
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pediatr., 21 January 2015
Sec. Pediatric Cardiology
Volume 2 - 2014 | https://doi.org/10.3389/fped.2014.00148
This article is part of the Research TopicFrontiers in Autoimmune Disease: Rheumatic Fever and Rheumatic Heart DiseaseView all 6 articles
 Marcia Holsbach Beltrame1
Marcia Holsbach Beltrame1 Sandra Jeremias Catarino1
Sandra Jeremias Catarino1 Isabela Goeldner1
Isabela Goeldner1 Angelica Beate Winter Boldt2
Angelica Beate Winter Boldt2 Iara José de Messias Reason1*
Iara José de Messias Reason1*The innate immune system is the first line of host defense against infection and is comprised of humoral and cellular mechanisms that recognize potential pathogens within minutes or hours of entry. The effector components of innate immunity include epithelial barriers, phagocytes, and natural killer cells, as well as cytokines and the complement system. Complement plays an important role in the immediate response against microorganisms, including Streptococcus sp. The lectin pathway is one of three pathways by which the complement system can be activated. This pathway is initiated by the binding of mannose-binding lectin (MBL), collectin 11 (CL-K1), and ficolins (Ficolin-1, Ficolin-2, and Ficolin-3) to microbial surface oligosaccharides and acetylated residues, respectively. Upon binding to target molecules, MBL, CL-K1, and ficolins form complexes with MBL-associated serine proteases 1 and 2 (MASP-1 and MASP-2), which cleave C4 and C2 forming the C3 convertase (C4b2a). Subsequent activation of complement cascade leads to opsonization, phagocytosis, and lysis of target microorganisms through the formation of the membrane-attack complex. In addition, activation of complement may induce several inflammatory effects, such as expression of adhesion molecules, chemotaxis and activation of leukocytes, release of reactive oxygen species, and secretion of cytokines and chemokines. In this chapter, we review the general aspects of the structure, function, and genetic polymorphism of lectin-pathway components and discuss most recent understanding on the role of the lectin pathway in the predisposition and clinical progression of Rheumatic Fever.
The high complexity of the human immune system provides not only effective defense against an impressive number of pathogens but also protection to undesirable response against self-components. The immune system is classically divided in two parts, the innate and the adaptative, which are wide-ranging and interconnected. The innate immune system provides an immediate and non-specific first line defense through humoral, cellular, and mechanical processes, playing a vital role in protection against pathogenic challenge (1). The complement system is an essential part of the innate immune system, with three overlapping roles: defense against infection, clearance of immune complexes and cell debris, and link between innate and adaptative immunity (2).
The complement system consists of more than 35 plasma proteins and cell surface complement receptors and regulatory proteins. Most of the soluble proteins circulate in functionally inactive forms as proenzymes or zymogens (3). Upon proteolytic cleavage, inactive molecules become activated resulting in a proteolytic cascade that elicits a number of effector functions including phagocytosis, inflammation, cell lysis, and guidance of the adaptative immune response (Figure 1) (4). Since activation of complement leads to potentially destructive effects, several inhibitors tightly regulate this system in order to protect host tissues (5). Thus, an effective performance of the complement system depends on balancing regulatory and activation mechanisms, focused on destroying invading microorganisms and limiting damage of host cells and tissues. The imbalance on this fine equilibrium leads to harmful effects to the host, with potentially severe outcomes (2).
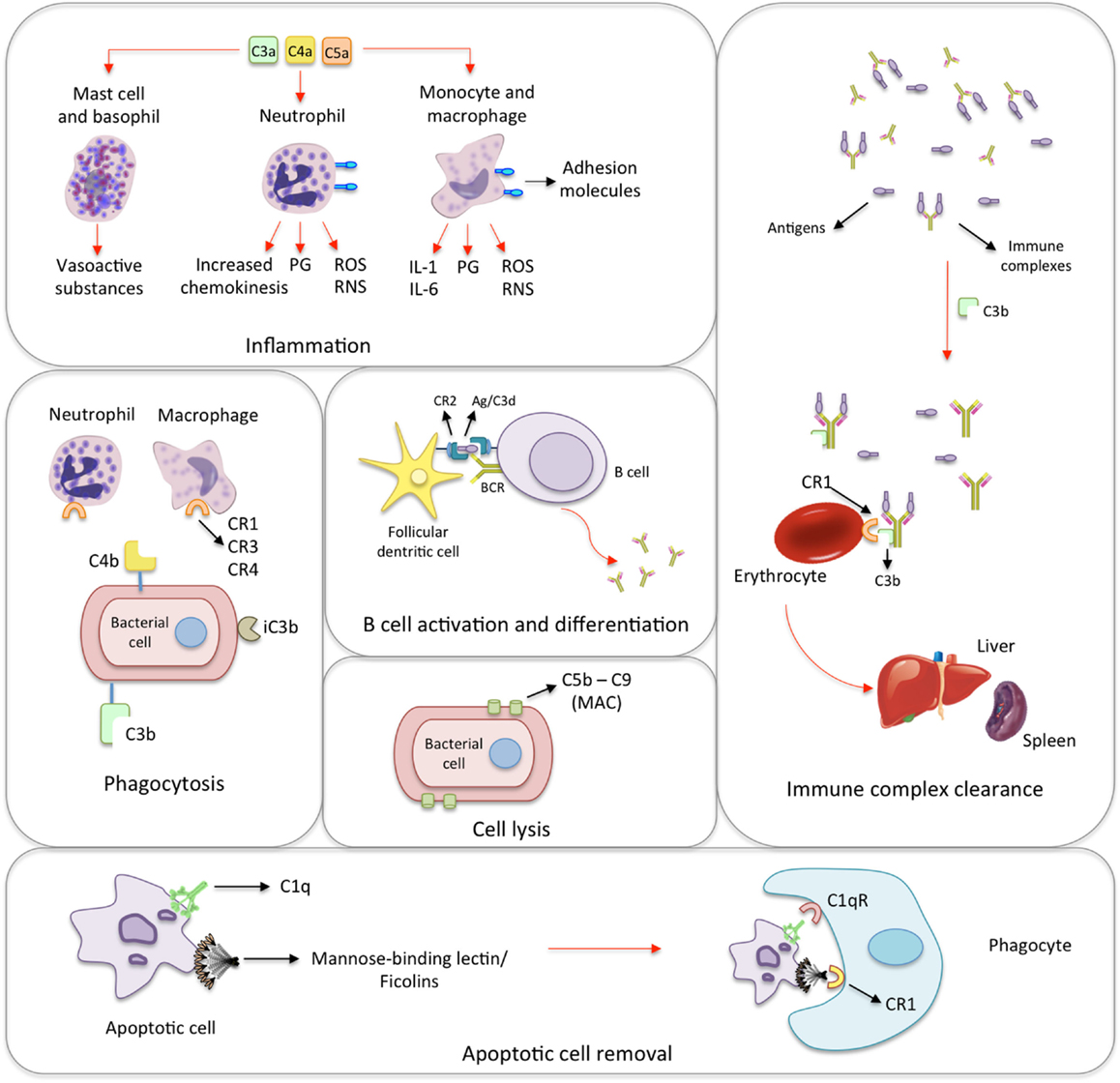
Figure 1. Biological functions of the complement system. Inflammation: the activation of the complement system generates many anaphylatoxins, among which C3a, C4a, and C5a. The binding of C3a, C4a, and C5a to receptors on mast cells and basophils leads to the release of histamine and other vasoactive mediators. In response to the activation by anaphylatoxins, neutrophils release prostaglandins (PG), reactive oxygen and nitrogen species (ROS and RNS, respectively) as well as increase the expression of adhesion molecules, and chemokinesis. Monocytes and macrophages show similar response and secrete interleukins 1 and 6 (IL-1 and IL-6). Phagocytosis: iC3b, C4b, and mainly C3b coat microorganisms and immune complexes, having opsonizing activity. Neutrophils and macrophages express complement receptors (CR1, CR2, and CR4), which bind C3b, C4b, and iC3b. This promotes the adherence of the microorganism to phagocytic host cells leading to phagocytosis. B cell activation and differentiation: the recognition of C3-tagged antigen plays an important role in B cell activation and differentiation. Cross-linking between complement receptor 2 (CR2) and B cell receptor (BCR) through C3d–antigen complexes lowers the threshold of B cell activation leading to migration, T cell/B cell interaction and antibody class-switch. Cell lysis: specific antibodies, MBL/ficolins, and spontaneous hydrolysis of C3 activate the complement on the surface of infectious microorganisms and lead to the formation of membrane-attack complexes (MAC), which cause their lysis. Immune complex clearance: immune complexes activate the complement system. The generated C3b binds to the complexes and to CR1 present on the surface of erythrocytes. During erythrocyte traffic through sinusoids in liver and spleen, resident phagocytes remove bound immune complexes leading to their clearance. Apoptotic cell removal: Mannose-binding lectin, ficolins and C1q bind debris of apoptotic cells, which are subsequently removed through binding to the C1qR and CR1 receptors on phagocytic cells.
The activation of complement can take place on the surface of pathogens or damaged/infected cells by three distinct but converging cascade pathways: classical, alternative, and lectin. All three pathways are initiated by multiple stimuli independently from each other and subsequently the proteolytic cascades converge toward the activation of the major component C3, which results in the assembly of the membrane-attack complex (MAC) (Figure 2) (5). The activation of the classical pathway is initiated on immune complexes by the binding of C1q to Fc portion of IgM or IgG (6, 7). On the other hand, the activation of the alternative pathway occurs by spontaneous hydrolysis of C3 in plasma (6). Similarly to the alternative pathway, the lectin pathway may be activated in the absence of immune complexes. It is initiated by the binding of pattern-recognition plasma molecules such as mannose-binding lectin (MBL), collectin 11 (CL-K1), or ficolins to carbohydrates or acetylated residues present on microorganisms or to aberrant glycocalyx patterns on apoptotic, necrotic, or malignant cells (7). The lectin pathway also plays a role in the coagulation system through the binding of MBL–MBL-associated serine proteases (MASPs) or ficolin–MASPs complexes to fibrinogen or fibrin (8).
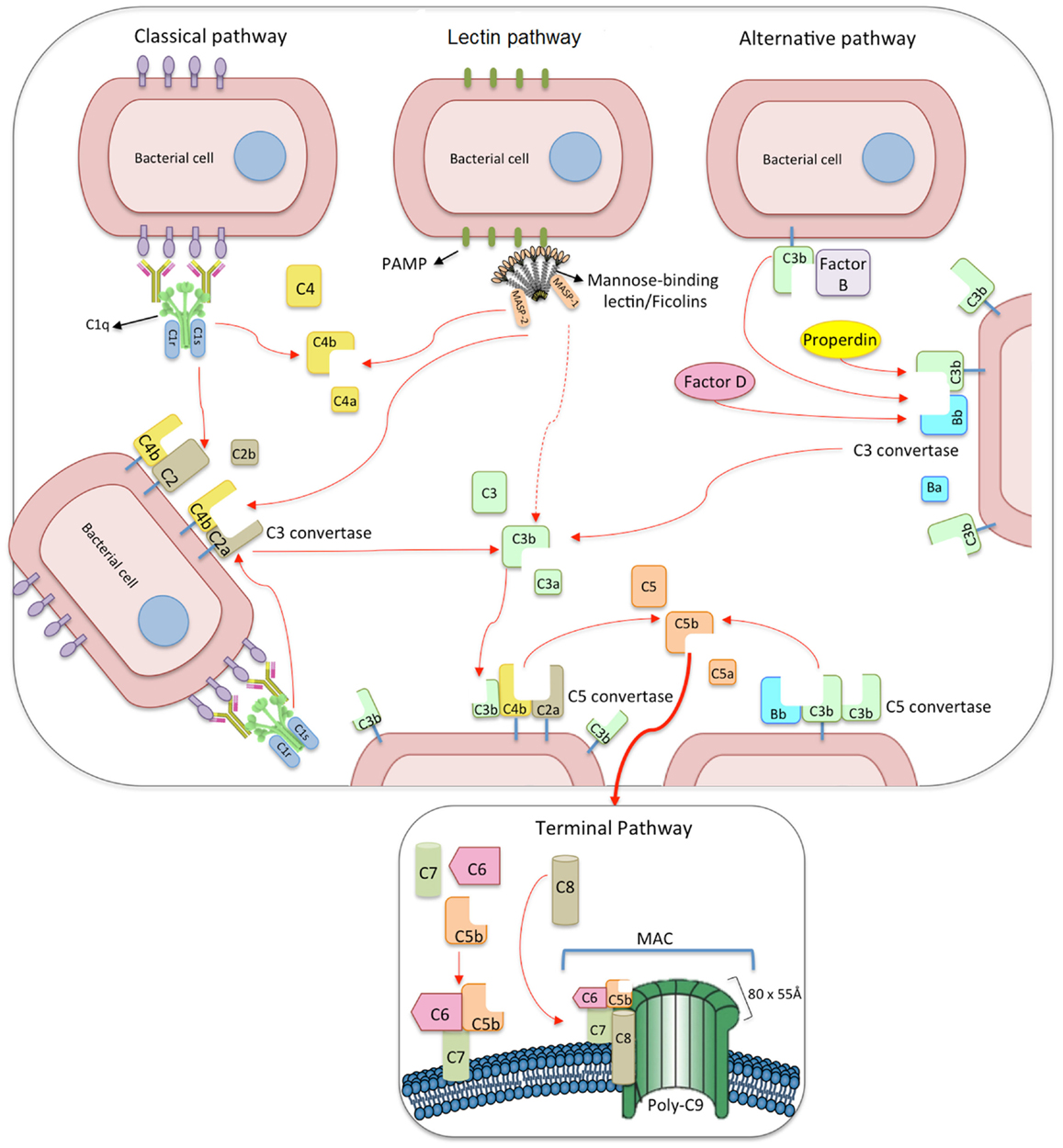
Figure 2. The three pathways of complement activation: classical, lectin, and alternative pathways. The classical pathway is initiated via binding of C1 complex (which consists of C1q, C1r, and C1s molecules) through its recognition molecule C1q to antibody complexes on the surface of pathogens. Subsequently, C1s cleaves C4, which binds covalently to the pathogen surface, and then cleaves C2, leading to the formation of C4b2a complex, the C3 convertase of the classical pathway. Activation of the lectin pathway occurs through the binding of the complex of mannose-binding lectin (MBL), CL-K1 or ficolins, and MBL-associated serine proteases 1 and 2 (MASP-1 and MASP-2, respectively) to various carbohydrates or acetylated residues on the surface of pathogens (PAMP, pathogen-associated molecular pattern). Like C1s, MASP-2 leads to the formation of the C3 convertase, C4b2a, but its activation is dependent on MASP-1. MASP-1 also cleaves C2 and C3. Activation of the alternative pathway depends on spontaneous low-grade hydrolysis of C3 in plasma leading to the formation of C3b. This C3b binds factor B (homologous to C2) to form a C3bB complex. The cleavage of factor B by factor D form the alternative pathway C3 convertase, C3bBb. Properdin stabilizes this complex. The C3 convertases cleave C3 to C3b, which binds covalently next to the site of complement activation (opsonization). This amplifies the cascade and mediates phagocytosis, as well as adaptative immune responses. The addition of further C3b molecules to the C3 convertase forms C5 convertases (C3bBbC3b for the alternative pathway or C4bC2aC3b for both classical and lectin pathways), initiating the assembly of the membrane-attack complex (MAC) by cleavage of C5 to C5a and C5b. Whereas C5a functions as a potent anaphylatoxin, C5b forms a complex with C6 and C7, which is inserted in the cell membrane. Thereafter, C8 and 10–18 C9 molecules (80 × 55 Å each) bind to this complex, resulting in a fully functional MAC (C5b-9). The three pathways converge to this common terminal pathway, culminating with cell lysis and death.
The lectin pathway is initiated when pattern-recognition molecules (MBL, CL-K1, and ficolins) bind to the so-called pathogen-associated molecular patterns (PAMPs) (D-mannose, N-acetyl-D-glucosamine, or acetyl groups), on the surface of pathogens or to apoptotic or necrotic cells (9).
Circulating MBL, CL-K1, and ficolins form complexes with two dimers of MASPs, MASP-1 and MASP-2. After the binding of MBL/MASPs, CL-K1/MASPs, or ficolin/MASPs complexes to their targets, MASP-1 can auto-activate and trigger MASP-2 (10), leading to C4 and C2 cleavage. This allows the assembly of the C3 and C5 convertases, with subsequent activation of C3 and C5, respectively, and generation of C3a and C5a, two pro-inflammatory anaphylatoxins that increase the inflammatory response. The fragment C3b binds covalently to hydroxyl and amino groups on the surface of target molecules of all three pathways. In the absence of complement regulatory proteins, a powerful amplification in the number of surface-bound C3b molecules takes place through the alternative pathway. In this amplification loop, factor B binds to the attached C3b and is cleaved by factor D generating the alternative pathway C3 convertase C3bBb, which leads to accelerated C3b formation (11, 12). C3b tags antigens/pathogens for opsonization and antigen presentation or killing by phagocytes through the interaction with complement membrane receptors CR1, CR2, CR3, and CR4, and the immunoglobulin superfamily member CRIg (13). Finally, the complement cascade culminates with the formation of the multiprotein complex (C5b, C6, C7, C8, and C9n) known as terminal complement complex or MAC, which are inserted as pores of up to 11 nm into the cell membrane inducing loss of membrane integrity and ultimately cell death (14, 15) (Figure 2).
The following topics will cover the main components of the lectin pathway, their functions, polymorphisms, and relevance on the susceptibility to rheumatic fever (RF) and rheumatic heart disease (RHD).
Mannose-binding lectin is a central recognition molecule of the lectin pathway, synthesized in liver cells and secreted into bloodstream as high molecular weight multimeric complexes (16). It is a member of the collectin family of proteins, sharing collagen, and carbohydrate-recognition domains (CRD). MBL is known as a C-type lectin due to the ability to recognize sugar moieties in a Ca2+-dependent manner, and is also referred as “defense collagen” because of the important role in the innate immunity and pathogen clearance (17).
Mannose-binding lectin is basically formed by a trimer of identical polypeptide chains, each containing a cysteine-rich N-terminal domain, a collagen-like region, an alpha-helical coiled-coil neck domain, and a C-terminal CRD (18). The three chains are associated by disulfide bonds and form the structural unit of MBL, which, in turn, polymerize into higher-order MBL oligomers (19) (Figure 3). Single MBL trimers are not fully functional, in contrast to dimers and higher-order oligomers, with tetramers predominating in the circulation (20). The recognition and binding of MBL to its ligands occurs through the CRD domain, and the oligomeric configuration confers multivalent and high avidity binding to targets (21, 22).
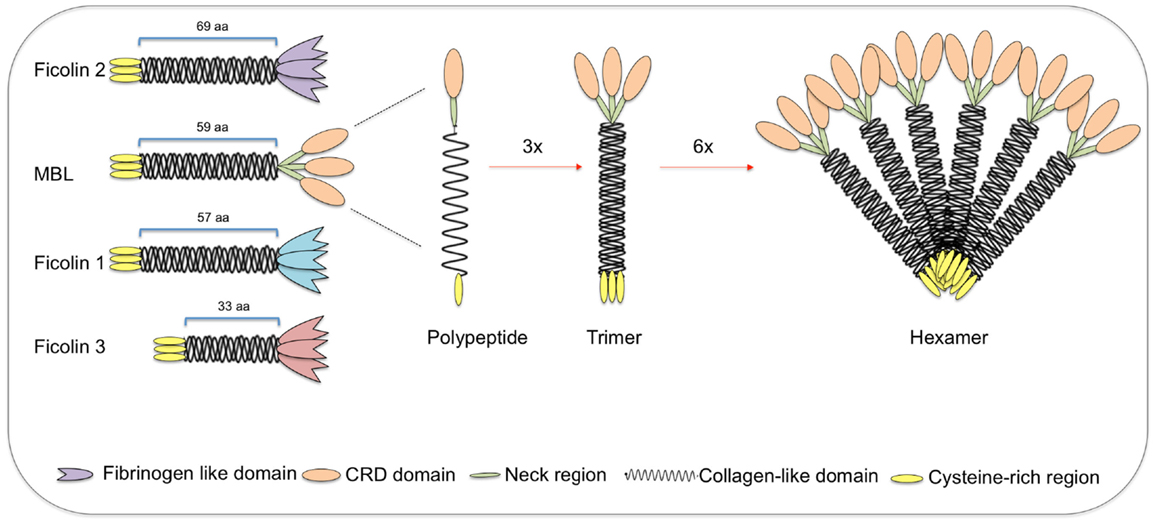
Figure 3. Structural subunits of mannan-binding lectin (MBL) and ficolins. Both MBL and ficolins contain a short N-terminal cysteine-rich region followed by a collagen-like sequence [length given in number of amino acids (aa)]. The C-terminal region is a carbohydrate-recognition binding domain for MBL (shown as oval forms) and a fibrinogen-like domain for ficolins (shown as tulip forms). The polypeptides interact through its collagen-like region forming triple helices (trimers), which further associate into higher oligomeric arrangements (tetramers to hexamers). Despite the very different structures, ficolin polypeptides and trimers interact in the same way as MBL, forming high oligomeric forms (tetramers). MBL-associated serine proteases interact with the collagen-like region, thereby activating the lectin pathway of complement.
Mannose-binding lectin recognizes repetitive arrays of carbohydrate structures on pathogenic organisms such as viruses, bacteria, fungi, protozoans, and multicellular parasites as well as on apoptotic/tumoral cells (23–27). And despite its name, MBL does not bind selectively only mannose or its multimers, but rather recognizes sugars with 3- and 4-OH groups placed in equatorial plane of the sugar ring, which include glucose, L-fucose, N-acetylmannosamine (ManNAc), and N-acetylglucosamine (GlcNAc), but not galactose (28). MBL can further bind phospholipids (29), nucleic acids (30), and non-glycosylated proteins (31).
After binding to targets, MBL induces several biological effects such as complement activation by the lectin pathway, opsonophagocytosis, modulation of inflammation, and recognition of altered self-structures (32). In addition, MBL may modulate cytokine production at both mRNA and protein levels (33).
Mannose-binding lectin also plays a role in the clearance of apoptotic cells by recognizing damage-associated molecular patterns (DAMPs) (34). By binding to terminal sugars of cytoskeletal proteins in apoptotic cells, MBL mediates their recognition and phagocytosis by macrophages leading to their clearance. In fact, both C1q and MBL facilitate the binding of apoptotic cells to immature dendritic cells as well as to macrophages (27). Defects in the clearance of apoptotic cells have been implicated in the pathogenesis of some autoimmune conditions, although the precise role of MBL in this process remains unknown. Experiments using MBL-deficient mice showed impaired removal of apoptotic cells but no relation to autoimmune disease (35).
Mannose-binding lectin is encoded by the MBL2 gene, located on the long arm of chromosome 10 (10q11.2–q21) (36). It is considered an acute-phase reactant (37), whose levels can increase up to threefold during the acute-phase response, mainly due to up-regulation by acute-phase mediators (38). MBL serum levels range from a few nanogram per milliliter to more than 10 μg/ml, with a mean value of around 0.8 μg/ml (39). However, MBL levels are largely dependent on MBL2 genetic polymorphisms, which are responsible for inter-individual variations of up to 10-fold on circulating MBL levels (40, 41). Besides genetic variation, MBL levels may also vary significantly during lifetime (42–44).
Mannose-binding lectin deficiency is fairly common, affecting approximately 8–10% of individuals and usually defined as ≤100 ng/ml in the circulation (45, 46). MBL deficiency is more harmful when there are additional co-existing immune defects (47), since the majority of MBL-deficient individuals are essentially healthy (48). MBL deficiency has been associated with upper respiratory tract infections in young children and with the susceptibility to severe infections in patients receiving chemotherapy (45). However, it may be beneficial in infections due to intracellular pathogens, such as Mycobacterium spp. and Leishmania chagasi, which use C3 opsonization and C3 receptors to invade host cells (49–52).
MBL2 is a highly polymorphic gene, exhibiting variants responsible for large variations in both MBL levels and functional activity (53–57). These variants include SNPs located in the first exon of MBL2 gene, being at least one synonymous SNP (on codon 44 for asparagine) and eight non-synonymous variants (including B, C, and D, which are detailed in the next paragraph). At least other three SNPs located in the promoter region of the MBL2 gene also have influence on MBL levels, called MBL2*H and L alleles (due to a polymorphism located at −550 bp), X and Y alleles (a SNP at −221 bp) and P and Q alleles (a non-coding SNP at +4 bp), all positions counted from the transcription start site (58, 59).
In 1991, Sumiya et al. sequenced the complete MBL2 gene in three British children with recurrent bacterial infections and low MBL levels. All of them had the B allele (an exon 1 point mutation at codon 54, changing GGC to GAC and causing an amino acid change of glycine to aspartic acid – p.Gly54Asp) (60). Others subsequently found two other deficiency-causing common substitutions, allele D in codon 52 (CGT to TGT), changing arginine to cysteine (p.Arg52Cys) and allele C, in codon 57 (GGA to GAA), substituting glycine for glutamic acid (p.Gly57Glu) (61) (Figure 4). Exon 1 mutations dramatically reduce protein assembly and stability, increasing the amount of poorly oligomerized MBL with reduced capacity of complement activation and ligand binding in homozygous (e.g., B/B) or compound heterozygous (e.g., B/C) carriers (18). The wild allele at these loci is called A, whereas D, B, and C alleles have been collectively called 0. While 0/0 individuals have near undetectable levels of high-order MBL oligomers, A/0 individuals may present reduced plasma protein levels (61, 62). In addition, a promoter variant 221 bp before the start of transcription site, with X and Y alleles (g.602G > C), markedly decreases levels of otherwise fully functional MBL proteins.
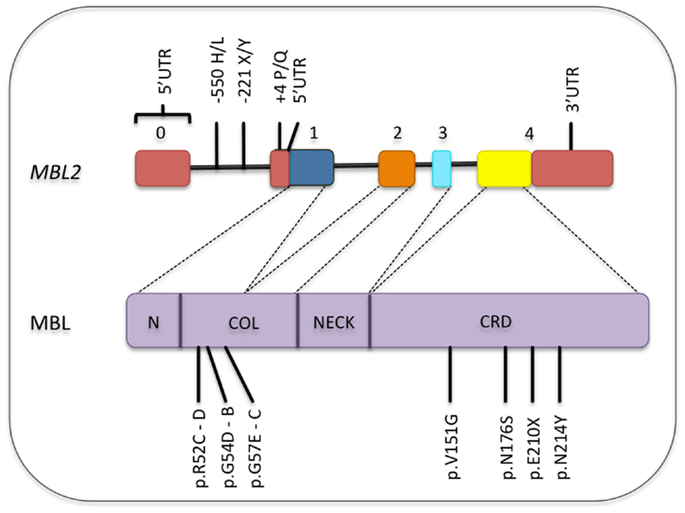
Figure 4. Common polymorphisms in the MBL2 gene and its corresponding locations in the MBL protein. Only the functional polymorphisms in the promoter and non-synonymous mutations are shown [SNP database and Boldt et al. (161)]. Exons are numbered. Exons, introns, and protein domains are not in scale. N, N-terminal region; COL, collagen-like region; CRD, carbohydrate-recognition domain.
Similarly to MBL, ficolins are pattern-recognition receptors that are able to associate with MASPs and activate the complement system through the lectin pathway, having an essential role in the immune defense against clinically important pathogens. Besides activating complement, they limit infection by stimulating the secretion of interferon gamma (IFN-γ), IL-17, IL-6, tumor necrosis factor alpha (TNF-α), and nitric oxide (NO) by macrophages (63).
Ficolins form oligomers of four structural subunits joined together via disulfide bridges at the N-terminal regions, similarly to MBL, but higher or smaller oligomers seem to be less common for ficolins (21). They should not be referred as lectins (meaning that carbohydrates are the preferred ligands for lectins), since they target acetylated compounds relatively independently of the structure of the acetylated molecule (64).
Three human ficolins have been described, each encoded by its own gene. Ficolin-1 can be found attached to cell membrane or soluble in plasma, with a concentration of 0.05–1.0 μg/ml. It is also found in the secretory granules of monocytes, gelatinase granules of neutrophils, and type II alveolar epithelial cells (65–68). Ficolin-1 recognizes common acetylated compounds, including GlcNAc and GalNAc (63, 67, 69, 70) binding to several microorganisms including Gram-positive (Staphylococcus aureus) and Gram-negative bacteria (Salmonella typhimurium LT2) (67, 69). It is the only human ficolin able to bind to sialic acid, found on capsular polysaccharides of some pathogens such as Streptococcus agalactiae as well as on the surface of immune cells. Thus, Ficolin-1 is supposed to play a role in the modulation of immune cell interaction (71) and in blood coagulation and/or fibrinolysis (72). Importantly, Ficolin-1 and pentraxin-3 heterocomplexes act as non-inflammatory signals, promoting clearance of altered self-cells and modulating IL-8 production (73).
FCN1 gene is located on chromosome 9q34 (74) and contains nine exons. Among the several SNPs described for the FCN1 gene, at least eight are associated with Ficolin-1 levels, four of them located in the promoter and in the first exon (74). These polymorphisms are partly responsible for the wide range (up to 15-fold) of inter-individual variability in Ficolin-1 plasma concentrations (66). FCN1 polymorphisms were associated with increased fatal outcome in patients with systemic inflammation (75) and to the susceptibility to rheumatoid arthritis (76). Low Ficolin-1 levels have been associated with a 12-fold increased risk of fatal necrotizing enterocolitis, and with need for mechanical ventilation (77), as well as with occurrence of severe infections in cancer patients undergoing chemotherapy (78).
Ficolin-2 is a plasma protein, which is mainly produced in the liver (79), but low mRNA levels were also found in the bone marrow, intestine, tonsils, and fetal lung (80). In Europeans, the median plasma concentration is around 5 μg/ml, with inter-individual variations ranging from 1 to 12 μg/ml (81). Ficolin-2 is able to bind N-acetylated molecules, such as Acetyl-d-glucosamine (GlcNAc), N-acetylgalactosamine, and N-acetyllactosamine (71, 82), as well as artificially acetylated compounds (83). It binds also N-acetylneuraminic acid present on encapsulated opportunistic pathogens such as Group B streptococci (Streptococcus agalactiae) (84), bacterial peptidoglycan (PGN), fungal 1,3-beta-D-glucan (85), and envelope glycoproteins of hepatitis C virus (86). In addition, Ficolin-2 was shown to bind to Mycobacterium bovis (87) and flagellated protozoa including Giardia intestinalis (88) and Trypanosoma cruzi (89) and to interact with C-reactive protein, stabilizing its binding to bacteria and thereby activating complement (63, 90).
FCN2 gene is located on chromosome 9q34.3 (79), with three SNPs in the promoter region and one in exon 8 being associated with variation in Ficolin-2 plasma levels: −986G > A, −602G > A, and −4A > G and p.Ala258Ser, while two other SNPs, at positions −557 and −64, appear not to influence gene expression (91, 92). Ficolin-2 insufficiency (<1,200 ng/ml) (93) has been associated with bronchiectasis and respiratory infection especially when co-existing with atopic disorders (94–96), but did not affect susceptibility to invasive pneumococcal disease (97). Low Ficolin-2 levels were also related with prematurity, low birth weight, and perinatal infections in neonates from Poland (98) and with the susceptibility to chronic Chagas disease (99). And although not associated with the development of malaria, children with severe malaria presented higher levels of Ficolin-2 than children with the mild form of the disease (100). On the other hand, FCN2 polymorphisms associated with normal Ficolin-2 levels had a protective effect against the susceptibility to leprosy (101).
Ficolin-3 is the most abundant recognition molecule of the lectin-pathway with a mean plasma concentration around 18.4 μg/ml (102), varying approximately 10-fold among individuals (3–54 μg/ml) (103). Ficolin-3 was found highly expressed in the liver and lung tissues, indicating its significance in both activation of the lectin pathway and pulmonary host defense (80, 104). For that reason, Ficolin-3 is considered to play an important role in both systemic and local innate immune responses (80, 105, 106). Ficolin-3 was shown to recognize acetyl groups present in a wide range of microorganisms, including Salmonella typhimurium, Salmonella minnesota, Escherichia coli, and Aerococcus viridans (107, 108). It was also shown to share binding sites with Ficolin-2 and MBL on the surface of Giardia intestinalis (88). Furthermore, Ficolin-3 may mediate the clearance of late apoptotic cells and may have a beneficial role against autoimmunity (109, 110).
FCN3 gene is located on chromosome 1p36.11 and is highly conserved in humans. Five amino acid exchanges were described, all with allele frequencies below 5%: p.Leu12Val, p.Leu117fs, p.Thr125Ala, p.Glu166Asp, and p.Val287Ala (80). This high conservation indicates that Ficolin-3 might exert crucial function in the immune response. Indeed, Ficolin-3 insufficiency is extremely rare (111) and was found associated with necrotizing enterocolitis in premature neonates (112).
MBL-associated serine proteases act as activators of the lectin pathway upon binding of MBL, ficolins, and CL-K1 to carbohydrates or acetyl groups on the surface of pathogens or altered self-tissues (4). So far, five proteins have been identified, including three MASP enzymes (MASP-1, MASP-2, and MASP-3) and two truncated proteins, MAp19 and MAp44, which lacks the serine protease domain and consequently, functional activity (113, 114). All MASPs are capable to associate with MBL, ficolins, and CL-K1 in the presence of Ca2+, forming a proteolytic complex (39).
Both MASP-1 and MASP-2 play a crucial role in the activation of lectin pathway. Recent studies showed that MASP-1 can auto-activate and lead to MASP-2 activation (115, 116). MASP-2 can also auto-activate, but under physiological conditions, MASP-1 is the essential MASP-2 activator (39). MASP-2 is a protease that cleaves very efficiently C4 and C2, generating C3 convertase (113, 117). On the other hand, MASP-3 seems to attenuate the lectin-pathway activity due to competition for MASP binding sites on the recognition molecules (118). In addition, MASP-3 occurs predominantly complexed with Ficolin-3 and is thought to have an inhibitory effect on complement activation mediated by Ficolin-3 (119). MASP-3 also participate on developmental processes (120). The roles of MAp19 and MAp44 are still not well understood, but MAp44 was shown to negatively regulate the lectin pathway by competing for the same binding sites of MASP-2 and MASP-1 (121, 122).
MASP-1 was the first MASP to be reported (123). While MASP-1 and MASP-2 are produced mainly in the liver and present in plasma at 11 and 0.4 μg/ml, respectively (124, 125), MASP-3 is produced in several other tissues besides the liver (118). All three MASPs are structurally similar to each other and to both C1r and C1s. MASP-1, MASP-3, and MAp44 are codified by MASP1 gene on chromosome 3q27–q28 and MASP-2 and MAp19 are encoded by MASP2 gene located on chromosome 1p36.23–31 (119, 121, 126, 127). Some polymorphisms on MASP1 and MASP2 genes lead to changes in serum levels and functions of MASPs, thereby influencing complement activation by the lectin pathway (128, 129). Ammitzbøll et al. found 10 SNPs in the MASP1 gene that were associated with serum levels of MASP-1, MASP-3, and MAp44 (130).
MASP1 polymorphisms were associated with 3MC syndrome (131–133) and to Pseudomonas aeruginosa colonization in cystic fibrosis patients (134). Moreover, MASP levels (MASP-1, MASP-2, and MASP-3) were shown as a predictor for infection and prolonged dependency of intensive care in critically ill children (135). On the other hand, MASP2 polymorphisms were associated with the susceptibility to leprosy (136), human T lymphotropic virus infection (137), malaria (138), Chagas disease (139), bacterial infections (140), and hepatitis C (141). MASP-2 levels have been related also with a number of diseases, including schizophrenia (142), septic shock (143), acute lymphoblastic leukemia, non-Hodgkin lymphoma, central nervous system tumors (144), colorectal cancer (145, 146), among others. Taken together, these studies have provided evidence for an emerging and important biological role of MASPs in human diseases.
The lectin pathway appears to be involved in the pathophysiology of different cardiac conditions (147), sometimes with opposite roles or even ambiguous functions. Although low MBL producing genotypes were associated with coronary artery disease in American Indians (148), elevated MBL levels were associated with an increased risk of future coronary artery disease in men, but not in women, in the United Kingdom (149). In addition, MBL was shown to have an ambiguous role in the development of coronary artery lesions in Kawasaki disease, being protective in infants but potentially harmful in older patients (150). A protective role for high MBL levels were reported in myocardial infarction, particularly in diabetes (151), and MBL2 variants related to functional MBL deficiency were shown to increase by twofold the risk of myocardial infarction in healthy individuals (152). Also, MBL levels measured 1 month after acute myocardial infarction were inversely associated with the incidence of reinfarction, suggesting that low MBL levels could predispose to ischemic events (153). However, experimental studies showed involvement of MBL in ischemia/reperfusion injury, probably due to its ability to recognize altered self-structures, thereby mediating complement activation (154, 155). Absence of MBL/MASP pathway activation has protected against tissue damage and preserve cardiac function in these disease models (154). In addition, inhibition of MBL by monoclonal antibodies decreased significantly the infarcted size and tissue injury by limiting neutrophil infiltration and gene expression of pro-inflammatory mediators (155). In fact, increasing evidence is indicating a pro-inflammatory role for MBL in chronic diseases and situations where there is undesirable complement activation and tissue injury (49–51).
Ficolins and MASPs were found to play a role as well in myocardial infarction. Ficolin-2 and Ficolin-3 levels were associated with left ventricular dilatation after myocardial infarction (156) and with advanced heart failure and outcome, respectively (157). Recent study involving all MASPs showed that MASP-1 levels were highest in subacute myocardial infarction and lowest in acute stroke patients, while MASP-2 levels were low in both conditions and MASP-3 and MAp44 levels did not differ between the groups. On the other hand, MASPs and MAp44 levels were associated with cardiovascular risk factors including dyslipidemia, obesity, and hypertension in patients with stable coronary artery disease (158). Moreover, MASP-2 levels were found significantly reduced in myocardial infarction, probably due to the activation of the lectin pathway during acute myocardial ischemia (159).
Despite the important role of the lectin pathway in complement activation and host defense against infection and autoimmunity, studies on the significance of components of this pathway in RF and RHD are still scarce. Different groups investigated the role of MBL and ficolins in the innate immunity against streptococcal infection. MBL binds strongly to N-acetyl-β D-glucosamine on the streptococcal cell wall and thereby promotes complement deposition and opsonization in vitro (160). Thus, MBL could play an essential role in the first steps of immune defense against streptococci infection, leading to complement activation, and pathogen phagocytosis (161). In addition, Ficolin-2 binds molecular patterns such as lipoteichoic acid on Gram-positive bacteria cell wall, including Streptococcus pyogenes (162), also mediating bacteria opsonization and elimination (79). Moreover, a critical role for ficolins in the protection against Streptococcus pneumoniae infection was shown in experimental models using ficolin deficient mice, supporting the contribution of these pattern-recognition molecules in the immune defense during streptococci infection (163).
Inherited MBL insufficiency, which leads to impaired innate immune function and enhanced susceptibility to infection, is essentially caused by three structural variants in exon 1 of MBL2 gene. These polymorphisms include the previously mentioned variations in the collagenous tail, with the alleles being designated, respectively, D, B, and C (collectively termed “0”). In addition, another promoter variant identified as X/Y (g.602G > C) is known to significantly reduce circulating levels of functional MBL. In an initial study in 2001, there was no association between the A, B, C, and D alleles and RHD in Chinese patients (164). However, the authors suggested a putative role for MBL deficiency in the progression of RHD, by considering the age of onset of heart disease. The mean age of onset of cardiac symptoms of patients with the deficient B allele was significantly lower compared with patients with MBL2 genotype A/A (30 ± 14 vs. 37 ± 11 years, p < 0.05). These results supported the hypothesis that MBL deficiency caused by the B allele could facilitate the development of RHD in younger people and accelerate the progression of RHD. Another study reported lower MBL serum levels in RHD patients from Yemen, compared with blood donors (165).
On the other hand, Schafranski et al. showed that MBL levels were significantly high in patients with RHD from South Brazil and that MBL deficiency was more prevalent among controls. The authors suggested that high MBL levels could be a cause of undesirable complement activation in RHD patients, contributing to the pathogenesis of rheumatic cardiomyopathy (166). Subsequent analysis on MBL2 polymorphisms in the same cohort of patients showed that MBL2 genotypes associated with high MBL levels were also significantly associated with RHD, if compared with controls, suggesting a role for high-producing MBL2 genotypes in the susceptibility to RHD. Moreover, MBL2 A/A genotypes were significantly associated with higher MBL levels in RHD patients and not in controls. Homozygosity for the MBL2*A allele as well as for haplotypes associated with high MBL levels were associated with increased risk of RHD. Conversely, the frequency of MBL2 variant alleles as well as 0/0 genotypes, which are associated with MBL deficiency, were lower in patients (167). These findings indicated that both MBL2 alleles and protein levels were associated with susceptibility to RHD. Since MBL is a key molecule in the innate host defense against bacterial infection, the authors postulated that MBL could be considered as a double-edged sword molecule in the physiopathology of RF and RHD, on one hand conferring protection against initial infection by rheumatogenic streptococci, but on the other hand eliciting inflammation and complement tissue damage in the chronic stage of the disease (167). Later on, these findings were confirmed by showing that high MBL levels (>2,800 ng/ml) increased the risk of RHD and that genotypes associated with high MBL production (YA/YA and YA/XA) were associated with both acute and chronic rheumatic carditis (168). The results led to the conclusion that high MBL levels and its associated genotypes could be involved in development of cardiac manifestation in RF, and the association was due to genetic influence of MBL2 polymorphisms rather than to an acute-phase reaction. The authors suggested that genotypes associated with high MBL production may represent a risk factor for development of acute and chronic rheumatic carditis in RF, and MBL could be involved in the ongoing inflammatory process and progression to the chronic cardiac form (168). Under stressful conditions such as inflammatory processes mediated by oxidative stress, self-cell surfaces may become glycosylated and thereby acting as targets for MBL/ficolin binding. This in turn, would lead to pathological complement activation and inflammation (169). In fact, myxomatous tissue rich in hyaluronic acid was observed in heart valves of RHD patients. Since N-acetyl-d-glucosamine is a ligand for MBL and a major constituent of hyaluronic acid in myxomatous tissue, activation of complement in RF may occur partially due to the binding of MBL to neo exposed ligands in the heart valves (170). Only the long-term follow-up of patients with acute RF without valve sequel would answer this question and confirm if in fact, high MBL levels and associated genotypes are risk factors to carditis in RF. Considering that no effective treatment is currently available for rheumatic carditis, studies exploring anti-MBL blocking in experimental models of RF and RHD could bring new insights in to this question and might unveil alternatives for future treatment of this disease.
Another study on patients with chronic severe aortic regurgitation (AR) of rheumatic etiology by Ramasawmy et al. showed that MBL2*D allele frequency was higher among AR patients than in controls from São Paulo, Brazil. The B and C alleles had similar frequencies in both groups, whereas the frequency of the 0/0 genotypes was significantly different between patients and controls, but similar in both groups for A/0 heterozygotes. Thus homozygous or compound heterozygous patients for defective MBL2 alleles (genotype 0/0) presented higher risk of developing chronic AR. In this case, the authors postulated that continuous exposure to Streptococcus group A antigens in MBL-deficient individuals would lead to subsequent abnormal immune response against heart proteins, leading to rheumatic AR (171). These results were different than previous reports (167, 168), in which MBL2 wild-type A allele increased the risk to RHD in patients with mitral valve lesion of rheumatic etiology, rather than aortic vulvar lesion, as reported for AR patients. It is worth mentioning that the ancestry of patients differed significantly between the studies, whereas the AR patients from São Paulo were of mixed Brazilian ethnicity, those from the South were mainly Euro-Brazilians.
Some polymorphisms in MASP2 gene were shown to impair the activation of the lectin pathway. A mutation in exon 3, g.5620A > G, leads to an amino acid substitution from aspartate to glycine (Asp120Gly or p.D120G) and disturbs calcium ion binding site in the first CUB1 domain of the mature protein. This defect causes loss of MASP-2 binding ability to MBL and ficolins, abolishing complement activation and decreasing MASP-2 circulating levels. The mutation p.D120G occurs in 0.15–0.3% of European individuals and has been reported to be the main cause for low MASP-2 levels in this population, with homozygosity resulting in MASP-2 deficiency (129, 172, 173).
In two independent studies, partial or total MASP-2 deficiencies resulting from p.D120G mutation were not associated with RF/RHD and severe chronic AR in Brazilian patients, respectively. No homozygotes but only heterozygotes for the G allele were found with no significant differences between patients and controls, indicating that the p.D120G variation in the MASP2 gene does not have a relevant role in the pathogenesis of RF (171, 174). However, one must consider that the G allele is rare and only studies with a large number of subjects would have the statistical power to indicate a possible role for p.D120G in the development of RF or RHD (171, 172, 174–177). Recently, a number of different MASP2 polymorphisms encompassing the promoter region to exon 12 and MASP-2 levels were investigated in RF and RHD patients. The authors found that low MASP-2 producing variants were associated with protection against the development of RHD and haplotypes sharing intron 9 – exon 12 polymorphisms increased the susceptibility to RHD, when compared to RF patients without cardiac disease. In addition, MASP-2 levels were lower in patients an associated with MASP2 haplotypes. The results suggest a role for MASP2 gene polymorphisms and protein levels in RHD (178).
The recognition molecules of the lectin pathway, including Ficolin-2, associate with MASPs in serum, forming complexes able to activate complement (179). Ficolin-2 presents a wide specificity for microorganisms, thereby having an important role in the first line of innate immune defense. Although clinical studies focusing on Ficolin-2 are still in their initial stages, there is evidence that Ficolin-2 deficiency might increase the risk of respiratory infections (180). So far, the only study on Ficolin-2 in RF/RHD investigated polymorphisms in the promoter region of the FCN2 gene (at positions -986, -602, and -4) in patients and controls from South Brazil. The haplotype-986/-602/-4 G/G/A, associated with low Ficolin-2 levels, was significantly associated with RHD when compared to controls, suggesting that this haplotype may play a role in the progression of RF to its chronic form. On the other hand, the frequency of the FCN2 haplotype-986/-602/-4 A/G/A was higher in the controls than in the patients (RF plus RHD), showing a protective effect against RF and RHD. This data led the authors to suggest that FCN2 promoter haplotypes are associated with the susceptibility to RF and its chronic form RHD, with -986/-602/-4 G/G/A haplotype representing a novel risk factor for the susceptibility and clinical progression of the disease (181). The putative role of MBL and Ficolin-2 in RF and RHD is summarized in Figure 5.
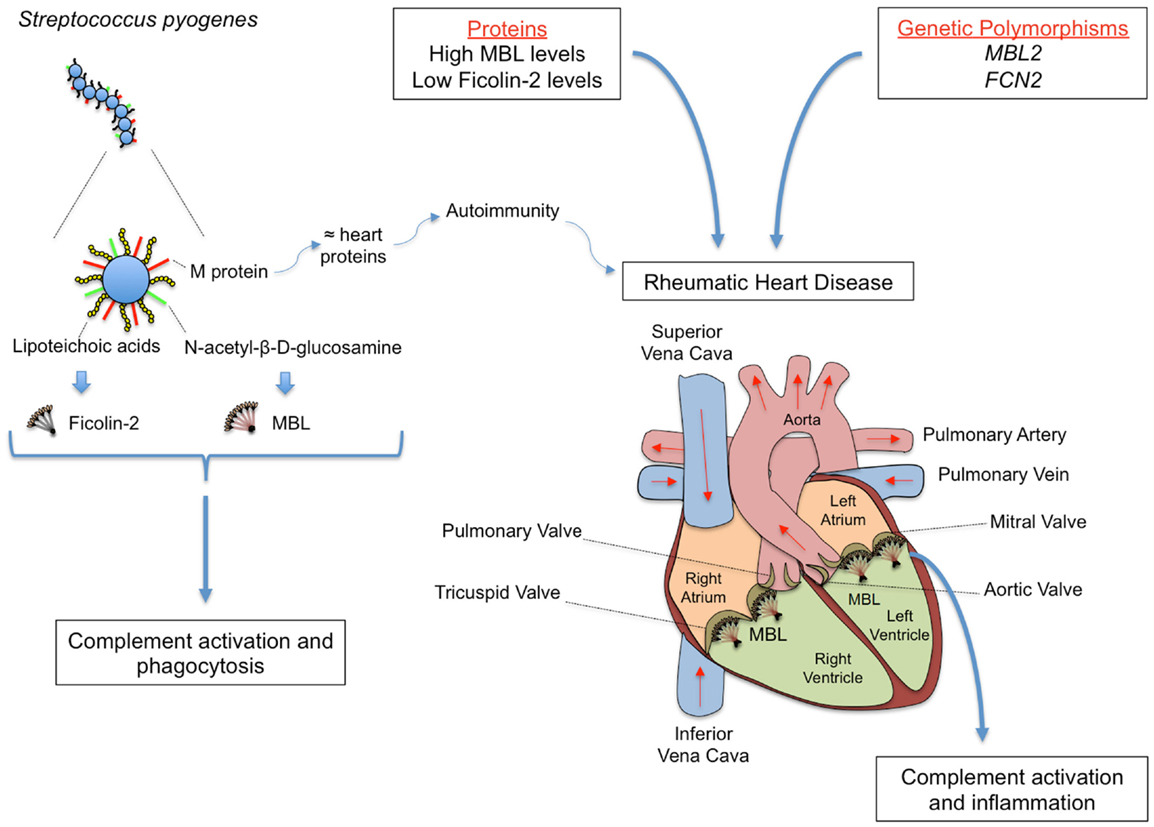
Figure 5. Rheumatic fever (RF) and its most severe sequel chronic rheumatic heart disease (RHD) are chronic inflammations that follow oropharynx infection by Streptococcus pyogenes, whose cell wall presents several PAMPs, including M protein, lipoteichoic acid, and N-acetil-β-d-glucosamine. M protein shares structural homology with heart proteins such as myosin and tropomyosin, leading to the formation of cross-reactive auto-antibodies. MBL and Ficolin-2 bind N-acetyl-β-d-glucosamine and lipoteichoic acid, respectively, inducing complement activation and phagocytosis. Although conferring protection against the initial infection, MBL may deposit on the altered valves, eliciting inflammation, and complement tissue damage in the chronic stage of the disease. The functional importance of the proteins may vary during infection and disease establishment, with MBL2 and FCN2 polymorphisms leading to high MBL and low Ficolin-2 levels, respectively, being associated with increased susceptibility to RHD.
In conclusion, the studies accomplished so far on the proteins and genes of the lectin pathway of complement pointed to an important role for the lectin pathway in the susceptibility to RF and clinical progression to RHD. However, future studies are required in order to clarify the role of the recognition molecules and serine proteases of the lectin pathway in RF and RHD.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Dunkelberger JR, Song W-C. Complement and its role in innate and adaptive immune responses. Cell Res (2010) 20:34–50. doi: 10.1038/cr.2009.139
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Walport MJ. Complement. First of two parts. N Engl J Med (2001) 344:1058–66. doi:10.1056/NEJM200104053441406
3. Degn SE, Thiel S. Humoral pattern recognition and the complement system. Scand J Immunol (2013) 78:181–93. doi:10.1111/sji.12070
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol (2010) 11:785–97. doi:10.1038/ni.1923
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Blom AM, Villoutreix BO, Dahlbäck B. Complement inhibitor C4b-binding protein-friend or foe in the innate immune system? Mol Immunol (2004) 40:1333–46. doi:10.1016/j.molimm.2003.12.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Chen M, Daha MR, Kallenberg CG. The complement system in systemic autoimmune disease. J Autoimmun (2010) 34:J276–86. doi:10.1016/j.jaut.2009.11.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Ali YM, Lynch NJ, Haleem KS, Fujita T, Endo Y, Hansen S, et al. The lectin pathway of complement activation is a critical component of the innate immune response to pneumococcal infection. PLoS Pathog (2012) 8:e1002793. doi:10.1371/journal.ppat.1002793
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Jensenius JC. The manna-binding lectin (MBL) pathway of complement activation: biochemistry, biology and clinical implications. Adv Exp Med Biol (2005) 564:21–2. doi:10.1007/0-387-25515-X_6
9. Degn SE, Jensenius JC, Bjerre M. The lectin pathway and its implications in coagulation, infections and auto-immunity. Curr Opin Organ Transplant (2011) 16:21–7. doi:10.1097/MOT.0b013e32834253df
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Héja D, Kocsis A, Dobó J, Szilágyi K, Szász R, Závodszky P, et al. Revised mechanism of complement lectin-pathway activation revealing the role of serine protease MASP-1 as the exclusive activator of MASP-2. Proc Natl Acad Sci U S A (2012) 109:10498–503. doi:10.1073/pnas.1202588109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Fishelson Z, Pangburn M, Muller-Eberhard H. Characterization of the initial C3 convertase of the alternative pathway of human complement. J Immunol (1984) 132:1430–4.
12. Lesavre PH, Hugli TE, Esser AF, Muller-Eberhard H, Müller-Eberhard HJ. The alternative pathway C3/C5 convertase: chemical basis of factor B activation. J Immunol (1979) 123:529–34.
13. He J, Wiesmann C, van Lookeren Campagne M. A role of macrophage complement receptor CRIg in immune clearance and inflammation. Mol Immunol (2008) 45:4041–7. doi:10.1016/j.molimm.2008.07.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Gulla KC, Gupta K, Hajela K. Functional estimation of mannose binding lectin associated serine protease (MBL-MASPs) in human serum. Indian J Med Res (2009) 130:428–32.
15. Fosbrink M, Niculescu F, Rus H. The role of c5b-9 terminal complement complex in activation of the cell cycle and transcription. Immunol Res (2005) 31:37–46. doi:10.1385/IR:31:1:37
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Jensen PH, Laursen I, Matthiesen F, Højrup P. Posttranslational modifications in human plasma MBL and human recombinant MBL. Biochim Biophys Acta (2007) 1774:335–44. doi:10.1016/j.bbapap.2006.12.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Bohlson SS, Fraser DA, Tenner AJ. Complement proteins C1q and MBL are pattern recognition molecules that signal immediate and long-term protective immune functions. Mol Immunol (2007) 44:33–43. doi:10.1016/j.molimm.2006.06.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Larsen F, Madsen HO, Sim RB, Koch C, Garred P. Disease-associated mutations in human mannose-binding lectin compromise oligomerization and activity of the final protein. J Biol Chem (2004) 279:21302–11. doi:10.1074/jbc.M400520200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Wallis R, Drickamer K. Molecular determinants of oligomer formation and complement fixation in mannose-binding proteins. J Biol Chem (1999) 274:3580–9. doi:10.1074/jbc.274.6.3580
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Chen CB, Wallis R. Stoichiometry of complexes between mannose-binding protein and its associated serine proteases. Defining functional units for complement activation. J Biol Chem (2001) 276:25894–902. doi:10.1074/jbc.M103539200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Holmskov U, Thiel S, Jensenius JC. Collections and ficolins: humoral lectins of the innate immune defense. Annu Rev Immunol (2003) 21:547–78. doi:10.1146/annurev.immunol.21.120601.140954
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Yokota Y, Arai T, Kawasaki T. Oligomeric structures required for complement activation of serum mannan-binding proteins. J Biochem (1995) 117:414–9. doi:10.1093/jb/117.2.414
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Ambrosio AR, De Messias-Reason IJ. Leishmania (Viannia) braziliensis: interaction of mannose-binding lectin with surface glycoconjugates and complement activation. An antibody-independent defence mechanism. Parasite Immunol (2005) 27:333–40. doi:10.1111/j.1365-3024.2005.00782.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Jack D, Turner M. Anti-microbial activities of mannose-binding lectin. Biochem Soc Trans (2003) 31:753–7. doi:10.1042/BST0310753
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Klabunde J, Berger J, Jensenius JC, Klinkert MQ, Zelck UE, Kremsner PG, et al. Schistosoma mansoni: adhesion of mannan-binding lectin to surface glycoproteins of cercariae and adult worms. Exp Parasitol (2000) 95:231–9. doi:10.1006/expr.2000.4539
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Klabunde J, Uhlemann A-C, Tebo AE, Kimmel J, Schwarz RT, Kremsner PG, et al. Recognition of plasmodium falciparum proteins by mannan-binding lectin, a component of the human innate immune system. Parasitol Res (2002) 88:113–7. doi:10.1007/s00436-001-0518-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Nauta AJ, Castellano G, Xu W, Woltman AM, Borrias MC, Daha MR, et al. Opsonization with C1q and mannose-binding lectin targets apoptotic cells to dendritic cells. J Immunol (2004) 173:3044–50. doi:10.4049/jimmunol.173.5.3044
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Weis WI, Drickamer K, Hendrickson WA. Structure of a C-type mannose-binding protein complexed with an oligosaccharide. Nature (1992) 360:127–34. doi:10.1038/360127a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Kilpatrick DC. Phospholipid-binding activity of human mannan-binding lectin. Immunol Lett (1998) 61:191–5. doi:10.1016/S0165-2478(98)00031-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Palaniyar N, Nadesalingam J, Reid KB. Innate immune collectins bind nucleic acids and enhance DNA clearance in vitro. Ann N Y Acad Sci (2003) 1010:467–70. doi:10.1196/annals.1299.084
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Estabrook MM, Jack DL, Klein NJ, Jarvis GA. Mannose-binding lectin binds to two major outer membrane proteins, opacity protein and porin, of Neisseria meningitidis. J Immunol (2004) 172:3784–92. doi:10.4049/jimmunol.172.6.3784
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Tenner AJ, Robinson SL, Ezekowitz RA. Mannose binding protein (MBP) enhances mononuclear phagocyte function via a receptor that contains the 126,000 M(r) component of the C1q receptor. Immunity (1995) 3:485–93. doi:10.1016/1074-7613(95)90177-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Kang HJ, Lee S-M, Lee H-H, Kim JY, Lee B-C, Yum J-S, et al. Mannose-binding lectin without the aid of its associated serine proteases alters lipopolysaccharide-mediated cytokine/chemokine secretion from human endothelial cells. Immunology (2007) 122:335–42. doi:10.1111/j.1365-2567.2007.02644.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Ogden CA, DeCathelineau A, Hoffmann PR, Bratton D, Ghebrehiwet B, Fadok VA, et al. C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J Exp Med (2001) 194:781–95. doi:10.1084/jem.194.6.781
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Stuart LM, Takahashi K, Shi L, Savill J, Ezekowitz RA. Mannose-binding lectin-deficient mice display defective apoptotic cell clearance but no autoimmune phenotype. J Immunol (2005) 174:3220–6. doi:10.4049/jimmunol.174.6.3220
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Sastry K, Herman GA, Day L, Deignan E, Bruns G, Morton CC, et al. The human mannose-binding protein gene. Exon structure reveals its evolutionary relationship to a human pulmonary surfactant gene and localization to chromosome 10. J Exp Med (1989) 170:1175–89. doi:10.1084/jem.170.4.1175
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Ezekowitz RA, Day LE, Herman GA. A human mannose-binding protein is an acute-phase reactant that shares sequence homology with other vertebrate lectins. J Exp Med (1988) 167:1034–46. doi:10.1084/jem.167.3.1034
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Arai T, Tabona P, Summerfield JA. Human mannose-binding protein gene is regulated by interleukins, dexamethasone and heat shock. Q J Med (1993) 86:575–82.
39. Kjaer TR, Thiel S, Andersen GR. Toward a structure-based comprehension of the lectin pathway of complement. Mol Immunol (2013) 56:413–22. doi:10.1016/j.molimm.2013.05.007
40. Thiel S, Holmskov U, Hviid L, Laursen SB, Jensenius JC. The concentration of the C-type lectin, mannan-binding protein, in human plasma increases during an acute phase response. Clin Exp Immunol (1992) 90:31–5. doi:10.1111/j.1365-2249.1992.tb05827.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Steffensen R, Thiel S, Varming K, Jersild C, Jensenius JC. Detection of structural gene mutations and promoter polymorphisms in the mannan-binding lectin (MBL) gene by polymerase chain reaction with sequence-specific primers. J Immunol Methods (2000) 241:33–42. doi:10.1016/S0022-1759(00)00198-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Aittoniemi J, Miettinen A, Laippala P, Isolauri E, Viikari J, Ruuska T, et al. Age-dependent variation in the serum concentration of mannan-binding protein. Acta Paediatr (1996) 85:906–9. doi:10.1111/j.1651-2227.1996.tb14182.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Terai I, Kobayashi K. Perinatal changes in serum mannose-binding protein (MBP) levels. Immunol Lett (1993) 38:185–7. doi:10.1016/0165-2478(93)90004-L
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Trégoat V, Montagne P, Béné M-C, Faure G. Changes in the mannan binding lectin (MBL) concentration in human milk during lactation. J Clin Lab Anal (2002) 16:304–7. doi:10.1002/jcla.10055
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Heitzeneder S, Seidel M, Förster-Waldl E, Heitger A. Mannan-binding lectin deficiency – good news, bad news, doesn’t matter? Clin Immunol (2012) 143:22–38. doi:10.1016/j.clim.2011.11.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Eisen DP, Osthoff M. If there is an evolutionary selection pressure for the high frequency of MBL2 polymorphisms, what is it? Clin Exp Immunol (2014) 176:165–71. doi:10.1111/cei.12241
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Aittoniemi J, Baer M, Soppi E, Vesikari T, Miettinen A. Mannan binding lectin deficiency and concomitant immunodefects. Arch Dis Child (1998) 78:245–8. doi:10.1136/adc.78.3.245
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Dahl M, Tybjaerg-Hansen A, Schnohr P, Nordestgaard BG. A population-based study of morbidity and mortality in mannose-binding lectin deficiency. J Exp Med (2004) 199:1391–9. doi:10.1084/jem.20040111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Dornelles LN, Pereira-Ferrari L, Messias-Reason I. Mannan-binding lectin plasma levels in leprosy: deficiency confers protection against the lepromatous but not the tuberculoid forms. Clin Exp Immunol (2006) 145:463–8. doi:10.1111/j.1365-2249.2006.03161.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. De Messias-Reason IJ, Boldt AB, Moraes Braga AC, Von Rosen Seeling Stahlke E, Dornelles L, Pereira-Ferrari L, et al. The association between mannan-binding lectin gene polymorphism and clinical leprosy: new insight into an old paradigm. J Infect Dis (2007) 196:1379–85. doi:10.1086/521627
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Santos IK, Costa CH, Krieger H, Feitosa MF, Zurakowski D, Fardin B, et al. Mannan-binding lectin enhances susceptibility to visceral leishmaniasis. Infect Immun (2001) 69:5212–5. doi:10.1128/IAI.69.8.5212-5215.2001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Søborg C, Madsen HO, Andersen AB, Lillebaek T, Kok-Jensen A, Garred P. Mannose-binding lectin polymorphisms in clinical tuberculosis. J Infect Dis (2003) 188:777–82. doi:10.1086/377183
53. Bernig T, Taylor JG, Foster CB, Staats B, Yeager M, Chanock SJ. Sequence analysis of the mannose-binding lectin (MBL2) gene reveals a high degree of heterozygosity with evidence of selection. Genes Immun (2004) 5:461–76. doi:10.1038/sj.gene.6364116
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Boldt AB, Messias-Reason IJ, Meyer D, Schrago CG, Lang F, Lell B, et al. Phylogenetic nomenclature and evolution of mannose-binding lectin (MBL2) haplotypes. BMC Genet (2010) 11:38. doi:10.1186/1471-2156-11-38
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Boldt AB, Culpi L, Tsuneto LT, de Souza IR, Kun JF, Petzl-Erler ML. Diversity of the MBL2 gene in various Brazilian populations and the case of selection at the mannose-binding lectin locus. Hum Immunol (2006) 67:722–34. doi:10.1016/j.humimm.2006.05.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Boldt AB, Luty A, Grobusch MP, Dietz K, Dzeing A, Kombila M, et al. Association of a new mannose-binding lectin variant with severe malaria in Gabonese children. Genes Immun (2006) 7:393–400. doi:10.1038/sj.gene.6364312
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Verdu P, Barreiro LB, Patin E, Gessain A, Cassar O, Kidd JR, et al. Evolutionary insights into the high worldwide prevalence of MBL2 deficiency alleles. Hum Mol Genet (2006) 15:2650–8. doi:10.1093/hmg/ddl193
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Sjöberg AP, Manderson GA, Mörgelin M, Day AJ, Heinegård D, Blom AM. Short leucine-rich glycoproteins of the extracellular matrix display diverse patterns of complement interaction and activation. Mol Immunol (2009) 46:830–9. doi:10.1016/j.molimm.2008.09.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Swingler TE, Waters JG, Davidson RK, Pennington CJ, Puente XS, Darrah C, et al. Degradome expression profiling in human articular cartilage. Arthritis Res Ther (2009) 11:R96. doi:10.1186/ar2741
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Sumiya M, Super M, Tabona P, Levinsky RJ, Arai T, Turner MW, et al. Molecular basis of opsonic defect in immunodeficient children. Lancet (1991) 337:1569–70. doi:10.1016/0140-6736(91)93263-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Madsen HO, Garred P, Thiel S, Kurtzhals JA, Lamm LU, Ryder LP, et al. Interplay between promoter and structural gene variants control basal serum level of mannan-binding protein. J Immunol (1995) 155:3013–20.
62. Madsen HO, Videm V, Svejgaard A, Svennevig JL, Garred P. Association of mannose-binding-lectin deficiency with severe atherosclerosis. Lancet (1998) 352:959–60. doi:10.1016/S0140-6736(05)61513-9
63. Ren Y, Ding Q, Zhang X. Ficolins and infectious diseases. Virol Sin (2014) 29:25–32. doi:10.1007/s12250-014-3421-2
64. Krarup A, Thiel S, Hansen A, Fujita T, Jensenius JC. L-ficolin is a pattern recognition molecule specific for acetyl groups. J Biol Chem (2004) 279:47513–9. doi:10.1074/jbc.M407161200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Rørvig S, Honore C, Larsson L-I, Ohlsson S, Pedersen CC, Jacobsen LC, et al. Ficolin-1 is present in a highly mobilizable subset of human neutrophil granules and associates with the cell surface after stimulation with fMLP. J Leukoc Biol (2009) 86:1439–49. doi:10.1189/jlb.1008606
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Wittenborn T, Thiel S, Jensen L, Nielsen HJ, Jensenius JC. Characteristics and biological variations of M-ficolin, a pattern recognition molecule, in plasma. J Innate Immun (2010) 2:167–80. doi:10.1159/000218324
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Liu Y, Endo Y, Iwaki D, Nakata M, Matsushita M, Wada I, et al. Human M-ficolin is a secretory protein that activates the lectin complement pathway. J Immunol (2005) 175:3150–6. doi:10.4049/jimmunol.175.5.3150
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Honoré C, Rørvig S, Munthe-Fog L, Hummelshøj T, Madsen HO, Borregaard N, et al. The innate pattern recognition molecule ficolin-1 is secreted by monocytes/macrophages and is circulating in human plasma. Mol Immunol (2008) 45:2782–9. doi:10.1016/j.molimm.2008.02.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Frederiksen PD, Thiel S, Larsen CB, Jensenius JC. M-ficolin, an innate immune defence molecule, binds patterns of acetyl groups and activates complement. Scand J Immunol (2005) 62:462–73. doi:10.1111/j.1365-3083.2005.01685.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Kjaer TR, Hansen AG, Sørensen UB, Nielsen O, Thiel S, Jensenius JC. Investigations on the pattern recognition molecule M-ficolin: quantitative aspects of bacterial binding and leukocyte association. J Leukoc Biol (2011) 90:425–37. doi:10.1189/jlb.0411201
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Gout E, Garlatti V, Smith DF, Lacroix M, Dumestre-Pérard C, Lunardi T, et al. Carbohydrate recognition properties of human ficolins: glycan array screening reveals the sialic acid binding specificity of M-ficolin. J Biol Chem (2010) 285:6612–22. doi:10.1074/jbc.M109.065854
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Tanio M, Kondo S, Sugio S, Kohno T. Trivalent recognition unit of innate immunity system: crystal structure of trimeric human M-ficolin fibrinogen-like domain. J Biol Chem (2007) 282:3889–95. doi:10.1074/jbc.M608627200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Ma YJ, Doni A, Romani L, Jürgensen HJ, Behrendt N, Mantovani A, et al. Ficolin-1-PTX3 complex formation promotes clearance of altered self-cells and modulates IL-8 production. J Immunol (2013) 191:1324–33. doi:10.4049/jimmunol.1300382
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Ammitzbøll CG, Kjær TR, Steffensen R, Stengaard-Pedersen K, Nielsen HJ, Thiel S, et al. Non-synonymous polymorphisms in the FCN1 gene determine ligand-binding ability and serum levels of M-ficolin. PLoS One (2012) 7:e50585. doi:10.1371/journal.pone.0050585
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Munthe-Fog L, Hummelshoj T, Honoré C, Moller ME, Skjoedt MO, Palsgaard I, et al. Variation in FCN1 affects biosynthesis of ficolin-1 and is associated with outcome of systemic inflammation. Genes Immun (2012) 13:515–22. doi:10.1038/gene.2012.27
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Vander Cruyssen B, Nuytinck L, Boullart L, Elewaut D, Waegeman W, Van Thielen M, et al. Polymorphisms in the ficolin 1 gene (FCN1) are associated with susceptibility to the development of rheumatoid arthritis. Rheumatology (Oxford) (2007) 46:1792–5. doi:10.1093/rheumatology/kem266
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Schlapbach LJ, Kessler U, Thiel S, Hansen AG, Nelle M, Ammann RA, et al. M-ficolin in the neonatal period: associations with need for mechanical ventilation and mortality in premature infants with necrotising enterocolitis. Mol Immunol (2009) 46:2597–603. doi:10.1016/j.molimm.2009.05.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Ameye L, Paesmans M, Thiel S, Jensenius JC, Aoun M. M-ficolin levels are associated with the occurrence of severe infections in patients with haematological cancer undergoing chemotherapy. Clin Exp Immunol (2012) 167:303–8. doi:10.1111/j.1365-2249.2011.04512.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Matsushita M, Endo Y, Taira S, Sato Y, Fujita T, Ichikawa N, et al. A novel human serum lectin with collagen- and fibrinogen-like domains that functions as an opsonin. J Biol Chem (1996) 271:2448–54. doi:10.1074/jbc.271.5.2448
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Hummelshoj T, Fog LM, Madsen HO, Sim RB, Garred P. Comparative study of the human ficolins reveals unique features of ficolin-3 (Hakata antigen). Mol Immunol (2008) 45:1623–32. doi:10.1016/j.molimm.2007.10.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Munthe-Fog L, Hummelshøj T, Hansen BE, Koch C, Madsen HO, Skjødt K, et al. The impact of FCN2 polymorphisms and haplotypes on the ficolin-2 serum levels. Scand J Immunol (2007) 65:383–92. doi:10.1111/j.1365-3083.2007.01915.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Le Y, Lee SH, Kon OL, Lu J. Human L-ficolin: plasma levels, sugar specificity, and assignment of its lectin activity to the fibrinogen-like (FBG) domain. FEBS Lett (1998) 425:367–70. doi:10.1016/S0014-5793(98)00267-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Faro J, Chen Y, Jhaveri P, Oza P, Spear GT, Lint TF, et al. L-ficolin binding and lectin pathway activation by acetylated low-density lipoprotein. Clin Exp Immunol (2008) 151:275–83. doi:10.1111/j.1365-2249.2007.03538.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Aoyagi Y, Adderson EE, Rubens CE, Bohnsack JF, Min JG, Matsushita M, et al. L-ficolin/mannose-binding lectin-associated serine protease complexes bind to group B streptococci primarily through N-acetylneuraminic acid of capsular polysaccharide and activate the complement pathway. Infect Immun (2008) 76:179–88. doi:10.1128/IAI.00837-07
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Ma YG, Cho MY, Zhao M, Park JW, Matsushita M, Fujita T, et al. Human mannose-binding lectin and L-ficolin function as specific pattern recognition proteins in the lectin activation pathway of complement. J Biol Chem (2004) 279:25307–12. doi:10.1074/jbc.M400701200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Liu J, Ali MA, Shi Y, Zhao Y, Luo F, Yu J, et al. Specifically binding of L-ficolin to N-glycans of HCV envelope glycoproteins E1 and E2 leads to complement activation. Cell Mol Immunol (2009) 6:235–44. doi:10.1038/cmi.2009.32
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Carroll MV, Lack N, Sim E, Krarup A, Sim RB. Multiple routes of complement activation by Mycobacterium bovis BCG. Mol Immunol (2009) 46:3367–78. doi:10.1016/j.molimm.2009.07.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Evans-Osses I, Ansa-Addo EA, Inal JM, Ramirez MI. Involvement of lectin pathway activation in the complement killing of Giardia intestinalis. Biochem Biophys Res Commun (2010) 395:382–6. doi:10.1016/j.bbrc.2010.04.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Cestari Idos S, Krarup A, Sim RB, Inal JM, Ramirez MI. Role of early lectin pathway activation in the complement-mediated killing of Trypanosoma cruzi. Mol Immunol (2009) 47:426–37. doi:10.1016/j.molimm.2009.08.030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Zhang J, Koh J, Lu J, Thiel S, Leong BS, Sethi S, et al. Local inflammation induces complement crosstalk which amplifies the antimicrobial response. PLoS Pathog (2009) 5:e1000282. doi:10.1371/journal.ppat.1000282
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Hummelshoj T, Munthe-Fog L, Madsen HO, Fujita T, Matsushita M, Garred P. Polymorphisms in the FCN2 gene determine serum variation and function of ficolin-2. Hum Mol Genet (2005) 14:1651–8. doi:10.1093/hmg/ddi173
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Herpers BL, Immink M-M, de Jong BA, van Velzen-Blad H, de Jongh BM, van Hannen EJ. Coding and non-coding polymorphisms in the lectin pathway activator L-ficolin gene in 188 Dutch blood bank donors. Mol Immunol (2006) 43:851–5. doi:10.1016/j.molimm.2005.06.035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Garred P, Honoré C, Ma YJ, Rørvig S, Cowland J, Borregaard N, et al. The genetics of ficolins. J Innate Immun (2010) 2:3–16. doi:10.1159/000242419
94. Kilpatrick DC, Chalmers JD, MacDonald SL, Murray M, Mohammed A, Hart SP, et al. Stable bronchiectasis is associated with low serum L-ficolin concentrations. Clin Respir J (2009) 3:29–33. doi:10.1111/j.1752-699X.2008.00105.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Atkinson AP, Cedzynski M, Szemraj J, St Swierzko A, Bak-Romaniszyn L, Banasik M, et al. L-ficolin in children with recurrent respiratory infections. Clin Exp Immunol (2004) 138:517–20. doi:10.1111/j.1365-2249.2004.02634.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Cedzynski M, Atkinson AP, St Swierzko A, MacDonald SL, Szala A, Zeman K, et al. L-ficolin (ficolin-2) insufficiency is associated with combined allergic and infectious respiratory disease in children. Mol Immunol (2009) 47:415–9. doi:10.1016/j.molimm.2009.08.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Chapman SJ, Vannberg FO, Khor CC, Segal S, Moore CE, Knox K, et al. Functional polymorphisms in the FCN2 gene are not associated with invasive pneumococcal disease. Mol Immunol (2007) 44:3267–70. doi:10.1016/j.molimm.2006.04.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Swierzko AS, Atkinson AP, Cedzynski M, Macdonald SL, Szala A, Domzalska- Popadiuk I, et al. Two factors of the lectin pathway of complement, l-ficolin and mannan-binding lectin, and their associations with prematurity, low birthweight and infections in a large cohort of Polish neonates. Mol Immunol (2009) 46:551–8. doi:10.1016/j.molimm.2008.07.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Luz PR, Boldt AB, Grisbach C, Kun JF, Velavan TP, Messias-Reason IJ. Association of L-ficolin levels and FCN2 genotypes with chronic Chagas disease. PLoS One (2013) 8:e60237. doi:10.1371/journal.pone.0060237
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Faik I, Oyedeji SI, Idris Z, de Messias-Reason IJ, Lell B, Kremsner PG, et al. Ficolin-2 levels and genetic polymorphisms of FCN2 in malaria. Hum Immunol (2011) 72:74–9. doi:10.1016/j.humimm.2010.10.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. De Messias-Reason I, Kremsner PG, Kun JF. Functional haplotypes that produce normal ficolin-2 levels protect against clinical leprosy. J Infect Dis (2009) 199:801–4. doi:10.1086/597070
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Krarup A, Sørensen UB, Matsushita M, Jensenius JC, Thiel S. Effect of capsulation of opportunistic pathogenic bacteria on binding of the pattern recognition molecules mannan-binding lectin, L-ficolin, and H-ficolin. Infect Immun (2005) 73:1052–60. doi:10.1128/IAI.73.2.1052-1060.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Munthe-Fog L, Hummelshøj T, Ma YJ, Hansen BE, Koch C, Madsen HO, et al. Characterization of a polymorphism in the coding sequence of FCN3 resulting in a ficolin-3 (Hakata antigen) deficiency state. Mol Immunol (2008) 45:2660–6. doi:10.1016/j.molimm.2007.12.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Akaiwa M, Yae Y, Sugimoto R, Suzuki SO, Iwaki T, Izuhara K, et al. Hakata antigen, a new member of the ficolin/opsonin p35 family, is a novel human lectin secreted into bronchus/alveolus and bile. J Histochem Cytochem (1999) 47:777–86. doi:10.1177/002215549904700607
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Matsushita M. Ficolins in complement activation. Mol Immunol (2013) 55:22–6. doi:10.1016/j.molimm.2012.08.017
107. Sugimoto R, Yae Y, Akaiwa M, Kitajima S, Shibata Y, Sato H, et al. Cloning and characterization of the Hakata antigen, a member of the ficolin/opsonin p35 lectin family. J Biol Chem (1998) 273:20721–7. doi:10.1074/jbc.273.33.20721
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Tsujimura M, Miyazaki T, Kojima E, Sagara Y, Shiraki H, Okochi K, et al. Serum concentration of Hakata antigen, a member of the ficolins, is linked with inhibition of Aerococcus viridans growth. Clin Chim Acta (2002) 325:139–46. doi:10.1016/S0009-8981(02)00274-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Kuraya M, Ming Z, Liu X, Matsushita M, Fujita T. Specific binding of L-ficolin and H-ficolin to apoptotic cells leads to complement activation. Immunobiology (2005) 209:689–97. doi:10.1016/j.imbio.2004.11.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Honoré C, Hummelshoj T, Hansen BE, Madsen HO, Eggleton P, Garred P. The innate immune component ficolin 3 (Hakata antigen) mediates the clearance of late apoptotic cells. Arthritis Rheum (2007) 56:1598–607. doi:10.1002/art.22564
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Thiel S. Complement activating soluble pattern recognition molecules with collagen-like regions, mannan-binding lectin, ficolins and associated proteins. Mol Immunol (2007) 44:3875–88. doi:10.1016/j.molimm.2007.06.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Schlapbach LJ, Thiel S, Kessler U, Ammann RA, Aebi C, Jensenius JC. Congenital H-ficolin deficiency in premature infants with severe necrotising enterocolitis. Gut (2011) 60:1438–9. doi:10.1136/gut.2010.226027
113. Thiel S, Vorup-Jensen T, Stover CM, Schwaeble W, Laursen S, Poulsen K, et al. A second serine protease associated with mannan-binding lectin that activates complement. Nature (1997) 386:506–10. doi:10.1038/386506a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Thiel S, Kolev M, Degn S, Steffensen R, Hansen AG, Ruseva M, et al. Polymorphisms in mannan-binding lectin (MBL)-associated serine protease 2 affect stability, binding to MBL, and enzymatic activity. J Immunol (2009) 182:2939–47. doi:10.4049/jimmunol.0802053
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Degn SE, Jensen L, Hansen AG, Duman D, Tekin M, Jensenius JC, et al. Mannan-binding lectin-associated serine protease (MASP)-1 is crucial for lectin pathway activation in human serum, whereas neither MASP-1 nor MASP-3 is required for alternative pathway function. J Immunol (2012) 189:3957–69. doi:10.4049/jimmunol.1201736
116. Héja D, Harmat V, Fodor K, Wilmanns M, Dobó J, Kékesi KA, et al. Monospecific inhibitors show that both mannan-binding lectin-associated serine protease-1 (MASP-1) and -2 are essential for lectin pathway activation and reveal structural plasticity of MASP-2. J Biol Chem (2012) 287:20290–300. doi:10.1074/jbc.M112.354332
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Møller-Kristensen M, Thiel S, Sjöholm A, Matsushita M, Jensenius JC. Cooperation between MASP-1 and MASP-2 in the generation of C3 convertase through the MBL pathway. Int Immunol (2007) 19:141–9. doi:10.1093/intimm/dxl131
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Degn SE, Jensen L, Gál P, Dobó J, Holmvad SH, Jensenius JC, et al. Biological variations of MASP-3 and MAp44, two splice products of the MASP1 gene involved in regulation of the complement system. J Immunol Methods (2010) 361:37–50. doi:10.1016/j.jim.2010.07.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Skjoedt M-O, Hummelshoj T, Palarasah Y, Honore C, Koch C, Skjodt K, et al. A novel mannose-binding lectin/ficolin-associated protein is highly expressed in heart and skeletal muscle tissues and inhibits complement activation. J Biol Chem (2010) 285:8234–43. doi:10.1074/jbc.M109.065805
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Degn SE, Jensenius JC, Thiel S. Disease-causing mutations in genes of the complement system. Am J Hum Genet (2011) 88:689–705. doi:10.1016/j.ajhg.2011.05.011
121. Degn SE, Hansen AG, Steffensen R, Jacobsen C, Jensenius JC, Thiel S. MAp44, a human protein associated with pattern recognition molecules of the complement system and regulating the lectin pathway of complement activation. J Immunol (2009) 183:7371–8. doi:10.4049/jimmunol.0902388
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
122. Degn SE, Thiel S, Nielsen O, Hansen AG, Steffensen R, Jensenius JC. MAp19, the alternative splice product of the MASP2 gene. J Immunol Methods (2011) 373:89–101. doi:10.1016/j.jim.2011.08.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Ikeda K, Sannoh T, Kawasaki N, Kawasaki T, Yamashina I. Serum lectin with known structure activates complement through the classical pathway. J Biol Chem (1987) 262:7451–4.
124. Thiel S, Jensen L, Degn SE, Nielsen HJ, Gál P, Dobó J, et al. Mannan-binding lectin (MBL)-associated serine protease-1 (MASP-1), a serine protease associated with humoral pattern-recognition molecules: normal and acute-phase levels in serum and stoichiometry of lectin pathway components. Clin Exp Immunol (2012) 169:38–48. doi:10.1111/j.1365-2249.2012.04584.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Ytting H, Christensen IJ, Thiel S, Jensenius JC, Svendsen MN, Nielsen L, et al. Biological variation in circulating levels of mannan-binding lectin (MBL) and MBL-associated serine protease-2 and the influence of age, gender and physical exercise. Scand J Immunol (2007) 66:458–64. doi:10.1111/j.1365-3083.2007.01991.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Dahl M, Thiel S, Matsushita M, Fujita T, Willis A, Christensen T, et al. MASP-3 and its association with distinct complexes of the mannan-binding lectin complement activation pathway. Immunity (2001) 15:127–35. doi:10.1016/S1074-7613(01)00161-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Stover CM, Schwaeble WJ, Lynch NJ, Thiel S, Speicher MR. Assignment of the gene encoding mannan-binding lectin-associated serine protease 2 (MASP2) to human chromosome 1p36.3 – >p36.2 by in situ hybridization and somatic cell hybrid analysis. Cytogenet Cell Genet (1999) 84:148–9. doi:10.1159/000015243
128. Sørensen R, Thiel S, Jensenius JC. Mannan-binding-lectin-associated serine proteases, characteristics and disease associations. Springer Semin Immunopathol (2005) 27:299–319. doi:10.1007/s00281-005-0006-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Thiel S, Steffensen R, Christensen IJ, Ip WK, Lau YL, Reason IJ, et al. Deficiency of mannan-binding lectin associated serine protease-2 due to missense polymorphisms. Genes Immun (2007) 8:154–63. doi:10.1038/sj.gene.6364373
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
130. Ammitzbøll CG, Steffensen R, Jørgen Nielsen H, Thiel S, Stengaard-Pedersen K, Bøgsted M, et al. Polymorphisms in the MASP1 gene are associated with serum levels of MASP-1, MASP-3, and MAp44. PLoS One (2013) 8:e73317. doi:10.1371/journal.pone.0073317
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Rooryck C, Diaz-Font A, Osborn DP, Chabchoub E, Hernandez-Hernandez V, Shamseldin H, et al. Mutations in the lectin complement pathway genes COLEC11 and MASP1 cause 3MC syndrome. Nat Genet (2011) 43:197–203. doi:10.1038/ng.757.Mutations
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
132. Yongqing T, Wilmann PG, Reeve SB, Coetzer TH, Smith AI, Whisstock JC, et al. The x-ray crystal structure of mannose-binding lectin-associated serine proteinase-3 reveals the structural basis for enzyme inactivity associated with the Carnevale, Mingarelli, Malpuech, and Michels (3MC) syndrome. J Biol Chem (2013) 288:22399–407. doi:10.1074/jbc.M113.483875
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
133. Sirmaci A, Walsh T, Akay H, Spiliopoulos M, Sakalar YB, Hasanefendioglu-Bayrak A, et al. MASP1 mutations in patients with facial, umbilical, coccygeal, and auditory findings of Carnevale, Malpuech, OSA, and Michels syndromes. Am J Hum Genet (2010) 87:679–86. doi:10.1016/j.ajhg.2010.09.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
134. Haerynck F, Van Steen K, Cattaert T, Loeys B, Van Daele S, Schelstraete P, et al. Polymorphisms in the lectin pathway genes as a possible cause of early chronic Pseudomonas aeruginosa colonization in cystic fibrosis patients. Hum Immunol (2012) 73:1175–83. doi:10.1016/j.humimm.2012.08.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
135. Ingels C, Vanhorebeek I, Steffensen R, Derese I, Jensen L, Wouters PJ, et al. Lectin pathway of complement activation and relation with clinical complications in critically ill children. Pediatr Res (2014) 75:99–108. doi:10.1038/pr.2013.180
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. Boldt AB, Goeldner I, Stahlke ER, Thiel S, Jensenius JC, de Messias-Reason IJ. Leprosy association with low MASP-2 levels generated by MASP2 haplotypes and polymorphisms flanking MAp19 exon 5. PLoS One (2013) 8:e69054. doi:10.1371/journal.pone.0069054
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
137. Coelho AV, Brandão LA, Guimarães RL, Loureiro P, de Lima Filho JL, de Alencar LC, et al. Mannose binding lectin and mannose binding lectin-associated serine protease-2 genes polymorphisms in human T-lymphotropic virus infection. J Med Virol (2013) 85:1829–35. doi:10.1002/jmv.23656
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
138. Holmberg V, Onkamo P, Lahtela E, Lahermo P, Bedu-Addo G, Mockenhaupt FP, et al. Mutations of complement lectin pathway genes MBL2 and MASP2 associated with placental malaria. Malar J (2012) 11:61. doi:10.1186/1475-2875-11-61
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
139. Boldt AB, Luz PR, Messias-Reason IJ. MASP2 haplotypes are associated with high risk of cardiomyopathy in chronic Chagas disease. Clin Immunol (2011) 140:63–70. doi:10.1016/j.clim.2011.03.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
140. De Rooij B-JF, van Hoek B, ten Hove WR, Roos A, Bouwman LH, Schaapherder AF, et al. Lectin complement pathway gene profile of donor and recipient determine the risk of bacterial infections after orthotopic liver transplantation. Hepatology (2010) 52:1100–10. doi:10.1002/hep.23782
141. Tulio S, Faucz FR, Werneck RI, Olandoski M, Alexandre RB, Boldt AB, et al. MASP2 gene polymorphism is associated with susceptibility to hepatitis C virus infection. Hum Immunol (2011) 72:912–5. doi:10.1016/j.humimm.2011.06.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
142. Mayilyan KR, Arnold JN, Presanis JS, Soghoyan AF, Sim RB. Increased complement classical and mannan-binding lectin pathway activities in schizophrenia. Neurosci Lett (2006) 404:336–41. doi:10.1016/j.neulet.2006.06.051
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
143. Charchaflieh J, Wei J, Labaze G, Hou YJ, Babarsh B, Stutz H, et al. The role of complement system in septic shock. Clin Dev Immunol (2012) 2012:407324. doi:10.1155/2012/407324
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
144. Fisch UP, Zehnder A, Hirt A, Niggli FK, Simon A, Ozsahin H, et al. Mannan-binding lectin (MBL) and MBL-associated serine protease-2 in children with cancer. Swiss Med Wkly (2011) 141:w13191. doi:10.4414/smw.2011.13191
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
145. Ytting H, Christensen IJ, Thiel S, Jensenius JC, Nielsen HJ. Serum mannan-binding lectin-associated serine protease 2 levels in colorectal cancer: relation to recurrence and mortality. Clin Cancer Res (2005) 11:1441–6. doi:10.1158/1078-0432.CCR-04-1272
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
146. Ytting H, Christensen IJ, Thiel S, Jensenius JC, Nielsen HJ. Pre- and postoperative levels in serum of mannan-binding lectin associated serine protease-2 – a prognostic marker in colorectal cancer. Hum Immunol (2008) 69:414–20. doi:10.1016/j.humimm.2008.05.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
147. Lappegård KT, Garred P, Jonasson L, Espevik T, Aukrust P, Yndestad A, et al. A vital role for complement in heart disease. Mol Immunol (2014) 61:126–34. doi:10.1016/j.molimm.2014.06.036
148. Best LG, Davidson M, North KE, MacCluer JW, Zhang Y, Lee ET, et al. Prospective analysis of mannose-binding lectin genotypes and coronary artery disease in American Indians: the Strong Heart Study. Circulation (2004) 109:471–5. doi:10.1161/01.CIR.0000109757.95461.10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
149. Keller TT, van Leuven SI, Meuwese MC, Wareham NJ, Luben R, Stroes ES, et al. Serum levels of mannose-binding lectin and the risk of future coronary artery disease in apparently healthy men and women. Arterioscler Thromb Vasc Biol (2006) 26:2345–50. doi:10.1161/01.ATV.0000240517.69201.77
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
150. Biezeveld MH, Geissler J, Weverling GJ, Kuipers IM, Lam J, Ottenkamp J, et al. Polymorphisms in the mannose-binding lectin gene as determinants of age-defined risk of coronary artery lesions in Kawasaki disease. Arthritis Rheum (2006) 54:369–76. doi:10.1002/art.21529
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
151. Saevarsdottir S, Oskarsson OO, Aspelund T, Eiriksdottir G, Vikingsdottir T, Gudnason V, et al. Mannan binding lectin as an adjunct to risk assessment for myocardial infarction in individuals with enhanced risk. J Exp Med (2005) 201:117–25. doi:10.1084/jem.20041431
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
152. Vengen IT, Madsen HO, Garred P, Platou C, Vatten L, Videm V. Mannose-binding lectin deficiency is associated with myocardial infarction: the HUNT2 study in Norway. PLoS One (2012) 7:e42113. doi:10.1371/journal.pone.0042113
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
153. Ueland T, Espevik T, Kjekshus J, Gullestad L, Omland T, Squire IB, et al. Mannose binding lectin and soluble toll-like receptor 2 in heart failure following acute myocardial infarction. J Card Fail (2006) 12:659–63. doi:10.1016/j.cardfail.2006.07.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
154. Walsh MC, Bourcier T, Takahashi K, Shi L, Busche MN, Rother RP, et al. Mannose-binding lectin is a regulator of inflammation that accompanies myocardial ischemia and reperfusion injury. J Immunol (2005) 175:541–6. doi:10.4049/jimmunol.175.1.541
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
155. Jordan JE, Montalto MC, Stahl GL. Inhibition of mannose-binding lectin reduces postischemic myocardial reperfusion injury. Circulation (2001) 104:1413–8. doi:10.1161/hc3601.095578
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
156. Schoos MM, Munthe-Fog L, Skjoedt M-O, Ripa RS, Lønborg J, Kastrup J, et al. Association between lectin complement pathway initiators, C-reactive protein and left ventricular remodeling in myocardial infarction-a magnetic resonance study. Mol Immunol (2013) 54:408–14. doi:10.1016/j.molimm.2013.01.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
157. Prohászka Z, Munthe-Fog L, Ueland T, Gombos T, Yndestad A, Förhécz Z, et al. Association of ficolin-3 with severity and outcome of chronic heart failure. PLoS One (2013) 8:e60976. doi:10.1371/journal.pone.0060976
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
158. Frauenknecht V, Thiel S, Storm L, Meier N, Arnold M, Schmid J-P, et al. Plasma levels of mannan-binding lectin (MBL)-associated serine proteases (MASPs) and MBL-associated protein in cardio- and cerebrovascular diseases. Clin Exp Immunol (2013) 173:112–20. doi:10.1111/cei.12093
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
159. Zhang M, Hou YJ, Cavusoglu E, Lee DC, Steffensen R, Yang L, et al. MASP-2 activation is involved in ischemia-related necrotic myocardial injury in humans. Int J Cardiol (2013) 166:499–504. doi:10.1016/j.ijcard.2011.11.032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
160. Neth O, Jack DL, Dodds AW, Holzel H, Klein NJ, Turner MW. Mannose-binding lectin binds to a range of clinically relevant microorganisms and promotes complement deposition. Infect Immun (2000) 68:688–93. doi:10.1128/IAI.68.2.688-693.2000
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
161. Boldt BW, Goeldner I, de Messias-Reason IJ. Relevance of the lectin pathway of complement in rheumatic diseases. Adv Clin Chem (2012) 56:105–53. doi:10.1016/B978-0-12-394317-0.00012-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
162. Lynch NJ, Roscher S, Hartung T, Morath S, Matsushita M, Maennel DN, et al. L-ficolin specifically binds to lipoteichoic acid, a cell wall constituent of Gram-positive bacteria, and activates the lectin pathway of complement. J Immunol (2004) 172:1198–202. doi:10.4049/jimmunol.172.2.1198
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
163. Endo Y, Takahashi M, Iwaki D, Ishida Y, Nakazawa N, Kodama T, et al. Mice deficient in ficolin, a lectin complement pathway recognition molecule, are susceptible to Streptococcus pneumoniae infection. J Immunol (2012) 189:5860–6. doi:10.4049/jimmunol.1200836
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
164. Jin Z, Ji Z, Hu J. Mannose-binding lectin gene site mutations and the susceptibility of rheumatic heart disease. Zhonghua Yi Xue Za Zhi (2001) 81:1284–6.
165. Scalzi V, Hadi HA, Alessandri C, Croia C, Conti V, Agati L, et al. Anti-endothelial cell antibodies in rheumatic heart disease. Clin Exp Immunol (2010) 161:570–5. doi:10.1111/j.1365-2249.2010.04207.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
166. Schafranski MD, Stier A, Nisihara R, Messias-Reason IJ. Significantly increased levels of mannose-binding lectin (MBL) in rheumatic heart disease: a beneficial role for MBL deficiency. Clin Exp Immunol (2004) 138:521–5. doi:10.1111/j.1365-2249.2004.02645.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
167. Messias Reason IJ, Schafranski MD, Jensenius JC, Steffensen R. The association between mannose-binding lectin gene polymorphism and rheumatic heart disease. Hum Immunol (2006) 67:991–8. doi:10.1016/j.humimm.2006.08.296
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
168. Schafranski MD, Pereira Ferrari L, Scherner D, Torres R, Jensenius JC, de Messias-Reason IJ. High-producing MBL2 genotypes increase the risk of acute and chronic carditis in patients with history of rheumatic fever. Mol Immunol (2008) 45:3827–31. doi:10.1016/j.molimm.2008.05.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
169. Collard CD, Väkevä A, Morrissey MA, Agah A, Rollins SA, Reenstra WR, et al. Complement activation after oxidative stress: role of the lectin complement pathway. Am J Pathol (2000) 156:1549–56. doi:10.1016/S0002-9440(10)65026-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
170. Nayar S, Nayar PG, Cherian KM. Heart valve structure: a predisposing factor for rheumatic heart disease. Heart (2006) 92:1151–2. doi:10.1136/hrt.2005.075358
171. Ramasawmy R, Spina GS, Fae KC, Pereira AC, Nisihara R, Messias Reason IJ, et al. Association of mannose-binding lectin gene polymorphism but not of mannose-binding serine protease 2 with chronic severe aortic regurgitation of rheumatic etiology. Clin Vaccine Immunol (2008) 15:932–6. doi:10.1128/CVI.00324-07
172. Stengaard-Pedersen K, Thiel S, Gadjeva M, Møller-Kristensen M, Sørensen R, Jensen LT, et al. Inherited deficiency of mannan-binding lectin-associated serine protease 2. N Engl J Med (2003) 349:554–60. doi:10.1056/NEJMoa022836
173. Vallès X, Sarrias M-R, Casals F, Farnós M, Piñer R, Suárez B, et al. Genetic and structural analysis of MBL2 and MASP2 polymorphisms in south-eastern African children. Tissue Antigens (2009) 74:298–307. doi:10.1111/j.1399-0039.2009.01328.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
174. Schafranski MD, Pereira Ferrari L, Scherner D, Torres R, de Messias-Reason IJ. Functional MASP2 gene polymorphism in patients with history of rheumatic fever. Hum Immunol (2008) 69:41–4. doi:10.1016/j.humimm.2007.11.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
175. García-Laorden MI, García-Saavedra A, de Castro FR, Violán JS, Rajas O, Blanquer J, et al. Low clinical penetrance of mannose-binding lectin-associated serine protease 2 deficiency. J Allergy Clin Immunol (2006) 118:1383–6. doi:10.1016/j.jaci.2006.08.004
176. Olesen H, Jensenius JC, Steffensen R, Thiel S, Schiotz P. The mannan-binding lectin pathway and lung disease in cystic fibrosis-disfunction of mannan-binding lectin-associated serine protease 2 (MASP-2) may be a major modifier. Clin Immunol (2006) 121:324–31. doi:10.1016/j.clim.2006.08.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
177. Miller C, Wilgenbusch S, Michaels M, Chi DS, Youngberg G, Krishnaswamy G. Molecular defects in the mannose binding lectin pathway in dermatological disease: case report and literature review. Clin Mol Allergy (2010) 8:6. doi:10.1186/1476-7961-8-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
178. Catarino SJDS, Boldt AB, Beltrame MH, Nisihara RM, Schafranski MD, de Messias-Reason IJ. Association of MASP2 polymorphisms and protein levels with rheumatic fever and rheumatic heart disease. Hum Immunol (2014) 75(12):1197–202. doi:10.1016/j.humimm.2014.10.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
179. Matsushita M, Thiel S, Jensenius JC, Terai I, Fujita T. Proteolytic activities of two types of mannose-binding lectin-associated serine protease. J Immunol (2000) 165:2637–42. doi:10.4049/jimmunol.165.5.2637
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
180. Kilpatrick DC, Chalmers JD. Human L-ficolin (ficolin-2) and its clinical significance. J Biomed Biotechnol (2012) 2012:138797. doi:10.1155/2012/138797
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
181. Messias-Reason IJ, Schafranski MD, Kremsner PG, Kun JF. Ficolin 2 (FCN2) functional polymorphisms and the risk of rheumatic fever and rheumatic heart disease. Clin Exp Immunol (2009) 157:395–9. doi:10.1111/j.1365-2249.2009.03975.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: lectin pathway, complement system, MBL, ficolins, gene polymorphisms
Citation: Beltrame MH, Catarino SJ, Goeldner I, Boldt ABW and de Messias-Reason IJ (2015) The lectin pathway of complement and rheumatic heart disease. Front. Pediatr. 2:148. doi: 10.3389/fped.2014.00148
Received: 28 July 2014; Paper pending published: 03 September 2014;
Accepted: 29 December 2014; Published online: 21 January 2015.
Edited by:
Luiza Guilherme, University of São Paulo, BrazilReviewed by:
Seppo Meri, University of Helsinki, FinlandCopyright: © 2015 Beltrame, Catarino, Goeldner, Boldt and de Messias-Reason. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Iara José de Messias Reason, Departamento de Patologia Clínica, Setor de Ciências da Saúde, Hospital das Clínicas, Universidade Federal do Paraná, Rua General Carneiro, 181, Curitiba 80060-900, PR, Brazil e-mail:aWFyYXJlYXNvbkBoYy51ZnByLmJy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.