 Shota Yamamoto
Shota Yamamoto Tomomi Hashidate-Yoshida1
Tomomi Hashidate-Yoshida1 Hideo Shindou
Hideo Shindou- 1Department of Lipid Life Science, National Center for Global Health and Medicine, Tokyo, Japan
- 2Institute of Microbial Chemistry, Tokyo, Japan
- 3Department of Lipid Signaling, National Center for Global Health and Medicine, Tokyo, Japan
- 4Department of Medical Lipid Science, Graduate School of Medicine, The University of Tokyo, Tokyo, Japan
Peripheral nerve injury (PNI) induces neuronal hyperexcitability, which underlies neuropathic pain. The emergence of RNA sequencing technologies has enabled profiling of transcriptional changes in pathological conditions. However, these approaches do not provide information regarding metabolites such as lipids that are not directly encoded by genes. Fatty acids (FAs) are some of the essential lipids in mammalian organisms and are mainly stored as membrane phospholipids. In response to various biological stimuli, FAs are rapidly released and converted into several mediators, such as eicosanoids and docosanoids. FAs themselves or their metabolites play important roles in physiology and pathology. In this study, using a comprehensive lipidomic analysis of FA metabolites, 152 species were measured in the dorsal root ganglia of mice at multiple time points after PNI. We found that PNI increased the ω-6 FA metabolites produced by cyclooxygenases but not those produced by lipoxygenases or cytochrome P450 enzymes in the dorsal root ganglia. In contrast, ω-3 FA metabolites biosynthesized by any enzyme transiently increased after nerve injury. Overall, these findings provide a new resource and valuable insights into PNI pathologies, including pain and nerve regeneration.
Introduction
Peripheral nerve injury (PNI) induces refractory pain syndrome, which is known as neuropathic pain. Neuropathic pain is often caused by damage to the nervous system resulting from diabetes, cancer, chemotherapy, viral infection, autoimmune disease, stroke, and trauma (1). Although neuropathic pain is estimated to affect 7–10% of the human population (2), there is currently no effective treatment. However, advances in understanding the pathology of neuropathic pain have revealed the potential for novel therapeutic strategies (3).
Recently, the emergence of RNA sequencing techniques and relevant technological improvements have greatly advanced life science research, including pain research (4–6). However, these approaches cannot provide information regarding small-molecule metabolites that are not directly encoded by genes. Among metabolites, lipids play pivotal roles in physiological and pathological conditions (7, 8). In particular, fatty acids (FAs) have diverse roles in mammalian organisms, and most of them are stored as glycerophospholipids (9) [side note, FAs can be illustrated as XX:Yω-Z (where XX, Y, and Z are carbon number, double bond number, and the position of the carbon having the first double bond from the methyl-end, respectively)]. Various biological stimuli can induce the release of FAs through phospholipase A1/2 reactions. These FAs can be further metabolized into diverse lipid mediators by three major enzymes, cyclooxygenases (COX1 and 2), lipoxygenases (LOX), and cytochrome P450 (CYP) enzymes (10). Lipid mediators exert their biological functions through G-protein coupled receptors, ion channels, and possibly nuclear receptors (10, 11).
Accumulating evidence has gradually clarified the role of lipid mediators in the pathophysiology of pain (12–14). For example, prostaglandin E2 (PGE2) and PGI2, which are COX-produced metabolites of arachidonic acid (AA, 20:4ω-6), induce sensitization of transient receptor potential vanilloid 1 (TRPV1) via intracellular protein kinase and are involved in inflammatory pain (15, 16). Moreover, 9-hydroxyoctadecadienoic acid (9-HODE) and 13-HODE, which are LOX-produced metabolites of linoleic acid (LA, 18:2ω-6), could also activate TRPV1 and cause inflammation-induced pain hypersensitivity (17, 18). In contrast to the roles of ω-6 FA in promoting pain, ω-3 FA metabolites, such as eicosapentaenoic acid (EPA, 20:5ω-3) and docosahexaenoic acid (DHA, 22:6ω-3), are reported to improve pain. These ω-3 FA-derived anti-inflammatory lipid mediators are coined “specialized pro-resolving lipid mediators” (SPMs) (19, 20). Recently, there have been increasing reports regarding the pharmacological efficacy of SPMs in the treatment of pain disorders (21–23), although the biosynthetic pathways, receptors, and in vivo presence of SPMs remain to be validated (24). Whereas previous studies have reported the pathophysiological roles of specific lipid mediators of interest, comprehensive lipidomic information on pain pathology is currently lacking.
To address this gap, we used a PNI-induced neuropathic pain model to conduct an extensive lipidomic analysis of FA metabolites in the dorsal root ganglion (DRG), the cell body assembly tissue of primary afferents. Moreover, we monitored the quantitative changes in 152 species of FA metabolites in the DRG after PNI over time, focusing on the metabolites of ω-6 FA, such as AA and LA.
Results
We performed lipidomic analysis of the lumbar DRG from mice after PNI (Figure 1A), over multiple time points ranging from 1 to 14 days after injury. First, we observed that PNI induced mechanical allodynia from day 3, which was suspended until day 14 (Figure 1B). Next, we monitored 152 species of FA metabolites and free FAs (AA, EPA, and DHA) using liquid chromatography-tandem mass spectrometry (LC-MS/MS), and used standard lipids to create a standard curve for data qualification and quantification (Supplementary Table 1). We analyzed pooled L4 DRG samples from 6 to 8 mice combined because we could constantly detect only approximately 10 species within the calibration range from the DRG tissue of one mouse. In the present study, 44 species were detected within the quantifiable range in most samples. We classified each metabolite by its synthetic pathway according to a previous study (25), even though some FA metabolites can be synthesized through more than 2 pathways. The results were expressed as the average of the quantitative values for each lipid molecule per DRG tissue (picogram/DRG), and the raw data are listed in Supplementary Table 2.
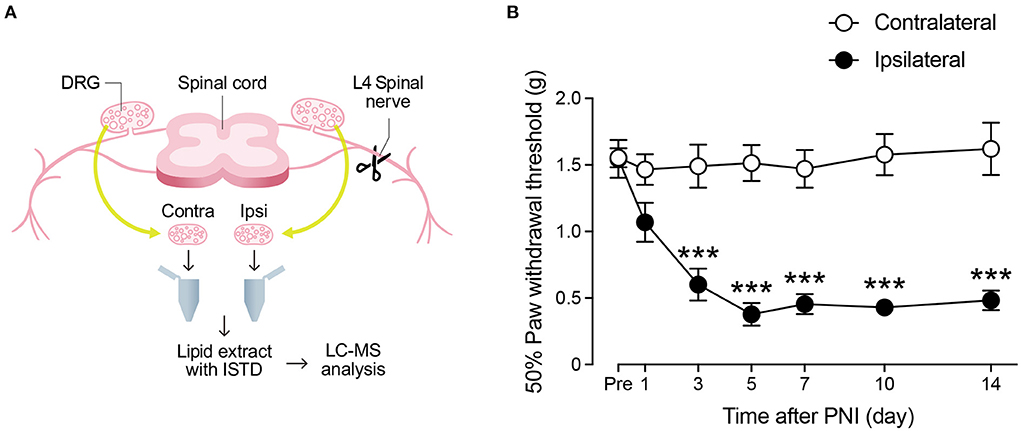
Figure 1. Schematic diagram of lipidomic analysis. (A) Schematic diagram of the experimental protocol for measuring fatty acid metabolites (Contra, contralateral; Ipsi, ipsilateral; ISTD, internal standard). See also Material and Methods. (B) Paw withdrawal threshold to mechanical stimuli before and after peripheral nerve injury (PNI) (n = 8; ***P < 0.001 vs. the contralateral side). Values are means ± s.e.m.
COX-produced metabolites are increased from 7 days after PNI
AA can be subjected to COX-mediated oxidization and is metabolized to PGH2, which is further metabolized to prostanoids, such as PGD2, PGE2, PGI2, and thromboxane A2 (TXA2), by terminal PG synthases (Figure 2). Among the 152 species of FA metabolites measured, we detected eight species of COX-produced AA metabolites: PGA2, PGD2, PGE2, PGF2α, 6-keto-PGF1α (a metabolite of PGI2), TXB2 (a metabolite of TXA2), 12-hydroxyheptadecatrienoic acid (12-HHT), and 11-hydroxyeicosatetraenoic acid (11-HETE) (Figure 2). We found that PNI did not alter the levels of these metabolites on days 1 and 3 after PNI, despite the detection of pain behavior on day 3. However, most of their levels, except those of PGA2 and 6-keto-PGF1α, gradually increased and remained constant until day 14. In addition, 11-HETE also showed the same alteration pattern as the downstream species of PGH2, even though it was not biosynthesized via PGH2. The levels of PGA2 and 6-keto-PGF1α were not changed by nerve injury (Figure 2). Furthermore, PNI did not alter the level of free AA in the DRG (Supplementary Figure 1A).
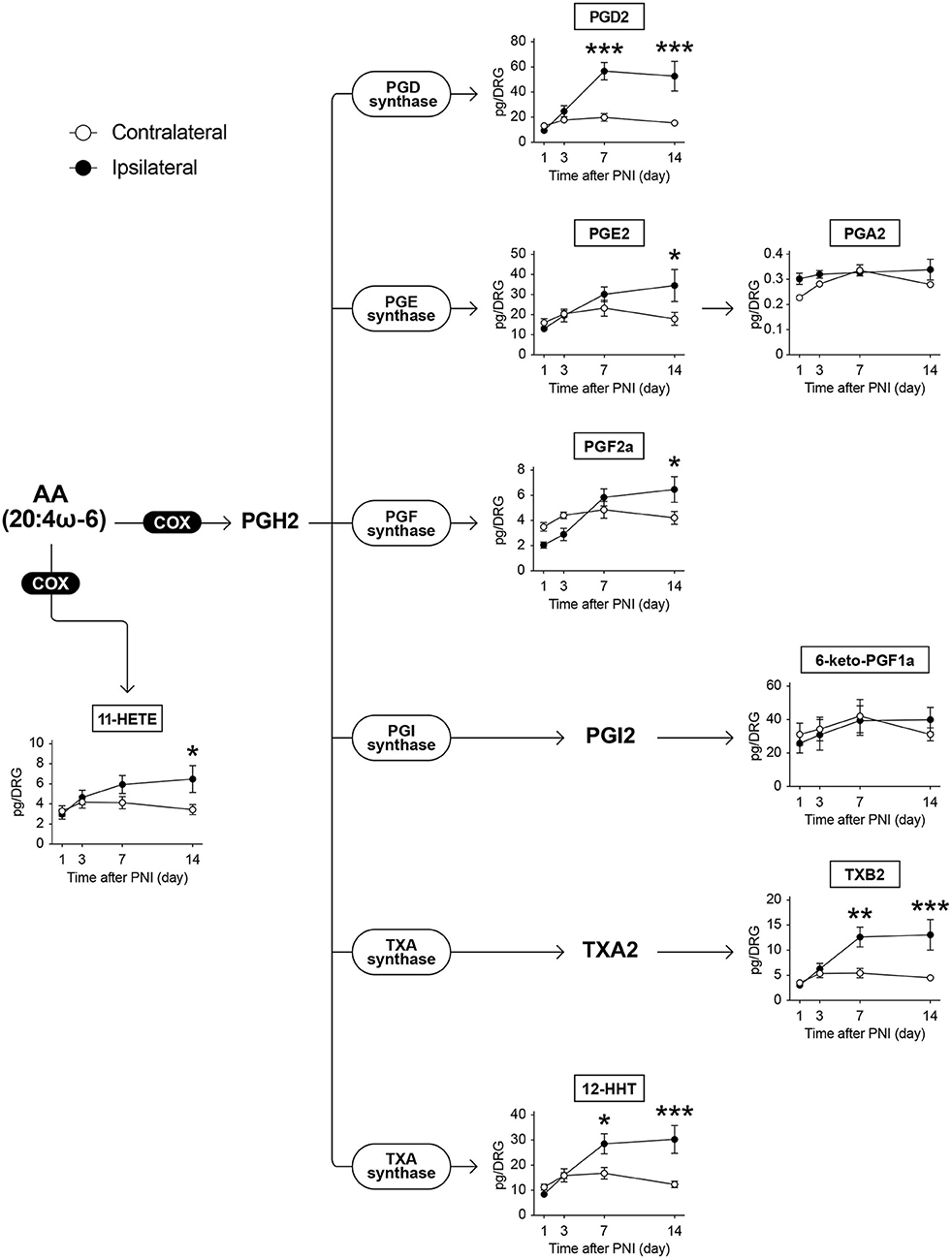
Figure 2. PNI increases AA-derived COX-produced metabolites in the DRG. The metabolic pathways of arachidonic acid (AA) after oxidation by cyclooxygenase (COX) enzymes; eight species could be quantified (n = 3–6; *P < 0.05, **P < 0.01, ***P < 0.001 vs. the contralateral side). Values are means ± s.e.m. The presented data are from one set of experiments (PGA2), or compiled from two sets of experiments (others). The raw data are listed in Supplementary Table 2.
In addition to the above-mentioned species, we detected PGD1 and PGE1 (metabolites of dihomo-γ-linolenic acid, 20:3ω-6), 1a,1b-dihomo-PGF2α (a metabolite of adrenic acid, 22:4ω-6), and PGE3 (a metabolite of EPA) (Figure 3). We found that these metabolites were increased in the same manner as AA metabolites after PNI (Figure 3). Although we also found that PNI increased the level of free EPA (Supplementary Figure 1B), which was not reflected in the level of PGE3. These results suggest that PNI increases most of the COX-produced lipid mediators in the DRG, regardless of whether they are derived from ω-6 or ω-3 FA, and that these increases correlate temporally with the development of mechanical allodynia from day 7 onward.
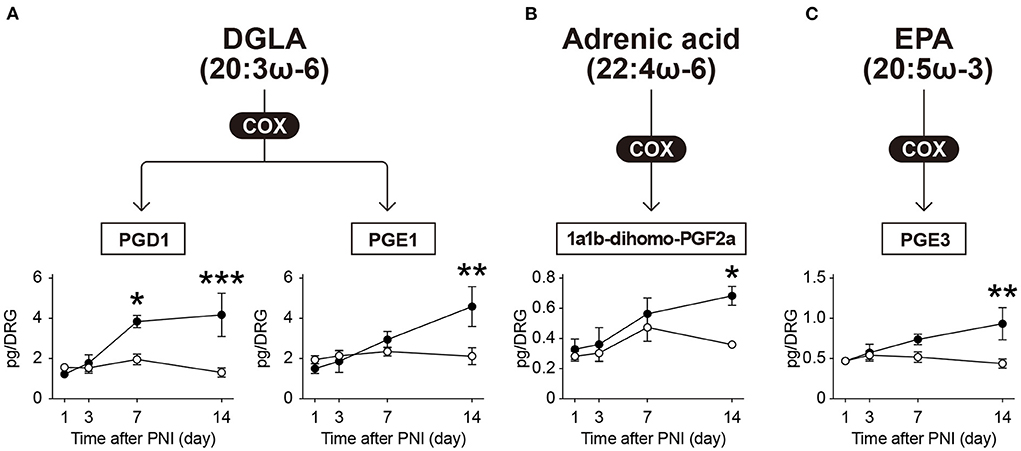
Figure 3. The levels of COX-produced metabolites (other than those derived from arachidonic acid) are elevated after PNI. The metabolic pathways of (A) dihomo-γ-linolenic acid (DGLA), (B) adrenic acid, and (C) eicosapentaenoic acid (EPA) after oxidation by cyclooxygenase (COX) enzymes; a total of four species could be quantified (n = 4–6; *P < 0.05, **P < 0.01, ***P < 0.001 vs. the contralateral side). Values are means ± s.e.m. The presented data are compiled from two sets of experiments. The raw data are listed in Supplementary Table 2.
PNI increases LOX-produced metabolites derived from ω-3 FA but not from ω-6 FA
There are several subtypes of LOX enzymes including 5-LOX, 12-LOXs, and 15-LOX. When AA is metabolized by 5-LOX, 5-hydroperoxyeicosatetraenoic acid (5-HpETE) is produced, which is further metabolized to leukotriene A4 (LTA4) via 5-LOX-mediated dehydration. In contrast, HETEs are produced when HpETEs are subjected to reaction with peroxidase, and further oxidation produces ox-ETEs (also known as KETEs) (Figure 4). In our measurement, we detected 16 and 5 species of LOX-produced metabolites derived from ω-6 and ω-3 FA, respectively.
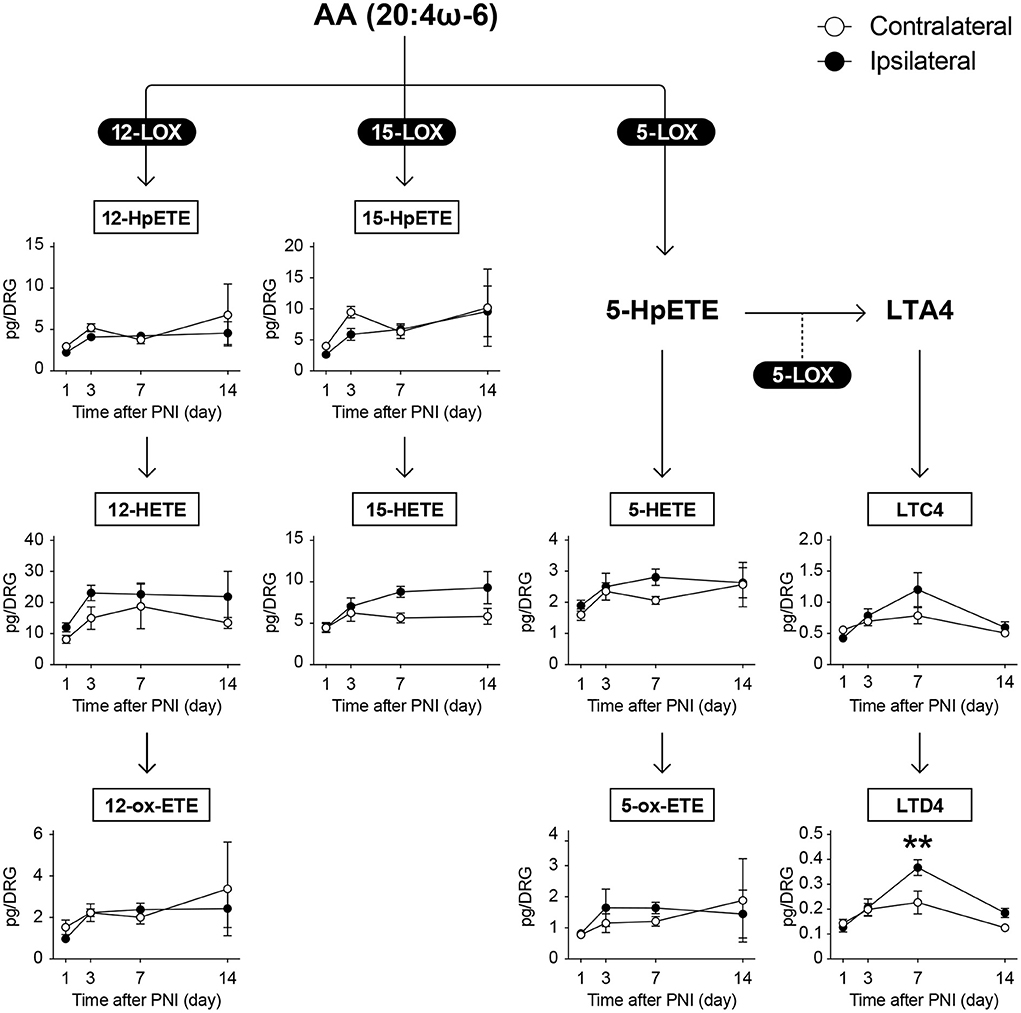
Figure 4. PNI does not affect most of the AA-derived LOX-produced metabolites in the DRG. The metabolic pathways of arachidonic acid (AA) after oxidation by lipoxygenase (LOX) enzymes; nine species could be quantified (n = 3–6; **P < 0.01 vs. the contralateral side). Values are means ± s.e.m. The presented data are from one set of experiments (5-ox-ETE, 12-HpETE, 12-ox-ETE, and 15-HpETE), or compiled from two sets of experiments (others). The raw data are listed in Supplementary Table 2.
We detected LTC4, LTD4, 5-HETE, 5-ox-ETE, 12-HpETE, 12-HETE, 12-ox-ETE, 15-HpETE, and 15-HETE as AA metabolites (Figure 4); 9-hydroperoxyoctadecadienoic acid (9-HpODE), 9-HODE, 9-ox-ODE (also known as 9-KODE), 13-HpODE, 13-HODE, and 13-ox-ODE as LA metabolites (Figure 5A); and 15-hydroxytrienoic acid (15-HETrE) as a metabolite of dihomo-γ-linolenic acid (Figure 5B). We found that PNI significantly increased the level of LTD4 on day 7 (Figure 4). Although some other species showed an increasing trend (15-HETE, P = 0.0827 on day 14; LTC4, P = 0.0848 on day 7), the amounts of other LOX-produced metabolites were not altered after nerve injury (Figures 4, 5A,B). Regarding ω-3 FA, we detected 12-hydroxy-EPA (12-HEPE) and 15-HEPE as EPA metabolites (Figure 5C), and 13-hydroxy-DHA (13-HDHA), 14-HDHA, and 17-HDHA as DHA metabolites (Figure 5D). In contrast to the metabolites from ω-6 FA, LOX-produced metabolites from ω-3 FA were significantly increased after PNI. Moreover, these species were increased from day 3 to 7 after nerve injury and returned to levels comparable with those of the contralateral side by day 14 (Figures 5C,D). Furthermore, these metabolites did not correlate with the amount of free fatty acids (Supplementary Figures 1B,C). These results indicate that LOX-produced metabolites derived from EPA and DHA in the DRG are lipid species that transiently increase during the onset of mechanical allodynia.
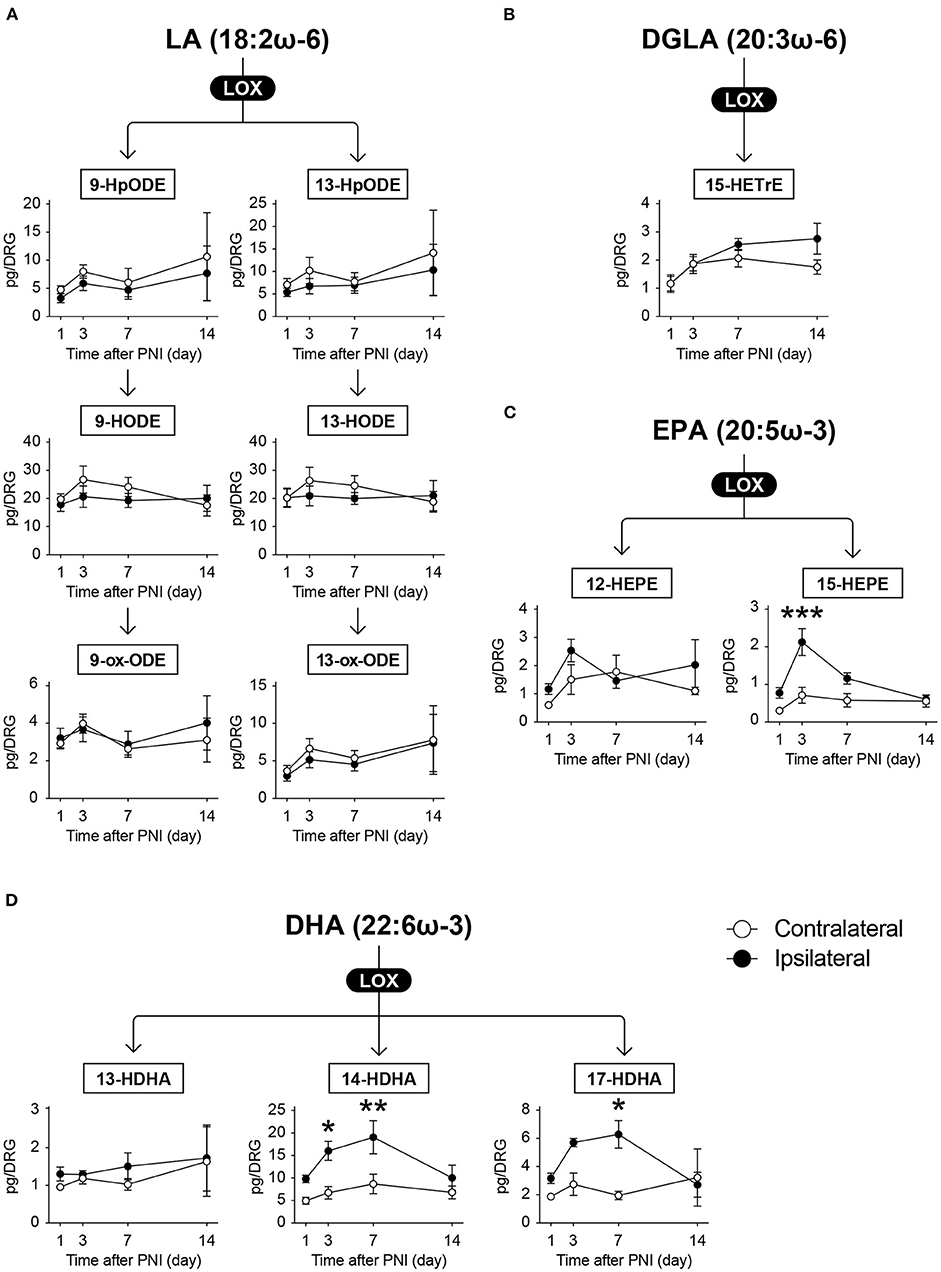
Figure 5. Omega 3 FA-derived metabolites synthesized by LOX enzymes are transiently increased after PNI. The metabolic pathways of (A) linoleic acid (LA), (B) dihomo-γ-linolenic acid (DGLA), (C) eicosapentaenoic acid (EPA), and (D) docosahexaenoic acid (DHA) after oxidation by lipoxygenase (LOX) enzymes. In total, six and five species could be quantified from ω-6 and ω-3 fatty acids (FAs), respectively (n = 3–6; *P < 0.05, **P < 0.01, ***P < 0.001 vs. the contralateral side). Values are means ± s.e.m. The presented data are from one set of experiments (9-HpODE, Day 1 of 13-HpODE, 15-HEPE, 13-HDHA, and 17-HDHA), or compiled from two sets of experiments (others). The raw data are listed in Supplementary Table 2.
CYP-produced metabolites show changes similar to LOX-produced metabolites
FAs can also be subjected to CYP-mediated oxidation and metabolized to HETEs or epoxyeicosatrienoic acids (EETs). In this study, we monitored 22 species of CYP-produced metabolites and detected five species within the quantifiable range (Figure 6): 16-HETE and 20-carboxy-AA as AA metabolites; 9,10-dihydroxyoctadecenoic acid (9,10-DiHOME) and 12,13-DiHOME as LA metabolites; and 17,18-dihydroxyeicosatetraenoic acid (17,18-DiHETE) as an EPA metabolite. We found that the amount of ω-6 FA metabolites produced by CYP enzymes did not change after PNI (Figures 6A,B). In contrast, PNI markedly increased the levels of 17,18-DiHETE on day 7 (Figure 6C). Thus, we found that these CYP-produced metabolites in the DRG showed changes similar to LOX-produced metabolites after PNI.
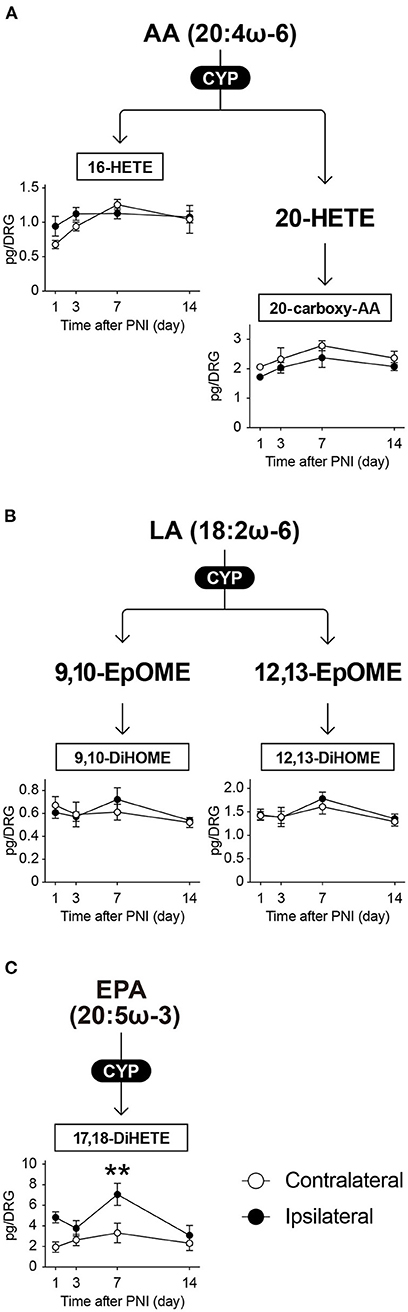
Figure 6. CYP-produced EPA metabolite 17,18-DiHETE is transiently increased after PNI. The metabolic pathways of (A) arachidonic acid (AA), (B) linoleic acid (LA), and (C) eicosapentaenoic acid (EPA) after oxidation by cytochrome-P450 (CYP) enzymes. In total, five species could be quantified (n = 3–6; **P < 0.01 vs. the contralateral side). Values are means ± s.e.m. The presented data are from one set of experiments (16-HETE and 20-carboxy-AA), or compiled from two sets of experiments (others). The raw data are listed in Supplementary Table 2.
PNI affects the metabolism of lipids other than the biosynthetic products by COX/LOX/CYP enzymes
Various endogenous lipid species are produced by non-enzymatic oxidation. In this study, we quantified four species (Figures 7A–C): 8-iso-PGE2 as an AA metabolite; 15-ox-EDE (also known as 15-KEDE) as a metabolite of eicosadienoic acid (20:2ω-6); and 16-HDHA and 20-HDHA as DHA metabolites. Among these, 8-iso-PGE2 level was significantly increased on day 14 after PNI (Figure 7A). However, the levels of the other three species were unaffected by nerve injury until day 14 (Figures 7B,C).
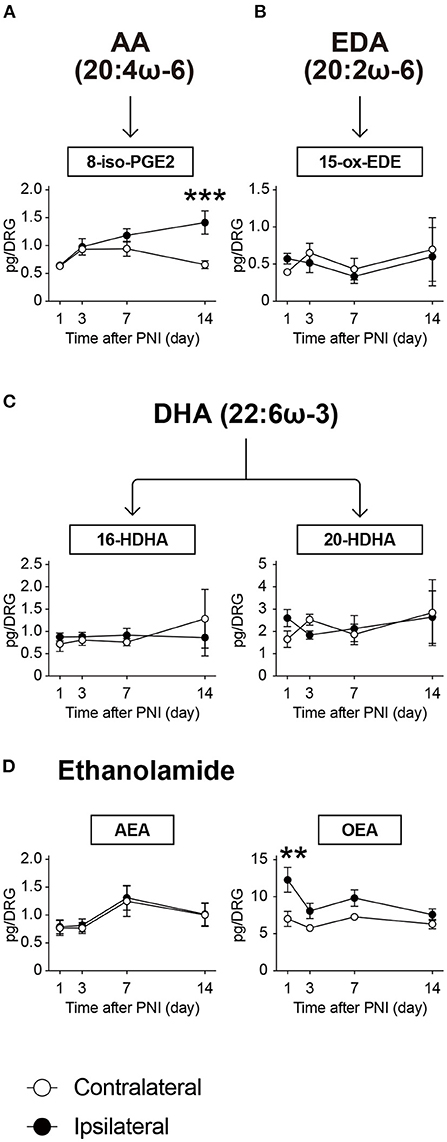
Figure 7. PNI affects the metabolism of isoprostane and ethanolamide in the DRG. The metabolic pathways of (A) arachidonic acid (AA), (B) eicosadienoic acid (EDA), (C) docosahexaenoic acid (DHA) after non-enzymatic oxidation, and (D) ethanolamide-related species. In total, six species could be quantified (n = 3–6; **P < 0.01, ***P < 0.001 vs. the contralateral side). Values are means ± s.e.m. The presented data are from one set of experiments (Day 1 of 8-iso-PGE2, Days 1, 3, and 14 of 15-ox-EDE, Day 1 of 16-HDHA, and Day 1 of 20-HDHA), or compiled from two sets of experiments (others). The raw data are listed in Supplementary Table 2.
We also measured ethanolamide-related species and detected arachidonoylethanolamide (AEA) and oleoylethanolamide (OEA) within the calibration range. We found that OEA transiently increased on the day after PNI, whereas AEA was not affected (Figure 7D). These results suggest that PNI affects lipid metabolism, including the products of major metabolic enzymes as well as non-enzymatic products or ethanolamides in the DRG.
Discussion
Lipids play crucial homeostatic roles in the healthy body and undergo qualitative and quantitative changes after injury and infection as well as in chronic diseases (26–28). Many studies have demonstrated that PNI alters the metabolism of FAs and other lipid classes, such as cholesterols, in the DRG, which are involved in pain and regeneration (14, 29–31). Although most previous studies have provided information regarding changes of particular lipid species or transcriptomic changes at a single time point, there is a lack of lipidomic information that can provide a comprehensive view of lipids in time dependent manner. Here, we showed how PNI alters FAs metabolism in the DRG on days 1, 3, 7, and 14.
Several groups have reported an increase in prostanoids, such as PGE2 and 6-keto-PGF1α, in the injured nerves (32–36); however little information is available regarding the DRG. Additionally, there are conflicting reports on changes in PGE2 levels after PNI, which either increased or remained unchanged (33, 35). In our study, most of the prostanoids and their derived species were significantly increased in the DRG at 14 days after PNI, regardless of whether they were derived from ω-6 or ω-3 FA; among them, PGD1, PGD2, TXB2, and 12-HHT were also increased on Day 7. COX-1 was reported to be expressed in small-diameter primary sensory neurons in the DRG, whereas COX-2 was not detected (37, 38). In contrast, non-neuronal cells were found to express both COX-1 and COX-2 (39). Furthermore, several recent studies using single-cell RNA sequencing (scRNA-seq) analysis have shown that terminal prostanoid synthases, such as PGD synthase, PGE synthase, and TXA synthase, are abundantly expressed in non-neuronal cells (5, 29, 40). These scRNA-seq resources have also shown that PNI upregulates the expression of terminal prostanoid synthases in macrophages, satellite glial cells (SGCs), endothelial cells, and fibroblasts in the DRG (5, 29). Given that various immune cells infiltrate, proliferate, and activate in response to nerve injury in the DRG (41, 42), the increased levels of prostanoids observed in this study might be of non-neuronal origin. However, we observed these increases only on days 7 and 14, despite the fact that the number of non-neuronal cells is increased and terminal PG synthases are upregulated by day 3 after nerve injury (5, 29, 43). Moreover, we observed the transient increase of free EPA levels after PNI, but not AA and DHA. As it was not consistent with the increasing time course of PGE3, the activity of metabolic enzymes may be more important in controlling prostanoid levels than the amounts of free FAs. These metabolic profiles could only be revealed by measuring the metabolites themselves, which are not encoded by genes; however, the related enzymes that regulate prostanoid levels, such as lysophospholipid acyltransferases (9), phospholipase A (44), and their catabolic enzymes, need to be evaluated.
We revealed that PNI induced differential metabolism of LOX-produced FA metabolites, compared with that of COX-produced metabolites. We found LTD4 to be an increased metabolite from ω-6 FA after PNI, whereas the others were unchanged. Increased LTD4 is thought to be derived from macrophages because both Alox5 (5-LOX) and Alox5ap (arachidonate 5-lipoxygenase activating protein, FLAP) are selectively expressed in macrophages in the DRG (5, 29). Although LA-derived LOX-produced metabolites, 9- and 13-HODEs, have been reported to be increased in the DRG in the spared nerve injury model or in the chemotherapy-induced peripheral neuropathy model (45, 46), we did not observe quantitative changes in these metabolites during our study. This discrepancy might be due to the differences in nerve injury models (spared nerve injury or spinal nerve injury), but not due to the strain and age of mice (almost the same as this study). Intriguingly, we found that PNI transiently increased the levels of ω-3 FA-derived LOX-produced metabolites (15-HEPE, 14-HDHA, and 17-HDHA) in the DRG on days 3 and 7, and these were returned to the same level as the contralateral side by day 14. It is reported that 15-HEPE and 17-HDHA are precursors of lipoxin A5 and D-series resolvins, respectively (19, 20), even though we could not detect these in the present study. Lipoxins and resolvins are well-known SPM family species, and many studies have demonstrated the anti-inflammatory and analgesic properties of their pharmacological treatment (20–23). The present study raises the following new questions: how are ω-3 FAs selectively subjected to LOX-mediated oxidation and why are ω-3 FA-derived metabolites transiently increased in a specified period? Further studies are needed to resolve these important questions and understand the pathology of nerve injuries.
Following PNI, the amount of 8-iso-PGE2, which is synthesized by non-enzymatic oxidation, was significantly increased. An isoprostane, 8-iso-PGE2 is a biomarker of oxidative stress; thus, it is indicated that PNI elevates oxidative stress. In fact, several reports have demonstrated elevated oxidative stress after nerve injury (31, 47). Therefore, our results are consistent with the previous reports.
Our lipidomic analysis revealed that PNI also affects the amount of ethanolamides in the DRG. Among ethanolamide-related species, AEA is one of the most studied endocannabinoids and acts on type 1 cannabinoid receptors (48). In this study, although AEA levels were not altered, PNI increased the amount of OEA, which was reported to activate peroxisome proliferator-activated receptor-α (PPARα), with a median effective concentration of 0.12 μM (49). Recently, PPARα is reported to be enriched in SGCs in the DRG, and the expression of PPARα target genes is upregulated 3 days after nerve injury (29). Moreover, activation of PPARα signaling promotes axonal regeneration in peripheral nerves (29). Although further investigation is needed, the increased OEA may be an acute response to promote axonal regeneration after nerve injury.
We detected only five CYP-produced metabolites in our measurement. Although the metabolites derived from LA, such as epoxyoctadecenoic acids and DiHOMEs, have been shown to be increased in the DRG in several pain models, including the spared nerve injury model (46, 50, 51); however, in this study, the spinal nerve injury procedure did not elicit the changes in DiHOMEs from LA. In addition, as for EETs that could not be detected in the present study, detection sensitivity can be improved by using more suitable methods for measuring EETs, and further analysis may reveal interesting phenomena.
As important evidence, it has been reported that excessive dietary ω-6 FA induces damages to the peripheral nervous systems and results in sensory abnormalities (52). Furthermore, sciatic nerve transection increases AA-containing phosphatidylcholine in the mouse DRG (53). These reports and the evidence provided from the present study imply the importance of quality of lipid in our health and disease. Given that most of FAs are stored as glycerophospholipids, the studies focused on glycerophospholipids will be needed to understand PNI pathologies.
In conclusion, we presented the temporal changes in FA metabolism in the DRG after nerve injury. Moreover, we demonstrated the importance of focusing on metabolites themselves. It is becoming increasingly important to study biology through the lipidome or metabolome in combination with the transcriptome. However, because some FA metabolites and lipid species can be synthesized through several pathways, careful consideration should be used when focusing on such species. We expect that our findings will help identify novel principles of the pathology of nerve injury, contribute to pain alleviation, and promote nerve regeneration.
Materials and methods
Mice
C57BL/6 mice were purchased from CLEA Japan (Japan). All the mice used were 7–9 weeks old at the start of the experiments. Animals were housed in groups of 3–6 per cages with lights on from 8:00 to 20:00 and were provided food and water ad libitum. All animal experiments were conducted according to the guidelines of the Animal Research Committee of the National Center for Global Health and Medicine, using protocols approved by the committee.
Peripheral nerve injury
We only used male mice for the spinal nerve injury procedure as a model of neuropathic pain (54). Briefly, mice were anesthetized with isoflurane and a small incision was made on the back. The L5 transverse process was then removed to expose the L4 spinal nerve. The exposed L4 spinal nerve was carefully cut, and then, the wound and skin were sutured with 5-0 silk.
Behavioral test
Mechanical sensitivity was assessed using the von Frey test. Each mouse was placed in a wire mesh cage and habituated for more an hour before testing. Calibrated von Frey filaments (0.02–2.0 g) were applied to the mid-plantar skin of each hind paw. The 50% paw withdrawal threshold was determined using the up-down method (54).
Sample preparation for lipid measurement
Mice were deeply anesthetized and perfused with ice-cold saline through the left ventricle. The L4 DRG were quickly removed and frozen in liquid nitrogen. Frozen tissues were pulverized and the lipid components were extracted for 60 min at 4 °C in methanol spiked with a deuterium-labeled internal standard mixture, which included PGD2-d4, PGE2-d4, PGF2α-d9, 6-keto-PGF1α-d4, TXB2-d4, LTB4-d4, LTC4-d5, LTD4-d5, LTE4-d5, 5-HETE-d8, 12-HETE-d8, 15-HETE-d8, AEA-d4, OEA-d4, tetranor-PGEM-d6, AA-d8, EPA-d5, and DHA-d5. The supernatants were collected after centrifugation at 15,000 × g for 10 min at 4 °C. The methanol extracts were purified by a solid-phase method using an Oasis HLB column (Waters).
Lipid measurement
Endogenous FA metabolites were measured using a triple quadrupole mass spectrometer LCMS-8060 (Shimadzu) as previously described (25, 55). Briefly, a reverse-phase column (Kinetex C8, 2.1 × 150 mm, 2.6 μm, Phenomenex) was used for chromatographic separation with a binary mobile phase comprising 0.1% formic acid/water (mobile phase A) and acetonitrile (mobile phase B). The gradient of the mobile phase (%A/%B) was programmed as follows: 0 min (90/10), 5 min (75/25), 10 min (65/35), 20 min (25/75), 20.1–28 min (5/95), 28.1–30 min (90/10). The flow rate was 0.4 ml/min, and the column temperature was 40 °C. The selected reaction-monitoring transitions are listed in Supplementary Table 1. Raw data were analyzed by using LabSolutions Insight (Shimadzu). The signals were compared with the standard curves for quantification. Although each sample containing six (respectively 8) DRGs was analyzed using LC-MS, the graphs show the values of metabolites calculated per one DRG.
Statistical analyses
Data were analyzed using two-way ANOVA with a post-hoc Bonferroni test using GraphPad Prism 7 software to determine differences among groups. Statistical significance was set at P < 0.05.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary materials, further inquiries can be directed to the corresponding authors.
Ethics statement
The animal study was reviewed and approved by the Animal Research Committee of the National Center for Global Health and Medicine.
Author contributions
SY designed and performed most of the experiments, analyzed the data, and wrote the manuscript. TH-Y performed LC-MS analysis and analyzed the data. TS supervised the study. HS conceived the study, supervised the overall project, and wrote the manuscript. All the authors have read, discussed the manuscript, and contributed to the article and approved the submitted version.
Funding
This work was supported by JSPS KAKENHI Grant Numbers JP19K16938 and JP21K15309 (to SY), the Core Research for Evolutional Science and Technology (CREST) from the Japan Agency for Medical Research and Development (AMED) under Grant Number 22gm0910011 (to HS), the Project for Cancer Research and Therapeutic Evolution (P-CREATE) from AMED under Grant Number 21cm0106116 (to HS), by The Nakatomi Foundation (to SY), and by Takeda Science Foundation 15668360 (to TS). SY was a research fellow of the JSPS (21J00759).
Acknowledgments
The authors are grateful to Kanako Ashikari-Yamamoto for providing an excellent illustration (Figure 1A). We thank Dr. Keizo Waku (Teikyo University, Japan) for constructive comments and all members of our laboratory for their advice, discussion, and technical supports. We would like to thank Editage (www.editage.com) for English language editing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Department of Lipid Signaling and Lipid Life Science, National Center for Global Health and Medicine, is financially supported by Ono Pharmaceutical Co., Ltd.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpain.2022.948689/full#supplementary-material
Abbreviations
AA, arachidonic acid; AEA, arachidonoyl ethanolamide; COX, cyclooxygenase; CYP, cytochrome-P450; DHA, docosahexaenoic acid; DiHETE, dihydroxyeicosatetraenoic acid; DiHOME, dihydroxyoctadecenoic acid; DRG, dorsal root ganglion; EET, epoxyeicosatrienoic acid; EPA, eicosapentaenoic acid; FA, fatty acid; HDHA, hydroxydocosahexaenoic acid; HEPE, hydroxyeicosapentaenoic acid; HETE, hydroxyeicosatetraenoic acid; HODE, hydroxyoctadecadienoic acid; HpETE, hydroperoxyeicosatetraenoic acid; HpODE, hydroperoxyoctadecadienoic acid; LA, linoleic acid; LC-MS/MS, liquid chromatography-mass spectrometry; LOX, lipoxygenase; LT, leukotriene; OEA, oleoyl ethanolamide; PG, prostaglandin; PNI, peripheral nerve injury; PPARα, peroxisome proliferator-activated receptor-α; scRNA-seq, single cell RNA-sequencing; SGC, satellite glial cell; SPM, specialized pro-resolving lipid mediator; TRPV1, transient receptor potential vanilloid 1; TX, thromboxane; 12-HHT, 12-hydroxyheptadecatrienoic acid; 15-HETrE, 15-hydroxytrienoic acid.
References
1. Colloca L, Ludman T, Bouhassira D, Baron R, Dickenson AH, Yarnitsky D, et al. Neuropathic Pain. Nat Rev Dis Primers. (2017) 3:17002. doi: 10.1038/nrdp.2017.2
2. van Hecke O, Austin SK, Khan RA, Smith BH, Torrance N. Neuropathic pain in the general population: a systematic review of epidemiological studies. Pain. (2014) 155:654–62. doi: 10.1016/j.pain.2013.11.013
3. Yekkirala AS, Roberson DP, Bean BP, Woolf CJ. Breaking Barriers to Novel Analgesic Drug Development. Nat Rev Drug Discov. (2017) 16:545–64. doi: 10.1038/nrd.2017.87
4. Niehaus JK, Taylor-Blake B, Loo L, Simon JM, Zylka MJ. Spinal macrophages resolve nociceptive hypersensitivity after peripheral injury. Neuron. (2021) 109:1274–82 e6. doi: 10.1016/j.neuron.2021.02.018
5. Renthal W, Tochitsky I, Yang L, Cheng YC, Li E, Kawaguchi R, et al. Transcriptional reprogramming of distinct peripheral sensory neuron subtypes after axonal injury. Neuron. (2020) 108:128–44 e9. doi: 10.1016/j.neuron.2020.07.026
6. Nguyen MQ, Le Pichon CE, Ryba N. Stereotyped transcriptomic transformation of somatosensory neurons in response to injury. Elife. (2019) 8:e49679. doi: 10.7554/eLife.49679
7. Kuivenhoven JA, Hegele RA. Mining the genome for lipid genes. Biochim Biophys Acta. (2014) 1842:1993–2009. doi: 10.1016/j.bbadis.2014.04.028
8. Lamari F, Mochel F, Sedel JM F. Saudubray Disorders of phospholipids, sphingolipids and fatty acids biosynthesis: toward a new category of inherited metabolic diseases. J Inherit Metab Dis. (2013) 36:411–25. doi: 10.1007/s10545-012-9509-7
9. Valentine WJ, Yanagida K, Kawana H, Kono N, Noda NN, Aoki J, et al. Update and nomenclature proposal for mammalian lysophospholipid acyltransferases, which create membrane phospholipid diversity. J Biol Chem. (2022) 298:101470. doi: 10.1016/j.jbc.2021.101470
10. Shimizu T. Lipid mediators in health and disease: enzymes and receptors as therapeutic targets for the regulation of immunity and inflammation. Annu Rev Pharmacol Toxicol. (2009) 49:123–50. doi: 10.1146/annurev.pharmtox.011008.145616
11. Harayama T, Shimizu T. Roles of polyunsaturated fatty acids, from mediators to membranes. J Lipid Res. (2020) 61:1150–60. doi: 10.1194/jlr.R120000800
12. Yamamoto S, Egashira N. Lipid signaling in chemotherapy-induced peripheral neuropathy. Curr Opin Toxicol. (2021) 28:1–6. doi: 10.1016/j.cotox.2021.08.007
13. Osthues T, Sisignano M. Oxidized lipids in persistent pain states. Front Pharmacol. (2019) 10:1147. doi: 10.3389/fphar.2019.01147
14. Sisignano M, Bennett DL, Geisslinger G, Scholich K. Trp-channels as key integrators of lipid pathways in nociceptive neurons. Prog Lipid Res. (2014) 53:93–107. doi: 10.1016/j.plipres.2013.11.002
15. Moriyama T, Higashi T, Togashi K, Iida T, Segi E, Sugimoto Y, et al. Sensitization of Trpv1 by Ep1 and Ip reveals peripheral nociceptive mechanism of prostaglandins. Mol Pain. (2005) 1:3. doi: 10.1186/1744-8069-1-3
16. Southall MD, Vasko MR. Prostaglandin receptor subtypes, Ep3c and Ep4, mediate the prostaglandin E2-induced camp production and sensitization of sensory neurons. J Biol Chem. (2001) 276:16083–91. doi: 10.1074/jbc.M011408200
17. Patwardhan AM, Akopian AN, Ruparel NB, Diogenes A, Weintraub ST, Uhlson C, et al. Heat generates oxidized linoleic acid metabolites that activate Trpv1 and produce pain in rodents. J Clin Invest. (2010) 120:1617–26. doi: 10.1172/JCI41678
18. Patwardhan AM, Scotland PE, Akopian AN, Hargreaves KM. Activation of Trpv1 in the spinal cord by oxidized linoleic acid metabolites contributes to inflammatory hyperalgesia. Proc Natl Acad Sci U S A. (2009) 106:18820–4. doi: 10.1073/pnas.0905415106
19. Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature. (2014) 510:92–101. doi: 10.1038/nature13479
20. Serhan CN, Levy BD. Resolvins in inflammation: emergence of the pro-resolving superfamily of mediators. J Clin Invest. (2018) 128:2657–69. doi: 10.1172/JCI97943
21. Fattori V, Zaninelli TH, Rasquel-Oliveira FS, Casagrande R, Verri WA Jr. Specialized pro-resolving lipid mediators: a new class of non-immunosuppressive and non-opioid analgesic drugs. Pharmacol Res. (2020) 151:104549. doi: 10.1016/j.phrs.2019.104549
22. Leuti A, Fava M, Pellegrini N, Maccarrone M. Role of specialized pro-resolving mediators in neuropathic pain. Front Pharmacol. (2021) 12:717993. doi: 10.3389/fphar.2021.717993
23. Tao X, Lee MS, Donnelly CR, Ji RR. Neuromodulation, specialized proresolving mediators, and resolution of pain. Neurotherapeutics. (2020) 17:886–99. doi: 10.1007/s13311-020-00892-9
24. Schebb NH, Kuhn H, Kahnt AS, Rund KM, O'Donnell VB, Flamand N, et al. Formation, signaling and occurrence of specialized pro-resolving lipid mediators-what is the evidence so far? Front Pharmacol. (2022) 13:838782. doi: 10.3389/fphar.2022.838782
25. Yamada M, Kita Y, Kohira T, Yoshida K, Hamano F, Tokuoka SM, et al. A Comprehensive quantification method for eicosanoids and related compounds by using liquid chromatography/mass spectrometry with high speed continuous ionization polarity switching. J Chromatogr B Analyt Technol Biomed Life Sci. (2015) 995–6:74–84. doi: 10.1016/j.jchromb.2015.05.015
26. Piomelli D, Astarita G, Rapaka R. A neuroscientist's guide to lipidomics. Nat Rev Neurosci. (2007) 8:743–54. doi: 10.1038/nrn2233
27. Harayama T, Riezman H. Understanding the diversity of membrane lipid composition. Nat Rev Mol Cell Biol. (2018) 19:281–96. doi: 10.1038/nrm.2017.138
28. Piomelli D, Sasso O. Peripheral gating of pain signals by endogenous lipid mediators. Nat Neurosci. (2014) 17:164–74. doi: 10.1038/nn.3612
29. Avraham O, Deng PY, Jones S, Kuruvilla R, Semenkovich CF, Klyachko VA, et al. Satellite glial cells promote regenerative growth in sensory neurons. Nat Commun. (2020) 11:4891. doi: 10.1038/s41467-020-18642-y
30. Lee J, Shin JE, Lee B, Kim H, Jeon Y, Ahn SH, et al. The stem cell marker Prom1 promotes axon regeneration by down-regulating cholesterol synthesis via Smad signaling. Proc Natl Acad Sci U S A. (2020) 117:15955–66. doi: 10.1073/pnas.1920829117
31. Petho G, Reeh PW. Sensory and signaling mechanisms of bradykinin, eicosanoids, platelet-activating factor, and nitric oxide in peripheral nociceptors. Physiol Rev. (2012) 92:1699–775. doi: 10.1152/physrev.00048.2010
32. Mahmoud MF, Rezq S, Alsemeh AE, Abdelfattah MAO, El-Shazly AM, Daoud R, et al. Potamogeton Perfoliatus L. Extract attenuates neuroinflammation and neuropathic pain in sciatic nerve chronic constriction injury-induced peripheral neuropathy in rats. Front Pharmacol. (2021) 12:799444. doi: 10.3389/fphar.2021.799444
33. Schafers M, Marziniak M, Sorkin LS, Yaksh TL, Sommer C. Cyclooxygenase inhibition in nerve-injury- and tnf-induced hyperalgesia in the rat. Exp Neurol. (2004) 185:160–8. doi: 10.1016/j.expneurol.2003.09.015
34. St-Jacques B, Ma W. Role of prostaglandin E2 in the synthesis of the pro-inflammatory cytokine interleukin-6 in primary sensory neurons: an in vivo and in vitro study. J Neurochem. (2011) 118:841–54. doi: 10.1111/j.1471-4159.2011.07230.x
35. Treutlein EM, Kern K, Weigert A, Tarighi N, Schuh CD, Nusing RM, et al. The Prostaglandin E2 Receptor Ep3 Controls Cc-Chemokine Ligand 2-mediated neuropathic pain induced by mechanical nerve damage. J Biol Chem. (2018) 293:9685–95. doi: 10.1074/jbc.RA118.002492
36. Schuh CD, Pierre S, Weigert A, Weichand B, Altenrath K, Schreiber Y, et al. Prostacyclin mediates neuropathic pain through interleukin 1beta-expressing resident macrophages. Pain. (2014) 155:545–55. doi: 10.1016/j.pain.2013.12.006
37. Chopra B, Giblett S, Little JG, Donaldson LF, Tate S, Evans RJ, et al. Cyclooxygenase-1 is a marker for a subpopulation of putative nociceptive neurons in rat dorsal root ganglia. Eur J Neurosci. (2000) 12:911–20. doi: 10.1046/j.1460-9568.2000.00979.x
38. Hasegawa S, Kohro Y, Shiratori M, Ishii S, Shimizu T, Tsuda M, et al. Role of paf receptor in proinflammatory cytokine expression in the dorsal root ganglion and tactile allodynia in a rodent model of neuropathic pain. PLoS ONE. (2010) 5:e10467. doi: 10.1371/journal.pone.0010467
39. Jang Y, Kim M, Hwang SW. Molecular mechanisms underlying the actions of arachidonic acid-derived prostaglandins on peripheral nociception. J Neuroinflammation. (2020) 17:30. doi: 10.1186/s12974-020-1703-1
40. Avraham O, Feng R, Ewan EE, Rustenhoven J, Zhao G, Cavalli V. Profiling sensory neuron microenvironment after peripheral and central axon injury reveals key pathways for neural repair. Elife. (2021) 10:e68457. doi: 10.7554/eLife.68457
41. Austin PJ, Moalem-Taylor G. The neuro-immune balance in neuropathic pain: involvement of inflammatory immune cells, immune-like glial cells and cytokines. J Neuroimmunol. (2010) 229:26–50. doi: 10.1016/j.jneuroim.2010.08.013
42. Ji RR, Chamessian A, Zhang YQ. Pain regulation by non-neuronal cells and inflammation. Science. (2016) 354:572–7. doi: 10.1126/science.aaf8924
43. Kim CF, Moalem-Taylor G. Detailed Characterization of Neuro-Immune Responses Following Neuropathic Injury in Mice. Brain Res. (2011) 1405:95–108. doi: 10.1016/j.brainres.2011.06.022
44. Murakami M, Sato H, Taketomi Y. Updating phospholipase A2 biology. Biomolecules. (2020) 10:1457. doi: 10.3390/biom10101457
45. Hohmann SW, Angioni C, Tunaru S, Lee S, Woolf CJ, Offermanns S, et al. The G2a Receptor (Gpr132) contributes to oxaliplatin-induced mechanical pain hypersensitivity. Sci Rep. (2017) 7:446. doi: 10.1038/s41598-017-00591-0
46. Osthues T, Zimmer B, Rimola V, Klann K, Schilling K, Mathoor P, et al. The lipid receptor G2a (Gpr132) mediates macrophage migration in nerve injury-induced neuropathic pain. Cells. (2020) 9:1740. doi: 10.3390/cells9071740
47. Conforti L, Gilley J, Coleman MP. Wallerian degeneration: an emerging axon death pathway linking injury and disease. Nat Rev Neurosci. (2014) 15:394–409. doi: 10.1038/nrn3680
48. Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, et al. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. (1992) 258:1946–9. doi: 10.1126/science.1470919
49. Fu J, Gaetani S, Oveisi F, Lo Verme J, Serrano A, Rodriguez De Fonseca F, et al. Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor Ppar-Alpha. Nature. (2003) 425:90–3. doi: 10.1038/nature01921
50. Sisignano M, Angioni C, Park CK, Meyer Dos Santos S, Jordan H, Kuzikov M, et al. Targeting Cyp2j to reduce paclitaxel-induced peripheral neuropathic pain. Proc Natl Acad Sci U S A. (2016) 113:12544–9. doi: 10.1073/pnas.1613246113
51. Zimmer B, Angioni C, Osthues T, Toewe A, Thomas D, Pierre SC, et al. The Oxidized linoleic acid metabolite 12,13-dihome mediates thermal hyperalgesia during inflammatory pain. Biochim Biophys Acta Mol Cell Biol Lipids. (2018) 1863:669–78. doi: 10.1016/j.bbalip.2018.03.012
52. Boyd JT, LoCoco PM, Furr AR, Bendele MR, Tram M, Li Q, et al. Elevated dietary omega-6 polyunsaturated fatty acids induce reversible peripheral nerve dysfunction that exacerbates comorbid pain conditions. Nat Metab. (2021) 3:762–73. doi: 10.1038/s42255-021-00410-x
53. Mihara Y, Horikawa M, Sato S, Eto F, Hanada M, Banno T, et al. Lysophosphatidic Acid precursor levels decrease and an arachidonic acid-containing phosphatidylcholine level increases in the dorsal root ganglion of mice after peripheral nerve injury. Neurosci Lett. (2019) 698:69–75. doi: 10.1016/j.neulet.2018.12.035
54. Kohro Y, Matsuda T, Yoshihara K, Kohno K, Koga K, Katsuragi R, et al. Spinal astrocytes in superficial laminae gate brainstem descending control of mechanosensory hypersensitivity. Nat Neurosci. (2020) 23:1376–87. doi: 10.1038/s41593-020-00713-4
Keywords: dorsal root ganglion, fatty acid, leukotrien, lipid mediator, pain, peripheral nerve injury, prostaglandin
Citation: Yamamoto S, Hashidate-Yoshida T, Shimizu T and Shindou H (2022) Profiling of fatty acid metabolism in the dorsal root ganglion after peripheral nerve injury. Front. Pain Res. 3:948689. doi: 10.3389/fpain.2022.948689
Received: 20 May 2022; Accepted: 06 July 2022;
Published: 29 July 2022.
Edited by:
Isaura Tavares, University of Porto, PortugalReviewed by:
Karen Wagner, University of California, Davis, United StatesAubin Moutal, Saint Louis University, United States
Copyright © 2022 Yamamoto, Hashidate-Yoshida, Shimizu and Shindou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shota Yamamoto, c3l5YW1hbW90b0ByaS5uY2dtLmdvLmpw; Hideo Shindou, aHNoaW5kb3VAcmkubmNnbS5nby5qcA==