 Fiorela N. Hernandez Tejada
Fiorela N. Hernandez Tejada Jorge R. Galvez Silva
Jorge R. Galvez Silva Patrick A. Zweidler-McKay
Patrick A. Zweidler-McKay- Department of Pediatrics, University of Texas M. D. Anderson Cancer Center, Houston, TX, USA
Notch signaling can play oncogenic and tumor suppressor roles depending on cell type. Hematologic malignancies encompass a wide range of transformed cells, and consequently the roles of Notch are diverse in these diseases. For example Notch is a potent T-cell oncogene, with >50% of T-cell acute lymphoblastic leukemia (T-ALL) cases carry activating mutations in the Notch1 receptor. Targeting Notch signaling in T-ALL with gamma-secretase inhibitors, which prevent Notch receptor activation, has shown pre-clinical activity, and is under evaluation clinically. In contrast, Notch signaling inhibits acute myeloblastic leukemia growth and survival, and although targeting Notch signaling in AML with Notch activators appears to have pre-clinical activity, no Notch agonists are clinically available at this time. As such, despite accumulating evidence about the biology of Notch signaling in different hematologic cancers, which provide compelling clinical promise, we are only beginning to target this pathway clinically, either on or off. In this review, we will summarize the evidence for oncogenic and tumor suppressor roles of Notch in a wide range of leukemias and lymphomas, and describe therapeutic opportunities for now and the future.
Introduction
“Notch” is a critical developmental pathway, which controls cell fate, differentiation, proliferation, and survival, and is critical in numerous developmental processes including neurogenesis, angiogenesis, and hematopoiesis, among others (1). The ability of Notch signaling to inhibit or induce differentiation, drive or impair proliferation, and promote survival or induce apoptosis in a cell-specific manner provides the opportunity for Notch signaling to contribute to or impede oncogenesis in multiple cell types (2).
The role of Notch has been studied in a wide variety of hematological malignancies including T and B leukemias and lymphomas as well as myeloid leukemias (3–5). However, the consequences of Notch signaling in these diseases varies significantly, and thus targeting Notch, requires understanding the biology of Notch signaling on a disease-by-disease basis (Table 1).
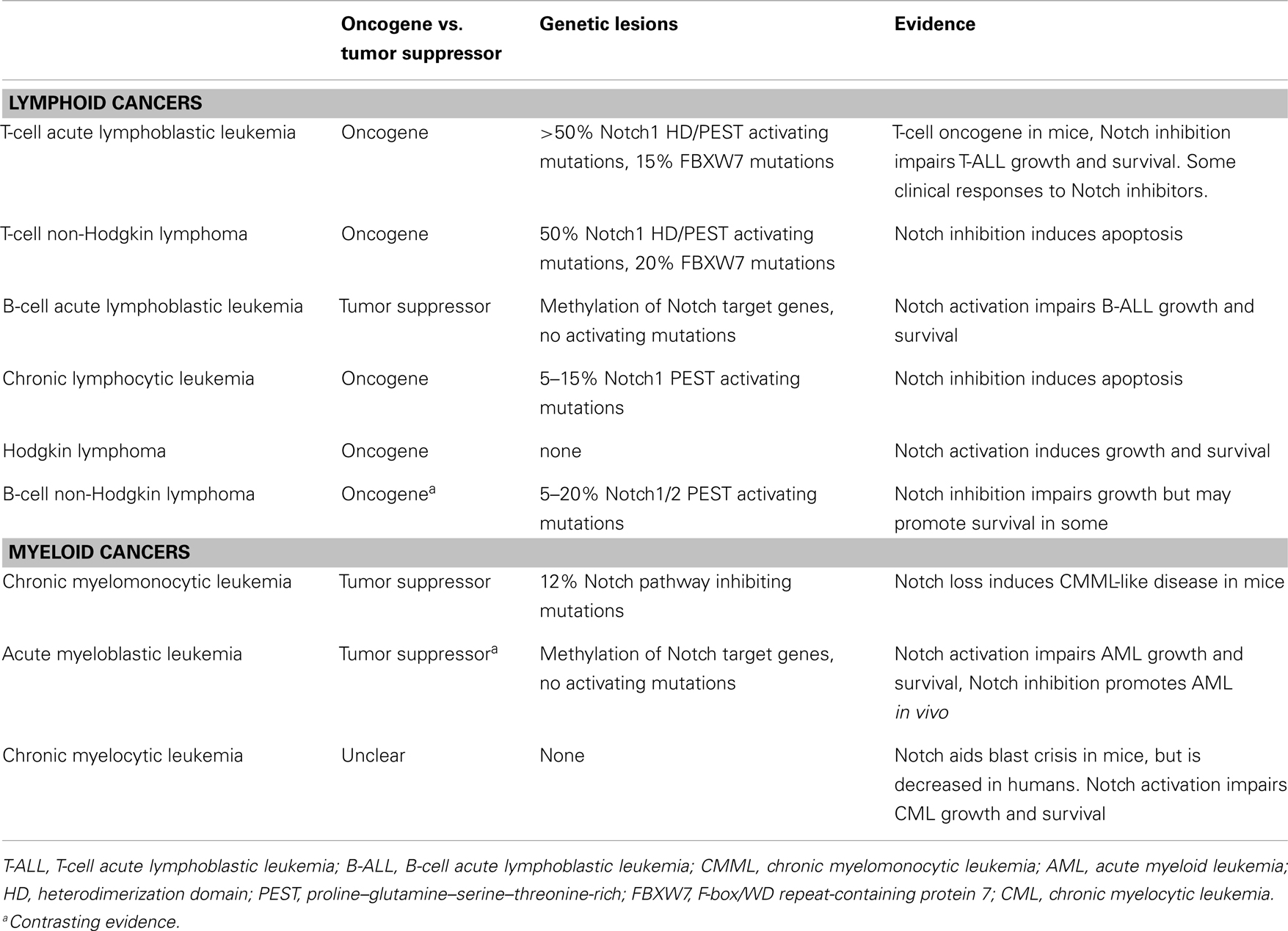
Table 1. The Notch pathway by cancer type.
In this review, we will outline the roles of the Notch pathway in a wide variety of leukemias and lymphomas, describe potential targeted therapies and discuss future directions.
Notch Pathway
In mammals, the Notch signaling pathway consists of five cell membrane-based ligands [Jagged1, Jagged2, Delta-like ligand-1 (DLL1), DLL3, and DLL4], each of which bind to and activate four cell membrane-based Notch receptors (Notch1–4) present on neighboring cells. Receptor interaction with ligand classically requires cell–cell interaction and induces enzymatic cleavage of the Notch receptors at their transmembrane domain by both metalloproteinases and the gamma-secretase complex (6). This releases the intracellular domain of Notch (ICN) from the membrane, which then translocates into the nucleus and associates with a common transcription factor RBPjk (also known as CSL), leading to the expression of Notch target genes, e.g., the HES/HEY family, which can vary depending on cell type (7). Thus, Notch signaling can occur in a variety of circumstances based on the presence of the five different ligands in the microenvironment, expression of metalloproteinase, and gamma-secretase complex enzymes, and the expression of the four Notch receptors, yielding a large number of potential variations on the Notch pathway.
Notch in Lymphoid Leukemias and Lymphomas
The normal developmental roles of the Notch pathway in lymphopoiesis have led to both similarities and contrasting findings across the range of lymphoid malignancies. For example, Notch has well-defined roles in inducing commitment, differentiation, and function in the T-cell lineage, while impairing early B-cell development, and inducing more mature marginal zone B-cell differentiation. Thus, in normal development Notch appears to generally support T-cell growth, differentiation, and survival, while effects in B cells appear to depend on the stage of differentiation, inhibiting immature B cells, and supporting at least a subset of more mature B cells (8).
T-Cell Acute Lymphoblastic Leukemia
Aberrant Notch activation was first linked to cancer in 1991 when Notch1 was identified as part of a t(7:9)(q34;q34) translocation in patients with T-cell acute lymphoblastic leukemia (T-ALL) (9). This translocation leads to high levels of truncated, constitutively active intracellular Notch1 (ICN1), implicating Notch as a T-cell oncogene. However, this translocation was subsequently found in <1% of human T-ALL. This discovery however initiated numerous studies of Notch signaling in normal T-cell development and leukemogenesis, where Notch was found to be a critical T-cell pathway and a potent T-cell oncogene in mice (10, 11). Over a decade later, it was shown that >50% of T-ALL cases have activating mutations in the Notch1 gene, revealing Notch1 mutation as the most common oncogenic lesion in T-ALL (12). Activation of Notch1 is found in all subtypes of T-ALL, including TAL1, HOX11, HOX11L2, LYL1, MLL-ENL, and AF10-CALM, suggesting that Notch1 activation is a fundamental event in T-cell transformation. Interestingly, two major mutational hot spots were characterized in T-ALL, with missense mutations in the heterodimerization (HD) domain in 30–45% of T-ALL cases, leading to lower protection from cleavage of Notch, resulting in potent Notch activation, and nonsense or missense mutations in the proline–glutamate–serine–threonine-rich (PEST) degradation domain in 20–25% of T-ALL, allowing for prolonged but less potent Notch signaling. In addition, Notch signaling is aberrantly activated through mutation of the Notch1-targeting E3 ligase FBXW7 in 10–15% of T-ALL cases, which leads to prolonged Notch activation through a similar mechanism to PEST domain mutation (13, 14). The relative strength of Notch signaling induced by the different mutations may provide insight into oncogenic mechanisms and may potentially be useful in selecting patients for Notch inhibitor therapies.
In patients with T-ALL, Notch1 and FBXW7 mutations have generally been associated with favorable prognosis and lower minimal residual disease (MRD) levels (15–17). However, Notch mutations have also been associated with higher rates of CNS relapse and poor outcome in Notch mutated patients with high end-induction MRD (18). Another study suggested that Notch/FBXW7 mutations predict better outcome only in the absence of RAS or PTEN alterations (19). Interestingly, Notch1 mutations occur at lower frequency in early thymic progenitor (ETP) T-ALL (20), suggesting alternate mechanisms of Notch activation, or distinct leukemogenic mechanisms. Given the overall high frequency of Notch activating lesions, inhibiting Notch in T-ALL has been an attractive therapeutic opportunity.
T-Cell Non-Hodgkin Lymphoma
Similar to T-ALL, T-NHL can carry activating Notch1 and/or inhibiting FBXW7 mutations in up to 50% (7/14) of cases (21), though the frequency may be lower in more mature T-NHL (22). Interestingly Notch1 is frequently expressed in anaplastic large cell lymphoma (ALCL) cells (23), and Notch signaling appears to induce proliferation and survival (24).
B-Cell Acute Lymphoblastic Leukemia
The role of Notch signaling in B-cell acute lymphoblastic leukemia (B-ALL) is less clear. In B-ALL, a disease of immature B-precursor cells, no Notch mutations have been found, and although Notch receptors are expressed, Notch signaling does not appear to be constitutively activated, in contrast to T-ALL (25, 26). Methylation studies reveal that several of the Notch pathway target genes are silenced in B-ALL, and re-expression inhibits growth and survival (27). Several studies have shown that induced activation of the Notch pathway in human B-ALL cells leads to growth arrest and apoptosis, suggesting a tumor suppressor role for Notch (26–28). In contrast, another report suggests that Notch signaling supports B-ALL in the bone marrow niche (29). Although the roles for Notch signaling in B-ALL are not yet fully defined, there may be an opportunity to pharmacologically induce Notch signaling in B-precursor ALL as a therapeutic approach.
Chronic Lymphocytic Leukemia
Chronic lymphocytic leukemia is a disease of mature B cells, which occurs in older adults. Recent sequencing data revealed that 5–15% of CLL samples carry apparent activating mutations in Notch1 (30–33). These were enriched in chemo-refractory disease (21%) and in patients whose CLL had undergone Richter’s transformation (31%) (33). These mutations occur almost exclusively in the PEST domain of Notch1, not the HD domain as seen in T-ALL, and >80% are a single recurrent 2-bp deletion (7544_7545 delCT), suggesting a related but distinct mutational profile to Notch1 mutations in T-ALL. In vitro, Notch signaling appears to prevent CLL apoptosis (34). In patients, Notch1 mutation is associated with poor outcome and resistance to fludarabine treatment (30, 31, 35, 36).
Hodgkin Lymphoma
Expression of Notch1 and ligand Jagged1 were shown in classical HL Reed-Sternberg cells (37). Activation of Notch signaling in vitro induced proliferation and survival in HL cells (24). Conversely, Notch inhibition led to decrease in NF-kB activity, supporting an oncogenic role for Notch in HL (38). Interestingly, it has been suggested that Notch signaling in HL leads to the loss of B-cell markers through repression of critical B-cell genes E2A and EBF (39).
B-Cell Non-Hodgkin Lymphoma
In B-cell NHL, evidence for Notch activation occurs in a subset of lymphoma subtypes. Mutations are found either in Notch1 or Notch2, and occur in the PEST domain, but not the HD domain, similar to CLL, but in contrast to T-ALL. In typically MYC-driven Burkitt lymphoma, 7% (5/70) carry Notch1 mutations (40), 8% (5/63) of BCL2-associated diffuse large B-cell lymphoma (DLBCL) carry similar PEST mutations of Notch2, and 6% (2/35) had amplification of the Notch2 locus (41). Marginal zone lymphomas also carry 5% (2/41) to 20% mutated Notch2, in addition to rare Notch1, SPEN, and DTX1 mutations (42, 43). Finally, >12% Notch1 mutations were found in mantle cell lymphomas and were associated with poor survival (44). Notably, Notch activating mutations have not been found in B-cell lymphoblastic and follicular lymphomas. These studies reveal that subsets of several mature B-NHLs carry Notch1/2 PEST mutations, suggesting an oncogenic role for Notch in these cancers.
Notch in Myeloid Leukemias
In myeloid cells, Notch may have a range of effects including inhibiting or promoting differentiation and stimulating or impairing growth and survival, depending on the cell type studied. Importantly, genetic inhibition of Notch signaling in murine models can lead to increased myeloid cells and myeloproliferation, suggesting that Notch may generally inhibit myeloid development (45–47). However, the roles of Notch in different myeloid leukemias have not been fully characterized.
Chronic Myelomonocytic Leukemia
Chronic myelomonocytic leukemia is a rare myeloproliferative and myelodysplastic leukemia, which typically occurs in older adults. However, a recent study found that inactivation of Notch signaling in murine bone marrow led to a myeloproliferative disease, and identified inactivating mutations in Notch pathway genes (NCSTN, APH1, MAML1, and NOTCH2) in 12% (5/42) CMML patient samples, implicating a tumor suppressor role for Notch in this disease (48).
Acute Myeloblastic Leukemia
With the unclear roles of Notch in myelopoiesis, murine models were used to investigate whether loss of Notch would alter myeloid leukemogenesis. Indeed loss of Notch in combination with loss of the myeloid tumor suppressor TET2 led to an AML-like disease in mice, suggesting a formal tumor suppressor role for Notch in AML (49). Consistent with this, human AML samples do not carry activating mutations in Notch pathway genes, except in rare cases of recurrent T-myeloid leukemias, which can carry Notch1 activating mutations from the initial T-ALL. AML cells do express Notch receptors on their surface, however, they lack constitutive Notch signaling and demonstrate methylation Notch pathway genes, similar to B-ALL (25, 49). In some studies, activation of Notch signaling in AML cells led to growth arrest, apoptosis, and differentiation, while inhibition of Notch led to increased aggressiveness in vivo, suggesting a tumor suppressing effect in AML (49–51). The mechanism of this effect may involve Notch-mediated suppression of CEBPA, Pu.1, BCL2, and the stabilization of p53 expression. In contrast, in some studies Notch signaling has variable effects on AML growth and survival, depending on the individual AML sample (52). Finally, Notch1, Jagged1, and DLL1 expression in patient samples were associated with poor outcome though the activity of the Notch pathway was not measured (53). Thus, there is generally evidence for a tumor suppressing effect in AML, however additional studies are needed to better characterize the roles of Notch in AML.
Chronic Myelogenous Leukemia
Chronic myelogenous leukemia is a mature myeloproliferative disease driven by the expression of BCR-ABL1 as a consequence of t(9;22) translocation. BCR-ABL1 appears to enhance Notch1 expression and activation, leading to decreased MYC expression and colony formation (54). In a murine model, expression of the Notch target gene HES1 was shown to cooperate with BCR-ABL1 expression to promote CML blast crisis (55). In contrast, HES1 downregulation was associated with blast crisis in human patient samples (56). Induced activation of Notch signaling in CML-derived cell lines reveal growth inhibition, suggesting an inhibiting role for Notch in CML (50, 57, 58). Thus, murine leukemogenesis studies suggest an oncogenic role while human studies suggest a tumor suppressor role, leaving the role of Notch in CML unclear.
Notch Targeting Therapeutics
Given the emerging data demonstrating roles for Notch signaling in a wide variety of leukemias and lymphomas, targeting the Notch pathway either with inhibitors or activators is a very compelling therapeutic possibility. However, caution should be used when targeting this pathway as disease-specific responses to Notch modulation may be counter-therapeutic. Several Notch inhibiting strategies are being tested in clinical trials, while Notch activators are still in the early pre-clinical stage (Figure 1).
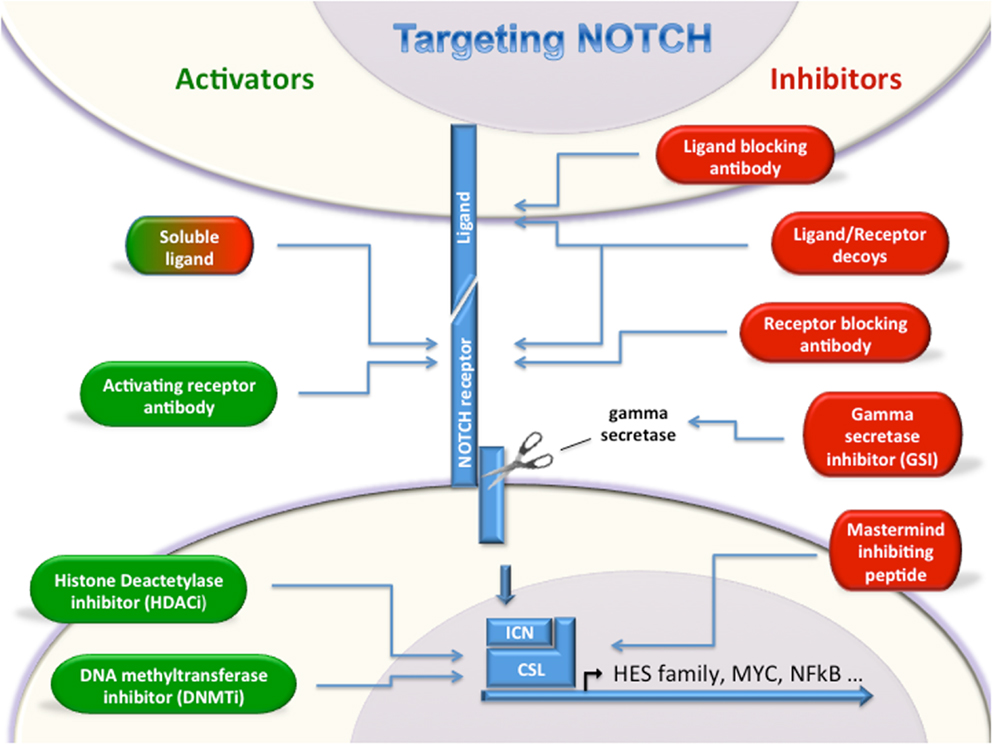
Figure 1. Targeting Notch. Multiple strategies for either activating or inhibiting Notch have been described. Notch activators (green, left) and Notch inhibitors (red, right) will allow modulation of Notch depending on oncogenic or tumor suppressor roles in a given cancer type. Soluble ligands can either activate or inhibit Notch signaling.
Notch Inhibiting Strategies
Gamma-Secretase Inhibitors
Interaction between Notch ligands and receptors induces a conformational change in the Notch receptors’ HD domain, which allows for enzymatic access to the transmembrane region. Following ligand interaction, the receptors are cleaved by ADAM/TACE metalloproteinases, and subsequently cleaved again by the gamma-secretase complex, which is a crucial step in the activation of Notch signaling. It is this step, which is targeted by GSIs [reviewed in Ref. (59)].
The potent role of Notch signaling in T-cell leukemogenesis and >50% presence of activating Notch1 gene mutation in T-ALL has prompted the testing of GSIs in multiple clinical trials, though most results are unpublished, BMS-906024 (NCT01363817), RO4929097 (NCT01088763), MK0752 [NCT00100152 (60)], PF03084014 (NCT00878189), and LY3039478 (NCT01158404). However, early trials were hampered by excessive toxicity from on-target effects on the intestinal epithelial differentiation, resulting in dose-limiting diarrhea (60, 61). Alternative schedules and dosing have been sought to ameliorate these symptoms with some success. One attractive combination is the use of glucocorticoids and GSI. In a murine study, it was demonstrated that steroids can ameliorate the GSI-induced gut toxicity in vivo, protecting the animals from developing intestinal goblet cell metaplasia (62). In addition, GSI treatment induces glucocorticoid receptor expression and restores steroid sensitivity (63). As single agent Phase I trials continue to address toxicity and activity, multiple mechanisms of resistance have been identified. For example, PTEN loss has been associated with GSI resistance (64). Also, GSI resistant cells appear to have distinct epigenetic status, and BRD4-inhibiting JQ1 has been shown to synergize with GSI (65). For future trials several classes of agents, including PI3K/mTOR, histone deacetylase, and proteasome inhibition, have been shown to enhance GSI effects in T-ALL (66–68).
The use of GSI in T-cell NHL, including ALK+ ALCL, has shown some pre-clinical promise inducing growth inhibition and caspase-mediated apoptosis with downregulation of cyclin D1, Bcl-XL, and XIAP (23, 69). Similarly, GSIs show pre-clinical promise for CLL, demonstrating decreased NF-kB, XIAP, c-IAP2 levels (34), though some data suggest Notch-independent mechanisms (70). GSIs have also been effective in two mantle cell lymphoma lines (44) and one DLBCL line without Notch mutations (71).
In contrast, pre-clinical testing of GSIs in some B-NHLs has shown a lack of efficacy and even perhaps pro-survival effects (72). And while some data suggest pre-clinical efficacy of GSIs in AML (73), other studies found that GSIs unexpectedly increased HES1 expression in GSI-sensitive B-NHL and AML lines (74), calling into question the rationale and mechanism for GSI effects in these diseases.
Monoclonal Antibodies
Another class of agents under development is the mAb targeted against Notch receptors or ligands, or the gamma-secretase complex. The anti-receptor antibodies inhibit the production of cleaved “activated” Notch receptors, e.g., Notch1 OMP52M51 (75), Notch2 OMP-59R5, Aveo anti-Notch1 or Notch3 (76), and Genentech anti-Notch1 or Notch2 (77). Experimental evidence demonstrates that Notch inhibition by either mAb against Notch1 or Notch2 appears to have anti-tumor and anti-angiogenic effects with limited gastrointestinal toxicities while simultaneous inhibition of Notch1 and 2 lead to gastrointestinal toxicity, as seen with many GSIs (77–79). Anti-ligand antibodies targeting DLL4 (REGN421, OMP-21M18), which block the ability of ligand to activate Notch receptors, have been shown to induce disorganized angiogenesis, reduce perfusion, and impair solid tumor growth while sparing intestinal toxicities in vivo (80, 81). Finally, an antibody against the gamma-secretase complex (A5226A) has shown pre-clinical activity against T-ALL (82).
It is hoped that this category of drugs could reduce or spare some of the toxicities associated with pan-Notch inhibition by GSIs, though this has not yet been confirmed clinically. These antibodies have not all been tested in hematologic cancers.
Decoys
Additional approaches to inhibit Notch signaling come from the use of proteins, fragments, or peptides, which inhibit Notch signaling. First, soluble Notch pathway proteins have been shown to inhibit Notch signaling through saturation of the Notch receptors with soluble ligand DLL4-Fc (81, 83), Jagged1 (84), DLK1 (85), EGFL7 (86), or through binding of ligands through soluble Notch1 receptor extracellular domain (87). Another decoy approach that has been developed is a Mastermind inhibiting peptide, which mimics the critical interaction domain of Mastermind-like1 (MAML1) blocking the interaction of MAML with the Notch intracellular domain (88). The success of these therapies may rely on the specific biology of a given tumor and the breadth and potency of Notch inhibition achieved. Although, these approaches may provide more options to inhibit Notch signaling, their protein/peptide structure makes them somewhat difficult to transform into a reliable clinical therapeutics.
Notch Activating Strategies
Ligand-Mimicking Proteins/Peptides
Soluble Notch ligands are generally thought to inhibit Notch receptor cleavage, however, several studies have demonstrated the feasibility of using such Notch ligands and ligand-mimicking proteins as agonists, e.g., clustered DLL1 extracellular domains (89), DLL1 DSL domain (90), Jagged1 DSL peptide (50, 91, 92), DNER (93), TSP2 (94), CCN3 (95), YB-1 (96), NB-3 (97), and periostin (98). Although, all of these proteins have been shown to activate Notch signaling in at least one model system, the role of most of these Notch agonists in cancer remains to be evaluated. Importantly, a Jagged1 DSL peptide has been shown to be effective in vitro against B-ALL (28) and AML (50), suggesting potential of Notch agonists as cancer therapeutics depending on tumor type.
Notch Receptor Activating Antibodies
Monoclonal antibodies have been developed, which are capable of specifically inducing cleavage of the Notch2 and Notch3 receptors (99, 100). These activating antibodies have the advantage of selectively inducing cleavage of one of the Notch receptors, allowing one to choose the best target in a given disease and avoiding global Notch activation, when desired. Notch receptor activating antibodies have not been evaluated in cancer models.
Inducers of Notch
In addition to targeted agents, which induce Notch signaling through Notch ligands or receptors, epigenetic modifiers, e.g., histone deacetylase inhibitors and DNA methyltransferase inhibitors, can induce Notch signaling in cancers where Notch plays a tumor suppressor role (27, 101–103).
Future Directions
Currently, several different GSIs and Notch inhibiting antibodies are in clinical trials, with additional Notch inhibiting approaches in near clinical development. With these agents, our greatest challenges are to overcome the intestinal toxicity caused by continuous Notch inhibition, identify patients who are likely to respond to Notch inhibition, and determine what combinations hold the most promise for diseases where the oncogenic role of Notch is fairly clear, i.e., T-ALL, CLL, and T-NHL, some mature B-NHL, perhaps HL. Looking toward the future, we hope to have Notch activators in clinical trial in the next few years so that we may target Notch in diseases where Notch is likely a tumor suppressor, i.e., CMML, AML, and B-ALL.
The Notch pathway is complex and the wide range of consequences in different cancer subtypes makes targeting this pathway challenging. However, as we learn more about the mechanisms and consequences of Notch signaling in the range of leukemia and lymphoma subtypes, we will be able to target Notch signaling to specifically impair the growth, survival, and/or differentiation of that disease while reducing the toxicities. With a growing number of Notch modulating therapeutic agents, we may have the tools to customize Notch targeting in the near future.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Penton AL, Leonard LD, Spinner NB. Notch signaling in human development and disease. Semin Cell Dev Biol (2012) 23(4):450–7. doi: 10.1016/j.semcdb.2012.01.010
2. Ntziachristos P, Lim JS, Sage J, Aifantis I. From fly wings to targeted cancer therapies: a centennial for Notch signaling. Cancer Cell (2014) 25(3):318–34. doi:10.1016/j.ccr.2014.02.018
3. Lobry C, Oh P, Mansour MR, Look AT, Aifantis I. Notch signaling: switching an oncogene to a tumor suppressor. Blood (2014) 123(16):2451–9. doi:10.1182/blood-2013-08-355818
4. Mirandola L, Comi P, Cobos E, Kast WM, Chiriva-Internati M, Chiaramonte R. Notch-ing from T-cell to B-cell lymphoid malignancies. Cancer Lett (2011) 308(1):1–13. doi:10.1016/j.canlet.2011.05.009
5. Aster JC, Blacklow SC, Pear WS. Notch signalling in T-cell lymphoblastic leukaemia/lymphoma and other haematological malignancies. J Pathol (2011) 223(2):262–73. doi:10.1002/path.2789
6. Guruharsha KG, Kankel MW, Artavanis-Tsakonas S. The Notch signalling system: recent insights into the complexity of a conserved pathway. Nat Rev Genet (2012) 13(9):654–66. doi:10.1038/nrg3272
7. Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell (2009) 137(2):216–33. doi:10.1016/j.cell.2009.03.045
8. Radtke F, MacDonald HR, Tacchini-Cottier F. Regulation of innate and adaptive immunity by Notch. Nat Rev Immunol (2013) 13(6):427–37. doi:10.1038/nri3445
9. Ellisen LW, Bird J, West DC, Soreng AL, Reynolds TC, Smith SD, et al. TAN-1, the human homolog of the Drosophila Notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell (1991) 66(4):649–61. doi:10.1016/0092-8674(91)90111-B
10. Pui JC, Allman D, Xu L, DeRocco S, Karnell FG, Bakkour S, et al. Notch1 expression in early lymphopoiesis influences B versus T lineage determination. Immunity (1999) 11(3):299–308. doi:10.1016/S1074-7613(00)80105-3
11. Pear WS, Aster JC, Scott ML, Hasserjian RP, Soffer B, Sklar J, et al. Exclusive development of T cell neoplasms in mice transplanted with bone marrow expressing activated Notch alleles. J Exp Med (1996) 183(5):2283–91. doi:10.1084/jem.183.5.2283
12. Weng AP, Ferrando AA, Lee W, Morris JPT, Silverman LB, Sanchez-Irizarry C, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science (2004) 306(5694):269–71. doi:10.1126/science.1102160
13. O’Neil J, Grim J, Strack P, Rao S, Tibbitts D, Winter C, et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. J Exp Med (2007) 204(8):1813–24. doi:10.1084/jem.20070876
14. Thompson BJ, Buonamici S, Sulis ML, Palomero T, Vilimas T, Basso G, et al. The SCFFBW7 ubiquitin ligase complex as a tumor suppressor in T cell leukemia. J Exp Med (2007) 204(8):1825–35. doi:10.1084/jem.20070872
15. Breit S, Stanulla M, Flohr T, Schrappe M, Ludwig WD, Tolle G, et al. Activating NOTCH1 mutations predict favorable early treatment response and long-term outcome in childhood precursor T-cell lymphoblastic leukemia. Blood (2006) 108(4):1151–7. doi:10.1182/blood-2005-12-4956
16. Bedard PW, Clerin V, Sushkova N, Tchernychev B, Antrilli T, Resmini C, et al. Characterization of the novel P-selectin inhibitor PSI-697 [2-(4-chlorobenzyl)-3-hydroxy-7,8,9,10-tetrahydrobenzo[h] quinoline-4-carboxylic acid] in vitro and in rodent models of vascular inflammation and thrombosis. J Pharmacol Exp Ther (2008) 324(2):497–506. doi:10.1124/jpet.107.128124
17. Asnafi V, Buzyn A, Le Noir S, Baleydier F, Simon A, Beldjord K, et al. NOTCH1/FBXW7 mutation identifies a large subgroup with favorable outcome in adult T-cell acute lymphoblastic leukemia (T-ALL): a Group for Research on Adult Acute Lymphoblastic Leukemia (GRAALL) study. Blood (2009) 113(17):3918–24. doi:10.1182/blood-2008-10-184069
18. Clappier E, Collette S, Grardel N, Girard S, Suarez L, Brunie G, et al. NOTCH1 and FBXW7 mutations have a favorable impact on early response to treatment, but not on outcome, in children with T-cell acute lymphoblastic leukemia (T-ALL) treated on EORTC trials 58881 and 58951. Leukemia (2010) 24(12):2023–31. doi:10.1038/leu.2010.205
19. Trinquand A, Tanguy-Schmidt A, Ben Abdelali R, Lambert J, Beldjord K, Lengline E, et al. Toward a NOTCH1/FBXW7/RAS/PTEN-based oncogenetic risk classification of adult T-cell acute lymphoblastic leukemia: a Group for Research in Adult Acute Lymphoblastic Leukemia study. J Clin Oncol (2013) 31(34):4333–42. doi:10.1200/JCO.2012.48.5292
20. Neumann M, Heesch S, Gokbuget N, Schwartz S, Schlee C, Benlasfer O, et al. Clinical and molecular characterization of early T-cell precursor leukemia: a high-risk subgroup in adult T-ALL with a high frequency of FLT3 mutations. Blood Cancer J (2012) 2(1):e55. doi:10.1038/bcj.2011.49
21. Park MJ, Taki T, Oda M, Watanabe T, Yumura-Yagi K, Kobayashi R, et al. FBXW7 and NOTCH1 mutations in childhood T cell acute lymphoblastic leukaemia and T cell non-Hodgkin lymphoma. Br J Haematol (2009) 145(2):198–206. doi:10.1111/j.1365-2141.2009.07607.x
22. Shimizu D, Taki T, Utsunomiya A, Nakagawa H, Nomura K, Matsumoto Y, et al. Detection of NOTCH1 mutations in adult T-cell leukemia/lymphoma and peripheral T-cell lymphoma. Int J Hematol (2007) 85(3):212–8. doi:10.1532/IJH97.06165
23. Kamstrup MR, Biskup E, Gjerdrum LM, Ralfkiaer E, Niazi O, Gniadecki R. The importance of Notch signaling in peripheral T-cell lymphomas. Leuk Lymphoma (2014) 55(3):639–44. doi:10.3109/10428194.2013.807510
24. Jundt F, Anagnostopoulos I, Forster R, Mathas S, Stein H, Dorken B. Activated Notch1 signaling promotes tumor cell proliferation and survival in Hodgkin and anaplastic large cell lymphoma. Blood (2002) 99(9):3398–403. doi:10.1182/blood.V99.9.3398
25. Chiaramonte R, Basile A, Tassi E, Calzavara E, Cecchinato V, Rossi V, et al. A wide role for NOTCH1 signaling in acute leukemia. Cancer Lett (2005) 219(1):113–20. doi:10.1016/j.canlet.2004.07.022
26. Zweidler-McKay PA, He Y, Xu L, Rodriguez CG, Karnell FG, Carpenter AC, et al. Notch signaling is a potent inducer of growth arrest and apoptosis in a wide range of B-cell malignancies. Blood (2005) 106(12):3898–906. doi:10.1182/blood-2005-01-0355
27. Kuang SQ, Fang Z, Zweidler-McKay PA, Yang H, Wei Y, Gonzalez-Cervantes EA, et al. Epigenetic inactivation of Notch-Hes pathway in human B-cell acute lymphoblastic leukemia. PLoS One (2013) 8(4):e61807. doi:10.1371/journal.pone.0061807
28. Kannan S, Fang W, Song G, Mullighan CG, Hammitt R, McMurray J, et al. Notch/HES1-mediated PARP1 activation: a cell type-specific mechanism for tumor suppression. Blood (2011) 117(10):2891–900. doi:10.1182/blood-2009-12-253419
29. Nwabo Kamdje AH, Mosna F, Bifari F, Lisi V, Bassi G, Malpeli G, et al. Notch-3 and Notch-4 signaling rescue from apoptosis human B-ALL cells in contact with human bone marrow-derived mesenchymal stromal cells. Blood (2011) 118(2):380–9. doi:10.1182/blood-2010-12-326694
30. Sportoletti P, Baldoni S, Cavalli L, Del Papa B, Bonifacio E, Ciurnelli R, et al. NOTCH1 PEST domain mutation is an adverse prognostic factor in B-CLL. Br J Haematol (2010) 151(4):404–6. doi:10.1111/j.1365-2141.2010.08368.x
31. Di Ianni M, Baldoni S, Rosati E, Ciurnelli R, Cavalli L, Martelli MF, et al. A new genetic lesion in B-CLL: a NOTCH1 PEST domain mutation. Br J Haematol (2009) 146(6):689–91. doi:10.1111/j.1365-2141.2009.07816.x
32. Puente XS, Pinyol M, Quesada V, Conde L, Ordonez GR, Villamor N, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature (2011) 475(7354):101–5. doi:10.1038/nature10113
33. Fabbri G, Rasi S, Rossi D, Trifonov V, Khiabanian H, Ma J, et al. Analysis of the chronic lymphocytic leukemia coding genome: role of NOTCH1 mutational activation. J Exp Med (2011) 208(7):1389–401. doi:10.1084/jem.20110921
34. Rosati E, Sabatini R, Rampino G, Tabilio A, Di Ianni M, Fettucciari K, et al. Constitutively activated Notch signaling is involved in survival and apoptosis resistance of B-CLL cells. Blood (2009) 113(4):856–65. doi:10.1182/blood-2008-02-139725
35. Rossi D, Rasi S, Fabbri G, Spina V, Fangazio M, Forconi F, et al. Mutations of NOTCH1 are an independent predictor of survival in chronic lymphocytic leukemia. Blood (2012) 119(2):521–9. doi:10.1182/blood-2011-09-379966
36. Oscier DG, Rose-Zerilli MJ, Winkelmann N, Gonzalez de Castro D, Gomez B, Forster J, et al. The clinical significance of NOTCH1 and SF3B1 mutations in the UK LRF CLL4 trial. Blood (2013) 121(3):468–75. doi:10.1182/blood-2012-05-429282
37. Jundt F, Schwarzer R, Dorken B. Notch signaling in leukemias and lymphomas. Curr Mol Med (2008) 8(1):51–9. doi:10.2174/156652408783565540
38. Schwarzer R, Dorken B, Jundt F. Notch is an essential upstream regulator of NF-kappaB and is relevant for survival of Hodgkin and Reed-Sternberg cells. Leukemia (2012) 26(4):806–13. doi:10.1038/leu.2011.265
39. Anderson LJ, Longnecker R. Epstein-Barr virus latent membrane protein 2A exploits Notch1 to alter B-cell identity in vivo. Blood (2009) 113(1):108–16. doi:10.1182/blood-2008-06-160937
40. Love C, Sun Z, Jima D, Li G, Zhang J, Miles R, et al. The genetic landscape of mutations in Burkitt lymphoma. Nat Genet (2012) 44(12):1321–5. doi:10.1038/ng.2468
41. Lee SY, Kumano K, Nakazaki K, Sanada M, Matsumoto A, Yamamoto G, et al. Gain-of-function mutations and copy number increases of Notch2 in diffuse large B-cell lymphoma. Cancer Sci (2009) 100(5):920–6. doi:10.1111/j.1349-7006.2009.01130.x
42. Troen G, Wlodarska I, Warsame A, Hernandez Llodra S, De Wolf-Peeters C, Delabie J. NOTCH2 mutations in marginal zone lymphoma. Haematologica (2008) 93(7):1107–9. doi:10.3324/haematol.11635
43. Rossi D, Trifonov V, Fangazio M, Bruscaggin A, Rasi S, Spina V, et al. The coding genome of splenic marginal zone lymphoma: activation of NOTCH2 and other pathways regulating marginal zone development. J Exp Med (2012) 209(9):1537–51. doi:10.1084/jem.20120904
44. Kridel R, Meissner B, Rogic S, Boyle M, Telenius A, Woolcock B, et al. Whole transcriptome sequencing reveals recurrent NOTCH1 mutations in mantle cell lymphoma. Blood (2012) 119(9):1963–71. doi:10.1182/blood-2011-11-391474
45. Zhou L, Li LW, Yan Q, Petryniak B, Man Y, Su C, et al. Notch-dependent control of myelopoiesis is regulated by fucosylation. Blood (2008) 112(2):308–19. doi:10.1182/blood-2007-11-115204
46. Yao D, Huang Y, Huang X, Wang W, Yan Q, Wei L, et al. Protein O-fucosyltransferase 1 (Pofut1) regulates lymphoid and myeloid homeostasis through modulation of Notch receptor ligand interactions. Blood (2011) 117(21):5652–62. doi:10.1182/blood-2010-12-326074
47. Yan Q, Yao D, Wei LL, Huang Y, Myers J, Zhang L, et al. O-fucose modulates Notch-controlled blood lineage commitment. Am J Pathol (2010) 176(6):2921–34. doi:10.2353/ajpath.2010.090702
48. Klinakis A, Lobry C, Abdel-Wahab O, Oh P, Haeno H, Buonamici S, et al. A novel tumour-suppressor function for the Notch pathway in myeloid leukaemia. Nature (2011) 473(7346):230–3. doi:10.1038/nature09999
49. Lobry C, Ntziachristos P, Ndiaye-Lobry D, Oh P, Cimmino L, Zhu N, et al. Notch pathway activation targets AML-initiating cell homeostasis and differentiation. J Exp Med (2013) 210(2):301–19. doi:10.1084/jem.20121484
50. Kannan S, Sutphin RM, Hall MG, Golfman LS, Fang W, Nolo RM, et al. Notch activation inhibits AML growth and survival: a potential therapeutic approach. J Exp Med (2013) 210(2):321–37. doi:10.1084/jem.20121527
51. Chadwick N, Fennessy C, Nostro MC, Baron M, Brady G, Buckle AM. Notch induces cell cycle arrest and apoptosis in human erythroleukaemic TF-1 cells. Blood Cells Mol Dis (2008) 41(3):270–7. doi:10.1016/j.bcmd.2008.06.003
52. Tohda S, Kogoshi H, Murakami N, Sakano S, Nara N. Diverse effects of the Notch ligands Jagged1 and Delta1 on the growth and differentiation of primary acute myeloblastic leukemia cells. Exp Hematol (2005) 33(5):558–63. doi:10.1016/j.exphem.2005.01.010
53. Xu X, Zhao Y, Xu M, Dai Q, Meng W, Yang J, et al. Activation of Notch signal pathway is associated with a poorer prognosis in acute myeloid leukemia. Med Oncol (2011) 28(Suppl 1):S483–9. doi:10.1007/s12032-010-9667-0
54. Suresh S, McCallum L, Crawford LJ, Lu WH, Sharpe DJ, Irvine AE. The matricellular protein CCN3 regulates NOTCH1 signalling in chronic myeloid leukaemia. J Pathol (2013) 231(3):378–87. doi:10.1002/path.4246
55. Nakahara F, Sakata-Yanagimoto M, Komeno Y, Kato N, Uchida T, Haraguchi K, et al. Hes1 immortalizes committed progenitors and plays a role in blast crisis transition in chronic myelogenous leukemia. Blood (2010) 115(14):2872–81. doi:10.1182/blood-2009-05-222836
56. Sengupta A, Banerjee D, Chandra S, Banerji SK, Ghosh R, Roy R, et al. Deregulation and cross talk among Sonic hedgehog, Wnt, Hox and Notch signaling in chronic myeloid leukemia progression. Leukemia (2007) 21(5):949–55. doi:10.1038/sj.leu.2404657
57. Yang Z, Yang C, Zhang S, Li Y, Chen J. Notch2 inhibits proliferation of chronic myeloid leukemia cells. Oncol Lett (2013) 5(4):1390–4. doi:10.3892/ol.2013.1159
58. Yin DD, Fan FY, Hu XB, Hou LH, Zhang XP, Liu L, et al. Notch signaling inhibits the growth of the human chronic myeloid leukemia cell line K562. Leuk Res (2009) 33(1):109–14. doi:10.1016/j.leukres.2008.06.023
59. Olsauskas-Kuprys R, Zlobin A, Osipo C. Gamma secretase inhibitors of Notch signaling. Onco Targets Ther (2013) 6:943–55. doi:10.2147/ott.s33766
60. Deangelo DJ, Stone RM, Silverman LB, Stock W, Attar EC, Fearin I. A phase I clinical trial of the Notch inhibitor MK-0752 in patients with T-cell acute lymphoblastic leukemia/lymphoma (T-ALL) and other leukemias. J Clin Onclo (2006). 2006 ASCO Annual Meeting Proceedings Part I. 24:6585.
61. Fre S, Huyghe M, Mourikis P, Robine S, Louvard D, Artavanis-Tsakonas S. Notch signals control the fate of immature progenitor cells in the intestine. Nature (2005) 435(7044):964–8. doi:10.1038/nature03589
62. Real PJ, Ferrando AA. NOTCH inhibition and glucocorticoid therapy in T-cell acute lymphoblastic leukemia. Leukemia (2009) 23(8):1374–7. doi:10.1038/leu.2009.75
63. Samon JB, Castillo-Martin M, Hadler M, Ambesi-Impiobato A, Paietta E, Racevskis J, et al. Preclinical analysis of the gamma-secretase inhibitor PF-03084014 in combination with glucocorticoids in T-cell acute lymphoblastic leukemia. Mol Cancer Ther (2012) 11(7):1565–75. doi:10.1158/1535-7163.MCT-11-0938
64. Palomero T, Dominguez M, Ferrando AA. The role of the PTEN/AKT pathway in NOTCH1-induced leukemia. Cell Cycle (2008) 7(8):965–70. doi:10.4161/cc.7.8.5753
65. Knoechel B, Roderick JE, Williamson KE, Zhu J, Lohr JG, Cotton MJ, et al. An epigenetic mechanism of resistance to targeted therapy in T cell acute lymphoblastic leukemia. Nat Genet (2014) 46:364–70. doi:10.1038/ng.2913
66. Sanda T, Li X, Gutierrez A, Ahn Y, Neuberg DS, O’Neil J, et al. Interconnecting molecular pathways in the pathogenesis and drug sensitivity of T-cell acute lymphoblastic leukemia. Blood (2010) 115(9):1735–45. doi:10.1182/blood-2009-07-235143
67. Cullion K, Draheim KM, Hermance N, Tammam J, Sharma VM, Ware C, et al. Targeting the Notch1 and mTOR pathways in a mouse T-ALL model. Blood (2009) 113(24):6172–81. doi:10.1182/blood-2008-02-136762
68. Shepherd C, Banerjee L, Cheung CW, Mansour MR, Jenkinson S, Gale RE, et al. PI3K/mTOR inhibition upregulates NOTCH-MYC signalling leading to an impaired cytotoxic response. Leukemia (2013) 27(3):650–60. doi:10.1038/leu.2012.285
69. Ramakrishnan V, Ansell S, Haug J, Grote D, Kimlinger T, Stenson M, et al. MRK003, a gamma-secretase inhibitor exhibits promising in vitro pre-clinical activity in multiple myeloma and non-Hodgkin’s lymphoma. Leukemia (2012) 26(2):340–8. doi:10.1038/leu.2011.192
70. Rosati E, Sabatini R, De Falco F, Del Papa B, Falzetti F, Di Ianni M, et al. γ-Secretase inhibitor I induces apoptosis in chronic lymphocytic leukemia cells by proteasome inhibition, endoplasmic reticulum stress increase and Notch down-regulation. Int J Cancer (2013) 132(8):1940–53. doi:10.1002/ijc.27863
71. Tohda S, Sato T, Kogoshi H, Fu L, Sakano S, Nara N. Establishment of a novel B-cell lymphoma cell line with suppressed growth by gamma-secretase inhibitors. Leuk Res (2006) 30(11):1385–90. doi:10.1016/j.leukres.2006.05.003
72. Hajdu M, Kopper L, Sebestyen A. Notch-regulation upon Dll4-stimulation of TGFb-induced apoptosis and gene expression in human B-cell non-Hodgkin lymphomas. Scand J Immunol (2010) 71(1):29–37. doi:10.1111/j.1365-3083.2009.02346.x
73. Li GH, Fan YZ, Liu XW, Zhang BF, Yin DD, He F, et al. Notch signaling maintains proliferation and survival of the HL60 human promyelocytic leukemia cell line and promotes the phosphorylation of the Rb protein. Mol Cell Biochem (2010) 340(1–2):7–14. doi:10.1007/s11010-010-0394-9
74. Kogoshi H, Sato T, Koyama T, Nara N, Tohda S. Gamma-secretase inhibitors suppress the growth of leukemia and lymphoma cells. Oncol Rep (2007) 18(1):77–80. doi:10.3892/or.18.1.77
75. Agnusdei V, Minuzzo S, Frasson C, Grassi A, Axelrod F, Satyal S, et al. Therapeutic antibody targeting of Notch1 in T-acute lymphoblastic leukemia xenografts. Leukemia (2014) 28(2):278–88. doi:10.1038/leu.2013.183
76. Okamura H, Lorusso J, Bell A, Perino S, Keane D, Brodeur J, et al. Monoclonal antibodies to Notch receptors inhibit tumor maintenance. American Association for Cancer Research (AACR) 101st American Association for Cancer Research Annual Meeting. Washington, DC (2010).
77. Wu Y, Cain-Hom C, Choy L, Hagenbeek TJ, de Leon GP, Chen Y, et al. Therapeutic antibody targeting of individual Notch receptors. Nature (2010) 464(7291):1052–7. doi:10.1038/nature08878
78. Aste-Amezaga M, Zhang N, Lineberger JE, Arnold BA, Toner TJ, Gu M, et al. Characterization of Notch1 antibodies that inhibit signaling of both normal and mutated Notch1 receptors. PLoS One (2010) 5(2):e9094. doi:10.1371/journal.pone.0009094
79. Falk R, Falk A, Dyson MR, Melidoni AN, Parthiban K, Young JL, et al. Generation of anti-Notch antibodies and their application in blocking Notch signalling in neural stem cells. Methods (2012) 58(1):69–78. doi:10.1016/j.ymeth.2012.07.008
80. Ridgway J, Zhang G, Wu Y, Stawicki S, Liang WC, Chanthery Y, et al. Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature (2006) 444(7122):1083–7. doi:10.1038/nature05313
81. Noguera-Troise I, Daly C, Papadopoulos NJ, Coetzee S, Boland P, Gale NW, et al. Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature (2006) 444(7122):1032–7. doi:10.1038/nature05355
82. Hayashi I, Takatori S, Urano Y, Miyake Y, Takagi J, Sakata-Yanagimoto M, et al. Neutralization of the gamma-secretase activity by monoclonal antibody against extracellular domain of nicastrin. Oncogene (2012) 31(6):787–98. doi:10.1038/onc.2011.265
83. Liu R, Li X, Tulpule A, Zhou Y, Scehnet JS, Zhang S, et al. KSHV-induced Notch components render endothelial and mural cell characteristics and cell survival. Blood (2010) 115(4):887–95. doi:10.1182/blood-2009-08-236745
84. Xiao Y, Gong D, Wang W. Soluble JAGGED1 inhibits pulmonary hypertension by attenuating Notch signaling. Arterioscler Thromb Vasc Biol (2013) 33(12):2733–9. doi:10.1161/ATVBAHA.113.302062
85. Sanchez-Solana B, Nueda ML, Ruvira MD, Ruiz-Hidalgo MJ, Monsalve EM, Rivero S, et al. The EGF-like proteins DLK1 and DLK2 function as inhibitory non-canonical ligands of NOTCH1 receptor that modulate each other’s activities. Biochim Biophys Acta (2011) 1813(6):1153–64. doi:10.1016/j.bbamcr.2011.03.004
86. Schmidt MH, Bicker F, Nikolic I, Meister J, Babuke T, Picuric S, et al. Epidermal growth factor-like domain 7 (EGFL7) modulates Notch signalling and affects neural stem cell renewal. Nat Cell Biol (2009) 11(7):873–80. doi:10.1038/ncb1896
87. Funahashi Y, Hernandez SL, Das I, Ahn A, Huang J, Vorontchikhina M, et al. A Notch1 ectodomain construct inhibits endothelial Notch signaling, tumor growth, and angiogenesis. Cancer Res (2008) 68(12):4727–35. doi:10.1158/0008-5472.CAN-07-6499
88. Moellering RE, Cornejo M, Davis TN, Del Bianco C, Aster JC, Blacklow SC, et al. Direct inhibition of the NOTCH transcription factor complex. Nature (2009) 462(7270):182–8. doi:10.1038/nature08543
89. Huang Y, Lin L, Shanker A, Malhotra A, Yang L, Dikov MM, et al. Resuscitating cancer immunosurveillance: selective stimulation of DLL1-Notch signaling in T cells rescues T-cell function and inhibits tumor growth. Cancer Res (2011) 71(19):6122–31. doi:10.1158/0008-5472.CAN-10-4366
90. Zhao XC, Dou GR, Wang L, Liang L, Tian DM, Cao XL, et al. Inhibition of tumor angiogenesis and tumor growth by the DSL domain of human Delta-like 1 targeted to vascular endothelial cells. Neoplasia (2013) 15(7):815–25. doi:10.1593/neo.13550
91. Dontu G, Jackson KW, McNicholas E, Kawamura MJ, Abdallah WM, Wicha MS. Role of Notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast Cancer Res (2004) 6(6):R605–15. doi:10.1186/bcr920
92. Nickoloff BJ, Qin JZ, Chaturvedi V, Denning MF, Bonish B, Miele L. Jagged-1 mediated activation of Notch signaling induces complete maturation of human keratinocytes through NF-kappaB and PPARgamma. Cell Death Differ (2002) 9(8):842–55. doi:10.1038/sj.cdd.4401036
93. Eiraku M, Tohgo A, Ono K, Kaneko M, Fujishima K, Hirano T, et al. DNER acts as a neuron-specific Notch ligand during Bergmann glial development. Nat Neurosci (2005) 8(7):873–80. doi:10.1038/nn1492
94. Meng H, Zhang X, Hankenson KD, Wang MM. Thrombospondin 2 potentiates Notch3/jagged1 signaling. J Biol Chem (2009) 284(12):7866–74. doi:10.1074/jbc.M803650200
95. Sakamoto K, Yamaguchi S, Ando R, Miyawaki A, Kabasawa Y, Takagi M, et al. The nephroblastoma overexpressed gene (NOV/ccn3) protein associates with Notch1 extracellular domain and inhibits myoblast differentiation via Notch signaling pathway. J Biol Chem (2002) 277(33):29399–405. doi:10.1074/jbc.M203727200
96. Rauen T, Raffetseder U, Frye BC, Djudjaj S, Muhlenberg PJ, Eitner F, et al. YB-1 acts as a ligand for Notch-3 receptors and modulates receptor activation. J Biol Chem (2009) 284(39):26928–40. doi:10.1074/jbc.M109.046599
97. Cui XY, Hu QD, Tekaya M, Shimoda Y, Ang BT, Nie DY, et al. NB-3/Notch1 pathway via Deltex1 promotes neural progenitor cell differentiation into oligodendrocytes. J Biol Chem (2004) 279(24):25858–65. doi:10.1074/jbc.M313505200
98. Tkatchenko TV, Moreno-Rodriguez RA, Conway SJ, Molkentin JD, Markwald RR, Tkatchenko AV. Lack of periostin leads to suppression of Notch1 signaling and calcific aortic valve disease. Physiol Genomics (2009) 39(3):160–8. doi:10.1152/physiolgenomics.00078.2009
99. Tanaka E, Asanuma K, Kim E, Sasaki Y, Oliva Trejo JA, Seki T, et al. Notch2 activation ameliorates nephrosis. Nat Commun (2014) 5:3296. doi:10.1038/ncomms4296
100. Li K, Li Y, Wu W, Gordon WR, Chang DW, Lu M, et al. Modulation of Notch signaling by antibodies specific for the extracellular negative regulatory region of NOTCH3. J Biol Chem (2008) 283(12):8046–54. doi:10.1074/jbc.M800170200
101. Zage PE, Nolo R, Fang W, Stewart J, Garcia-Manero G, Zweidler-McKay PA. Notch pathway activation induces neuroblastoma tumor cell growth arrest. Pediatr Blood Cancer (2012) 58(5):682–9. doi:10.1002/pbc.23202
102. Dumont AG, Yang Y, Reynoso D, Katz D, Trent JC, Hughes DP. Anti-tumor effects of the Notch pathway in gastrointestinal stromal tumors. Carcinogenesis (2012) 33(9):1674–83. doi:10.1093/carcin/bgs221
Keywords: Notch signaling, leukemia, lymphoma, oncogenes, tumor suppressor
Citation: Hernandez Tejada FN, Galvez Silva JR and Zweidler-McKay PA (2014) The challenge of targeting Notch in hematologic malignancies. Front. Pediatr. 2:54. doi: 10.3389/fped.2014.00054
Received: 24 March 2014; Accepted: 21 May 2014;
Published online: 10 June 2014.
Edited by:
Alan Wayne, Children’s Hospital Los Angeles, USAReviewed by:
Christian Capitini, University of Wisconsin, USAChintan Parekh, University of Southern California, USA
Copyright: © 2014 Hernandez Tejada, Galvez Silva and Zweidler-McKay. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Patrick A. Zweidler-McKay, Department of Pediatrics, University of Texas M. D. Anderson Cancer Center, 1515 Holcombe Blvd, Unit 853, Houston, TX 77030, USA e-mail:cHp3ZWlkbGVyQG1kYW5kZXJzb24ub3Jn