 Roland Staud
Roland Staud Melyssa M. Godfrey1
Melyssa M. Godfrey1 Patrick W. Stroman
Patrick W. Stroman- 1Division of Rheumatology and Clinical Immunology, University of Florida, Gainesville, FL, United States
- 2Center for Neuroscience Studies, Queen’s University, Kingston, ON, Canada
Widespread pain and hyperalgesia are characteristics of chronic musculoskeletal pain conditions, including fibromyalgia syndrome (FM). Despite mixed evidence, there is increasing consensus that these characteristics depend on abnormal pain augmentation and dysfunctional pain inhibition. Our recent investigations of pain modulation with individually adjusted nociceptive stimuli have confirmed the mechanical and thermal hyperalgesia of FM patients but failed to detect abnormalities of pain summation or descending pain inhibition. Furthermore, our functional magnetic resonance imaging evaluations of spinal and brainstem pain processing during application of sensitivity-adjusted heat stimuli demonstrated similar temporal patterns of spinal cord activation in FM and HC participants. However, detailed modeling of brainstem activation showed that BOLD activity during “pain summation” was increased in FM subjects, suggesting differences in brain stem modulation of nociceptive stimuli compared to HC. Whereas these differences in brain stem activation are likely related to the hypersensitivity of FM patients, the overall central pain modulation of FM showed no significant abnormalities. These findings suggest that FM patients are hyperalgesic but modulate nociceptive input as effectively as HC.
Introduction
Pain was defined by the International Association for the Study of Pain (IASP) as an unpleasant sensory and emotional experience associated with, or resembling that associated with, actual or potential tissue damage (1). Nociceptive pain depends on activation of nociceptors in peripheral tissues to direct individuals’ attention on impending injury and to elicit adaptive behaviors. Pain can be divided into acute pain, which is usually due to a recent or pending injury, and chronic pain which lasts for more than 3 months (2). Pain was labelled as chronic primary pain by the International Classification of Diseases (ICD-11) (3) when pain persists for more than 3 months and is associated with significant emotional distress and/or functional disability, and the pain is not better accounted for by another condition. Examples of chronic primary pain include chronic primary headache, complex regional pain syndrome (CRPS), chronic orofacial pain, chronic primary visceral pain, chronic widespread pain, and fibromyalgia (FM). These chronic pain conditions also fulfill the nociplastic pain classification criteria proposed by the IASP (4).
Nociplastic pain
The peripheral and central nervous system constantly adapts to intrinsic and extrinsic challenges. This neuronal plasticity can result in increased or reduced responsiveness of neurons to peripheral nociceptive input. Strong or prolonged nociceptive input can lead to peripheral and/or central sensitization which is characterized by increased responding of nociceptive neurons in the peripheral and central nervous system (CNS), respectively (4).
In 2021, the International Association for the Study of Pain created the new term “nociplastic pain” for chronic musculoskeletal pain that appears to be out of proportion to detectable tissue injuries. Patients with nociplastic pain often report insomnia, excessive fatigue, and cognitive abnormalities, as well as hypersensitivity to sound, light, taste, and odor (5). Some of the most prevalent nociplastic pain conditions include tension-type headache, chronic pelvic pain, chronic low back pain and FM. Although the exact mechanisms of nociplastic pain is unclear many investigators consider central pain processing abnormalities including abnormal pain modulation, as relevant (6). Furthermore, numerous findings of brain abnormalities have been associated with nociplastic pain, including changes in the grey matter volume and altered functional connectivity of brain regions involved in pain and sensory processing (7). Increasing evidence also suggests that neuroinflammation, characterized by glial activation, production of neurotransmitters, and proinflammatory cytokines, may also play an important role for nociplastic pain (8–10).
Although the term “nociplastic” pain was meant to describe complex chronic pain conditions like FM, irritable bowel syndrome, temporomandibular disorder, and chronic back pain (11), it is controversial. Neuroplasticity is not a novel concept as both peripheral and central sensitization depend on neuroplasticity associated with multiple translational and transcriptional changes of neurons in the peripheral and central nervous system (CNS) (4). Whereas the evidence for peripheral and central sensitization is well established in inflammatory or neuropathic pain conditions the mechanisms of nociplastic pain are not as well understood. We have recently shown that the pain of FM patients (or nociplastic pain) is due to hypersensitivity of central nervous system pathways and not to augmented pain processing in the dorsal horn of the spinal cord or brain as previously thought (12, 13).
Pain modulation during acute or chronic musculoskeletal pain
Peripheral sensitization
Although not considered a major contributor to chronic pain, peripheral sensitization can frequently be detected in patients with chronic musculoskeletal pain syndromes (14, 15) After repeated or intense noxious stimuli, sensitization of peripheral afferents can occur, resulting in decreased activation thresholds and amplified responses to subsequent inputs (16–18). Without continuing nociceptive input, increased pain sensitivity usually normalizes over time, making this phenomenon sometimes long lasting but reversible. Besides increased excitability of primary sensory neurons, increased expression of ion channels on nerve endings and axons can occur, including potassium channels (19), voltage-gated sodium (Nav) channels (20), voltage-gated calcium (Cav) channels (21), acid-sensing ion channels (ASICs) (22). and transient receptor potential (TRP) channels (23, 24).
Central sensitization
Central sensitization occurs mostly at the dorsal horn neuron level and is usually associated with acute tissue injury. It is a complex neurophysiological process that has been thought to be responsible for the increased pain sensitivity of many patients with chronic pain syndromes, including FM. It involves changes in the function and connectivity of dorsal horn neurons of the spinal cord, which play a critical role in processing and transmitting painful signals to the brain (25). Central sensitization involves an increase in the excitability of nociceptive neurons and this heightened excitability can be attributed to various mechanisms, including changes in the ion channels, particularly after activation of N-Methyl-D-Aspartate (NMDA) receptors. Central sensitization can also lead to the expansion of the receptive fields of dorsal horn neurons. Normally, these neurons respond to a specific area of the body, but in central sensitization, their responses can spread to adjacent or even distant regions. This can result in pain spreading beyond the initial injury site.
The lack of consistent evidence for a major role of peripheral sensitization in chronic musculoskeletal pain syndromes has focused increased attention on central pain amplification. Central sensitization has provided a mechanistic explanation for some of the temporal, spatial, and sensitivity changes in acute and chronic pain syndromes like FM and has focused attention on the important contribution of the central nervous system to the increased pain sensitivity of chronic pain patients. Whereas peripheral sensitization is characterized by reduced pain thresholds and hypersensitivity of local nociceptors (26–28), central sensitization causes pain hypersensitivity in areas outside of tissue injury and requires little peripheral input to maintain high pain sensitivity even after tissue healing has occurred. Although central sensitization clearly plays an important role for pain reported after tissue injury, its particular role in many patients with “nociplastic” pain syndromes, like FM is unclear.
Animal experiments have identified two major forms of pain sensitivity augmentation: central sensitization (CS) which occurs primarily in the spinal cord and long-term potentiation (LTP) which affects preferentially brain regions. CS is frequently present after tissue injury and LTP is a critical process for memory formation but also for chronic pain. CS comprises augmented responses of dorsal horn neurons after electrical nerve stimulation associated with long-lasting aftersensations (14). Its neural correlate in human subjects is “windup” which is associated with altered functional connectivity of regions involved in pain and sensory processing (7, 29). Spinal modulation of nociceptive input occurs in the dorsal horn of the spinal cord where incoming signals are transmitted to interneurons in Rexed laminae before they are projected to the brain. Central sensitivity changes can involve excitatory neurotransmitter release comprising glutamate and aspartate, or inhibitory neurotransmitters including GABA and glycine (30).
Long-term potentiation
The central nervous system is a complex network and its functioning depends on the excitability of individual neurons but the strength of their synaptic connections can vary (31). Such neuronal plasticity is crucial for effective nervous system functioning including memory formation (32) and chronic pain (33). Some forms of neuronal plasticity are due to long-term potentiation (LTP) which plays not only an important role in memory consolidation (34), but also for pain (35). Usually LTP of spinal neurons occurs during high frequency electrical stimulation (33, 36), but can also be induced by tissue inflammation (37), and peripheral nerve injury (38).
Descending pain modulation
The dorsal horn of the spinal cord comprises 2nd order neurons which project to the brainstem and brain. Descending modulation from the midbrain periaqueductal gray and the rostral ventromedial medulla (RVM) affects the activity of these neurons as either pain inhibitory or facilitatory (39) through release of multiple neurotransmitters including serotonin, noradrenaline, and dopamine (40). Abnormalities of descending pain modulation, including pain facilitation and pain inhibition can be evaluated by quantitative sensory tests like temporal summation of pain (TSP) (41, 42) and conditioned pain modulation (CPM), respectively (43).
It is well known that cognitions and feelings can influence pain perception (44). Previous research has demonstrated that distractions can reduce pain intensity, whereas attention to a nociceptive stimulus will increase the pain perceived (45). Furthermore, interventions that elicit positive affect may result in reduced pain perception, whereas the presence of negative affect may increase pain intensity (46). Previous findings seemed to indicate that descending pain modulation is dysfunctional in chronic pain conditions such as FM, temporomandibular disorder, irritable bowel syndrome and chronic headaches (47). Therefore, dysfunctional pain modulation seemed to contribute to the development and maintenance of chronic pain syndromes (48). Descending pain control from the brain stem affects the entire body (49) and can dramatically increase the pain sensitivity of individuals with chronic pain (50, 51). However, not all studies of chronic pain patients reported abnormal descending pain modulation. Normal pain modulation was reported in some studies of patients with chronic low back pain (52, 53).
Neuro-inflammation
Glial cells are critical for structure and function of the brain (54). Proinflammatory cytokine signaling between glial cells provides protection from infection and plays an important role in tissue repair and recovery (55). Glia can be activated by many cytokines, chemokines, proteases, and growth factors, that not only play an important role for chronic pain (56) but are also important for memory (57) and pain modulation (58). For example, cytokine inhibition has been found to improve osteoarthritis pain (59), low back pain (60), and rheumatoid arthritis pain (61). Therefore, excessive glial activation (neuroinflammation) may play an important role in chronic pain states and decreasing neuroinflammation may be a promising treatment target for patients with such conditions (62). Previous positron emission tomography (PET) studies of chronic pain patients reported widespread cortical enhancements consistent with neuroinflammation. For example, when the [11C]PBR28 PET ligand which binds to the translocator protein (TSPO) on activated microglia or astrocytes (63), was used in FM patients neuroinflammation could be observed in M1, S1, precuneus, superior parietal lobe, insula, and thalamus (64, 65). In another study of FM patients, increased TSPO binding could be demonstrated in the medial and lateral areas of the frontal and parietal lobe (66) and fatigue ratings of FM patients correlated with [11C]PBR28 binding in the anterior and posterior middle cingulate cortices.
Nociplastic pain in FM and other chronic musculoskeletal pain syndromes
Pain is a multifactorial experience associated with memory as well as psycho-social factors (67, 68). While acute pain will focus the individual on the noxious stimuli thus preventing further tissue damage (69), chronic pain is considered “maladaptive” and almost always associated with neuronal plasticity. In chronic musculoskeletal pain conditions, like FM or low back pain, painful sensations are considered nociplastic and strongly associated with hyperalgesia/allodynia (11).
Other views of chronic (nociplastic) pain are centered on stress evoked, sympathetically maintained, neuropathic pain which depends on dorsal root ganglia abnormalities as critical for its pathogenesis (70). This hypothesis is based on evidence that nociplastic pain has some neuropathic features, including paresthesias and allodynia. In some studies, nearly half of FM patients demonstrated evidence of peripheral nerve damage including small nerve fiber pathology (71). However, this hypothesis fails to explain the chronic pain mechanisms in the majority of FM patients.
Pain facilitation and inhibition are not abnormal in FM patients
FM is a complex syndrome that is comprised of widespread pain, fatigue, insomnia and cognitive difficulties. Its pathogenesis as a central nervous system (CNS) disorder is widely accepted and patients with this syndrome demonstrate hypersensitivity to painful and non-painful stimuli. The symptoms of FM are unspecific and can be found in many other chronic pain disorders including migraine, temporo-mandibular disorder (TMD) and chronic low back pain (72). Enhanced pain facilitation, together with other factors like negative affect, may also help explain the known discordance between the severity of tissue abnormalities and clinical pain associated with FM, OA, and low back pain (73, 74). Deficient descending pain inhibition (Conditioned Pain Modulation: CPM) has been reported in FM (75) but also in other chronic pain conditions, like IBS (76), migraine (47), and low back pain (77). However, high variability of the CPM efficacy of chronic pain patients was noted in previous studies which may be related to the experimental conditions used, including the application of non-standardized conditioning (78) and test (79) stimuli.
Although dysfunctional pain modulation has been proposed as a relevant pathogenetic mechanism of FM, this view has recently become controversial. Therefore, we examined pain modulation of FM patients in several studies, using only experimental stimuli that were adjusted to each participant's pain sensitivity thus eliminating ceiling and floor effects associated with non-standardized stimuli (12, 13, 80).
One of these studies (participants n = 51) (12) examined the pain inhibitory and facilitatory capacity of FM patients (Figures 1, 2), using such standardized pain stimuli (12). This study demonstrated effective pain inhibition in 87% of FM patients similar to HC. Only 13% of participants failed to show pain reductions or demonstrated pain facilitation. During testing of pain facilitation, FM patients showed normal TSP similar to HC (p > 0.05). These results differ from the findings of some other FM studies of pain modulation (75, 81). One reason for the difference of our results in regards to several previous FM studies (52, 82) may be that we applied only test stimuli that were carefully adjusted to each participant's pain sensitivity, thus avoiding ceiling and floor effects. These results provided strong evidence that FM patients can effectively modulate experimental pain similar to HC.
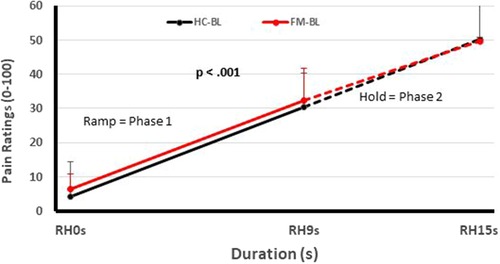
Figure 1. Testing of pain facilitation of HC and FM participants. All subjects received sensitivity-adjusted Ramp&Hold (RH) stimuli at the hand. Pain ratings of the second phase of RH (dotted lines) depend on each individual's pain summation ability. Although pain ratings of FM participant and HC increase significantly during RH (p < 0.001), their rate of temporal summation in phase 2 was not significantly different from each other (p > 0.05). (RH, ramp & hold; s, seconds; HC, healthy controls; FM, fibromyalgia) [Reproduced with permission (12)].
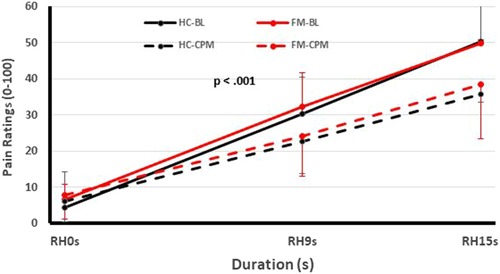
Figure 2. Testing of pain inhibition of FM participants and HC using heat Ramp&Hold (RH) applications to one hand (test stimuli) during immersion of the other hand in a cold water bath (conditioning stimulus). Time course of Ramp&Hold (RH) ratings of FM and HC during baseline (solid lines) and CPM (dotted lines). During baseline (solid lines), the RH ratings of both groups increased significantly (p < 0.001) but this increase was not different for HC and FM participants (p > 0.05). During CPM (dotted lines), the RH ratings increased significantly less than during baseline testing in HC and FM subjects (p < 0.03), but there was no significant difference of CPM efficiency between groups noted (p > 0.05). (FM, fibromyalgia; HC, healthy control; CPM, conditioned pain modulation; RH, ramp & hold; BL, baseline; s, second) [Reproduced with permission (12)].
Pain hypersensitivity is a key feature of FM
Over the last decade, our understanding of chronic pain has significantly advanced thanks to behavioral and brain imaging studies (83). Some trials suggested that FM is associated with abnormal resting-state functional connectivity (84) and decreased gray matter of pain processing brain regions (85). Our understanding of pain advanced significantly with the discovery of several brainstem regions that can control pain-related activity in the spinal cord (SC) (50, 83). Several brain and brain stem regions can modulate the activity of dorsal horn neurons through descending modulation, resulting in either pain inhibition or facilitation (86). These areas comprise the periaqueductal gray matter (PAG) and the rostral ventromedial medulla (RVM), which connect with dorsal horn neurons of the SC (87). Because our understanding of pain modulatory processes, particularly in the brainstem and spinal cord, was mostly derived from animal studies (88, 89), we examined the pain modulation of chronic pain patients using functional magnetic resonance imaging of the spinal cord. Our studies (participants n = 59) showed that FM patients not only failed to activate some regions of the descending pain modulatory system but also demonstrated lower connectivity to other pain modulatory areas including the amygdala, hippocampus, and brainstem (80, 90). However, when we tested spinal cord activity of FM patients during the application of standardized experimental pain stimuli, the spinal cord BOLD responses of FM patients were similar to those of HC, indicating that pain related spinal cord activity of FM patients was similar to HC (83, 91). Statistical modeling of pain related CNS activation demonstrated different connectivity of FM patients to/from the PAG compared to HC and was associated with participants’ pain ratings. Thus, despite demonstrating similar efficacy of pain facilitation and inhibition compared to HC, FM participants showed some differences of descending pain regulation via the PAG-RVM-spinal cord pathway. At least some of these differences may be associated with the hyperalgesia of FM participants.
Caveats
Experimental pain studies play a crucial role in advancing our understanding of pain modulation and its relevance to chronic pain. While these studies provide valuable insights, researchers must continue to bridge the gap between experimental findings and the complex pain experiences of chronic pain patients to develop more effective and tailored pain management strategies. Interdisciplinary approaches that combine neurobiology, psychology, and clinical practice are key to this endeavor.
Conclusions
The presumption that abnormal pain facilitation and inhibition are major contributors to FM pathogenesis remains controversial. No significant differences in pain facilitation or inhibition could be detected between FM patients and HC in one of our recent investigations when quantitative sensory testing was performed using standardized nociceptive stimuli (12). Given that the detection of effective pain modulation by CPM seems to be dependent on stimulus modality, heat vs. pressure (79), our investigation using predominantly heat stimuli may have facilitated our ability to detect normal pain modulation of FM patients. Our conclusions are also supported by several brainstem and spinal cord imaging investigations of FM patients and HC which demonstrated similar spinal cord activity in both groups during pain facilitation with standardized heat pain stimuli (13, 80). This lack of spinal cord functional abnormalities in FM seems to emphasize brain and brainstem functional alterations as more important for the pain and hyperalgesia of these patients and not abnormal pain modulation. There is increasing evidence for such functional brain abnormalities in FM (92–95) but the specificity of these findings is still unclear. Overall, the results of our studies seem to suggest that not only pain facilitation but also pain inhibition are normal in FM patients. Our findings also indicate that one of the hallmarks of FM and possibly of other chronic musculoskeletal pain syndrome is hyperalgesia and not abnormal pain modulation. Future studies need to focus on CNS mechanisms that are responsible for FM patients’ hypersensitivity to sensory stimuli which is not limited to nociceptive input but extends to most other sensory domains including sound, light, and sense of smell.
Author contributions
RS: Investigation, Writing – original draft. MG: Conceptualization, Investigation, Writing – review & editing. PS: Investigation, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Raja SN, Carr DB, Cohen M, Finnerup NB, Flor H, Gibson S, et al. The revised international association for the study of pain definition of pain: concepts, challenges, and compromises. Pain. (2020) 161:1976–82. doi: 10.1097/j.pain.0000000000001939
2. Treede RD, Rief W, Barke A, Aziz Q, Bennett MI, Benoliel R, et al. A classification of chronic pain for ICD-11. Pain. (2015) 156:1003–7. doi: 10.1097/j.pain.0000000000000160
3. Nicholas M, Vlaeyen JWS, Rief W, Barke A, Aziz Q, Benoliel R, et al. The IASP classification of chronic pain for ICD-11: chronic primary pain. Pain. (2019) 160:28–37. doi: 10.1097/j.pain.0000000000001390
4. Kosek E, Clauw D, Nijs J, Baron R, Gilron I, Harris RE, et al. Chronic nociplastic pain affecting the musculoskeletal system: clinical criteria and grading system. Pain. (2021) 162:2629–34. doi: 10.1097/j.pain.0000000000002324
5. Staud R, Godfrey MM, Robinson ME. Fibromyalgia patients are not only hypersensitive to painful stimuli but also to acoustic stimuli. J. Pain. (2021) 22:914–25. doi: 10.1016/j.jpain.2021.02.009
6. Kosek E, Cohen M, Baron R, Gebhart GF, Mico JA, Rice ASC, et al. Do we need a third mechanistic descriptor for chronic pain states? Pain. (2016) 157:1382–6. doi: 10.1097/j.pain.0000000000000507
7. Smallwood RF, Laird AR, Ramage AE, Parkinson AL, Lewis J, Clauw DJ, et al. Structural brain anomalies and chronic pain: a quantitative meta-analysis of gray matter volume. J. Pain. (2013) 14:663–75. doi: 10.1016/j.jpain.2013.03.001
8. Schwartz M, Deczkowska A. Neurological disease as a failure of brain-immune crosstalk: the multiple faces of neuroinflammation. Trends Immunol. (2016) 37:668–79. doi: 10.1016/j.it.2016.08.001
9. Gui WS, Wei X, Mai CL, Murugan M, Wu LJ, Xin WJ, et al. Interleukin-1β overproduction is a common cause for neuropathic pain, memory deficit, and depression following peripheral nerve injury in rodents. Mol. Pain. (2016) 12:1–15. doi: 10.1177/1744806916646784
10. Guimaraes MR, Soares AR, Cunha AM, Esteves M, Borges S, Magalhaes R, et al. Evidence for lack of direct causality between pain and affective disturbances in a rat peripheral neuropathy model. Genes Brain Behav. (2019) 18:e12542. doi: 10.1111/gbb.12542
11. Fitzcharles MA, Cohen SP, Clauw DJ, Littlejohn G, Usui C, Hauser W. Nociplastic pain: towards an understanding of prevalent pain conditions. Lancet. (2021) 397:2098–110. doi: 10.1016/S0140-6736(21)00392-5
12. Staud R, Godfrey MM, Riley JL, Fillingim RB. Efficiency of pain inhibition and facilitation of fibromyalgia patients is not different from healthy controls: relevance of sensitivity-adjusted test stimuli. Br J Pain. (2023) 17:182–94. doi: 10.1177/20494637221138318
13. Bosma RL, Ameli Mojarad E, Leung L, Pukall C, Staud R, Stroman PW. Neural correlates of temporal summation of second pain in the human brainstem and spinal cord. Hum Brain Mapp. (2015) 36:5038–50. doi: 10.1002/hbm.22993
14. Woolf CJ. Evidence for a central component of post-injury pain hypersensitivity. Nature. (1983) 306:686–8. doi: 10.1038/306686a0
15. Scholz J, Woolf CJ. Can we conquer pain? Nat Neurosci. (2002) 5(Suppl 1):1062–7. doi: 10.1038/nn942
16. Ji RR, Kohno T, Moore KA, Woolf CJ. Central sensitization and LTP: do pain and memory share similar mechanisms? Trends Neurosci. (2003) 26:696–705. doi: 10.1016/j.tins.2003.09.017
17. Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. (2000) 288:1765–9. doi: 10.1126/science.288.5472.1765
18. Woolf CJ, Walters ET. Common patterns of plasticity contributing to nociceptive sensitization in mammals and aplysia. Trends Neurosci. (1991) 14:74–8. doi: 10.1016/0166-2236(91)90024-O
19. Smith PA. K(+) channels in primary afferents and their role in nerve injury-induced pain. Front Cell Neurosci. (2020) 14:566418. doi: 10.3389/fncel.2020.566418
20. Dib-Hajj SD, Waxman SG. Sodium channels in human pain disorders: genetics and pharmacogenomics. Annu Rev Neurosci. (2019) 42:87–106. doi: 10.1146/annurev-neuro-070918-050144
21. Yang J, Xie MX, Hu L, Wang XF, Mai JZ, Li YY, et al. Upregulation of N-type calcium channels in the soma of uninjured dorsal root ganglion neurons contributes to neuropathic pain by increasing neuronal excitability following peripheral nerve injury. Brain Behav Immun. (2018) 71:52–65. doi: 10.1016/j.bbi.2018.04.016
22. Wemmie JA, Taugher RJ, Kreple CJ. Acid-sensing ion channels in pain and disease. Nat Rev Neurosci. (2013) 14:461–71. doi: 10.1038/nrn3529
23. Iannone LF, De Logu F, Geppetti P, De Cesaris F. The role of TRP ion channels in migraine and headache. Neurosci Lett. (2022) 768:136380. doi: 10.1016/j.neulet.2021.136380
24. Go EJ, Ji J, Kim YH, Berta T, Park CK. Transient receptor potential channels and Botulinum neurotoxins in chronic pain. Front Mol Neurosci. (2021) 14:772719. doi: 10.3389/fnmol.2021.772719
25. Woolf CJ. Central sensitization—uncovering the relation between pain and plasticity. Anesthesiology. (2007) 106:864–7. doi: 10.1097/01.anes.0000264769.87038.55
26. Chen X, Tanner K, Levine JD. Mechanical sensitization of cutaneous C-fiber nociceptors by prostaglandin E2 in the rat. Neurosci Lett. (1999) 267:105–8. doi: 10.1016/S0304-3940(99)00345-6
27. Guenther S, Reeh PW, Kress M. Rises in (Ca2+)i mediate capsaicin- and proton-induced heat sensitization of rat primary nociceptive neurons. Eur J Neurosci. (1999) 11:3143–50. doi: 10.1046/j.1460-9568.1999.00734.x
28. Hucho T, Levine JD. Signaling pathways in sensitization: toward a nociceptor cell biology. Neuron. (2007) 55:365–76. doi: 10.1016/j.neuron.2007.07.008
29. Staud R, Craggs JG, Perlstein WM, Robinson ME, Price DD. Brain activity associated with slow temporal summation of C-fiber evoked pain in fibromyalgia patients and healthy controls. Eur J Pain. (2008) 12:1078–89. doi: 10.1016/j.ejpain.2008.02.002
30. Schaible HG, Ebersberger A, Von Banchet GS. Mechanisms of pain in arthritis. Ann N Y Acad Sci. (2002) 966:343–54. doi: 10.1111/j.1749-6632.2002.tb04234.x
31. Holtmaat A, Svoboda K. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci. (2009) 10:647–58. doi: 10.1038/nrn2699
32. Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. (1993) 361:31–9. doi: 10.1038/361031a0
33. Liu XG, Sandkuhler J. Long-term potentiation of C-fiber-evoked potentials in the rat spinal dorsal horn is prevented by spinal N-methyl-D-aspartic acid receptor blockage. Neurosci Lett. (1995) 191:43–6. doi: 10.1016/0304-3940(95)11553-0
34. Neves G, Cooke SF, Bliss TV. Synaptic plasticity, memory and the hippocampus: a neural network approach to causality. Nat Rev Neurosci. (2008) 9:65–75. doi: 10.1038/nrn2303
35. Ruscheweyh R, Wilder-Smith O, Drdla R, Liu XG, Sandkuhler J. Long-term potentiation in spinal nociceptive pathways as a novel target for pain therapy. Mol Pain. (2011) 7:20. doi: 10.1186/1744-8069-7-20
36. Liu X, Sandkuhler J. Characterization of long-term potentiation of C-fiber-evoked potentials in spinal dorsal horn of adult rat: essential role of NK1 and NK2 receptors. J Neurophysiol. (1997) 78:1973–82. doi: 10.1152/jn.1997.78.4.1973
37. Ikeda H, Stark J, Fischer H, Wagner M, Drdla R, Jager T, et al. Synaptic amplifier of inflammatory pain in the spinal dorsal horn. Science. (2006) 312:1659–62. doi: 10.1126/science.1127233
38. Zhou LJ, Ren WJ, Zhong Y, Yang T, Wei XH, Xin WJ, et al. Limited BDNF contributes to the failure of injury to skin afferents to produce a neuropathic pain condition. Pain. (2010) 148:148–57. doi: 10.1016/j.pain.2009.10.032
39. Heinricher MM, Tavares I, Leith JL, Lumb BM. Descending control of nociception: specificity, recruitment and plasticity. Brain Res Rev. (2009) 60:214–25. doi: 10.1016/j.brainresrev.2008.12.009
40. Wood PB. Role of central dopamine in pain and analgesia. Expert Rev Neurother. (2008) 8:781–97. doi: 10.1586/14737175.8.5.781
41. Staud R, Vierck CJ, Cannon RL, Mauderli AP, Price DD. Abnormal sensitization and temporal summation of second pain (wind-up) in patients with fibromyalgia syndrome. Pain. (2001) 91:165–75. doi: 10.1016/S0304-3959(00)00432-2
42. Vierck CJ Jr, Cannon RL, Fry G, Maixner W, Whitsel BL. Characteristics of temporal summation of second pain sensations elicited by brief contact of glabrous skin by a preheated thermode. J Neurophysiol. (1997) 78:992–1002. doi: 10.1152/jn.1997.78.2.992
43. Nir RR, Yarnitsky D. Conditioned pain modulation. Curr Opin Support Palliat Care. (2015) 9:131–7. doi: 10.1097/SPC.0000000000000126
44. McPhee ME, Graven-Nielsen T. Positive affect and distraction enhance whereas negative affect impairs pain modulation in patients with recurrent low back pain and matched controls. Pain. (2022) 163:887–96. doi: 10.1097/j.pain.0000000000002442
45. Moore DJ, Eccleston C, Keogh E. Cognitive load selectively influences the interruptive effect of pain on attention. Pain. (2017) 158:2035–41. doi: 10.1097/j.pain.0000000000001011
46. Meagher MW, Arnau RC, Rhudy JL. Pain and emotion: effects of affective picture modulation. Psychosom Med. (2001) 63:79–90. doi: 10.1097/00006842-200101000-00010
47. Lewis GN, Rice DA, McNair PJ. Conditioned pain modulation in populations with chronic pain: a systematic review and meta-analysis. J Pain. (2012) 13:936–44. doi: 10.1016/j.jpain.2012.07.005
48. Yarnitsky D. Conditioned pain modulation (the diffuse noxious inhibitory control-like effect): its relevance for acute and chronic pain states. Curr Opin Anaesthesiol. (2010) 23:611–5. doi: 10.1097/ACO.0b013e32833c348b
49. Talbot JD, Duncan GH, Bushnell MC, Boyer M. Diffuse noxious inhibitory controls (DNICs): psychophysical evidence in man for intersegmental suppression of noxious heat perception by cold pressor pain. Pain. (1987) 30:221–32. doi: 10.1016/0304-3959(87)91078-5
50. Millan MJ. Descending control of pain. Prog Neurobiol. (2002) 66:355–474. doi: 10.1016/S0301-0082(02)00009-6
51. Gerhardt A, Hartmann M, Tesarz J, Janke S, Leisner S, Seidler G, et al. Subgroups of musculoskeletal pain patients and their psychobiological patterns—the login study protocol. BMC Musculoskelet Disord. (2012) 13:136. doi: 10.1186/1471-2474-13-136
52. Julien N, Goffaux P, Arsenault P, Marchand S. Widespread pain in fibromyalgia is related to a deficit of endogenous pain inhibition. Pain. (2005) 114:295–302. doi: 10.1016/j.pain.2004.12.032
53. Gerhardt A, Eich W, Treede RD, Tesarz J. Conditioned pain modulation in patients with nonspecific chronic back pain with chronic local pain, chronic widespread pain, and fibromyalgia. Pain. (2017) 158:430–9. doi: 10.1097/j.pain.0000000000000777
54. Allen NJ, Lyons DA. Glia as architects of central nervous system formation and function. Science. (2018) 362:181–5. doi: 10.1126/science.aat0473
55. Dave VA, Klein RS. The multitaskers of the brain: glial responses to viral infections and associated post-infectious neurologic sequelae. Glia. (2023) 71:803–18. doi: 10.1002/glia.24294
56. Kosek E, Altawil R, Kadetoff D, Finn A, Westman M, Le Maitre E, et al. Evidence of different mediators of central inflammation in dysfunctional and inflammatory pain—interleukin-8 in fibromyalgia and interleukin-1 beta in rheumatoid arthritis. J Neuroimmunol. (2015) 280:49–55. doi: 10.1016/j.jneuroim.2015.02.002
57. Habibi L, Ebtekar M, Jameie SB. Immune and nervous systems share molecular and functional similarities: memory storage mechanism. Scand J Immunol. (2009) 69:291–301. doi: 10.1111/j.1365-3083.2008.02215.x
58. Milligan ED, Watkins LR. Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci. (2009) 10:23–36. doi: 10.1038/nrn2533
59. Miller RE, Miller RJ, Malfait AM. Osteoarthritis joint pain: the cytokine connection. Cytokine. (2014) 70:185–93. doi: 10.1016/j.cyto.2014.06.019
60. Dimitroulas T, Lambe T, Raphael JH, Kitas GD, Duarte RV. Biologic drugs as analgesics for the management of low back pain and sciatica. Pain Med. (2018) 20(9):1678–86.
61. Krock E, Jurczak A, Svensson CI. Pain pathogenesis in rheumatoid arthritis-what have we learned from animal models? Pain. (2018) 159(Suppl 1):S98–109. doi: 10.1097/j.pain.0000000000001333
62. Craft JM, Watterson DM, Van Eldik LJ. Neuroinflammation: a potential therapeutic target. Expert Opin Ther Targets. (2005) 9:887–900. doi: 10.1517/14728222.9.5.887
63. Nakatomi Y, Mizuno K, Ishii A, Wada Y, Tanaka M, Tazawa S, et al. Neuroinflammation in patients with chronic fatigue syndrome/myalgic encephalomyelitis: an (1)(1)C-(R)-PK11195 PET study. J Nucl Med. (2014) 55:945–50. doi: 10.2967/jnumed.113.131045
64. Jung YH, Kim H, Seo S, Lee D, Lee JY, Moon JY, et al. Central metabolites and peripheral parameters associated neuroinflammation in fibromyalgia patients: a preliminary study. Medicine (Baltimore). (2023) 102:e33305. doi: 10.1097/MD.0000000000033305
65. Theoharides TC, Tsilioni I, Bawazeer M, Cells M. Neuroinflammation and pain in fibromyalgia syndrome. Front Cell Neurosci. (2019) 13:353. doi: 10.3389/fncel.2019.00353
66. Albrecht DS, Forsberg A, Sandstrom A, Bergan C, Kadetoff D, Protsenko E, et al. Brain glial activation in fibromyalgia—a multi-site positron emission tomography investigation. Brain Behav Immun. (2019) 75:72–83. doi: 10.1016/j.bbi.2018.09.018
67. Lumley MA, Cohen JL, Borszcz GS, Cano A, Radcliffe AM, Porter LS, et al. Pain and emotion: a biopsychosocial review of recent research. J Clin Psychol. (2011) 67:942–68. doi: 10.1002/jclp.20816
68. Gilam G, Gross JJ, Wager TD, Keefe FJ, Mackey SC. What is the relationship between pain and emotion? Bridging constructs and communities. Neuron. (2020) 107:17–21. doi: 10.1016/j.neuron.2020.05.024
69. Melzack R. Pain and the neuromatrix in the brain. J Dent Educ. (2001) 65:1378–82. doi: 10.1002/j.0022-0337.2001.65.12.tb03497.x
70. Martinez-Lavin M. Dorsal root ganglia: fibromyalgia pain factory? Clin Rheumatol. (2021) 40:783–7. doi: 10.1007/s10067-020-05528-z
71. Martinez-Lavin M. Fibromyalgia and small fiber neuropathy: the plot thickens! Clin Rheumatol. (2018) 37:3167–71. doi: 10.1007/s10067-018-4300-2
72. Clauw DJ. Fibromyalgia and related conditions. Mayo Clin Proc. (2015) 90:680–92. doi: 10.1016/j.mayocp.2015.03.014
73. Bedson J, Croft PR. The discordance between clinical and radiographic knee osteoarthritis: a systematic search and summary of the literature. BMC Musculoskelet Disord. (2008) 9:116. doi: 10.1186/1471-2474-9-116
74. Finan PH, Buenaver LF, Bounds SC, Hussain S, Park RJ, Haque UJ, et al. Discordance between pain and radiographic severity in knee osteoarthritis: findings from quantitative sensory testing of central sensitization. Arthritis Rheum. (2013) 65:363–72. doi: 10.1002/art.34646
75. O'Brien AT, Deitos A, Trinanes Pego Y, Fregni F, Carrillo-de-la-Pena MT. Defective endogenous pain modulation in fibromyalgia: a meta-analysis of temporal summation and conditioned pain modulation paradigms. J Pain. (2018) 19:819–36. doi: 10.1016/j.jpain.2018.01.010
76. Albusoda A, Friss K, Gysan M, Ruffle J, Aziz Q, Farmer A. Conditioned pain modulation in functional gastrointestinal disorders: systematic review & meta-analysis. Gut. (2018) 67:A207–A207. doi: 10.1136/gutjnl-2018-BSGAbstracts.414
77. Mlekusch S, Neziri AY, Limacher A, Juni P, Arendt-Nielsen L, Curatolo M. Conditioned pain modulation in patients with acute and chronic low back pain. Clin J Pain. (2016) 32:116–21. doi: 10.1097/AJP.0000000000000238
78. Nuwailati R, Curatolo M, LeResche L, Ramsay DS, Spiekerman C, Drangsholt M. Reliability of the conditioned pain modulation paradigm across three anatomical sites. Scand J Pain. (2020) 20:283–96. doi: 10.1515/sjpain-2019-0080
79. Horn-Hofmann C, Kunz M, Madden M, Schnabel EL, Lautenbacher S. Interactive effects of conditioned pain modulation and temporal summation of pain-the role of stimulus modality. Pain. (2018) 159:2641–8. doi: 10.1097/j.pain.0000000000001376
80. Bosma RL, Mojarad EA, Leung L, Pukall C, Staud R, Stroman PW. FMRI of spinal and supra-spinal correlates of temporal pain summation in fibromyalgia patients. Hum Brain Mapp. (2016) 37:1349–60. doi: 10.1002/hbm.23106
81. Potvin S, Marchand S. Pain facilitation and pain inhibition during conditioned pain modulation in fibromyalgia and in healthy controls. Pain. (2016) 157:1704–10. doi: 10.1097/j.pain.0000000000000573
82. Brietzke AP, Antunes LC, Carvalho F, Elkifury J, Gasparin A, Sanches PRS, et al. Potency of descending pain modulatory system is linked with peripheral sensory dysfunction in fibromyalgia: an exploratory study. Medicine (Baltimore). (2019) 98:e13477. doi: 10.1097/MD.0000000000013477
83. Stroman PW, Bosma RL, Cotoi AI, Leung RH, Kornelsen J, Lawrence-Dewar JM, et al. Continuous descending modulation of the spinal cord revealed by functional MRI. PLoS One. (2016) 11:e0167317. doi: 10.1371/journal.pone.0167317
84. Napadow V, LaCount L, Park K, As-Sanie S, Clauw DJ, Harris RE. Intrinsic brain connectivity in fibromyalgia is associated with chronic pain intensity. Arthritis Rheum. (2010) 62:2545–55. doi: 10.1002/art.27497
85. Burgmer M, Gaubitz M, Konrad C, Wrenger M, Hilgart S, Heuft G, et al. Decreased gray matter volumes in the cingulo-frontal cortex and the amygdala in patients with fibromyalgia. Psychosom Med. (2009) 71:566–73. doi: 10.1097/PSY.0b013e3181a32da0
86. Watkins LR, Wiertelak EP, McGorry M, Martinez J, Schwartz B, Sisk D, et al. Neurocircuitry of conditioned inhibition of analgesia: effects of amygdala, dorsal raphe, ventral medullary, and spinal cord lesions on antianalgesia in the rat. Behav Neurosci. (1998) 112:360–78. doi: 10.1037/0735-7044.112.2.360
87. Ossipov MH, Morimura K, Porreca F. Descending pain modulation and chronification of pain. Curr Opin Support Palliat Care. (2014) 8:143–51. doi: 10.1097/SPC.0000000000000055
88. Jensen KB, Kosek E, Petzke F, Carville S, Fransson P, Marcus H, et al. Evidence of dysfunctional pain inhibition in fibromyalgia reflected in rACC during provoked pain. Pain. (2009) 144:95–100. doi: 10.1016/j.pain.2009.03.018
89. Truini A, Tinelli E, Gerardi MC, Calistri V, Iannuccelli C, La Cesa S, et al. Abnormal resting state functional connectivity of the periaqueductal grey in patients with fibromyalgia. Clin Exp Rheumatol. (2016) 34:129–33.
90. Ioachim G, Warren HJM, Powers JM, Staud R, Pukall CF, Stroman PW. Altered pain in the brainstem and spinal cord of fibromyalgia patients during the anticipation and experience of experimental pain. Front Neurol. (2022) 13:862976. doi: 10.3389/fneur.2022.862976
91. Staud R, Boissoneault J, Lai S, Mejia MS, Ramanlal R, Godfrey MM, et al. Spinal cord neural activity of patients with fibromyalgia and healthy controls during temporal summation of pain: an fMRI study. J Neurophysiol. (2021) 126:946–56. doi: 10.1152/jn.00276.2021
92. Lopez-Sola M, Pujol J, Wager TD, Garcia-Fontanals A, Blanco-Hinojo L, Garcia-Blanco S, et al. Altered functional magnetic resonance imaging responses to nonpainful sensory stimulation in fibromyalgia patients. Arthritis Rheumatol. (2014) 66:3200–9. doi: 10.1002/art.38781
93. Cagnie B, Coppieters I, Denecker S, Six J, Danneels L, Meeus M. Central sensitization in fibromyalgia? A systematic review on structural and functional brain MRI. Semin Arthritis Rheum. (2014) 44:68–75. doi: 10.1016/j.semarthrit.2014.01.001
94. Harris RE, Sundgren PC, Craig AD, Kirshenbaum E, Sen A, Napadow V, et al. Elevated insular glutamate in fibromyalgia is associated with experimental pain. Arthritis Rheum. (2009) 60:3146–52. doi: 10.1002/art.24849
Keywords: chronic pain, fibromyalgia, central sensitization, QST, conditioned pain modulation
Citation: Staud R, Godfrey MM and Stroman PW (2023) Fibromyalgia is associated with hypersensitivity but not with abnormal pain modulation: evidence from QST trials and spinal fMRI. Front. Pain Res. 4:1284103. doi: 10.3389/fpain.2023.1284103
Received: 27 August 2023; Accepted: 22 November 2023;
Published: 5 December 2023.
Edited by:
Marco L. Loggia, Massachusetts General Hospital and Harvard Medical School, United StatesReviewed by:
Akiko Okifuji, The University of Utah, United StatesPablo de la Coba, University of Extremadura, Spain
© 2023 Staud, Godfrey and Stroman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roland Staud c3RhdWRyQHVmbC5lZHU=