 Rahul Chanchlani1,2,3
Rahul Chanchlani1,2,3 Rulan S. Parekh1,2,4*
Rulan S. Parekh1,2,4*
- 1Division of Pediatric Nephrology, Hospital for Sick Children, Toronto, ON, Canada
- 2Child Health Evaluative Sciences, Research Institute, Hospital for Sick Children, Toronto, ON, Canada
- 3Division of Pediatric Nephrology, McMaster Children’s Hospital, Hamilton, ON, Canada
- 4Department of Medicine, Division of Nephrology, University Health Network, Toronto, ON, Canada
Nephrotic syndrome is a common glomerular disease in children with significant variability in both incidence and steroid responsiveness among various ethnic groups. The average incidence of nephrotic syndrome is 2–16.9 per 100,000 children worldwide. Understanding the variability by ethnicity may point to potential factors leading to nephrotic syndrome, which remains elusive, and may highlight factors accounting for differences in medication response. The emerging role of genetic factors associated with steroid responsive and steroid-resistant forms of nephrotic syndrome within an ethnic group can provide insight into potential biological mechanisms leading to disease. For example, among African-Americans, the risk variants in APOL1 are associated with a more than 10-fold increase in risk of focal segmental glomerulosclerosis and high-risk carriers have a twofold greater risk of progression to end-stage renal disease. Ongoing collaborative studies should consider capturing data on self-reported ethnicity to understand differences in incidence and outcomes. In the future, the availability of whole-genome data will provide an excellent opportunity for new clinical and translational research in childhood nephrotic syndrome and lead to a better understanding of the disease.
Introduction
Nephrotic syndrome is a common childhood kidney disease characterized by a constellation of heavy proteinuria (urine protein to creatinine ratio >200 mg/mmol or ≥3 + proteinuria on urine dipstick) resulting in hypoalbuminemia (<25 g/L), hyperlipidemia, and peripheral edema (1). Despite treating nephrotic syndrome with prednisone for over 50 years, the exact mechanism of disease remains unclear. Most studies focus on podocyte injury (2), immunological (3), and environmental factors (4) as potential culprits in the pathogenesis of nephrotic syndrome, but causal factors remain elusive.
Corticosteroids remain the mainstay for treatment of nephrotic syndrome. Based on the response to corticosteroids, children with nephrotic syndrome segregate into a steroid-sensitive group that has a good long-term prognosis, but risk of frequent relapses, and a steroid-resistant group with higher risk of developing chronic kidney disease. Response to medications is quite variable with some children requiring further courses of steroid-sparing agents, while others achieve complete remission after the first course of prednisone. Steroid-sparing agents, such as cyclophosphamide, mycophenolate mofetil, calcineurin inhibitors, and rituximab, are often used to induce or maintain remission with mixed results. It is unclear what leads to this individual variability in drug response and the future risk of relapses.
Minimal change disease (MCD) is the most common histological variant of nephrotic syndrome and accounts for approximately 80% of cases in children based on historical data from 1967 to 1976 (5). Focal segmental glomerulosclerosis (FSGS) is less common but can be progressive with poor long-term outcomes. Data suggest differences in the incidence of FSGS among various ethnic groups, for example, a study involving 86 children from Kansas city, MO, USA, reported the annual incidence of FSGS of 1.6 per 100,000 in African-Americans compared to 0.3 per 100,000 in European Americans (6).
With recent advancements and improved understanding of the human genome, 45 genes are found to be associated with familial and sporadic nephrotic syndrome that primarily affect podocyte structure and function (7). Most recently identified, the gene APOL1 is a major risk factor for FSGS among African-Americans accounting for progressive kidney disease. The influence of genetic factors among other ethnic groups is less clear.
This review focuses on the current understanding of ethnic differences in childhood nephrotic syndrome, discusses ongoing studies and role of ethnicity, and the influence of genetics within specific ethnic groups.
Ethnic Differences in Nephrotic Syndrome
Based on our review of the literature, the average incidence of nephrotic syndrome is 4.7 (range 1.15–16.9) per 100,000 persons in studies reported from 1946 to 2014, and the proportion with steroid resistance is 12.4% (range 2.1–27.3%) from 1986 to 2014 (Figures 1A,B). Hence, there is a considerable variation in disease burden by country of origin and steroid responsiveness, suggesting the potential role of ethnicity in susceptibility to disease (8–23).
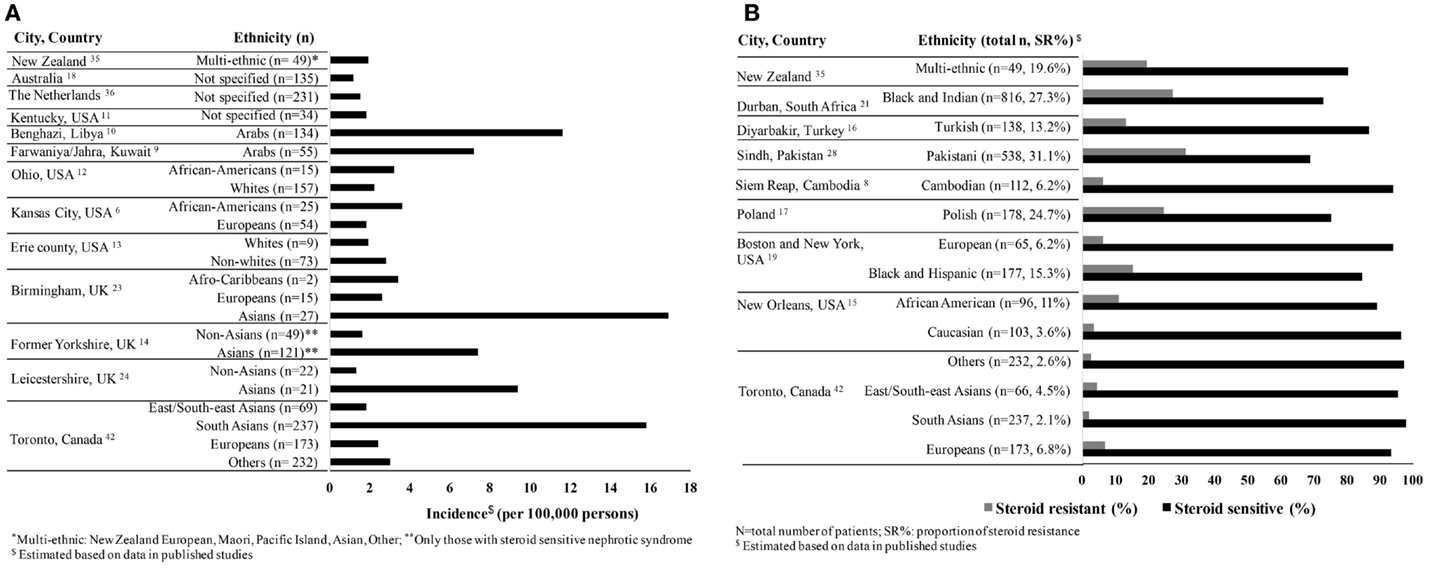
Figure 1. (A) Incidence of childhood nephrotic syndrome per 100,000 persons by ethnicity reported from 1946 to 2014. (B) Variability of steroid responsiveness by ethnicity among children with nephrotic syndrome in reported studies from 1986 to 2014.
In studies, where there are large diaspora populations, such as the United Kingdom, South Asians are reported to have a higher incidence of nephrotic syndrome ranging from 7.4 to 16.9 per 100,000 persons compared to Europeans (14, 24, 25). Studies from the US report a higher estimated incidence among children of African compared to European descent (Figure 1A) (6, 13). The proportion of steroid resistance also varies by ethnicity from 20% among Europeans, 16–27% among Africans, 27–54% among Asians, and 20–39% among South Asians (Figure 1B) (26–29). It is possible that these differences are at least partially attributable to variations in clinical management, selection bias, and definitions of outcomes. African-American children are more likely to have biopsy-proven FSGS (42–72%) with worse outcomes as compared to European children and commonly progress to end-stage renal disease (ESRD) (19, 26, 27, 30–32). Comparatively, the proportion of FSGS in India ranges from 15.3 to 39.1% (33, 34). All these reports have differing definitions, inconsistent inclusion, and selection criteria at tertiary centers and variable access to health care, which impact comparison of incidence rates and outcomes such as steroid responsiveness. In addition, most studies combine both steroid resistant and FSGS as a single category, thereby leading to further heterogeneity in study populations and impacting outcomes. On the other hand, registries from New Zealand and Netherlands did not show differences in disease burden by ethnicity primarily due to lack of power or reported ethnic diversity (35, 36). These registries or case series have short follow-up and limited clinical information; hence, differences in treatment response by ethnicity are not well reported. To address these limitations, a number of ongoing registries and cohort studies conducted worldwide deserve mention. These studies include children with various types of nephrotic syndrome with the overall goal to identify clinical, histological and genomic predictors of nephrotic syndrome. The highlights of each study are shown in Table 1.
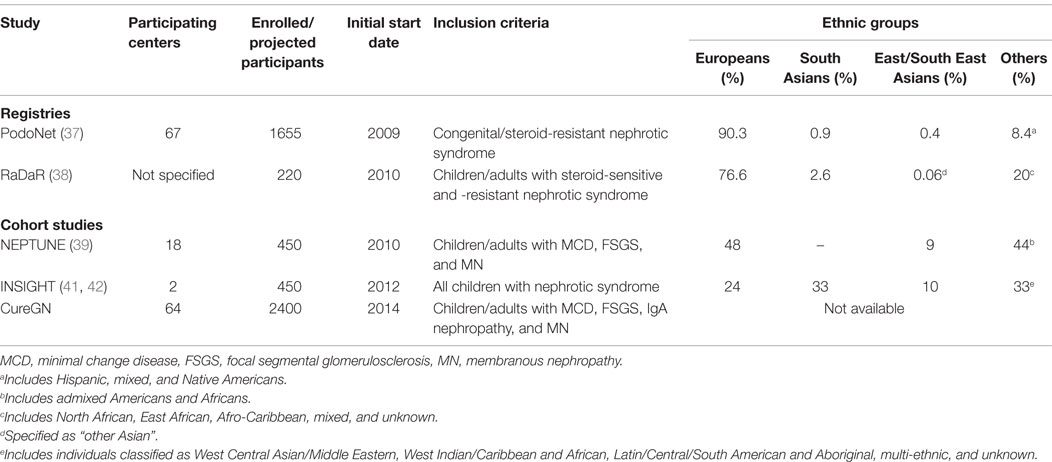
Table 1. Distribution of ethnic groups in ongoing glomerular disease registries and prospective studies.
PodoNet is an international registry that includes children with steroid-resistant disease and congenital nephrotic syndrome recruited predominantly from European countries having 90.3% participants of European descent (37). Registry for Rare Kidney Diseases (RaDaR), recently renamed the National Study for Nephrotic Syndrome (NephroS) study, is a web-based UK registry that aims to capture clinical information on all children and adults with nephrotic syndrome and studies to date have focused on genetic testing of steroid-resistant disease primarily involving Europeans (76.6%) (38).
Nephrotic Syndrome Study Network (NEPTUNE) and the Cure Glomerulonephropathy Network (CureGN) are both North American multicenter collaborative longitudinal cohort studies, which enroll both children and adults with biopsy-proven disease (MCD and FSGS). NEPTUNE recruits an incident cohort at the time of first biopsy and follows them closely for up to 5 years (39). It comprises a diverse ethnic cohort based on genetically derived ancestry. CureGN is a new multicenter study enrolling up to 2400 children and adults with incident or prevalent MCD and FSGS. All of these studies to date have limited enrollment of either South Asians or East/Southeast Asians as many may not receive a biopsy or fulfill entry criteria, thus limiting their generalizability to other patient populations.
Canadian Childhood Nephrotic Syndrome (CHILDNEPH) is enrolling children across Canada to study treatment strategies in nephrotic syndrome and system factors driving treatment variation (40). Insight into Nephrotic Syndrome: Investigating Genes, Health, and Therapeutics (INSIGHT) is an ongoing longitudinal study, initially from Toronto, ON, Canada, that aims to detect factors that affect disease susceptibility and treatment response among children with nephrotic syndrome (41). The preliminary results from this study (42) demonstrate that South Asians have approximately 6 times higher incidence of nephrotic syndrome but significantly lower odds of frequently relapsing disease as compared to Europeans.
There is a clear geographical variation in the incidence of nephrotic syndrome. It is possible that the differences in incidence by ethnicity are due to referral patterns at tertiary care centers or publication bias. The reason for variation in response to steroids and other immunosuppressive medications among specific ethnic groups has not been well documented. The results from registries and prospective studies will provide an excellent resource for future clinical and genetic research to understand disease pathogenesis and differences by ancestry.
A major limitation in understanding ethnic differences is the lack of detailed self-reported ethnicity data collected in most studies. Moreover, due to admixture it can be difficult to establish ethnicity among multi-ethnic children. Additionally, various cultural, environmental, and socioeconomic factors also act as important confounders and may influence individual variability to medication response and frequency of relapses. We provide a few suggestions for future studies in terms of collecting and reporting ethnicity data in children with nephrotic syndrome until genetic ancestry is available in some studies:
1. Ethnicity should be self-reported;
2. Ethnicity of two or more generations should be captured for multigenerational admixture;
3. A uniform definition of ethnic groups should be used to compare across studies;
4. Collaborative studies, both national and international, should include diverse ethnicities.
Role of Ethnicity and Genetics in Childhood Nephrotic Syndrome
There is an evolving role of genetic risk and development of nephrotic syndrome in children. The discovery of NPHS1 and NPHS2 genes leading to congenital nephrotic syndrome provided the first evidence of a genetic cause of steroid-resistant disease. Since then, 45 genes have been associated with monogenic forms of nephrotic syndrome and highlight abnormalities in podocyte structure and function leading to disease (7). The current known genes associated with nephrotic syndrome account for only 20–30% of hereditary and 10–20% of sporadic cases. The majority of these genes especially NPHS2 have been identified in European (43, 44), Japanese (45), and Turkish (46) families but have not been studied across broad ancestral groups where the allele frequency may vary and additional variants may be important. With advancement in genetic analyses, an increasing number of polymorphisms have been detected in children with nephrotic syndrome. It is important to note, however, that some variants are of unknown significance. It is difficult to determine whether these variants are common and/or pathogenic by ethnic or ancestral groups until genetic databases include more comprehensive information across many ethnic groups. Understanding the epidemiological differences in kidney disease by ethnicity may suggest possible genetic risk. For example, African-Americans are known to have a 7.5% lifetime risk of reaching end-stage kidney disease, which is significantly higher than approximately 2% in European Americans (47). In 2008, studies using mapping admixture by linkage disequilibrium identified loci on chromosome 22 that explained the higher incidence of development of FSGS and ESRD among Africans Americans compared to European Americans. Initially, genetic risk was attributed to variants in the MYH9 gene (48, 49). With new data available in the HapMAP, the major source of genetic risk for African-American non-diabetic ESRD and FSGS was localized to APOL1, encoding apolipoprotein L1 (ApoL1), which is only 14 kb from the gene MYH9 (50, 51). Risk of advanced kidney disease is two to seven times greater for those carrying risk alleles of APOL1, as compared to controls. The spectrum of APOL1-associated kidney disease is quite diverse and includes nephrotic syndrome (FSGS), non-diabetic chronic kidney disease secondary to hypertension, HIV-associated collapsing glomerulopathy (52), sickle cell nephropathy (53), and lupus nephritis (54). A recent study also demonstrated that the risk of progression of chronic kidney disease is approximately two times higher in African-Americans with APOL1 variants despite adequate blood pressure control (55). Interestingly, some individuals carrying APOL1 risk alleles do not develop kidney disease highlighting the role of additional genetic or environmental interactions (56). To further understand the association of APOL1 variants with different rates of chronic kidney disease across various African countries and potential environmental interaction, the Kidney Disease Network of Human Hereditary and Health in Africa (H3Africa) consortium was established (57).
Recently, a study among 214 children with steroid-sensitive nephrotic syndrome from South Asia, specifically Sri Lanka, reported HLA-DQA1 missense coding variants as possible candidate loci based on exome array in case–control study compared to 149 healthy controls. The study findings were then confirmed among a replication sample of 100 European children with steroid-sensitive nephrotic syndrome and 205 European controls. This study supports the contributory role of immune response in the pathogenesis of nephrotic syndrome (58) and will need to be tested in additional cohorts with diverse ethnic groups. Previous smaller studies in Chinese and Japanese children have also reported association between variants in HLA-DQ3, HLA-DQ8, HLA-DR, HLADQW2, HLA-DQA1, and HLA-DQB1 in nephrotic syndrome (59–62).
Ethnic Differences in Other Glomerular Diseases
In specific ethnic groups, genome-wide association studies have identified susceptibility loci for membranous and IgA nephropathies. HLA-DQA1 allele on chromosome 6p21 is most closely associated with idiopathic membranous nephropathy in persons of European ancestry (63). IgA nephropathy, the most common cause of glomerulonephritis in adults, has a higher prevalence in Asians as compared to North-Americans or Europeans (64). Recently, in a large diverse cohort of patients with IgA nephropathy, individuals with Pacific Asian origin were shown to have a higher risk of progression to ESRD (65). Studies among East Asians with IgA nephropathy have identified susceptibility loci with the strongest association in the region that include the HLA-DRB1, -DQA1, and -DQB1 genes (66). Similar studies are quite scarce in children with nephrotic syndrome.
Conclusion
In children with nephrotic syndrome, incidence and response to treatment varies by ethnicity. It is likely that genetic and environmental risk factors play a substantial role in explaining these ethnic differences and need further study.
Author Contributions
RC drafted the manuscript; RP conceptualized the work and critically revised the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Authors would like to acknowledge Tonny Banh, Rawan Rumman, Esther Kim, and Jovanka Vasilevska-Ristovska for providing useful suggestions in the preparation of manuscript.
References
2. Schonenberger E, Ehrich JH, Haller H, Schiffer M. The podocyte as a direct target of immunosuppressive agents. Nephrol Dial Transplant (2011) 26(1):18–24. doi: 10.1093/ndt/gfq617
3. Sahali D, Sendeyo K, Mangier M, Audard V, Zhang SY, Lang P, et al. Immunopathogenesis of idiopathic nephrotic syndrome with relapse. Semin Immunopathol (2014) 36(4):421–9. doi:10.1007/s00281-013-0415-3
4. de Burbure C, Buchet JP, Leroyer A, Nisse C, Haguenoer JM, Mutti A, et al. Renal and neurologic effects of cadmium, lead, mercury, and arsenic in children: evidence of early effects and multiple interactions at environmental exposure levels. Environ Health Perspect (2006) 114(4):584–90. doi:10.1289/ehp.8202
5. The primary nephrotic syndrome in children. Identification of patients with minimal change nephrotic syndrome from initial response to prednisone. A report of the International Study of Kidney Disease in Children. J Pediatr (1981) 98(4):561–4. doi:10.1016/S0022-3476(81)80760-3
6. Srivastava T, Simon SD, Alon US. High incidence of focal segmental glomerulosclerosis in nephrotic syndrome of childhood. Pediatr Nephrol (1999) 13(1):13–8. doi:10.1007/s004670050555
7. Bierzynska A, Soderquest K, Koziell A. Genes and podocytes – new insights into mechanisms of podocytopathy. Front Endocrinol (2014) 5:226. doi:10.3389/fendo.2014.00226
8. Copelovitch L, Sam Ol O, Taraquinio S, Chanpheaktra N. Childhood nephrotic syndrome in Cambodia: an association with gastrointestinal parasites. J Pediatr (2010) 156(1):76–81. doi:10.1016/j.jpeds.2009.06.049
9. Zaki M, Helin I, Manandhar DS, Hunt MC, Khalil AF. Primary nephrotic syndrome in Arab children in Kuwait. Pediatr Nephrol (1989) 3(2):218–20; discussion 21. doi:10.1007/BF00852914
10. Elzouki AY, Amin F, Jaiswal OP. Primary nephrotic syndrome in Arab children. Arch Dis Child (1984) 59(3):253–5. doi:10.1136/adc.59.3.253
11. Wyatt RJ, Marx MB, Kazee M, Holland NH. Current estimates of the incidence of steroid responsive idiopathic nephrosis in Kentucky children 1-9 years of age. Int J Pediatr Nephrol (1982) 3(2):63–5.
12. Rothenberg MB, Heymann W. The incidence of the nephrotic syndrome in children. Pediatrics (1957) 19(3):446–52.
13. Schlesinger ER, Sultz HA, Mosher WE, Feldman JG. The nephrotic syndrome. Its incidence and implications for the community. Am J Dis Child (1968) 116(6):623–32. doi:10.1001/archpedi.1968.02100020627009
14. McKinney PA, Feltbower RG, rocklebank JT, Fitzpatrick MM. Time trends and ethnic patterns of childhood nephrotic syndrome in Yorkshire, UK. Pediatr Nephrol (2001) 16(12):1040–4. doi:10.1007/s004670100021
15. Kim JS, Bellew CA, Silverstein DM, Aviles DH, Boineau FG, Vehaskari VM. High incidence of initial and late steroid resistance in childhood nephrotic syndrome. Kidney Int (2005) 68(3):1275–81. doi:10.1111/j.1523-1755.2005.00524.x
16. Bircan Z, Yavuz Yilmaz A, Katar S, Vitrinel A, Yildirim M. Childhood idiopathic nephrotic syndrome in Turkey. Pediatr Int (2002) 44(6):608–11. doi:10.1046/j.1442-200X.2002.01628.x
17. Banaszak B, Banaszak P. The increasing incidence of initial steroid resistance in childhood nephrotic syndrome. Pediatr Nephrol (2012) 27(6):927–32. doi:10.1007/s00467-011-2083-7
18. Hodson ECJ, Willis N. Congenital and idiopathic nephrotic syndrome. In: Elliott E, editor. Ninth Annual Report Australian Paediatric Surveillance Unit. Parramatta: Snap Printing (2002). p. 6–8.
19. Ingulli E, Tejani A. Racial differences in the incidence and renal outcome of idiopathic focal segmental glomerulosclerosis in children. Pediatr Nephrol (1991) 5(4):393–7. doi:10.1007/BF01453661
20. Eddy AA, Symons JM. Nephrotic syndrome in childhood. Lancet (2003) 362(9384):629–39. doi:10.1016/S0140-6736(03)14184-0
21. Bhimma R, Adhikari M, Asharam K. Steroid-resistant nephrotic syndrome: the influence of race on cyclophosphamide sensitivity. Pediatr Nephrol (2006) 21(12):1847–53. doi:10.1007/s00467-006-0276-2
22. Chang JW, Tsai HL, Yang LY, Chen TJ. Epidemiology and predictors of end-stage renal disease in Taiwanese children with idiopathic nephrotic syndrome. J Epidemiol (2012) 22(6):517–22. doi:10.2188/jea.JE20120033
23. Sharples PM, Poulton J, White RH. Steroid responsive nephrotic syndrome is more common in Asians. Arch Dis Child (1985) 60(11):1014–7. doi:10.1136/adc.60.11.1014
24. Feehally J, Burden AC, Mayberry JF, Probert CS, Roshan M, Samanta AK, et al. Disease variations in Asians in Leicester. Q J Med (1993) 86(4):263–9.
25. Feehally J, Kendell NP, Swift PG, Walls J. High incidence of minimal change nephrotic syndrome in Asians. Arch Dis Child (1985) 60(11):1018–20. doi:10.1136/adc.60.11.1018
26. Boyer O, Moulder JK, Somers MJ. Focal and segmental glomerulosclerosis in children: a longitudinal assessment. Pediatr Nephrol (2007) 22(8):1159–66. doi:10.1007/s00467-007-0493-3
27. Bonilla-Felix M, Parra C, Dajani T, Ferris M, Swinford RD, Portman RJ, et al. Changing patterns in the histopathology of idiopathic nephrotic syndrome in children. Kidney Int (1999) 55(5):1885–90. doi:10.1046/j.1523-1755.1999.00408.x
28. Mubarak M, Kazi JI, Shakeel S, Lanewala A, Hashmi S. The spectrum of histopathological lesions in children presenting with steroid-resistant nephrotic syndrome at a single center in Pakistan. ScientificWorldJournal (2012) 2012:681802. doi:10.1100/2012/681802
29. Andreoli SP. Racial and ethnic differences in the incidence and progression of focal segmental glomerulosclerosis in children. Adv Ren Replace Ther (2004) 11(1):105–9. doi:10.1053/j.arrt.2003.10.015
30. Warady BA, Chadha V. Chronic kidney disease in children: the global perspective. Pediatr Nephrol (2007) 22(12):1999–2009. doi:10.1007/s00467-006-0410-1
31. Chernin G, Heeringa SF, Gbadegesin R, Liu J, Hinkes BG, Vlangos CN, et al. Low prevalence of NPHS2 mutations in African American children with steroid-resistant nephrotic syndrome. Pediatr Nephrol (2008) 23(9):1455–60. doi:10.1007/s00467-008-0861-7
32. Sorof JM, Hawkins EP, Brewer ED, Boydstun II, Kale AS, Powell DR. Age and ethnicity affect the risk and outcome of focal segmental glomerulosclerosis. Pediatr Nephrol (1998) 12(9):764–8. doi:10.1007/s004670050542
33. Das U, Dakshinamurty KV, Prayaga A. Pattern of biopsy-proven renal disease in a single center of south India: 19 years experience. Indian J Nephrol (2011) 21(4):250–7. doi:10.4103/0971-4065.85482
34. Gulati S, Sharma AP, Sharma RK, Gupta A. Changing trends of histopathology in childhood nephrotic syndrome. Am J Kidney Dis (1999) 34(4):646–50. doi:10.1016/S0272-6386(99)70388-4
35. Wong W. Idiopathic nephrotic syndrome in New Zealand children, demographic, clinical features, initial management and outcome after twelve-month follow-up: results of a three-year national surveillance study. J Paediatr Child Health (2007) 43(5):337–41. doi:10.1111/j.1440-1754.2007.01077.x
36. El Bakkali L, Rodrigues Pereira R, Kuik DJ, Ket JC, van Wijk JA. Nephrotic syndrome in the Netherlands: a population-based cohort study and a review of the literature. Pediatr Nephrol (2011) 26(8):1241–6. doi:10.1007/s00467-011-1851-8
37. Trautmann A, Bodria M, Ozaltin F, Gheisari A, Melk A, Azocar M, et al. Spectrum of steroid-resistant and congenital nephrotic syndrome in children: the PodoNet registry cohort. Clin J Am Soc Nephrol (2015) 10(4):592–600. doi:10.2215/CJN.06260614
38. Ding WY, Koziell A, McCarthy HJ, Bierzynska A, Bhagavatula MK, Dudley JA, et al. Initial steroid sensitivity in children with steroid-resistant nephrotic syndrome predicts post-transplant recurrence. J Am Soc Nephrol (2014) 25(6):1342–8. doi:10.1681/ASN.2013080852
39. Gadegbeku CA, Gipson DS, Holzman LB, Ojo AO, Song PX, Barisoni L, et al. Design of the Nephrotic Syndrome Study Network (NEPTUNE) to evaluate primary glomerular nephropathy by a multidisciplinary approach. Kidney Int (2013) 83(4):749–56. doi:10.1038/ki.2012.428
40. Samuel S, Scott S, Morgan C, Dart A, Mammen C, Parekh R, et al. The Canadian Childhood Nephrotic Syndrome (CHILDNEPH) Project: overview of design and methods. Can J Kidney Health Dis (2014) 1:17. doi:10.1186/2054-3581-1-17
41. Hussain N, Zello JA, Vasilevska-Ristovska J, Banh TM, Patel VP, Patel P, et al. The rationale and design of Insight into Nephrotic Syndrome: investigating genes, health and therapeutics (INSIGHT): a prospective cohort study of childhood nephrotic syndrome. BMC Nephrol (2013) 14:25. doi:10.1186/1471-2369-14-25
42. Banh TM, Vasilevska-Ristovska J, Hussain N, Hebert D, Licht C, Pearl RJ, et al. Ethnic differences in incidence and outcomes of nephrotic syndrome. Poster Presentation at the Pediatric Academic Society, Vancouver (2014).
43. Ruf RG, Lichtenberger A, Karle SM, Haas JP, Anacleto FE, Schultheiss M, et al. Patients with mutations in NPHS2 (podocin) do not respond to standard steroid treatment of nephrotic syndrome. J Am Soc Nephrol (2004) 15(3):722–32. doi:10.1097/01.ASN.0000113552.59155.72
44. Machuca E, Hummel A, Nevo F, Dantal J, Martinez F, Al-Sabban E, et al. Clinical and epidemiological assessment of steroid-resistant nephrotic syndrome associated with the NPHS2 R229Q variant. Kidney Int (2009) 75(7):727–35. doi:10.1038/ki.2008.650
45. Maruyama K, Iijima K, Ikeda M, Kitamura A, Tsukaguchi H, Yoshiya K, et al. NPHS2 mutations in sporadic steroid-resistant nephrotic syndrome in Japanese children. Pediatr Nephrol (2003) 18(5):412–6. doi:10.1007/s00467-003-1120-6
46. Berdeli A, Mir S, Yavascan O, Serdaroglu E, Bak M, Aksu N, et al. NPHS2 (podicin) mutations in Turkish children with idiopathic nephrotic syndrome. Pediatr Nephrol (2007) 22(12):2031–40. doi:10.1007/s00467-007-0595-y
47. Kiberd BA, Clase CM. Cumulative risk for developing end-stage renal disease in the US population. J Am Soc Nephrol (2002) 13(6):1635–44. doi:10.1097/01.ASN.0000014251.87778.01
48. Kopp JB, Smith MW, Nelson GW, Johnson RC, Freedman BI, Bowden DW, et al. MYH9 is a major-effect risk gene for focal segmental glomerulosclerosis. Nat Genet (2008) 40(10):1175–84. doi:10.1038/ng.226
49. Kao WH, Klag MJ, Meoni LA, Reich D, Berthier-Schaad Y, Li M, et al. MYH9 is associated with nondiabetic end-stage renal disease in African Americans. Nat Genet (2008) 40(10):1185–92. doi:10.1038/ng.232
50. Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, Freedman BI, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science (2010) 329(5993):841–5. doi:10.1126/science.1193032
51. Tzur S, Rosset S, Shemer R, Yudkovsky G, Selig S, Tarekegn A, et al. Missense mutations in the APOL1 gene are highly associated with end stage kidney disease risk previously attributed to the MYH9 gene. Hum Genet (2010) 128(3):345–50. doi:10.1007/s00439-010-0861-0
52. Kopp JB, Nelson GW, Sampath K, Johnson RC, Genovese G, An P, et al. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol (2011) 22(11):2129–37. doi:10.1681/ASN.2011040388
53. Ashley-Koch AE, Okocha EC, Garrett ME, Soldano K, De Castro LM, Jonassaint JC, et al. MYH9 and APOL1 are both associated with sickle cell disease nephropathy. Br J Haematol (2011) 155(3):386–94. doi:10.1111/j.1365-2141.2011.08832.x
54. Freedman BI, Langefeld CD, Andringa KK, Croker JA, Williams AH, Garner NE, et al. End-stage renal disease in African Americans with lupus nephritis is associated with APOL1. Arthritis Rheumatol (2014) 66(2):390–6. doi:10.1002/art.38220
55. Parsa A, Kao WH, Xie D, Astor BC, Li M, Hsu CY, et al. APOL1 risk variants, race, and progression of chronic kidney disease. N Engl J Med (2013) 369(23):2183–96. doi:10.1056/NEJMoa1310345
56. Larsen CP, Freedman BI. Apolipoprotein L1-associated nephropathy and the future of renal diagnostics. J Am Soc Nephrol (2015) 26(6):1232–5. doi:10.1681/ASN.2014101052
57. Osafo C, Raji YR, Burke D, Tayo BO, Tiffin N, Moxey-Mims MM, et al. Human Heredity and Health (H3) in Africa kidney disease research network: a focus on methods in sub-saharan Africa. Clin J Am Soc Nephrol (2015) 10(12):2279–87. doi:10.2215/CJN.11951214
58. Gbadegesin RA, Adeyemo A, Webb NJ, Greenbaum LA, Abeyagunawardena A, Thalgahagoda S, et al. HLA-DQA1 and PLCG2 are candidate risk loci for childhood-onset steroid-sensitive nephrotic syndrome. J Am Soc Nephrol (2015) 26(7):1701–10. doi:10.1681/ASN.2014030247
59. Zhou GP, Guo YQ, Ji YH, Zhang GL. Major histocompatibility complex class II antigens in steroid-sensitive nephrotic syndrome in Chinese children. Pediatr Nephrol (1994) 8(2):140–1. doi:10.1007/BF00865460
60. Konrad M, Mytilineos J, Bouissou F, Scherer S, Gulli MP, Meissner I, et al. HLA class II associations with idiopathic nephrotic syndrome in children. Tissue Antigens (1994) 43(5):275–80. doi:10.1111/j.1399-0039.1994.tb02340.x
61. Kobayashi T, Ogawa A, Takahashi K, Uchiyama MHLA-. DQB1 allele associates with idiopathic nephrotic syndrome in Japanese children. Acta Paediatr Jpn (1995) 37(3):293–6. doi:10.1111/j.1442-200X.1995.tb03317.x
62. Abe KK, Michinaga I, Hiratsuka T, Ogahara S, Naito S, Arakawa K, et al. Association of DQB1*0302 alloantigens in Japanese pediatric patients with steroid-sensitive nephrotic syndrome. Nephron (1995) 70(1):28–34. doi:10.1159/000188540
63. Stanescu HC, Arcos-Burgos M, Medlar A, Bockenhauer D, Kottgen A, Dragomirescu L, et al. Risk HLA-DQA1 and PLA(2)R1 alleles in idiopathic membranous nephropathy. N Engl J Med (2011) 364(7):616–26. doi:10.1056/NEJMoa1009742
64. Donadio JV, Grande JP. IgA nephropathy. N Engl J Med (2002) 347(10):738–48. doi:10.1056/NEJMra020109
65. Barbour SJ, Cattran DC, Kim SJ, Levin A, Wald R, Hladunewich MA, et al. Individuals of Pacific Asian origin with IgA nephropathy have an increased risk of progression to end-stage renal disease. Kidney Int (2013) 84(5):1017–24. doi:10.1038/ki.2013.210
Keywords: steroid resistant, steroid dependent, minimal change disease, focal segmental glomerulosclerosis, nephrotic syndrome
Citation: Chanchlani R and Parekh RS (2016) Ethnic Differences in Childhood Nephrotic Syndrome. Front. Pediatr. 4:39. doi: 10.3389/fped.2016.00039
Received: 02 December 2015; Accepted: 04 April 2016;
Published: 19 April 2016
Edited by:
Robert P. Woroniecki, State University of New York, USAReviewed by:
Kimberly Jean Reidy, Albert Einstein College of Medicine, USAIbrahim F. Shatat, Medical University of South Carolina, USA
Michelle Rheault, University of Minnesota, USA
Copyright: © 2016 Chanchlani and Parekh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rulan S. Parekh, cnVsYW4ucGFyZWtoQHNpY2traWRzLmNh