 Peter A. Smith
Peter A. Smith
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pain Res. , 22 September 2023
Sec. Neuropathic Pain
Volume 4 - 2023 | https://doi.org/10.3389/fpain.2023.1220034
This article is part of the Research Topic Insights in Pain Mechanisms: 2022 View all 6 articles
Neuropathic pain can result from injury to, or disease of the nervous system. It is notoriously difficult to treat. Peripheral nerve injury promotes Schwann cell activation and invasion of immunocompetent cells into the site of injury, spinal cord and higher sensory structures such as thalamus and cingulate and sensory cortices. Various cytokines, chemokines, growth factors, monoamines and neuropeptides effect two-way signalling between neurons, glia and immune cells. This promotes sustained hyperexcitability and spontaneous activity in primary afferents that is crucial for onset and persistence of pain as well as misprocessing of sensory information in the spinal cord and supraspinal structures. Much of the current understanding of pain aetiology and identification of drug targets derives from studies of the consequences of peripheral nerve injury in rodent models. Although a vast amount of information has been forthcoming, the translation of this information into the clinical arena has been minimal. Few, if any, major therapeutic approaches have appeared since the mid 1990's. This may reflect failure to recognise differences in pain processing in males vs. females, differences in cellular responses to different types of injury and differences in pain processing in humans vs. animals. Basic science and clinical approaches which seek to bridge this knowledge gap include better assessment of pain in animal models, use of pain models which better emulate human disease, and stratification of human pain phenotypes according to quantitative assessment of signs and symptoms of disease. This can lead to more personalized and effective treatments for individual patients. Significance statement: There is an urgent need to find new treatments for neuropathic pain. Although classical animal models have revealed essential features of pain aetiology such as peripheral and central sensitization and some of the molecular and cellular mechanisms involved, they do not adequately model the multiplicity of disease states or injuries that may bring forth neuropathic pain in the clinic. This review seeks to integrate information from the multiplicity of disciplines that seek to understand neuropathic pain; including immunology, cell biology, electrophysiology and biophysics, anatomy, cell biology, neurology, molecular biology, pharmacology and behavioral science. Beyond this, it underlines ongoing refinements in basic science and clinical practice that will engender improved approaches to pain management.
Diseases or lesions that affect the somatosensory system often elicit long lasting neuropathic pain. The signs and symptoms in each individual depend strongly on the nature of the injury as well as their sex, age, ethnicity, genetic predisposition, intestinal microbiome, possible exposure to prior neonatal injury, personality and cultural and environmental factors (1–11). The predominant signs and symptoms include bouts of spontaneous “electric shock-like” pain, the generation of pain by non-noxious touch or cold (mechanical or thermal allodynia) as well as hyperalgesia and sensory disturbances. The latter may present as paresthesias, described as a crawling or pricking sensation or tingling (12). Some patients experience anesthesia dolorosa where the site of injury is painful yet insensitive to touch (13). Others experience the persistent burning pain of causalgia (14). Neuropathic pain is often intractable (15), insensitive to the actions of NSAID's and resistant to the actions of opioids (16, 17). Unlike nociceptive pain, which alerts and protects an individual from actual or potential tissue injury, neuropathic pain persists long after damaged tissue has healed and recovered (18, 19). Since it appears to serve no obvious biological purpose, neuropathic pain has long been assumed to be maladaptive (20–23).
Maladaptive or not, neuropathic pain afflicts 5%–10% of the world's population (15, 24, 25) and frequently presents with co-morbidities such as anxiety, depression, irritability and sleep disorders (12, 26).
Such high prevalence reflects the association of neuropathic pain with a broad range of injuries and/or maladies (12, 14, 27). These not only include peripheral nerve trauma (13, 23, 28, 29), amputation (30), brain trauma (14, 20) or spinal cord injury (31, 32). Neuropathic pain may also occur as a result of multiple sclerosis (33, 34), stroke (14, 35), fibromyalgia (36, 37), small fiber neuropathy (38), post herpetic or trigeminal neuralgia (14, 39), migraine (40), osteoarthritis (41, 42), complex regional pain syndromes I and II (43, 44), rheumatoid arthritis (45), painful diabetic neuropathy (46, 47), autoimmune disease (48), viral infections such as HIV (49–51) or COVID 19 (52) and neuropathies associated with cancer per se (47) and/or chemotherapy (53–55). Neuropathic pain is also prevalent in individuals afflicted with posttraumatic stress disorder (56) and is a positive sign of rare yet debilitating Na+ channelopathies (57–59). In view of the prevalence of this frequently intractable condition, there is a clear and increasingly urgent need to develop new therapeutic approaches (14, 17, 22).
Despite the heterogeneity of the patient population and the association of neuropathic pain with multiple clinical conditions (27), much of the present understanding derives from studies using peripheral nerve injury models in rodents (47, 60, 61). Frequently used models include chronic constriction injury of the sciatic nerve (CCI), spared nerve injury (SNI) of sciatic nerve branches, spinal nerve ligation (SNL), chronic constriction of dorsal root ganglia (CCDRG) and partial nerve ligation or the Seltzer model (PNL) (60–63). This multidisciplinary review will present a synopsis of these findings showing how they have led to a very general understanding of pain aetiology and to the identification of numerous potential drug targets. Despite this, translation between the laboratory and clinic has met with very limited success (10, 25, 64). The extent of the misalignment between preclinical pain research and the clinical population is becoming increasingly clear (25, 65). In view of this, clinical and basic science strategies that seek to bridge this knowledge gap will be presented.
Peripheral nerve injury capable of causing neuropathic pain does not usually kill peripheral neurons (66). It does however promote Wallerian degeneration of severed axons. This is driven by activation of Schwann cells, fibroblasts, mast cells, keratinocytes, epithelial cells at the site of injury as well as neutrophil, macrophage and T- lymphocyte invasion. This is accompanied by activation of satellite glial cells and resident macrophages within the dorsal root ganglia (DRG) (18, 67–72).
Once activated, each of these immunocompetent cell types generate and release an assortment of pro-inflammatory primary mediators (Table 1 and Figure 1). These include interleukins 1α, 1β, 6, 8, 15, 17 and 18 (IL-1α, IL-1β, IL-6, IL-8, IL-15, IL-17 and IL-18) (73–84), tumor necrosis factor α (TNF-α) (81, 85, 86), leukemia inhibitory factor (LIF) (87), oncostatin M (OSM) (88), nerve growth factor (NGF) (18, 89, 90), serotonin, histamine, and substance P (91–94), the secreted glycoproteins Wnt3a and Wnt5a (wingless-type mammary tumor virus integration site family, members 3A and 5A) (95, 96) and the chemokines CCL-2 (97–99), CXCL-1 (70, 100), CXCL-4 (101) and CXCL-12 (102–104) (Table 1). Generation of primary mediators is accompanied by the production of reactive oxygen and nitrogen species (ROS and NOS) such as peroxynitrite and hydrogen peroxide (105–107). These damage mitochondria causing them to leak ROS and components of damage associated molecular patterns (DAMPs) (27, 108). Mitochondrial dysfunction is emerging as a key process in pain etiology (55, 109).
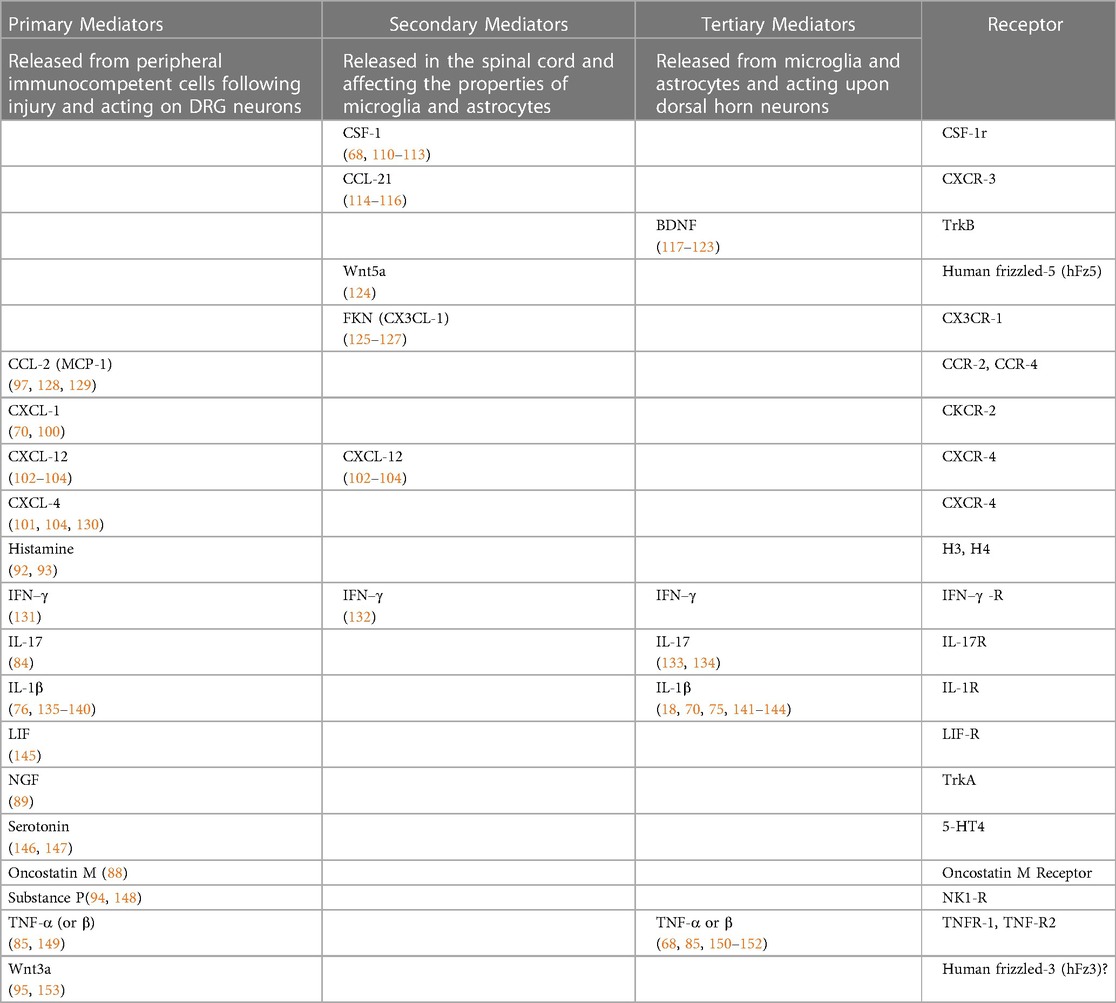
Table 1. List of primary, secondary and tertiary mediators involved in the onset and maintenance of neuropathic pain in response to peripheral nerve injury.
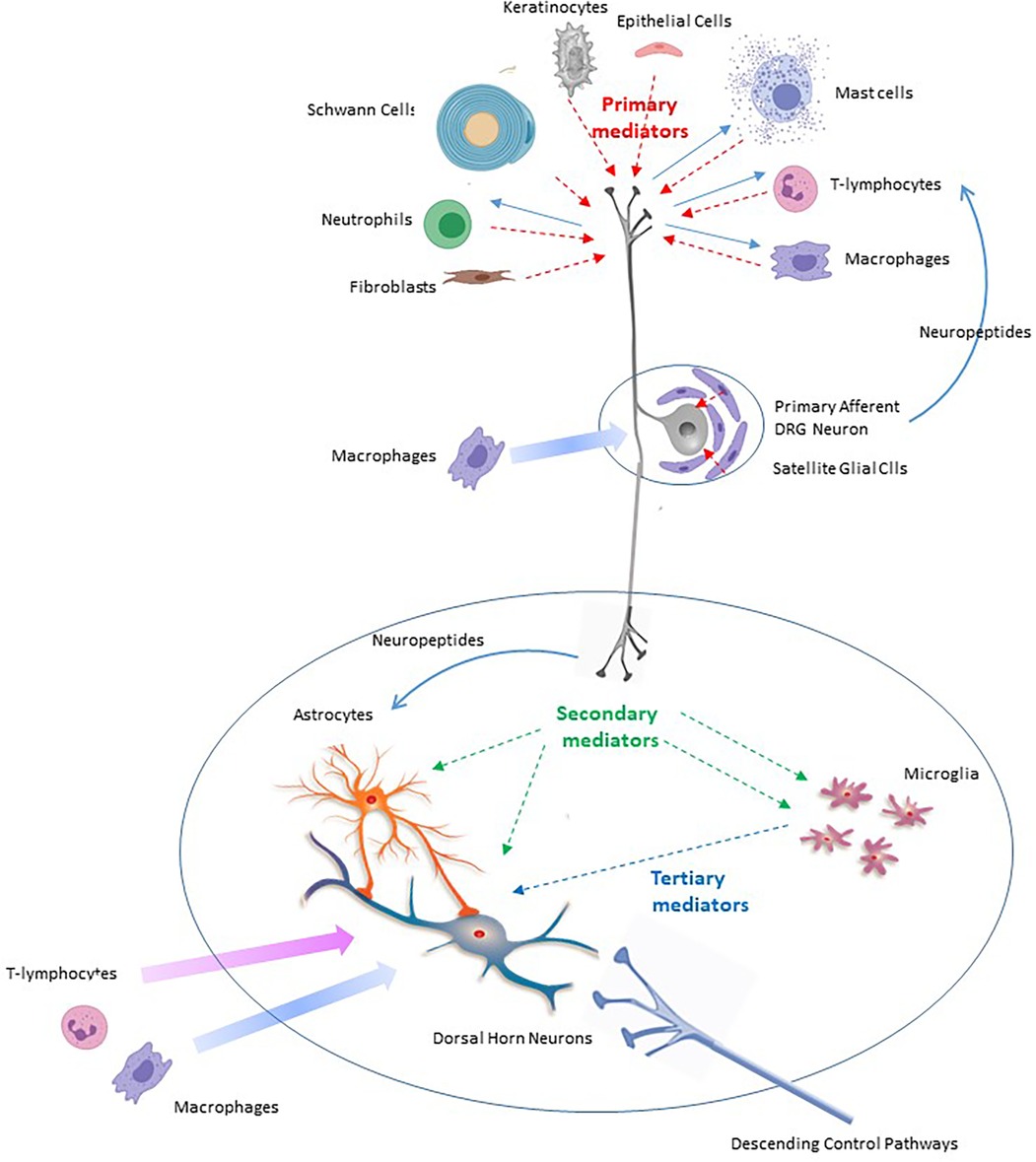
Figure 1. Scheme to illustrate the roles of primary, secondary and tertiary mediators in the onset and maintenance of neuropathic pain in response to peripheral nerve injury.
Whilst some primary mediators have predominantly localized actions, others are released into the systemic circulation (38, 82).
Schwann cell derived IL-1α and TNF-α serve as very early mediators in the response of axons to injury. They recruit macrophages and initiate molecular and cellular events in Wallarian degeneration such as the production of additional cytokines and NGF (81).
Generation of IL-1β is brought about by activation of the Nod-like receptor family pyrin domain containing 3 (NLRP3) inflammasone (33, 154–156). NLRP3 is activated following the release of DAMPs and their interaction with pattern recognition receptors (PRRs) (157), such as Toll-like receptors (TLRs) (158, 159) and with purinergic P2X7 receptors (159). IL-1β is released as a pro-protein and processed into its mature bioactive form by caspase-1 (160) or by metalloproteases 2 and 9 (MMP2 and MMP9) (161). Release of IL-1β from macrophages, dendritic cells and neutrophils, may be brought about via the formation of gasdermin D pores in the cell membrane (160, 162, 163). Alternatively IL-1β release may involve its exocytosis via panexin channels (164).
Metalloproteases also cleave the membrane bound form of TNF-α into the mature 17-kDa form (165). This and their ability to also cleave IL-1β and to produce pain when administered intrathecally has led to the suggestion that MMP antagonists may be useful in pain management (161). There are however no reports of use of metalloprotease inhibitors in pain management in the clinic. This may be due, in part, to the observation that activated MMP2 and MMP9 cleave the mature form of NGF into biologically inactive products (166). The effect of MMP blockade here would be to preserve the presence of pro-inflammatory NGF. This possible proinflammatory action of MMP blockers would tend to restrain any anti-inflammatory/analgesic action.
The overall response of neuronal tissue to inflammatory mediators is described as “neuroinflammation” (167–170). It is characterized by glial cell proliferation and modulation of their phenotype as well as increased neuronal activity. Although the same mediators are responsible for both phenomena, neuroinflammation should not be confused with classical inflammation of whole tissues which is associated with redness (rubor), swelling (tumour), heat (calor) and pain (dolor).
Administration of primary mediators in vivo promotes pain in uninjured animals (70, 74, 135, 165) and perturbation of their actions in nerve-injured animals abrogates or attenuates signs of neuropathic pain (73, 75, 76, 79, 89, 92, 93, 98, 101, 104, 114, 171–173).
Primary mediators such as IL-1β, IL-17, TNF-α, CCL-2, CXCL-12 or type 1 interferons (IFN-1) (131) interact with their cognate receptors on primary afferent neurons to promote extensive changes in genes coding for chemokines, cytokines, eicosanoids, receptors, neuropeptides, signal transduction molecules, synaptic vesicle proteins and ion channels (174, 175). They also affect the expression of long non-coding RNA's (176) and microRNA's (mIR) (177–185). The latter post-transcriptionally regulate the protein expression of hundreds of genes in a sequence-specific manner (186–188) to orchestrate both immune and neuronal processes (189). The observation that extracellular release of mIRs from rodent DRG is increased after CCI (190) is consistent with their suggested role in pain etiology.
It should be noted that the actions of primary mediators are not restricted to peripheral nociceptors (136–138). Tactile information from fast conducting Aβ fibres is processed exclusively within the deep dorsal horn. After peripheral inflammation however, inhibitory spinal circuits are compromised so that innocuous tactile information finds its way to pain processing neurons in the substantia gelatinosa (191). In other words, types of afferent that do not convey pain under normal circumstances start to signal information that is interpreted as pain after nerve injury. This likely contributes to the phenomenon of allodynia. Actions of primary mediators on any type of sensory neuron may therefore have relevance to the onset of pain.
Primary mediators that enter the blood stream promote plasma extravasation and increased permeability of the blood-brain barrier (192) and the blood-nerve barrier in the periphery (193). This and the chemoattractant properties of mediators such as TNF-α (81) facilitate the continuing recruitment of immunocompetent macrophages, leucocytes and lymphocytes to the site of nerve injury as well as to the spinal cord, DRG and supra-spinal structures (70, 77, 78, 194, 195).
Although these findings point to numerous drug targets, clinical trials that involve the perturbation of the action of chemokines, cytokines and other primary mediators have failed to bring forth new and effective therapeutic entities (17, 141, 196).
Peripheral nerve injury, via the actions of primary mediators, leads to ectopic spontaneous activity in primary afferents that is crucial for the onset and persistence of neuropathic pain in humans and signs of such pain in rodent models (19, 30, 35, 197–206). Thus, suppression of aberrant peripheral nerve activity in animal models in vivo by either optogenetic or pharmacological methodologies (205, 207) leads to attenuation of hyperalgesia and abatement of injury-induced allodynia.
In general, peripheral nerve injury, by the action of primary mediators, decreases K+ channel function and increases that of voltage-gated Na+ and Ca2+ channels, TRP channels and HCN channels in DRG neurons (208–213). Injury-induced changes in ion channels can also provoke bursting activity in sensory neurons (214) that may relate to release of ATP and its interaction with P2X3 receptors at the site of injury and the initiation synchronous oscillations in primary afferents (206). In addition, altered excitability may be a consequence of mitochondrial dysfunction and chronic energy deficit (215).
Peripheral activity after injury may affect the whole somatosensory system. It may provoke enduring low frequency cortical oscillations and synaptic remodeling in S1 somatosensory cortex as well as for inducing animals' pain-like behaviors (206). This is supported by the observation that enhancing the synchrony of DRG neuronal activity causes synaptic changes in S1 and pain-like behaviors similar to those seen after spared nerve injury (SNI).
An overview of the actions of IL-1β, TNF-α, Wnt ligands, chemokines and other primary mediators on peripheral neurons is presented in the succeeding sections.
Acute application of IL-1β increases the excitability of DRG neurons by relieving slow inactivation of tetrodotoxin (TTX)-resistant voltage-gated sodium channels (135). IL-1β levels peak at 1 d after injury and remain elevated for ∼7 d (139) and investigations of its longer term actions following 5 d–6 d exposure reveal different effects on different neuronal subpopulations (136). These are observed at remarkably low concentrations (216).
The long term effects of IL-1β on small IB4 -positive neurons (most of which are non-peptidergic, low threshold mechanoceptors) include a reversible increase in action potential (AP) amplitude as a result of increased tetrodotoxin (TTX)-sensitive Na+ current and an irreversible increase in AP duration as result of decreased Ca2+- sensitive K+ conductance (138).
The effects of IL-1β on medium sized neurons, which are the cell bodies of Aδ fibres, are dominated by decreases in K+ currents (137). Although the precise ionic mechanisms differ, IL-1β increases the excitability of both small-diameter IB4-positive neurons and medium-diameter neurons. By contrast, large neurons which are the cell bodies of fast conducting Aβ fibres and IB4-negative neurons, which are predominantly peptidergic nociceptors, are little affected (136).
Macrophage and Schwann cell derived TNF-α is upregulated at the site of injury following CCI (85) and its peripheral application promotes ectopic activity in nociceptors in vivo (217). This effect is enhanced after SNL injury (173). Microinjection of TNF-α lowers mechanical pain threshold in nerve-injured animals in a similar fashion to IL-1β. Most actions of TNF-α in DRG involve modifications of Na+ channel function (218) rather than effects on K+ channels (210). For example, TNF-α upregulates Nav1.7 (219) as well as slow persistent TTX-resistant Na+ channel currents (149).
Intraplantar injection of Wnt3a promotes mechanical hypersensitivity and thermal hyperalgesia in uninjured animals. It also upregulates the ionotropic ATP receptor P2X3 as well as TRPA1 receptor channels. P2X3 receptors may be activated by the passive release of ATP from damaged cells leading to increased sensory neuron excitability (153). Wnt3a also stimulates production of TNF-α and IL-18, thereby augmenting the overall inflammatory response
Several chemokines excite DRG neurons (97, 220).
CCL-2 signals through CCR-2 to increase nociceptor excitability (97, 128, 221, 222). Its effectiveness is increased after DRG compression (CCDRG) (223). CCL-2 is expressed by DRG neurons where it is packaged into large dense-core vesicles. Release of vesicles can be induced by depolarization in a Ca2+-dependent manner (224). This autocrine function could thereby amplify injury-induced excitatory processes evoked in DRG.
CXCL-12 signalling through its cognate receptor, CXCR-4 increases excitability of Nav1.8-positive DRG neurons and this plays a role in the generation of mechanical allodynia as well as small-fiber degeneration in a mouse model of peripheral diabetic neuropathy (101). CXCL-12 and CXCR-4 are upregulated after CCDRG. In addition, intrathecal injection of a CXCL-12 antagonist or a CXCL-12 neutralizing antibody reverse allodynia after SNI or CCDRG (103, 104, 130). These findings suggest that peripheral CXCL-12/CXCR-4 signaling contributes to pain after damage to the DRG per se (104).
In addition to secreted proteins, chemokines, cytokines and growth factors, several small molecules produced at the site of injury act as primary mediators. These include prostaglandin E2, bradykinin, serotonin (146) and histamine (92); all of which increase the excitability of DRG neurons (131, 147). Actions of both serotonin and PGE2 involve augmentation of TTX-resistant INa in nociceptors (225).
As already mentioned, manipulation of the actions of cytokines, chemokines or other primary mediators has so far failed to bring forth any promising therapeutic approaches. On the other hand, the crucial role of primary afferent hyperexcitability and spontaneous activity in pain etiology (30, 199, 202, 205) draws attention to the potential use of ion channels as therapeutic targets (59, 208–210, 226).
DRG neurons express a variety of K+ channel subtypes including delayed rectifiers (Kv1.1, 1.2), A-channels (Kv1.4, 3.3, 3.4, 4.1, 4.2, and 4.3), KCNQ or M-channels (Kv7.2, 7.3, 7.4, and 7.5), ATP-sensitive K+ channels (KIR6.2), Ca2+-activated K+ channels (KCa1.1, 2.1, 2.2, 2.3, and 3.1), Na+ -activated K+ channels (KCa4.1and 4.2) and two pore domain leak channels (K2p; TWIK related channels). These channel subtypes are preferentially and differentially expressed in various neuronal subpopulations and attempts to restore K+ channel function have involved the use of channel activators (210). Although Kv7 activators are quite effective in rodent models (227, 228),, the anticonvulsant, retigabine failed to reach its efficacy endpoint in a trial for post herpetic neuralgia (17).. Nevertheless, as will be outlined below, better phenotypical stratification of patents into clusters on the basis of quantitative measurements of their pathophysiology may reveal clinical efficacy of drugs that failed to demonstrate effectiveness in large groups of patients (8). In the case of K+ channel activators, over 200 new molecules are currently under investigation (227).
Mechanisms that control K+ channel expression and function may present additional therapeutic targets. For example, the expression of Kv7.2, Kv1.4 and KCa1.1 is controlled by the histone methyltranferase G9a (229). Pharmacological inhibition of G9a attenuates neuropathic pain in rodent models (230, 231). Although there is considerable interest in developing histone methyltransferase inhibitors in cancer treatment (232), none have been examined for treatment of neuropathic pain.
A variety of Na+ channel blockers show promise as therapeutic agents; inhibition of Nav1.7, 1.8 or 1.9 seems particularly effective (208, 233, 234). Because it is not found to any great extent in non-neuronal vital tissue such as heart or skeletal muscle, Nav1.7 represents an especially attractive target for therapeutic manipulation (59, 208). Indeed, some level of success has been realized in phase II clinical trials for trigeminal and diabetic neuralgia with the Nav1.7 blocker, vixotrigine (235, 236) but phase III trials remain at the planning stage (235).
Expression of Nav1.8 in DRG neurons is controlled by NGF (237) and the NGF binding antibody tanezumab is effective in various human pain states (238). Small molecule, peripherally-acting TrkA inhibitors have also been identified (239–241).
DRG neurons express high voltage-activated (HVA) Ca2+ channels; Cav2.2 (N-type) as Cav2.1 (P/Q-type) and Cav1.2 (L-type) (242). Low voltage-activated (LVA) channels (T-type) are also present, notably Cav3.2 and 3.3 (243–245).
Because Cav2.1 (P/Q type) and Cav2.2 (N-type) Ca2+ channels contain a synaptic protein interaction site (246) they are closely associated with the synaptic vesicles that govern neurotransmitter release. In view of this, the role of Ca2+ channels in controlling neuronal excitability and reports of upregulation of both HVA and LVA Ca2+ channels by injury (247–249), Ca2+ channels emerge as an important therapeutic target for pain management (208, 209, 226, 250–252). This potential has been realized by the use of the of N-type Ca2+ channel blocker ziconotide as a last resort for pain that is refractory to all other treatments (253). The drawback is that ziconotide needs to be delivered directly to the spinal cord via the intrathecal route (254). In view of this, there is strong interest in developing orally effective N-channel blockers (209, 226, 251, 253) and although several promising agents have appeared in the last five years, the ubiquitous distribution of N-type channels throughout the nervous system means that side effects of such agents may present a serious barrier to drug development.
The function of N-type Ca2+ channels is modulated by Gi/o coupled agonists (255, 256) but the clinical efficacy of the α2-adrenoceptor agonist, clonidine is limited to subsets of patients within the postherpetic neuralgia, complex regional pain syndrome or diabetic neuropathy cohorts (257). Nevertheless, this documented efficacy of clonidine has led to an extensive in silico modelling study. Compounds with nanomolar affinities for the α2a-adrenoceptors and limited ability to recruit arrestin β have been identified and tested in animal models where they behave as non-sedating, orally effective agents that attenuate signs of neuropathic, inflammatory and acute pain (258). The potent α2-adrenoceptor agonist, xylazine has been available for over 30 years, but its use has been restricted to pain management in veterinary medicine as it promotes severe hypotension and dangerous bradycardia in humans (259). It also has documented abuse potential (260).
In addition, the therapeutically important gabapentinoids (16, 261) modulate HVA Ca2+ channel function by binding to their α2δ–1 regulatory subunits (262). Gabapentinoids may antagonise the actions of the endogenous ligand thrombospondin (263). This means that perturbation of thrombospondin expression and/or function may present a novel therapeutic route to pain management. The α2δ–1 subunit plays a major role in Ca2+ channel trafficking, expression and function (22, 248, 264, 265) and deletion of the α2δ–1 gene delays development of mechanical hypersensitivity that follows peripheral nerve damage (262). α2δ–1 is also implicated in controlling the expression of Ca2+ permeable AMPA channels (266) and NMDA receptor channels (267). It is likely therefore that the therapeutic benefits of gabapentinoids involve interactions with several channel types.
LVA T-channels control nociceptor excitability (226, 268–270) and are involved in transmitter release from primary afferent terminals (271, 272). In some patients, gain of function mutations of Cav3.1 contribute to trigeminal neuralgia (273). Peripheral nerve injury (CCI or diabetic neuropathy model) increases function of Cav3.2, in rodent DRG neurons (249, 269) and specific knockdown of Cav3.2 induces marked analgesia in vivo (270).
Although several small molecule Cav3.2 blockers have shown promise in preclinical studies (274, 275) most have failed to exert a significant effects in cohorts of pain patients (208, 276). On the other hand, the high-affinity T-type channel blocker Z944 is especially effective in murine pain models and this may reflect selective blockade of Cav3.1, Cav3.2, and Cav3.3 (277) in peripheral, spinal and thalamic neurons (278, 279). Preliminary results of phase 1 and phase 2 trials with Z944 also appear promising (280).
Some neuropathic pain patients respond favorably to cannabinoids (281) and this may be ascribed to inhibition of Cav3.1 and/or Cav3.2 Ca2+ channels (282, 283) as well as inhibition of N-type Ca2+ channels (284), augmentation of BK type K+ channel currents (285) and stabilization of an inactivated state of Nav1.8 channels (286). There has been considerable interest in NMP-7 and Compound 9 which affect Cav3.2 channels by interactions CB1 and/or CB2 receptors. Although these compounds seem highly effective in animal models, they do not appear to have been tested in the clinic (287–289).
Rather than direct channel block or inhibition by the action of Gi/o coupled agonists, there is considerable interest in modulating Cav3 channel activity by targeting the molecular mechanisms that regulate them.
For example, upregulation of the deubiquitinase, USP5 by IL-1β impairs Cav3.2 ubiquitination thereby protecting it from proteasomal degradation and prolonging its surface expression (140, 272, 290, 291). USP5 knockdown thus increases Cav3.2 ubiquitination, reduces its surface expression leading to reduction of Cav3.2 whole-cell currents. This in turn, leads to attenuation of mechanical hypersensitivity in murine models of both inflammatory and neuropathic pain. As shown in Figure 2A, Cav3.2/USP5 interactions are interrupted by a novel bioactive rhodanine compound (292), by the antiparasitic agent, suramin, and by a TAT-cUBP1-USP5 peptide. Each of these substances attenuate surface expression of Cav3.2 and show analgesic activity in neuropathic and inflammatory pain models (292–294). These observations may lead to the development of new therapeutic approaches (292).
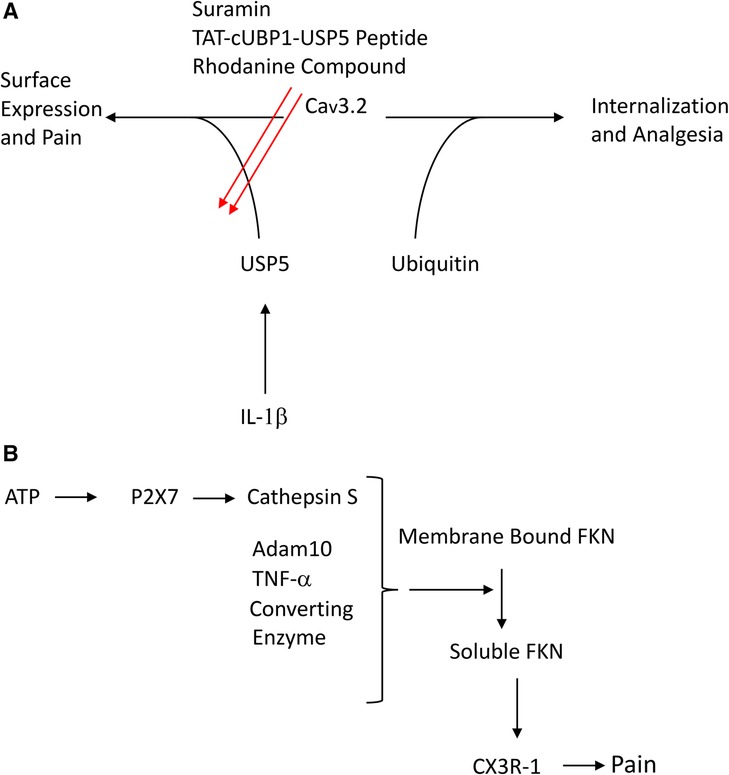
Figure 2. (A) Scheme to illustrate control of Cav3.2 expression by ubiquitin and the deubiquitinase USP5 (B) scheme to illustrate role of cathespin and FKN in neuropathic pain.
Nerve injury or long-term exposure to IL-1β increases HCN channel function in DRG (137, 295). This increase drives spontaneous activity (296, 297) and increases the release of neurotransmitter from primary afferents terminals (298, 299). HCN channel blockers thus supress signs of neuropathic pain in rodent models (300, 301) and selective deletion of HCN2 in nociceptive neurons prevents the development of neuropathic and inflammatory pain (296). Because the HCN2 channel subtype is mainly expressed in neurons as opposed to other excitable tissues (302), HCN2 blockers abrogate DRG hyperexcitability without affecting the HCN1 channels that control cardiac rhythmicity (303). In the clinic, the non-selective HCN blocker, ivabradine which is approved for treatment of heart failure, has a beneficial effect in painful diabetic neuropathy but only a weak effect in other forms of neuropathic pain (304).
TRPV1 receptor channels in nociceptors are upregulated following SNL injury (305) and sensitized by the action of inflammatory mediators (211). Unfortunately, the clinical effectiveness of TRPV1 blockers is limited by the presence of undesirable side effects (306). By contrast, transdermal patches containing a high concentration of the TRPV1 agonist, capsaicin have a role pain management (16). They are applied for 60 min in combination with regional anesthesia. This high level of capsaicin destroys the terminals of TRPV1 expressing nociceptors. This may include those that have sprouted into areas previously occupied by low threshold mechanoceptors (29).
In animal models, combining local anesthetics with capsaicin is especially effective in attenuating signs of pain. Molecules such as lidocaine pass freely through the pore of activated TRPV1 channels and thereby gain access to their intracellular binding site on the Na+ channel. The local anesthetic thus directly and selectively targets TRPV1 expressing nociceptors (307).
In addition to generation and release of inflammatory primary mediators, peripheral nerve injury alters expression of neuropeptides and their cognate receptors in primary afferent neurons (308–311). Neuronal activity promotes the release of neuropeptides such as CGRP and substance P from peripheral nerve endings, DRG cell bodies (312, 313) and primary afferent terminals (314, 315). They modulate sensory neuron activity by excitatory actions in DRG (148, 316, 317) and by their participation in axon reflexes at peripheral nerve endings (318, 319). Although increased effect of CGRP and substance P thus likely contributes to increased excitability and spontaneous activity of peripheral nerve, substance P antagonists are not effective in pain management in the clinical setting (320).
Erenumab, a monoclonal antibody raised against CGRP is available for the management of migraine (321) and recent evidence support the use of CGRP antagonists in the management of trigeminal neuralgia (321, 322). CGRP antagonism, both in the clinic and in animal models is less effective in males than in females (323, 324).
Neuronal activity produces enduring changes in immune and glial cell function (18, 27, 68, 125, 325–327). This process has been termed neurogenic neuroinflammation (328, 329).
Injury-induced upregulation and release of neuropeptides is one of several mechanisms that effects transmission from neurons to glia and immune cells. For example, CGRP, substance P and vasoactive intestinal peptide (VIP) act on their cognate receptors on immune cells and vasculature to promote inflammation (318). CGRP regulates spinal microglial activation in a rodent model of neuropathic pain (330) and substance P regulates expression of IL-1β in keratinocytes (331).
Neuron-immune cell interactions can also be brought about by the synthesis and release of cytokines (110) and chemokines (224) from neurons per se.
By contrast with neurogenic neuroinflammation which is a consequence of injury, essentially the reverse effect; suppression of immune system activity by neuronal activity, characterises a well-defined immune reflex. This contributes to the resolution of inflammation following injury (327). The best characterized part of this reflex involves the vagal release of acetylcholine which acts on the nicotinic acetylcholine receptor subunit α7 (α7nAChR) on innate immune cells to supress cytokine generation and release (327). Activation of β2 adrenoceptors is also immunosuppressant and this is thought to involve downregulation of the TNF-α signaling pathway within the DRG. This may contribute to the efficacy of serotonin - noradrenaline re uptake inhibitors (SNRI's) in neuropathic pain (332). This is because invading sympathetic fibres following nerve injury (44) provide a source of noradrenaline to active immunosuppressant β2 adrenoceptors and noradrenaline abundance is increased by the action of the SNRI, duloxetine. It has also been reported that activation of β2 adrenoceptors on microglia attenuates signs of neuropathic pain in a mouse model (333).
In addition to altered neuronal signalling, neuroinflammation, hyperexcitability, modulation of glial phenotypes and altered expression and function of numerous proteins, neuropathic pain is often associated with enduring structural changes in the peripheral, central and autonomic nervous systems (29, 44, 64, 334, 335).
Neuropathic pain generated by peripheral nerve injury may involve sprouting of nociceptors into denervated territories such as skeletal muscle and skin. Here they replace the initial map and configuration of low threshold sensory axons that do not regenerate. Genetic ablation of nociceptors fully abrogates this type of re-innervation allodynia. These results reveal the emergence of a component of neuropathic pain that is driven by structural plasticity of peripheral sensory nerves, abnormal terminal connectivity and malfunction of nociceptors during reinnervation (29).
Peripheral nerve injury provokes sprouting of perivascular sympathetic axons and appearance of ectopic excitatory α-adrenoceptors on the cell bodies of primary afferent neurons and on their terminals at the site of injury (44, 255). This sprouting may be driven by the neurotrophic action of LIF or NGF (145, 336, 337) and/or may be a consequence of spontaneous afferent activity (338). This is yet another means by which nerve injury increases primary afferent excitability (44, 255, 339–341), leading to signs of neuropathic pain in animal models (342) and to the development of complex regional pain syndromes in humans (343).
The chronic nature neuropathic pain (14, 18) contrasts with nociceptive pain and inflammation that are usually short-lasting or acute. This is because identified “off signals” actively supress the classical signs of inflammation that follow injury to non-neuronal tissue (344, 345). It is not yet understood why these signals fail to activate in neuropathic pain. “Off signals” include lipid-derived specialized pro-resolving mediators (SPMs) and anti-inflammatory cytokines such as IL-10 (346–348) and perhaps IL-6 (349, 350). Subtypes of immune cells such as antinociceptive (M2) macrophages, pain-resolving microglia and regulatory T-cells and modulators of the gut microbiota-immune system are also involved (11).
As emphasised above, spontaneous and ectopic activity in primary afferent fibres is crucial for the maintenance and persistence of signs of neuropathic pain (19, 30, 35, 197–205). Excessive neuronal activity as seen in neurogenic inflammation alters the phenotype of glia and immune cells to provoke the generation of inflammatory mediators (329). It is possible that incessant neurogenic neuroinflammation overcomes the resolution processes that normally terminate inflammation thereby contributing to the indefinite persistence of neuropathic pain.
In addition, the injury-induced structural changes in peripheral afferent (29) and sympathetic nerves (44) and in higher brain structures may be irreversible (64, 335). These enduring changes also contribute to the chronic nature of neuropathic pain (170).
As mentioned already, nerve injury, via the action of primary mediators, upregulates mRNA for a variety of proteins and their receptors in primary afferent neurons (68, 351, 352). These include the secreted proteins CSF-1 (72, 110, 111, 353), CCL-2 (224, 354–356), TNF-α (357), IL-1β and IL-10 (354, 358), CXCL-12 (103, 104), CCL-21 (175, 352), Wnt5a (124) as well as neuropeptides such as CGRP (315) and NPY (351). These act as secondary mediators (68) that alert spinal microglia and astrocytes to the presence of peripheral nerve injury (Table 1 and Figure 1).
The best characterized secondary mediators include the cytokine CSF-1, the chemokines CCL-21 and CXCL-12 as well as Wnt5 and CGRP. Secondary mediators affect the properties of spinal microglia and astrocytes which in turn release tertiary mediators (68) (Table 1 and Figure 1). As will be described below, glial-derived tertiary mediators such as IL-1β and BDNF (117) act on neurons to bring about misprocessing of sensory information and increased activity and excitability leading to central sensitisation (359) (Table 1 and Figure 1).
Although microglia play a predominant role in central sensitization in males, invading macrophages and T-lymphocytes are predominant in females (360–362). Spinal signalling mechanisms invoked in males are therefore very different from those invoked in females (10, 352, 361). Lines of investigation initiated over 20 years ago have been directed towards understanding the numerous cellular and molecular processes that underlie this difference (68, 360, 362–369) and relevant and important differences will be outlined in the succeeding sections.
Injury-induced release of inflammatory primary mediators such as interleukin 1β from macrophages and satellite glial cells in DRG promote de novo synthesis of CSF-1 in primary afferent neurons (68, 71, 72, 353, 370) (Table 1 and Figures 1, 3).
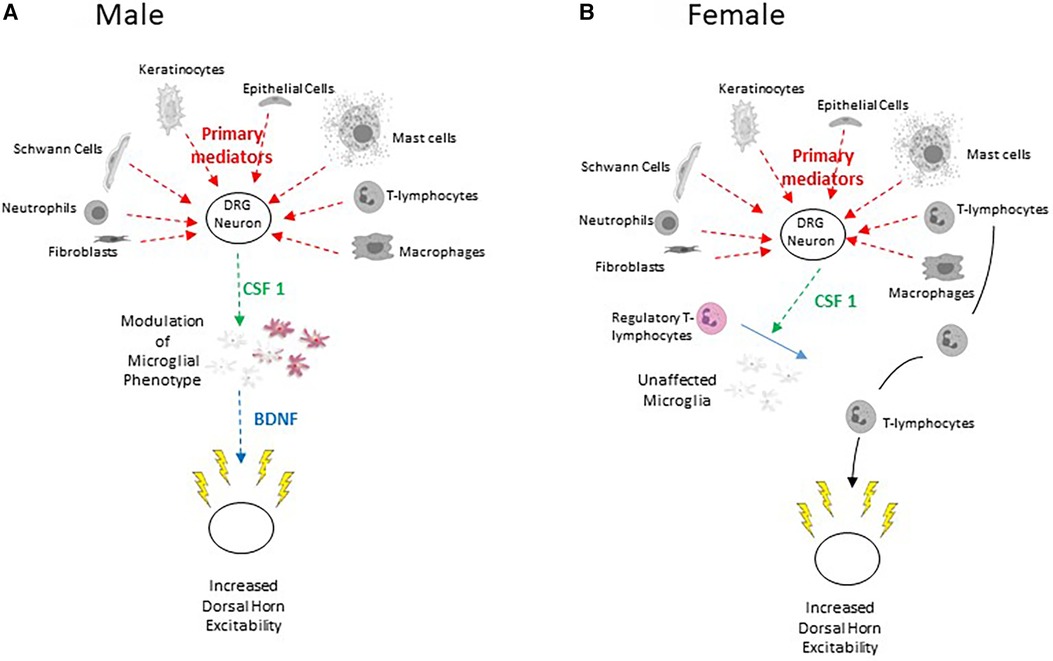
Figure 3. (A) Scheme to illustrate the sensory neuron – CSF-1 – microglia – BDNF pathway characterized in male rodents. Lightly coloured microglia represent those in the resting state and the purple colour represents their transformation to the P2X4 expressing phenotype (B) Scheme to illustrate train of events leading to injury-induced increased dorsal horn excitability in females.
CSF-1 induces phenotypic modulation of spinal microglia and stmulates their proliferation and renewal. Intrathecal injection of this cytokine promotes mechanical allodynia in naïve male rodents but not in females (110, 112, 371, 372). Selective depletion of the Csf1 gene from sensory neurons abrogates nerve injury-induced mechanical hypersensitivity and attenuates proliferation and phenotypic modulation of spinal microglial (71). Nerve-injury also increases mRNA for the CSF-1 receptor in microglia (112, 373) of male rodents. This activation persists for more than 6 weeks after injury (353).
As a corollary of this, it has been shown that alleviation of neuropathic pain by spinal cord stimulation involves a reduction in CSF-1 levels in DRG and spinal cord (373). Other work showed that following injury, the spinal invasion of regulatory T-lymphocytes (suppressor T-cells) attenuate modulation of microglial phenotype in females only. This is supported by the observation that female mice engineered to lack regulatory T-lymphocytes show increased injury induced CSF1-induced microglial modulation and pain hypersensitivity similar to that seen in males (374) (Figure 3B).
In male mice, a major consequence of the release of CSF-1 from primary afferent terminals is promotion of the expression of the ionotropic ATP receptor, P2X4 in spinal microglia (110, 112, 113). ATP-derived from dorsal horn neurons activates these receptors, promoting Ca2+ influx and release of the tertiary mediator BDNF (Figure 3A) (22, 118–120, 375–379). This mechanism is crucial to microglial signalling and the development of central sensitization in males (376, 380) but not in females (362, 381).
Taken together with the observation that exposure of dorsal horn neurons to CSF-1 increases their excitability via a BDNF-dependent process (113), these data strongly support the role of CSF-1 as a secondary mediator signalling between injured primary afferents and microglia (68) (Figure 3A).
In addition to CSF-1, several lines of evidence support the role of CXCL-12 (C-X-C motif chemokine 12) in signalling between injured sensory neurons and astrocytes (102, 104). CXCL-12 and its cognate receptor, CXCR-4 are constitutively expressed in spinal astrocytes and microglia of male rodents (102, 382).
Peripheral nerve injury upregulates CXCL-12 in DRG and CXCR-4 in spinal cord astrocytes (103, 104, 382–384) as a possible consequence of miR-130a-5p downregulation (385) and/or the action of TNF-α (103). As already mentioned, intrathecal administration of CXCL-12 induces hypersensitivity in naive male mice (382). In addition, CXCL-12 antagonists transiently reverse allodynia after DRG crush in male mice (104).
CXCL-12 is thus involved in signaling from injured primary afferents to astrocytes (385). In addition, by virtue of the presence CXCR-4 on microglia, it is also involved in signalling between astrocytes and microglia (382). The CXCL-12/CXCR4 system may also be involved in hyperalgesic priming (386). Hyperalgesic priming describes enhancement of responses to potentially painful stimuli following repetitive stimulation (369, 387, 388). CXCL-12 thus functions as both a secondary mediator between primary afferents and spinal glial cells as a primary mediator between activated immune cells and primary afferents (see Figure 1, Table 1 and above).
Intrathecal administration of CCL-21 (chemokine C-C motif ligand 21) produces pain-like behaviour in naive male mice and CCL-21 neutralizing antibodies or blockade of its cognate CXCR-3 receptor attenuates nerve injury-induced pain (114). The failure of CCL-21 deficient male mice to display tactile allodynia following nerve injury (389) is attributed to failure of microglia to upregulate the purinergic P2X4 receptor (115, 175). CCL-21 is upregulated in DRG following nerve injury, vesicles containing CCL-21 are preferentially transported into axons (390), and it can be released from terminals of injured neurons (116, 391). These findings identify CCL-21 as a third, pro-inflammatory secondary mediator between injured primary afferents and microglia in male mice (68, 175, 383).
CCL-21 also signals to astrocytes where it triggers intracellular Ca2+ transients (385, 392). Despite these findings which were made in male rodents, RNA profiling of the DRGs of humans with neuropathic pain suggests, that CCL-21 may only be involved in female patients (352). These findings underline the importance of both sex and species dependencies of pain etiology.
Stimulation of primary afferents with capsaicin promotes CGRP release in the spinal dorsal horn and this release is increased following nerve injury (315). Since CGRP also affects microglia function (330) it, like other secondary messengers, alerts microglia to the presence of peripheral injury.
In the spared nerve injury (SNI) model, there is a transient effect of CGRP antagonists on mechanical hypersensitivity in female mice only. Consistent with these findings, intrathecally administered CGRP causes a long-lasting, mechanical hypersensitivity in female mice but more transient effects in males. In addition, hyperalgesic priming in female, but not in male rodents is blocked and reversed by intrathecal injection of CGRP antagonists. Systemic administration of a CGRP antibody, blocks hyperalgesic priming specifically in female rodents yet fails to reverse it once it is established. As will be mentioned below, part of the action of CGRP may involve direct modulation of spinal neurons without the intervention of microglia or astrocytes (323).
Unilateral CCI (chronic constriction injury) increases IL-6 mRNA and protein bilaterally in both neurons and satellite glial cells of the DRG (83). IL-6 promotes hyperalgesic priming in rodents (323) and conditional knockout of its cognate gp130 receptor in nociceptors abrogates pain in inflammatory and tumor-induced pain models (393). Although these results are consistent with a secondary mediator role for IL-6, other work suggests that it may have an anti-nociceptive action both in the periphery and at the spinal level following SNI (spared nerve injury) in rodents (349, 350) and may be capable of inducing a desensitized microglial phenotype (394).
IFN-γ alters spinal microglial function and induces tactile allodynia. Genetic ablation of the interferon receptor (IFN-γR) impairs nerve injury-evoked allodynia and prevents phenotypic modulation of spinal microglia (395). The P2X4 receptor is upregulated in IFN-γ stimulated - microglia and, as mentioned already, these purinergic receptors play a crucial role in the onset of neuropathic pain in males (118, 120, 375, 377, 378). IFN-γ also increases dorsal horn excitability (396, 397) and facilitates synaptic transmission between C-fibres and Lamina 1 neurons via a microglial-dependent mechanism (132). Although IFN-γ is found in DRG neurons (398) and the level of IFN-γ is increased in spinal cord following peripheral nerve injury (399) this may originate from invading T-lymphocytes. However, given the role of T-lymphocytes in females (361, 362), IFN-γ may be important in pain aetiology in women.
The lysosomal cysteine protease, cathepsin S is released from microglia by a P2X7-dependent mechanism (400). Cathepsin S, as well as the metalloproteinase ADAM10 and TNF-α converting enzyme liberate the soluble form of the chemokine, fractalkine (FKN; CX3CL-1) from dorsal horn neurons (125, 126, 401) (Figure 2B).
The transmembrane form of FKN and its cognate receptor (CX3CR-1) are expressed constitutively in spinal cord neurons (402, 403). CX3CR-1 which is strongly expressed in dorsal horn microglia (125, 403, 404), is upregulated after nerve injury. In naïve animals, intrathecal injection of FKN produces mechanical allodynia and thermal hyperalgesia whereas injection of an antibody raised against CX3CR-1 attenuates signs of neuropathic pain in animal models (404). This is consistent with the observation that peripheral nerve injury fails to provoke allodynia in mice lacking CX3CR-1 (405).
Spinal nerve ligation (SNL) also increases the level of the soluble form of FKN in cerebrospinal fluid (401) and such release appears to be obligatory for the expression of neuropathic pain (127, 383, 406). Soluble FKN modulates microglial phenotype leading to the generation of tertiary mediators such as TNF and IL-1β (404, 407).
Antibodies raised against CX3CR-1 reduce nociceptive responses when administered as long as 5–7 days after CCI suggesting that the prolonged release of FKN contributes to the maintenance as opposed to the onset of neuropathic pain. This may also relate to the observation that SNL provokes de novo expression of FKN in dorsal horn astrocytes (403).
In addition to producing synaptic potentials in almost all CNS neurons, glutamate affects astrocytes, T-cells, endothelial cells, microglia and vascular cells by interaction with mGluRs (329, 408, 409). These actions are predominantly anti-inflammatory (410). For example, mGluR5 activation in spinal microglia inhibits the release of inflammatory mediators both in vitro (410) and in vivo (411). Also, activation of group I mGluRs in astrocytes leads to increased glutamate and potassium uptake (412). These actions may thus be associated with offset of neuroinflammation rather than its onset.
The secondary mediator CSF-1 interacts with CSF-1R on spinal microglia (353). This leads to increased expression of the tertiary mediator BDNF as a result of up regulation of the Bndf gene (413). As illustrated in Figure 3, the release of BDNF plays an indispensable role in the onset and maintenance of neuropathic pain in male but not in female rodents (22, 68, 113, 117, 119, 121–123, 414–420). BDNF acts primarily via TrkB to increase dorsal horn excitability (113).
Exposure of dorsal horn neurons to CSF-1 also increases the frequency and amplitude of sEPSC's (spontaneous excitatory postsynaptic currents) and this effect is abrogated by the BDNF binding protein TrkB-fc (113). These findings underline the importance of the sensory neuron- CSF-1 -microglia - BDNF signalling process in the aetiology of neuropathic pain (14, 22, 68, 110, 183, 421) (Figure 3A).
ATP activation of microglial P2X4 receptors leading to the release of BDNF is involved in the aetiology of neuropathic pain in males, but not in females. This is congruent with the absence of functional P2X receptors on microglia of female rodents (364). There is also evidence for a role of microglial metabotropic P2Y6, 11, 12, 13 and 14 receptors in the onset of neuropathic pain (68, 422–427). Primary afferent neurons are not the primary source of ATP following peripheral nerve injury. It may rather derive from neurons in the superficial dorsal horn itself (428) as well as from microglia themselves (429). BDNF release from neurons is vesicular and dependent on extracellular Ca2+ (118, 119, 375, 377, 378).
The action of ATP on microglia is not the sole mechanism for promoting BDNF release. Wnt proteins that are upregulated in the spinal cord in various pain models (50, 124, 429–431) activate “frizzled” receptors (432) on microglia to increase expression of BDNF and promote its release (420, 433). This phenomenon has been examined in models of HIV pain which involve exposure of sensory neurons to toxic viral coat proteins such as Vpr1 (49) or gp120 (433, 434). The latter promotes allodynia and increases glutamatergic neuronal activity leading to NMDA receptor activation and increases the level of intraneuronal Ca2+. This, in turn promotes Wnt protein synthesis and release (435, 436).
Phenotypic modulation of microglial function in rodent dorsal horn persists for more than 3 months after injury (437). Thus sequestration of BDNF with TrkBFc (438) or selective depletion of spinal microglia with the targeted immunotoxin Mac1-saporin almost completely reverses mechanical and thermal allodynia up to 3 months after injury. By contrast, intrathecal injection of a cocktail of antibodies against IL-1β, TNF-α, and IL-6 significantly attenuates tactile and cold allodynia at 2 weeks but not at 3 months after injury. These findings suggest that different mediators should be targeted in the short vs. long term management of neuropathic pain (437).
The tertiary mediator, IL-1β is produced and released from macrophages, astrocytes and microglia (18, 439, 440). Release of IL-1β from microglia is a consequence of activation of P2X7 receptors (164, 380, 441, 442) and may be provoked by the action of FKN (407). In agreement with this, it has been reported that the Cav1 channel blocker, cilnidipine which also blocks microglial P2X7 receptors, impairs IL-1β release and reverses SNL-induced mechanical hypersensitivity (142). It has also been suggested that P2X4 receptors interact intracellularly with P2X7 receptors to augment P2X7 receptor-mediated IL-1β release (442).
In addition to the extracellular actions of BDNF and IL-1β, cell-to-cell transport of material via exosomes or extracellular vesicles is now believed to contribute to the development of central sensitisation (183, 443–449). Extracellular vesicles are released from both microglia (450) and astrocytes (451) and are taken up by neurons (447). They may serve as a conduit for the transfer of microRNA's between cell types (452). For example, Nav1.7 protein may be transported from primary afferents to the dendrites of lamina II neurons; a process which may be effected by transfer of exosomes (449).
The cellular mechanisms that are involved in actions of microglial-derived BDNF include enhancement of excitatory processes and attenuation of inhibition (22, 170). In addition to actions on neurons, BDNF also activates astrocytes (453) which release additional mediators such as FKN (403) and IL-1β (18).
Exposure of rat substantia gelatinosa neurons to BDNF increases excitatory synaptic drive to excitatory neurons and decreases excitatory drive to inhibitory neurons (122, 414). In mice, effects of BDNF are dominated by increased excitatory drive to excitatory neurons (113). Whilst resting potential, rheobase, input resistance and excitability are little affected in rat neurons (113, 122, 414), the altered synaptic activity increases spontaneous AP discharge in excitatory neurons whilst reducing it in inhibitory neurons (414).
Several observations show that these actions of BDNF are relevant to injury-CSF-1-microglia-BDNF evoked central sensitization (Figure 3A). Firstly BDNF-induced changes in synaptic transmission and its lack of effect on the intrinsic excitability the cell bodies of lamina II neurons very much parallel those invoked by peripheral nerve injury (122, 454–456). Secondly, Ca2+ responses evoked by neuronal depolarization are enhanced by BDNF and by conditioned medium from lipopolysaccharide-activated microglia. The effect of this conditioned medium is attenuated by sequestering BDNF with TrkBd5 (122). Thirdly, the secondary mediator CSF-1 increases synaptic excitation of excitatory lamina II neurons in mice and this effect is abrogated by sequestering BDNF with TrkBfc (113).
It should be noted however that mitochondrial dysfunction following peripheral nerve injury and the resultant high levels of superoxide may also contribute to increased excitatory synaptic strength in dorsal horn neurons and neuropathic mechanical hypersensitivity (457).
CCI of the sciatic nerve reduces expression of the potassium-chloride exporter (KCC2) in lamina 1 neurons of the dorsal horn (458, 459). The resulting intracellular accumulation of Cl− reverses the Cl− concentration gradient such that normally outward, inhibitory GABAergic synaptic currents mediated by Cl− influx become inward excitatory currents mediated by Cl− efflux (458–460). Knockdown of KCC2 in uninjured rats reduces pain thresholds and induces neuropathic pain-like behaviors. By contrast, rescue of KCC2 expression abrogates signs of neuropathic pain in nerve injured animals (461, 462). Taken together and as illustrated in Figure 4, these findings strongly implicate perturbation of the Cl− gradient and the phenomenon of disinhibition in the pathophysiology of central sensitization (121, 458).
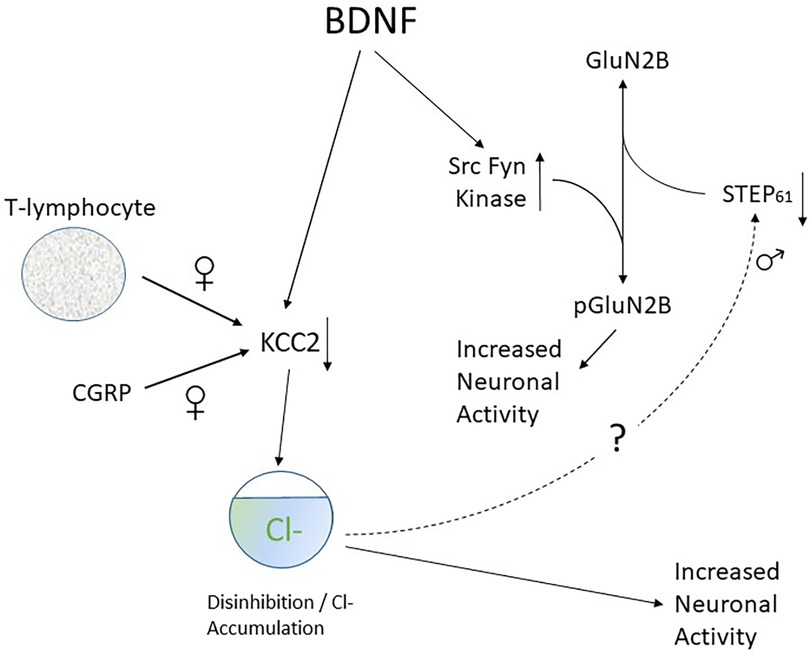
Figure 4. Scheme to illustrate connection between KCC2 downregulation and GluN2B phosphorylation and its sex dependence.
BDNF is responsible for downregulation of KCC2 protein levels in male rats (121, 419). Thus, administration of ATP-activated microglia reproduces the shift in anion gradient seen after nerve injury in the same way as BDNF. Also, blocking TrkB or using interfering RNA against BDNF reverses both injury-induced pain behaviors and the shift in Cl− gradient (121). Changes in KCC2 expression in deep dorsal horn neurons are confined to nociceptive neurons that project via the spinothalamic tract whereas wide dynamic range (WDR) neurons that are activated by a variety of sensory modalities are unaffected (461). It has also been shown that neurons in lamina I are more susceptible to changes in Cl− gradient than those in lamina II (459) and biophysical and modelling analysis shows this loss is especially effective in promoting increased neuronal firing (463). These are important observations as lamina I and deep dorsal horn nociceptive neurons are the primary site for relay of nociceptive information to the brain (464–466). Loss of GABAergic inhibition enables non-noxious Aβ fiber-mediated excitatory transmission to access and excite the pain transmitting neurons of the superficial spinal dorsal horn. Thus, as already mentioned, tactile activation of Aβ fibres is perceived as pain and this process plays a role in the establishment of mechanical allodynia (467–469).
Descending serotonergic inhibition of nociceptive processing from the nucleus raphe magnus becomes excitatory and proalgesic in rats subject to spared nerve injury (SNI). This change is also dependent on collapse of the Cl− gradient following KCC2 hypofunction in the dorsal horn as the KCC2 enhancer CLP290 restores both 5-HT–mediated descending inhibition and analgesia (470).
KCC2 downregulation also contributes to pain hypersensitivity in females (363). Whereas this is mediated by release of BDNF from microglia in males, it involves activation and invasion of adaptive immune cells such as T-lymphocytes in females (362, 381) (Figure 3) as well as downregulation of KCC2 expression by CGRP (323).
BDNF enhances excitatory responses to NMDA in rat spinal cord in vitro (471). In male rodents, this potentiation is dependent on BDNF-mediated GABA disinhibition. By processes yet to be discovered, KCC2-dependent disinhibition promotes downregulation of the tyrosine phosphatase STEP61. Loss of function of STEP61 phosphatase then clears the way for phosphorylation of GluN2B subunits by the Src family kinase Fyn (472). As illustrated in Figure 4, synaptic NMDAR responses are therefore enhanced and neuronal excitability is increased. Decreased activity of STEP61 is both necessary and sufficient to affect GluN2B function (473), This sequence of events is supported by the observation that blocking of KCC2-mediated disinhibition with acetazolamide (474) reverses the downregulation of STEP61 and attenuates behavioural hypersensitivity generated by chronic inflammation.
In female rats however BDNF fails to downregulate KCC2 and STEP61 and to upregulate pFyn, GluN2B and its phosphorylated form GluN2B. This means that BDNF fails to affect synaptic NMDAR responses in lamina I neurons of females. Ovariectomy recapitulates the male pathological pain neuronal phenotype in female rats, with BDNF driving coupling between disinhibition and NMDAR potentiation in lamina I neurons following the elimination of sex hormones (475).
This sex difference in spinal pain processing in rodents is conserved in humans. Thus ex vivo spinal treatment with BDNF downregulates KCC2 and STEP61 and upregulates markers of facilitated excitation in superficial dorsal horn neurons from male but not female human organ donors (475).
In addition to the postsynaptic effects described above (121, 473, 475), BDNF activation of TrkB increases the function of presynaptic NMDA receptors on primary afferent terminals (476). This leads to the potentiation of glutamate release from primary afferents that is observed after SNL (477) and may account for the increased frequency of sEPSC's seen in some dorsal horn neurons in the presence of BDNF(414). Functional upregulation of GluN2B subunits of NMDA receptors (478) may also account for the observation that long term potentiation (LTP) of synaptic transmission of C-fibre responses is enhanced by BDNF (479).
The level of IL-1β is elevated in the cerebrospinal fluid (CSF) of patients with complex regional pain syndrome (480) and in spinal cords obtained post-mortem from patients with painful HIV related neuropathy (50). As already mentioned, activation of P2X7 receptors promotes release of IL-1β from microglia (142, 164, 380, 441) and this is amplified by the action of FKN (407).
Microglial derived IL-1β stimulates astrocytic production of TNF-α well as IL-1β itself (440, 481) thereby amplifying the overall IL-1β signal. IL-1β promotes internalization of the astrocytic glutamate transporter (EAAT2) thereby reducing the capacity of astrocytes to take up glutamate (482, 483). Loss of EAAT2 function thus augments excitatory synaptic transmission and induces hyperalgesia and increased sensitivity of dorsal horn neurons to primary afferent stimulation (484, 485). Activated astrocytes also release CSF-1 (187) thereby amplifying signaling via the CSF-1-microglia-BDNF cascade (Figure 3A). Astrocytes also release the NMDA receptor co-agonist D-serine (486) thereby further augmenting overall dorsal horn excitability.
In a similar fashion to BDNF, IL-1β increases glutamate release from primary afferents and augments excitatory synaptic transmission between primary afferent C-fibres and lamina 1 neurons. It also amplifies Ca2+ responses evoked by exposure of neurons to 20 mM K+ (143, 407, 483).
Like BDNF, IL-1β also does not affect the membrane potential or rheobase of lamina II neurons, suggesting that most of its effect on dorsal horn excitability can be ascribed to changes in synaptic transmission (143, 144). Exposure of rat spinal cord to IL-1β for 6–8 d increases the amplitude of spontaneous EPSC's (sEPSC) in putative excitatory ‘delay’ neurons, and decreases the frequency of spontaneous IPSC's (sIPSC). These actions are similar but not identical to those seen with BDNF or peripheral nerve injury (414, 454, 455). Acute application of IL-1β increases the amplitude of AMPA and NMDA currents dorsal horn neurons (487). Its effect on glutamate release can be ascribed to augmentation of presynaptic NMDA receptor function (483) where signaling between IL-1r and NMDA involves the sphingomyelinase/ceramide pathway (477, 483).
Taken together, all of these actions of IL-1β would be expected to increase dorsal horn excitability and to facilitate the transfer of nociceptive information.
TNF-α decreases the excitability of a subset of spinal GABAergic neurons by suppression of current through HCN channels (488). These effects diminish with time suggesting TNF-α may be primarily involved with the induction rather than the persistence of neuropathic pain (489). As might be expected, blockade of TNF receptor 1 attenuates signs of neuropathic pain in the CCI model but this only occurs in males and not in female rodents (150).
Although FKN action on microglia and potentiation of synaptic transmission in the dorsal horn involves IL-1β but not TNF-α (407), it does appear to be inolved in the generation of a phenomenon named “gliomic LTP” (151, 490). By contrast with classical LTP which is highly localized, “gliomic LTP” spreads extensivly throughout the dorsal horn by the action of TNF-α and of the NMDA co-agonist D-serine (490).
IL-17 is expressed in spinal astrocytes and its cognate receptor is expressed in neurons, especially by those expressing somatostatin (133). SNI-induced static and dynamic allodynia are prevented by intrathecal injection of IL-17 neutralizing antibody and attenuated in IL-17a mutant mice. IL-17 neutralizing antibodies supress LTP of C-fiber evoked field potentials in spinal cord and intrathecal injection of IL-17 or its overexpression in astrocytes produces mechanical allodynia and facilitates spinal LTP (134). IL-17 also supresses inhibitory transmission and enhances excitatory transmission in spinal lamina IIo (133). It may thus serve both as a primary and tertiary mediator (Table 1) but the mechanism of its release from astrocytes is yet to be determined.
Inasmuch as injury-increased peripheral hyperexcitability leads to enduring changes in the dorsal horn, increased dorsal excitability contributes to alterations in supraspinal structures.
Several detailed reviews address supra-spinal changes associated with neuropathic pain (491, 492).
Blood borne inflammatory mediators (38, 493) generated at the site of injury open tight junctions between capillary endothelial cells leading to increased permeability of the blood-brain barrier (192). This allows supra-spinal neurons to interact with blood cells and respond to the cytokines and chemokines they produce (195). Following peripheral injury, afferent information is modulated in various thalamic nuclei (494), somatosensory cortex (495), insular and anterior cingulate cortex (491, 496), nucleus accumbens, and amygdala (497–500). Ascending pathways also interact with the mesolimbic dopamine system (501),
Peripheral nerve injury changes the properties of microglia in the contralateral thalamus, sensory cortex and amygdala as might be expected from the known anatomical arrangement of ascending sensory fibres. Brain regions not directly involved in either sensory or affective aspects of pain such as the motor cortex, do not display altered microglial function (497). This selective modulation of microglia and immune cells in nociceptive pathways (497) may be a consequence of localized neurogenic neuroinflammation as a result of enduring intense activity (329).
Cortico-cortical or cortico-subcortical interactions contribute to the co-morbidies seen in some patients. For example, one form of long-term potentiation (LTP) in the anterior cingulate cortex (ACC) which is triggered by the activation of NMDA receptors and expressed by an increase in AMPA-receptor function, sustains the affective component of the pain state. Another form of LTP in the ACC, which is triggered by the activation of kainate receptors and expressed by an increase in glutamate release, may contribute to pain-related anxiety (491).
There are several parallels between injury-induced cellular changes in higher centres and those seen in the periphery or spinal cord. For example, peripheral neuropathy induces HCN channel dysfunction in medial prefrontal cortex (502) and thalamus (503, 504) and Nav1.3 function is altered in thalamic neurons (505, 506). Both channel types are similarly affected by peripheral nerve injury (208). These findings are fortuitous in terms of drug action and identification of therapeutic targets; drugs developed to act peripherally may also exert beneficial effects as a result of similar central actions.
Cortical structures modulate nociception through descending control of spinal circuitry (507). This occurs by direct corticospinal projections as well as activation of structures in the brainstem such as the periaqueductal grey matter, locus coeruleus, raphe nuclei and rostroventral medulla (492). Descending inhibition of spinal nociceptive processing is mediated via 5HT7 receptors and α2 adrenoceptors whereas serotonergic activation of metabotropic 5HT2 receptors and ionotropic 5HT3 receptors facilitates transmission (508–512). This explains the effectiveness of noradrenaline-serotonin reuptake inhibitors (SNRI) in pain management (16) and the limited efficacy of selective serotonin reuptake inhibitors.
There is normally a balance between descending inhibition and excitation but after peripheral nerve injury the excitatory processes gain the upper hand (470, 513). These changes have been associated with the persistence as opposed to the onset of pain (514, 515).
Peripheral nerve injury impairs dopamine release in the reward circuitry associated with the mesolimbic system (497, 501). This may also relate to the changes in affect (anxiety, depression) experienced by neuropathic pain patients (516). Peripheral nerve injury selectively increases excitability of the nucleus accumbens indirect pathway spiny projection neurons and alters their synaptic connectivity. In addition, tactile allodynia can be reversed by inhibiting and exacerbated by exciting these neurons. This suggests that neurons in the nucleus accumbens not only participate in the central representation of pain, but that they may gate activity in ascending pathways associated with expression of pain in higher centres (517).
Management of neuropathic pain in the clinic involves serotonin-noradrenaline reuptake inhibitors (SNRI), gabapentinoids, capsaicin patches, classical tricyclic antidepressants such as amityptyline, high dose opioids as well as tramadol and botulinum toxin (12, 14, 16, 22). Although the effectiveness of these drugs is limited, extensive preclinical research as outlined above has failed to reveal any effective therapies since the approval of tramadol, a mild opioid with SNRI properties, in the mid 1990′s. To put this into perspective, hundreds of drug targets have been identified over the years; a perfunctory examination of publications appearing in the first 4 months of 2023, identified about 650 papers that dealt with neuropathic pain. Of these, 28 studies identified a “magic molecule”, that was implicated pain etiology in an animal model. Despite this proliferation of potential drug targets, no new drugs have appeared.
What can be done? How can the data gap between animal studies and clinical practice be bridged?
Classical rodent pain models such as SNI (spared nerve injury), CCI (chronic constriction injury), SNL (spinal nerve ligation) or CCDRG (chronic constriction of DRG) have revealed general principles that help to explain the aetilogy of neuropathic pain. These include the identification of various chemokines, cytokines, neuropeptides and growth factors as primary, secondary or tertiary mediators, the concept of neuroinflammation and bidirectional signalling between neurons and immune cells, alterations in synaptic transmission, ion channels and descending modulation, the roles of microglia and astrocytes, central sensitization and role of peripheral spontaneous activity (12, 15, 20, 22, 27, 47, 68, 183, 208, 210). These findings fall short of addressing the multiplicity of chronic pain presentations in the clinic (8, 47) as even in animal models, different types of nerve injury provoke distinct behavioral, physiological and cellular responses.
For example, mechanical allodynia produced by CCI is short-lived and recovery is seen in about 4 weeks whereas that produced by SNI persists for 7 weeks or more (61, 72). Similarly, changes in synaptic transmission in lamina II neurons are more robust after sciatic CCI than after complete sciatic nerve section (axotomy) (455). These findings relate to the observation that CCI promotes stronger and more long lasting upregulation of the inflammatory mediators IL-1β, TNF-α, IL-10, MCP-1/CCL-2 in nerve stumps than nerve crush (354), Recent work has also shown that glycine inputs onto radial neurons in spinal lamina II are reduced following partial nerve ligation (PNL) of the sciatic nerve, this finding was not seen in animals subject to CCI (63).
Whilst neuropathic pain associated with multiple sclerosis is characterized by loss of spinal neurons (371), this is not seen with CCI (518, 519). Although the NGF binding antibody tanezumab is effective in some pain patients (238), studies in animal models suggest that NGF itself may be effective in management of pain and neuropathy associated with HIV infection (520).
The nature of peripheral injury also dictates the precise spinal circuitry involved in the generation of mechanical allodynia (521). Thus nerve injuries generate allodynia by activation of excitatory neurons that express protein kinase C gamma (PKCγ) (522) whereas mechanical allodynia induced by inflammation involves excitatory neurons that are calretinin positive (523). Cholecystokinin (CCK) positive neurons are important in both situations. Punctate allodynia as produced by Von Frey filaments is distinct from dynamic allodynia that is produced by brushing a cotton swab across the hindpaw skin (521). A subset of CCK positive neurons are primarily involved in conveying dynamic rather than punctate allodynia.
Work using knockout mice has shown that deficiency of CCL19/21 attenuates nerve injury evoked pain but not the hyperalgesia observed in an animal model of multiple sclerosis (116).
This issue of injury-specific mechanisms is starting to be resolved as basic scientists have increasingly turned their attention to disease models rather than classical neuropathic pain models such as CCI and SNI. There are now reliable animal models for diabetic neuropathy (524), multiple sclerosis (34), phantom limb pain (30), chemotherapy induced pain (53, 129, 525), spinal cord injury (31) and trigeminal neuralgia (526).
In the situation of inflammatory as opposed to neuropathic pain, it has recently been reported that nociceptor-neuroimmune interactomes reveal cell type- and injury-specific pathways in three different inflammatory models (527). The availability of a similar database in the neuropathic pain field would be of great advantage to developing specific treatments.
Another major step forward from the basic science perspective is the ongoing improvement in pain assessment in animal models. Regardless of the type of nerve injury used, preclinical effectiveness of therapeutic interventions has classically been assessed in rodent models by examining drugs’ ability to attenuate withdrawal responses to stimuli that would normally be innocuous (47, 60, 61). This typically involves measurements of mechanical or thermal withdrawal thresholds to quantify hyperalgesia or allodynia. Such responses are difficult to quantify as they may be influenced by the subjective impressions of the investigator as well as the olfactory signals they emit. For example, male investigators promote analgesia in female mice (528). In addition, withdrawal responses to innocuous stimuli in injured animal may simply reflect activation of spinal reflexes (529, 530) rather than bona fide manifestations of pain. This may help to explain why classical rodent models have limited ability to predict clinical efficacy (17, 47, 529, 531). In view of this, non-invasive models for objective assessment of chronic pain have been developed. These involve assessment of hypersonic vocalisation, facial grimace score, quantification of social interaction, rearing and nest-building (47, 532–537) and the use of operant models in which the animal is required to make a decision based on the cortical processing of a noxious stimulus (538–540).
The use of operant and non-invasive protocols to effect translation between preclinical observations and development of effective therapeutic approaches may be further refined by combining findings from as many as 6 operant protocols (534).
Advances in technology now permit the use of human nerves in the laboratory (475). Because this has identified the cellular basis for differences in nociceptive processing between humans and rodents (541), the use of such models may be a way forward for identification of more relevant therapeutic targets. Human nociceptors are more heterogeneous than those in rodents and there are also differences in ion channel function and expression leading to differences in cellular excitability (19, 542, 543). Most human DRG neurons exhibit TRPV1 receptor channels but these are expressed exclusively in peptidergic nociceptors in rodents (544). A subpopulation of human DRG neurons display a relatively large constitutive Ca2+ channel current and although HVA Ca2+ current density is significantly smaller in human than in rodent DRG, the proportion of nifedipine-sensitive (Cav1.2) currents is much greater (543). Although this identifies dihydropyridines as a potential therapeutic approach to some types of neuropathic pain, their further development is limited by their propensity to produce postural hypotension (545).
Contemporary methodologies that allow the collection of data from human nerves include observation of nociceptor morphology in skin biopsy samples (546) and use of explant cultures of DRG neurons from aborted fetuses (49). Human DRG's have also been acutely isolated from organ donors or cadavers or from patients undergoing surgical treatment for spinal reconstruction (475, 543, 547).
The use of human induced pluripotent stem cells (hiPSC) differentiated into nociceptive sensory neurons may provide a means to address the limited availability of human DRG neurons (548–554). The use of hiPSC has the advantage of providing large numbers of human neurons, glia and immunocompetent cells (555). This in turn allows the application of high throughput technologies to screen small molecule therapeutic agents to modify nociceptor function (556).
In recent years, considerable attention has been paid to analysis of molecular mechanisms of pain in male vs. female rodents (10, 362, 381, 475). As already emphasized, microglia are not required for mechanical sensitivity to pain in female mice as they require activation of adaptive immune cells such as T-lymphocytes (362, 381). The difference may result from a lack of P2X4 receptors in the microglia of females (364, 376). Despite this, behavioral responses to nerve injury in female rats are similar to those seen in males and both involve downregulation of KCC2 and perturbation of Cl- gradients (363). Because BDNF is not necessary for the development of allodynia in females (362), the mediator released from adaptive immune cells remains to be determined. The possible involvement of IFN-γ has already been alluded to.
Numerous differences in pain mechanisms in males vs. females have emerged over recent years (10). For example loss of GABAA receptors containing the α6 subunit plays a predominant role in female rodents (557). The relative importance of CGRP in females (323), the role of macrophage derived IL-23 (368), and the absence of functional P2X receptors on microglia of female rodents have already been alluded to (364).
This realization has obvious implications for the design of clinical trials (10); potential new therapies must be evaluated in women and men as separate subgroups of patients.
As mentioned in the introduction, patients with neuropathic pain are heterogeneous in clinical presentation, pathophysiology, aetilogy, causative injury, genetics and prior life experience (5). This is reflected in a large variability in their response to treatment (8, 25).
One way forward from the clinical perspective is the quantitative, phenotypical stratification of patent types in order to delineate responders from non-responders. This statistical subgrouping of patients can have a role in determining treatment (12, 25, 558, 559). Several tools are available for patient stratification.
Firstly, quantitative sensory testing (QST) enables identification of various subtypes of neuropathic pain by formalization and quantification of an existing battery of neurological tests, such as response to von Frey filaments, vibration, heat, pressure and cold as well as wind-up ratio and dynamic allodynia (5, 25, 559, 560). By comparing responses with large datasets that represent normal responses to sensory tests, neuropathic pain patients can be grouped into clusters based on their sensory profiles (5). The validity of this type of approach is supported by the observation that post-hoc analysis of responders to treatments in clinical trials suggest that clinical effectiveness may cluster according to pain phenotype (559).
Secondly, human microneurography techniques can now distinguish mechanosensitive C-fibres from non-mechanoceptive fibres in a given patient (542). It can also be used to detect spontaneous activity in nocceptors (561).
The multidimensional expereince of pain is a result of spatiotempotral patterns of brain activity wherein afferent nociceptive information is modulated by cognitive factors and from which descending modulatory systems control spinal level processing (25, 508). Analysis of electroencephalographic patterns (562) and non-invasive neuroimaging techniques such as paradigm free functional magnetic resonance imaging (fMRI) therefore offer the opportunity to examine the experience and chronification of pain in individual patients. Similarly, the use of positron emission tomography (PET) with radioligands targeting the 18 kDa translocator protein SPO has recently emerged as a technique for observing neuroinflammation and glial activation in patients in vivo (27).
Lastly, examination of expression of miRNAs in epidermis strongly discriminating pain patients from healthy individuals (563). In addition to providing a means to stratify patients prior to treatment, the above techniques also provide information concerning the effectiveness of therapeutic intervention.
The different mechanisms underlying neuropathic pain in females vs. males his has obvious therapeutic implications; drugs which are effective in men may be less effective in women and vice-versa (10, 361, 362, 365, 366, 564–569).
In support of this, recent work using resting-state magnetoencephalography has identified differential changes in patterns of brain oscillatory activity in males vs. females (570). Also a genome wide association study identified 123 single nucleotide polymorphisms (SNP) at 5 independent loci that are significantly associated with chronic pain in men. By contrast, 286 SNP's at 10 independent loci were identified in women (571). Gene-level analyses revealed sex-specific associations with chronic pain with 31 genes in females and 37 genes in males. All 37 chronic pain associated genes in men and 30/31 genes in women were expressed in DRG (571). In an extension of this, analysis of altered mRNA expression in the DRG of neuropathic pain patients also revealed profound sex differences in differentially expressed genes. Thus, message for IL-1β, TNF, CXCL-14 and OSM (Oncostatin M) were increased in males whereas CCL-1, CCL-21, PENK (proenkephalin A) and TLR3 (toll-like receptor 3) were increased in females. Cytokine signalling pathways associated with neuropathic pain in males included OSM, LIF, and SOCS1 (suppressor of cytokine signalling 1) whereas CCL-1, CCL-19 and CCL-21 were involved in females. Moreover,components of the JUN-FOS signalling pathway were enriched in males whereas genes coding centromere proteins were enriched in females (352).
Given the drawbacks of classical therapeutic approaches; drug toxicity, off target effects, drug interactions and in some cases drug dependence, there is a movement in all fields of medicine towards genetic rather than pharmacological approaches to disease management. Pain management is no exception to this trend (572).
By way of demonstration of the principle, CRISPR (clustered regularly interspaced short palindromic repeats) technology has been used in a mouse model to prevent expression of Nav1.7 by editing a regulatory sequence (573). This technology might have therapeutic potential in management of persistent pain states.in the clinic.
This review underlines the difficulty in translation between animal studies and the treatment of pain in the clinic. Although classical animal models have revealed many of the essential biological mechanisms that underlie neuropathic pain, such as peripheral and central sensitization and some of the molecular and cellular mechanisms involved, animal models do not adequately model the multiplicity of disease states or injuries that may bring about pain in humans. In terms of pharmacological management “one size does not fit all”, perhaps there will never be a panacea for neuropathic pain in the same way as opioids serve for most forms of nociceptive pain. Despite this, there is some cause for optimising treatment for individual patients by careful evaluation of the pain phenotype and delivering treatment accordingly.
PS was responsible for all aspects of writing the manuscript.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Mogil JS. Sources of individual differences in pain. Ann Rev Neurosci. (2021) 44(1):1–25. doi: 10.1146/annurev-neuro-092820-105941
2. Moriarty O, Tu Y, Sengar AS, Salter MW, Beggs S, Walker SM. Priming of adult incision response by early-life injury: neonatal microglial inhibition has persistent but sexually dimorphic effects in adult rats. J Neurosci. (2019) 39(16):3081–93. doi: 10.1523/JNEUROSCI.1786-18.2019
3. Dworsky-Fried Z, Kerr BJ, Taylor AMW. Microbes, microglia, and pain. Neurobiol Pain. (2020) 7:100045. doi: 10.1016/j.ynpai.2020.100045
4. Fitzgerald M, McKelvey R. Nerve injury and neuropathic pain—A question of age. Exp Neurol. (2016) 275(Pt 2):296–302. doi: 10.1016/j.expneurol.2015.07.013
5. Baron R, Maier C, Attal N, Binder A, Bouhassira D, Cruccu G, et al. Peripheral neuropathic pain: a mechanism-related organizing principle based on sensory profiles. Pain. (2017) 158(2):261–72. doi: 10.1097/j.pain.0000000000000753
6. Hastie BA, Riley JL III, Kaplan L, Herrera DG, Campbell CM, Virtusio K, et al. Ethnicity interacts with the OPRM1 gene in experimental pain sensitivity. Pain. (2012) 153(8):1610–9. doi: 10.1016/j.pain.2012.03.022
7. Ding W, You Z, Chen Q, Yang L, Doheny J, Zhou X, et al. Gut Microbiota influences neuropathic pain through modulating proinflammatory and anti-inflammatory T cells. Anesth Analg. (2021) 132(4):1146–55. doi: 10.1213/ANE.0000000000005155
8. Edwards RR, Schreiber KL, Dworkin RH, Turk DC, Baron R, Freeman R, et al. Optimizing and accelerating the development of precision pain treatments for chronic pain: IMMPACT review and recommendations. J Pain. (2023) 24(2):204–25. doi: 10.1016/j.jpain.2022.08.010
9. Lee J, Lee G, Ko G, Joong LS. Nerve injury-induced gut dysbiosis contributes to spinal cord TNF-alpha expression and nociceptive sensitization. Brain Behav Immun. (2023) 110:155–61. doi: 10.1016/j.bbi.2023.03.005
10. Ghazisaeidi S, Muley MM, Salter MW. Neuropathic pain: mechanisms, sex differences, and potential therapies for a global problem. Annu Rev Pharmacol Toxicol. (2023) 63:565–83. doi: 10.1146/annurev-pharmtox-051421-112259
11. Fiore NT, Debs SR, Hayes JP, Duffy SS, Moalem-Taylor G. Pain-resolving immune mechanisms in neuropathic pain. Nat Rev Neurol. (2023) 19(4):199–220. doi: 10.1038/s41582-023-00777-3
12. Bannister K, Sachau J, Baron R, Dickenson AH. Neuropathic pain: mechanism-based therapeutics. Annu Rev Pharmacol Toxicol (2020) 60:257–74. doi: 10.1146/annurev-pharmtox-010818-021524
13. Wall PD, Devor M, Inbal R, Scadding JW, Schonfeld D, Seltzer Z, et al. Autotomy following peripheral nerve lesions: experimental anaesthesia dolorosa. Pain. (1979) 7:103–13. doi: 10.1016/0304-3959(79)90002-2
14. Finnerup NB, Kuner R, Jensen TS. Neuropathic pain: from mechanisms to treatment. Physiol Rev. (2021) 101(1):259–301. doi: 10.1152/physrev.00045.2019
15. Colloca L, Ludman T, Bouhassira D, Baron R, Dickenson AH, Yarnitsky D, et al. Neuropathic pain. Nat Rev Dis Primers. (2017) 3:17002. doi: 10.1038/nrdp.2017.2
16. Finnerup NB, Attal N, Haroutounian S, McNicol E, Baron R, Dworkin RH, et al. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol. (2015) 14:162–73. doi: 10.1016/S1474-4422(14)70251-0
17. Yekkirala AS, Roberson DP, Bean BP, Woolf CJ. Breaking barriers to novel analgesic drug development. Nat Rev Drug Discov. (2017) 16:545–64. doi: 10.1038/nrd.2017.87
18. Scholz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci. (2007) 10(11):1361–8. doi: 10.1038/nn1992
19. Walters ET, Crook RJ, Neely GG, Price TJ, Smith ESJ. Persistent nociceptor hyperactivity as a painful evolutionary adaptation. Trends Neurosci. (2023) 46(3):211–27. doi: 10.1016/j.tins.2022.12.007
20. Costigan M, Scholz J, Woolf CJ. Neuropathic pain: a maladaptive response of the nervous system to damage. Annu Rev Neurosci. (2009) 32:1–32. doi: 10.1146/annurev.neuro.051508.135531
21. Iadarola MJ, Caudle RM. Good pain, bad pain. Science. (1997) 278:239–40. doi: 10.1126/science.278.5336.239
22. Alles SRA, Smith PA. The etiology and pharmacology of neuropathic pain. Pharmacol Rev. (2018) 70:315–47. doi: 10.1124/pr.117.014399
23. Gold MS, Gebhart GF. Nociceptor sensitization in pain pathogenesis. Nat Med. (2010) 16(11):1248–57. doi: 10.1038/nm.2235
24. Schwartz SM, Barpujari A, Finnerup NB, Raja SN. Pharmacologic therapies for neuropathic pain: an assessment of reporting biases in randomized controlled trials. Pain. (2022) 163(4):795–804. doi: 10.1097/j.pain.0000000000002426
25. Soliman N, Kersebaum D, Lawn T, Sachau J, Sendel M, Vollert J. Improving neuropathic pain treatment - by rigorous stratification from bench to bedside. J Neurochem. (2023). doi: 10.1111/jnc.15798. [Epub ahead of print] 36852505
26. Gormsen L, Rosenberg R, Bach FW, Jensen TS. Depression, anxiety, health-related quality of life and pain in patients with chronic fibromyalgia and neuropathic pain. Eur J Pain. (2010) 14(2):127–8. doi: 10.1016/j.ejpain.2009.03.010
27. Grace PM, Tawfik VL, Svensson CI, Burton MD, Loggia ML, Hutchinson MR. The neuroimmunology of chronic pain: from rodents to humans. J Neurosci. (2021) 41(5):855. doi: 10.1523/JNEUROSCI.1650-20.2020
28. Waxman SG. Peripheral afferents and the pain experience. Pain. (2019) 160(7):1487–8. doi: 10.1097/j.pain.0000000000001527
29. Gangadharan V, Zheng H, Taberner FJ, Landry J, Nees TA, Pistolic J, et al. Neuropathic pain caused by miswiring and abnormal end organ targeting. Nature. (2022) 606(7912):137–45. doi: 10.1038/s41586-022-04777-z
30. Vaso A, Adahan HM, Gjika A, Zahaj S, Zhurda T, Vyshka G, et al. Peripheral nervous system origin of phantom limb pain. Pain. (2014) 155(7):1384–91. doi: 10.1016/j.pain.2014.04.018
31. Carlton SM, Du J, Tan HY, Nesic O, Hargett GL, Bopp AC, et al. Peripheral and central sensitization in remote spinal cord regions contribute to central neuropathic pain after spinal cord injury. Pain. (2009) 147(1–3):265–76. doi: 10.1016/j.pain.2009.09.030
32. Wu J, Renn CL, Faden AI, Dorsey SG. TrkB.T1 contributes to neuropathic pain after spinal cord injury through regulation of cell cycle pathways. J Neurosci. (2013) 33(30):12447–63. doi: 10.1523/JNEUROSCI.0846-13.2013
33. Yousuf MS, Noh MC, Friedman TN, Zubkow K, Johnson JC, Tenorio G, et al. Sensory neurons of the dorsal root ganglia become hyperexcitable in a T-cell-mediated MOG-EAE model of multiple sclerosis. eNeuro. (2019) 6(2):ENEURO.0024–19.2019. doi: 10.1523/ENEURO.0024-19.2019
34. Olechowski CJ, Truong JJ, Kerr BJ. Neuropathic pain behaviours in a chronic-relapsing model of experimental autoimmune encephalomyelitis (EAE). Pain. (2009) 141(1–2):156–64. doi: 10.1016/j.pain.2008.11.002
35. Haroutounian S, Ford AL, Frey K, Nikolajsen L, Finnerup NB, Neiner A, et al. How central is central poststroke pain? The role of afferent input in poststroke neuropathic pain: a prospective, open-label pilot study. Pain. (2018) 159(7):1317–24. doi: 10.1097/j.pain.0000000000001213
36. Staud R. Fibromyalgia pain: do we know the source? Curr Opin Rheumatol. (2004) 16(2):157–63. doi: 10.1097/00002281-200403000-00016
37. Sumpton JE, Moulin DE. Fibromyalgia. Handb Clin Neurol. (2014) 119:513–27. doi: 10.1016/B978-0-7020-4086-3.00033-3
38. Kress L, Egenolf N, Sommer C, Uceyler N. Cytokine expression profiles in white blood cells of patients with small fiber neuropathy. BMC Neurosci. (2023) 24(1):1. doi: 10.1186/s12868-022-00770-4
39. Zak-Prelich M, McKenzie RC, Sysa-Jedrzejowska A, Norval M. Local immune responses and systemic cytokine responses in zoster: relationship to the development of postherpetic neuralgia. Clin Exp Immunol. (2003) 131(2):318–23. doi: 10.1046/j.1365-2249.2003.02061.x
40. Cregg R, Momin A, Rugiero F, Wood JN, Zhao J. Pain channelopathies. J Physiol. (2010) 588(Pt 11):1897–904. doi: 10.1113/jphysiol.2010.187807
41. Lu VB, Smith PA, Rashiq S. The excitability of dorsal horn neurons is affected by cerebrospinal fluid from humans with osteoarthritis. Can J Physiol Pharmacol. (2012) 90(6):783–90. doi: 10.1139/y2012-014
42. Rahman W, Patel R, Dickenson AH. Electrophysiological evidence for voltage-gated calcium channel 2 (Cav2) modulation of mechano- and thermosensitive spinal neuronal responses in a rat model of osteoarthritis. Neuroscience. (2015) 305:76–85. doi: 10.1016/j.neuroscience.2015.07.073
43. Jensen MP, Chodroff MJ, Dworkin RH. The impact of neuropathic pain on health-related quality of life: review and implications. Neurology. (2007) 68(15):1178–82. doi: 10.1212/01.wnl.0000259085.61898.9e
44. McLachlan EM, Janig W, Michalis M. Peripheral nerve injury triggers noradrenergic sprouting within dorsal root ganglia. Nature. (1993) 363:543–6. doi: 10.1038/363543a0
45. Rifbjerg-Madsen S, Christensen AW, Christensen R, Hetland ML, Bliddal H, Kristensen LE, et al. Pain and pain mechanisms in patients with inflammatory arthritis: a danish nationwide cross-sectional DANBIO registry survey. PLoS One. (2017) 12(7):e0180014. doi: 10.1371/journal.pone.0180014
46. Zochodne DW. Neurotrophins and other growth factors in diabetic neuropathy. Semin Neurol. (1996) 16(2):153–61. doi: 10.1055/s-2008-1040971
47. Bouali-Benazzouz R, Landry M, Benazzouz A, Fossat P. Neuropathic pain modeling: focus on synaptic and ion channel mechanisms. Prog Neurobiol. (2021) 201:102030. doi: 10.1016/j.pneurobio.2021.102030
48. Mifflin KA, Kerr BJ. Pain in autoimmune disorders. J Neurosci Res. (2017) 95(6):1282–94. doi: 10.1002/jnr.23844
49. Acharjee S, Noorbakhsh F, Stemkowski PL, Olechowski C, Cohen EA, Ballanyi K, et al. HIV-1 viral protein R causes peripheral nervous system injury associated with in vivo neuropathic pain. FASEB J. (2010) 24(11):4343–53. doi: 10.1096/fj.10-162313
50. Shi Y, Gelman BB, Lisinicchia JG, Tang SJ. Chronic-pain-associated astrocytic reaction in the spinal cord dorsal horn of human immunodeficiency virus-infected patients. J Neurosci. (2012) 32(32):10833–40. doi: 10.1523/JNEUROSCI.5628-11.2012
51. Ru W, Liu X, Bae C, Shi Y, Walikonis R, Mo CJ, et al. Microglia mediate HIV-1 gp120-induced synaptic degeneration in spinal pain neural circuits. J Neurosci. (2019) 39(42):8408–21. doi: 10.1523/JNEUROSCI.2851-18.2019
52. Attal N, Martinez V, Bouhassira D. Potential for increased prevalence of neuropathic pain after the COVID-19 pandemic. Pain Rep. (2021) 6(1):e884. doi: 10.1097/PR9.0000000000000884
53. Descoeur J, Pereira V, Pizzoccaro A, Francois A, Ling B, Maffre V, et al. Oxaliplatin-induced cold hypersensitivity is due to remodelling of ion channel expression in nociceptors. EMBO Mol Med. (2011) 3(5):266–78. doi: 10.1002/emmm.201100134
54. Xiao W, Boroujerdi A, Bennett GJ, Luo ZD. Chemotherapy-evoked painful peripheral neuropathy: analgesic effects of gabapentin and effects on expression of the alpha-2-delta type-1 calcium channel subunit. Neuroscience. (2007) 144(2):714–20. doi: 10.1016/j.neuroscience.2006.09.044
55. Flatters SJL, Bennett GJ. Studies of peripheral sensory nerves in paclitaxel-induced painful peripheral neuropathy: evidence for mitochondrial dysfunction. Pain. (2006) 122(3):245–57. doi: 10.1016/j.pain.2006.01.037
56. Gibson CA. Review of posttraumatic stress disorder and chronic pain: the path to integrated care. J Rehabil Res Dev. (2012) 49(5):753–76. doi: 10.1682/JRRD.2011.09.0158
57. Cummins TR, Dib-Hajj SD, Waxman SG. Electrophysiological properties of mutant Nav1.7 sodium channels in a painful inherited neuropathy. J Neurosci. (2004) 24(38):8232–6. doi: 10.1523/JNEUROSCI.2695-04.2004
58. Dib-Hajj SD, Rush AM, Cummins TR, Hisama FM, Novella S, Tyrrell L, et al. Gain-of-function mutation in Nav1.7 in familial erythromelalgia induces bursting of sensory neurons. Brain. (2005) 128(Pt 8):1847–54. doi: 10.1093/brain/awh514
59. Dib-Hajj SD, Waxman SG. Sodium channels in human pain disorders: genetics and pharmacogenomics. Annu Rev Neurosci. (2019) 42:87–106. doi: 10.1146/annurev-neuro-070918-050144
60. Stemkowski PL, Smith PA. An overview of animal models of neuropathic pain. In: Toth C, Moulin DE, editors. Neuropathic pain, causes, management and understanding. Cambridge: Cambridge University Press (2013). p. 33–50.
61. Decosterd I, Woolf CJ. Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain. (2000) 87(2):149–58. doi: 10.1016/S0304-3959(00)00276-1
62. Kim KJ, Yoon YW, Chung JM. Comparison of three rodent models of neuropathic pain. Exp Brain Res. (1997) 113:200–6. doi: 10.1007/BF02450318
63. Natale CA, Christie MJ, Aubrey KR. Spinal glycinergic currents are reduced in a rat model of neuropathic pain following partial nerve ligation but not chronic constriction injury. J Neurophysiol. (2023) 129(2):333–41. doi: 10.1152/jn.00451.2022
64. Price TJ, Basbaum AI, Bresnahan J, Chambers JF, De KY, Edwards RR, et al. Transition to chronic pain: opportunities for novel therapeutics. Nat Rev Neurosci. (2018) 19(7):383–4. doi: 10.1038/s41583-018-0012-5
65. Rice ASC, Finnerup NB, Kemp HI, Currie GL, Baron R. Sensory profiling in animal models of neuropathic pain: a call for back-translation. Pain. (2018) 159(5):819–24. doi: 10.1097/j.pain.0000000000001138
66. Schulte A, Lohner H, Degenbeck J, Segebarth D, Rittner HL, Blum R, et al. Unbiased analysis of the dorsal root anglion after peripheral nerve injury: no neuronal loss, no gliosis, but satellite glial cell plasticity. Pain. (2023) 164(4):728–40. doi: 10.1097/j.pain.0000000000002758
67. Radtke C, Vogt PM, Devor M, Kocsis JD. Keratinocytes acting on injured afferents induce extreme neuronal hyperexcitability and chronic pain. Pain. (2010) 148(1):94–102. doi: 10.1016/j.pain.2009.10.014
68. Boakye PA, Tang SJ, Smith PA. Mediators of neuropathic pain; focus on spinal microglia, CSF-1, BDNF, CCL21, TNF-alpha, Wnt Ligands, and Interleukin 1-beta. Front Pain Res. (2021) 2:698157. doi: 10.3389/fpain.2021.698157
69. McMahon SB, Cafferty WB, Marchand F. Immune and glial cell factors as pain mediators and modulators. Exp Neurol. (2005) 192(2):444–62. doi: 10.1016/j.expneurol.2004.11.001
70. Moalem G, Tracey DJ. Immune and inflammatory mechanisms in neuropathic pain. Brain Res Rev. (2006) 51(2):240–64. doi: 10.1016/j.brainresrev.2005.11.004
71. Yu X, Liu H, Hamel KA, Morvan MG, Yu S, Leff J, et al. Dorsal root ganglion macrophages contribute to both the initiation and persistence of neuropathic pain. Nat Commun. (2020) 11(1):264. doi: 10.1038/s41467-019-13839-2
72. Noh MC, Mikler B, Joy T, Smith PA. Time course of inflammation in dorsal root ganglia correlates with differential reversibility of mechanical allodynia. Neuroscience. (2020) 428:199–216. doi: 10.1016/j.neuroscience.2019.12.040
73. Sommer C, Petrausch S, Lindenlaub T, Toyka KV. Neutralizing antibodies to interleukin 1-receptor reduce pain associated behavior in mice with experimental neuropathy. Neurosci Lett. (1999) 270(1):25–8. doi: 10.1016/S0304-3940(99)00450-4
74. Zelenka M, Schafers M, Sommer C. Intraneural injection of interleukin-1beta and tumor necrosis factor-alpha into rat sciatic nerve at physiological doses induces signs of neuropathic pain. Pain. (2005) 116(3):257–63. doi: 10.1016/j.pain.2005.04.018
75. Wolf G, Gabay E, Tal M, Yirmiya R, Shavit Y. Genetic impairment of interleukin-1 signaling attenuates neuropathic pain, autotomy, and spontaneous ectopic neuronal activity, following nerve injury in mice. Pain. (2006) 120(3):315–24. doi: 10.1016/j.pain.2005.11.011
76. Gabay E, Wolf G, Shavit Y, Yirmiya R, Tal M. Chronic blockade of interleukin-1 (IL-1) prevents and attenuates neuropathic pain behavior and spontaneous ectopic neuronal activity following nerve injury. Eur J Pain. (2011) 15(3):242–8. doi: 10.1016/j.ejpain.2010.07.012
77. Kleinschnitz C, Hofstetter HH, Meuth SG, Braeuninger S, Sommer C, Stoll G. T cell infiltration after chronic constriction injury of mouse sciatic nerve is associated with interleukin-17 expression. Exp Neurol. (2006) 200(2):480–5. doi: 10.1016/j.expneurol.2006.03.014
78. Gomez-Nicola D, Valle-Argos B, Suardiaz M, Taylor JS, Nieto-Sampedro M. Role of IL-15 in spinal cord and sciatic nerve after chronic constriction injury: regulation of macrophage and T-cell infiltration. J Neurochem. (2008) 107(6):1741–52. doi: 10.1111/j.1471-4159.2008.05746.x
79. Kim CF, Moalem-Taylor G. Interleukin-17 contributes to neuroinflammation and neuropathic pain following peripheral nerve injury in mice. J Pain. (2011) 12(3):370–83. doi: 10.1016/j.jpain.2010.08.003
80. Vasudeva K, Vodovotz Y, Azhar N, Barclay D, Janjic JM, Pollock JA. In vivo and systems biology studies implicate IL-18 as a central mediator in chronic pain. J Neuroimmunol. (2015) 283:43–9. doi: 10.1016/j.jneuroim.2015.04.012
81. Shamash S, Reichert F, Rotshenker S. The cytokine network of wallerian degeneration: tumor necrosis factor-alpha, interleukin-1alpha, and interleukin-1beta. J Neurosci. (2002) 22(8):3052–60. doi: 10.1523/JNEUROSCI.22-08-03052.2002
82. Khan J, Hassun H, Zusman T, Korczeniewska O, Eliav E. Interleukin-8 levels in rat models of nerve damage and neuropathic pain. Neurosci Lett. (2017) 657:106–12. doi: 10.1016/j.neulet.2017.07.049
83. Dubovy P, Klusakova I, Svizenska I, Brazda V. Satellite glial cells express IL-6 and corresponding signal-transducing receptors in the dorsal root ganglia of rat neuropathic pain model. Neuron Glia Biol. (2010) 6(1):73–83. doi: 10.1017/S1740925X10000074
84. Day YJ, Liou JT, Lee CM, Lin YC, Mao CC, Chou AH, et al. Lack of interleukin-17 leads to a modulated micro-environment and amelioration of mechanical hypersensitivity after peripheral nerve injury in mice. Pain. (2014) 155(7):1293–302. doi: 10.1016/j.pain.2014.04.004
85. Leung L, Cahill CM. TNF-alpha and neuropathic pain–a review. J Neuroinflammation. (2010) 7:27. doi: 10.1186/1742-2094-7-27
86. Bastien D, Lacroix S. Cytokine pathways regulating glial and leukocyte function after spinal cord and peripheral nerve injury. Exp Neurol. (2014) 258:62–77. doi: 10.1016/j.expneurol.2014.04.006
87. Thompson SW, Dray A, Urban L. Leukemia inhibitory factor induces mechanical allodynia but not thermal hyperalgesia in the juvenile rat. Neuroscience. (1996) 71(4):1091–4. doi: 10.1016/0306-4522(95)00537-4
88. Mwirigi J, Franco-Enzastiga UM, Sankaranarayanan I, Tavares-Ferreira D, Shiers SI, Ray P, et al. Oncostatin M induces nociceptive signaling in human dorsal root ganglia. J Pain. (2023) 24(4, Supplement):16. doi: 10.1016/j.jpain.2023.02.061
89. Pezet S, McMahon SB. Neurotrophins: mediators and modulators of pain. Ann Rev Neurosci. (2006) 29(1):507–38. doi: 10.1146/annurev.neuro.29.051605.112929
90. Tanaka T, Okuda H, Isonishi A, Terada Y, Kitabatake M, Shinjo T, et al. Dermal macrophages set pain sensitivity by modulating the amount of tissue NGF through an SNX25-Nrf2 pathway. Nat Immunol. (2023) 24(3):439–51. doi: 10.1038/s41590-022-01418-5
91. Kaur G, Singh N, Jaggi AS. Mast cells in neuropathic pain: an increasing spectrum of their involvement in pathophysiology. Rev Neurosci. (2017) 28(7):759–66. doi: 10.1515/revneuro-2017-0007
92. Obara I, Telezhkin V, Alrashdi I, Chazot PL. Histamine, histamine receptors, and neuropathic pain relief. Br J Pharmacol. (2020) 177(3):580–99. doi: 10.1111/bph.14696
93. Khalilzadeh E, Azarpey F, Hazrati R, Vafaei SG. Evaluation of different classes of histamine H1 and H2 receptor antagonist effects on neuropathic nociceptive behavior following tibial nerve transection in rats. Eur J Pharmacol. (2018) 834:221–9. doi: 10.1016/j.ejphar.2018.07.011
94. Wei T, Guo TZ, Li WW, Hou S, Kingery WS, Clark JD. Keratinocyte expression of inflammatory mediators plays a crucial role in substance P-induced acute and chronic pain. J Neuroinflammation. (2012) 9:181. doi: 10.1038/s41590-022-01418-5
95. Simonetti M, Agarwal N, Stosser S, Bali KK, Karaulanov E, Kamble R, et al. Wnt-Fzd signaling sensitizes peripheral sensory neurons via distinct noncanonical pathways. Neuron. (2014) 83(1):104–21. doi: 10.1016/j.neuron.2014.05.037
96. van Vliet AC, Lee J, van der Poel M, Mason MRJ, Noordermeer JN, Fradkin LG, et al. Coordinated changes in the expression of wnt pathway genes following human and rat peripheral nerve injury. PLoS One. (2021) 16(4):e0249748. doi: 10.1371/journal.pone.0249748
97. White FA, Wilson NM. Chemokines as pain mediators and modulators. Curr Opin Anaesthesiol. (2008) 21(5):580–5. doi: 10.1097/ACO.0b013e32830eb69d
98. Pawlik K, Piotrowska A, Kwiatkowski K, Ciapala K, Popiolek-Barczyk K, Makuch W, et al. The blockade of CC chemokine receptor type 1 influences the level of nociceptive factors and enhances opioid analgesic potency in a rat model of neuropathic pain. Immunology. (2020) 159(4):413–28. doi: 10.1111/imm.13172
99. Xian H, Jiang Y, Zhang H, Ma SB, Zhao R, Cong R. CCL2-CCR2 Axis potentiates NMDA receptor signaling to aggravate neuropathic pain induced by brachial Plexus avulsion. Neuroscience. (2020) 425:29–38. doi: 10.1016/j.neuroscience.2019.11.012
100. Moalem G, Grafe P, Tracey DJ. Chemical mediators enhance the excitability of unmyelinated sensory axons in normal and injured peripheral nerve of the rat. Neuroscience. (2005) 134(4):1399–411. doi: 10.1016/j.neuroscience.2005.05.046
101. Jayaraj ND, Bhattacharyya BJ, Belmadani AA, Ren D, Rathwell CA, Hackelberg S, et al. Reducing CXCR4-ediated nociceptor hyperexcitability reverses painful diabetic neuropathy. J Clin Invest. (2018) 128(6):2205–25. doi: 10.1172/JCI92117
102. Dubovy P, Klusakova I, Svizenska I, Brazda V. Spatio-temporal changes of SDF1 and its CXCR4 receptor in the dorsal root ganglia following unilateral sciatic nerve injury as a model of neuropathic pain. Histochem Cell Biol. (2010) 133(3):323–37. doi: 10.1007/s00418-010-0675-0
103. Bai L, Wang X, Li Z, Kong C, Zhao Y, Qian JL, et al. Upregulation of chemokine CXCL12 in the dorsal root ganglia and spinal cord contributes to the development and maintenance of neuropathic pain following spared nerve injury in rats. Neurosci Bull. (2016) 32(1):27–40. doi: 10.1007/s12264-015-0007-4
104. Yu Y, Huang X, Di Y, Qu L, Fan N. Effect of CXCL12/CXCR4 signaling on neuropathic pain after chronic compression of dorsal root ganglion. Sci Rep. (2017) 7(1):5707. doi: 10.1038/s41598-017-05954-1
105. Kawano T, Zoga V, Kimura M, Liang MY, Wu HE, Gemes G, et al. Nitric oxide activates ATP-sensitive potassium channels in mammalian sensory neurons: action by direct S-nitrosylation. Mol Pain. (2009) 5:12. doi: 10.1186/1744-8069-5-12
106. Renganathan M, Cummins TR, Waxman SG. Nitric oxide blocks fast, slow, and persistent na+ channels in C-type DRG neurons by S-nitrosylation. J Neurophysiol. (2002) 87(2):761–75. doi: 10.1152/jn.00369.2001
107. Renganathan M, Cummins TR, Hormuzdiar WN, Black JA, Waxman SG. Nitric oxide is an autocrine regulator of na(+) currents in axotomized C-type DRG neurons. J Neurophysiol. (2000) 83(4):2431–42. doi: 10.1152/jn.2000.83.4.2431
108. Grace PM, Gaudet AD, Staikopoulos V, Maier SF, Hutchinson MR, Salvemini D, et al. Nitroxidative signaling mechanisms in pathological pain. Trends Neurosci. (2016) 39(12):862–79. doi: 10.1016/j.tins.2016.10.003
109. George DS, Hackelberg S, Jayaraj ND, Ren D, Edassery SL, Rathwell CA, et al. Mitochondrial calcium uniporter deletion prevents painful diabetic neuropathy by restoring mitochondrial morphology and dynamics. Pain. (2022) 163(3):560–78. doi: 10.1097/j.pain.0000000000002391
110. Guan Z, Kuhn JA, Wang X, Colquitt B, Solorzano C, Vaman S, et al. Injured sensory neuron-derived CSF1 induces microglial proliferation and DAP12-dependent pain. Nat Neurosci. (2016) 19(1):94–101. doi: 10.1038/nn.4189
111. Chitu V, Gokhan S, Nandi S, Mehler MF, Stanley ER. Emerging roles for CSF-1 receptor and its ligands in the nervous system. Trends Neurosci. (2016) 39(6):378–93. doi: 10.1016/j.tins.2016.03.005
112. Okubo M, Yamanaka H, Kobayashi K, Dai Y, Kanda H, Yagi H, et al. Macrophage-Colony stimulating factor derived from injured primary afferent induces proliferation of spinal microglia and neuropathic pain in rats. PLoS One. (2016) 11(4):e0153375. doi: 10.1371/journal.pone.0153375
113. Boakye PA, Rancic V, Whitlock KH, Simmons D, Longo FM, Ballanyi K, et al. Receptor dependence of BDNF actions in superficial dorsal horn: relation to central sensitization and actions of macrophage colony stimulating factor 1. J Neurophysiol. (2019) 121(6):2308–22. doi: 10.1152/jn.00839.2018
114. Piotrowska A, Rojewska E, Pawlik K, Kreiner G, Ciechanowska A, Makuch W, et al. Pharmacological blockade of CXCR3 by (+/-)-NBI-74330 reduces neuropathic pain and enhances opioid effectiveness - evidence from in vivo and in vitro studies. Biochim Biophys Acta Mol Basis Dis. (2018) 1864(10):3418–37. doi: 10.1016/j.bbadis.2018.07.032
115. Biber K, Tsuda M, Tozaki-Saitoh H, Tsukamoto K, Toyomitsu E, Masuda T, et al. Neuronal CCL21 up-regulates microglia P2X4 expression and initiates neuropathic pain development. EMBO J. (2011) 30(9):1864–73. doi: 10.1038/emboj.2011.89
116. Schmitz K, Pickert G, Wijnvoord N, Haussler A, Tegeder I. Dichotomy of CCL21 and CXCR3 in nerve injury-evoked and autoimmunity-evoked hyperalgesia. Brain Behav Immun. (2013) 32:186–200. doi: 10.1016/j.bbi.2013.04.011
117. Biggs JE, Lu VB, Stebbing MJ, Balasubramanyan S, Smith PA. Is BDNF sufficient for information transfer between microglia and dorsal horn neurons during the onset of central sensitization? Mol Pain. (2010) 6:44. doi: 10.1186/1744-8069-6-44
118. Trang T, Beggs S, Wan X, Salter MW. P2X4-receptor-mediated synthesis and release of brain-derived neurotrophic factor in microglia is dependent on calcium and p38-mitogen-activated protein kinase activation. J Neurosci. (2009) 29(11):3518–28. doi: 10.1523/JNEUROSCI.5714-08.2009
119. Trang T, Beggs S, Salter MW. Brain-derived neurotrophic factor from microglia: a molecular substrate for neuropathic pain. Neuron Glia Biol. (2011) 7(1):99–108. doi: 10.1017/S1740925X12000087
120. Ulmann L, Hatcher JP, Hughes JP, Chaumont S, Green PJ, Conquet F, et al. Up-regulation of P2X4 receptors in spinal microglia after peripheral nerve injury mediates BDNF release and neuropathic pain. J Neurosci. (2008) 28(44):11263–8. doi: 10.1523/JNEUROSCI.2308-08.2008
121. Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, et al. BDNF From microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. (2005) 438(7070):1017-21. doi: 10.1038/nature04223
122. Lu VB, Biggs JE, Stebbing MJ, Balasubramanyan S, Todd KG, Lai AY, et al. BDNF Drives the changes in excitatory synaptic transmission in the rat superficial dorsal horn that follow sciatic nerve injury. J Physiol (Lond). (2009) 587:1013–32. doi: 10.1113/jphysiol.2008.166306
123. Smith PA. BDNF: no gain without pain? Neuroscience. (2014) 283:107–23. doi: 10.1016/j.neuroscience.2014.05.044
124. Simonetti M, Kuner R. Spinal Wnt5a plays a key role in spinal dendritic spine remodeling in neuropathic and inflammatory pain models and in the proalgesic effects of peripheral Wnt3a. J Neurosci. (2020) 40(35):6664. doi: 10.1523/JNEUROSCI.2942-19.2020
125. Silva R, Malcangio M. Fractalkine/CX3CR1 pathway in neuropathic pain: an update. Front Pain Res. (2021) 2:35. doi: 10.3389/fpain.2021.684684
126. Clark AK, Malcangio M. Microglial signalling mechanisms: cathepsin S and fractalkine. Exp Neurol. (2012) 234(2):283–92. doi: 10.1016/j.expneurol.2011.09.012
127. Clark AK, Malcangio M. Fractalkine/CX3CR1 signaling during neuropathic pain. Front Cell Neurosci (2014)8:121. doi: 10.3389/fncel.2014.00121
128. Jung H, Bhangoo S, Banisadr G, Freitag C, Ren D, White FA, et al. Visualization of chemokine receptor activation in transgenic mice reveals peripheral activation of CCR2 receptors in states of neuropathic pain. J Neurosci. (2009) 29(25):8051–62. doi: 10.1523/JNEUROSCI.0485-09.2009
129. Al-Mazidi S, Alotaibi M, Nedjadi T, Chaudhary A, Alzoghaibi M, Djouhri L. Blocking of cytokines signalling attenuates evoked and spontaneous neuropathic pain behaviours in the paclitaxel rat model of chemotherapy-induced neuropathy. Eur J Pain. (2018) 22(4):810–21. doi: 10.1002/ejp.1169
130. Luo X, Tai WL, Sun L, Qiu Q, Xia Z, Chung SK, et al. Central administration of C-X-C chemokine receptor type 4 antagonist alleviates the development and maintenance of peripheral neuropathic pain in mice. PLoS One. (2014) 9(8):e104860. doi: 10.1371/journal.pone.0104860
131. Barragan-Iglesias P, Franco-Enzastiga U, Jeevakumar V, Shiers S, Wangzhou A, Granados-Soto V, et al. Type I interferons act directly on nociceptors to produce pain sensitization: implications for viral infection-induced pain. J Neurosci. (2020) 40(18):3517–32. doi: 10.1523/JNEUROSCI.3055-19.2020
132. Reischer G, Heinke B, Sandkuhler J. Interferon gamma facilitates the synaptic transmission between primary afferent C-fibres and lamina I neurons in the rat spinal dorsal horn via microglia activation. Mol Pain. (2020) 16:1744806920917249. doi: 10.1177/1744806920917249
133. Luo H, Liu HZ, Zhang WW, Matsuda M, Lv N, Chen G, et al. Interleukin-17 regulates neuron-glial communications, synaptic transmission, and neuropathic pain after chemotherapy. Cell Rep. (2019) 29(8):2384–97. doi: 10.1016/j.celrep.2019.10.085
134. Sun JL, Dai WJ, Shen XY, Lu N, Zhang YQ. Interleukin-17 is involved in neuropathic pain and spinal synapse plasticity on mice. J Neuroimmunol. (2023) 377:578068. doi: 10.1016/j.jneuroim.2023.578068
135. Binshtok AM, Wang H, Zimmermann K, Amaya F, Vardeh D, Shi L, et al. Nociceptors are interleukin-1{beta} sensors. J Neurosci. (2008) 28(52):14062–73. doi: 10.1523/JNEUROSCI.3795-08.2008
136. Stemkowski PL, Smith PA. Long-term IL-1beta exposure causes subpopulation-dependent alterations in rat dorsal root ganglion neuron excitability. J Neurophysiol. (2012) 107(6):1586–97. doi: 10.1152/jn.00587.2011
137. Stemkowski PL, Noh MC, Chen Y, Smith PA. Increased excitability of medium-sized dorsal root ganglion neurons by prolonged interleukin-1beta exposure is K(+) channel dependent and reversible. J Physiol. (2015) 593(16):3739–55. doi: 10.1113/JP270905
138. Noh MC, Stemkowski PL, Smith PA. Long-term actions of interleukin-1beta on K(+), Na(+) and Ca(2+) channel currents in small, IB4-positive dorsal root ganglion neurons; possible relevance to the etiology of neuropathic pain. J Neuroimmunol. (2019) 332:198–211. doi: 10.1016/j.jneuroim.2019.05.002
139. Nadeau S, Filali M, Zhang J, Kerr BJ, Rivest S, Soulet D, et al. Functional recovery after peripheral nerve injury is dependent on the pro-inflammatory cytokines IL-1beta and TNF: implications for neuropathic pain. J Neurosci. (2011) 31(35):12533–42. doi: 10.1523/JNEUROSCI.2840-11.2011
140. Stemkowski PL, Garcia-Caballero A, Gadotti VM, MΓÇÖDahoma S, Chen L, Souza IA, et al. Identification of interleukin-1 beta as a key mediator in the upregulation of Cav3.2ΓÇôUSP5 interactions in the pain pathway. Mol Pain. (2017) 13:1744806917724698. doi: 10.1177/1744806917724698
141. Webster CI, Hatcher J, Burrell M, Thom G, Thornton P, Gurrell I, et al. Enhanced delivery of IL-1 receptor antagonist to the central nervous system as a novel anti-transferrin receptor-IL-1RA fusion reverses neuropathic mechanical hypersensitivity. Pain. (2017) 158(4):660–8. doi: 10.1097/j.pain.0000000000000810
142. Yamashita T, Kamikaseda S, Tanaka A, Tosaki-Saitoh H, Caaveiro J, Inoue K, et al. New inhibitory effects of cilnidipine on microglial P2X7 receptors and IL-1 beta release: an involvement in its alleviating effect on neuropathic pain. Cells. (2021) 10:434.33670748
143. Gustafson-Vickers SL, Lu VB, Lai AY, Todd KG, Ballanyi K, Smith PA. Long-term actions of interleukin-1beta on delay and tonic firing neurons in rat superficial dorsal horn and their relevance to central sensitization. Mol Pain. (2008) 4:63. doi: 10.1186/1744-8069-4-63
144. Kawasaki Y, Zhang L, Cheng JK, Ji RR. Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha in regulating synaptic and neuronal activity in the superficial spinal cord. J Neurosci. (2008) 28(20):5189–94. doi: 10.1523/JNEUROSCI.3338-07.2008
145. Thompson SW, Majithia AA. Leukemia inhibitory factor induces sympathetic sprouting in intact dorsal root ganglia in the adult rat in vivo. J Physiol (Lond). (1998) 506 (Pt 3):809–16. doi: 10.1111/j.1469-7793.1998.809bv.x
146. Lopez ER, Carbajal AG, Tian JB, Bavencoffe A, Zhu MX, Dessauer CW, et al. Serotonin enhances depolarizing spontaneous fluctuations, excitability, and ongoing activity in isolated rat DRG neurons via 5-HT4 receptors and cAMP-dependent mechanisms. Neuropharmacology. (2021) 184:108408. doi: 10.1016/j.neuropharm.2020.108408
147. Ma C, Greenquist KW, LaMotte RH. Inflammatory mediators enhance the excitability of chronically compressed dorsal root ganglion neurons. J Neurophysiol. (2006) 95(4):2098–107. doi: 10.1152/jn.00748.2005
148. Abdulla FA, Stebbing MJ, Smith PA. Effects of substance P on excitability and ionic currents of normal and axotomized rat dorsal root ganglion neurons. Eur J Neurosci. (2001) 13:545–52. doi: 10.1046/j.0953-816x.2000.01429.x
149. Gudes S, Barkai O, Caspi Y, Katz B, Lev S, Binshtok AM. The role of slow and persistent TTX-resistant sodium currents in acute tumor necrosis factor-alpha-mediated increase in nociceptors excitability. J Neurophysiol. (2015) 113(2):601–19. doi: 10.1152/jn.00652.2014
150. del Rivero T, Fischer R, Yang F, Swanson KA, Bethea JR. Tumor necrosis factor receptor 1 inhibition is therapeutic for neuropathic pain in males but not in females. Pain. (2019) 160(4):922–31. doi: 10.1097/j.pain.0000000000001470
151. Gruber-Schoffnegger D, Drdla-Schutting R, Honigsperger C, Wunderbaldinger G, Gassner M, Sandkuhler J. Induction of thermal hyperalgesia and synaptic long-term potentiation in the spinal cord Lamina I by TNF-alpha and IL-1beta is mediated by glial cells. J Neurosci. (2013) 33(15):6540–51. doi: 10.1523/JNEUROSCI.5087-12.2013
152. Kanda H, Kobayashi K, Yamanaka H, Okubo M, Noguchi K. Microglial TNFalpha induces COX2 and PGI2 synthase expression in spinal endothelial cells during neuropathic pain. eNeuro. (2017) 4(2):ENEURO.0064–17.2017. doi: 10.1523/ENEURO.0064-17.2017
153. Liu S, Liu YP, Huang ZJ, Zhang YK, Song AA, Ma PC, et al. Wnt/Ryk signaling contributes to neuropathic pain by regulating sensory neuron excitability and spinal synaptic plasticity in rats. Pain. (2015) 156(12):2572–84. doi: 10.1097/j.pain.0000000000000366
154. Yuan Y, Zhao Y, Shen M, Wang C, Dong B, Xie K, et al. Spinal NLRP3 inflammasome activation mediates IL-1beta release and contributes to remifentanil-induced postoperative hyperalgesia by regulating NMDA receptor NR1 subunit phosphorylation and GLT-1 expression in rats. Mol Pain. (2022) 17448069221093016. doi: 10.1177/17448069221093016
155. Sun X, Cao L, Ge JL, Ge JY, Yang XF, Du BX, et al. The NLRP3-related inflammasome modulates pain behavior in a rat model of trigeminal neuropathic pain. Life Sci. (2021) 277:119489. doi: 10.1016/j.lfs.2021.119489
156. Jia M, Wu C, Gao F, Xiang H, Sun N, Peng P, et al. Activation of NLRP3 inflammasome in peripheral nerve contributes to paclitaxel-induced neuropathic pain. Mol Pain. (2017) 13:1744806917719804. doi: 10.1177/1744806917719804
157. Lee MS, Kim YJ. Pattern-recognition receptor signaling initiated from extracellular, membrane, and cytoplasmic space. Mol Cells. (2007) 23(1):1–10.17464205
158. Albiger B, Dahlberg S, Henriques-Normark B, Normark S. Role of the innate immune system in host defence against bacterial infections: focus on the toll-like receptors. J Intern Med. (2007) 261(6):511–28. doi: 10.1111/j.1365-2796.2007.01821.x
159. Grace PM, Strand KA, Galer EL, Rice KC, Maier SF, Watkins LR. Protraction of neuropathic pain by morphine is mediated by spinal damage associated molecular patterns (DAMPs) in male rats. Brain Behav Immun. (2018) 72:45–50. doi: 10.1016/j.bbi.2017.08.018
160. Heilig R, Dick MS, Sborgi L, Meunier E, Hiller S, Broz P. The gasdermin-D pore acts as a conduit for IL-1beta secretion in mice. Eur J Immunol. (2018) 48(4):584–92. doi: 10.1002/eji.201747404
161. Kawasaki Y, Xu ZZ, Wang X, Park JY, Zhuang ZY, Tan PH, et al. Distinct roles of matrix metalloproteases in the early- and late-phase development of neuropathic pain. Nat Med. (2008) 14(3):331–6. doi: 10.1038/nm1723
162. Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC. The pore-forming protein gasdermin D regulates interleukin-1 secretion from living macrophages. Immunity. (2018) 48(1):35–44. doi: 10.1016/j.immuni.2017.11.013
163. Orning P, Lien E, Fitzgerald KA. Gasdermins and their role in immunity and inflammation. J Exp Med. (2019) 216(11):2453–65. doi: 10.1084/jem.20190545
164. Mousseau M, Burma NE, Lee KY, Leduc-Pessah H, Kwok CHT, Reid AR, et al. Microglial pannexin-1 channel activation is a spinal determinant of joint pain. Sci Adv. (2018) 4(8):eaas9846. doi: 10.1126/sciadv.aas9846
165. Sommer C, Schmidt C, George A, Toyka KV. A metalloprotease-inhibitor reduces pain associated behavior in mice with experimental neuropathy. Neurosci Lett. (1997) 237(1):45–8. doi: 10.1016/S0304-3940(97)00813-6
166. Kucharczyk M, Kurek A, Detka J, Slusarczyk J, Papp M, Tota K, et al. Chronic mild stress influences nerve growth factor through a matrix metalloproteinase-dependent mechanism. Psychoneuroendocrinology. (2016) 66:11–21. doi: 10.1016/j.psyneuen.2015.12.019
167. DiSabato DJ, Quan N, Godbout JP. Neuroinflammation: the devil is in the details. J Neurochem. (2016) 139(S2):136–53. doi: 10.1111/jnc.13607
168. Milatovic D, Zaja-Milatovic S, Breyer RM, Aschner M, Montine TJ. Chapter 55 - neuroinflammation and oxidative injury in developmental neurotoxicity. In: Gupta RC, editor. Reproductive and developmental toxicology. 2nd edn. Cambridge, Massachusetts: Academic Press (2017). p. 1051–61.
169. Saab CY, Waxman SG, Hains BC. Alarm or curse? The pain of neuroinflammation. Brain Res Rev. (2008) 58(1):226–35. doi: 10.1016/j.brainresrev.2008.04.002
170. Smith PA. The known biology of neuropathic pain and its relevance to pain management. Can J Neurol Sci. (2023) 1–8. [Epub ahead of print]
171. Bogacka J, Ciapala K, Pawlik K, Kwiatkowski K, Dobrogowski J, Przeklasa-Muszynska A, et al. CCR4 Antagonist (C021) administration diminishes hypersensitivity and enhances the analgesic potency of morphine and buprenorphine in a mouse model of neuropathic pain. Front Immunol. (2020) 11:1241. doi: 10.3389/fimmu.2020.01241
172. Bogacka J, Ciapala K, Pawlik K, Dobrogowski J, Przeklasa-Muszynska A, Mika J. Blockade of CCR4 diminishes hypersensitivity and enhances opioid analgesia - evidence from a mouse model of diabetic neuropathy. Neuroscience. (2020) 441:77–92. doi: 10.1016/j.neuroscience.2020.06.025
173. Schafers M, Lee DH, Brors D, Yaksh TL, Sorkin LS. Increased sensitivity of injured and adjacent uninjured rat primary sensory neurons to exogenous tumor necrosis factor-alpha after spinal nerve ligation. J Neurosci. (2003) 23(7):3028–38. doi: 10.1523/JNEUROSCI.23-07-03028.2003
174. Zhang X, Xiao HS. Gene array analysis to determine the components of neuropathic pain signaling. Curr Opin Mol Ther. (2005) 7(6):532–7.16370375
175. Biber K, Boddeke E. Neuronal CC chemokines: the distinct roles of CCL21 and CCL2 in neuropathic pain. Front Cell Neurosci. (2014) 8:210. doi: 10.3389/fncel.2014.00210
176. Baskozos G, Dawes JM, Austin JS, Antunes-Martins A, McDermott L, Clark AJ, et al. Comprehensive analysis of long noncoding RNA expression in dorsal root ganglion reveals cell-type specificity and dysregulation after nerve injury. Pain. (2019) 160(2):463–85. doi: 10.1097/j.pain.0000000000001416
177. Favereaux A, Thoumine O, Bouali-Benazzouz R, Roques V, Papon MA, Salam SA, et al. Bidirectional integrative regulation of Cav1.2 calcium channel by microRNA miR-103: role in pain. EMBO J. (2011) 30(18):3830–41. doi: 10.1038/emboj.2011.249
178. Su S, Shao J, Zhao Q, Ren X, Cai W, Li L, et al. MiR-30b attenuates neuropathic pain by regulating voltage-gated sodium channel Nav1.3 in rats. Front Mol Neurosci. (2017) 10:126. doi: 10.3389/fnmol.2017.00126
179. Ye G, Zhang Y, Zhao J, Chen Y, Kong L, Sheng C, et al. Mir-384-5p ameliorates neuropathic pain by targeting SCN3A in a rat model of chronic constriction injury. Neurol Res. (2020) 42(4):299–307. doi: 10.1080/01616412.2020.1723313
180. Sakai A, Saitow F, Miyake N, Miyake K, Shimada T, Suzuki H. miR-7a alleviates the maintenance of neuropathic pain through regulation of neuronal excitability. Brain. (2013) 136(Pt 9):2738–50. doi: 10.1093/brain/awt191
181. Sakai A, Suzuki H. Emerging roles of microRNAs in chronic pain. Neurochem Int. (2014) 77:58–67. doi: 10.1016/j.neuint.2014.05.010
182. Sakai A, Saitow F, Maruyama M, Miyake N, Miyake K, Shimada T, et al. MicroRNA cluster miR-17-92 regulates multiple functionally related voltage-gated potassium channels in chronic neuropathic pain. Nat Commun. (2017) 8:16079. doi: 10.1093/brain/awt191
183. Malcangio M. Role of the immune system in neuropathic pain. Scand J Pain. (2020) 20(1):33–7. doi: 10.1515/sjpain-2019-0138
184. Gada Y, Pandey A, Jadhav N, Ajgaonkar S, Mehta D, Nair S. New vistas in microRNA regulatory interactome in neuropathic pain. Front Pharmacol. (2021) 12:778014. doi: 10.3389/fphar.2021.778014
185. Chen HP, Zhou W, Kang LM, Yan H, Zhang L, Xu BH, et al. Intrathecal miR-96 inhibits Nav1.3 expression and alleviates neuropathic pain in rat following chronic construction injury. Neurochem Res. (2014) 39(1):76–83. doi: 10.1007/s11064-013-1192-z
186. Qiu S, Liu B, Mo Y, Wang X, Zhong L, Han X, et al. MiR-101 promotes pain hypersensitivity in rats with chronic constriction injury via the MKP-1 mediated MAPK pathway. J Cell Mol Med. (2020) 24(16):8986–97. doi: 10.1111/jcmm.15532
187. Liu L, Xu D, Wang T, Zhang Y, Yang X, Wang X, et al. Epigenetic reduction of miR-214-3p upregulates astrocytic colony-stimulating factor-1 and contributes to neuropathic pain induced by nerve injury. Pain. (2020) 161(1):96–108. doi: 10.1097/j.pain.0000000000001681
188. Zhao YY, Wu ZJ, Zhu LJ, Niu TX, Liu B, Li J. Emerging roles of miRNAs in neuropathic pain: from new findings to novel mechanisms. Front Mol Neurosci. (2023) 16:1110975. doi: 10.3389/fnmol.2023.1110975
189. Kalpachidou T, Kummer KK, Kress M. Non-coding RNAs in neuropathic pain. Neuronal Signal. (2020) 4(1):NS20190099. doi: 10.1042/NS20190099
190. Ikuma Y, Sakai A, Sakamoto A, Suzuki H. Increased extracellular release of microRNAs from dorsal root ganglion cells in a rat model of neuropathic pain caused by peripheral nerve injury. PLoS One. (2023) 18(1):e0280425. doi: 10.1371/journal.pone.0280425
191. Baba H, Doubell TP, Woolf CJ. Peripheral inflammation facilitates aβ fiber-mediated synaptic input to the substantia gelatinosa of the adult rat spinal cord. J Neurosci. (1999) 19:859–67. doi: 10.1523/JNEUROSCI.19-02-00859.1999
192. Xanthos DN, Pungel I, Wunderbaldinger G, Sandkuhler J. Effects of peripheral inflammation on the blood-spinal cord barrier. Mol Pain. (2012) 8:44. doi: 10.1186/1744-8069-8-44
193. Lim TKY, Shi XQ, Martin HC, Huang H, Luheshi G, Rivest S, et al. Blood-nerve barrier dysfunction contributes to the generation of neuropathic pain and allows targeting of injured nerves for pain relief. Pain. (2014) 155(5):954–67. doi: 10.1016/j.pain.2014.01.026
194. DeLeo JA, Sorkin LS, Watkins LR. Immune and glial regulation of pain. Seattle: IASP Press (2007).
195. Greenhalgh AD, David S, Bennett FC. Immune cell regulation of glia during CNS injury and disease. Nat Rev Neurosci. (2020) 21(3):139–52. doi: 10.1038/s41583-020-0263-9
196. Hefti F. Pharmacology of nerve growth factor and discovery of tanezumab, an anti-nerve growth factor antibody and pain therapeutic. Pharmacol Res. (2020) 154:104240. doi: 10.1016/j.phrs.2019.04.024
197. North RY, Odem MA, Li Y, Tatsui CE, Cassidy RM, Dougherty PM, et al. Electrophysiological alterations driving pain-associated spontaneous activity in human sensory neuron somata parallel alterations described in spontaneously active rodent nociceptors. J Pain. (2022) 23(8):1343–57. doi: 10.1016/j.jpain.2022.02.009
198. Haroutounian S, Nikolajsen L, Bendtsen TF, Finnerup NB, Kristensen AD, Hasselstrom JB, et al. Primary afferent input critical for maintaining spontaneous pain in peripheral neuropathy. Pain. (2014) 155(7):1272–9. doi: 10.1016/j.pain.2014.03.022
199. Pitcher GM, Henry JL. Governing role of primary afferent drive in increased excitation of spinal nociceptive neurons in a model of sciatic neuropathy. Exp Neurol. (2008) 214(2):219–28. doi: 10.1016/j.expneurol.2008.08.003
200. Devor M. Centralization, central sensitization and neuropathic pain. Focus on “sciatic chronic constriction injury produces cell-type-specific changes in the electrophysiological properties of rat substantia gelatinosa neurons”. J Neurophysiol. (2006) 96(2):522–3. doi: 10.1152/jn.00365.2006
201. Devor M, Vaso A, Adahan HM, Vyshka G. PNS Origin of phantom limb sensation and pain: reply to letter to the editor regarding foell et al., peripheral origin of phantom limb pain: is it all resolved? Pain (2014) 155(10):2207–8. doi: 10.1016/j.pain.2014.08.018
202. Koplovitch P, Devor M. Dilute lidocaine suppresses ectopic neuropathic discharge in dorsal root ganglia without blocking axonal propagation: a new approach to selective pain control. Pain. (2018) 159(7):1244–56. doi: 10.1097/j.pain.0000000000001205
203. Liu CN, Raber P, Ziv-Sefer S, Devor M. Hyperexcitability in sensory neurons of rats selected for high versus low neuropathic pain phenotype. Neuroscience. (2001) 105(1):265–75. doi: 10.1016/S0306-4522(01)00161-0
204. Sukhotinsky I, Ben Dor E, Raber P, Devor M. Key role of the dorsal root ganglion in neuropathic tactile hypersensibility. Eur J Pain. (2004) 8(2):135–43. doi: 10.1016/S1090-3801(03)00086-7
205. Yatziv SL, Devor M. Suppression of neuropathic pain by selective silencing of dorsal root ganglion ectopia using nonblocking concentrations of lidocaine. Pain. (2019) 160(9):2105–14. doi: 10.1097/j.pain.0000000000001602
206. Chen C, Sun L, Adler A, Zhou H, Zhang L, Zhang L, et al. Synchronized activity of sensory neurons initiates cortical synchrony in a model of neuropathic pain. Nat Commun. (2023) 14(1):689. doi: 10.1038/s41467-023-36093-z
207. Daou I, Beaudry H, Ase AR, Wieskopf JS, Ribeiro-da-Silva A, Mogil JS, et al. Optogenetic silencing of Nav1.8-positive afferents alleviates inflammatory and neuropathic pain. eNeuro. (2016) 3(1):ENEURO.0140–15.2016. doi: 10.1523/ENEURO.0140-15.2016
208. Alles SRA, Smith PA. Peripheral voltage-gated cation channels in neuropathic pain and their potential as therapeutic targets. Front Pain Res. (2021) 2:750583. doi: 10.3389/pain.2021.750583
209. Waxman SG, Zamponi GW. Regulating excitability of peripheral afferents: emerging ion channel targets. Nat Neurosci. (2014) 17(2):153–63. doi: 10.1038/nn.3602
210. Smith PA. K+ channels in primary afferents and their role in pain produced by peripheral nerev injury. Front Cell Neurosci. (2020) 14:294. doi: 10.3389/fncel.2020.566418
211. Basso L, Altier C. Transient receptor potential channels in neuropathic pain. Curr Opin Pharmacol. (2017) 32:9–15. doi: 10.1016/j.coph.2016.10.002
212. Du X, Gamper N. Potassium channels in peripheral pain pathways: expression, function and therapeutic potential. Curr Neuropharmacol. (2013) 11(6):621–40. doi: 10.2174/1570159X113119990042
213. Chaplan SR, Guo HQ, Lee DH, Luo L, Liu C, Kuei C, et al. Neuronal hyperpolarization-activated pacemaker channels drive neuropathic pain. J Neurosci. (2003) 23(4):1169–78. doi: 10.1523/JNEUROSCI.23-04-01169.2003
214. Amir R, Michaelis M, Devor M. Burst discharge in primary sensory neurons: triggered by subthreshold oscillations, maintained by depolarizing afterpotentials. J Neurosci. (2002) 22(3):1187–98. doi: 10.1523/JNEUROSCI.22-03-01187.2002
215. Bennett GJ, Doyle T, Salvemini D. Mitotoxicity in distal symmetrical sensory peripheral neuropathies. Nat Rev Neurol. (2014) 10(6):326–36. doi: 10.1038/nrneurol.2014.77
216. Stemkowsi PL, Bukhanova-Schulz N, Baldwin T, de Chaves E Posse, Smith P.A. Are sensory neurons exquisitely sensitive to interleukin 1β? J Neuroimmunol. (2021) 354:577529. doi: 10.1016/j.jneuroim.2021.577529
217. Sorkin LS, Xiao WH, Wagner R, Myers RR. Tumour necrosis factor-alpha induces ectopic activity in nociceptive primary afferent fibres. Neuroscience. (1997) 81(1):255–62. doi: 10.1016/S0306-4522(97)00147-4
218. Jin X, Gereau RW IV. Acute p38-mediated modulation of tetrodotoxin-resistant sodium channels in mouse sensory neurons by tumor necrosis factor-{alpha}. J Neurosci. (2006) 26(1):246–55. doi: 10.1523/JNEUROSCI.3858-05.2006
219. de Macedo FHP, Aires RD, Fonseca EG, Ferreira RCM, Machado DPD, Chen L, et al. TNF-alpha mediated upregulation of NaV1.7 currents in rat dorsal root ganglion neurons is independent of CRMP2 SUMOylation. Mol Brain. (2019) 12(1):117. doi: 10.1186/s13041-019-0538-0
220. White FA, Feldman P, Miller RJ. Chemokine signaling and the management of neuropathic pain. Mol Interv. (2009) 9(4):188–95. doi: 10.1124/mi.9.4.7
221. Oh SB, Tran PB, Gillard SE, Hurley RW, Hammond DL, Miller RJ. Chemokines and glycoprotein120 produce pain hypersensitivity by directly exciting primary nociceptive neurons. J Neurosci. (2001) 21(14):5027–35. doi: 10.1523/JNEUROSCI.21-14-05027.2001
222. Sun JH, Yang B, Donnelly DF, Ma C, LaMotte RH. MCP-1 enhances excitability of nociceptive neurons in chronically compressed dorsal root ganglia. J Neurophysiol. (2006) 96(5):2189–99. doi: 10.1152/jn.00222.2006
223. White FA, Sun J, Waters SM, Ma C, Ren D, Ripsch M, et al. Excitatory monocyte chemoattractant protein-1 signaling is up-regulated in sensory neurons after chronic compression of the dorsal root ganglion. Proc Natl Acad Sci U S A. (2005) 102(39):14092–7. doi: 10.1073/pnas.0503496102
224. Jung H, Toth PT, White FA, Miller RJ. Monocyte chemoattractant protein-1 functions as a neuromodulator in dorsal root ganglia neurons. J Neurochem. (2008) 104(1):254–63. doi: 10.1111/j.1471-4159.2007.04969.x
225. Gold MS, Reichling DB, Shuster MJ, Levine JD. Hyperalgesic agents increase a tetrodotoxin-resistant na+ current in nociceptors. Proc Natl Acad Sci U S A. (1996) 93(3):1108–12. doi: 10.1073/pnas.93.3.1108
226. Zamponi GW, Striessnig J, Koschak A, Dolphin AC. The Physiology, Pathology, And pharmacology of voltage-gated calcium channels and their future therapeutic potential. Pharmacol Rev. (2015) 67(4):821–70. doi: 10.1124/pr.114.009654
227. Du X, Gao H, Jaffe D, Zhang H, Gamper N. M-type K(+) channels in peripheral nociceptive pathways. Br J Pharmacol. (2018) 175(12):2158–72. doi: 10.1111/bph.13978
228. Jones F, Gamper N, Gao H. Kv7 channels and excitability disorders. Handb Exp Pharmacol. (2021) 267:185–230. doi: 10.1007/164_2021_457
229. Laumet G, Garriga J, Chen SR, Zhang Y, Li DP, Smith TM, et al. G9a is essential for epigenetic silencing of K+ channel genes in acute-to-chronic pain transition. Nat Neurosci. (2015) 18(12):1746–55. doi: 10.1038/nn.4165
230. Liang L, Zhao JY, Kathryn T, Bekker A, Tao YX. BIX01294, A G9a inhibitor, alleviates nerve injury-induced pain hypersensitivities during both development and maintenance periods. Transl Perioper Pain Med. (2019) 6(4):106–14. doi: 10.31480/2330-4871/097
231. Wang X, Shen X, Ma S, Liu Y, Xu S, Wu S, et al. Threshold effect of G9a/glp on peripheral nerve injury induced hypersensitivity. Mol Pain. (2017) 13:1744806917729305. doi: 10.1177/1744806917729305
232. Rugo HS, Jacobs I, Sharma S, Scappaticci F, Paul TA, Jensen-Pergakes K, et al. The promise for histone methyltransferase inhibitors for epigenetic therapy in clinical oncology: a narrative review. Adv Ther. (2020) 37(7):3059–82. doi: 10.1007/s12325-020-01379-x
233. Dib-Hajj SD, Yang Y, Black JA, Waxman SG. The na(V)1.7 sodium channel: from molecule to man. Nat Rev Neurosci. (2013) 14(1):49–62. doi: 10.1038/nrn3404
234. Dib-Hajj SD, Black JA, Waxman SG. Nav1.9: a sodium channel linked to human pain. Nat Rev Neurosci. (2015) 16(9):511–9. doi: 10.1038/nrn3977
235. Kotecha M, Cheshire WP, Finnigan H, Giblin K, Naik H, Palmer J, et al. Design of phase 3 studies evaluating vixotrigine for treatment of trigeminal neuralgia. J Pain Res. (2020) 13:1601–9. doi: 10.2147/JPR.S247182
236. Zakrzewska JM, Palmer J, Morisset V, Giblin GM, Obermann M, Ettlin DA, et al. Safety and efficacy of a Nav1.7 selective sodium channel blocker in patients with trigeminal neuralgia: a double-blind, placebo-controlled, randomised withdrawal phase 2a trial. Lancet Neurol. (2017) 16(4):291–300. doi: 10.1016/S1474-4422(17)30005-4
237. Dib-Hajj SD, Black JA, Cummins TR, Kenney AM, Kocsis JD, Waxman SG. Rescue of alpha-SNS sodium channel expression in small dorsal root ganglion neurons after axotomy by nerve growth factor in vivo. J Neurophysiol. (1998) 79(5):2668–76. doi: 10.1152/jn.1998.79.5.2668
238. Patel MK, Kaye AD, Urman RD. Tanezumab: therapy targeting nerve growth factor in pain pathogenesis. J Anaesthesiol Clin Pharmacol. (2018) 34(1):111–6. doi: 10.4103/joacp.JOACP_389_15
239. Bagal SK, Andrews M, Bechle BM, Bian J, Bilsland J, Blakemore DC, et al. Discovery of potent, selective, and peripherally restricted pan-trk kinase inhibitors for the treatment of pain. J Med Chem. (2018) 61(15):6779–800. doi: 10.1021/acs.jmedchem.8b00633
240. Bagal SK, Omoto K, Blakemore DC, Bungay PJ, Bilsland JG, Clarke PJ, et al. Discovery of Allosteric, Potent, Subtype Selective, And peripherally restricted TrkA kinase inhibitors. J Med Chem. (2019) 62(1):247–65. doi: 10.1021/acs.jmedchem.8b00280
241. Tang S, Xue Y, Dengqi X, Shao L. Design, development and evaluation of a prodrug-type TrkA-selective inhibitor with antinociceptive effects in vivo. Eur J Med Chem. (2023) 245(Pt 2):114901. doi: 10.1016/j.ejmech.2022.114901
242. Ramachandra R, Hassan B, McGrew SG, Dompor J, Farrag M, Ruiz-Velasco V, et al. Identification of CaV channel types expressed in muscle afferent neurons. J Neurophysiol. (2013) 110(7):1535–43. doi: 10.1152/jn.00069.2013
243. Rose KE, Lunardi N, Boscolo A, Dong X, Erisir A, Jevtovic-Todorovic V, et al. Immunohistological demonstration of CaV3.2 T-type voltage-gated calcium channel expression in soma of dorsal root ganglion neurons and peripheral axons of rat and mouse. Neuroscience. (2013) 250:263–74. doi: 10.1016/j.neuroscience.2013.07.005
244. Talley EM, Cribbs LL, Lee JH, Daud A, Perez-Reyes E, Bayliss DA. Differential distribution of three members of a gene family encoding low voltage-activated (T-type) calcium channels. J Neurosci. (1999) 19(6):1895–911. doi: 10.1523/JNEUROSCI.19-06-01895.1999
245. Altier C, Zamponi GW. Targeting Ca2+ channels to treat pain: t-type versus N-type. Trends Pharmacol Sci. (2004) 25(9):465–70. doi: 10.1016/j.tips.2004.07.004
246. Rettig J, Sheng ZH, Kim DK, Hodson CD, Snutch TP, Catterall WA. Isoform-specific interaction of the alpha1A subunits of brain Ca2+ channels with the presynaptic proteins syntaxin and SNAP-25. Proc Natl Acad Sci U S A. (1996) 93(14):7363–8. doi: 10.1073/pnas.93.14.7363
247. Yang J, Xie MX, Hu L, Wang XF, Mai JZ, Li YY, et al. Upregulation of N-type calcium channels in the soma of uninjured dorsal root ganglion neurons contributes to neuropathic pain by increasing neuronal excitability following peripheral nerve injury. Brain Behav Immun. (2018) 71:52–65. doi: 10.1016/j.bbi.2018.04.016
248. Alles SR, Garcia E, Balasubramanyan S, Jones K, Tyson JR, Joy T, et al. Peripheral nerve injury increases contribution of L-type calcium channels to synaptic transmission in spinal lamina II: role of alpha2delta-1 subunits. Mol Pain. (2018) 14:1744806918765806. doi: 10.1177/1744806918765806
249. Jagodic MM, Pathirathna S, Joksovic PM, Lee W, Nelson MT, Naik AK, et al. Upregulation of the T-type calcium current in small rat sensory neurons after chronic constrictive injury of the sciatic nerve. J Neurophysiol. (2008) 99(6):3151–6. doi: 10.1152/jn.01031.2007
250. Bourinet E, Altier C, Hildebrand ME, Trang T, Salter MW, Zamponi GW. Calcium-permeable ion channels in pain signaling. Physiol Rev. (2014) 94(1):81–140. doi: 10.1152/physrev.00023.2013
251. Patel R, Montagut-Bordas C, Dickenson AH. Calcium channel modulation as a target in chronic pain control. Br J Pharmacol. (2018) 175(12):2173–84. doi: 10.1111/bph.13789
252. Tibbs GR, Posson DJ, Goldstein PA. Voltage-gated ion channels in the PNS: novel therapies for neuropathic pain? Trends Pharmacol Sci. (2016) 37(7):522–42. doi: 10.1016/j.tips.2016.05.002
253. Zamponi GW. Targeting voltage-gated calcium channels in neurological and psychiatric diseases. Nat Rev Drug Discov. (2016) 15(1):19–34. doi: 10.1038/nrd.2015.5
254. Matis G, De NP, Dupoiron D, Likar R, Zuidema X, Rasche D. Intrathecal pain management with ziconotide: time for consensus? Brain Behav. (2021)11(Suppl 1):e02055. doi: 10.1002/brb3.2055
255. Abdulla FA, Smith PA. Ectopic α2-adrenoceptors couple to N-type Ca2+ channels in axotomized rat sensory neurons. J Neurosci. (1997) 17(5):1633–41. doi: 10.1523/JNEUROSCI.17-05-01633.1997
256. Zamponi GW, Snutch TP. Modulation of voltage-dependent calcium channels by G proteins. Curr Opin Neurobiol. (1998) 8(3):351–6. doi: 10.1016/S0959-4388(98)80060-3
257. Kirkpatrick AF, Derasari M, Glodek JA, Piazza PA. Postherpetic neuralgia: a possible application for topical clonidine. Anesthesiology. (1992) 76(6):1065–6. doi: 10.1097/00000542-199206000-00044
258. Fink EA, Xu J, Hubner H, Braz JM, Seemann P, Avet C, et al. Structure-based discovery of nonopioid analgesics cting through the alpha2A-adrenergic receptor. Science. (2022) 377(6614):eabn7065. doi: 10.1126/science.abn7065
259. Greene SA, Thurmon JC. Xylazine: a review of its pharmacology and use in veterinary medicine. J Vet Pharmacol Ther. (1988) 11(4):295–313. doi: 10.1111/j.1365-2885.1988.tb00189.x
260. Friedman J, Montero F, Bourgois P, Wahbi R, Dye D, Goodman-Meza D, et al. Xylazine spreads across the US: a growing component of the increasingly synthetic and polysubstance overdose crisis. Drug Alcohol Depend. (2022) 233:109380. doi: 10.1016/j.drugalcdep.2022.109380
261. Moore RA, Wiffen PJ, Derry S, Toelle T, Rice AS. Gabapentin for chronic neuropathic pain and fibromyalgia in adults. Cochrane Database Syst Rev. (2014) 4:CD007938. doi: 10.1002/14651858.CD007938.pub2
262. Dolphin AC. Calcium channel auxiliary alpha2delta and beta subunits: trafficking and one step beyond. Nat Rev Neurosci. (2012) 13(8):542–55. doi: 10.1038/nrn3311
263. Eroglu C, Allen NJ, Susman MW, O’Rourke NA, Park CY, Ozkan E, et al. Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell. (2009) 139(2):380–92. doi: 10.1016/j.cell.2009.09.025
264. Hoppa MB, Lana B, Margas W, Dolphin AC, Ryan TA. Alpha2delta expression sets presynaptic calcium channel abundance and release probability. Nature. (2012) 486(7401):122–5. doi: 10.1038/nature11033
265. Field MJ, Cox PJ, Stott E, Melrose H, Offord J, Su TZ, et al. Identification of the {alpha}2-{delta}-1 subunit of voltage-dependent calcium channels as a molecular target for pain mediating the analgesic actions of pregabalin. PNAS. (2006) 103(46):17537–42. doi: 10.1073/pnas.0409066103
266. Li L, Chen SR, Zhou MH, Wang L, Li DP, Chen H, et al. Alpha 2 Delta 1 switches the phenotype of synaptic AMPA receptors by physically disrupting heteromeric subunit assembly. Cell Rep. (2021) 36(3):109396. doi: 10.1016/j.celrep.2021.109396
267. Chen J, Li L, Chen SR, Chen H, Xie JD, Sirrieh RE, et al. The alpha2delta-1-NMDA receptor Complex is critically involved in neuropathic pain development and gabapentin therapeutic actions. Cell Rep. (2018) 22(9):2307–21. doi: 10.1016/j.celrep.2018.02.021
268. Abdulla FA, Smith PA. Axotomy- and autotomy-induced changes in Ca2+ and K+ channel currents of rat dorsal root ganglion neurons. J Neurophysiol. (2001) 85:644–58. doi: 10.1152/jn.2001.85.2.644
269. Jagodic MM, Pathirathna S, Nelson MT, Mancuso S, Joksovic PM, Rosenberg ER, et al. Cell-Specific alterations of T-type calcium current in painful diabetic neuropathy enhance excitability of sensory neurons. J Neurosci. (2007) 27(12):3305–16. doi: 10.1523/JNEUROSCI.4866-06.2007
270. Francois A, Schuetter N, Laffray S, Sanguesa J, Pizzoccaro A, Dubel S, et al. The low-threshold calcium channel Cav3.2 determines low-threshold mechanoreceptor function. Cell Rep. (2015) 10(3):370–82. doi: 10.1016/j.celrep.2014.12.042
271. Jacus MO, Uebele VN, Renger JJ, Todorovic SM. Presynaptic CaV3.2 channels regulate excitatory neurotransmission in nociceptive dorsal horn neurons. J Neurosci. (2012) 32(27):9374–82. doi: 10.1523/JNEUROSCI.0068-12.2012
272. Garcia-Caballero A, Gadotti VM, Stemkowski P, Weiss N, Souza IA, Hodgkinson V, et al. The deubiquitinating enzyme USP5 modulates neuropathic and inflammatory pain by enhancing Cav3.2 channel activity. Neuron. (2014) 83(5):1144–58. doi: 10.1016/j.neuron.2014.07.036
273. Alaklabi AM, Gambeta E, Zamponi GW. Electrophysiological characterization of a CaV3.1 calcium channel mutation linked to trigeminal neuralgia. Pflugers Arch. (2023) 475:711–8. doi: 10.1007/s00424-023-02808-w
274. Choe W, Messinger RB, Leach E, Eckle VS, Obradovic A, Salajegheh R, et al. TTA-P2 is a potent and selective blocker of T-type calcium channels in rat sensory neurons and a novel antinociceptive agent. Mol Pharmacol. (2011) 80(5):900–10. doi: 10.1124/mol.111.073205
275. Francois A, Kerckhove N, Meleine M, Alloui A, Barrere C, Gelot A, et al. State-dependent properties of a new T-type calcium channel blocker enhance ca(V)3.2 selectivity and support analgesic effects. Pain. (2013) 154(2):283–93. doi: 10.1016/j.pain.2012.10.023
276. Snutch TP, Zamponi GW. Recent advances in the development of T-type calcium channel blockers for pain intervention. Br J Pharmacol. (2017) 175:2375–83. doi: 10.1111/bph.13906
277. Tringham E, Powell KL, Cain SM, Kuplast K, Mezeyova J, Weerapura M, et al. T-type calcium channel blockers that attenuate thalamic burst firing and suppress absence seizures. Sci Transl Med. (2012) 4(121):121ra19. doi: 10.1126/scitranslmed.3003120
278. Harding EK, Dedek A, Bonin RP, Salter MW, Snutch TP, Hildebrand ME. The T-type calcium channel antagonist, Z944, reduces spinal excitability and pain hypersensitivity. Br J Pharmacol. (2021) 178:3517–32. doi: 10.1111/bph.15498
279. Zamponi GW, Lewis RJ, Todorovic SM, Arneric SP, Snutch TP. Role of voltage-gated calcium channels in ascending pain pathways. Brain Res Rev. (2009) 60(1):84–9. doi: 10.1016/j.brainresrev.2008.12.021
280. Nam G. T-type calcium channel blockers: a patent review (2012-2018). Null. (2018) 28(12):883–901.
281. Hauser W, Petzke F, Fitzcharles MA. Efficacy, tolerability and safety of cannabis-based medicines for chronic pain management - an overview of systematic reviews. Eur J Pain. (2018) 22(3):455–70. doi: 10.1002/ejp.1118
282. Ross HR, Napier I, Connor M. Inhibition of recombinant human T-type calcium channels by Delta9-tetrahydrocannabinol and cannabidiol. J Biol Chem. (2008) 283(23):16124–34. doi: 10.1074/jbc.M707104200
283. Bladen C, Mirlohi S, Santiago M, Longworth M, Kassiou M, Banister S, et al. Modulation of human T-type calcium channels by synthetic cannabinoid receptor agonists in vitro. Neuropharmacology. (2021) 187:108478. doi: 10.1016/j.neuropharm.2021.108478
284. Mackie K, Hille B. Cannabinoids inhibit N-type calcium channels in neuroblastoma-glioma cells. Proc Natl Acad Sci U S A. (1992) 89(9):3825–9. doi: 10.1073/pnas.89.9.3825
285. Li Y, Zhang L, Wu Y, Zheng Q, Chen M, Qian Z, et al. Cannabinoids-induced peripheral analgesia depends on activation of BK channels. Brain Res. (2019) 1711:23–8. doi: 10.1016/j.brainres.2019.01.007
286. Zhang HXB, Bean BP. Cannabidiol inhibition of murine primary nociceptors: tight binding to slow inactivated states of Nav1.8 channels. J Neurosci. (2021) 41(30):6371. doi: 10.1523/JNEUROSCI.3216-20.2021
287. You H, Altier C, Zamponi GW. CCR2 Receptor ligands inhibit Cav3.2 T-type calcium channels. Mol Pharmacol. (2010) 77(2):211–7. doi: 10.1124/mol.109.059022
288. You H, Gadotti VM, Petrov RR, Zamponi GW, Diaz P. Functional characterization and analgesic effects of mixed cannabinoid receptor/T-type channel ligands. Mol Pain. (2011) 7:89. doi: 10.1124/mol.109.059022
289. Rangel-Galvan M, Castro ME, Perez-Aguilar JM, Caballero NA, Rangel-Huerta A, Melendez FJ. Theoretical study of the structural stability, chemical reactivity, and protein interaction for NMP compounds as modulators of the endocannabinoid system. Molecules. (2022) 27(2):414. doi: 10.3390/molecules27020414
290. Tomita S, Sekiguchi F, Kasanami Y, Naoe K, Tsubota M, Wake H, et al. Cav3.2 overexpression in L4 dorsal root ganglion neurons after L5 spinal nerve cutting involves egr-1, USP5 and HMGB1 in rats: An emerging signaling pathway for neuropathic pain. Eur J Pharmacol. (2020) 888:173587. doi: 10.1016/j.ejphar.2020.173587
291. Ali MY, Gadotti VM, Huang S, Garcia-Caballero A, Antunes FTT, Jung HA, et al. Icariside II, a prenyl-flavonol, Alleviates Inflammatory and Neuropathic Pain by Inhibiting T-Type Calcium Channels and USP5–Cav3.2 Interactions. ACS Chem Neurosci. (2023) 14(10):1859–69. doi: 10.1021/acschemneuro.3c00083
292. Garcia-Caballero A, Gadotti VM, Ali MY, Bladen C, Gambeta E, Van Humbeck JF, et al. A synthetically accessible small-molecule inhibitor of USP5-Cav3.2 calcium channel interactions with analgesic properties. ACS Chem Neurosci. (2022) 13(4):524–36. doi: 10.1021/acschemneuro.1c00765
293. Gadotti VM, Caballero AG, Berger ND, Gladding CM, Chen L, Pfeifer TA, et al. Small organic molecule disruptors of Cav3.2 - USP5 interactions reverse inflammatory and neuropathic pain. Mol Pain. (2015) 11:12. doi: 10.1186/s12990-015-0011-8
294. Garcia-Caballero A, Gadotti VM, Chen L, Zamponi GW. A cell-permeant peptide corresponding to the cUBP domain of USP5 reverses inflammatory and neuropathic pain. Mol Pain. (2016) 12:1744806916642444. doi: 10.1177/1744806916642444
295. Smith T, Al OM, Sathish J, Djouhri L. Increased expression of HCN2 channel protein in L4 dorsal root ganglion neurons following axotomy of L5- and inflammation of L4-spinal nerves in rats. Neuroscience. (2015) 295:90–102. doi: 10.1016/j.neuroscience.2015.03.041
296. Emery EC, Young GT, Berrocoso EM, Chen L, McNaughton PA. HCN2 ion channels play a central role in inflammatory and neuropathic pain. Science. (2011) 333(6048):1462–6. doi: 10.1126/science.1206243
297. Djouhri L, Smith T, Ahmeda A, Alotaibi M, Weng X. Hyperpolarization-activated cyclic nucleotide-gated channels contribute to spontaneous activity in L4 C-fiber nociceptors, but not abeta-non-nociceptors, after axotomy of L5-spinal nerve in the rat in vivo. Pain. (2018) 159(7):1392–402. doi: 10.1097/j.pain.0000000000001224
298. Antal M, Papp I, Bahaerguli N, Veress G, Vereb G. Expression of hyperpolarization-activated and cyclic nucleotide-gated cation channel subunit 2 in axon terminals of peptidergic nociceptive primary sensory neurons in the superficial spinal dorsal horn of rats. Eur J Neurosci. (2004) 19(5):1336–42. doi: 10.1111/j.1460-9568.2004.03235.x
299. Papp I, Szucs P, Hollo K, Erdelyi F, Szabo G, Antal M. Hyperpolarization-activated and cyclic nucleotide-gated cation channel subunit 2 ion channels modulate synaptic transmission from nociceptive primary afferents containing substance P to secondary sensory neurons in laminae I-IIo of the rodent spinal dorsal horn. Eur J Neurosci. (2006) 24(5):1341–52. doi: 10.1111/j.1460-9568.2006.05013.x
300. Young GT, Emery EC, Mooney ER, Tsantoulas C, McNaughton PA. Inflammatory and neuropathic pain are rapidly suppressed by peripheral block of hyperpolarisation-activated cyclic nucleotide-gated ion channels. Pain. (2014) 155(9):1708–19. doi: 10.1016/j.pain.2014.05.021
301. Noh S, Kumar N, Bukhanova N, Chen Y, Stemkowsi PL, Smith PA. The heart-rate-reducing agent, ivabradine, reduces mechanical allodynia in a rodent model of neuropathic pain. Eur J Pain. (2014) 18:1139–47. doi: 10.1002/j.1532-2149.2014.00460.x
302. Santoro B, Shah MM. Hyperpolarization-Activated cyclic nucleotide-gated channels as drug targets for neurological disorders. Annu Rev Pharmacol Toxicol. (2020) 60:109–31. doi: 10.1146/annurev-pharmtox-010919-023356
303. Tsantoulas C, Mooney ER, McNaughton PA. HCN2 ion channels: basic science opens up possibilities for therapeutic intervention in neuropathic pain. Biochem J. (2016) 473(18):2717–36. doi: 10.1042/BCJ20160287
304. Bernard Healey SA, Scholtes I, Abrahams M, McNaughton PA, Menon DK, Lee MC. Role of hyperpolarization-activated cyclic nucleotide-gated ion channels in neuropathic pain: a proof-of-concept study of ivabradine in patients with chronic peripheral neuropathic pain. Pain Rep. (2021) 6(4):e967. doi: 10.1097/PR9.0000000000000967
305. Vilceanu D, Honore P, Hogan QH, Stucky CL. Spinal nerve ligation in mouse upregulates TRPV1 heat function in injured IB4-positive nociceptors. J Pain. (2010) 11(6):588–99. doi: 10.1016/j.jpain.2009.09.018
306. Iftinca M, Defaye M, Altier C. TRPV1-Targeted Drugs in development for human pain conditions. Drugs. (2020) 81:7–27. doi: 10.1007/s40265-020-01429-2
307. Binshtok AM, Bean BP, Woolf CJ. Inhibition of nociceptors by TRPV1-mediated entry of impermeant sodium channel blockers. Nature. (2007) 449(7162):607–10. doi: 10.1038/nature06191
308. Landry M, Holmberg K, Zhang X, Hokfelt T. Effect of axotomy on expression of NPY, galanin, and NPY Y1 and Y2 receptors in dorsal root ganglia and the superior cervical ganglion studied with double-labeling in situ hybridization and immunohistochemistry. Exp Neurol. (2000) 162(2):361–84. doi: 10.1006/exnr.1999.7329
309. Wakisaka S, Kajander KC, Bennett GJ. Increased neuropeptide Y (NPY)-like immunoreactivity in rat sensory neurons following peripheral axotomy. Neurosci Lett. (1991) 124:200–3. doi: 10.1016/0304-3940(91)90093-9
310. Noguchi K, De Leon M, Nahin RL, Senba E, Ruda MA. Quantification of axotomy-induced alteration of neuropeptide mRNAs in dorsal root ganglion neurons with special reference to neuropeptide Y mRNA and the effects of neonatal capsaicin treatment. J Neurosci Res. (1993) 35(1):54–66. doi: 10.1002/jnr.490350108
311. Abdulla FA, Smith PA. Nerve injury increases an excitatory action of neuropeptide Y and Y2- agonists on dorsal root ganglion neurons. Neuroscience. (1999) 89(1):43–60. doi: 10.1016/S0306-4522(98)00443-6
312. Huang LY, Neher E. Ca(2+)-dependent exocytosis in the somata of dorsal root ganglion neurons. Neuron. (1996) 17(1):135–45. doi: 10.1016/S0896-6273(00)80287-1
313. Eberhardt M, Hoffmann T, Sauer SK, Messlinger K, Reeh PW, Fischer MJ. Calcitonin gene-related peptide release from intact isolated dorsal root and trigeminal ganglia. Neuropeptides. (2008) 42(3):311–7. doi: 10.1016/j.npep.2008.01.002
314. Sauer SK, Reeh PW, Bove GM. Noxious heat-induced CGRP release from rat sciatic nerve axons in vitro. Eur J Neurosci. (2001) 14(8):1203–8. doi: 10.1046/j.0953-816x.2001.01741.x
315. Gardell LR, Vanderah TW, Gardell SE, Wang R, Ossipov MH, Lai J, et al. Enhanced evoked excitatory transmitter release in experimental neuropathy requires descending facilitation. J Neurosci. (2003) 23(23):8370–9. doi: 10.1523/JNEUROSCI.23-23-08370.2003
316. Dray A, Pinnock RD. Effects of substance P on adult rat sensory ganglion neurones in vitro. Neurosci Lett. (1982) 33(1):61–6. doi: 10.1016/0304-3940(82)90130-6
317. Natura G, von Banchet GS, Schaible HG. Calcitonin gene-related peptide enhances TTX-resistant sodium currents in cultured dorsal root ganglion neurons from adult rats. Pain. (2005) 116(3):194–204. doi: 10.1016/j.pain.2005.04.002
318. McMahon SB, La Russa F., Bennett DL. Crosstalk between the nociceptive and immune systems in host defence and disease. Nat Rev Neurosci. (2015) 16(7):389–402. doi: 10.1038/nrn3946
319. Matsuka Y, Afroz S, Dalanon JC, Iwasa T, Waskitho A, Oshima M. The role of chemical transmitters in neuron-glia interaction and pain in sensory ganglion. Neurosci Biobehav Rev. (2020) 108:393–9. doi: 10.1016/j.neubiorev.2019.11.019
320. Goldstein DJ, Wang O, Gitter BD, Iyengar S. Dose-response study of the analgesic effect of lanepitant in patients with painful diabetic neuropathy. Clin Neuropharmacol. (2001) 24(1):16–22. doi: 10.1097/00002826-200101000-00004
321. Parascandolo E, Levinson K, Rizzoli P, Sharon R. Efficacy of erenumab in the treatment of trigeminal neuralgia: a retrospective case series. Neurol Clin Pract. (2021) 11(3):227–31. doi: 10.1212/CPJ.0000000000001075
322. Schott Andersen AS, Maarbjerg S, Noory N, Heinskou TB, Forman JL, Cruccu G, et al. Safety and efficacy of erenumab in patients with trigeminal neuralgia in Denmark: a double-blind, randomised, placebo-controlled, proof-of-concept study. Lancet Neurol. (2022) 21(11):994–1003. doi: 10.1016/S1474-4422(22)00294-0
323. Paige C, Plasencia-Fernandez I, Kume M, Papalampropoulou-Tsiridou M, Lorenzo LE, David ET, et al. A female-specific role for calcitonin gene-related peptide (CGRP) in rodent pain models. J Neurosci. (2022) 42(10):1930–44. doi: 10.1523/JNEUROSCI.1137-21.2022
324. Ahlstrom FHG, Matlik K, Viisanen H, Blomqvist KJ, Liu X, Lilius TO, et al. Spared nerve injury causes sexually dimorphic mechanical allodynia and differential gene expression in spinal cords and dorsal root ganglia in rats. Mol Neurobiol. (2021) 58(10):5396–419. doi: 10.1007/s12035-021-02447-1
325. Chiu IM, von Hehn CA, Woolf CJ. Neurogenic inflammation and the peripheral nervous system in host defense and immunopathology. Nat Neurosci. (2012) 15(8):1063–7. doi: 10.1038/nn.3144
326. Talbot S, Foster SL, Woolf CJ. Neuroimmunity: physiology and pathology. Annu Rev Immunol. (2016) 34:421–47. doi: 10.1146/annurev-immunol-041015-055340
327. Tracey KJ. Reflex control of immunity. Nat Rev Immunol. (2009) 9(6):418–28. doi: 10.1038/nri2566
328. Chavan SS, Pavlov VA, Tracey KJ. Mechanisms and therapeutic relevance of neuro-immune communication. Immunity. (2017) 46(6):927–42. doi: 10.1016/j.immuni.2017.06.008
329. Xanthos DN, Sandkuhler J. Neurogenic neuroinflammation: inflammatory CNS reactions in response to neuronal activity. Nat Rev Neurosci. (2014) 15(1):43–53. doi: 10.1038/nrn3617
330. An Q, Sun C, Li R, Chen S, Gu X, An S, et al. Calcitonin gene-related peptide regulates spinal microglial activation through the histone H3 lysine 27 trimethylation via enhancer of zeste homolog-2 in rats with neuropathic pain. J Neuroinflammation. (2021) 18(1):117. doi: 10.1186/s12974-021-02168-1
331. Shi X, Wang L, Li X, Sahbaie P, Kingery WS, Clark JD. Neuropeptides contribute to peripheral nociceptive sensitization by regulating interleukin-1beta production in keratinocytes. Anesth Analg. (2011) 113(1):175–83. doi: 10.1213/ANE.0b013e31821a0258
332. Kremer M, Yalcin I, Goumon Y, Wurtz X, Nexon L, Daniel D, et al. A dual noradrenergic mechanism for the relief of neuropathic allodynia by the antidepressant drugs duloxetine and amitriptyline. J Neurosci. (2018) 38(46):9934–54. doi: 10.1523/JNEUROSCI.1004-18.2018
333. Damo E, Agarwal A, Simonetti M. Activation of beta2-adrenergic receptors in microglia alleviates neuropathic hypersensitivity in mice. Cells. (2023) 12(2):284. doi: 10.3390/cells12020284
334. Bailey AL, Ribeiro-da-Silva A. Transient loss of terminals from non-peptidergic nociceptive fibers in the substantia gelatinosa of spinal cord following chronic constriction injury of the sciatic nerve. Neuroscience. (2006) 138(2):675–90. doi: 10.1016/j.neuroscience.2005.11.051
335. Yousefpour N, Locke S, Deamond H, Wang C, Marques L, St-Louis M, et al. Time-dependent and selective microglia-mediated removal of spinal synapses in neuropathic pain. Cell Rep. (2023) 42(1):112010. doi: 10.1016/j.celrep.2023.112010
336. Cheng CF, Cheng JK, Chen CY, Rau RH, Chang YC, Tsaur ML. Nerve growth factor-induced synapse-like structures in contralateral sensory ganglia contribute to chronic mirror-image pain. Pain. (2015) 156(11):2295–309. doi: 10.1097/j.pain.0000000000000280
337. Smithson LJ, Krol KM, Kawaja MD. Neuronal degeneration associated with sympathosensory plexuses in the trigeminal ganglia of aged mice that overexpress nerve growth factor. Neurobiol Aging. (2014) 35(12):2812–21. doi: 10.1016/j.neurobiolaging.2014.06.014
338. Xie W, Strong JA, Li H, Zhang JM. Sympathetic sprouting near sensory neurons after nerve injury occurs preferentially on spontaneously active cells and is reduced by early nerve block. J Neurophysiol. (2007) 97(1):492–502. doi: 10.1152/jn.00899.2006
339. Yen LD, Bennett GJ, Ribeiro-da-Silva A. Sympathetic sprouting and changes in nociceptive sensory innervation in the glabrous skin of the rat hind paw following partial peripheral nerve injury. J Comp Neurol. (2006) 495(6):679–90. doi: 10.1002/cne.20899
340. Ramer MS, Bisby MA. Sympathetic axons surround neuropeptide-negative axotomized sensory neurons. Neuroreport. (1998) 9(13):3109–13. doi: 10.1097/00001756-199809140-00035
341. Devor M, Janig W, Michaelis M. Modulation of activity in dorsal root ganglion neurons by sympathetic activation in nerve-injured rats. J Neurophysiol. (1994) 71:38–47. doi: 10.1152/jn.1994.71.1.38
342. Pertin M, Allchorne AJ, Beggah AT, Woolf CJ, Decosterd I. Delayed sympathetic dependence in the spared nerve injury (SNI) model of neuropathic pain. Mol Pain. (2007) 3:21. doi: 10.1186/1744-8069-3-21
343. Rho RH, Brewer RP, Lamer TJ, Wilson PR. Complex regional pain syndrome. Mayo Clin Proc. (2002) 77(2):174–80. doi: 10.1016/S0025-6196(11)62332-X
344. Ji RR, Xu ZZ, Strichartz G, Serhan CN. Emerging roles of resolvins in the resolution of inflammation and pain. Trends Neurosci. (2011) 34(11):599–609. doi: 10.1016/j.tins.2011.08.005
345. Ji RR. Specialized pro-resolving mediators as resolution pharmacology for the control of pain and itch. Annu Rev Pharmacol Toxicol. (2022) 63:273–93. doi: 10.1146/annurev-pharmtox-051921-084047
346. Buckley CD, Gilroy DW, Serhan CN. Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity. (2014) 40(3):315–27. doi: 10.1016/j.immuni.2014.02.009
347. Buckley CD, Gilroy DW, Serhan CN, Stockinger B, Tak PP. The resolution of inflammation. Nat Rev Immunol. (2013) 13(1):59–66. doi: 10.1038/nri3362
348. Sankaranarayanan I, Tavares-Ferreira D, Mwirigi JM, Mejia GL, Burton MD, Price TJ. Inducible co-stimulatory molecule (ICOS) alleviates paclitaxel-induced neuropathic pain via an IL-10-mediated mechanism in female mice. J Neuroinflammation. (2023) 20(1):32. doi: 10.1186/s12974-023-02719-8
349. Flatters SJ, Fox AJ, Dickenson AH. Nerve injury alters the effects of interleukin-6 on nociceptive transmission in peripheral afferents. Eur J Pharmacol. (2004) 484(2–3):183–91. doi: 10.1016/j.ejphar.2003.11.013
350. Flatters SJ, Fox AJ, Dickenson AH. Spinal interleukin-6 (IL-6) inhibits nociceptive transmission following neuropathy. Brain Res. (2003) 984(1–2):54–62. doi: 10.1016/S0006-8993(03)03092-0
351. Chalaki M, Cruz LJ, van Neerven SGA, Verhaagen J, Dahan A, Malessy MJA. Molecular changes in the dorsal root ganglion during the late phase of peripheral nerve injury-induced pain in rodents: a systematic review. Anesthesiology. (2022) 136(2):362–88. doi: 10.1097/ALN.0000000000004092
352. Ray PR, Shiers S, Caruso JP, Tavares-Ferreira D, Sankaranarayanan I, Uhelski ML, et al. RNA profiling of human dorsal root ganglia reveals sex-differences in mechanisms promoting neuropathic pain. Brain. (2023) 146(2):749–66. doi: 10.1093/brain/awac266
353. Yu X, Basbaum A, Guan Z. Contribution of colony-stimulating factor 1 to neuropathic pain. Pain Rep. (2021) 6(1):e883. doi: 10.1097/PR9.0000000000000883
354. Kleinschnitz C, Brinkhoff J, Zelenka M, Sommer C, Stoll G. The extent of cytokine induction in peripheral nerve lesions depends on the mode of injury and NMDA receptor signaling. J Neuroimmunol. (2004) 149(1–2):77–83. doi: 10.1016/j.jneuroim.2003.12.013
355. Chun S, Kwon YB. The CCL2 elevation in primary afferent fibers produces zymosan-induced hyperalgesia through microglia-mediated neuronal activation in the spinal dorsal horn. Brain Res Bull. (2019) 149:53–9. doi: 10.1016/j.brainresbull.2019.04.014
356. Hamed EA, Mohamed Farghaly HS, Abdel Mola AF, Fahmi MK, Makhlouf MM, Balfas MA. Role of monocyte chemoattractant protein-1, stromal derived factor-1 and retinoic acid in pathophysiology of neuropathic pain in rats. J Basic Clin Physiol Pharmacol. (2016) 27(4):411–24. doi: 10.1515/jbcpp-2015-0105
357. Sacerdote P, Franchi S, Trovato AE, Valsecchi AE, Panerai AE, Colleoni M. Transient early expression of TNF-alpha in sciatic nerve and dorsal root ganglia in a mouse model of painful peripheral neuropathy. Neurosci Lett. (2008) 436(2):210–3. doi: 10.1016/j.neulet.2008.03.023
358. Lee HL, Lee KM, Son SJ, Hwang SH, Cho HJ. Temporal expression of cytokines and their receptors mRNAs in a neuropathic pain model. Neuroreport. (2004) 15(18):2807–11.15597059
359. Woolf CJ. Evidence for a central component of post-injury pain hypersensitivity. Nature. (1983) 306(5944):686–8.6656869
360. Halievski K, Ghazisaeidi S, Salter MW. Sex-Dependent mechanisms of chronic pain: a focus on microglia and P2X4R. J Pharmacol Exp Ther. (2020) 375(1):202–9. doi: 10.1124/jpet.120.265017
361. Mapplebeck JC, Beggs S, Salter MW. Molecules in pain and sex: a developing story. Mol Brain. (2017) 10(1):9. doi: 10.1186/s13041-017-0289-8
362. Sorge RE, Mapplebeck JC, Rosen S, Beggs S, Taves S, Alexander JK, et al. Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat Neurosci. (2015) 18(8):1081–3. doi: 10.1038/nn.4053
363. Mapplebeck JCS, Lorenzo LE, Lee KY, Gauthier C, Muley MM, De KY, et al. Chloride dysregulation through downregulation of KCC2 mediates neuropathic pain in both sexes. Cell Rep. (2019) 28(3):590–6. doi: 10.1016/j.celrep.2019.06.059
364. Mapplebeck JCS, Dalgarno R, Tu Y, Moriarty O, Beggs S, Kwok CHT, et al. Microglial P2X4R-evoked pain hypersensitivity is sexually dimorphic in rats. Pain. (2018) 159(9):1752–63. doi: 10.1097/j.pain.0000000000001265
365. Mogil JS. Sex differences in pain and pain inhibition: multiple explanations of a controversial phenomenon. Nat Rev Neurosci. (2012) 13(12):859–66. doi: 10.1038/nrn3360
366. Mogil JS. Qualitative sex differences in pain processing: emerging evidence of a biased literature. Nat Rev Neurosci. (2020) 21(7):353–65. doi: 10.1038/s41583-020-0310-6
367. Mifflin KA, Frieser E, Benson C, Baker G, Kerr BJ. Voluntary wheel running differentially affects disease outcomes in male and female mice with experimental autoimmune encephalomyelitis. J Neuroimmunol. (2017) 305:135–44. doi: 10.1016/j.jneuroim.2017.02.005
368. Luo X, Chen O, Wang Z, Bang S, Ji J, Lee SH, et al. IL-23/IL-17A/TRPV1 axis produces mechanical pain via macrophage-sensory neuron crosstalk in female mice. Neuron. (2021) 109:2691–706. doi: 10.1016/j.neuron.2021.06.015
369. Paige C, Barba-Escobedo PA, Mecklenburg J, Patil M, Goffin V, Grattan DR, et al. Neuroendocrine mechanisms governing sex differences in hyperalgesic priming involve prolactin receptor sensory neuron signaling. J Neurosci. (2020) 40(37):7080–90. doi: 10.1523/JNEUROSCI.1499-20.2020
370. Lim H, Lee H, Noh K, Lee SJ. IKK/NF-kappaB-dependent satellite glia activation induces spinal cord microglia activation and neuropathic pain after nerve injury. Pain. (2017) 158(9):1666–77. doi: 10.1097/j.pain.0000000000000959
371. Gushchina S, Pryce G, Yip PK, Wu D, Pallier P, Giovannoni G, et al. Increased expression of colony-stimulating factor-1 in mouse spinal cord with experimental autoimmune encephalomyelitis correlates with microglial activation and neuronal loss. Glia. (2018) 66(10):2108–25. doi: 10.1002/glia.23464
372. Lee J, Hwang H, Lee SJ. Distinct roles of GT1b and CSF-1 in microglia activation in nerve injury-induced neuropathic pain. Mol Pain. (2021) 17:17448069211020918. doi: 10.1177/17448069211020918
373. Sun C, Tao X, Wan C, Zhang X, Zhao M, Xu M, et al. Spinal cord stimulation alleviates neuropathic pain by attenuating microglial activation via reducing colony-stimulating factor 1 levels in the spinal cord in a rat model of chronic constriction injury. Anesth Analg. (2022) 135(1):178–90. doi: 10.1213/ANE.0000000000006016
374. Kuhn JA, Vainchtein ID, Braz J, Hamel K, Bernstein M, Craik V, et al. Regulatory T-cells inhibit microglia-induced pain hypersensitivity in female mice. Elife. (2021) 10:e69056. doi: 10.7554/eLife.69056
375. Beggs S, Trang T, Salter MW. P2x4r+ microglia drive neuropathic pain. Nat Neurosci. (2012) 15(8):1068–73. doi: 10.1038/nn.3155
376. Tam TH, Salter MW. Purinergic signalling in spinal pain processing. Purinergic Signal. (2021) 17(1):49–54. doi: 10.1007/s11302-020-09748-5
377. Trang T, Beggs S, Salter MW. ATP receptors gate microglia signaling in neuropathic pain. Exp Neurol. (2012) 234(2):354–61. doi: 10.1016/j.expneurol.2011.11.012
378. Trang T, Salter MW. P2x4 purinoceptor signaling in chronic pain. Purinergic Signal. (2012) 8(3):621–8. doi: 10.1007/s11302-012-9306-7
379. Tsuda M, Shigemoto-Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter MW, et al. P2x(4) receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature. (2003) 424:778–83. doi: 10.1038/nature01786
380. Malcangio M. Spinal mechanisms of neuropathic pain: is there a P2X4-BDNF controversy? Neurobiol Pain. (2017) 1:1–5. doi: 10.1016/j.ynpai.2017.04.001
381. Sorge RE, Totsch SK. Sex differences in pain. J Neurosci Res. (2016) 95:1271–81. doi: 10.1002/jnr.23841
382. Luo X, Tai WL, Sun L, Pan Z, Xia Z, Chung SK, et al. Crosstalk between astrocytic CXCL12 and microglial CXCR4 contributes to the development of neuropathic pain. Mol Pain. (2016) 12:1744806916636385. doi: 10.1177/1744806916636385
383. Old EA, Malcangio M. Chemokine mediated neuron-glia communication and aberrant signalling in neuropathic pain states. Curr Opin Pharmacol. (2012) 12(1):67–73. doi: 10.1016/j.coph.2011.10.015
384. Liu ZY, Song ZW, Guo SW, He JS, Wang SY, Zhu JG, et al. CXCL12/CXCR4 signaling contributes to neuropathic pain via central sensitization mechanisms in a rat spinal nerve ligation model. CNS Neurosci Ther. (2019) 25(9):922–36. doi: 10.1111/cns.13128
385. Dong J, Xu C, Xia R, Zhang Z. Upregulating miR-130a-5p relieves astrocyte over activation-induced neuropathic pain through targeting C-X-C motif chemokine receptor 12/C-X-C motif chemokine receptor 4 axis. Neuroreport. (2021) 32(2):135–43. doi: 10.1097/WNR.0000000000001573
386. Yang F, Sun W, Luo WJ, Yang Y, Yang F, Wang XL, et al. SDF1-CXCR4 signaling contributes to the transition from acute to chronic pain state. Mol Neurobiol. (2017) 54(4):2763–75. doi: 10.1007/s12035-016-9875-5
387. Hendrich J, Alvarez P, Joseph EK, Chen X, Bogen O, Levine JD. Electrophysiological correlates of hyperalgesic priming in vitro and in vivo. Pain. (2013) 154(10):2207–15. doi: 10.1016/j.pain.2013.07.004
388. Ferrari LF, Bogen O, Levine JD. Nociceptor subpopulations involved in hyperalgesic priming. Neuroscienc. (2010) 165(3):896–901. doi: 10.1016/j.neuroscience.2009.11.029
389. Honjoh K, Nakajima H, Hirai T, Watanabe S, Matsumine A. Relationship of inflammatory cytokines from M1-type microglia/macrophages at the injured site and lumbar enlargement with neuropathic pain after spinal cord injury in the CCL21 knockout (plt) mouse. Front Cell Neurosci. (2019) 13:525. doi: 10.3389/fncel.2019.00525
390. de Jong EK, Vinet J, Stanulovic VS, Meijer M, Wesseling E, Sjollema K, et al. Expression, transport, and axonal sorting of neuronal CCL21 in large dense-core vesicles. FASEB J. (2008) 22(12):4136–45. doi: 10.1096/fj.07-101907
391. de Jong EK, Dijkstra IM, Hensens M, Brouwer N, van Amerongen M, Liem RS, et al. Vesicle-mediated transport and release of CCL21 in endangered neurons: a possible explanation for microglia activation remote from a primary lesion. J Neurosci. (2005) 25(33):7548–57. doi: 10.1523/JNEUROSCI.1019-05.2005
392. van Weering HR, de Jong AP, de Haas AH, Biber KP, Boddeke HW. CCL21-induced Calcium transients and proliferation in primary mouse astrocytes: cXCR3-dependent and independent responses. Brain Behav Immun. (2010) 24(5):768–75. doi: 10.1016/j.bbi.2009.04.007
393. Andratsch M, Mair N, Constantin CE, Scherbakov N, Benetti C, Quarta S, et al. A key role for gp130 expressed on peripheral sensory nerves in pathological pain. J Neurosci. (2009) 29(43):13473–83. doi: 10.1523/JNEUROSCI.1822-09.2009
394. Recasens M, Almolda B, Perez-Clausell J, Campbell IL, Gonzalez B, Castellano B. Chronic exposure to IL-6 induces a desensitized phenotype of the microglia. J Neuroinflammation. (2021) 18(1):31. doi: 10.1186/s12974-020-02063-1
395. Tsuda M, Masuda T, Kitano J, Shimoyama H, Tozaki-Saitoh H, Inoue K. IFN-gamma receptor signaling mediates spinal microglia activation driving neuropathic pain. Proc Natl Acad Sci U S A. (2009) 106(19):8032–7. doi: 10.1073/pnas.0810420106
396. Vikman KS, Siddall PJ, Duggan AW. Increased responsiveness of rat dorsal horn neurons in vivo following prolonged intrathecal exposure to interferon-[gamma]. Neuroscience. (2005) 135(3):969–77. doi: 10.1016/j.neuroscience.2005.06.059
397. Vikman KS, Hill RH, Backstrom E, Robertson B, Kristensson K. Interferon-gamma induces characteristics of central sensitization in spinal dorsal horn neurons in vitro. Pain. (2003) 106(3):241–51. doi: 10.1016/S0304-3959(03)00262-8
398. Neumann H, Schmidt H, Wilharm E, Behrens L, Wekerle H. Interferon gamma gene expression in sensory neurons: evidence for autocrine gene regulation. J Exp Med. (1997) 186(12):2023–31. doi: 10.1084/jem.186.12.2023
399. Costigan M, Moss A, Latremoliere A, Johnston C, Verma-Gandhu M, Herbert TA, et al. T-cell infiltration and signaling in the adult dorsal spinal cord is a major contributor to neuropathic pain-like hypersensitivity. J Neurosci. (2009) 29(46):14415–22. doi: 10.1523/JNEUROSCI.4569-09.2009
400. Clark AK, Wodarski R, Guida F, Sasso O, Malcangio M. Cathepsin S release from primary cultured microglia is regulated by the P2X7 receptor. Glia. (2010) 58(14):1710–26. doi: 10.1002/glia.21042
401. Clark AK, Yip PK, Malcangio M. The liberation of fractalkine in the dorsal horn requires microglial cathepsin S. J Neurosci. (2009) 29(21):6945–54. doi: 10.1523/JNEUROSCI.0828-09.2009
402. Verge GM, Milligan ED, Maier SF, Watkins LR, Naeve GS, Foster AC. Fractalkine (CX3CL1) and fractalkine receptor (CX3CR1) distribution in spinal cord and dorsal root ganglia under basal and neuropathic pain conditions. Eur J Neurosci. (2004) 20(5):1150–60. doi: 10.1111/j.1460-9568.2004.03593.x
403. Lindia JA, McGowan E, Jochnowitz N, Abbadie C. Induction of CX3CL1 expression in astrocytes and CX3CR1 in microglia in the spinal cord of a rat model of neuropathic pain. J Pain. (2005) 6(7):434–8. doi: 10.1016/j.jpain.2005.02.001
404. Milligan ED, Zapata V, Chacur M, Schoeniger D, Biedenkapp J, O’connor KA, et al. Evidence that exogenous and endogenous fractalkine can induce spinal nociceptive facilitation in rats. Eur J Neurosci. (2004) 20(9):2294–302. doi: 10.1111/j.1460-9568.2004.03709.x
405. Staniland AA, Clark AK, Wodarski R, Sasso O, Maione F, D’Acquisto F, et al. Reduced inflammatory and neuropathic pain and decreased spinal microglial response in fractalkine receptor (CX3CR1) knockout mice. J Neurochem. (2010) 114(4):1143–57. doi: 10.1111/j.1471-4159.2010.06837.x
406. Clark AK, Yip PK, Grist J, Gentry C, Staniland AA, Marchand F, et al. Inhibition of spinal microglial cathepsin S for the reversal of neuropathic pain. Proc Natl Acad Sci U S A. (2007) 104(25):10655–60. doi: 10.1073/pnas.0610811104
407. Clark AK, Gruber-Schoffnegger D, Drdla-Schutting R, Gerhold KJ, Malcangio M, Sandkuhler J. Selective activation of microglia facilitates synaptic strength. J Neurosci. (2015) 35(11):4552–70. doi: 10.1523/JNEUROSCI.2061-14.2015
408. Biber K, Laurie DJ, Berthele A, Sommer B, Tolle TR, Gebicke-Harter PJ, et al. Expression and signaling of group I metabotropic glutamate receptors in astrocytes and microglia. J Neurochem. (1999) 72(4):1671–80. doi: 10.1046/j.1471-4159.1999.721671.x
409. Byrnes KR, Loane DJ, Faden AI. Metabotropic glutamate receptors as targets for multipotential treatment of neurological disorders. Neurotherapeutics. (2009) 6(1):94–107. doi: 10.1016/j.nurt.2008.10.038
410. Byrnes KR, Stoica B, Loane DJ, Riccio A, Davis MI, Faden AI. Metabotropic glutamate receptor 5 activation inhibits microglial associated inflammation and neurotoxicity. Glia. (2009) 57(5):550–60. doi: 10.1002/glia.20783
411. Byrnes KR, Stoica B, Riccio A, Pajoohesh-Ganji A, Loane DJ, Faden AI. Activation of metabotropic glutamate receptor 5 improves recovery after spinal cord injury in rodents. Ann Neurol. (2009) 66(1):63–74. doi: 10.1002/ana.21673
412. Devaraju P, Sun MY, Myers TL, Lauderdale K, Fiacco TA. Astrocytic group I mGluR-dependent potentiation of astrocytic glutamate and potassium uptake. J Neurophysiol. (2013) 109(9):2404–14. doi: 10.1152/jn.00517.2012
413. Yang G, Tan Q, Li Z, Liu K, Wu J, Ye W, et al. The AMPK pathway triggers autophagy during CSF1-induced microglial activation and may be implicated in inducing neuropathic pain. J Neuroimmunol. (2020) 345:577261. doi: 10.1016/j.jneuroim.2020.577261
414. Lu VB, Ballanyi K, Colmers WF, Smith PA. Neuron type-specific effects of brain-derived neurotrophic factor in rat superficial dorsal horn and their relevance to “central sensitization”. J Physiol. (2007) 584:543–63. doi: 10.1113/jphysiol.2007.141267
415. Ha SO, Kim JK, Hong HS, Kim DS, Cho HJ. Expression of brain-derived neurotrophic factor in rat dorsal root ganglia, spinal cord and gracile nuclei in experimental models of neuropathic pain. Neuroscience. (2001) 107(2):301–9. doi: 10.1016/S0306-4522(01)00353-0
416. Miletic G, Miletic V. Increases in the concentration of brain derived neurotrophic factor in the lumbar spinal dorsal horn are associated with pain behavior following chronic constriction injury in rats. Neurosci Lett. (2002) 319(3):137–40. doi: 10.1016/S0304-3940(01)02576-9
417. Garraway SM, Petruska JC, Mendell LM. BDNF sensitizes the response of lamina II neurons to high threshold primary afferent inputs. Eur J Neurosci. (2003) 18(9):2467–76. doi: 10.1046/j.1460-9568.2003.02982.x
418. Bardoni R, Ghirri A, Salio C, Prandini M, Merighi A. BDNF-mediated modulation of GABA and glycine release in dorsal horn lamina II from postnatal rats. Dev Neurobiol. (2007) 67(7):960–75. doi: 10.1002/dneu.20401
419. Ferrini F, De Koninck Y. Microglia control neuronal network excitability via BDNF signalling. Neural Plast. (2013) 2013:429815. doi: 10.1155/2013/429815
420. Zhang W, Shi Y, Peng Y, Zhong L, Zhu S, Zhang W, et al. Neuron activity-induced wnt signaling up-regulates expression of brain-derived neurotrophic factor in the pain neural circuit. J Biol Chem. (2018) 293(40):15641–51. doi: 10.1074/jbc.RA118.002840
421. Biggs JE, Boakye PA, Ganesan N, Stemkowski PL, Lantero A, Ballanyi K, et al. Analysis of the long-term actions of gabapentin and pregabalin in dorsal root ganglia and substantia gelatinosa. J Neurophysiol. (2014) 112(10):2398–412. doi: 10.1152/jn.00168.2014
422. Huang D, Yang J, Liu X, He L, Luo X, Tian H, et al. P2y6 receptor activation is involved in the development of neuropathic pain induced by chronic constriction injury of the sciatic nerve in rats. J Clin Neurosci. (2018) 56:156–62. doi: 10.1016/j.jocn.2018.07.013
423. Zhang X, Li G. P2y receptors in neuropathic pain. Pharmacol Biochem Behav. (2019) 186:172788. doi: 10.1016/j.pbb.2019.172788
424. Inoue K, Koizumi S, Tsuda M. The role of nucleotides in the neuron–glia communication responsible for the brain functions. J Neurochem. (2007) 102(5):1447–58. doi: 10.1111/j.1471-4159.2007.04824.x
425. Barragan-Iglesias P, Pineda-Farias JB, Cervantes-Duran C, Bravo-Hernandez M, Rocha-Gonzalez HI, Murbartian J, et al. Role of spinal P2Y6 and P2Y11 receptors in neuropathic pain in rats: possible involvement of glial cells. Mol Pain. (2014) 10:29. doi: 10.1186/1744-8069-10-29
426. Kobayashi K, Yamanaka H, Fukuoka T, Dai Y, Obata K, Noguchi K. P2y12 receptor upregulation in activated microglia is a gateway of p38 signaling and neuropathic pain. J Neurosci. (2008) 28(11):2892–902. doi: 10.1523/JNEUROSCI.5589-07.2008
427. Kobayashi K, Yamanaka H, Yanamoto F, Okubo M, Noguchi K. Multiple P2Y subtypes in spinal microglia are involved in neuropathic pain after peripheral nerve injury. Glia. (2012) 60(10):1529–39. doi: 10.1002/glia.22373
428. Masuda T, Ozono Y, Mikuriya S, Kohro Y, Tozaki-Saitoh H, Iwatsuki K, et al. Dorsal horn neurons release extracellular ATP in a VNUT-dependent manner that underlies neuropathic pain. Nat Commun. (2016) 7:12529. doi: 10.1038/ncomms12529
429. Chu J, Yang J, Zhou Y, Chen J, Chen KH, Zhang C, et al. ATP-releasing SWELL1 channel in spinal microglia contributes to neuropathic pain. Sci Adv. (2023) 9(13):eade9931. doi: 10.1126/sciadv.ade9931
430. Shi Y, Shu J, Gelman BB, Lisinicchia JG, Tang SJ. Wnt signaling in the pathogenesis of human HIV-associated pain syndromes. J Neuroimmune Pharmacol. (2013) 8(4):956–64. doi: 10.1007/s11481-013-9474-4
431. Zhang YK, Huang ZJ, Liu S, Liu YP, Song AA, Song XJ. WNT signaling underlies the pathogenesis of neuropathic pain in rodents. J Clin Invest. (2013) 123(5):2268–86. doi: 10.1172/JCI65364
432. Wang HY, Liu T, Malbon CC. Structure-function analysis of frizzleds. Cell Signal. (2006) 18(7):934–41. doi: 10.1016/j.cellsig.2005.12.008
433. Huang J, Bloe CB, Zhou X, Wu S, Zhang W. The role of the spinal wnt signaling pathway in HIV-related neuropathic pain. Cell Mol Neurobiol. (2020) 40(7):1075–85. doi: 10.1007/s10571-020-00805-6
434. Zhou X, Tao L, Zhao M, Wu S, Obeng E, Wang D, et al. Wnt/beta-catenin signaling regulates brain-derived neurotrophic factor release from spinal microglia to mediate HIV1 gp120-induced neuropathic pain. Mol Pain. (2020) 16:1744806920922100. doi: 10.1177/1744806920922100
435. Li Y, Li B, Wan X, Zhang W, Zhong L, Tang SJ. NMDA Receptor activation stimulates transcription-independent rapid wnt5a protein synthesis via the MAPK signaling pathway. Mol Brain. (2012) 5:1. doi: 10.1186/1756-6606-5-1
436. Chen J, Park CS, Tang SJ. Activity-dependent synaptic wnt release regulates hippocampal long term potentiation. J Biol Chem. (2006) 281(17):11910–6. doi: 10.1074/jbc.M511920200
437. Echeverry S, Shi XQ, Yang M, Huang H, Wu Y, Lorenzo LE, et al. Spinal microglia are required for long-term maintenance of neuropathic pain. Pain. (2017) 158(9):1792–801. doi: 10.1097/j.pain.0000000000000982
438. Croll SD, Chesnutt CR, Rudge JS, Acheson A, Ryan TE, Siuciak JA, et al. Co-infusion with a TrkB-fc receptor body carrier enhances BDNF distribution in the adult rat brain. Exp Neurol. (1998) 152(1):20–33. doi: 10.1006/exnr.1998.6836
439. Clark AK, D’Aquisto F, Gentry C, Marchand F, McMahon SB, Malcangio M. Rapid co-release of interleukin 1beta and caspase 1 in spinal cord inflammation. J Neurochem. (2006) 99(3):868–80. doi: 10.1111/j.1471-4159.2006.04126.x
440. Gajtko A, Bakk E, Hegedus K, Ducza L, Hollo K. IL-1beta induced cytokine expression by spinal astrocytes can play a role in the maintenance of chronic inflammatory pain. Front Physiol. (2020) 11:543331. doi: 10.3389/fphys.2020.543331
441. Clark AK, Staniland AA, Marchand F, Kaan TKY, McMahon SB, Malcangio M. P2X7-dependent release of interleukin-1{beta} and nociception in the spinal cord following lipopolysaccharide. J Neurosci. (2010) 30(2):573–82. doi: 10.1523/JNEUROSCI.3295-09.2010
442. Kohno K, Tsuda M. Role of microglia and P2X4 receptors in chronic pain. Pain Rep. (2021) 6(1):e864. doi: 10.1097/PR9.0000000000000864
443. Simeoli R, Montague K, Jones HR, Castaldi L, Chambers D, Kelleher JH, et al. Exosomal cargo including microRNA regulates sensory neuron to macrophage communication after nerve trauma. Nat Commun. (2017) 8(1):1778. doi: 10.1038/s41467-017-01841-5
444. D’Agnelli S, Gerra MC, Bignami E, Arendt-Nielsen L. Exosomes as a new pain biomarker opportunity. Mol Pain. (2020) 16:1744806920957800. doi: 10.1177/1744806920957800
445. Hori N, Narita M, Yamashita A, Horiuchi H, Hamada Y, Kondo T, et al. Changes in the expression of IL-6-mediated MicroRNAs in the dorsal root ganglion under neuropathic pain in mice. Synapse. (2016) 70(8):317–24. doi: 10.1002/syn.21902
446. McDonald MK, Tian Y, Qureshi RA, Gormley M, Ertel A, Gao R, et al. Functional significance of macrophage-derived exosomes in inflammation and pain. Pain. (2014) 155(8):1527–39. doi: 10.1016/j.pain.2014.04.029
447. Yu X, Abdul M, Fan BQ, Zhang L, Lin X, Wu Y, et al. The release of exosomes in the medial prefrontal cortex and nucleus accumbens brain regions of chronic constriction injury (CCI) model mice could elevate the pain sensation. Neurosci Lett. (2020) 723:134774. doi: 10.1016/j.neulet.2020.134774
448. Morteza BH, Ahmadi S, Tarighat F, Rahbarghazi R, Soleimanpour H. Interplay between exosomes and autophagy machinery in pain management: state of the art. Neurobiol Pain. (2022) 12:100095. doi: 10.1016/j.ynpai.2022.100095
449. Alles SRA, Nascimento F, Lujan R, Luiz AP, Millet Q, Bangash MA, et al. Sensory neuron-derived na(V)1.7 contributes to dorsal horn neuron excitability. Sci Adv. (2020) 6(8):eaax4568.32128393
450. Paolicelli RC, Bergamini G, Rajendran L. Cell-to-cell communication by extracellular vesicles: focus on microglia. Neuroscience. (2019) 405:148–57. doi: 10.1016/j.neuroscience.2018.04.003
451. Gosselin RD, Meylan P, Decosterd I. Extracellular microvesicles from astrocytes contain functional glutamate transporters: regulation by protein kinase C and cell activation. Front Cell Neurosci. (2013) 7:251. doi: 10.3389/fncel.2013.00009
452. Zhang YU, Ye G, Zhao J, Chen Y, Kong L, Sheng C, et al. Exosomes carried miR-181c-5p alleviates neuropathic pain in CCI rat models. An Acad Bras Cienc. (2022) 94(3):e20210564. doi: 10.1590/0001-3765202220210564
453. Zhang X, Wang J, Zhou Q, Xu Y, Pu S, Wu J, et al. Brain-derived neurotrophic factor-activated astrocytes produce mechanical allodynia in neuropathic pain. Neuroscience. (2011) 199:452–60. doi: 10.1016/j.neuroscience.2011.10.017
454. Balasubramanyan S, Stemkowski PL, Stebbing MJ, Smith PA. Sciatic chronic constriction injury produces cell-type specific changes in the electrophysiological properties of rat Substantia Gelatinosa neurons. J Neurophysiol. (2006) 96:579–90. doi: 10.1152/jn.00087.2006
455. Chen Y, Balasubramanyan S, Lai AY, Todd KG, Smith P.A. Effects of sciatic nerve axotomy on excitatory synaptic transmission in rat substantia Gelatinosa. J Neurophysiol. (2009) 102:3203–15. doi: 10.1152/jn.00296.2009
456. Leitner J, Westerholz S, Heinke B, Forsthuber L, Wunderbaldinger G, Jager T, et al. Impaired excitatory drive to spinal GABAergic neurons of neuropathic mice. PLoS One. (2013) 8(8):e73370. doi: 10.1371/journal.pone.0073370
457. Bae C, Wang J, Shim HS, Tang SJ, Chung JM, La JH. Mitochondrial superoxide increases excitatory synaptic strength in spinal dorsal horn neurons of neuropathic mice. Mol Pain. (2018) 14:1744806918797032. doi: 10.1177/1744806918797032
458. Coull JA, Boudreau D, Bachand K, Prescott SA, Nault F, Sik A, et al. Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature. (2003) 424(6951):938–42. doi: 10.1038/nature01868
459. Ferrini F, Perez-Sanchez J, Ferland S, Lorenzo LE, Godin AG, Plasencia-Fernandez I, et al. Differential chloride homeostasis in the spinal dorsal horn locally shapes synaptic metaplasticity and modality-specific sensitization. Nat Commun. (2020) 11(1):3935. doi: 10.1038/s41467-020-17824-y
460. Prescott SA, Sejnowski TJ, de Koninck Y. Reduction of anion reversal potential subverts the inhibitory control of firing rate in spinal lamina I neurons: towards a biophysical basis for neuropathic pain. Mol Pain. (2006) 2:32. doi: 10.1186/1744-8069-2-32
461. Lavertu G, Côté S, De Koninck Y. Enhancing K–cl co-transport restores normal spinothalamic sensory coding in a neuropathic pain model. Brain. (2014) 137(3):724–38. doi: 10.1093/brain/awt334
462. Yeo M, Chen Y, Jiang C, Chen G, Wang K, Chandra S, et al. Repurposing cancer drugs identifies kenpaullone which ameliorates pathologic pain in preclinical models via normalization of inhibitory neurotransmission. Nat Commun. (2021) 12(1):6208. doi: 10.1038/s41467-021-26270-3
463. Prescott SA, de Koninck Y, Sejnowski TJ. Biophysical basis for three distinct dynamical mechanisms of action potential initiation. PLoS Comput Biol. (2008) 4:e1000198. doi: 10.1371/journal.pcbi.1000198
464. Todd AJ. Neuronal circuitry for pain processing in the dorsal horn. Nat Rev Neurosci. (2010) 11(12):823–36. doi: 10.1038/nrn2947
465. Peirs C, Seal RP. Neural circuits for pain: recent advances and current views. Science. (2016) 354(6312):578–84. doi: 10.1126/science.aaf8933
466. Peirs C, Dallel R, Todd AJ. Recent advances in our understanding of the organization of dorsal horn neuron populations and their contribution to cutaneous mechanical allodynia. J Neural Transm (Vienna). (2020) 127(4):505–25. doi: 10.1007/s00702-020-02159-1
467. Baba H, Ji RR, Kohno T, Moore KA, Ataka T, Wakai A, et al. Removal of GABAergic inhibition facilitates polysynaptic A fiber-mediated excitatory transmission to the superficial spinal dorsal horn. Mol Cell Neurosci. (2003) 24(3):818–30. doi: 10.1016/S1044-7431(03)00236-7
468. Prescott SA, Ma Q, De KY. Normal and abnormal coding of somatosensory stimuli causing pain. Nat Neurosci. (2014) 17(2):183–91. doi: 10.1038/nn.3629
469. Price TJ, Prescott SA. Inhibitory regulation of the pain gate and how its failure causes pathological pain. Pain. (2015) 156(5):789–92. doi: 10.1097/j.pain.0000000000000139
470. Aby F, Lorenzo LE, Grivet Z, Bouali-Benazzouz R, Martin H, Valerio S, et al. Switch of serotonergic descending inhibition into facilitation by a spinal chloride imbalance in neuropathic pain. Sci Adv. (2022) 8(30):eabo0689. doi: 10.1126/sciadv.abo0689
471. Kerr BJ, Bradbury EJ, Bennett DL, Trivedi PM, Dassan P, French J, et al. Brain-Derived neurotrophic factor modulates nociceptive sensory inputs and NMDA-evoked responses in the rat spinal cord. J Neurosci. (1999) 19(12):5138–48. doi: 10.1523/JNEUROSCI.19-12-05138.1999
472. Hildebrand ME, Xu J, Dedek A, Li Y, Sengar AS, Beggs S, et al. Potentiation of synaptic GluN2B NMDAR currents by Fyn kinase is gated through BDNF-mediated disinhibition in spinal pain processing. Cell Rep. (2016) 17(10):2753–65. doi: 10.1016/j.celrep.2016.11.024
473. Dedek A, Xu J, Kandegedara CM, Lorenzo LE, Godin AG, De KY, et al. Loss of STEP61 couples disinhibition to N-methyl-d-aspartate receptor potentiation in rodent and human spinal pain processing. Brain. (2019) 142(6):1535–46. doi: 10.1093/brain/awz105
474. Lee KY, Prescott SA. Chloride dysregulation and inhibitory receptor blockade yield equivalent disinhibition of spinal neurons yet are differentially reversed by carbonic anhydrase blockade. Pain. (2015) 156(12):2431–7. doi: 10.1097/j.pain.0000000000000301
475. Dedek A, Xu J, Lorenzo LE, Godin AG, Kandegedara CM, Glavina G, et al. Sexual dimorphism in a neuronal mechanism of spinal hyperexcitability across rodent and human models of pathological pain. Brain. (2022) 145(3):1124–38. doi: 10.1093/brain/awab408
476. Chen W, Walwyn W, Ennes HS, Kim H, McRoberts JA, Marvizon JC. BDNF Released during neuropathic pain potentiates NMDA receptors in primary afferent terminals. Eur J Neurosci. (2014) 39(9):1439–54. doi: 10.1111/ejn.12516
477. Yan X, Jiang E, Gao M, Weng HR. Endogenous activation of presynaptic NMDA receptors enhances glutamate release from the primary afferents in the spinal dorsal horn in a rat model of neuropathic pain. J Physiol. (2013) 591(7):2001–19. doi: 10.1113/jphysiol.2012.250522
478. Ding X, Cai J, Li S, Liu XD, Wan Y, Xing GG. BDNF Contributes to the development of neuropathic pain by induction of spinal long-term potentiation via SHP2 associated GluN2B-containing NMDA receptors activation in rats with spinal nerve ligation. Neurobiol Dis. (2015) 73:428–51. doi: 10.1016/j.nbd.2014.10.025
479. Li S, Cai J, Feng ZB, Jin ZR, Liu BH, Zhao HY, et al. BDNF Contributes to spinal long-term potentiation and mechanical hypersensitivity via fyn-mediated phosphorylation of NMDA receptor GluN2B subunit at tyrosine 1472 in rats following spinal nerve ligation. Neurochem Res. (2017) 42(10):2712–29. doi: 10.1007/s11064-017-2274-0
480. Alexander GM, van Rijn MA, van Hilten JJ, Perreault MJ, Schwartzman RJ. Changes in cerebrospinal fluid levels of pro-inflammatory cytokines in CRPS. Pain. (2005) 116(3):213–9. doi: 10.1016/j.pain.2005.04.013
481. Pineau I, Lacroix S. Proinflammatory cytokine synthesis in the injured mouse spinal cord: multiphasic expression pattern and identification of the cell types involved. J Comp Neurol. (2007) 500(2):267–85. doi: 10.1002/cne.21149
482. Yan X, Li F, Maixner DW, Yadav R, Gao M, Ali MW, et al. Interleukin-1beta released by microglia initiates the enhanced glutamatergic activity in the spinal dorsal horn during paclitaxel-associated acute pain syndrome. Glia. (2019) 67(3):482–97. doi: 10.1002/glia.23557
483. Yan X, Weng HR. Endogenous interleukin-1beta in neuropathic rats enhances glutamate release from the primary afferents in the spinal dorsal horn through coupling with presynaptic N-methyl-D-aspartic acid receptors. J Biol Chem. (2013) 288(42):30544–57. doi: 10.1074/jbc.M113.495465
484. Weng HR, Chen JH, Cata JP. Inhibition of glutamate uptake in the spinal cord induces hyperalgesia and increased responses of spinal dorsal horn neurons to peripheral afferent stimulation. Neuroscience. (2006) 138(4):1351–60. doi: 10.1016/j.neuroscience.2005.11.061
485. Weng HR, Chen JH, Pan ZZ, Nie H. Glial glutamate transporter 1 regulates the spatial and temporal coding of glutamatergic synaptic transmission in spinal lamina II neurons. Neuroscience. (2007) 149(4):898–907. doi: 10.1016/j.neuroscience.2007.07.063
486. Miraucourt LS, Peirs C, Dallel R, Voisin DL. Glycine inhibitory dysfunction turns touch into pain through astrocyte-derived D-serine. Pain. (2011) 152(6):1340–8. doi: 10.1016/j.pain.2011.02.021
487. Liu T, Jiang CY, Fujita T, Luo SW, Kumamoto E. Enhancement by interleukin-1beta of AMPA and NMDA receptor-mediated currents in adult rat spinal superficial dorsal horn neurons. Mol Pain. (2013) 9:16. doi: 10.1186/1744-8069-9-16
488. Zhang H, Nei H, Dougherty PM. A p38 mitogen-activated protein kinase-dependent mechanism of disinhibition in spinal synaptic transmission induced by tumor necrosis factor-alpha. J Neurosci. (2010) 30(38):12844–55. doi: 10.1523/JNEUROSCI.2437-10.2010
489. Zhang H, Zhang H, Dougherty PM. Dynamic effects of TNF-α on synaptic transmission in mice over time following sciatic nerve chronic constriction injury. J Neurophysiol. (2013) 110(7):1663–71. doi: 10.1152/jn.01088.2012
490. Kronschlager MT, Drdla-Schutting R, Gassner M, Honsek SD, Teuchmann HL, Sandkuhler J. Gliogenic LTP spreads widely in nociceptive pathways. Science. (2016) 354(6316):1144–8. doi: 10.1126/science.aah5715
491. Bliss TV, Collingridge GL, Kaang BK, Zhuo M. Synaptic plasticity in the anterior cingulate cortex in acute and chronic pain. Nat Rev Neurosci. (2016) 17(8):485–96. doi: 10.1038/nrn.2016.68
492. Gamal-Eltrabily M, Martinez-Lorenzana G, Gonzalez-Hernandez A, Condes-Lara M. Cortical modulation of nociception. Neuroscience. (2021) 458:256–70. doi: 10.1016/j.neuroscience.2021.01.001
493. Sandy-Hindmarch O, Bennett DL, Wiberg A, Furniss D, Baskozos G, Schmid AB. Systemic inflammatory markers in neuropathic pain, nerve injury, and recovery. Pain. (2022) 163(3):526–37. doi: 10.1097/j.pain.0000000000002386
494. Zhang WT, Sha WL, Zhu Q, Wu XB, He C. Plasticity of neuronal excitability and synaptic balance in the anterior nucleus of paraventricular thalamus after nerve injury. Brain Res Bull. (2022) 188:1–10. doi: 10.1016/j.brainresbull.2022.07.008
495. Song ZH, Song XJ, Yang CL, Cao P, Mao Y, Jin Y, et al. Up-regulation of microglial chemokine CXCL12 in anterior cingulate cortex mediates neuropathic pain in diabetic mice. Acta Pharmacol Sin. (2023) 44(7):1337–49. doi: 10.1038/s41401-022-01046-7
496. Zhuo M. Contribution of synaptic plasticity in the insular cortex to chronic pain. Neuroscience. (2016) 338:220–9. doi: 10.1016/j.neuroscience.2016.08.014
497. Taylor AM, Mehrabani S, Liu S, Taylor AJ, Cahill CM. Topography of microglial activation in sensory- and affect-related brain regions in chronic pain. J Neurosci Res. (2016) (2017) 95(6):1330–5. doi: 10.1002/jnr.23883
498. Fiore NT, Austin PJ. Peripheral nerve injury triggers neuroinflammation in the medial prefrontal Cortex and ventral hippocampus in a subgroup of rats with coincident affective behavioural changes. Neuroscience. (2019) 416:147–67. doi: 10.1016/j.neuroscience.2019.08.005
499. Wu XB, Zhu Q, Gao YJ. CCL2/CCR2 contributes to the altered excitatory-inhibitory synaptic balance in the nucleus Accumbens shell following peripheral nerve injury-induced neuropathic pain. Neurosci Bull. (2021) 37(7):921–33. doi: 10.1007/s12264-021-00697-6
500. Lu JS, Yang L, Chen J, Xiong FF, Cai P, Wang XY, et al. Basolateral amygdala astrocytes modulate diabetic neuropathic pain and may be a potential therapeutic target for koumine. Br J Pharmacol. (2023) 180(10):1408–28. doi: 10.1111/bph.16011
501. Taylor AM, Castonguay A, Taylor AJ, Murphy NP, Ghogha A, Cook C, et al. Microglia disrupt mesolimbic reward circuitry in chronic pain. J Neurosci. (2015) 35(22):8442–50. doi: 10.1523/JNEUROSCI.4036-14.2015
502. Cordeiro MS, Zhang Z, Seguela P. Peripheral neuropathy induces HCN channel dysfunction in pyramidal neurons of the medial prefrontal Cortex. J Neurosci. (2015) 35(38):13244–56. doi: 10.1523/JNEUROSCI.0799-15.2015
503. Ding W, You Z, Shen S, Chen L, Zhu S, Mao J. Inhibition of HCN channel activity in the thalamus attenuates chronic pain in rats. Neurosci Lett. (2016) 631:97–103. doi: 10.1016/j.neulet.2016.08.021
504. Yan Y, Zhu M, Cao X, Xu G, Shen W, Li F, et al. Thalamocortical circuit controls neuropathic pain via up-regulation of HCN2 in the ventral posterolateral thalamus. Neurosci Bull. (2023) 39(5):774–92. doi: 10.1007/s12264-022-00989-5
505. Hains BC, Saab CY, Waxman SG. Alterations in burst firing of thalamic VPL neurons and reversal by na(v)1.3 antisense after spinal cord injury. J Neurophysiol. (2006) 95(6):3343–52. doi: 10.1152/jn.01009.2005
506. Hains BC, Saab CY, Waxman SG. Changes in electrophysiological properties and sodium channel Nav1.3 expression in thalamic neurons after spinal cord injury. Brain. (2005) 128(Pt 10):2359–71. doi: 10.1093/brain/awh623
507. Bannister K, Dickenson AH. The plasticity of descending controls in pain: translational probing. J Physiol. (2017) 595(13):4159–66. doi: 10.1113/JP274165
508. Bannister K, Patel R, Goncalves L, Townson L, Dickenson AH. Diffuse noxious inhibitory controls and nerve injury: restoring an imbalance between descending monoamine inhibitions and facilitations. Pain. (2015) 156(9):1803–11. doi: 10.1097/j.pain.0000000000000240
509. Bannister K, Dickenson AH. What do monoamines do in pain modulation? Curr Opin Support Palliat Care. (2016) 10(2):143–8. doi: 10.1097/SPC.0000000000000207
510. Bannister K, Dickenson AH. What the brain tells the spinal cord. Pain. (2016) 157(10):2148–51. doi: 10.1097/j.pain.0000000000000568
511. Bannister K, Lockwood S, Goncalves L, Patel R, Dickenson AH. An investigation into the inhibitory function of serotonin in diffuse noxious inhibitory controls in the neuropathic rat. Eur J Pain. (2017) 21(4):750–60. doi: 10.1002/ejp.979
512. Bannister K, Qu C, Navratilova E, Oyarzo J, Xie JY, King T, et al. Multiple sites and actions of gabapentin-induced relief of ongoing experimental neuropathic pain. Pain. (2017) 158:2386–95. doi: 10.1097/j.pain.0000000000001040
513. Frezel N, Ranucci M, Foster E, Wende H, Pelczar P, Mendes R, et al.. c-Maf-positive spinal cord neurons are critical elements of a dorsal horn circuit for mechanical hypersensitivity in neuropathy. Cell Rep. (2023) 42(4):112295. doi: 10.1016/j.celrep.2023.112295
514. Bee LA, Dickenson AH. Rostral ventromedial medulla control of spinal sensory processing in normal and pathophysiological states. Neuroscience. (2007) 147(3):786–93. doi: 10.1016/j.neuroscience.2007.05.004
515. De Felice M, Sanoja R, Wang R, Vera-Portocarrero L, Oyarzo J, King T, et al. Engagement of descending inhibition from the rostral ventromedial medulla protects against chronic neuropathic pain. Pain. (2011) 152(12):2701–9. doi: 10.1016/j.pain.2011.06.008
516. Mitsi V, Zachariou V. Modulation of pain, nociception, and analgesia by the brain reward center. Neuroscience. (2016) 338:81–92. doi: 10.1016/j.neuroscience.2016.05.017
517. Ren W, Centeno MV, Berger S, Wu Y, Na X, Liu X, et al. The indirect pathway of the nucleus accumbens shell amplifies neuropathic pain. Nat Neurosci. (2016) 19(2):220–2. doi: 10.1038/nn.4199
518. Polgar E, Hughes DI, Riddell JS, Maxwell DJ, Puskar Z, Todd AJ. Selective loss of spinal GABAergic or glycinergic neurons is not necessary for development of thermal hyperalgesia in the chronic constriction injury model of neuropathic pain. Pain. (2003) 104(1–2):229–39. doi: 10.1016/S0304-3959(03)00011-3
519. Polgar E, Gray S, Riddell JS, Todd AJ. Lack of evidence for significant neuronal loss in laminae I-III of the spinal dorsal horn of the rat in the chronic constriction injury model. Pain. (2004) 111(1–2):144–50. doi: 10.1016/j.pain.2004.06.011
520. Webber CA, Salame J, Luu GL, Acharjee S, Ruangkittisakul A, Martinez JA, et al. Nerve growth factor acts through the TrkA receptor to protect sensory neurons from the damaging effects of the HIV-1 viral protein, Vpr. Neuroscience. (2013) 252:512–25. doi: 10.1016/j.neuroscience.2013.07.046
521. Peirs C, Williams SG, Zhao X, Arokiaraj CM, Ferreira DW, Noh MC, et al. Mechanical allodynia circuitry in the dorsal horn is defined by the nature of the injury. Neuron. (2021) 109(1):73–90. doi: 10.1016/j.neuron.2020.10.027
522. Polgar E, Fowler JH, McGill MM, Todd AJ. The types of neuron which contain protein kinase C gamma in rat spinal cord. Brain Res. (1999) 833(1):71–80. doi: 10.1016/S0006-8993(99)01500-0
523. Smith KM, Browne TJ, Davis OC, Coyle A, Boyle KA, Watanabe M, et al. Calretinin positive neurons form an excitatory amplifier network in the spinal cord dorsal horn. Elife. (2019) 8:e49190. doi: 10.7554/eLife.49190
524. Malcangio M, Tomlinson DR. A pharmacologic analysis of mechanical hyperalgesia in streptozotocin/diabetic rats. Pain. (1998) 76(1–2):151–7. doi: 10.1016/S0304-3959(98)00037-2
525. Yilmaz E, Gold MS. Paclitaxel-induced increase in NCX activity in subpopulations of nociceptive afferents: a protective mechanism against chemotherapy-induced peripheral neuropathy? Cell Calcium. (2016) 60(1):25–31. doi: 10.1016/j.ceca.2016.04.009
526. Idanpaan-Heikkila JJ, Guilbaud G. Pharmacological studies on a rat model of trigeminal neuropathic pain: baclofen, but not carbamazepine, morphine or tricyclic antidepressants, attenuates the allodynia-like behaviour. Pain. (1999) 79(2–3):281–90. doi: 10.1016/S0304-3959(98)00172-9
527. Jain A, Gyori BM, Hakim S, Bunga S, Taub DG, Ruiz-Cantero MC, et al. Nociceptor neuroimmune interactomes reveal cell type- and injury-specific inflammatory pain pathways. BioRxiv. (2023) 2023.02.01.526526. doi: 10.1101/2023.02.01.526526. Preprint.
528. Sorge RE, Martin LJ, Isbester KA, Sotocinal SG, Rosen S, Tuttle AH, et al. Olfactory exposure to males, including men, causes stress and related analgesia in rodents. Nat Methods. (2014) 11(6):629–32. doi: 10.1038/nmeth.2935
529. Mogil JS. Animal models of pain: progress and challenges. Nat Rev Neurosci. (2009) 10(4):283–94. doi: 10.1038/nrn2606
530. Mogil JS, Crager SE. What should we be measuring in behavioral studies of chronic pain in animals? Pain. (2004) 112:12–5. doi: 10.1016/j.pain.2004.09.028
531. Sexton JE, Cox JJ, Zhao J, Wood JN. The genetics of pain: implications for therapeutics. Annu Rev Pharmacol Toxicol. (2017) 58:123–42. doi: 10.1146/annurev-pharmtox-010617-052554
532. Turner PV, Pang DS, Lofgren JL. A review of pain assessment methods in laboratory rodents. Comp Med. (2019) 69(6):451–67. doi: 10.30802/AALAS-CM-19-000042
533. Sotocinal SG, Sorge RE, Zaloum A, Tuttle AH, Martin LJ, Wieskopf JS, et al. The rat grimace scale: a partially automated method for quantifying pain in the laboratory rat via facial expressions. Mol Pain. (2011) 7:55. doi: 10.1186/1744-8069-7-55
534. Diester CM, Santos EJ, Moerke MJ, Negus SS. Behavioral battery for testing candidate analgesics in mice. I. validation with positive and negative controls. J Pharmacol Exp Ther. (2021) 377(2):232–41.33622770
535. Diester CM, Lichtman AH, Negus SS. Behavioral battery for testing candidate analgesics in mice. II. Effects of Endocannabinoid Catabolic Enzyme Inhibitors and 9–Tetrahydrocannabinol. J Pharmacol Exp Ther. (2021) 377(2):242–53. doi: 10.1124/jpet.120.000464
536. Andrade-Gonzalez RD, Perrusquia-Hernandez E, Montes-Angeles CD, Castillo-Diaz LA, Hernandez Campos ME, Perez-Martinez IO. Encoding signs of orofacial neuropathic pain from facial expressions in mice. Arch Oral Biol. (2022) 135:105369. doi: 10.1016/j.archoralbio.2022.105369
537. Zylka MJ, McCoy ES, Park SK, Patel RP, Ryan DF, Mullen ZJ, et al. Development and validation of painface, a software platform that simplifies and standardizes mouse grimace analyses. J Pain. (2023) 24(4, Supplement):35–6. doi: 10.1016/j.jpain.2023.02.113
538. Harte SE, Meyers JB, Donahue RR, Taylor BK, Morrow TJ. Mechanical conflict system: a novel operant method for the assessment of nociceptive behavior. PLoS One. (2016) 11(2):e0150164. doi: 10.1371/journal.pone.0150164
539. Mauderli AP, Acosta-Rua A, Vierck CJ. An operant assay of thermal pain in conscious, unrestrained rats. J Neurosci Methods. (2000) 97(1):19–29. doi: 10.1016/S0165-0270(00)00160-6
540. Negus SS, Vanderah TW, Brandt MR, Bilsky EJ, Becerra L, Borsook D. Preclinical assessment of candidate analgesic drugs: recent advances and future challenges. J Pharmacol Exp Ther. (2006) 319(2):507–14. doi: 10.1124/jpet.106.106377
541. Rostock C, Schrenk-Siemens K, Pohle J, Siemens J. Human vs. Mouse nociceptors - similarities and differences. Neuroscience. (2018) 387:13–27. doi: 10.1016/j.neuroscience.2017.11.047
542. Middleton SJ, Barry AM, Comini M, Li Y, Ray PR, Shiers S, et al. Studying human nociceptors: from fundamentals to clinic. Brain. (2021) 144:1312–36. doi: 10.1093/brain/awab048
543. Hartung JE, Moy JK, Loeza-Alcocer E, Nagarajan V, Jostock R, Christoph T, et al. Voltage gated calcium channels in human dorsal root ganglion neurons. Pain. (2021) (2022) 163(6):e774–85. doi: 10.1097/j.pain.0000000000002465
544. Shiers S, Klein RM, Price TJ. Quantitative differences in neuronal subpopulations between mouse and human dorsal root ganglia demonstrated with RNAscope in situ hybridization. Pain. (2020) 161(10):2410–24. doi: 10.1097/j.pain.0000000000001973
545. Muizelaar JP, Kleyer M, Hertogs IA, DeLange DC. Complex regional pain syndrome (reflex sympathetic dystrophy and causalgia): management with the calcium channel blocker nifedipine and/or the alpha-sympathetic blocker phenoxybenzamine in 59 patients. Clin Neurol Neurosurg. (1997) 99(1):26–30. doi: 10.1016/S0303-8467(96)00594-X
546. Cheng C, Guo GF, Martinez JA, Singh V, Zochodne DW. Dynamic plasticity of axons within a cutaneous milieu. J Neurosci. (2010) 30(44):14735–44. doi: 10.1523/JNEUROSCI.2919-10.2010
547. Renthal W, Chamessian A, Curatolo M, Davidson S, Burton M, Dib-Hajj S, et al. Human cells and networks of pain: transforming pain target identification and therapeutic development. Neuron. (2021) 109(9):1426–9. doi: 10.1016/j.neuron.2021.04.005
548. McDermott LA, Weir GA, Themistocleous AC, Segerdahl AR, Blesneac I, Baskozos G, et al. Defining the functional role of NaV1.7 in human nociception. Neuron. (2019) 101(5):905–19. doi: 10.1016/j.neuron.2019.01.047
549. Meents JE, Bressan E, Sontag S, Foerster A, Hautvast P, Rosseler C, et al. The role of Nav1.7 in human nociceptors: insights from human induced pluripotent stem cell-derived sensory neurons of erythromelalgia patients. Pain. (2019) 160(6):1327–41. doi: 10.1097/j.pain.0000000000001511
550. Chambers SM, Qi Y, Mica Y, Lee G, Zhang XJ, Niu L, et al. Combined small-molecule inhibition accelerates developmental timing and converts human pluripotent stem cells into nociceptors. Nat Biotechnol. (2012) 30(7):715–20. doi: 10.1038/nbt.2249
551. Young GT, Gutteridge A, Fox H, Wilbrey AL, Cao L, Cho LT, et al. Characterizing human stem cell-derived sensory neurons at the single-cell level reveals their ion channel expression and utility in pain research. Mol Ther. (2014) 22(8):1530–43. doi: 10.1038/mt.2014.86
552. Lampert A, Bennett DL, McDermott LA, Neureiter A, Eberhardt E, Winner B, et al. Human sensory neurons derived from pluripotent stem cells for disease modelling and personalized medicine. Neurobiol Pain. (2020) 8:100055. doi: 10.1016/j.ynpai.2020.100055
553. Labau JIR, Andelic M, Faber CG, Waxman SG, Lauria G, Dib-Hajj SD. Recent advances for using human induced-pluripotent stem cells as pain-in-a-dish models of neuropathic pain. Exp Neurol. (2022) 358:114223. doi: 10.1016/j.expneurol.2022.114223
554. Alsaloum M, Labau JIR, Liu S, Effraim PR, Waxman SG. Stem cell-derived sensory neurons modelling inherited erythromelalgia: normalization of excitability. Brain. (2023) 146(1):359–71. doi: 10.1093/brain/awac031
555. Haile Y, Nakhaei-Nejad M, Boakye PA, Baker G, Smith PA, Murray AG, et al. Reprogramming of HUVECs into induced pluripotent stem cells (HiPSCs), generation and characterization of HiPSC-derived neurons and astrocytes. PLoS One. (2015) 10(3):e0119617. doi: 10.1371/journal.pone.0119617
556. Vojnits K, Mahammad S, Collins TJ, Bhatia M. Chemotherapy-Induced neuropathy and drug discovery platform using human sensory neurons converted directly from adult peripheral blood. Stem Cells Transl Med. (2019) 8(11):1180–91. doi: 10.1002/sctm.19-0054
557. Rodriguez-Palma EJ, De la Luz-Cuellar YE, Islas-Espinoza AM, Felix-Leyva AE, Shiers SI, Garcia G, et al. Activation of alpha 6 -containing GABA A receptors induces antinociception under physiological and pathological conditions. Pain. (2023) 164(5):948–66. doi: 10.1097/j.pain.0000000000002763
558. Backonja MM, Attal N, Baron R, Bouhassira D, Drangholt M, Dyck PJ, et al. Value of quantitative sensory testing in neurological and pain disorders: neuPSIG consensus. Pain. (2013) 154(9):1807–19. doi: 10.1016/j.pain.2013.05.047
559. Vollert J, Maier C, Attal N, Bennett DLH, Bouhassira D, Enax-Krumova EK, et al. Stratifying patients with peripheral neuropathic pain based on sensory profiles: algorithm and sample size recommendations. Pain. (2017) 158(8):1446–55. doi: 10.1097/j.pain.0000000000000935
560. Rolke R, Baron R, Maier C, Tolle TR, Treede DR, Beyer A, et al. Quantitative sensory testing in the German research network on neuropathic pain (DFNS): standardized protocol and reference values. Pain. (2006) 123(3):231–43. doi: 10.1016/j.pain.2006.01.041
561. Serra J, Bostock H, Sola R, Aleu J, Garcia E, Cokic B, et al. Microneurographic identification of spontaneous activity in C-nociceptors in neuropathic pain states in humans and rats. Pain. (2012) 153(1):42–55. doi: 10.1016/j.pain.2011.08.015
562. Pinheiro ES, de Queiros FC, Montoya P, Santos CL, do Nascimento MA, Ito CH, et al. Electroencephalographic Patterns in Chronic Pain: A Systematic Review of the Literature. PLoS One. (2016) 11(2):e0149085. doi: 10.1371/journal.pone.0149085
563. Andelic M, Salvi E, Marcuzzo S, Marchi M, Lombardi R, Cartelli D, et al. Integrative miRNA-mRNA profiling of human epidermis: unique signature of SCN9A painful neuropathy. Brain. (2023) (2023) 146(7):3049–62. doi: 10.1093/brain/awad025
564. North RY, Li Y, Ray P, Rhines LD, Tatsui CE, Rao G, et al. Electrophysiological and transcriptomic correlates of neuropathic pain in human dorsal root ganglion neurons. Brain. (2019) 142(5):1215–26. doi: 10.1093/brain/awz063
565. Fillingim RB, King CD, Ribeiro-Dasilva MC, Rahim-Williams B, Riley JL III. Sex, gender, and pain: a review of recent clinical and experimental findings. J Pain. (2009) 10(5):447–85. doi: 10.1016/j.jpain.2008.12.001
566. Martin LJ, Acland EL, Cho C, Gandhi W, Chen D, Corley E, et al. Male-Specific conditioned pain hypersensitivity in mice and humans. Curr Biol. (2019) 29(2):192–201. doi: 10.1016/j.cub.2018.11.030
567. Hendrich J, Alvarez P, Joseph EK, Ferrari LF, Chen X, Levine JD. In vivo and in vitro comparison of female and male nociceptors. J Pain. (2012) 13(12):1224–31. doi: 10.1016/j.jpain.2012.09.009
568. Shansky RM, Murphy AZ. Considering sex as a biological variable will require a global shift in science culture. Nat Neurosci. (2021) 24(4):457–64. doi: 10.1038/s41593-021-00806-8
569. Mifflin KA, Kerr BJ. Sex-related differences in acute and chronic pain: a bench to bedside perspective. Can J Anaesth. (2013) 60(3):221–6. doi: 10.1007/s12630-012-9881-7
570. Fauchon C, Kim JA, El-Sayed R, Osborne NR, Rogachov A, Cheng JC, et al. Exploring sex differences in alpha brain activity as a potential neuromarker associated with neuropathic pain. Pain. (2022) 163(7):1291–302. doi: 10.1097/j.pain.0000000000002491
571. Johnston KJA, Ward J, Ray PR, Adams MJ, McIntosh AM, Smith BH, et al. Sex-stratified genome-wide association study of multisite chronic pain in UK biobank. PLoS Genet. (2021) 17(4):e1009428. doi: 10.1371/journal.pgen.1009428
572. Ovsepian SV, Waxman SG. Gene therapy for chronic pain: emerging opportunities in target-rich peripheral nociceptors. Nat Rev Neurosci. (2023) 24(4):252–65. doi: 10.1038/s41583-022-00673-7
Keywords: neurogenic neuroinflammation, allodynia, dorsal horn, dorsal root ganglia, neuropathy, nerve injury, neuroinflammation, neuroimmunology
Citation: Smith PA (2023) Neuropathic pain; what we know and what we should do about it. Front. Pain Res. 4:1220034. doi: 10.3389/fpain.2023.1220034
Received: 9 May 2023; Accepted: 5 September 2023;
Published: 22 September 2023.
Edited by:
Laiche Djouhri, Qatar University, QatarReviewed by:
Alfredo Ribeiro-da-Silva, McGill University, Canada© 2023 Smith. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter A. Smith cGFzM0B1YWxiZXJ0YS5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.