increased longevity in culture (17, 18). BCL-2 overexpression in these animals leads to autoimmunity similar to that measured in patients with systemic lupus erythematosus (SLE) (17). In contrast, global deletion of Bcl-2 leads to a significant decrease in the number of double-positive (DP) thymocytes and peripheral (splenic) B and T cells. TUNEL staining of the spleens and thymi of aged Bcl-2 knockout mice reveals a significant increase in apoptotic cells. Thymocytes lacking BCL-2 are also more susceptible to a wide range of apoptotic stimuli (19).
MCL-1 plays a significant role in immune ontogeny and maintenance. Global deletion of Mcl-1 is embryonic lethal; therefore, conditional knockout models have been utilized to determine the role of MCL-1 in various immune cell subsets (20). Globally, MCL-1, and not BCL-2, expression is critical for maintaining hematopoietic stem cell survival. Inducible deletion of Mcl-1 causes rapid bone marrow depletion and mice become moribund within several weeks. These animals rapidly develop severely reduced numbers of hematopoietic stem cells and other bone marrow progenitor populations (21). T cell-specific Mcl-1 deletion leads to a significant reduction in T lymphocytes, as well as an apparent blockade at the DN2/3 stage of thymocyte development (22). Unlike BCL-2, MCL-1 expression remains constant or is slightly upregulated upon T cell receptor (TCR) stimulation (23, 24). Another distinctive feature of MCL-1 is that it plays a key role in the maintenance of immunosuppressive regulatory T cells (Tregs). Mice with Treg-specific MCL-1 deletion experience weight loss, inflammation, and death due to global autoimmunity within 4–8 weeks (25). Additionally, B cell-specific MCL-1 deletion leads to impaired B cell development beginning at the pro-B cell stage (22). MCL-1 is also essential for the formation of germinal centers and the survival of plasma cells (26, 27).
The remaining antiapoptotic proteins have not been as well characterized in immune system homeostasis. Loss of BCL-XL has a minimal effect on overall T cell survival. While mice lacking BCL-XL have reduced numbers of DP thymocytes, they have normal peripheral lymphocyte numbers indicating that BCL-XL alone is not critical for lymphocyte homeostasis (28). Deletion of Bfl-1 causes a decrease in DP thymocytes and an increase in double-negative (DN) and CD8+ single-positive thymocytes. However, BFL-1-deficient lymphocytes have no significant increase in resistance to apoptotic stimuli (29). Deletion of Bfl-1 in the myeloid lineage causes a decrease in the granulocyte population and causes increased levels of spontaneous apoptosis in both granulocytes and neutrophils in culture (29, 30). BFL-1 has also been shown to be elevated in several human malignancies, including B cell chronic lymphocytic leukemia (CLL) and familial SLE (31, 32).
Based on the differential reliance on specific antiapoptotic BCL-2 proteins in immune control, it may be feasible to target exclusive subsets of lymphocytes with agents having specificity to the BH3-binding domains of these proteins (Figure 2). Careful consideration will be needed in balancing BH3 mimetic dosing if given together with other chemotherapeutic agents that may lower the therapeutic threshold of immune cells or even alter their antiapoptotic dependency during treatment. The long- and short-term effects of antiapoptotic protein targeting of the immune system is unknown and how other proteins in this subclass may compensate is unclear. Rapid upregulation of non-targeted antiapoptotic proteins has been found to occur in lymphocytes when treated with BH3 mimetics (33). How this will impact immune effects clinically has yet to be determined.
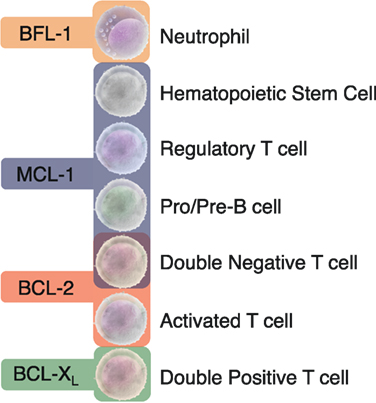
Figure 2. Distinct dependencies of antiapoptotic BCL-2 proteins exist in specific immune cell subsets. Animal models with global or conditional deletion of single antiapoptotic proteins have demonstrated that these proteins are essential for unique subsets of immune cells. Most studies have focused on the importance of the BCL-2 family in the lymphoid lineage in both developing and mature B and T cells. The differential dependencies of immune cells on unique antiapoptotic proteins may allow for the targeted drugging of specific immune cell subsets.
Most data regarding the role of BH3-only proteins has involved studying the direct activator proteins BIM, BID, and PUMA though genetic deletion in murine models. BIM is considered the master regulator of immune cell homeostasis. Bim deletion causes increased numbers of lymphoid and myeloid cells, defects in thymocyte development, and global lymphocyte resistance to multiple apoptotic stimuli (34, 35). BIM is essential for negative selection of immature T cells in the thymus, and mice lacking BIM have shorter lifespans due to the development of fatal autoimmunity (34, 36). BIM also plays an important role in regulating the survival of CD4+ T cells in the periphery. Decreased BIM expression with age leads to longer-lived CD4+ T cells that are prone to functional defects and become increasingly unable to properly respond to pathogens (37). Deletion of additional BH3-only proteins exacerbates the immune dysfunction seen in Bim knockout animals. Combined deletion of Bim and Puma causes increased resistance to a wide range of apoptotic stimuli, and a subset of these mice develop spontaneous follicular B cell lymphoma (38). Triple knockout of Bim, Puma, and Bid leads to increased lymphocytosis and profound, yet not complete, apoptotic resistance (39). The prevalence of hematological malignancies has not yet been characterized in these triple knockout mice (39).
BID is a unique BH3-only protein because its structure is more similar to the multidomain antiapoptotics and it provides functional cross talk between the intrinsic and extrinsic apoptotic pathways. BID is cleaved to its functional form, tBID, by activated caspase-8, downstream of plasma membrane death receptor activation (40). Young mice deficient in BID have no major hematological defects, whereas aged mice develop neutrophilia and many succumb to a hematopoietic malignancy resembling human chronic myelomonocytic leukemia (CMML) (41). BID may also play a key role in survival of Langerhans cells, a unique subset of dendritic cells, as BID-deficient Langerhans cells are more resistant to CD4+ T cell-mediated apoptosis (42).
PUMA is regulated by the tumor suppressor p53 and has been implicated as a mediator of apoptosis in several subsets of immune cells (43, 44). PUMA cooperates with BIM to regulate activated T cell contraction following an immune response. Mice lacking PUMA accumulate activated CD8+ T cells in their spleen following herpes simplex virus (HSV-1) infection, and these cells are more resistant to cytokine deprivation in culture (45). PUMA is also upregulated in activated B cells and Puma deletion leads to apoptotic resistance and B cell accumulation in addition to increased levels of memory B cells following antigen stimulation (46). In the myeloid compartment, PUMA deficiency has been shown to impair the regulation of neutrophil contraction following an immune response, thus compromising the ability to properly respond to bacterial infections, which can lead to lethal sepsis (47).
Similar to PUMA, the indirect activator NOXA is regulated by p53 (48). Noxa knockout mice have no aberrations in thymocyte development; however, NOXA may play a minor role in regulating the formation and maintenance of effector memory T cells during and following an immune response (49, 50). NOXA has also been shown to play a key role in B cell activation and efficient generation of high-affinity antibody clones. Loss of Noxa leads to an accumulation of low affinity B cells due to dysregulated apoptosis of these cells during immune response initiation (51).
Normal B and T cell development is maintained in cells lacking BAD and lymphocytes from BAD-deficient animals retain normal sensitivity to apoptotic stimuli (52). Currently, BAD appears most important in B cell ontogeny and maturation. Although incompletely understood, Bad knockout mice have reduced IgG production after lipopolysaccharide (LPS) stimulation and aged mice develop diffuse large B cell lymphoma (DLBCL) that is increasingly penetrant following ionizing radiation (52).
Although not as extensively studied as other BH3-only proteins, Bmf knockout mice maintain normal overall T cell counts and have no known abnormalities in thymocyte development but do experience B cell hyperplasia. BMF-deficient T- and pre-B cells are resistant to apoptosis in response to glucocorticoids or HDAC inhibition (53). Mice lacking BMF also develop thymic lymphomas following exposure to γ-irradiation (53).
Extensive characterization of the BH3-only proteins in the immune system has revealed both overlapping and non-redundant roles for many of these proteins in specific immune cell subsets. Major focus has been placed on designing therapeutics that mimic the binding of these proteins to multidomain apoptotic effectors. As development of these compounds increases for use as single agent or combination anti-cancer therapeutics, it will be essential to continue defining the exact roles of the BH3-only proteins in the ontogeny and maintenance of clinically relevant anti-viral, anti-bacterial, and antitumor immune responses. How these proteins are displaced following treatment and sequestered by expressed antiapoptotics in immune cell subtypes is of particular interest. Harnessing their immune-based “off-target” effects could allow for powerful modulation of the immune system alone or in concert with other emerging immune-based therapies.
The BCL-2 family of proteins are heavily implicated in tumorigenesis and targeting their interactions shows great promise in overcoming apoptotic resistance in a vast array of malignancies (54, 55). Given the importance of BH3-only proteins in regulating the cross talk between the anti- and proapoptotic multidomain BCL-2 proteins, most drug development has centered on recapitulating their mechanism(s) of action in cells. These so called “BH3 mimetics” encompass an array of natural products, small molecules, and peptide therapeutics that mimic the BH3-domain-directed binding of BCL-2 proteins to either lower the apoptotic threshold or directly initiate the intrinsic apoptotic cascade. Most work has focused on the role of these compounds as anticancer therapeutics as described below. Less well understood is how these compounds can be used to modulate normal immune responses. It is important to consider that cancer cells are “primed to die” due to their extreme dependence on thwarting apoptosis given their genetic and metabolic abnormalities. In contrast, although susceptible to these compounds, normal lymphocytes may have different sensitivities to these drugs as compared to malignant cells. It is critical that these potential differences are further elucidated in order to generate the most effective strategies for immune modulation.
Oblimersen Sodium (Genasense)
Over 25 years ago, it was demonstrated that an antisense oligonucleotide targeting BCL-2 could abrogate in vivo tumor growth (56). These studies led to the development of the optimized antisense oligonucleotide oblimersen sodium (G3139; genasense; augmerosen), which demonstrated in vitro and in vivo efficacy against multiple hematological malignancies, including non-Hodgkin’s lymphoma (NHL), EBV-associated lymphoproliferative disorders, and Philadelphia chromosome-positive leukemia (57–59). After showing promise in multiple preclinical models, oblimersen was tested in clinical trials for both hematological malignancies and solid tumors. Phase I and II clinical trials were encouraging against a wide range of cancer types, including hormone-refractory prostate cancer, CLL, and NHL (60–62). Unfortunately, despite initial promise, several phase III studies have found that oblimersen does not improve the response rate seen with the current standards of care (63, 64). Oblimersen has not been FDA approved and subsequent attention has turned to the development of small molecules and peptide therapeutics that directly disrupt intracellular BCL-2 family protein:protein interactions.
ABT-737 and ABT-263 (Navitoclax)
The first small molecule to effectively mimic the interaction between antiapoptotics and BH3-only proteins was the BAD-derived BH3-only mimetic ABT-737 (65). Discovered using a nuclear magnetic resonance (NMR)-based screening method, this small molecule has high affinity for the BH3-binding pocket of BCL-2, BCL-XL, and BCL-W (65). ABT-737 has been shown to disrupt BAX/BCL-2 complexes, leading to the release of cytochrome c and the initiation of the caspase cascade (66, 67). BAX/BAK double knockout cells administered ABT-737 experience no significant decrease in viability, indicating that ABT-737 functions through on-target binding to antiapoptotic proteins to induce the intrinsic mitochondrial apoptotic pathway (67). ABT-737 has potent in vitro activity against a wide range of hematological malignancies, including acute myeloid leukemia (AML), multiple myeloma (MM), and acute lymphoblastic leukemia (ALL) (67–69). Subsequent studies in xenograft models of adult and pediatric hematological diseases have confirmed the on-target potency of ABT-737 against malignant cells (70–72). The oral analog of ABT-737, ABT-263 (navitoclax), has been tested in clinical trials for both hematological and solid tumors (73). However, patients experienced dose-limiting thrombocytopenia due to the dependency of platelets on BCL-XL and BAK (74, 75). Another important caveat is that many cancers are refractory or become resistant to ABT-737 or ABT-263 due to upregulation of antiapoptotic proteins (e.g., MCL-1, BFL-1) that lack specificity to either compound (67, 76, 77). It will therefore be imperative to consider which antiapoptotic proteins target cells express and understand their real-time compensatory capacity for apoptotic resistance before treatment with BH3 mimetics having limited antiapoptotic protein specificity.
ABT-199 (Venetoclax)
To overcome the thrombocytopenia caused by ABT-263, a new BCL-2-specific BH3 mimetic was derived based on the X-ray crystal structure of BCL-2 and ABT-263 (78). ABT-199 binds BCL-2 with subnanomolar affinity (78). ABT-199 has demonstrated effective in vitro and in vivo cell killing in a range of cancers, including chronic myelogenous leukemia (CML), AML, and T-cell ALL (79–81). Based on its efficacy in clinical studies, ABT-199 has recently gained FDA approval for the treatment of refractory 17-p-deleted CLL, making it the first clinically approved small molecule targeting intracellular protein:protein interactions (82). Interestingly, there are reports describing ABT-199 inducing cell death in normal immune cell subsets in addition to its desired anticancer activity as was found in the case of normal mature B cells isolated from patients with CLL (83).
GX15-070 (Obatoclax)
The first pan inhibitor of the antiapoptotic BCL-2 proteins was GX15-070 (84). GX15-070 binds all antiapoptotics with nanomolar to low micromolar affinity and, importantly, is able to overcome ABT-737 apoptotic resistance in cells with high MCL-1 expression (85, 86). GX15-070 has efficacy against a range of solid tumors and hematological malignancies (86–88). GX15-070 was well tolerated in phase I clinical trials for patients with CLL, refractory leukemia, and myelodysplasia (89, 90). Unfortunately, in phase II and phase I/II clinical trials, GX15-070 did not improve outcomes in patients with myelofibrosis, mantle cell lymphoma, or AML (91–93). On target specificity has been questioned regarding the mechanism of action of GX15-070, as BAX/BAK double knockout cells die when treated with this compound (87). In fact, multiple modes of cell death have been measured in response to GX15-070 treatment. Canonical features of apoptosis, necrosis, and autophagy are seen in infant ALL (MLL-rearranged ALL) patient samples following treatment, indicating activation of related but not overlapping cell death mechanisms (94).
Gossypol Family
Gossypol is a natural phenolic pigment isolated from cottonseed. Its negative enantiomer R-(−)-gossypol, or AT-101, binds BCL-2, BCL-XL, BCL-W, and MCL-1 and may have efficacy as an anticancer therapeutic (95, 96). Like GX15-070, gossypol may not induce cell death exclusively via the intrinsic apoptotic pathway because BAX/BAK double knockout cells die following treatment (97). Despite potential off-target effects, gossypol has shown efficacy against several hematological malignancies, including DLBCL, MM, and CLL (98–100). Gossypol has entered several clinical trials, including a phase II trial for small cell lung cancer; however, the results have not been promising (101). Subsequent studies have focused on the development of small molecules derived from gossypol in order to improve its potency and potential clinical efficacy (102, 103). Interestingly, it has been demonstrated that gossypol induces apoptosis in polymorphonuclear leukocytes and monocytes isolated from healthy donors, suggesting a future potential as an immune modulator (104).
The promising clinical results of the aforementioned BCL-2 modulators have driven the discovery of a diverse range of small molecules and peptide therapeutics currently in clinical and preclinical development (Table 1). New small molecules have been designed as specific inhibitors of single or multiple antiapoptotic proteins. Single protein inhibitors will be useful for targeting malignant cells that are highly dependent on one antiapoptotic protein with theoretically minimal off-target effects. However, malignant cells not initially killed have been shown to rapidly upregulate antiapoptotic proteins lying outside of the primary compound’s binding profile. Pan apoptotic inhibitors may have increased potency but run the risk of greater off-target cell killing. In the context of immune modulation, targeting single antiapoptotics may be more desirable in manipulating the immune response over time and may allow for greater therapeutic dissection of specific myeloid, T, and B cell subpopulations.
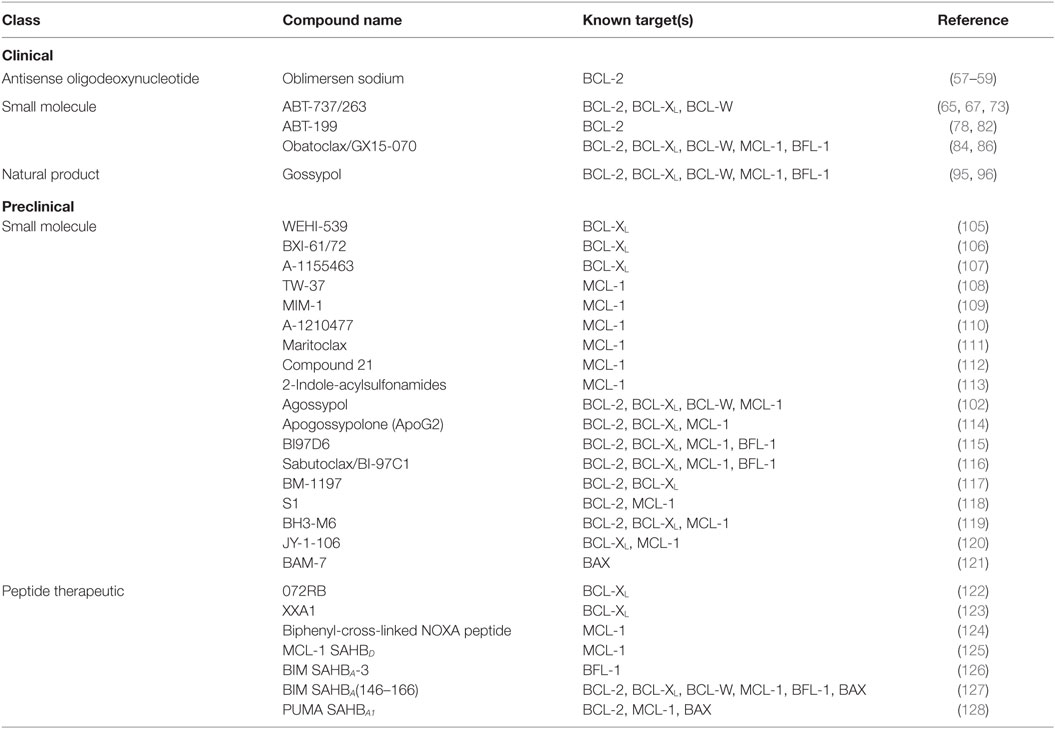
Table 1. Clinical and preclinical BH3 mimetics.
In addition to small molecules, there is increasing interest in the use of peptide-based BCL-2 therapeutics that mimic the binding interface of specific BH3-only:antiapoptotic complexes. Isolated native BH3 helices are not attractive pharmaceutical compounds as they do not typically maintain their helical structure, are quickly degraded, and lack cellular penetrability (129). Therefore, it is necessary to chemically modify these peptides in order to maintain their secondary structure, binding affinity, and protease resistance as has been done through chemical hydrocarbon stapling, chemical cross-linking, or peptide amphiphile/micelle incorporation (124, 129). While BCL-2 peptides have yet to reach clinical testing, the ability to target larger surface areas of protein:protein interactions is a promising and highly specific targeting strategy.
Most studies to date measuring the immune effects from BCL-2 modulation have tested ABT-737 in the context of autoimmunity or transplant tolerance and emphasize the compound’s effects on lymphocytes (T and B cells). One of the first studies to examine the potential for BH3 mimetics to target the immune system found that treatment with ABT-737 induces apoptosis in lymphocytes and reduces the severity of disease in several murine models of autoimmunity (130). Specifically, treatment with ABT-737 significantly reduces paw swelling in mice with collagen-induced arthritis and improves overall survival and renal function in mice with a SLE-like syndrome (130, 131). ABT-737 also suppresses immune responses to immunization against keyhole limpet hemocyanin (KLH), as T cells isolated and restimulated from ABT-737-treated mice have a significant reduction in proliferation upon re-exposure to KLH (130). Subsequent studies on the effects of ABT-737 on normal hematopoietic compartments have shown that treatment causes a significant reduction in CD4+ and CD8+ T cells, B cells, and some subsets of dendritic cells, thus perhaps diminishing proper antigen presentation and T and B cell expansion (132). In fact, treatment with ABT-737 leads to prolonged pancreatic islet allograft survival in a murine model of spontaneous diabetes, and animals are able to maintain normal long-term control of blood glucose levels compared to vehicle-treated controls (132). While ABT-737 treatment alone is able to suppress allogeneic T cell responses in vitro, treatment with ABT-737 in vivo acts synergistically with cyclosporine A to reduce skin graft rejection in a MHC mismatched transplant model (133). Interestingly, ABT-737 has also been shown to preferentially induce apoptosis in conventional T cells (Tcons) leading to a relative enrichment of immunosuppressive Tregs. This enrichment was found to slow progression in a murine model of graft-versus-host disease (GVHD) and improve overall survival following hematopoietic stem cell transplantation (134). In addition to potential benefits in transplantation and autoimmunity, ABT-737 may be useful in the mediation of inflammatory diseases. ABT-737 induces apoptosis in T lymphocytes and lamina propria mononuclear cells in a BIM-dependent manner, which is able to reduce levels of inflammation in spontaneous (IL-10−/−) and acute models of colitis (135).
In addition to lymphocytes, ABT-737 also has the potential to affect mature cells of other hematopoietic lineages. Mast cells are specialized myeloid cells that sense pathogens and initiate inflammatory responses. Their dysregulation can lead to aberrant inflammation and allergic reactions (136). Different mast cell populations are sensitive to ABT-737 at varying dosages (137). As expected, human and murine mast cell resistance to ABT-737 correlates with decreased BCL-2 and increased MCL-1 expression (137). Interestingly, in vivo analysis indicates that while mast cells were highly sensitive to ABT-737, T cells isolated from the peritoneum of treated mice are unaffected by ABT-737. Other studies have demonstrated a marked reduction in T cells isolated from lymphoid organs and peripheral blood following ABT-737 administration, indicating that immune cell localization and tissue environment may be a critical factor in determining sensitivity to BH3 mimetics (132, 133). These parameters should be carefully considered when testing the in vivo efficacy of these compounds prior to clinical translation for immune control.
Because of apparent Treg dependency on MCL-1, recent work has used GX15-070 to differentially target Tregs over Tcons. T cell sensitivity to GX15-070 appears to vary in vitro depending on a T cell’s activation status. Mature human T cells that have undergone prolonged activation are more resistant to GX15-070 compared to lymphocytes in the early stages of activation (138). This sensitivity profile extends to peripheral blood mononuclear cells (PBMCs) isolated from patients with ovarian cancer. In vitro treatment leads to significantly increased CD8+:Treg and CD4+:Treg ratios, indicating that GX15-070 preferentially induces apoptosis in the Treg subpopulation (138). Depletion of Tregs while preserving Tcons is a promising therapeutic strategy for amplifying the antitumor immune response. These results support the finding that in vivo treatment with GX15-070 following vaccination leads to decreased lung metastases in a murine model of lung adenocarcinoma (139).
These studies emphasize the promising immunomodulatory potential of BH3 mimetics. More extensive testing using a wider range of small molecules and peptide therapeutics with varying antiapoptotic specificities will provide a fuller understanding of the mechanisms responsible for clinically effective immune control.
Although most published clinical reports using BCL-2 therapeutics have concentrated on their antitumor effects, BH3 mimetic-induced manipulation of immune surveillance and activation could have profound ramifications for people suffering from a myriad of immunologically mediated conditions. Understanding the specific effects on leukocyte subsets of patients treated with these compounds will be paramount for their effective clinical translation. Beyond the results from testing these compounds in vitro and in preclinical animal models, most of what we currently know about the effects of these compounds on the human immune system must be extrapolated from oncology-based clinical trials.
Scrutinizing these studies indicate that targeted effects on the immune system include significant lymphopenias and neutropenias. Lymphopenia is a desirable effect in many cases, especially when treating hematologic malignancies like CLL. Patients with relapsed or refractory CLL treated with ABT-199 all had a >50% reduction in their absolute lymphocyte counts (ALC) and the majority had a 100% reduction. Neutropenia (grades 3 or 4) was reported in 35% of the patients with 6% of the patients experiencing serious infections and 2.5% developing autoimmune neutropenia (140). While ABT-263 induces on-target thrombocytopenia, it also elicits neutropenia 28% of the time with many patients developing subsequent infectious sequelae (141, 142). Similar findings have been observed in patients treated with other BH3 mimetics at antitumor dosing, including AT-101 and GX15-070 (89, 143).
Phase I clinical trials using the BCL-2 mRNA-targeting oligonucleotide oblimersen found that treatment with this compound led to global myelosuppression including neutropenia and lymphopenia (144, 145). However, a caveat to these and other trials is that these studies included concurrent cytotoxic chemotherapy making it difficult to specifically analyze the effects of BCL-2 targeting alone on the immune system. Of note, oblimersen has been tested in pediatric patients with neuroblastoma making it the first compound targeting BCL-2 in children (61, 144, 145). Dose escalation phase I studies evaluating the safety of the BCL-2 deoxyribonucleic acid inhibitor (DNAi) PNT2258 in patients with advanced solid tumors found rapid (within hours) decrease in lymphocytes with most patients having a >50% reduction. This phenomenon was dose dependent and used as a surrogate for efficacy (146).
Outside of the oncologic arena, BCL-2 family modulation using ABT-199 has been tested in patients with SLE. Most data remains preliminary, such as those presented at a recent American College of Rheumatology Annual Meeting (2015) demonstrating a dose-dependent reduction in total lymphocytes and B cells in particular. Neutropenia also occurred but was less consistent and correlated to different dosing thresholds (www.clinicaltrials.gov identifier NCT01686555) (147).
Clinical trials using direct BCL-2 modulation are on the rise. Searching www.clinicaltrials.gov as of August, 2016 found a number of active trials testing ABT-199 (15 trials), ABT-263 (4 trials), and gossypol/AT-101 (3 trials). Searching “oblimersen”/“genasense” or “obatoclax”/“GX15-070” revealed no entries. However, all of these active trials are testing these drugs as anticancer agents and none are directly investigating their effects on immune function or autoimmune disease. As the number of clinically available BH3 mimetics continues to grow and we further expand our knowledge regarding how these compounds affect specific immune cell subsets, we can expect a robust increase in the clinical trials evaluating BH3 mimetics in the context of immune modulation.
BH3 mimetics represent a unique arsenal of compounds with a wide range of potential therapeutic interventions beyond that of directly inducing malignant cell death. Determining the effects of long-term selective pressure on the immune system is gaining significance as patients are increasingly treated with this class of drugs. BH3 mimetics could potentially be used alone or in combination with classic immunotherapies such as stem cell transplantation and donor lymphocyte infusions, or with other more advanced therapies such as checkpoint inhibition, cancer vaccines, antibody-based therapies, chimeric antigen receptor (CAR) T-cells, and bispecific T cell engagers (BiTEs) antibodies (148) (Figure 3).
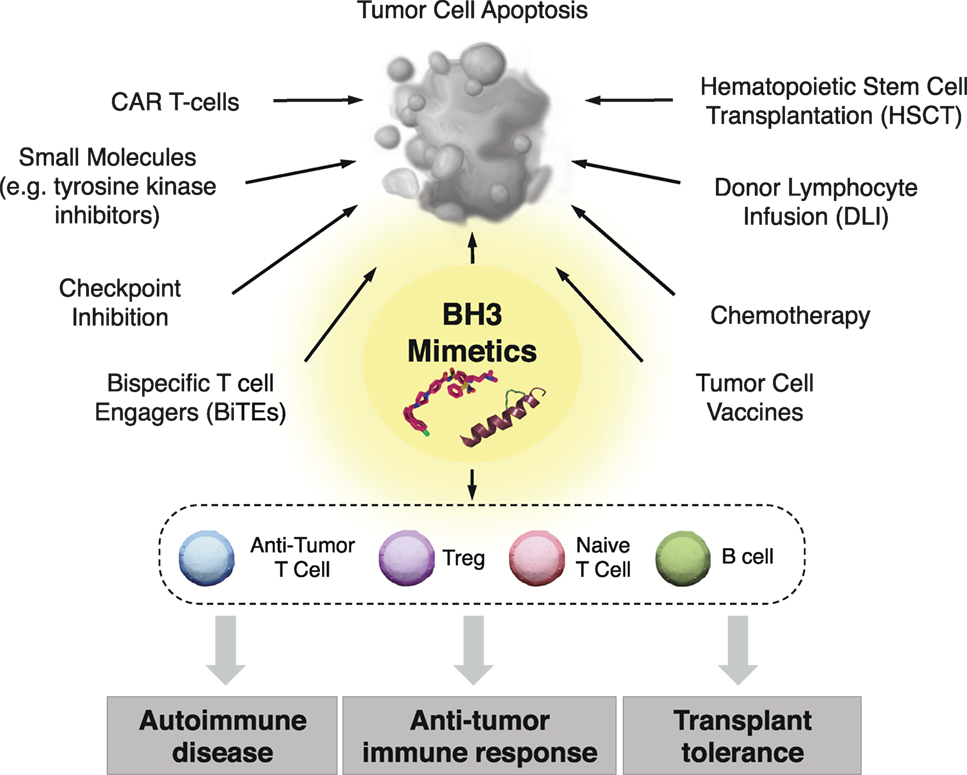
Figure 3. Clinical implementation of BH3 mimetics for immune modulation. In addition to their use as anticancer therapeutics, BH3 mimetics have promise for targeting specific immune cells subsets, which may provide therapeutic benefit in the context of antitumor immune responses, transplantation tolerance, and autoimmune diseases. Additionally, BH3 mimetics may be combined with both classical and cutting edge chemo- and immunotherapeutics to improve the standards of care in patients with a wide range of hematological malignancies.
However, there are clinical considerations and unanswered mechanistic questions that should be addressed as BH3 mimetics become effective clinical immunomodulators. First, while several studies with ABT-737 and GX15-070 have been performed to determine how they affect immune responses, there is an ever-expanding range of BH3 mimetics with diverse BCL-2 protein specificities. How these agents affect immune cell subsets as single agents or in combination is unclear. Second, resistance to a single BH3 mimetic is a reoccurring problem in cancer cells due to the ability of antiapoptotic proteins to rapidly compensate for functional loss of another (33). Determining if this occurs in non-malignant immune cells, the kinetics of change, and the functional significance of BCL-2 repertoire shifts in immune cell subsets in response to BH3 mimetic treatments will be critical in order to determine if resistance to these therapies will develop over time. BH3 peptide profiling is a powerful method that allows for the determination of the antiapoptotic proteins upon which a cell is dependent (149, 150). However, whether performed on isolated mitochondria or semi-intact cells, this method alone does not conclusively determine BH3 dependency or sequestration in real time. Use of BH3 profiling in conjunction with other described methods such as qRT-PCR, quantitative fluorescence cytometry, and BIM:antiapoptotic dissociation analysis should allow for a more complete assessment of apoptotic dependency throughout treatment (151, 152).
Other treatment-related questions include timing, dosing schedule, and combination therapy. The ideal timing of BH3 treatment in conjunction with other immunotherapies will need to be delineated. Single dosing versus metronomic dosing may have a major impact on the effectiveness of the treatment, severity of off-target effects, and emergence of resistance. Assessing initial dependency and following this long term under constant pressure from mimetic treatment may allow for increasingly thoughtful decisions regarding the best way to implement these compounds in the clinic. In fact, it may be possible to “prime” cells to die by treating with BH3 mimetics at lower doses, thus causing upregulation of potential resistance factors (for example, treating with ABT-737 may cause MCL-1 or BFL-1 upregulation). Subsequent treatment with a complementary compound targeting these resistance factors (an MCL-1 or BFL-1-specific compound in this example) may lead to even more potent cell killing or sequential immune cell modulation. The potential efficacies of these strategies remain largely unexplored.
Finally, combining BH3 mimetics with conventional chemotherapy, DNA/histone-modifying drugs (e.g., HDAC inhibitors, methyltransferase inhibitors), and/or small molecules (e.g., tyrosine kinase inhibitors) may offer even further differential immune effects when used at lower doses, thus decreasing overall therapeutic toxicities. Such combination therapy may also prevent evolution of BCL-2 resistance within the immune system as well as within the primary malignancy. Combining BH3 mimetics with other immune-directed therapies such as rituximab, checkpoint inhibitors, ibrutinib, calcineurin, and mTOR inhibitors may allow for even greater immunological fine-tuning, as the apoptotic repertoires and BH3 mimetic sensitivities in specific immune subsets are likely to change in response to these drugs. Overall, as greater preclinical and clinical understanding of the effects of BH3 mimetics on the immune system expands, we can expect to usher in new and exciting immunomodulatory capacities of this drug class either when used alone or in conjunction with other treatments.
LL, MN, AH, and JL: all the authors listed have made substantial, direct, and intellectual contribution to the work and approved it for publication. LL and JL drafted the final work and revised it critically for content.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Strasser A, Harris AW, Huang DC, Krammer PH, Cory S. Bcl-2 and Fas/APO-1 regulate distinct pathways to lymphocyte apoptosis. EMBO J (1995) 14(24):6136–47.
2. Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol (2013) 15(1):49–63. doi:10.1038/nrm3722
3. Itoh N, Yonehara S, Ishii A, Yonehara M, Mizushima S, Sameshima M, et al. The polypeptide encoded by the cDNA for human cell surface antigen Fas can mediate apoptosis. Cell (1991) 66(2):233–43. doi:10.1016/0092-8674(91)90614-5
4. Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science (1998) 281(5381):1305–8. doi:10.1126/science.281.5381.1305
5. Fukuhara S, Rowley JD. Chromosome 14 translocations in non-Burkitt lymphomas. Int J Cancer (1978) 22(1):14–21. doi:10.1002/ijc.2910220105
6. Tsujimoto Y, Croce CM. Analysis of the structure, transcripts, and protein products of bcl-2, the gene involved in human follicular lymphoma. Proc Natl Acad Sci U S A (1986) 83(14):5214–8. doi:10.1073/pnas.83.14.5214
7. Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature (1988) 335(6189):440–2. doi:10.1038/335440a0
8. Shamas-Din A, Brahmbhatt H, Leber B, Andrews DW. BH3-only proteins: orchestrators of apoptosis. Biochim Biophys Acta (2011) 1813(4):508–20. doi:10.1016/j.bbamcr.2010.11.024
9. Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell (2005) 17(3):393–403. doi:10.1016/j.molcel.2004.12.030
10. Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR, et al. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell (2005) 17(4):525–35. doi:10.1016/j.molcel.2005.02.003
11. Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, et al. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell (2000) 102(1):43–53. doi:10.1016/S0092-8674(00)00009-X
12. Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell (2000) 102(1):33–42. doi:10.1016/S0092-8674(00)00008-8
13. Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell (1997) 91(4):479–89. doi:10.1016/S0092-8674(00)80434-1
14. Renault TT, Chipuk JE. Getting away with murder: how does the BCL-2 family of proteins kill with immunity? Ann N Y Acad Sci (2013) 1285(1):59–79. doi:10.1111/nyas.12045
15. Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, et al. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol Cell (2000) 6(6):1389–99. doi:10.1016/S1097-2765(00)00136-2
16. Biswas S, Shi Q, Matise L, Cleveland S, Dave U, Zinkel S. A role for proapoptotic Bax and Bak in T-cell differentiation and transformation. Blood (2010) 116(24):5237–46. doi:10.1182/blood-2010-04-279687
17. Strasser A, Whittingham S, Vaux DL, Bath ML, Adams JM, Cory S, et al. Enforced BCL2 expression in B-lymphoid cells prolongs antibody responses and elicits autoimmune disease. Proc Natl Acad Sci U S A (1991) 88(19):8661–5. doi:10.1073/pnas.88.19.8661
18. Strasser A, Harris AW, Cory S. bcl-2 transgene inhibits T cell death and perturbs thymic self-censorship. Cell (1991) 67(5):889–99. doi:10.1016/0092-8674(91)90362-3
19. Veis DJ, Sorenson CM, Shutter JR, Korsmeyer SJ. Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell (1993) 75(2):229–40. doi:10.1016/0092-8674(93)80065-M
20. Rinkenberger JL, Horning S, Klocke B, Roth K, Korsmeyer SJ. Mcl-1 deficiency results in peri-implantation embryonic lethality. Genes Dev (2000) 14(1):23–7. doi:10.1101/gad.14.1.23
21. Opferman JT, Iwasaki H, Ong CC, Suh H, Mizuno S-I, Akashi K, et al. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science (2005) 307(5712):1101–4. doi:10.1126/science.1106114
22. Opferman JT, Letai A, Beard C, Sorcinelli MD, Ong CC, Korsmeyer SJ. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature (2003) 426(6967):671–6. doi:10.1038/nature02067
23. Hildeman D, Jorgensen T, Kappler J, Marrack P. Apoptosis and the homeostatic control of immune responses. Curr Opin Immunol (2007) 19(5):516–21. doi:10.1016/j.coi.2007.05.005
24. Dzhagalov I, Dunkle A, He Y-W. The anti-apoptotic Bcl-2 family member Mcl-1 promotes T lymphocyte survival at multiple stages. J Immunol (2008) 181(1):521–8. doi:10.4049/jimmunol.181.1.521
25. Pierson W, Cauwe B, Policheni A, Schlenner SM, Franckaert D, Berges J, et al. Antiapoptotic Mcl-1 is critical for the survival and niche-filling capacity of Foxp3+ regulatory T cells. Nat Immunol (2013) 14(9):959–65. doi:10.1038/ni.2649
26. Vikstrom I, Carotta S, Lüthje K, Peperzak V, Jost PJ, Glaser S, et al. Mcl-1 is essential for germinal center formation and B cell memory. Science (2010) 330(6007):1095–9. doi:10.1126/science.1191793
27. Peperzak V, Vikstrom I, Walker J, Glaser SP, LePage M, Coquery CM, et al. Mcl-1 is essential for the survival of plasma cells. Nat Immunol (2013) 14(3):290–7. doi:10.1038/ni.2527
28. Ma A, Pena JC, Chang B, Margosian E, Davidson L, Alt FW, et al. Bclx regulates the survival of double-positive thymocytes. Proc Natl Acad Sci U S A (1995) 92(11):4763–7. doi:10.1073/pnas.92.11.4763
29. Ottina E, Grespi F, Tischner D, Soratroi C, Geley S, Ploner A, et al. Targeting antiapoptotic A1/Bfl-1 by in vivo RNAi reveals multiple roles in leukocyte development in mice. Blood (2012) 119(25):6032–42. doi:10.1182/blood-2011-12-399089
30. Hamasaki A, Sendo F, Ishida N, Negishi I, Nakayama K, Hatakeyama S, et al. Accelerated neutrophil apoptosis in mice lacking A1-a, a subtype of the bcl-2-related A1 gene. J Exp Med (1998) 188(11):1985–92. doi:10.1084/jem.188.11.1985
31. Morales AA, Olsson A, Celsing F, Osterborg A, Jondal M, Osorio LM. High expression of bfl-1 contributes to the apoptosis resistant phenotype in B-cell chronic lymphocytic leukemia. Int J Cancer (2005) 113(5):730–7. doi:10.1002/ijc.20614
32. Andre JM, Cimaz R, Ranchin B, Galambrun C, Bertrand Y, Bouvier R, et al. Overexpression of the antiapoptotic gene Bfl-1 in B cells from patients with familial systemic lupus erythematosus. Lupus (2007) 16(2):95–100. doi:10.1177/0961203306075382
33. Cippà PE, Kraus AK, Lindenmeyer MT, Chen J, Guimezanes A, Bardwell PD, et al. Resistance to ABT-737 in activated T lymphocytes: molecular mechanisms and reversibility by inhibition of the calcineurin-NFAT pathway. Cell Death Dis (2012) 3(4):e299. doi:10.1038/cddis.2012.38
34. Bouillet P, Metcalf D, Huang DC, Tarlinton DM, Kay TW, Köntgen F, et al. Proapoptotic Bcl-2 relative bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science (1999) 286(5445):1735–8. doi:10.1126/science.286.5445.1735
35. Herold MJ, Stuchbery R, Merino D, Willson T, Strasser A, Hildeman D, et al. Impact of conditional deletion of the pro-apoptotic BCL-2 family member BIM in mice. Cell Death Dis (2014) 5(10):e1446–7. doi:10.1038/cddis.2014.409
36. Bouillet P, Purton JF, Godfrey DI, Zhang L-C, Coultas L, Puthalakath H, et al. BH3-only Bcl-2 family member Bim is required for apoptosis of autoreactive thymocytes. Nature (2002) 415(6874):922–6. doi:10.1038/415922a
37. Tsukamoto H, Huston GE, Dibble J, Duso DK, Swain SL. Bim dictates naive CD4 T cell lifespan and the development of age-associated functional defects. J Immunol (2010) 185(8):4535–44. doi:10.4049/jimmunol.1001668
38. Erlacher M, Labi V, Manzl C, Böck G, Tzankov A, Häcker G, et al. Puma cooperates with Bim, the rate-limiting BH3-only protein in cell death during lymphocyte development, in apoptosis induction. J Exp Med (2006) 203(13):2939–51. doi:10.1084/jem.20061552
39. Ren D, Cheng EH, Kim H, Wang GX, Bean GR, Takeuchi O, et al. BID, BIM, and PUMA are essential for activation of the BAX- and BAK-dependent cell death program. Science (2010) 330(6009):1390–3. doi:10.1126/science.1190217
40. Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell (1998) 94(4):481–90. doi:10.1016/S0092-8674(00)81589-5
41. Zinkel SS, Ong CC, Ferguson DO, Iwasaki H, Akashi K, Bronson RT, et al. Proapoptotic BID is required for myeloid homeostasis and tumor suppression. Genes Dev (2003) 17(2):229–39. doi:10.1101/gad.1045603
42. Pradhan S, Genebriera J, Denning WL, Felix K, Elmets CA, Timares L. CD4 T cell-induced, bid-dependent apoptosis of cutaneous dendritic cells regulates T cell expansion and immune responses. J Immunol (2006) 177(9):5956–67. doi:10.4049/jimmunol.177.9.5956
43. Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell (2001) 7(3):683–94. doi:10.1016/S1097-2765(01)00214-3
44. Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell (2001) 7(3):673–82. doi:10.1016/S1097-2765(01)00213-1
45. Fischer SF, Belz GT, Strasser A. BH3-only protein Puma contributes to death of antigen-specific T cells during shutdown of an immune response to acute viral infection. Proc Natl Acad Sci U S A (2008) 105(8):3035–40. doi:10.1073/pnas.0706913105
46. Clybouw C, Fischer S, Auffredou MT, Hugues P, Alexia C, Bouillet P, et al. Regulation of memory B-cell survival by the BH3-only protein Puma. Blood (2011) 118(15):4120–8. doi:10.1182/blood-2011-04-347096
47. Garrison SP, Thornton JA, Häcker H, Webby R, Rehg JE, Parganas E, et al. The p53-target gene puma drives neutrophil-mediated protection against lethal bacterial sepsis. Isberg RR, editor. PLoS Pathog (2010) 6(12):e1001240. doi:10.1371/journal.ppat.1001240
48. Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, et al. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science (2000) 288(5468):1053–8. doi:10.1126/science.288.5468.1053
49. Wensveen FM, van Gisbergen KP, Derks IA, Gerlach C, Schumacher TN, van Lier RA, et al. Apoptosis threshold set by Noxa and Mcl-1 after T cell activation regulates competitive selection of high-affinity clones. Immunity (2010) 32(6):754–65. doi:10.1016/j.immuni.2010.06.005
50. Wensveen FM, Klarenbeek PL, van Gisbergen KP, Pascutti MF, Derks IA, van Schaik BD, et al. Pro-apoptotic protein Noxa regulates memory T cell population size and protects against lethal immunopathology. J Immunol (2013) 190(3):1180–91. doi:10.4049/jimmunol.1202304
51. Wensveen FM, Derks IA, van Gisbergen KP, de Bruin AM, Meijers JC, Yigittop H, et al. BH3-only protein Noxa regulates apoptosis in activated B cells and controls high-affinity antibody formation. Blood (2012) 119(6):1440–9. doi:10.1182/blood-2011-09-378877
52. Ranger AM, Zha J, Harada H, Datta SR, Danial NN, Gilmore AP, et al. Bad-deficient mice develop diffuse large B cell lymphoma. Proc Natl Acad Sci U S A (2003) 100(16):9324–9. doi:10.1073/pnas.1533446100
53. Labi V, Erlacher M, Kiessling S, Manzl C, Frenzel A, O’Reilly L, et al. Loss of the BH3-only protein Bmf impairs B cell homeostasis and accelerates gamma irradiation-induced thymic lymphoma development. J Exp Med (2008) 205(3):641–55. doi:10.1084/jem.20071658
54. Placzek WJ, Wei J, Kitada S, Zhai D, Reed JC, Pellecchia M. A survey of the anti-apoptotic Bcl-2 subfamily expression in cancer types provides a platform to predict the efficacy of Bcl-2 antagonists in cancer therapy. Cell Death Dis (2010) 1(5):e40. doi:10.1038/cddis.2010.18
55. Opferman JT. Attacking cancer’s Achilles heel: antagonism of anti-apoptotic BCL-2 family members. FEBS J (2015) 283(14):2661–75. doi:10.1111/febs.13472
56. Reed JC, Cuddy M, Haldar S, Croce C, Nowell P, Makover D, et al. BCL2-mediated tumorigenicity of a human T-lymphoid cell line: synergy with MYC and inhibition by BCL2 antisense. Proc Natl Acad Sci U S A (1990) 87(10):3660–4. doi:10.1073/pnas.87.10.3660
57. Klasa RJ, Bally MB, Ng R, Goldie JH, Gascoyne RD, Wong FM. Eradication of human non-Hodgkin’s lymphoma in SCID mice by BCL-2 antisense oligonucleotides combined with low-dose cyclophosphamide. Clin Cancer Res (2000) 6(6):2492–500.
58. Guinness ME, Kenney JL, Reiss M, Lacy J. Bcl-2 antisense oligodeoxynucleotide therapy of Epstein-Barr virus-associated lymphoproliferative disease in severe combined immunodeficient mice. Cancer Res (2000) 60(19):5354–8.
59. Tauchi T, Sumi M, Nakajima A, Sashida G, Shimamoto T, Ohyashiki K. BCL-2 antisense oligonucleotide genasense is active against imatinib-resistant BCR-ABL-positive cells. Clin Cancer Res (2003) 9(11):4267–73.
60. Tolcher AW, Chi K, Kuhn J, Gleave M, Patnaik A, Takimoto C, et al. A phase II, pharmacokinetic, and biological correlative study of oblimersen sodium and docetaxel in patients with hormone-refractory prostate cancer. Clin Cancer Res (2005) 11(10):3854–61. doi:10.1158/1078-0432.CCR-04-2145
61. O’Brien SM, Cunningham CC, Golenkov AK, Turkina AG, Novick SC, Rai KR. Phase I to II multicenter study of oblimersen sodium, a Bcl-2 antisense oligonucleotide, in patients with advanced chronic lymphocytic leukemia. J Clin Oncol (2005) 23(30):7697–702. doi:10.1200/JCO.2005.02.4364
62. Pro B, Leber B, Smith M, Fayad L, Romaguera J, Hagemeister F, et al. Phase II multicenter study of oblimersen sodium, a Bcl-2 antisense oligonucleotide, in combination with rituximab in patients with recurrent B-cell non-Hodgkin lymphoma. Br J Haematol (2008) 143(3):355–60. doi:10.1111/j.1365-2141.2008.07353.x
63. Chanan-Khan AA, Niesvizky R, Hohl RJ, Zimmerman TM, Christiansen NP, Schiller GJ, et al. Phase III randomised study of dexamethasone with or without oblimersen sodium for patients with advanced multiple myeloma. Leuk Lymphoma (2009) 50(4):559–65. doi:10.1080/10428190902748971
64. Bedikian AY, Bedikian AY, Millward M, Millward M, Pehamberger H, Pehamberger H, et al. Bcl-2 antisense (oblimersen sodium) plus dacarbazine in patients with advanced melanoma: the Oblimersen Melanoma Study Group. J Clin Oncol (2006) 24(29):4738–45. doi:10.1200/JCO.2006.06.0483
65. Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature (2005) 435(7042):677–81. doi:10.1038/nature03579
66. Chauhan D, Velankar M, Brahmandam M, Hideshima T, Podar K, Richardson P, et al. A novel Bcl-2/Bcl-X(L)/Bcl-w inhibitor ABT-737 as therapy in multiple myeloma. Oncogene (2007) 26(16):2374–80. doi:10.1038/sj.onc.1210028
67. Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell (2006) 10(5):375–88. doi:10.1016/j.ccr.2006.10.006
68. Kline MP, Rajkumar SV, Timm MM, Kimlinger TK, Haug JL, Lust JA, et al. ABT-737, an inhibitor of Bcl-2 family proteins, is a potent inducer of apoptosis in multiple myeloma cells. Leukemia (2007) 21(7):1549–60. doi:10.1038/sj.leu.2404719
69. Del Gaizo Moore V, Schlis KD, Sallan SE, Armstrong SA, Letai A. BCL-2 dependence and ABT-737 sensitivity in acute lymphoblastic leukemia. Blood (2008) 111(4):2300–9. doi:10.1182/blood-2007-06-098012
70. Mason KD, Vandenberg CJ, Scott CL, Wei AH, Cory S, Huang DCS, et al. In vivo efficacy of the Bcl-2 antagonist ABT-737 against aggressive Myc-driven lymphomas. Proc Natl Acad Sci U S A (2008) 105(46):17961–6. doi:10.1073/pnas.0809957105
71. Ishitsuka K, Kunami N, Katsuya H, Nogami R, Ishikawa C, Yotsumoto F, et al. Targeting Bcl-2 family proteins in adult T-cell leukemia/lymphoma: in vitro and in vivo effects of the novel Bcl-2 family inhibitor ABT-737. Cancer Lett (2012) 317(2):218–25. doi:10.1016/j.canlet.2011.11.030
72. High LM, Szymanska B, Wilczynska-Kalak U, Barber N, O’Brien R, Khaw SL, et al. The Bcl-2 homology domain 3 mimetic ABT-737 targets the apoptotic machinery in acute lymphoblastic leukemia resulting in synergistic in vitro and in vivo interactions with established drugs. Mol Pharmacol (2010) 77(3):483–94. doi:10.1124/mol.109.060780
73. Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res (2008) 68(9):3421–8. doi:10.1158/0008-5472.CAN-07-5836
74. Wilson WH, O’Connor OA, Czuczman MS, LaCasce AS, Gerecitano JF, Leonard JP, et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: a phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol (2010) 11(12):1149–59. doi:10.1016/S1470-2045(10)70261-8
75. Kodama T, Takehara T, Hikita H, Shimizu S, Li W, Miyagi T, et al. Thrombocytopenia exacerbates cholestasis-induced liver fibrosis in mice. Gastroenterology (2010) 138(7):.e1–7. doi:10.1053/j.gastro.2010.02.054
76. van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell (2006) 10(5):389–99. doi:10.1016/j.ccr.2006.08.027
77. Yecies D, Carlson NE, Deng J, Letai A. Acquired resistance to ABT-737 in lymphoma cells that up-regulate MCL-1 and BFL-1. Blood (2010) 115(16):3304–13. doi:10.1182/blood-2009-07-233304
78. Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med (2013) 19(2):202–8. doi:10.1038/nm.3048
79. Vogler M, Dinsdale D, Dyer MJS, Cohen GM. ABT-199 selectively inhibits BCL2 but not BCL2L1 and efficiently induces apoptosis of chronic lymphocytic leukaemic cells but not platelets. Br J Haematol (2013) 163(1):139–42. doi:10.1111/bjh.12457
80. Pan R, Hogdal LJ, Benito JM, Bucci D, Han L, Borthakur G, et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov (2014) 4(3):362–75. doi:10.1158/2159-8290.CD-13-0609
81. Peirs S, Matthijssens F, Goossens S, Van de Walle I, Ruggero K, de Bock CE, et al. ABT-199 mediated inhibition of BCL-2 as a novel therapeutic strategy in T-cell acute lymphoblastic leukemia. Blood (2014) 124(25):3738–47. doi:10.1182/blood-2014-05-574566
82. Green DR. A BH3 mimetic for killing cancer cells. Cell (2016) 165(7):1560. doi:10.1016/j.cell.2016.05.080
83. Khaw SL, Merino D, Anderson MA, Glaser SP, Bouillet P, Roberts AW, et al. Both leukaemic and normal peripheral B lymphoid cells are highly sensitive to the selective pharmacological inhibition of prosurvival Bcl-2 with ABT-199. Leukemia (2014) 28(6):1207–15. doi:10.1038/leu.2014.1
84. Shore GC, Viallet J. Modulating the bcl-2 family of apoptosis suppressors for potential therapeutic benefit in cancer. Hematology Am Soc Hematol Educ Program (2005) 2005(1):226–30. doi:10.1182/asheducation-2005.1.226
85. Nguyen M, Marcellus RC, Roulston A, Watson M, Serfass L, Murthy Madiraju SR, et al. Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc Natl Acad Sci U S A (2007) 104(49):19512–7. doi:10.1073/pnas.0709443104
86. Trudel S, Li ZH, Rauw J, Tiedemann RE, Wen XY, Stewart AK. Preclinical studies of the pan-Bcl inhibitor obatoclax (GX015-070) in multiple myeloma. Blood (2007) 109(12):5430–8. doi:10.1182/blood-2006-10-047951
87. Konopleva M, Watt J, Contractor R, Tsao T, Harris D, Estrov Z, et al. Mechanisms of antileukemic activity of the novel Bcl-2 homology domain-3 mimetic GX15-070 (obatoclax). Cancer Res (2008) 68(9):3413–20. doi:10.1158/0008-5472.CAN-07-1919
88. Pérez-Galán P, Roué G, López-Guerra M, Nguyen M, Villamor N, Montserrat E, et al. BCL-2 phosphorylation modulates sensitivity to the BH3 mimetic GX15-070 (Obatoclax) and reduces its synergistic interaction with bortezomib in chronic lymphocytic leukemia cells. Leukemia (2008) 22(9):1712–20. doi:10.1038/leu.2008.175
89. O’Brien SM, Claxton DF, Crump M, Faderl S, Kipps T, Keating MJ, et al. Phase I study of obatoclax mesylate (GX15-070), a small molecule pan-Bcl-2 family antagonist, in patients with advanced chronic lymphocytic leukemia. Blood (2009) 113(2):299–305. doi:10.1182/blood-2008-02-137943
90. Schimmer AD, O’Brien S, Kantarjian H, Brandwein J, Cheson BD, Minden MD, et al. A phase I study of the pan bcl-2 family inhibitor obatoclax mesylate in patients with advanced hematologic malignancies. Clin Cancer Res (2008) 14(24):8295–301. doi:10.1158/1078-0432.CCR-08-0999
91. Parikh SA, Kantarjian H, Schimmer A, Walsh W, Asatiani E, El-Shami K, et al. Phase II study of obatoclax mesylate (GX15-070), a small-molecule BCL-2 family antagonist, for patients with myelofibrosis. Clin Lymphoma Myeloma Leuk (2010) 10(4):285–9. doi:10.3816/CLML.2010.n.059
92. Goy A, Hernandez-Ilzaliturri FJ, Kahl B, Ford P, Protomastro E, Berger M. A phase I/II study of the pan Bcl-2 inhibitor obatoclax mesylate plus bortezomib for relapsed or refractory mantle cell lymphoma. Leuk Lymphoma (2014) 55(12):2761–8. doi:10.3109/10428194.2014.907891
93. Schimmer AD, Raza A, Carter TH, Claxton D, Erba H, DeAngelo DJ, et al. A multicenter phase I/II study of obatoclax mesylate administered as a 3- or 24-hour infusion in older patients with previously untreated acute myeloid leukemia. PLoS One (2014) 9(10):e108694. doi:10.1371/journal.pone.0108694
94. Urtishak KA, Edwards AYZ, Wang L-S, Hudome A, Robinson BW, Barrett JS, et al. Potent obatoclax cytotoxicity and activation of triple death mode killing across infant acute lymphoblastic leukemia. Blood (2013) 121(14):2689–703. doi:10.1182/blood-2012-04-425033
95. Zhang M, Liu H, Guo R, Ling Y, Wu X, Li B, et al. Molecular mechanism of gossypol-induced cell growth inhibition and cell death of HT-29 human colon carcinoma cells. Biochem Pharmacol (2003) 66(1):93–103. doi:10.1016/S0006-2952(03)00248-X
96. Etxebarria A, Landeta O, Antonsson B, Basañez G. Regulation of antiapoptotic MCL-1 function by gossypol: mechanistic insights from in vitro reconstituted systems. Biochem Pharmacol (2008) 76(11):1563–76. doi:10.1016/j.bcp.2008.08.003
97. Lei X, Chen Y, Du G, Yu W, Wang X, Qu H, et al. Gossypol induces Bax/Bak-independent activation of apoptosis and cytochrome c release via a conformational change in Bcl-2. FASEB J (2006) 20(12):2147–9. doi:10.1096/fj.05-5665fje
98. Mohammad RM, Wang S, Aboukameel A, Chen B, Wu X, Chen J, et al. Preclinical studies of a nonpeptidic small-molecule inhibitor of Bcl-2 and Bcl-X(L) [(-)-gossypol] against diffuse large cell lymphoma. Mol Cancer Ther (2005) 4(1):13–21. doi:10.1186/1476-4598-4-13
99. Kline MP, Rajkumar SV, Timm MM, Kimlinger TK, Haug JL, Lust JA, et al. R-(-)-gossypol (AT-101) activates programmed cell death in multiple myeloma cells. Exp Hematol (2008) 36(5):568–76. doi:10.1016/j.exphem.2008.01.003
100. Balakrishnan K, Wierda WG, Keating MJ, Gandhi V. Gossypol, a BH3 mimetic, induces apoptosis in chronic lymphocytic leukemia cells. Blood (2008) 112(5):1971–80. doi:10.1182/blood-2007-12-126946
101. Baggstrom MQ, Qi Y, Koczywas M, Argiris A, Johnson EA, Millward MJ, et al. A phase II study of AT-101 (Gossypol) in chemotherapy-sensitive recurrent extensive-stage small cell lung cancer. J Thorac Oncol (2011) 6(10):1757–60. doi:10.1097/JTO.0b013e31822e2941
102. Becattini B, Kitada S, Leone M, Monosov E, Chandler S, Zhai D, et al. Rational design and real time, in-cell detection of the proapoptotic activity of a novel compound targeting Bcl-X(L). Chem Biol (2004) 11(3):389–95. doi:10.1016/j.chembiol.2004.02.020
103. Dash R, Azab B, Quinn BA, Shen X, Wang X-Y, Das SK, et al. Apogossypol derivative BI-97C1 (Sabutoclax) targeting Mcl-1 sensitizes prostate cancer cells to mda-7/IL-24-mediated toxicity. Proc Natl Acad Sci U S A (2011) 108(21):8785–90. doi:10.1073/pnas.1100769108
104. Barba-Barajas M, Hernández-Flores G, Lerma-Díaz JM, Ortiz-Lazareno PC, Domínguez-Rodríguez JR, Barba-Barajas L, et al. Gossypol induced apoptosis of polymorphonuclear leukocytes and monocytes: involvement of mitochondrial pathway and reactive oxygen species. Immunopharmacol Immunotoxicol (2009) 31(2):320–30. doi:10.1080/08923970902718049
105. Lessene G, Czabotar PE, Sleebs BE, Zobel K, Lowes KN, Adams JM, et al. Structure-guided design of a selective BCL-X(L) inhibitor. Nat Chem Biol (2013) 9(6):390–7. doi:10.1038/nchembio.1246
106. Park D, Magis AT, Li R, Owonikoko TK, Sica GL, Sun S-Y, et al. Novel small-molecule inhibitors of Bcl-XL to treat lung cancer. Cancer Res (2013) 73(17):5485–96. doi:10.1158/0008-5472.CAN-12-2272
107. Tao Z-F, Hasvold L, Wang L, Wang X, Petros AM, Park CH, et al. Discovery of a potent and selective BCL-X L inhibitor with in vivo activity. ACS Med Chem Lett (2014) 5(10):1088–93. doi:10.1021/ml5001867
108. Wang G, Nikolovska-Coleska Z, Yang C-Y, Wang R, Tang G, Guo J, et al. Structure-based design of potent small-molecule inhibitors of anti-apoptotic Bcl-2 proteins. J Med Chem (2006) 49(21):6139–42. doi:10.1021/jm060460o
109. Cohen NA, Stewart ML, Gavathiotis E, Tepper JL, Bruekner SR, Koss B, et al. A competitive stapled peptide screen identifies a selective small molecule that overcomes MCL-1-dependent leukemia cell survival. Chem Biol (2012) 19(9):1175–86. doi:10.1016/j.chembiol.2012.07.018
110. Leverson JD, Zhang H, Chen J, Tahir SK, Phillips DC, Xue J, et al. Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax). Cell Death Dis (2015) 6(1):e1590. doi:10.1038/cddis.2014.561
111. Doi K, Li R, Sung S-S, Wu H, Liu Y, Manieri W, et al. Discovery of marinopyrrole A (maritoclax) as a selective Mcl-1 antagonist that overcomes ABT-737 resistance by binding to and targeting Mcl-1 for proteasomal degradation. J Biol Chem (2012) 287(13):10224–35. doi:10.1074/jbc.M111.334532
112. Abulwerdi FA, Liao C, Mady AS, Gavin J, Shen C, Cierpicki T, et al. 3-Substituted-N-(4-hydroxynaphthalen-1-yl)arylsulfonamides as a novel class of selective Mcl-1 inhibitors: structure-based design, synthesis, SAR, and biological evaluation. J Med Chem (2014) 57(10):4111–33. doi:10.1021/jm500010b
113. Pelz NF, Bian Z, Zhao B, Shaw S, Tarr JC, Belmar J, et al. Discovery of 2-indole-acylsulfonamide myeloid cell leukemia 1 (Mcl-1) inhibitors using fragment-based methods. J Med Chem (2016) 59(5):2054–66. doi:10.1021/acs.jmedchem.5b01660
114. Arnold AA, Aboukameel A, Chen J, Yang D, Wang S, Al-Katib A, et al. Preclinical studies of apogossypolone: a new nonpeptidic pan small-molecule inhibitor of Bcl-2, Bcl-XL and Mcl-1 proteins in follicular small cleaved cell lymphoma model. Mol Cancer (2008) 7(1):20. doi:10.1186/1476-4598-7-20
115. Wei J, Stebbins JL, Kitada S, Dash R, Zhai D, Placzek WJ, et al. An optically pure apogossypolone derivative as potent pan-active inhibitor of anti-apoptotic bcl-2 family proteins. Front Oncol (2011) 1:28. doi:10.3389/fonc.2011.00028
116. Wei J, Stebbins JL, Kitada S, Dash R, Placzek W, Rega MF, et al. BI-97C1, an optically pure Apogossypol derivative as pan-active inhibitor of antiapoptotic B-cell lymphoma/leukemia-2 (Bcl-2) family proteins. J Med Chem (2010) 53(10):4166–76. doi:10.1021/jm1001265
117. Bai L, Chen J, McEachern D, Liu L, Zhou H, Aguilar A, et al. BM-1197: a novel and specific Bcl-2/Bcl-xL inhibitor inducing complete and long-lasting tumor regression in vivo. PLoS One (2014) 9(6):e99404. doi:10.1371/journal.pone.0099404
118. Zhang Z, Song T, Zhang T, Gao J, Wu G, An L, et al. A novel BH3 mimetic S1 potently induces Bax/Bak-dependent apoptosis by targeting both Bcl-2 and Mcl-1. Int J Cancer (2011) 128(7):1724–35. doi:10.1002/ijc.25484
119. Kazi A, Sun J, Doi K, Sung S-S, Takahashi Y, Yin H, et al. The BH3 alpha-helical mimic BH3-M6 disrupts Bcl-X(L), Bcl-2, and MCL-1 protein-protein interactions with Bax, Bak, Bad, or Bim and induces apoptosis in a Bax- and Bim-dependent manner. J Biol Chem (2011) 286(11):9382–92. doi:10.1074/jbc.M110.203638
120. Cao X, Yap JL, Newell-Rogers M, Newell-Rogers MK, Peddaboina C, Jiang W, et al. The novel BH3 α-helix mimetic JY-1-106 induces apoptosis in a subset of cancer cells (lung cancer, colon cancer and mesothelioma) by disrupting Bcl-xL and Mcl-1 protein-protein interactions with Bak. Mol Cancer (2013) 12(1):42. doi:10.1186/1476-4598-12-42
121. Gavathiotis E, Reyna DE, Bellairs JA, Leshchiner ES, Walensky LD. Direct and selective small-molecule activation of proapoptotic BAX. Nat Chem Biol (2012) 8(7):639–45. doi:10.1038/nchembio.995
122. Ponassi R, Biasotti B, Tomati V, Bruno S, Poggi A, Malacarne D, et al. A novel Bim-BH3-derived Bcl-XL inhibitor: biochemical characterization, in vitro, in vivo and ex-vivo anti-leukemic activity. Cell Cycle (2014) 7(20):3211–24. doi:10.4161/cc.7.20.6830
123. Dutta S, Ryan J, Chen TS, Kougentakis C, Letai A, Keating AE. Potent and specific peptide inhibitors of human pro-survival protein Bcl-xL. J Mol Biol (2015) 427(6 Pt B):1241–53. doi:10.1016/j.jmb.2014.09.030
124. Muppidi A, Doi K, Edwardraja S, Drake EJ, Gulick AM, Wang H-G, et al. Rational design of proteolytically stable, cell-permeable peptide-based selective Mcl-1 inhibitors. J Am Chem Soc (2012) 134(36):14734–7. doi:10.1021/ja306864v
125. Stewart ML, Fire E, Keating AE, Walensky LD. The MCL-1 BH3 helix is an exclusive MCL-1 inhibitor and apoptosis sensitizer. Nat Chem Biol (2010) 6(8):595–601. doi:10.1038/nchembio.391
126. Huhn AJ, Guerra RM, Harvey EP, Bird GH, Walensky LD. Selective covalent targeting of anti-apoptotic BFL-1 by cysteine-reactive stapled peptide inhibitors. Cell Chem Biol (2016) 23(9):1123–34. doi:10.1016/j.chembiol.2016.07.022
127. LaBelle JL, Katz SG, Bird GH, Gavathiotis E, Stewart ML, Lawrence C, et al. A stapled BIM peptide overcomes apoptotic resistance in hematologic cancers. J Clin Invest (2012) 122(6):2018–31. doi:10.1172/JCI46231
128. Edwards AL, Gavathiotis E, LaBelle JL, Braun CR, Opoku-Nsiah KA, Bird GH, et al. Multimodal interaction with BCL-2 family proteins underlies the proapoptotic activity of PUMA BH3. Chem Biol (2013) 20(7):888–902. doi:10.1016/j.chembiol.2013.06.007
129. Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, et al. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science (2004) 305(5689):1466–70. doi:10.1126/science.1099191
130. Bardwell PD, Gu J, McCarthy D, Wallace C, Bryant S, Goess C, et al. The Bcl-2 family antagonist ABT-737 significantly inhibits multiple animal models of autoimmunity. J Immunol (2009) 182(12):7482–9. doi:10.4049/jimmunol.0802813
131. Lawlor KE, Smith SD, van Nieuwenhuijze A, Huang DCS, Wicks IP. Evaluation of the Bcl-2 family antagonist ABT-737 in collagen-induced arthritis. J Leukoc Biol (2011) 90(4):819–29. doi:10.1189/jlb.0311174
132. Carrington EM, Vikstrom IB, Light A, Sutherland RM, Londrigan SL, Mason KD, et al. BH3 mimetics antagonizing restricted prosurvival Bcl-2 proteins represent another class of selective immune modulatory drugs. Proc Natl Acad Sci U S A (2010) 107(24):10967–71. doi:10.1073/pnas.1005256107
133. Cippà PE, Kraus AK, Edenhofer I, Segerer S, Chen J, Hausmann M, et al. The BH3-mimetic ABT-737 inhibits allogeneic immune responses. Transpl Int (2011) 24(7):722–32. doi:10.1111/j.1432-2277.2011.01272.x
134. Gabriel SS, Bon N, Chen J, Wekerle T, Bushell A, Fehr T, et al. Distinctive expression of Bcl-2 factors in regulatory T cells determines a pharmacological target to induce immunological tolerance. Front Immunol (2016) 7(1):73. doi:10.3389/fimmu.2016.00073
135. Lutz C, Mozaffari M, Tosevski V, Caj M, Cippà P, McRae BL, et al. Increased lymphocyte apoptosis in mouse models of colitis upon ABT-737 treatment is dependent upon BIM expression. Clin Exp Immunol (2015) 181(2):343–56. doi:10.1111/cei.12635
136. Metz M, Maurer M. Mast cells – key effector cells in immune responses. Trends Immunol (2007) 28(5):234–41. doi:10.1016/j.it.2007.03.003
137. Karlberg M, Ekoff M, Huang DCS, Mustonen P, Harvima IT, Nilsson G. The BH3-mimetic ABT-737 induces mast cell apoptosis in vitro and in vivo: potential for therapeutics. J Immunol (2010) 185(4):2555–62. doi:10.4049/jimmunol.0903656
138. Kim PS, Jochems C, Grenga I, Donahue RN, Tsang KY, Gulley JL, et al. Pan-Bcl-2 inhibitor, GX15-070 (obatoclax), decreases human T regulatory lymphocytes while preserving effector T lymphocytes: a rationale for its use in combination immunotherapy. J Immunol (2014) 192(6):2622–33. doi:10.4049/jimmunol.1301369
139. Farsaci B, Sabzevari H, Higgins JP, Di Bari MG, Takai S, Schlom J, et al. Effect of a small molecule BCL-2 inhibitor on immune function and use with a recombinant vaccine. Int J Cancer (2010) 127(7):1603–13. doi:10.1002/ijc.25177
140. Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF, et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med (2016) 374(4):311–22. doi:10.1056/NEJMoa1513257
141. Roberts AW, Seymour JF, Brown JR, Wierda WG, Kipps TJ, Khaw SL, et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. J Clin Oncol (2012) 30(5):488–96. doi:10.1200/JCO.2011.34.7898
142. Roberts AW, Advani RH, Kahl BS, Persky D, Sweetenham JW, Carney DA, et al. Phase 1 study of the safety, pharmacokinetics, and antitumour activity of the BCL2 inhibitor navitoclax in combination with rituximab in patients with relapsed or refractory CD20+ lymphoid malignancies. Br J Haematol (2015) 170(5):669–78. doi:10.1111/bjh.13487
143. Swiecicki PL, Bellile E, Sacco AG, Pearson AT, Taylor JMG, Jackson TL, et al. A phase II trial of the BCL-2 homolog domain 3 mimetic AT-101 in combination with docetaxel for recurrent, locally advanced, or metastatic head and neck cancer. Invest New Drugs (2016) 34(4):481–9. doi:10.1007/s10637-016-0364-5
144. Rheingold SR, Hogarty MD, Blaney SM, Zwiebel JA, Sauk-Schubert C, Chandula R, et al. Phase I trial of G3139, a bcl-2 antisense oligonucleotide, combined with doxorubicin and cyclophosphamide in children with relapsed solid tumors: a Children’s Oncology Group Study. J Clin Oncol (2007) 25(12):1512–8. doi:10.1200/JCO.2006.09.5125
145. Jansen B, Wacheck V, Heere-Ress E, Schlagbauer-Wadl H, Hoeller C, Lucas T, et al. Chemosensitisation of malignant melanoma by BCL2 antisense therapy. Lancet (2000) 356(9243):1728–33. doi:10.1016/S0140-6736(00)03207-4
146. Tolcher AW, Rodrigueza WV, Rasco DW, Patnaik A, Papadopoulos KP, Amaya A, et al. A phase 1 study of the BCL2-targeted deoxyribonucleic acid inhibitor (DNAi) PNT2258 in patients with advanced solid tumors. Cancer Chemother Pharmacol (2014) 73(2):363–71. doi:10.1007/s00280-013-2361-0
147. Lu P, Fleischmann R, Curtis C, Ignatenko S, Desai M, Wong SL, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of the BCL-2 inhibitor venetoclax (ABT-199) in a phase 1 single and multiple ascending dose study in female patients with systemic lupus erythematosus. ACR ARHP Annual Meeting. San Francisco, CA (2015).
148. Karlsson H, Karlsson SCH, Lindqvist AC, Fransson M, Paul-Wetterberg G, Nilsson B, et al. Combining CAR T cells and the Bcl-2 family apoptosis inhibitor ABT-737 for treating B-cell malignancy. Cancer Gene Ther (2013) 20(7):386–93. doi:10.1038/cgt.2013.35
149. Ryan J, Letai A. BH3 profiling in whole cells by fluorimeter or FACS. Methods (2013) 61(2):156–64. doi:10.1016/j.ymeth.2013.04.006
150. Deng J, Carlson N, Takeyama K, Dal Cin P, Shipp M, Letai A. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell (2007) 12(2):171–85. doi:10.1016/j.ccr.2007.07.001
151. Xiao Y, Nimmer P, Sheppard GS, Bruncko M, Hessler P, Lu X, et al. MCL-1 is a key determinant of breast cancer cell survival: validation of MCL-1 dependency utilizing a highly selective small molecule inhibitor. Mol Cancer Ther (2015) 14(8):1837–47. doi:10.1158/1535-7163.MCT-14-0928
Keywords: BCL-2, lymphocytes, BH3 mimetic, apoptosis, cell death, immune system, small molecules, immunotherapy
Citation: Ludwig LM, Nassin ML, Hadji A and LaBelle JL (2016) Killing Two Cells with One Stone: Pharmacologic BCL-2 Family Targeting for Cancer Cell Death and Immune Modulation. Front. Pediatr. 4:135. doi: 10.3389/fped.2016.00135
Received: 11 October 2016; Accepted: 30 November 2016;
Published: 21 December 2016
Edited by:
Peter Michael Gordon, University of Minnesota, USAReviewed by:
Christopher C. Porter, University of Colorado Denver School of Medicine, USACopyright: © 2016 Ludwig, Nassin, Hadji and LaBelle. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James L. LaBelle, amxhYmVsbGVAcGVkcy5ic2QudWNoaWNhZ28uZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.