 Sascha R. A. Alles
Sascha R. A. Alles Peter A. Smith
Peter A. Smith- 1Department of Anesthesiology and Critical Care Medicine, University of New Mexico School of Medicine, Albuquerque, NM, United States
- 2Department of Pharmacology, Neuroscience and Mental Health Institute, University of Alberta, Edmonton, AB, Canada
The persistence of increased excitability and spontaneous activity in injured peripheral neurons is imperative for the development and persistence of many forms of neuropathic pain. This aberrant activity involves increased activity and/or expression of voltage-gated Na+ and Ca2+ channels and hyperpolarization activated cyclic nucleotide gated (HCN) channels as well as decreased function of K+ channels. Because they display limited central side effects, peripherally restricted Na+ and Ca2+ channel blockers and K+ channel activators offer potential therapeutic approaches to pain management. This review outlines the current status and future therapeutic promise of peripherally acting channel modulators. Selective blockers of Nav1.3, Nav1.7, Nav1.8, Cav3.2, and HCN2 and activators of Kv7.2 abrogate signs of neuropathic pain in animal models. Unfortunately, their performance in the clinic has been disappointing; some substances fail to meet therapeutic end points whereas others produce dose-limiting side effects. Despite this, peripheral voltage-gated cation channels retain their promise as therapeutic targets. The way forward may include (i) further structural refinement of K+ channel activators such as retigabine and ASP0819 to improve selectivity and limit toxicity; use or modification of Na+ channel blockers such as vixotrigine, PF-05089771, A803467, PF-01247324, VX-150 or arachnid toxins such as Tap1a; the use of Ca2+ channel blockers such as TTA-P2, TTA-A2, Z 944, ACT709478, and CNCB-2; (ii) improving methods for assessing “pain” as opposed to nociception in rodent models; (iii) recognizing sex differences in pain etiology; (iv) tailoring of therapeutic approaches to meet the symptoms and etiology of pain in individual patients via quantitative sensory testing and other personalized medicine approaches; (v) targeting genetic and biochemical mechanisms controlling channel expression using anti-NGF antibodies such as tanezumab or re-purposed drugs such as vorinostat, a histone methyltransferase inhibitor used in the management of T-cell lymphoma, or cercosporamide a MNK 1/2 inhibitor used in treatment of rheumatoid arthritis; (vi) combination therapy using drugs that are selective for different channel types or regulatory processes; (vii) directing preclinical validation work toward the use of human or human-derived tissue samples; and (viii) application of molecular biological approaches such as clustered regularly interspaced short palindromic repeats (CRISPR) technology.
Introduction
Whilst opioids are extremely effective in managing deep and nociceptive pain, the drugs available for treatment of neuropathic pain display limited effectiveness (1, 2). Sites of action of anti-allodynic agents such gabapentinoids, tricyclic antidepressants, and noradrenaline-serotonin uptake inhibitors such as duloxetine or venlafaxine reside predominantly within the spinal cord and at other central loci (2–5). Because the persistence of aberrant and spontaneous activity in injured peripheral neurons is imperative for the development and persistence of many forms of neuropathic pain (2, 6–14), the peripheral nervous system offers a range of actual and potential drug targets. It has been argued that targeting the peripheral nervous system with substances that do not readily cross the blood-brain barrier, may circumvent the dose-limiting side effects seen with centrally acting agents (15). For example, adverse centrally-mediated effects of gabapentin include dizziness, somnolence, fatigue, ataxia, and nystagmus (16). This review thus outlines the current status and future promise of peripherally-acting agents; focusing on those that interact with cation channels in primary afferent neurons.
Peripheral nerve injury promotes Wallerian degeneration of severed axons, Schwann cell activation and the generation and release of chemokines, cytokines, and growth factors. These sensitize sensory nerve endings, attract macrophages and lymphocytes, alter gene expression, promote post-translational modification of proteins and alter ion channel function (17–23). The activity and/or expression of voltage-gated TTX-sensitive Na+ channels, voltage-gated Ca2+ channels, ASIC channels, TRP channels, and HCN channels is increased (24–27) whereas that of K+ channels is decreased (28). These peripheral ion channels thus present a viable target for therapeutic intervention (24, 28) as alterations in their activity underlies the increased excitability of primary afferents (11, 12, 29–35). In the interest of brevity, this review is confined to description of injury-induced changes in voltage-gated cation channels in primary afferent neurons and their potential as therapeutic targets. Information on ligand-gated channels which includes purinergic P2X3 channels, acid sensing ion channels (ASIC), and various types of TRP channel may be found in recent publications and reviews (3, 25, 36–40).
A summary of viable therapeutic approaches to the management of neuropathic pain by modulation of function or expression of voltage-gated cation channels is presented in Table 1.
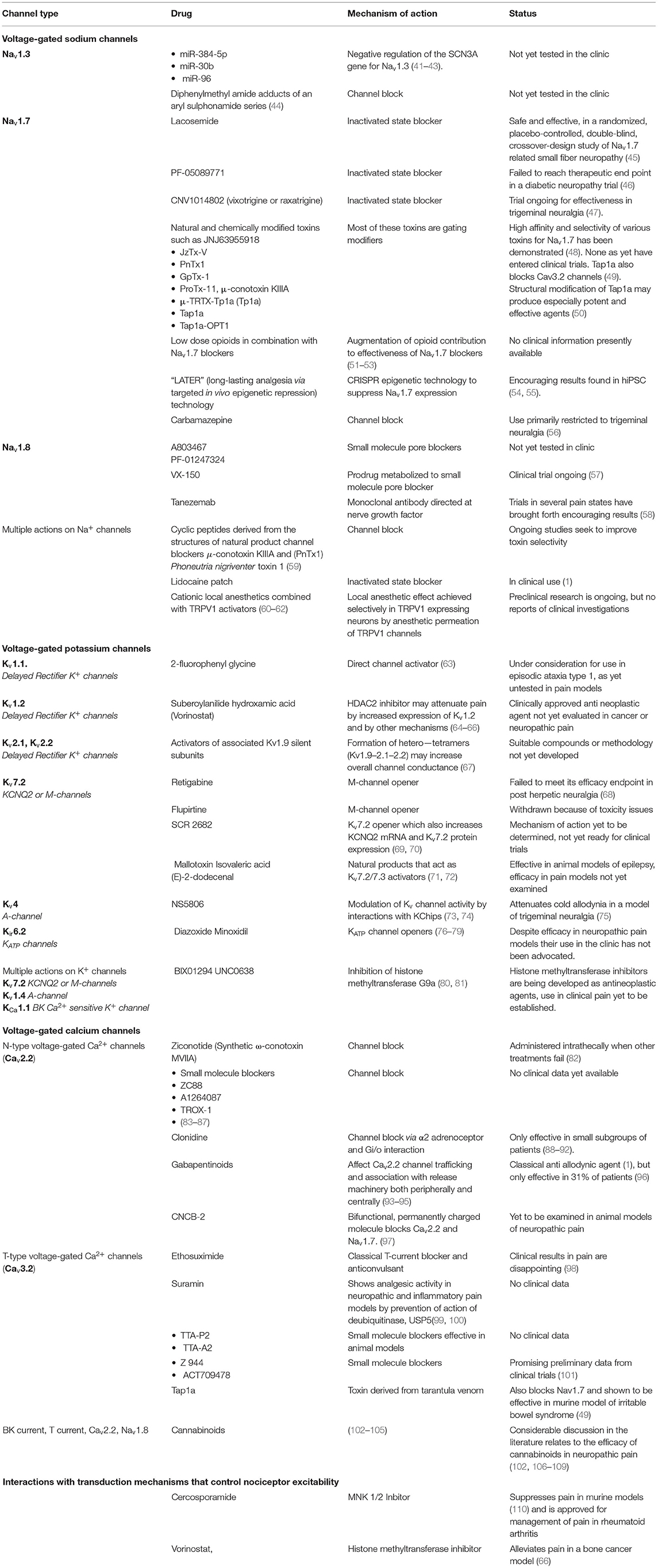
Table 1. Potential and actual therapeutic candidates.
Voltage-Gated Na+ Channels
Injury-induced increases in Na+ channel function were first described over 20 years ago (111–113). They reflect altered expression of channel protein and/or its accessory subunits, altered trafficking or post-translational modification and/or modulation (114, 115).
The genetic and structural definitions of Nav1.1–Nav1.9 channel subtypes was also established many years ago (116–118) and this has led to a mechanistic and molecular understanding of injury-induced changes (8, 114). This has paved the way for selective targeting of TTX-sensitive Nav1.3, 1.6, and 1.7 channels and TTX-resistant Nav1.8 channels as these are particularly important in the generation and maintenance of neuropathic pain (114, 119–122).
As described below, different Nav channel subtypes in different neuronal populations are involved in different types of neuropathic and nociceptive pain (114, 123–125).
Expression and Therapeutic Modulation of TTX-Sensitive Na+ Channels
Role of Nav1.3 in Neuropathic Pain
Nav1.3 channels were previously known as type III Na+ channels. They are TTX-sensitive products of the SCN3A gene and are found in neurons and cardiac myocytes with the highest level in embryonic and early postnatal animals (117, 126, 127). In DRG neurons, they exhibit rapid recovery from inactivation or “repriming,” thereby enhancing repetitive discharge (128). Their involvement in neuropathic pain is supported by the attenuation of allodynia seen with intra-ganglionic injection of adeno-associated virus expressing small hairpin RNA targeting Nav1.3 (129). Nerve injury upregulates and promotes re-expression of Nav1.3 in adult DRG neurons (127, 130, 131) as well as in spinal dorsal horn and thalamus (132, 133). This may reflect removal of suppression of the SCN3A gene by microRNAs such as miR-384-5p, mir-96 and/or miR-30b suggesting that their targeted delivery may be of use in pain management (41–43).
Pharmacological Manipulation of Nav1.3
Because Nav1.3 is mainly present in embryonic and early neonatal animals and because nerve injury promotes selective upregulation of Nav1.3 in nociceptive pathways of adults, there is considerable interest in developing Nav1.3 blockers. Structure activity studies starting with a diphenylmethyl amide adduct of an aryl sulphonamide has led to the development of compounds with good selectivity for Nav1.3 as well as favorable pharmacokinetics (44).
Role of Nav1.6 in Neuropathic Pain
Nav1.6 is another TTX-sensitive Na+ channel. It is the product of the SCN8A gene (117) and was previously known as PN4. Nav1.6 channels are expressed along the whole length of sensory unmyelinated axons (134) and are clustered at nodes of Ranvier in myelinated fibers where they participate in “saltatory” conduction (135).
The observation that knockout of Nav1.6 reduces injury-induced pain behaviors and sensory neuron excitability (136–138) implicates it in the etiology of neuropathic pain. It has recently been implicated in a model of vincristine-induced chemotherapy induced peripheral neuropathy (CIPN) and allodynia (139) and is upregulated in the DRG in a model of diabetic neuropathy (140). These findings are corroborated by the description of a gain-of-function mutation in Nav1.6 in a case of trigeminal neuralgia (141). Since its role in in pain etiology was established relatively recently (114, 142), there have been as yet no attempts to modulate Nav1.6 channel activity either in animal models or in the clinic.
Role of Nav1.7 in Neuropathic Pain
The TTX-sensitive Nav1.7 channel is involved in a multiplicity of neuropathic and nociceptive pain states (8, 48, 54, 114, 123, 143–146). It is the product of the SCN9A gene and was previously known as PN1. Nav1.7 is the dominant voltage-gated Na+ channel in peripheral sympathetic neurons and in all types of DRG neuron (117, 147). Its expression extends from peripheral nerve endings in the skin and viscera to primary afferent terminals in the dorsal horn (148) where it is especially concentrated (147). Nav1.7 is preferentially expressed in small diameter nociceptors including both the CGRP-positive subcategory and the non-peptidergic subcategory that bind the plant lectin IB4 from Griffonia simplicifolia (114). It is also found in olfactory sensory neurons, magnocellular neurosecretory cells of the hypothalamic supraoptic nucleus and in vagal afferents (51, 149–151). Because it is not found to any great extent in vital non-neuronal tissue such as heart or skeletal muscle (114, 147), Nav1.7 represents a specially attractive target for therapeutic manipulation. Although it is found in pancreatic alpha and beta cells it may be inactivated at their normal resting potential (152).
Immunohistochemical studies first demonstrated Nav1.7 upregulation in severed axons within human painful neuromas (122, 153) and Nav1.7 has been shown to be necessary for the release of the pain modulator substance P from primary afferent terminals (124).
Despite this, Nav1.7 does not appear to be involved in all manifestations of neuropathic pain. For example oxaliplatin-induced pain and cancer-induced bone pain do not require the presence of Nav1.7 or the Nav1.8-positive nociceptors in which Nav1.7 is enriched (123). By contrast, paclitaxel-induced CIPN involves the direction of Nav1.7 to cell membranes and axons of primary afferent fibers (154). Also, neuropathic pain produced by constriction injury (CCI) is abolished when Nav1.7 is selectively deleted in murine sensory neurons and although spinal nerve transection or tight ligation (SNL) also produces cold and mechanical allodynia this is not affected by selective knockout of Nav1.7 in DRG neurons. By contrast, knockout of Nav1.7 in both sympathetic and sensory fibers attenuates both forms of allodynia (123). This is because SNL involves sprouting of Nav1.7 expressing perivascular sympathetic fibers (155, 156) and their ectopic interaction with DRG neurons (157–159).
Patients with a rare, chronic pain conditions such as primary erythromelalgia or paroxysmal extreme pain disorder exhibit gain of function mutations in SCN9A (8, 146, 160–163). As of 2019, 30 mutations in SCN9A genes had been described in inherited erythromelalgia and 13 in paroxysmal extreme pain disorder (114). In the case of inherited erythromelalgia, isoleucine 848 is replaced by threonine. This I848T mutation increases the amplitude of current produced by Nav1.7 in response to slow, small depolarizations as a result of a hyperpolarizing shift in activation and slowed deactivation (161). Recently, protein kinase C has been found to be responsible for the phosphorylation of T848 found in mutant channels and this accounts for the shift in activation (164). Meents et al. (165) have differentiated human induced pluripotent stem cells (hiPSC) from erythromelalgia patients into sensory nociceptors. This will provide an extensive supply of human nociceptors for further study of erythromelalgia. Mutations seen in Nav1.7 channels of erythromelagia patients also occur in those with paroxysmal extreme pain disorder with an additional suppression of fast inactivation (163). Gain of function mutations of SCN9A also worsen neuropathic pain in a small cohort of patients with painful diabetic neuropathy (166).
Although some patients with small fiber neuropathy display the I228M gain-of-function mutation in Nav1.7, a pain phenotype does not appear until they reach adulthood (167). Expression of this same mutation in mice promotes increased DRG excitability without the appearance of a measurable pain phenotype. It is suggested that some compensatory mechanism may restrain the development of pain in the mouse model and the possible existence of a similar process in humans may delay the development of a pain phenotype until adulthood (168).
Patients with a rare congenital insensitivity to pain (CIP) express a loss of function mutation in Nav1.7 (169) and global knockout of Nav1.7 in mice recapitulates this human phenotype (170). Differentiation of hiPSC's from CIP patients into sensory nociceptors, produced cells where Nav1.7 was appropriately expressed and trafficked to the cell membrane. Since these cells failed to respond to depolarizing stimuli, CIP can be attributed to changes in the function of the channels per se rather than defects in their expression or trafficking (54). These results also provide new evidence for a role of Nav1.7 in human nociception. As of 2019, 26 mutations in SCN9A have been reported to contribute to CIP.
In addition to its role in controlling neuronal excitability and neurotransmitter release, Nav1.7 directly or indirectly affects gene expression (51, 52, 171). Nav1.7 deletion, leads to upregulation of Penk mRNA for the enkephalin precursor proenkephalin in DRG as well as met-enkephalin protein. Since a similar effect is seen with TTX, the upregulation of endogenous opioid function may be contingent on decreased levels of intracellular Na+ (52). These authors also showed that blockade of opioid receptors with naloxone reduces the analgesia seen in both male and female Nav1.7-null mutant mice and in a human patient with Nav1.7 dependent congenital insensitivity to pain [see also (51)]. The relationship between increased opioid function and decreased Nav1.7 function is supported by the observation that the analgesic effect of a selective Nav1.7 blocker, μ-theraphotoxin-Pn3a (from the tarantula Pamphobeteus nigricolor), is augmented by administration with sub-effective doses of opioids or with an enkephalinase inhibitor (172). Further analysis of this effect showed that Nav1.7 knockout mice have normal peripheral nociceptor activity but synaptic transmission from nociceptor central terminals is greatly reduced in an opioid-dependent fashion. Analgesia was reversed substantially by central but not peripheral application of opioid antagonists (51). These authors thus concluded inhibition of neurotransmitter release is the principal mechanism of analgesia in mouse and human Nav1.7-null mutants.
Second order sensory neurons in the spinal dorsal horn express few transcripts of Nav1.7 mRNA. Despite this, immunoreactivity for channel protein is abundant yet is reduced following rhizotomy (173). This suggests that sensory neurons are the source of Nav1.7 in spinal dorsal horn neurons and that intercellular transport of the protein occurs between these two neuronal populations. This conclusion was supported by the observation that selective deletion of Nav1.7 in peripheral neurons reduced the intrinsic excitability of dorsal horn neurons.
Pharmacological Manipulation of Nav1.7
Although it may not be involved in all types of neuropathic pain (123) it is absence from non-neuronal tissue such as heart or skeletal muscle (114, 147). Nav1.7 is therefore clearly an attractive target for therapeutic intervention (48, 114, 145, 154, 174). Moreover, the anticonvulsant lacosamide, which is an inactivated state blocker of Na+ channels (175, 176) has been found to be safe and effective, in a randomized, placebo-controlled, double-blind, crossover-design study of Nav1.7 related small fiber neuropathy [(45), Table 1]. Also, the effectiveness of carbamazepine which is used to treat trigeminal neuralgia (56) may in part reflect its affinity for Nav1.7 (177).
There is also considerable interest in various sulfonamide analogs which display selectivity toward Nav1.7 and are effective in pain mitigation in animal models [(178–183); see Table 1]. Therapeutic concentrations of the inactivated state blocker PF-05089771 increase the rheobase of control neurons, but not that of Nav1.7 knock-out neurons. Despite this selectivity for Nav1.7 and its effectiveness in animal models in vivo (54), a clinical study of PF-05089771 in subjects with painful diabetic peripheral neuropathy failed to meet defined efficacy criteria (46).
Another broad spectrum non-sulfonamide Nav blocker, vixotrigine, which was previously known as raxatrigine, or CNV1014802, BIIB074, or GSK-1014802 (184), has shown effectiveness in animal models of Nav1.7-dependent pain. Its safety in human patients has been established (185). A phase III clinical trial for effectiveness in trigeminal neuralgia and phase II trial for small fiber neuropathy are presently ongoing (47).
Nav1.7 and Natural Toxins
Another approach to therapeutic modulation of Nav1.7 activity involves potential use and/or structural modification of natural toxins (48, 186–190). These are typically gating modifiers as opposed to simple pore blockers so some natural toxins increase channel function whereas others attenuate it [(48); Table 2]. Starting points include the cone snail toxin, μ-conotoxin KIIIA, and PnTx1 (Phoneutria nigriventer toxin 1) from a Brazilian spider. Although structure activity studies of small cyclic peptides derived from the structure of these toxins has not as yet revealed Na+ channels subtype ligands, the analgesic effect of many of the ligands involves modulation Nav1.7 channel function. This result was achieved by observing attenuation of pain produced by the Nav1.7 selective activator α-scorpion toxin OD1 [(191); Table 2]. Further modifications of small cyclic peptides may reveal more subtype selective ligands with appropriate pharmacokinetics in vivo and improved bioavailability (59).

Table 2. List of toxins that modulate Nav1.7 channel activity.
Studies and modification of arachnoid toxins which display natural selectivity toward Nav1.7 may also lead to development of effective agents (190). As listed in Table 2, there are several examples.
Venom from the tarantula Grammostola porteri contains the 34-residue peptide, GpTx-1, with high and selective affinity for Nav1.7 (IC50 = 10 nM). Structural modifications of this peptide led to the identification of [Ala5, Phe6, Leu26, Arg28] GpTx-1 (also known as GpTx-1-71) IC50 = 1.6 nM (192). Both peptides exert powerful antinociception in mouse models of acute, visceral, inflammatory and neuropathic pain without impairment of motor co-ordination or development of tolerance (144). Another modified toxin derived from JzTx-V (from venom of the Chinese tarantula Chilobrachys jingzhao) has a 100-fold improved efficacy compared to GP-Tx-1-71 (193).
Studies of the venom from the Peruvian green-velvet tarantula Thrixopelma pruriens revealed a 33 residue peptide termed μ-TRTX-Tp1a (Tp1a or ProTx-III) with high selectivity and affinity for Nav1.7 (194). Unlike other spider toxins that inhibit the function of Nav channels, Tp1a inhibited hNaV1.7 without significantly altering the voltage-dependence of activation or inactivation. Like PnTx1, the analgesic effect of Tp1a was demonstrated by its ability to reverse spontaneous pain induced in mice by intraplantar injection of the Nav1.7 activator OD1 (194).
Recently another peptide toxin named Tap1a from the Venezuelan tarantula Theraphosa apophysis was shown to reverse colonic mechanical hypersensitivity in a mouse model of irritable bowel syndrome. The toxin's efficacy was shown to reflect selective targeting of Nav1.7 as well as the T-type Ca2+ channel Cav3.2 (49).
High-throughput screening has also identified μ-TRTX-Df1a (Df1a) from the venom of the spider Davus fasciatus as an Nav modulator. This 34-residue peptide inhibits responses mediated by Nav1.7 that is endogenously expressed in the human neuroblastoma cell line SH-SY5Y. It also inhibits T-type calcium (Cav3.1 and Cav3.3) currents and other Nav currents expressed in HEK 293 cells but has no effect on the voltage-gated potassium channel [Kv2.1; (195)]. Df1a is active in vivo and reverses the spontaneous pain behaviors induced by the scorpion venom Nav activator OD1.
Other investigations have used the venom-peptide ProTX-II (Protoxin II) from the Peruvian green velvet tarantula (Thrixopelma pruriens) as a scaffold, to engineer a library of over 1,500 peptides. This identified JNJ63955918 as a potent, highly selective, closed-state Nav1.7 blocking peptide which induces insensitivity to pain that closely recapitulates key features of the Nav1.7-null phenotype seen in mice and humans (196).
More recently attention has been drawn to huwentoxin-IV, from the Chinese bird spider Haplopelma schmidti. Because it has high affinity for sodium channels it is an attractive scaffold for engineering Nav1.7-selective molecules and several new ligands with high affinity and selectivity have been identified (197).
Other natural products which block Nav1.7 channels include HNTX I and III from the spider Ornithoctonus hainana (198, 199), bulleyaconitine from aconitum bulleyanum plants (200) and the Japanese traditional medicine goshajinkigan (2, 201).
Clinical Status of Nav1.7 Blockers
In general, despite intensive pre-clinical studies with Nav1.7 blockers, tests of their efficacy in the clinic has yielded rather disappointing results [(48), Table 1] and to the best of our knowledge no studies of tarantula and other toxins in the clinic have appeared. Nevertheless, the continued study of toxins, small molecule blockers and monoclonal antibodies (202) should and will continue (2, 8). In particular, further structural modification of small molecule blockers such as CNV1014802 (vixotrigine) and PF-05089771 as well as chemical modification of natural toxins (48, 50) may provide a route to the development of more efficacious therapeutic entities. The tarantula toxin Tap1a shows particular promise as it appears to selectively target both Nav1.7 and Cav3.2 (49).
Since the consequences of Nav1.7 blockade are mediated at least in part by endogenous opioids (51, 52), benefit may be obtained by combining small molecule blockers or toxins with low doses of opioids (48, 53).
The development of monoclonal antibodies and the delivery of the inhibitory micoRNA miR-182 (203) or modifiers of Na+ channel β subunits (204) may reveal additional therapeutic approaches. This approach may be especially attractive as three different types of β subunits are differentially and selectively expressed in small, medium, and large diameter DRG neurons (205, 206).
An approach that has proved particularly effective for targeting Nav1.7 uses CRISPR-dCas9 technology (clustered regularly interspaced short palindromic repeats) (55). Epigenome engineering platforms were introduced intrathecally in mice via adeno-associated viruses. A novel approach that prevented expression of Nav1.7 by editing a regulatory sequence successfully repressed Nav1.7 expression in lumbar DRG, reduced thermal hyperalgesia in inflammatory pain models and decreased tactile allodynia in the neuropathic pain models without affecting normal motor function. It is anticipated that this “LATER” (long-lasting analgesia via targeted in vivo epigenetic repression technology) might have therapeutic potential in management of persistent pain states. This is important in practical terms as chronic pain patents usually present in the clinic when they have suffered for many months. The technology can of course be easily modified to control expression of any potential or central drug target.
Expression and Therapeutic Modulation of TTX-Resistant Na+ Channels
Role of Nav1.8 in Neuropathic Pain
The TTX-resistant Nav1.8 channel is predominant in small DRG neurons (124, 207–210) but its selective association with nociceptors has been questioned (211). It was originally known as SNS or PN3 and is encoded by the SCN10A gene (117). It is characterized by its high threshold for activation and its slow rate of inactivation at depolarized potentials (210). These properties enable it to generate a slow persistent inward current (212).
Although peripheral nerve injury attenuates Nav1.8 function in injured DRG neurons (213–215) it is thought to accumulate in uninjured neurons (216) and in neuromas that develop at sites of nerve injury (217). Selective blockade of Nav1.8 function promotes hypoalgesia (213), gain of function mutations of SCN10A in humans can promote painful neuropathy (218) and its optogenetic silencing in DRG attenuates neuropathic pain (219).
Pharmacological Manipulation of NaV1.8
The selective Nav1.8 blockers A803467 and PF-01247324 are being developed as potential antidysrhythmic agents (220). Although both are reported to attenuate allodynia in a rodent model (221, 222), they have yet to be used in clinical studies (223). Encouraging results have been seen with the pro-drug VX-150 which exhibits analgesic activity in healthy volunteers (57), but preclinical literature in support of these studies are not available online. The μO-conotoxins, MrVIA, MrVIB, and MfVIA block Nav1.8 and ongoing analysis seeks to increase their affinity by structural modifications (224).
Unlike the situation with Nav1.7, analgesia produced with blockade of Nav1.8 is not opioid-dependent (52) and may be attributable to decreased excitability of peripheral afferents and their central terminals (225).
Although the efficacy of the non-psychoactive cannabinoid, cannabidiol in management of neuropathic pain remains to be established (106), it was recently reported to decrease the excitability of DRG neurons by binding to the slow inactivated state of Nav1.8 channels (102).
Expression of Nav1.8 in peptidergic DRG neurons is controlled by nerve growth factor (NGF) (215) whereas its expression in non-peptidergic neurons is controlled by glial colony derived neurotrophic factor (GDNF) (226). This may account in part for the effectiveness of the NGF antagonist tanezumab in various pain states (58). In fact, its safety and efficacy in humans identifies tanezumab as one of more the promising new drug candidates for chronic and neuropathic pain (see Clinical Trials Government Identifiers: NCT02528188 and NCT02528188).
Role of Nav1.9 in Inflammatory Pain but Not in Neuropathic Pain
Nav1.9 is also TTX-resistant (227) and is encoded by the SCN11A gene. It was previously known as NaN. Unlike genes encoding other voltage-gated Na+ channels, murine SCN11A is only 75% identical to the human gene (114). Nav1.9 was previously known as NaN or SNS-2 (117) and because it inactivates extremely slowly, it is capable of producing a persistent inward current (228). This means that gain of function mutation of Nav1.9 causes decreased excitability because other voltage-gated Na+ channels are inactivated by persistent Nav1.9 mediated depolarization (8). In the peripheral nervous system, NaN/Nav1.9 was first detected in small DRG neurons of SNS/Nav1.8—null mice (228) where it is preferentially expressed in non-peptidergic neurons which bind the plant lectin IB4 (229). Channels are found in free nerve endings, along axons, in DRG cell bodies and in primary afferent terminals in spinal lamina II (substantia gelatinosa) (230). Unlike Nav1.7, sciatic injury reduces expression of mRNA and channel protein for Nav1.9 (231) and this may be attributable to loss of trophic support by GDNF (226). Since Nav1.9 knockout mice continue to display allodynia following nerve injury (232), this channel is unlikely to play a role in injury-induced neuropathic pain. This contrasts with the situation for inflammatory pain where a role for Nav1.9 is well-established (114, 232).
Selective Modulation of Na+ Channels in TRPV1 Nociceptors
The local anesthetic, lidocaine acts in its cationic form to block all types of Na+ channels from the cytoplasmic side of the membrane. Although the topical application of lidocaine by means of a transdermal patch continues to be used in clinical pain management (1), disturbance of other aspects of sensory transmission by local anesthetics necessitates the development of more refined approaches. An ingenious approach has been used to selectively target lidocaine to TRPV1 expressing nociceptors (60, 233). The quaternary analog of lidocaine, QX314 is unable to permeate the cell membrane. It is therefore ineffective when applied extracellularly but is an effective local anesthetic when applied to the cytoplasmic side of the cell membrane. The pore of open TRPV1 channels is large enough to admit QX314, so their activation on nociceptors by capsaicin allows entry of QX314 and an anesthetic effect which is selective for this neuronal population. Although these findings have been repeated by others (234, 235) and the effectiveness of a more potent cationic anesthetic BW-031 described (61), this approach is yet to be exploited in a clinical situation.
Voltage-Gated K+ Channels
It is well-established that decreased function of voltage-gated K+ channels contributes to injury-induced increases in peripheral nerve excitability and activity (28, 236–245). As with Na+ channels, K+ channel function can be modified by altered expression of channel protein and/or its accessory subunits, altered trafficking or post-translational modification or modulation. Also, the establishment of genetic and structural definitions of a broad variety of K+ channel types (246–249) has led to improved mechanistic understanding of injury induced changes. Although the selective targeting of K+ channels has so far been less rewarding than targeting of voltage-gated Na+ channels, potential targets include Kv7.2 and the histone methyltransferase G9a which controls expression of several voltage-gated K+ channels, namely Kv7.2, Kv1.4 KCa1.1 [(250), Table 1].
Decreased Expression and Therapeutic Modulation of Delayed Rectifier K+ Channels
Sciatic nerve transection decreases functional expression of delayed rectifier K+ currents in DRG neurons (236–238, 251). Injury-induced changes may in part reflect post-translational processes such as phosphorylation, endocytosis and/or trafficking (245, 252, 253) that may be independent of any change in expression of K+ channel genes and their products as will be described in detail below. This possibility is underlined by the observation that delayed rectifier currents are substantially reduced in a rodent model of painful diabetic neuropathy but the mRNA levels for Kv1.1, Kv1.2, Kv2.1, and Kv2.2 are unchanged (254).
There are many types of delayed rectifier K+ channels in DRG neurons that assemble as hetero-tetramers or homo-tetramers of various Kv1, Kv2, and Kv3 subtypes (28). Although most types of Kv1 and Kv2 channels are affected by peripheral nerve injury, their ubiquitous distribution in both excitable and non-excitable tissues restricts the therapeutic potential of substances that augment the activity of delayed rectifier K+ channels.
Role of Kv1.1 in Neuropathic Pain
Protein and mRNA for Kv1.1 is reduced in DRG following sciatic nerve injury (238, 245, 255) and this is associated with redistribution of channels away from nodal regions of A-δ fiber axons (245). Although expression of a dominant negative phenotype of Kv1.1 causes allodynia in mice (256), certain glycine derivatives act as Kv1.1 channel openers (63), and substances have been identified that attenuate the time dependent inactivation of Kv1.1 (257), its ubiquitous distribution in brain, heart, retina, skeletal muscle and pancreatic islets (247) may preclude the use of Kv1.1 activators in pain management.
Role of Kv1.2 in Neuropathic Pain
Knockdown of Kv1.2 by siRNA induces mechanical and thermal hypersensitivity in naive rats (258). mRNA for Kv1.2 is also downregulated in several neuropathic pain models (28, 238, 255, 259, 260), and overexpression of Kv1.2 impairs neuropathic pain but does not attenuate acute pain in rats (261). These findings correlate with injury-induced reduction of whole-cell Kv1.2 current (260) and reduced channel protein expression as demonstrated by immunohistochemistry (261, 262) and/or immunoblot (245, 263).
Six different mechanisms have been hitherto suggested to underlie decreased Kv1.2 expression in DRG after peripheral nerve injury.
(i) Altered expression of histone deacetylase2 (HDAC2) (263) by NF-κB p65-dependent transcriptional regulation (264).
(ii) Increased expression of the canonical maintenance methyltransferase DNMT1 via a CREB (cAMP response element binding protein)—dependent process. Blockade of DNMT1 upregulation attenuates hyperexcitability in the injured DRG neurons and alleviated nerve injury-induced pain hypersensitivity (260, 265).
(iii) A pathway involving the methyl-CpG-binding domain protein 1 (MBD1), which binds to methylated sequences of DNA and attracts the DNA methylation protein DNMT3a. Overexpression of MBD1 leads to spontaneous pain and evoked pain hypersensitivities in wild type mice (266, 267).
(iv) Decreased expression of ten-eleven translocation methylcytosine dioxygenase 1 (TET1). This promotes DNA demethylation and its overexpression in the DRG of nerve injured animals alleviates pain hypersensitivities without altering acute pain (268).
(v) Kv1.2 function may be controlled by the non-coding miniature RNA miR-137. Because it impairs Kv1.2 function, experimental impairment of miR-137 function, rescues channel expression and function and attenuates allodynia in rats subject to CCI (258).
(vi) A long non-coding RNA (Kcna2 antisense RNA) contributes to neuropathic pain by silencing the KCNA2 gene and thereby reducing expression of Kv1.2 in primary afferents (259).
Limited Feasibility of Pharmacological Manipulation of Kv1.2
No small molecule activators of Kv1.2 have been identified (118) and given their documented presence throughout the brain, in spinal cord, mechanoreceptors and proprioceptors, Schwann cells, the heart, vascular smooth muscle and retina (247), direct pharmacological manipulation of these channels is not a viable means of treatment of neuropathic pain. There are some reports of alleviation of pain in animal models by attenuation of HDAC2 action (64, 65) but these may reflect modulation of its actions in the spinal cord as well as upregulation of Kv1.2 in the periphery. The HDAC inhibitor and antineoplastic agent, suberoylanilide hydroxamic acid (vorinostat) has been shown to alleviate pain in a bone cancer model (66) but to the best of our knowledge no trails of its efficacy in any form of neuropathic pain have as yet appeared.
Minimal Role of Kv1.3, 1.5, and 1.6 in Injury- Induced Pain
These channels which also exhibit delayed rectification are expressed at relatively low levels compared to Kv1.1 and 1.2 in naïve DRG (238, 245). mRNA for Kv1.3 is decreased but that for Kv1.5 and 1.6 is little affected by nerve injury (238, 255). In view of the relatively limited expression of these channels in DRG, augmentation of their function would not seem to be a desirable therapeutic strategy for pain mitigation.
A Role for Kv2.1, 2.2, and Kv9.1 in Injury-Induced Pain
Channel protein and mRNA are reduced by nerve injury as is Kv2 whole-cell current comprising Kv2.1 and 2.2 (262, 269). These changes may, in part, reflect the influence of the silent subunit Kv9.1 in hetero-tetramers with both Kv2.1 and Kv2.2 (67, 247, 270, 271). Nerve injury downregulates Kv9.1 in DRG neurons and this may alter behavior of Kv9.1~Kv2.1~Kv2.2 hetero-tetramers (270). Selective downregulation of the Kcns gene in DRG in vivo but not in other tissues, reduces Kv9.1 expression and promotes changes in pain behavior consistent with its role in onset of neuropathic pain (67, 270). This suggests that restoring Kcns1 activity in the periphery has therapeutic potential in chronic pain (67).
As seen with Kv2.1, nerve injury downregulates mRNA for Kv2.2 in DRG (255, 269). Since Kv2.2 currents are also affected by the presence of Kv9.1 in hetero-tetramers this give further credibility to potentiation of Kv9.1 as a therapeutic approach (Table 1).
No Role for Kv3.1 and 3.2 in Neuropathic Pain
Although immunohistochemical, biophysical and Western immunoblot studies have identified these isoforms in DRG (272), there is little or no evidence for injury-induced changes in their expression or function (255).
Decreased Expression and Therapeutic Modulation of Kv7.2/7.3 M- Channels
Role of Kv7.2/7.3 in Neuropathic Pain
M-channels are the Kv7.2 and Kv7.3 products of the KCNQ2/3 genes (273). They are activated by depolarization in a similar fashion to delayed rectifiers but do not inactivate over periods of many minutes. This and the fact that M-channels start to activate at normal resting potential means that they play an important role in determining neuronal excitability and accommodation of firing (274, 275). Whole-cell M-current is reduced in a model of bone cancer pain (276), selective knockdown of Kv7.2 in DRG causes hyperalgesia (277) and peripheral nerve injury induces substantial downregulation of Kv7.2 protein (239). The observation that the M-channel openers such as flupirtine and retigabine alleviate hyperalgesia in several rodent pain models (239, 278, 279) initiated considerable interest in the potential therapeutic use of this type of drug (280–285).
Pharmacological Manipulation of Kv7.2/7.3
Although a clinical study of retigabine in post herpetic neuralgia failed to meet its efficacy endpoint (68), at least 200 Kv activators are currently under development (285). It has also been observed that the natural products, mallotoxin (MTX) and isovaleric acid (IVA), act synergistically to open neuronal KCNQ channels. This combination has been shown to suppress pentylenetetrazole-induced tonic seizures in mice but has not yet been examined in pain models (71). Similar effects were seen with (E)-2-dodecenal (E-2-D), a natural product derived from cilantro leaves (72). It has been suggested that co-administering MTX, IVA or E-2-D with retigabine may be highly effective in opening of KCNQ2/3 channels (71) (Table 1).
A novel Kv7.2 activator known as SCR 2682 was described recently (69). Acute application of SCR 2682 augments M-currents in DRG neurons and alleviates nerve injury induced pain in vivo. Both effects are reversed by M-channel inhibitor XE991. SCR 2682 also increases KCNQ2 mRNA and Kv7.2 protein expression in a rodent model of neuropathic pain (70) but its exact mechanism of action is yet to be determined.
Kv7 thus retains its potential as a drug target for neuropathic pain (Table 1); chemical modification of the retigabine structure may provide new and effective therapeutic agents.
The effects of nerve injury on expression of KCNQ depend on the actions of inflammatory mediators (286) and/or inhibition of transcription by repressor element 1-silencing transcription factor (REST also known as neuron-restrictive silencing factor, NRSF) (239, 287). Overexpression of REST in DRG neurons strongly suppresses M-current density, increases excitability induces mechanical and thermal hyperalgesia (288). Specific knockout of REST in DRG prevents injury-induced downregulation of REST target genes and prevents the development of hyperalgesia in various models of neuropathic pain; an effect that can be restored by REST overexpression (288).
REST inhibits transcription by recruiting the co-repressor complexes SIN3A/B and REST corepressor 1; these complexes modify target gene regions through the action of HDAC1/2, the histone demethylase LSD1 and the histone methyltransferase G9a (289, 290). Inhibition or genetic deletion of G9a in DRG abolishes injury-induced down-regulation of Kv7.2 and reduces neuropathic hyperalgesia. G9a may have an important role in K+ channel regulation as it has also been implicated in injury induced suppression of Kv1.4, Kv4.2, and BK channels (KCa1.1) (250). Two small molecule inhibitors of G9a are available, namely BIX01294 and UNC0638, both of which attenuate neuropathic pain in rodent models (80, 81). Although there is considerable interest in developing histone methyltranferase inhibitors in cancer treatment (291), to the best of our knowledge neither BIX01294 nor UNC0638 have been examined for treatment of pain in the clinic. Further development of drugs of this type may lead to new approaches to pain management (Table 1).
Decreased Expression and Therapeutic Modulation of A-Channels
A-type potassium channels are largely inactivated at the normal resting potential of DRG neurons and this inactivation must be removed by hyperpolarization prior to depolarization to effect channel opening. Once activated, A-channels display profound and usually rapid inactivation. Despite the rather complex protocols required to activate A-currents in a voltage-clamp experiment, A-channels play a role in neuronal activity by modulating the shape of action potential afterhyperpolarizations, participating in action potential repolarization (247, 292) and increasing the latency of depolarization activated action potentials. There are several different types of A-current distinguished by their sensitivity to the channel blocker 4-aminopyridine (4-AP) and by their rate of inactivation. Nerve injury, including diabetic neuropathy decreases whole-cell A-current in DRG neurons (237, 238, 254, 293, 294). This reflects altered functionality of Kv1.4, Kv3.4, and Kv4's, which are the dominant A-current types in DRG (28, 244). A-channels seem especially sensitive to changes induced in models of diabetic neuropathy (254).
Role of Kv1.4 in Neuropathic Pain
mRNA for Kv1.4 is downregulated in several models of neuropathic pain, including a model of diabetic neuropathy (238, 250, 254, 255). Knockdown of Kv1.4 with siRNA causes allodynia (295) and miR-17-92 overexpression downregulates A-channels and promotes hyperalgesia (296). The molecular mechanism of altered Kv1.4 expression is similar to that for Kv7.2 described above (250). This means that the effectiveness of G9a inhibitors in inhibiting neuropathic pain (80, 81) may involve preservation of function of both Kv7.2 and Kv1.4 after injury (Table 1).
Role of Kv3.4 in Neuropathic Pain
Kv3.4 are high threshold A-channels that are particularly sensitive to 4-AP block. Nerve injury decreases expression of Kv3.4 immunoreactivity (297) and mRNA is reduced in a model of diabetic neuropathy (254). Kv3.4 antisense produces mechanical hypersensitivity (297). It has also been reported that injury to the spinal cord per se causes Kv3.4 dysfunction in DRG (298). This may reflect the action of excitatory mediators released from the spinal site of injury. This raises the possibility that therapeutic control of DRG function may not only be beneficial for peripheral neuropathy, it may also have benefit for managing pain originating from spinal cord injury.
Role of Kv4.1, 4.2, and 4.3 in Neuropathic Pain
Immunoreactivity and/or mRNA for all three Kv4 channels is found in DRG neurons (28, 255, 294, 297, 299) with differences in their distribution across different neuronal types (300, 301). Decreased function of all Kv4 channels occurs after peripheral nerve injury (75, 244, 254, 293, 297, 299, 302), and knockdown of Kv4.1 and its modulatory subunits or antisense to Kv4.3 causes mechanical hypersensitivity (297, 299). Taken together these observations strongly suggest malfunction of Kv4 channels in neuropathic pain.
The expression and function of Kv4 channels in DRG is controlled by signaling pathways such as MAPK (293), Kv4 channel interacting proteins (KChIPs) and dipeptidyl-peptidase-like proteins (DPPLs) (303–305). The aforementioned neuron restrictor silencer factor (REST), which controls expression Kv7.2, also effects suppression of transcription of the Kv4.3 gene (KCND3) after nerve injury (302).
Pharmacological Manipulation of Kv4
Since no activators of Kv4 channels are available, targeting accessory subunits of A-channels may provide an alternative strategy (244). DPPLs and KChIPs not only govern the biophysical properties of Kv channels. They also impact channel assembly, channel trafficking to and from the cellular surface, and targeting of channels to different cellular compartments (304). The compound NS5806 has been reported to potentiate Kv4 currents in a KChip dependent manner (73, 74) and has recently been shown to attenuate cold allodynia in a rodent model of trigeminal neuralgia [(75), Table 1].
Decreased Expression and Therapeutic Modulation of Ca2+-Sensitive K+ Channels
Ca2+-sensitive K+ channels fall into three broad categories; KCa1.1, also known as BK or maxi gK, Ca channels which are high conductance, voltage-sensitive and blocked by low concentrations of tetraethylammonium; KCa2.1,2.2 and 2.3 which are apamin sensitive, low conductance, and voltage-independent and KCa3.1 which are intermediate conductance and clortrimazole sensitive (246). In neurons, these channels play a major role in the determination of spike width, repolarization, after hyperpolarization amplitude and duration, repetitive discharge characteristics, accommodation and overall excitability. As with other K+ channel types, their potential as therapeutic targets is limited by their ubiquitous distribution and function in both excitable and non-excitable tissues (246).
Role of KCa1.1/BK Channels in Neuropathic Pain
BK channels are encoded by the KCNMA1 gene and are present in all DRG neurons (240, 306–308). Their functional expression is reduced by peripheral nerve injury (236, 240, 309). This is associated with decreased expression of KCNMA1 and channel protein (250, 310). Their involvement in generation of pain is suggested by the observation that overexpression of BK increases mechanical threshold in a rodent neuropathic pain model (311). Also, the KCa1.1. blocker, iberiotoxin reduces mechanical withdrawal threshold.
Pharmacological Manipulation of KCa1.1/BK Channels
Intrathecal injection of the KCa1.1 channel opener [1,3-dihydro-1-[2-hydroxy-5-(trifluoromethyl)phenyl]-5-(trifluoromethyl)-2H-benzi midazol-2-one] dose-dependently reverses allodynia and hyperalgesia in nerve-injured rats but had no significant effect on nociception in control rats (310). This substance is one of several BK activators available including the highly effective GoSlo-SR family of anthraquinone analogs (312). Others include NS1619 (313, 314), NS11021 (315, 316), NS13558 (317), and 12,14-dichlorodehydroabietic acid (diCl-DHAA) (318). Because these drugs have profound effects on tissues such as cardiac myocytes and certain smooth muscles, they are unlikely to be of practical use in pain management.
On the other hand, there is considerable discussion in the literature relating to the efficacy of cannabinoids in neuropathic pain (102, 106, 107) and it has been suggested that augmentation of BK function may contribute to their potential therapeutic effect (103).
As was described for Kv7.2 and Kv1.4, injury-induced downregulation of KCNMA1 in DRG is a result of G9a activation (250). This underlines the potential therapeutic application of G9a blockers such as BIX01294 and UNC0638 (Table 1).
Role of KCa2.1, 2.2, 2.3, and 3.1 in Neuropathic Pain and KCa3.1 as a Therapeutic Target
There is little information about the possible role of KCa2 channels in pain but several recent reports have drawn attention to the possible role of KCa3.1 (Table 1). Although KCa3.1 knockout-mice show increased sensitivity to noxious chemical stimuli they exhibit normal behavioral responses to acute nociceptive, persistent inflammatory, and persistent neuropathic pain (319). Despite this, the KCa3.1 channel opener, ASP0819, modulates nociceptive processing and in vivo action potential activity in peripheral nerves in an animal model of fibromyalgia (320) and preliminary investigation of its action in the clinic have provided evidence of efficacy with minimal side effects (321).
Decreased Expression and Therapeutic Modulation of Inwardly Rectifying K+ Channels
Although a variety of two transmembrane domain inwardly-rectifying K+ channels are found in DRG neurons (28), by far the most information of relevance to pain mechanism and potential management relates to findings on the KATP channel; Kir6.2 (243, 322, 323).
Role of Kir6.2/KATP Channels in Neuropathic Pain
KATP channels play an indispensable role in pancreatic insulin secretion as a result of their inhibition by intracellular ATP and their activation by ADP (248). Sulphonylurea receptors (SUR or ATP binding cassettes) co-assemble with channel proteins (324). KATP channel activation can be achieved by the anti-hypertensive agents, diazoxide and pinacidil and their anti-nociceptive actions have been recognized for many years (325). Nerve injury reduces KATP currents and channel activity in DRG neurons (323, 326) and although there are several reports of the efficacy of KATP openers in neuropathic pain models (76–79), these findings do not appear to have been exploited in the clinic.
Decreased Expression and Therapeutic Modulation Tandem Pore Domain K+ Channels
Downregulation of TRESK, TASK3, and TWIK1 by Nerve Injury and Relevance to Neuropathic Pain
Four transmembrane-domain tandem pore domain (K2p) channels account for K+ leak conductance and set the resting membrane potential of most excitable cells including DRG neurons (28, 249, 327). TRESK (k2p18) channels seem particularly important in this regard (328). Their potential relevance to neuropathic pain is supported by the observation that sciatic nerve transection reduces TRESK/(k2p18)/KCNK18 mRNA to a greater extent than other K2p channels in DRG and in vivo knock down decreases threshold to painful mechanical stimuli (329, 330). Other K2P channels such TASK3 (K2p9) and TWIK1 (K2P1) are also down-regulated by spared nerve injury (SNI) (331).
Therapeutic Modulation of Tandem Pore Domain K+ Channels
Although activation of K2P channels contributes to the therapeutic effectiveness of volatile anesthetics such as isoflurane (327, 332) it is obviously impractical to use these drugs for long term pain management. The novel TREK2/K2p10.1 activator GI-530159 decreases DRG excitability (333), but its possible effectiveness in pain models has not yet been reported.
Voltage-Gated Ca2+ Channels
Voltage-gated Ca2+ channels (VGCCs) have been studied for more than 20 years as potential therapeutic targets for chronic pain (93, 334, 335). They are subdivided into high-voltage activated (HVA) L-types (Cav1.1, Cav1.2, Cav1.3, and Cav1.4), P/Q-type (Cav2.1), N-type (Cav2.2), and R-type (Cav2.3) and low voltage activated (LVA) T-types (Cav3.1, Cav3.2, Cav3.3) (93, 336, 337). The distribution of channels in DRG muscle afferents is Cav2.2 (N-type) > Cav2.1 (P/Q-type) > Cav1.2 (L-type) (338). There is little or no evidence for the expression of Cav1.1, Cav1.3, and Cav1.4 in DRG as these are found mainly in heart, skeletal muscle, endocrine cells, smooth muscle and the vestibular system (93, 336). R-type Cav2.3 and P/Q type Cav2.1 also appear to be absent from DRG (338).
VGCC set DRG neuron excitability either by generating voltage-gated inward currents or by producing outward currents following the activation of Ca2+ sensitive K+ channels (236). Influx of Ca2+ through HVA channels triggers release of excitatory neurotransmitters from presynaptic vesicles and thereby determines dorsal horn excitability. The role of VGCC in neuropathic pain and pain therapeutics in general is well-established (24, 93, 236, 339–344). This is underlined by the therapeutic effectiveness of the N-type Ca2+ channel blocker ziconotide (339), the established use of gabapentinoids which bind to the α2δ-1 regulatory subunit of HVA Ca2+ channels (3, 345, 346) and the observation that N-type VGCC knockout mice exhibit reduced signs of both inflammatory and neuropathic pain (347). The α2δ-1 subunit plays a major role in the expression and function of VGCC (346, 348) and α2δ-1 gene deletion delays mechanical hypersensitivity in response to peripheral nerve damage (349).
Since VGCC are responsible for triggering release of neurotransmitter, blocking, or genetically deleting these channels in peripheral neurons reduces synaptic input to the spinal cord (93) and ω-conotoxin GVIA reduces synaptic potentials in the spinal cord (350).
Early experimental investigations of the effects of nerve injury on VGCC function were completed some years before the establishment of formal structural and genetic definitions of channel subtypes. Axotomy or chronic constriction injury reduced function of HVA channels in the cell bodies of DRG neurons (236, 342, 351) and there was no preferential loss of N-type vs. L-type channels (236). As with Na+ and K+ channels, the structural and genetic definition of VGCC subtypes (336) has refined descriptions of injury induced changes and enabled the logical development of current and potential therapeutic agents (93, 335, 339).
Therapeutic Modulation of HVA Ca2+ Channels
L-Type Cav1.2 Channels in Neuropathic Pain
Although these L-type VGCC are present in rodent DRG (338), gain of function mutations in humans do not express a pain phenotype (93). On the other hand, following CCI of the sciatic nerve, the “classical” dihydropyridine, nitrendipine reduces the frequency of spontaneous EPSC's in rat lamina II (substantia gelatinosa) neurons. It also, albeit rather weakly, attenuates mechanical allodynia. These effects have been attributed to injury-induced upregulation of α2δ-1 and increased expression of Cav1.2 after nerve injury (348). Anti-Cav1.2 siRNA or selective knockdown of Cav1.2 in the spinal dorsal horn but not in DRG has been shown to reverse the nerve injury associated mechanical hypersensitivity of dorsal horn neurons. This implies that postsynaptic effects such as CREB phosphorylation in the spinal dorsal horn may also contribute to the participation of Cav1.2 in neuropathic pain (352, 353). It may relate to the finding that α2δ-1 remodels Cav1.2 voltage sensors and allows Ca2+ influx at physiological resting potentials (354).
Pharmacological Manipulation of L-Type Cav1.2 Channels
Since we could only find one very old report of clinical effectiveness of classical dihydropyridine, nifedipine in complex regional pain syndrome (355), it is presently assumed that L-type Ca2+ channels play a far smaller role in the etiology of neuropathic pain than N- or T-types (see below). This position may however need revision in the light of recent descriptions of prevalent nifedipine sensitive channels in human DRG neurons (356).
Some novel benzodiazepines exhibit selective T-channel block (357) whereas others block both Cav1.2 L-type and Cav3.2 T-type calcium channels (358). To the best of knowledge there are no reports of the effectiveness of these agents in the clinic.
Role of Cav2 Channels in Neuropathic Pain
Cav2 channels are the main subtype found in primary afferent terminals (93, 359). Cav2.1 (N-type) and Cav2.2 (P/Q type) both contain a synaptic protein interaction site (synprint) that interacts with SNARE proteins (syntaxin and SNAP-25) (360, 361). By this mechanism, channels can be closely associated with synaptic vesicles that govern release of neurotransmitter from primary afferent terminals. Although suppression of N-type Ca2+ channel current increases the excitability of DRG cell bodies by concomitant decrease of BK function (236, 306), this effect is overridden in vivo by the actions of Cav2 blockers to prevent neurotransmitter release from primary afferent terminals.
Pharmacological Manipulation of Cav2 Channels
As already mentioned, the Cav2.2 blocker ziconotide which is a synthetic version of ω-conotoxin MVIIA from the cone snail Conus magnus (362) is employed in pain management. The main drawback is that it needs to be delivered directly to the spinal cord via an intrathecal drug delivery system. Zicononotide (Prialt) is usually only effective in patients with severe, intractable forms of chronic pain such as that associated with cancer (82, 363).
There is therefore considerable interest in developing small molecule blockers of Cav2 channels that may be effective orally or perhaps by intravenous injection (24, 93, 339, 344). In our previous review (3) we drew attention to the state-dependent Cav2 blockers ZC88 (83, 84), A-1264087 (85, 364), and TROX-1 (86, 87, 365). Although all of these drugs display anti-allodynic efficacy in rodent models of neuropathic pain (344), there is as yet no evidence of any clinical efficacy.
Two tetrahydroisoquinoline derivatives have also been shown to display effectiveness in animal models (366, 367) but again clinical efficacy has not yet been demonstrated.
A permanently charged cationic derivative of an N-type calcium channel-blocker was recently synthesized (97). These authors anticipated that this charged compound (known as CNCB-2) would only be effective when applied intracellularly by a mechanism analogous to QX-314 block of Na+ channels (60). Surprisingly, extracellular application of CNCB-2 was more effective than intracellular application in inhibiting Cav2.2 channels. Inhibition was achieved without channel opening. Moreover, and quite unexpectedly, the compound was also highly effective in inhibiting Nav1.7 when applied extracellularly. CNCB-2 reduced excitability of mouse DRG neurons and produced long lasting analgesia in several pain models. Given the seminal role of Nav1.7 in the etiology of many forms of neuropathic pain (8, 114), bifunctional compounds such as CNCB-2, show considerable promise as therapeutic agents (Table 1).
Cav2.2 interacts with collapsin response mediator protein 2 (CRMP2) which directs the channels to presynaptic terminals (368). Interestingly it has been reported that impairment of CRMP2 function using a homopolyarginine (R9)-conjugated CBD3-A6K peptide inhibits Cav2.2-CRMP2 interaction, diminishes surface expression of Cav2.2 and alleviates tactile allodynia and ongoing pain in a rodent model (369). This observation suggests that CRMP2 may be developed as a novel therapeutic target.
N-type Ca2+ channels are modulated by Gi/o coupled agonists (157, 370). The α2-adrenoceptor agonist, clonidine displays anti-allodynic actions in a rodent model (371) and meta-analysis of clinical trials reveals clinical efficacy (372). Effects of clonidine may be mediated by α2-adrenergic inhibition of neurotransmitter release leading to modulation of pain processing at the spinal level (5) and/or by attenuation of aberrant interactions between sympathetic and sensory nerves in the periphery (156, 157, 373). Its effectiveness is however limited to subsets of patients within the diabetic neuropathy, complex regional pain syndrome or postherpetic neuralgia cohorts (88–92). In view of the restricted effectiveness of clonidine, it does not meet the criteria for first line treatment of neuropathic pain (1) (Table 1).
Gabapentinoids on the other hand are relatively but not completely effective in a variety of manifestations of neuropathic pain; about 31% of patients see clear benefit (96). Their mechanism is still incompletely understood but clearly involves impediment of transport of Cav2 channels to nerve terminals and their uncoupling from the neurotransmitter release process following interaction with their α2δ-1 accessory subunits (3, 94, 374). This occurs in both primary afferents and dorsal horn (95). Apart from the introduction of pregabalin (375) and an enacarbil derivative of gabapentin with improved oral bioavailability (376), there have been no major developments in the pharmacology of α2δ-1 ligands since their introduction in the 1990's. Since gabapentinoids act intracellularly, we have suggested that their effectiveness may be increased by allowing them to enter neurons via the open pore of TRPV1 channels (377).
Since Cav2.2 channels are found in pancreatic β-cells and are involved in the secretion of insulin (378) it remains to be established whether Cav2.2 blockers have undesirable effects on blood glucose levels. On the other hand, Cav2.2 has been implicated in microglial function (379, 380). This raises the possibility that some of the beneficial effects of Cav2.2 blockers result from actions on microglia.
Therapeutic Modulation of LVA Ca2+ Channels (T-Channels)
Role of Cav3.2 in Neuropathic Pain
T-type, LVA, Ca2+ channels (Cav3.1, Cav3.2, Cav3.3) play important roles in setting neuronal excitability (93, 336, 381) and in transmitter release from primary afferent terminals (382, 383). As with Cav2 channels, this later function may involve interaction of Cav3 channels with the synaptic vesicle release proteins syntaxin 1A and SNAP25 (synprint) (384). DRG neurons express Cav3.2 and 3.3 but not 3.1 (385–387). T-type calcium currents are increased in rodent DRG neurons after peripheral nerve injury in a model of diabetic neuropathy and after injury to the spinal cord per se (383, 388–390).
Although there are no reported mutations of Cav3.2 that produce a painful phenotype in humans, most of the work relevant to pain mechanisms has involved this channel as opposed to Cav3.1 or Cav3.3 (93, 334, 335, 339, 391–393). Cav3.2 is expressed in low-threshold mechanoreceptors and conditional knockout of the channel in this neuronal subtype has implicated Cav3.2 in allodynia linked to neuropathic pain (394). Several mechanisms control the functional expression of Cav3.2 channels.
(i) Upregulation of the deubiquitinase, USP5 by the action of the inflammatory mediator interleukin-1β. This impairs Cav3.2 ubiquitination thereby protecting it from proteasomal degradation and prolonging its surface expression (383, 395, 396). Knockdown of USP5 in vitro increases Cav3.2 ubiquitination and reduces Cav3.2 whole-cell currents and since impairment of USP5 function in vivo attenuates mechanical hypersensitivity in both inflammatory and neuropathic mouse models, this enzyme may represent a future therapeutic target (335, 383). Progress in this direction involves the observations that Cav3.2/USP5 interactions are interrupted by the anti-parasitic agent, suramin and by a TAT-cUBP1-USP5 peptide and both substances show analgesic activity in neuropathic and inflammatory pain models (99, 100) (Table 1).
(ii) Glycosylation and enhancement of channel trafficking in diabetic pain (397, 398). Deglycosylation of Cav3.2 with neuramidase reverses hyperalgesia in a model of diabetic neuropathy (398) (Table 1).
(iii) BDNF stimulation of TrkB coupled to PI3K-p38-PKA signaling in trigeminal neurons (399). Although a range of small molecule TrkB inhibitors are available (400), the multiple biological actions of BDNF in the developing and mature nervous system, preclude the use of these agents in pain management (401).
(iv) Cav3.2 channels interact with the scaffold protein Rack-1 [receptor for activated C kinase 1 (402)]. Whole-cell Cav3.2 current and channel expression in the plasma membrane is reduced when Cav3.2 and Rack-1 are co-expressed in tsA-201 cells. Molecular interaction between the two proteins was demonstrated by co-immunoprecipitation. These findings assume special significance in the light of the suggested role for Rack-1 in neuropathic pain (403).
Pharmacological Manipulation of Cav3.2
Although T-type Ca2+ channel blockers such as the anticonvulsant ethosuximide increases withdrawal thresholds in nerve-injured rats (404), clinical studies of its effectiveness in pain management have been disappointing (98). A similar picture emerges for other small molecule blockers of Cav3.2, most of which showed considerable promise in preclinical studies yet failed to exert significant effects in cohorts of pain patients (334).
For example, ABT-639 showed promise in preclinical studies (405–407) but clinical results have been disappointing (86, 334); it did not treat pain in patients with diabetic neuropathy (408) and has now been discontinued.
Also, because TTA-P2 is a highly selective Cav3.2 channel blocker that has minimal effects on other cation channels, it is used extensively in laboratory investigations of T-channel function. Although it is effective in rodent models of chronic inflammatory pain and diabetic neuropathy (409) we could find no reports of its efficacy in the clinic.
Similarly, TTA-A2 is used extensively in laboratory investigations (395) as it has higher affinity for Cav3.2 than Cav31.1 (410). Although it is effective in rodent models of irritable bowel syndrome (410), no clinical studies appear to have been done.
Z944 is another high-affinity T-type channel blocker that is effective against Cav3.1, Cav3.2, and Cav3.3 with little affinity for other Ca2+ channel types (411). Its effectiveness in murine pain models may reflect it actions on spinal and thalamic neurons (412, 413). So far, the results of phase 1 and phase 2 trials appear promising (101) (Table 1). Although there does not seem to be any preclinical information regarding the effectiveness of the N-(1-benzyl-1H-pyrazol-3-yl)-2-phenylacetamide derivative ACT-709478 in animal models of neuropathic pain (414), it appears to be showing promise in phase 2 trials (101) (Table 1).
As already mentioned the has been shown to also selectively block Cav3.2 (49).
Cannabinoids, which are effective in some neuropathic pain cases (108, 109), inhibit recombinant human T-type (Cav 3.1, 3.2) Ca2+ channels (104) and as mentioned above, augment BK (KCa1.1) currents. Intrathecal injection of the CB1/CB2 receptor agonist NMP-7 inhibits injury-induced neuropathic pain in a rodent model. This effect involves CB2 receptors and Cav3.2 channels (415). To the best of our knowledge, NMP-7 has not yet progressed to clinical trials but its preclinical effectiveness led to the development of the derivative [N-((1-(2-(tertbutylamino)-2-oxoethyl)piperidin-4-yl)methyl)-9-pentyl-9Hcarbazole-3-carboxamide] (Compound 9) which displays remarkable effectiveness in murine models of inflammatory and neuropathic pain (105).
HCN-Channels
Role of HCN2 and 3 in Neuropathic Pain
There are 4 isoforms of hyperpolarization-activated cyclic nucleotide–gated (HCN) channels (416); HCN1, HCN2, HCN3, and HCN4 coded by HCN1, HCN2, HCN3, and HCN4 genes. HCN3 are distinguished by their relatively low sensitivity to intracellular cAMP (416). HCN channels underlie neuronal H-current (Ih).
Ih is upregulated in DRG after nerve injury (417) where it drives spontaneous activity (30, 418–421) and increases transmitter release from primary afferents (422, 423).
Whereas, HCN1 and HCN4 channels are primarily expressed in cardiac pacemakers, HCN2 channels are mainly expressed in neurons. They have emerged as a promising peripheral drug target for neuropathic as well as inflammatory pain (3, 27, 335, 418–420, 424–426).
HCN2 is expressed is expressed in about 50% of small somatosensory neurons, which are mainly nociceptors. It plays an important role in the control of firing frequency in response to noxious stimuli (420). Indeed deletion of HCN2 in nociceptive neurons prevents the development of inflammatory and neuropathic pain (420).
HCN3 is expressed in most DRG neurons and is persistently activated at their normal resting potential thereby contributing to membrane resistance. Neurons from HCN3-knockout mice exhibit increased input resistance and increased excitability, but experience similar levels of mechanical allodynia and thermal hyperalgesia to wild-types following nerve injury. This suggests that HCN3 plays little or no role in processing of neuropathic pain (427).
Pharmacological Manipulation of HCN Channels
Ivabradine which blocks HCN1, 2, and 4 (416) is used clinically to treat chronic angina and heart failure (335). It abrogates signs of neuropathic pain in animal models through peripheral action on small sensory neurons (418, 425). The effectiveness of ivabradine may be in part attributed to its ability to increase Kv7 channel activity (428) and perhaps actions at the thalamic level as seen with the classical Ih blocker ZD7288 (429). Although we found ivabradine administered to nerve injured rats at a dose that significantly reduced mechanical allodynia was without noticeable effect on arterial pressure and produced only a 15% reduction in heart rate, its cardiovascular actions have detracted from its use as an analgesic agent in the clinic (430).
More recent work has thus focused on the search for selective HCN2 blockers (431) that may abrogate hyperexcitability of DRG neurons without affecting the HCN1 channels that are responsible for controlling cardiac rhythmicity (27). However, to the best of our knowledge, no small molecule blockers are as yet available.
Discussion
Unlike morphine for nociceptive pain, there is no equivalent panacea for neuropathic pain. The well-tried therapeutic approaches to neuropathic pain (gabapentinoids, tricyclic antidepressants and serotonin-noradrenaline reuptake inhibitors) retain their position in the “winners circle” of effective agents (1, 2). They have not yet been superseded by any of the treatments or approaches listed herein (223). Although a variety of therapeutic approaches have been mentioned above, Table 1 lists only those compounds that show considerable promise as therapeutic agents.
In the final section of the review, we suggest future considerations and refinements that may enable the further development and usage of peripherally-acting drugs as possible therapeutic approaches to pain management.
Use and Structural Refinement of Promising Candidate Molecules
Many drugs that are effective in animal models fail to lead to useful clinical agents because of dose limiting toxicities, unfavorable pharmacokinetics or “off target effects.” Some of these issues can be minimized by chemical modification of safe pharmacological agents or drug repurposing.
Therapeutic Potential of Na+ Channel Blockers
Several Na+ channel blockers show promise as therapeutic agents or as lead compounds for structural refinements (Table 1).
The first is the Nav1.7 blocker, vixotrigine (CNV1014802, BIIB074, or GSK-1014802) (184, 185). The outcomes of a phase III clinical trial for effectiveness in trigeminal neuralgia (NCT03637387) and phase II trial for small fiber neuropathy are eagerly awaited (47).
The Nav1.7 blocker PF-05089771 failed to meet defined efficacy criteria in patients with painful diabetic peripheral neuropathy (46). Since its use in clinical trials would have been contingent on establishment of safety for use in humans, it may serve as a safe lead compound for the development of more effective agents.
Certain natural toxins, notably those from various types of tarantula venom show selectivity and high affinity for Nav1.7 as well as analgesic effects in various pain models. One of the most promising agents is Tap1a as this interacts with both Nav1.7 and the T-type Ca2+ channel, Cav3.2 (49, 50). Recent studies of Tap1a have shown that it interacts with voltage-sensor domain II of Nav channels with nanomolar affinity. Structural modification of Tap1a has produced two peptides Tap1a-OPT1 and Tap1a-OPT2 that exhibit increased affinity for Nav1.1, Nav1.2, Nav1.3, Nav1.6, and Nav1.7. Intraplantar injection of Tap1a-OPT1 reduces Nav1.7/OD1-induced spontaneous pain behaviors in a murine model. Moreover the anti-nociceptive effect of Tap1a-OPT1 is significantly greater than the native peptide (50).
Although the selective Nav1.8 blockers A803467 and PF-01247324 attenuate allodynia in a rodent model (221, 222), they have not yet been examined in the clinic (223). The pro-drug VX-150 is metabolized into a highly selective Nav1.8 blocker which exhibits analgesic activity in healthy volunteers (57). Expression of Nav1.8 in peptidergic DRG neurons is controlled by NGF (215) and the NGF binding antibody tanezumab is effective in various pain states (58). In fact, its safety and efficacy in humans identifies tanezumab as one of more the promising new drug candidates for chronic and neuropathic pain (see Clinical Trials Government Identifiers: NCT02528188 and NCT02528188). Small molecule peripherally acting TrkA inhibitors have recently been described (432, 433).
We have also described the idea of combining cationic local anesthetics with TRPV 1 activators (60, 61, 233), although this seems to work well in animal models, this approach has not yet been demonstrated in a clinical situation.
Therapeutic Potential of K+ Channel Activators
Although a clinical study of retigabine in post herpetic neuralgia failed to meet its efficacy endpoint (68), there is considerable interest in its structure as template for ligand-based drug design of Kv7.2/3 activators (434); at least 200 Kv activators are currently under development (285). Certain natural products augment Kv currents and it has been suggested that these might augment retigabine effectiveness (71).
The sulphonylurea compound NS5806 which augments Kv4.3 type A-currents in animal models (75) is yet to be examined in the clinic.
A phase 2a clinical trial of the KCa3.1 channel opener, ASP0819 for fibromyalgia (NCT03056690), has provided evidence of efficacy with minimal side effects (321). As mentioned above, little, or no success has been realized with other direct activators of Kv1, 2, 3 or 4 or KCa1 or 2.
Therapeutic Potential of Ca2+ Channel Blockers
N-type Cav2 channels have been recognized as targets for anti-allodynic drugs for many years. The limitations to the use of the channel blocker ziconotide and the α2-adrenoceptor ligand clonidine have already been alluded to (363). Although gabapentinoids interact indirectly with Cav2 via their α2δ-1 subunits they are neither universally effective or without undesirable adverse effects (96). As mentioned above, a few small molecule Cav2 blockers are in development but none have as yet been tested in a clinical situation. The compound CNCB-2 is of special interest as it blocks both Cav2.2 and Nav1.7 channels (97).
The potential role of Cav3 in neuropathic pain was established about 12 years ago (389, 413, 435) but the classical Cav3 blocker ethosuximide displays only limited effectiveness in the clinic (98). In the interim, several small molecule blockers have appeared such as TTA-P2 and TTA-A2 which are highly selective for Cav3.2. Clinical studies are yet to be initiated or reported. By contrast, phase 1 and 2 clinical studies with two compounds Z944 and ACT-70948 have yielded promising results (101). Interest in cannabinoid modulation of Cav3.1 and 3.2 has led to development of a series of small molecule channel blockers such as “compound 9,” although it is remarkably effective in preclinical studies clinical studies are yet to be initiated (105).
Improve Assessment of “Pain” as Opposed to Nociception in Rodent Models
Preclinical effectiveness of therapeutic intervention in neuropathic pain is often assessed by examination of drugs' ability to attenuate behavioral indices of pain induced by surgical or chemical lesions to peripheral nerves of experimental animals (436, 437). Typical measurements involve examination of mechanical or thermal withdrawal thresholds or presence of hyperalgesia and or touch or cold-induced pain (mechanical or thermal allodynia). It may be argued however that withdrawal of a foot or limb in response to a noxious stimulus may simply reflect activation of a spinal reflex (438). The inability to measure “pain” per se with both its nociceptive and emotional comments may underlie the limited ability of rodent models to predict clinical efficacy (68, 171, 439). In an attempt to assess true pain and its attenuation in rodent models, more recent non-invasive models for assessment of chronic pain involve quantification of indices such as facial grimace score as well as observation of social interaction and nest-building (Turner et al., 2019; Sotocinal et al., 2011) (437). This is complemented by the use operant models such as conditioned place preference protocols. In one version of this, rodents are required to make a conscious choice between being in a pain-inducing environment and an otherwise undesirable environment such as a brightly illuminated space (3, 440–442). The time spent in the undesirable brightly illuminated environment gives an index of the pain the animal is experiencing.
Translation between animal observations and development of effective human therapeutics may thus be improved by the use of these operant and non-invasive protocols.
Think About Sex
Women are more prone than men to develop neuropathic pain (12, 443–446). A recent genome wide association study revealed that 123 single nucleotide polymorphisms (SNP) at five independent loci were significantly associated with chronic pain in men whereas in women, 286 genome-wide SNPs were found at 10 independent loci (447). Gene-level analyses revealed sex-specific associations with chronic pain with 31 genes associated in females, 37 genes associated in males, and a single gene, DCC, which codes for the netrin 1 receptor associated in both sexes. Interestingly, all 37 chronic pain associated genes in men and 30/31 genes in women were found to be expressed in DRG (447). These findings match the documented, robust differences that exist in the genetic, molecular, cellular and systems-level mechanisms of acute and chronic pain processing that occur in male vs. female rodents and humans (12, 444, 446, 448–450). This means that preclinical studies previously done exclusively on male rodents need to be repeated in females. This is especially the case in the pain field because sexual convergence onto shared behavioral endpoints, such as allodynia or pain sensitivity, may also mask sex differences in underlying molecular and cellular mechanisms (448).
Among the cellular mechanisms so far identified, it has been reported that spinal microglia activation is required for injury-induced hypersensitivity in males whereas activation and invasion of adaptive immune cells such as T-lymphocytes is required in females (451, 452). Macrophage invasion of DRG is predominant in males and not in females (453) and nociception is regulated by spinal serotonin and noradrenaline in male but not in female mice (454). It has also recently been shown that ex vivo treatment of live human organ donor spinal cord tissue with BDNF downregulates markers of inhibition and upregulates markers of facilitated excitation in dorsal horn neurons from males but not females (455). Lastly, administration of IL-23 (Interleukin 23) produces mechanical allodynia in female but not male mice and chemotherapy-induced mechanical pain is selectively impaired in female mice lacking IL-23 or its cognate receptor. (456). These authors have suggested that the difference in response may be attributed to the function of sex hormones as IL-23-induced pain is suppressed by androgen and promoted by estrogen.
In the peripheral nervous system, blockade of Nav1.8 channels with A-803467 or Cav2.3 with SNX-482 is more effective in females than in males in various models of neuropathic pain (457, 458).
The realization that different mechanisms are engaged to generate pain in males vs. females has obvious therapeutic implications. If spinal serotonin and noradrenaline attenuate pain in male rather than female rodents (454), might SNRI's such as duloxetine and venlafaxine work better in men than in women? As already mentioned the Nav1.8 channel blocker A-803467 works better in woman than in men (457). The importance of the incorporation of sex as a variable in future studies cannot be over emphasized (447, 459).
Recognize Differences in Pain Etiology (Quantitative Sensory Testing and the Personalized Medicine Approach)
Patients with neuropathic pain are heterogeneous in pathophysiology, etiology and clinical presentation (460). Neuropathic pain can result from sources as varied as nerve compression, channelopathy, autoimmune disease, infection, disease or chemotherapy-induced neuropathy and the response of each individual is determined by a multiplicity of factors such as inherited genetic variants, sex, neonatal injury or maternal separation, age, ethnicity, intestinal microbiome, personality variables, and environmental factors (444, 461–467).
There are numerous examples of cellular mechanisms that may contribute to these differences. For example, adult pain responses are primed by neonatal pain experience and this is maintained by central neuroimmune activity (463). A-channels and HCN channels may be especially affected in diabetic neuropathy (26, 254); might A-current activators and HCN blockers be especially useful in this situation? Nav1.7 is found in both sensory and sympathetic nerve fibers, might Nav1.7 blockers be especially useful in complex regional pain syndromes? By contrast, in animal studies Nav1.7 does not appear to be involved in oxaliplatin-induced painful neuropathy (123) yet does appear to be involved in that seen with paclitaxel (154). Does this mean that Nav1.7 blockers might only be effective in subgroups of patients with chemotherapy induced neuropathy (CIPN)?
Perturbations of Nav1.6 function may contribute to trigeminal neuralgia (141), might Nav1.6 blockers be of special value in this situation? Beyond the peripheral nervous system, the neuronal subtypes in the dorsal horn that are involved in generation of mechanical allodynia is defined by the nature of peripheral nerve injury (468). This likely relates to the observation that CCI of the sciatic nerve produces transient allodynia in animal models whereas that produced by SNI is persistent (469, 470).
In the clinic, various subtypes of neuropathic pain may be identified using quantitative sensory testing (QST). This involves formalization and quantification of an existing battery of neurological tests, such as response to von Frey filaments, vibration, heat, pressure and cold as well as dynamic allodynia and wind-up ratio (460, 471). Findings are compared with large datasets that represent normal responses to sensory tests. Neuropathic pain patients can then be grouped into clusters based on their sensory profiles and that this may have a role in determining treatment (472, 473). Three distinct subgroups with characteristic sensory profiles have already been identified in patients with peripheral neuropathic pain (460). Cluster 1 showed a loss of small and large fiber function in combination with paradoxical heat sensations. Cluster 2 was characterized by preserved sensory functions in combination with heat and cold hyperalgesia and mild dynamic mechanical allodynia and Cluster 3 was characterized by a loss of small fiber function in combination with pinprick hyperalgesia and dynamic mechanical allodynia. The validity of QST is supported by the observation that post-hoc analysis of responders to treatments in clinical trials suggest that clinical effectiveness may cluster according to pain phenotype (472).
In view of this, can signs and symptoms observed in each individual patient in the clinic be traced back to underlying pathophysiology? This would permit a “personalized medicine approach” that would dictate the most appropriate therapeutic approach (437, 474, 475). Such an approach may necessitate better “harmonization” between preclinical studies and clinical observations. Thus, while studying chemotherapy-induced pain in rodents may be an appropriate model for understanding CIPN in the clinic, it is less clear how classical rodent pain models such as SNI or CCI relate to the multiplicity of chronic pain presentations in the clinic (437).
Target the Genetic and Biochemical Mechanisms That Control Channel Expression
As mentioned in the introduction, peripheral nerve injury or neuropathy is associated with the generation and release of a variety of inflammatory mediators (17–20). These mediators generally increase Na+, Ca2+ and HCN channel function and attenuate K+ channel function (8, 24, 27, 28, 114) thereby promoting the increase in primary afferent excitability which is crucial for the onset of and persistence of neuropathic pain (11, 12, 29–35). Despite the careful documentation of changes in peripheral ion channels associated with neuropathic pain, clinical results with K+ channel activators and novel Na+ or Ca2+ channel blockers have met with limited success.
One possible solution is to target the processes which control the function of multiple channel types. We have already mentioned the role of the histone methyltransferase G9a in controlling the expression of Kv7.2, Kv1.4, and KCa1.1 [(250), Table 1]. A G9a inhibitor, vorinostat is available for the management of cutaneous T-cell lymphoma. Perhaps repurposing this clinically-approved drug may lead to effective pain treatments.
The MNK-eIF4E signaling axis represents another potential drug target (110). These authors showed that a single phosphorylation site on S209 of the mRNA 5 cap-binding protein eIF4E is a critical mechanism for changes in nociceptor excitably. This is brought about by activation of mechanistic target of rapamycin (mTOR) and mitogen-activated protein kinases (MAPK) 1&2 which are downstream effectors of pro-nociceptive agents such as NGF (215) and IL-6 (476). MAPK 1 & 2 act through MAPK-interacting kinases (MNK) 1 & 2 and co-operates with mTOR to activate specific mRNA's. Nociceptor sensitization and pain behaviors are attenuated in neurons from eIF4E (S209A) mice where serine 209 is replaced by alanine, Mnk1/2 knockout mice and by the MNK1/2 inhibitor cercosporamide. These findings underline the idea that pathways that regulate mRNA translation are key factors in changes in injury-induces nociceptor excitability and in the maintenance and/or onset of neuropathic pain These findings beg the question of whether cercosporamide, which is already used to treat and control pain in rheumatoid arthritis (477) may also be useful in other forms neuropathic pain (Table 1).
Use the Right Combination
Combination therapy is a useful therapeutic technique that maximizes drug effects whilst limiting untoward effects. The use of low doses of two drugs that have different and possible synergistic mechanisms lessens their dose limiting side effects (478). A good example comes from the field of cardiovascular pharmacology. Both thiazide diuretics and angiotensin converting enzyme (ACE) inhibitors are useful in the management of hypertension. The combination of low doses of these drugs limits side effects. In this case, thiazides tend to lower blood K+ whereas ACE inhibitors tend to elevate it. In this case, the combination of drugs limits perturbation of blood K+ levels.
Combination therapy has been employed in pain management for many years (478), and in several cases increased therapeutic effects have been achieved using “add on” therapies which are not always based on rational application of known drug mechanisms. One logically derived combination therapy is the combination of opioids and Nav1.7 blockers (172) as endogenous opioids appear to be involved in their action (51, 52, 171). To best of our knowledge this type of drug combination has not yet been examined in the clinic.
Use Human Nerves
Several recent reviews have commented on the slow translation between animal studies and the development of new therapeutic agents for use in the clinic (48, 475, 479). This reflects the self-evident differences between the human and rodent nervous systems (480). It is already known that both rodent and human and nociceptors are more heterogeneous at a molecular level than previously appreciated, and although there are broad similarities between human and rodent nociceptors there are also important differences involving ion channel function, expression, and cellular excitability (356, 479). For example, murine SCN11A which codes for Nav1.9 is only 75% identical to the human gene (114). Differences in channel structure between humans and rodents may result in differences in pharmacology. Drugs identified to work well in rodent models may be less effective in humans.
Up until recently there were few feasible methodologies available for study of human nerves. However, recent advances in technology and methodology have increased the feasibility of human studies (356, 479). For example, nociceptor morphology can be observed using biopsy samples (481) and cultured human nociceptors (482). Acutely-isolated human DRG's have been obtained from donors undergoing surgical treatment that required ligation of spinal nerve roots for spinal reconstruction or to facilitate tumor resection (12) or from organ donors (356).
Amongst other differences, this has revealed that most human DRG neurons exhibit TRPV1 receptor channels whereas in rats, it is nearly exclusively expressed in peptidergic nociceptors (483). There are also pronounced differences between HVA Ca2+ currents in human DRG compared to rats. Thus, in human DRG, Ca2+ current density is significantly smaller, kinetics of activation, inactivation, and deactivation are slower but the proportion of nifedipine-sensitive currents is far greater (356). Perhaps this relates to the report that nifedipine may be effective in management of complex regional pain syndrome (355). A further difference between human and rat DRG neurons is that a subpopulation of human neurons display relatively large constitutive Ca2+ current inhibition as demonstrated by paired pulse facilitation in the absence of agonist (356).
The issue of limited availability of human DRG is also being addressed using human induced pluripotent stem cells (hiPSC) and differentiating them in into nociceptive sensory neurons (54, 165, 484–486). This type of system has the advantage of scalability (generation of large numbers of cells), investigation of multiple tissue types (generation of glial and immunocompetent cells) (487) and the application of high throughput technologies such as screening of small molecule therapeutic agents and gene therapy approaches to nociceptor function (488).
Microneurography which allows in vivo recording of nociceptor axonal electrical activity in humans has been available for many years (489). Technological improvements have shown that the specific C-fiber subpopulation affected (mechanoinsensitive vs. non-mechanoceptive) depends on the source of neuropathic pain and the type of neuropathy (479, 490) Modern microneurography approaches will thus play a role in the application of personalized medicine approaches to individual patients.
Find a CRISPR Solution
There is considerable interest in the application of molecular biological approaches such as use of CRISPR (clustered regularly interspaced short palindromic repeats) technology for the management of neuropathic pain. For example, McDermott et al. (54) used CRISPR technology to edit a Nav1.7 mutation to restore the pain phenotype in hiPSCs from patients with congenital insensitivity to pain (CIP). As already mentioned Moreno et al. (55) recently targeted Nav1.7 using CRISPR-dCas9 technology by using a novel approach that prevented expression of Nav1.7 by editing a regulatory sequence. These authors suggested that this “LATER” (long-lasting analgesia via targeted in vivo epigenetic repression) technology might have therapeutic potential in management of persistent pain states, including primary erythromelalgia or paroxysmal extreme pain disorder. The feasibility of this type of approach has recently been reviewed (491, 492).
Author Contributions
All authors were involved in the writing and/or review of the manuscript.
Funding
PS was supported by Canadian Institutes of Health Grant MOP 81089 and research supplements from the Faculty of Medicine and Dentistry, University of Alberta. SA receives funding from the Research Endowment fund of the Department of Anesthesiology and Critical Care Medicine, University of New Mexico School of Medicine and a US Department of Defense Chronic Pain Management Research Program Investigator-Initiated Research Award W81XWH-20-1-0930.
Conflict of Interest
SA is an inventor on two U.S. provisional patents (493, 494).
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Finnerup NB, Attal N, Haroutounian S, McNicol E, Baron R, Dworkin RH, et al. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol. (2015) 14:162–73. doi: 10.1016/S1474-4422(14)70251-0
2. Finnerup NB, Kuner R, Jensen TS. Neuropathic pain: from mechanisms to treatment. Physiol Rev. (2021) 101:259–301. doi: 10.1152/physrev.00045.2019
3. Alles SRA, Smith PA. The etiology and pharmacology of neuropathic pain. Pharmacol Rev. (2018) 70:315–47. doi: 10.1124/pr.117.014399
4. Bannister K, Qu C, Navratilova E, Oyarzo J, Xie JY, King T, et al. Multiple sites and actions of gabapentin-induced relief of ongoing experimental neuropathic pain. Pain. (2017) 158:2386–95. doi: 10.1097/j.pain.0000000000001040
5. Bannister K, Dickenson AH. The plasticity of descending controls in pain: translational probing. J Physiol. (2017) 595:4159–66. doi: 10.1113/JP274165
6. Vaso A, Adahan HM, Gjika A, Zahaj S, Zhurda T, Vyshka G, et al. Peripheral nervous system origin of phantom limb pain. Pain. (2014) 155:1384–91. doi: 10.1016/j.pain.2014.04.018
7. Yatziv SL, Devor M. Suppression of neuropathic pain by selective silencing of dorsal root ganglion ectopia using nonblocking concentrations of lidocaine. Pain. (2019) 160:2105–14. doi: 10.1097/j.pain.0000000000001602
8. Waxman SG. Peripheral afferents and the pain experience. Pain. (2019) 160:1487–8. doi: 10.1097/j.pain.0000000000001527
9. Gold MS, Gebhart GF. Nociceptor sensitization in pain pathogenesis. Nat Med. (2010) 16:1248–57. doi: 10.1038/nm.2235
10. Costigan M, Scholz J, Woolf CJ. Neuropathic pain: a maladaptive response of the nervous system to damage. Annu Rev Neurosci. (2009) 32:1–32. doi: 10.1146/annurev.neuro.051508.135531
11. Govrin-Lippmann R, Devor M. Ongoing activity in severed nerves: source and variation with time. Brain Res. (1978) 159:406–10. doi: 10.1016/0006-8993(78)90548-6
12. North RY, Li Y, Ray P, Rhines LD, Tatsui CE, Rao G, et al. Electrophysiological and transcriptomic correlates of neuropathic pain in human dorsal root ganglion neurons. Brain. (2019) 142:1215–26. doi: 10.1093/brain/awz063
13. Buch NS, Ahlburg P, Haroutounian S, Andersen NT, Finnerup NB, Nikolajsen L. The role of afferent input in postamputation pain: a randomized, double-blind, placebo-controlled crossover study. Pain. (2019) 160:1622–33. doi: 10.1097/j.pain.0000000000001536
14. Haroutounian S, Nikolajsen L, Bendtsen TF, Finnerup NB, Kristensen AD, Hasselstrøm Jr B, et al. Primary afferent input critical for maintaining spontaneous pain in peripheral neuropathy. Pain. (2014) 155:1272–9. doi: 10.1016/j.pain.2014.03.022
15. Sapio MR, Vazquez FA, Loydpierson AJ, Maric D, Kim JJ, LaPaglia DM, et al. Comparative analysis of dorsal root, nodose and sympathetic ganglia for the development of new analgesics. Front Neurosci. (2020) 14:615362. doi: 10.3389/fnins.2020.615362
16. Parsons B, Tive L, Huang S. Gabapentin: a pooled analysis of adverse events from three clinical trials in patients with postherpetic neuralgia. Am J Geriatr Pharmacother. (2004) 2:157–62. doi: 10.1016/j.amjopharm.2004.09.004
17. Boakye PA, Tang SJ, Smith PA. Mediators of neuropathic pain; focus on spinal microglia, CSF-1, BDNF, CCL21, TNF-alpha, Wnt ligands, and Interleukin 1-beta. Front Pain Res. (2021) 2:41. doi: 10.3389/fpain.2021.698157
18. Scholz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci. (2007) 10:1361–8. doi: 10.1038/nn1992
19. Moalem G, Tracey DJ. Immune and inflammatory mechanisms in neuropathic pain. Brain Res Rev. (2006) 51:240–64. doi: 10.1016/j.brainresrev.2005.11.004
20. Calvo M, Dawes JM, Bennett DL. The role of the immune system in the generation of neuropathic pain. Lancet Neurol. (2012) 11:629–42. doi: 10.1016/S1474-4422(12)70134-5
21. Stemkowski PL, Smith PA. Long-term IL-1beta exposure causes subpopulation-dependent alterations in rat dorsal root ganglion neuron excitability. J Neurophysiol. (2012) 107:1586–97. doi: 10.1152/jn.00587.2011
22. Stemkowski PL, Noh MC, Chen Y, Smith PA. Increased excitability of medium-sized dorsal root ganglion neurons by prolonged interleukin-1beta exposure is K(+) channel dependent and reversible. J Physiol. (2015) 593:3739–55. doi: 10.1113/JP270905
23. Noh M-C, Stemkowski PL, Smith PA. Long-term actions of interleukin-1β on K+, Na+ Ca2+ channel currents in small, IBsb4/s-positive dorsal root ganglion neurons possible relevance to the etiology of neuropathic pain. J Neuroimmunol.(2019) 332:198–211. doi: 10.1016/j.jneuroim.2019.05.002
24. Waxman SG, Zamponi GW. Regulating excitability of peripheral afferents: emerging ion channel targets. Nat Neurosci. (2014) 17:153–63. doi: 10.1038/nn.3602
25. Basso L, Altier C. Transient Receptor Potential Channels in neuropathic pain. Curr Opin Pharmacol. (2017) 32:9–15. doi: 10.1016/j.coph.2016.10.002
26. Tsantoulas C, Lainez S, Wong S, Mehta I, Vilar B, McNaughton PA. Hyperpolarization-activated cyclic nucleotide-gated 2 (HCN2) ion channels drive pain in mouse models of diabetic neuropathy. Sci Transl Med. (2017) 9:eaam6072. doi: 10.1126/scitranslmed.aam6072
27. Tsantoulas C, Mooney ER, McNaughton PA. HCN2 ion channels: basic science opens up possibilities for therapeutic intervention in neuropathic pain. Biochem J. (2016) 473:2717–36. doi: 10.1042/BCJ20160287
28. Smith PA. K+ channels in primary afferents and their role in pain produced by peripheral nerev injury. Front Cell Neurosci. (2020) 14:294. doi: 10.3389/fncel.2020.566418
29. Study RE, Kral MG. Spontaneous action potential activity in isolated dorsal root ganglion neurons from rats with a painful neuropathy. Pain. (1996) 65:235–42. doi: 10.1016/0304-3959(95)00216-2
30. Djouhri L, Smith T, Ahmeda A, Alotaibi M, Weng X. Hyperpolarization-activated cyclic nucleotide-gated channels contribute to spontaneous activity in L4 C-fiber nociceptors, but not Abeta-non-nociceptors, after axotomy of L5-spinal nerve in the rat in vivo. Pain. (2018) 159:1392–402. doi: 10.1097/j.pain.0000000000001224
31. Abdulla FA, Smith PA. Axotomy and autotomy-induced changes in the excitability of rat dorsal root ganglion neurons. J Neurophysiol. (2001) 85:630–43. doi: 10.1152/jn.2001.85.2.630
32. Liu CN, Raber P, Ziv-Sefer S, Devor M. Hyperexcitability in sensory neurons of rats selected for high versus low neuropathic pain phenotype. Neuroscience. (2001) 105:265–75. doi: 10.1016/S0306-4522(01)00161-0
33. Wall PD, Devor M, Inbal R, Scadding JW, Schonfeld D, Seltzer Z, et al. Autotomy following peripheral nerve lesions: experimental anaesthesia dolorosa. Pain. (1979) 7:103–13. doi: 10.1016/0304-3959(79)90002-2
34. Pitcher GM, Henry JL. Governing role of primary afferent drive in increased excitation of spinal nociceptive neurons in a model of sciatic neuropathy. Exp Neurol. (2008) 214:219–28. doi: 10.1016/j.expneurol.2008.08.003
35. Devor M. Ectopic discharge in Abeta afferents as a source of neuropathic pain. Exp Brain Res. (2009) 196:115–28. doi: 10.1007/s00221-009-1724-6
36. Staaf S, Oerther S, Lucas G, Mattsson JP, Ernfors P. Differential regulation of TRP channels in a rat model of neuropathic pain. Pain. (2009) 144:187–99. doi: 10.1016/j.pain.2009.04.013
37. Ikeda-Miyagawa Y, Kobayashi K, Yamanaka H, Okubo M, Wang S, Dai Y, et al. Peripherally increased artemin is a key regulator of TRPA1/V1 expression in primary afferent neurons. Mol Pain. (2015) 11:8. doi: 10.1186/s12990-015-0004-7
38. Wei S, Jin J, Liu T-T, Qiu C-Y, Hu W-P. TNF-alpha acutely enhances acid-sensing ion channel currents in rat dorsal root ganglion neurons via a p38 MAPK pathway. J Neuroinflamm. (2021) 18:92. doi: 10.1186/s12974-021-02151-w
39. Iftinca M, Defaye M, Altier C. TRPV1-targeted drugs in development for human pain conditions. Drugs. (2020) 81:7–27. doi: 10.1007/s40265-020-01429-2
40. Inoue K, Tsuda M. Nociceptive signaling mediated by P2X3, P2X4 and P2X7 receptors. Biochem Pharmacol. (2021) 187:114309. doi: 10.1016/j.bcp.2020.114309
41. Ye G, Zhang Y, Zhao J, Chen Y, Kong L, Sheng C, et al. miR-384-5p ameliorates neuropathic pain by targeting SCN3A in a rat model of chronic constriction injury. Neurol Res. (2020) 42:299–307. doi: 10.1080/01616412.2020.1723313
42. Su S, Shao J, Zhao Q, Ren X, Cai W, Li L, et al. MiR-30b attenuates neuropathic pain by regulating voltage-gated sodium channel Nav1.3 in rats. Front Mol Neurosci. (2017) 10:126. doi: 10.3389/fnmol.2017.00126
43. Chen HP, Zhou W, Kang LM, Yan H, Zhang L, Xu BH, et al. Intrathecal miR-96 inhibits Nav1.3 expression and alleviates neuropathic pain in rat following chronic construction injury. Neurochem Res. (2014) 39:76–83. doi: 10.1007/s11064-013-1192-z
44. Pryde DC, Swain NA, Stupple PA, West CW, Marron B, Markworth CJ, et al. The discovery of a potent Nav1.3 inhibitor with good oral pharmacokinetics. Medchemcomm. (2017) 8:1255–67. doi: 10.1039/C7MD00131B
45. de Greef BTA, Hoeijmakers JGJ, Geerts M, Oakes M, Church TJE, Waxman SG, et al. Lacosamide in patients with Nav1.7 mutations-related small fibre neuropathy: a randomized controlled trial. Brain. (2019) 142:263–75. doi: 10.1093/brain/awy329
46. McDonnell A, Collins S, Ali Z, Iavarone L, Surujbally R, Kirby S, et al. Efficacy of the Nav1.7 blocker PF-05089771 in a randomised, placebo-controlled, double-blind clinical study in subjects with painful diabetic peripheral neuropathy. Pain. (2018) 159:1465–76. doi: 10.1097/j.pain.0000000000001227
47. Kotecha M, Cheshire WP, Finnigan H, Giblin K, Naik H, Palmer J, et al. Design of phase 3 studies evaluating vixotrigine for treatment of trigeminal neuralgia. J Pain Res. (2020) 13:1601–9. doi: 10.2147/JPR.S247182
48. Eagles DA, Chow CY, King GF. Fifteen years of NaV 1.7 channels as an analgesic target: why has excellent in vitro pharmacology not translated into in vivo analgesic efficacy? Br J Pharmacol. (2020). doi: 10.1111/bph.15327. [Epub ahead of print].
49. Cardoso FC, Castro J, Grundy L, Schober G, Garcia-Caraballo S, Zhao T, et al. A spider-venom peptide with multitarget activity on sodium and calcium channels alleviates chronic visceral pain in a model of irritable bowel syndrome. Pain. (2021) 162:569–81. doi: 10.1097/j.pain.0000000000002041
50. Hu H, Mawlawi SE, Zhao T, Deuis JR, Jami S, Vetter I, et al. Engineering of a spider peptide via conserved structure-function traits optimizes sodium channel inhibition in vitro and anti-nociception in vivo. Front Mol Biosci. (2021) 8:742457. doi: 10.3389/fmolb.2021.742457
51. MacDonald DI, Sikandar S, Weiss J, Pyrski M, Luiz AP, Millet Q, et al. A central mechanism of analgesia in mice and humans lacking the sodium channel NaV1.7. Neuron. (2021) 109:1497–512. doi: 10.1016/j.neuron.2021.03.012
52. Minett MS, Pereira V, Sikandar S, Matsuyama A, Lolignier S, Kanellopoulos AH, et al. Endogenous opioids contribute to insensitivity to pain in humans and mice lacking sodium channel Nav17. Nat Commun. (2015) 6:8967. doi: 10.1038/ncomms9967
53. Mueller A, Starobova H, Morgan M, Dekan Z, Cheneval O, Schroeder CI, et al. Antiallodynic effects of the selective NaV1.7 inhibitor Pn3a in a mouse model of acute postsurgical pain: evidence for analgesic synergy with opioids and baclofen. Pain. (2019) 160:1766–80. doi: 10.1097/j.pain.0000000000001567
54. McDermott LA, Weir GA, Themistocleous AC, Segerdahl AR, Blesneac I, Baskozos G, et al. Defining the functional role of NaV1.7 in human nociception. Neuron. (2019) 101:905–19. doi: 10.1016/j.neuron.2019.01.047
55. Moreno AM, Aleman F, Catroli GF, Hunt M, Hu M, Dailamy A, et al. Long-lasting analgesia via targeted in situ repression of NaV1.7 in mice. Sci Transl Med. (2021) 13:584. doi: 10.1126/scitranslmed.aay9056
56. Demant DT, Lund K, Vollert J, Maier C, Segerdahl M, Finnerup NB, et al. The effect of oxcarbazepine in peripheral neuropathic pain depends on pain phenotype: a randomised, double-blind, placebo-controlled phenotype-stratified study. Pain. (2014) 155:2263–73. doi: 10.1016/j.pain.2014.08.014
57. Hijma HJH, Siebenga PSP, de Kam MLM, Groeneveld GJ. A phase 1, randomized, double-blind, placebo-controlled, crossover study to evaluate the pharmacodynamic effects of VX-150, a highly selective NaV1.8 inhibitor, in healthy male adults. Pain Med. (2021) 22:1814–26. doi: 10.1093/pm/pnab032
58. Patel MK, Kaye AD, Urman RD. Tanezumab: therapy targeting nerve growth factor in pain pathogenesis. J Anaesthesiol Clin Pharmacol. (2018) 34:111–6. doi: 10.4103/joacp.JOACP_155_18
59. Peigneur S, da Costa OC, de Sousa Fonseca FC, McMahon KL, Mueller A, Cheneval O, et al. Small cyclic sodium channel inhibitors. Biochem Pharmacol. (2021) 183:114291. doi: 10.1016/j.bcp.2020.114291
60. Binshtok AM, Gerner P, Oh SB, Puopolo M, Suzuki S, Roberson DP, et al. Coapplication of lidocaine and the permanently charged sodium channel blocker QX-314 produces a long-lasting nociceptive blockade in rodents. Anesthesiology. (2009) 111:127–37. doi: 10.1097/ALN.0b013e3181a915e7
61. Tochitsky I, Jo S, Andrews N, Kotoda M, Doyle B, Shim J, et al. Inhibition of inflammatory pain and cough by a novel charged sodium channel blocker. Br J Pharmacol. (2021) 178:M3905–23. doi: 10.1111/bph.15531
62. Puopolo M, Binshtok AM, Woolf CJ, Bean BP. Permeation and block of TRPV1 channels by the cationic lidocaine derivative QX-314. J Neurophysiol. (2013) 109:1704–12. doi: 10.1152/jn.00012.2013
63. Manville RW, Abbott GW. Isoform-selective KCNA1 potassium channel openers built from glycine. J Pharmacol Exp Ther. (2020) 373:391–401. doi: 10.1124/jpet.119.264507
64. Ouyang B, Chen D, Hou X, Wang T, Wang J, Zou W, et al. Normalizing HDAC2 levels in the spinal cord alleviates thermal and mechanical hyperalgesia after peripheral nerve injury and promotes GAD65 and KCC2 expression. Front Neurosci. (2019) 13:346. doi: 10.3389/fnins.2019.00346
65. Hou X, Weng Y, Wang T, Ouyang B, Li Y, Song Z, et al. Suppression of HDAC2 in spinal cord alleviates mechanical hyperalgesia and restores KCC2 expression in a rat model of bone cancer pain. Neuroscience. (2018) 377:138–49. doi: 10.1016/j.neuroscience.2018.02.026
66. He XT, Hu XF, Zhu C, Zhou KX, Zhao WJ, Zhang C, et al. Suppression of histone deacetylases by SAHA relieves bone cancer pain in rats via inhibiting activation of glial cells in spinal dorsal horn and dorsal root ganglia. J Neuroinflamm. (2020) 17:125. doi: 10.1186/s12974-020-01740-5
67. Tsantoulas C, Denk F, Signore M, Nassar MA, Futai K, McMahon SB. Mice lacking Kcns1 in peripheral neurons show increased basal and neuropathic pain sensitivity. Pain. (2018) 159:1641–51. doi: 10.1097/j.pain.0000000000001255
68. Yekkirala AS, Roberson DP, Bean BP, Woolf CJ. Breaking barriers to novel analgesic drug development. Nat Rev Drug Discov. (2017) 16:545–64. doi: 10.1038/nrd.2017.87
69. Liu Y, Bian X, Wang K. Pharmacological activation of neuronal voltage-gated Kv7/KCNQ/M-channels for potential therapy of epilepsy and pain. Handb Exp Pharmacol. (2021) 267:231–51. doi: 10.1007/164_2021_458
70. Wang J, Liu Y, Hu F, Yang J, Guo X, Hou X, et al. Activation of neuronal voltage-gated potassium Kv7/KCNQ/M-current by a novel channel opener SCR2682 for alleviation of chronic pain. J Pharmacol Exp Ther. (2021) 377:20–8. doi: 10.1124/jpet.120.000357
71. Manville RW, Abbott GW. Ancient and modern anticonvulsants act synergistically in a KCNQ potassium channel binding pocket. Nat Commun. (2018) 9:3845. doi: 10.1038/s41467-018-06339-2
72. Manville RW, Abbott GW. Cilantro leaf harbors a potent potassium channel-activating anticonvulsant. FASEB J. (2019) 33:11349–63. doi: 10.1096/fj.201900485R
73. Gonzalez WG, Pham K, Miksovska J. Modulation of the voltage-gated potassium channel (Kv4.3) and the auxiliary protein (KChIP3) interactions by the current activator NS5806. J Biol Chem. (2014) 289:32201–13. doi: 10.1074/jbc.M114.577528
74. Lundby A, Jespersen T, Schmitt N, Grunnet M, Olesen SP, Cordeiro JM, et al. Effect of the I(to) activator NS5806 on cloned K(V)4 channels depends on the accessory protein KChIP2. Br J Pharmacol. (2010) 160:2028–44. doi: 10.1111/j.1476-5381.2010.00859.x
75. Kanda H, Ling J, Chang YT, Erol F, Viatchenko-Karpinski V, Yamada A, et al. Kv4.3 channel dysfunction contributes to trigeminal neuropathic pain manifested with orofacial cold hypersensitivity in rats. J Neurosci. (2021) 41:2091–105. doi: 10.1523/JNEUROSCI.2036-20.2021
76. Koh WU, Shin JW, Bang JY, Kim SG, Song JG. The antiallodynic effects of nefopam are mediated by the adenosine triphosphate-sensitive potassium channel in a neuropathic pain model. Anesth Analg. (2016) 123:762–70. doi: 10.1213/ANE.0000000000001411
77. Gutierrez VP, Zambelli VO, Picolo G, Chacur M, Sampaio SC, Brigatte P, et al. The peripheral L-arginine-nitric oxide-cyclic GMP pathway and ATP-sensitive K(+) channels are involved in the antinociceptive effect of crotalphine on neuropathic pain in rats. Behav Pharmacol. (2012) 23:14–24. doi: 10.1097/FBP.0b013e32834eafbc
78. Wu XF, Liu WT, Liu YP, Huang ZJ, Zhang YK, Song XJ. Reopening of ATP-sensitive potassium channels reduces neuropathic pain and regulates astroglial gap junctions in the rat spinal cord. Pain. (2011) 152:2605–15. doi: 10.1016/j.pain.2011.08.003
79. Luu W, Bjork J, Salo E, Entenmann N, Jurgenson T, Fisher C, et al. Modulation of SUR1 KATP channel subunit activity in the peripheral nervous system reduces mechanical hyperalgesia after nerve injury in mice. Int J Mol Sci. (2019) 20:2251. doi: 10.3390/ijms20092251
80. Wang X, Shen X, Ma S, Liu Y, Xu S, Wu S, et al. Threshold effect of G9a/Glp on peripheral nerve injury induced hypersensitivity. Mol Pain. (2017) 13:1744806917729305. doi: 10.1177/1744806917729305
81. Liang L, Zhao JY, Kathryn T, Bekker A, Tao YX. BIX01294, a G9a inhibitor, alleviates nerve injury-induced pain hypersensitivities during both development and maintenance periods. Transl Perioper Pain Med. (2019) 6:106–14. doi: 10.31480/2330-4871/097
82. McGivern JG. Ziconotide: a review of its pharmacology and use in the treatment of pain. Neuropsychiatr Dis Treat. (2007) 3:69–85. doi: 10.2147/nedt.2007.3.1.69
83. Zhang S, Yang L, Zhang K, Liu X, Dai W, Zhang C, et al. ZC88, a novel N-type calcium channel blocker from 4-amino-piperidine derivatives state-dependent inhibits Cav2.2 calcium channels. Brain Res. (2015) 1605:12–21. doi: 10.1016/j.brainres.2015.01.054
84. Meng G, Wu N, Zhang C, Su RB, Lu XQ, Liu Y, et al. Analgesic activity of ZC88, a novel N-type voltage-dependent calcium channel blocker, and its modulation of morphine analgesia, tolerance and dependence. Eur J Pharmacol. (2008) 586:130–8. doi: 10.1016/j.ejphar.2008.02.066
85. Xu J, Chu KL, Zhu CZ, Niforatos W, Swensen A, Searle X, et al. A mixed Ca2+ channel blocker, A-1264087, utilizes peripheral and spinal mechanisms to inhibit spinal nociceptive transmission in a rat model of neuropathic pain. J Neurophysiol. (2014) 111:394–404. doi: 10.1152/jn.00463.2013
86. Patel R, Rutten K, Valdor M, Schiene K, Wigge S, Schunk S, et al. Electrophysiological characterization of activation state-dependent Ca(v)2 channel antagonist TROX-1 in spinal nerve injured rats. Neuroscience. (2015) 297:47–57. doi: 10.1016/j.neuroscience.2015.03.057
87. Abbadie C, McManus OB, Sun SY, Bugianesi RM, Dai G, Haedo RJ, et al. Analgesic effects of a substituted N-triazole oxindole (TROX-1), a state-dependent, voltage-gated calcium channel 2 blocker. J Pharmacol Exp Ther. (2010) 334:545–55. doi: 10.1124/jpet.110.166363
88. Kirkpatrick AF, Derasari M, Glodek JA, Piazza PA. Postherpetic neuralgia: a possible application for topical clonidine. Anesthesiology. (1992) 76:1065–6. doi: 10.1097/00000542-199206000-00044
89. Kocot-Kepska M, Zajaczkowska R, Mika J, Kopsky DJ, Wordliczek J, Dobrogowski J, et al. Topical treatments and their molecular/cellular mechanisms in patients with peripheral neuropathic pain-narrative review. Pharmaceutics. (2021) 13:450. doi: 10.3390/pharmaceutics13040450
90. Yoon SY, Roh DH, Yeo JH, Woo J, Han SH, Kim KS. Analgesic efficacy of alpha2 adrenergic receptor agonists depends on the chronic state of neuropathic pain: role of regulator of g protein signaling 4. Neuroscience. (2021) 455:177–94. doi: 10.1016/j.neuroscience.2020.12.021
91. Zeigler D, Lynch SA, Muir J, Benjamin J, Max MB. Transdermal clonidine versus placebo in painful diabetic neuropathy. Pain. (1992) 48:403–8. doi: 10.1016/0304-3959(92)90092-P
92. Zuniga RE, Perera S, Abram SE. Intrathecal baclofen: a useful agent in the treatment of well-established complex regional pain syndrome. Reg Anesth Pain Med. (2002) 27:90–3. doi: 10.1053/rapm.2002.29244
93. Zamponi GW, Striessnig J, Koschak A, Dolphin AC. The physiology, pathology, and pharmacology of voltage-gated calcium channels and their future therapeutic potential. Pharmacol Rev. (2015) 67:821–70. doi: 10.1124/pr.114.009654
94. Hoppa MB, Lana B, Margas W, Dolphin AC, Ryan TA. alpha2delta expression sets presynaptic calcium channel abundance and release probability. Nature. (2012) 486:122–5. doi: 10.1038/nature11033
95. Biggs JE, Boakye PA, Ganesan N, Stemkowski PL, Lantero A, Ballanyi K, et al. Analysis of the long-term actions of gabapentin and pregabalin in dorsal root ganglia and substantia gelatinosa. J Neurophysiol. (2014) 112:2398–412. doi: 10.1152/jn.00168.2014
96. Moore RA, Wiffen PJ, Derry S, McQuay HJ. Gabapentin for chronic neuropathic pain and fibromyalgia in adults. Cochrane Database Syst Rev. (2011) CD007938. doi: 10.1002/14651858.CD007938.pub2
97. Lee S, Jo S, Talbot S, Zhang HB, Kotoda M, Andrews NA, et al. Novel charged sodium and calcium channel inhibitor active against neurogenic inflammation. Elife. (2019) 8. doi: 10.7554/eLife.48118
98. Kerckhove N, Pereira B, Soriot-Thomas S, Alchaar H, Deleens R, Hieng VS, et al. Efficacy and safety of a T-type calcium channel blocker in patients with neuropathic pain: a proof-of-concept, randomized, double-blind and controlled trial. Eur J Pain. (2018) 22:1321–30. doi: 10.1002/ejp.1221
99. Garcia-Caballero A, Gadotti VM, Chen L, Zamponi GW. A cell-permeant peptide corresponding to the cUBP domain of USP5 reverses inflammatory and neuropathic pain. Mol Pain. (2016) 12. doi: 10.1177/1744806916642444
100. Gadotti VM, Caballero AG, Berger ND, Gladding CM, Chen L, Pfeifer TA, et al. Small organic molecule disruptors of Cav3.2 - USP5 interactions reverse inflammatory and neuropathic pain. Mol Pain. (2015) 11:12. doi: 10.1186/s12990-015-0011-8
101. Nam G. T-type calcium channel blockers: a patent review (2012-2018). Expert Opin Ther Pat. (2018) 28:883–901. doi: 10.1080/13543776.2018.1541982
102. Zhang HXB, Bean BP. Cannabidiol inhibition of murine primary nociceptors: tight binding to slow inactivated states of Nav1.8 channels. J Neurosci. (2021) 41:6371. doi: 10.1523/JNEUROSCI.3216-20.2021
103. Li Y, Zhang L, Wu Y, Zheng Q, Chen M, Qian Z, et al. Cannabinoids-induced peripheral analgesia depends on activation of BK channels. Brain Res. (2019) 1711:23–8. doi: 10.1016/j.brainres.2019.01.007
104. Ross HR, Napier I, Connor M. Inhibition of recombinant human T-type calcium channels by Delta9-tetrahydrocannabinol and cannabidiol. J Biol Chem. (2008) 283:16124–34. doi: 10.1074/jbc.M707104200
105. Bladen C, McDaniel SW, Gadotti VM, Petrov RR, Berger ND, Diaz P, et al. Characterization of novel cannabinoid based T-type calcium channel blockers with analgesic effects. ACS Chem Neurosci. (2015) 6:277–87. doi: 10.1021/cn500206a
106. Arout CA, Haney M, Herrmann ES, Bedi G, Cooper ZD. A placebo-controlled investigation of the analgesic effects, abuse liability, safety and tolerability of a range of oral cannabidiol doses in healthy humans. Br J Clin Pharmacol. (2021). doi: 10.1111/bcp.14973. [Epub ahead of print].
107. Cristino L, Bisogno T, Di MV. Cannabinoids and the expanded endocannabinoid system in neurological disorders. Nat Rev Neurol. (2020) 16:9–29. doi: 10.1038/s41582-019-0284-z
108. Moulin D, Boulanger A, Clark AJ, Clarke H, Dao T, Finley GA, et al. Pharmacological management of chronic neuropathic pain: revised consensus statement from the Canadian Pain Society. Pain Res Manag. (2014) 19:328–35. doi: 10.1155/2014/754693
109. Hauser W, Petzke F, Fitzcharles MA. Efficacy, tolerability and safety of cannabis-based medicines for chronic pain management - an overview of systematic reviews. Eur J Pain. (2017) 22:455–70. doi: 10.1002/ejp.1118
110. Moy JK, Khoutorsky A, Asiedu MN, Black BJ, Kuhn JL, Barragan-Iglesias P, et al. The MNK-eIF4E signaling axis contributes to injury-induced nociceptive plasticity and the development of chronic pain. J Neurosci. (2017) 37:7481–99. doi: 10.1523/JNEUROSCI.0220-17.2017
111. Abdulla FA, Smith PA. Changes in Na+ channel currents of rat dorsal root ganglion neurons following axotomy and axotomy-induced autotomy. J Neurophysiol. (2002) 88:2518–29. doi: 10.1152/jn.00913.2001
112. Dib-Hajj SD, Fjell J, Cummins TR, Zheng Z, Fried K, LaMotte R, et al. Plasticity of sodium channel expression in DRG neurons in the chronic constriction injury model of neuropathic pain. Pain. (1999) 83:591–600. doi: 10.1016/S0304-3959(99)00169-4
113. Everill B, Cummins TR, Waxman SG, Kocsis JD. Sodium currents of large (Abeta-type) adult cutaneous afferent dorsal root ganglion neurons display rapid recovery from inactivation before and after axotomy. Neuroscience. (2001) 106:161–9. doi: 10.1016/S0306-4522(01)00258-5
114. Bennett DL, Clark AJ, Huang J, Waxman SG, Dib-Hajj SD. The role of voltage-gated sodium channels in pain signaling. Physiol Rev. (2019) 99:1079–151. doi: 10.1152/physrev.00052.2017
115. Waxman SG, Dib-Hajj S, Cummins TR, Black JA. Sodium channels and their genes: dynamic expression in the normal nervous system, dysregulation in disease states(1). Brain Res. (2000) 886:5–14. doi: 10.1016/S0006-8993(00)02774-8
116. Goldin AL, Barchi RL, Caldwell JH, Hofmann F, Howe JR, Hunter JC, et al. Nomenclature of voltage-gated sodium channels. Neuron. (2000) 28:365–8. doi: 10.1016/S0896-6273(00)00116-1
117. Catterall WA, Goldin AL, Waxman SG. International union of pharmacology. XLVII Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol Rev. (2005) 57:397–409. doi: 10.1124/pr.57.4.4
118. Alexander SPH, Mathie A, Peters JA, Veale EL, Striessnig J, Kelly E, et al. The concise giude to pharmacology 2019/20: ion channels. Br J Pharmacol. (2019) 176(Suppl. 1):S142–228. doi: 10.1111/bph.14750
119. Dray A. Neuropathic pain: emerging treatments. Br J Anaesth. (2008) 101:48–58. doi: 10.1093/bja/aen107
120. Priest BT. Future potential and status of selective sodium channel blockers for the treatment of pain. Curr Opin Drug Discov Devel. (2009) 12:682–92.
121. Siqueira SR, Alves B, Malpartida HM, Teixeira MJ, Siqueira JT. Abnormal expression of voltage-gated sodium channels Nav1.7, Nav1.3 and Nav1.8 in trigeminal neuralgia. Neuroscience. (2009) 164:573–7. doi: 10.1016/j.neuroscience.2009.08.037
122. Black JA, Nikolajsen L, Kroner K, Jensen TS, Waxman SG. Multiple sodium channel isoforms and mitogen-activated protein kinases are present in painful human neuromas. Ann Neurol. (2008) 64:644–53. doi: 10.1002/ana.21527
123. Minett MS, Falk S, Santana-Varela S, Bogdanov YD, Nassar MA, Heegaard AM, et al. Pain without nociceptors? Nav1.7-independent pain mechanisms. Cell Rep. (2014) 6:301–12. doi: 10.1016/j.celrep.2013.12.033
124. Minett MS, Nassar MA, Clark AK, Passmore G, Dickenson AH, Wang F, et al. Distinct Nav1.7-dependent pain sensations require different sets of sensory and sympathetic neurons. Nat Commun. (2012) 3:791. doi: 10.1038/ncomms1795
125. Minett MS, Eijkelkamp N, Wood JN. Significant determinants of mouse pain behaviour. PLoS ONE. (2014) 9:e104458. doi: 10.1371/journal.pone.0104458
126. Beckh S, Noda M, Lubbert H, Numa S. Differential regulation of three sodium channel messenger RNAs in the rat central nervous system during development. EMBO J. (1989) 8:3611–6. doi: 10.1002/j.1460-2075.1989.tb08534.x
127. Waxman SG, Kocsis JD, Black JA. Type III sodium channels mRNA is expressed in embroyonic but not in adult spinal sensory neurons and is re-expressed following axotomy. J Neurophysiol. (1994) 72:466–70. doi: 10.1152/jn.1994.72.1.466
128. Cummins TR, Aglieco F, Renganathan M, Herzog RI, Dib-Hajj SD, Waxman SG. Nav1.3 sodium channels: rapid repriming and slow closed-state inactivation display quantitative differences after expression in a mammalian cell line and in spinal sensory neurons. J Neurosci. (2001) 21:5952–61. doi: 10.1523/JNEUROSCI.21-16-05952.2001
129. Samad OA, Tan AM, Cheng X, Foster E, Dib-Hajj SD, Waxman SG. Virus-mediated shRNA knockdown of Na(v)1.3 in rat dorsal root ganglion attenuates nerve injury-induced neuropathic pain. Mol Ther. (2013) 21:49–56. doi: 10.1038/mt.2012.169
130. Cummins TR, Waxman SG. Downregulation of tetrodotoxin-resistant sodium currents and upregulation of a rapidly repriming tetrodotoxin-sensitive sodium current in small spinal sensory neurons after nerve injury. J Neurosci. (1997) 17:3503–14. doi: 10.1523/JNEUROSCI.17-10-03503.1997
131. Hains BC, Waxman SG. Sodium channel expression and the molecular pathophysiology of pain after SCI. Prog Brain Res. (2007) 161:195–203. doi: 10.1016/S0079-6123(06)61013-3
132. Hains BC, Saab CY, Klein JP, Craner MJ, Waxman SG. Altered sodium channel expression in second-order spinal sensory neurons contributes to pain after peripheral nerve injury. J Neurosci. (2004) 24:4832–9. doi: 10.1523/JNEUROSCI.0300-04.2004
133. Hains BC, Saab CY, Waxman SG. Changes in electrophysiological properties and sodium channel Nav1.3 expression in thalamic neurons after spinal cord injury. Brain. (2005) 128:2359–71. doi: 10.1093/brain/awh623
134. Black JA, Renganathan M, Waxman SG. Sodium channel Na(v)1.6 is expressed along nonmyelinated axons and it contributes to conduction. Brain Res Mol Brain Res. (2002) 105:19–28. doi: 10.1016/S0169-328X(02)00385-6
135. Caldwell JH, Schaller KL, Lasher RS, Peles E, Levinson SR. Sodium channel Na(v)1.6 is localized at nodes of ranvier, dendrites, and synapses. Proc Natl Acad Sci USA. (2000) 97:5616–20. doi: 10.1073/pnas.090034797
136. Xie W, Strong JA, Ye L, Mao JX, Zhang JM. Knockdown of sodium channel NaV1.6 blocks mechanical pain and abnormal bursting activity of afferent neurons in inflamed sensory ganglia. Pain. (2013) 154:1170–80. doi: 10.1016/j.pain.2013.02.027
137. Chen L, Huang J, Zhao P, Persson AK, Dib-Hajj FB, Cheng X, et al. Conditional knockout of NaV16 in adult mice ameliorates neuropathic pain. Sci Rep. (2018) 8:3845. doi: 10.1038/s41598-018-22216-w
138. Xie W, Strong JA, Zhang JM. Local knockdown of the NaV1.6 sodium channel reduces pain behaviors, sensory neuron excitability, and sympathetic sprouting in rat models of neuropathic pain. Neuroscience. (2015) 291:317–30. doi: 10.1016/j.neuroscience.2015.02.010
139. Chen L, Huang J, Benson C, Lankford KL, Zhao P, Carrara J, et al. Sodium channel Nav1.6 in sensory neurons contributes to vincristine-induced allodynia. Brain. (2020) 143:2421–36. doi: 10.1093/brain/awaa208
140. Craner MJ, Klein JP, Renganathan M, Black JA, Waxman SG. Changes of sodium channel expression in experimental painful diabetic neuropathy. Ann Neurol. (2002) 52:786–92. doi: 10.1002/ana.10364
141. Tanaka BS, Zhao P, Dib-Hajj FB, Morisset V, Tate S, Waxman SG, et al. A gain-of-function mutation in Nav1.6 in a case of trigeminal neuralgia. Mol Med. (2016) 22:338–48. doi: 10.2119/molmed.2016.00131
142. Israel MR, Tanaka BS, Castro J, Thongyoo P, Robinson SD, Zhao P, et al. NaV 1.6 regulates excitability of mechanosensitive sensory neurons. J Physiol. (2019) 597:3751–68. doi: 10.1113/JP278148
143. Yeomans DC, Wilson SP. Herpes virus-based recombinant herpes vectors: gene therapy for pain and molecular tool for pain science. Gene Ther. (2009) 16:502–8. doi: 10.1038/gt.2009.25
144. Chen C, Xu B, Shi X, Zhang M, Zhang Q, Zhang T, et al. GpTx-1 and [Ala(5), Phe(6), Leu(26), Arg(28) ]GpTx-1, two peptide NaV 1.7 inhibitors: analgesic and tolerance properties at the spinal level. Br J Pharmacol. (2018) 175:3911–27. doi: 10.1111/bph.14461
145. Dib-Hajj SD, Yang Y, Black JA, Waxman SG. The Na(V)1.7 sodium channel: from molecule to man. Nat Rev Neurosci. (2013) 14:49–62. doi: 10.1038/nrn3404
146. Dib-Hajj SD, Waxman SG. Sodium channels in human pain disorders: genetics and pharmacogenomics. Annu Rev Neurosci. (2019) 42:87–106. doi: 10.1146/annurev-neuro-070918-050144
147. Toledo-Aral JJ, Moss BL, He ZJ, Koszowski AG, Whisenand T, Levinson SR, et al. Identification of PN1, a predominant voltage-dependent sodium channel expressed principally in peripheral neurons. Proc Natl Acad Sci USA. (1997) 94:1527–32. doi: 10.1073/pnas.94.4.1527
148. Black JA, Frezel N, Dib-Hajj SD, Waxman SG. Expression of Nav1.7 in DRG neurons extends from peripheral terminals in the skin to central preterminal branches and terminals in the dorsal horn. Mol Pain. (2012) 8:82. doi: 10.1186/1744-8069-8-82
149. Black JA, Hoeijmakers JG, Faber CG, Merkies IS, Waxman SG. NaV1.7: stress-induced changes in immunoreactivity within magnocellular neurosecretory neurons of the supraoptic nucleus. Mol Pain. (2013) 9:39. doi: 10.1186/1744-8069-9-39
150. Muroi Y, Ru F, Kollarik M, Canning BJ, Hughes SA, Walsh S, et al. Selective silencing of Na(V)1.7 decreases excitability and conduction in vagal sensory neurons. J Physiol. (2011) 589:5663–76. doi: 10.1113/jphysiol.2011.215384
151. Trombley PQ, Westbrook GL. Voltage-gated currents in identified rat olfactory receptor neurons. J Neurosci. (1991) 11:435–44. doi: 10.1523/JNEUROSCI.11-02-00435.1991
152. Zhang Q, Chibalina MV, Bengtsson M, Groschner LN, Ramracheya R, Rorsman NJ, et al. Na+ current properties in islet alpha- and beta-cells reflect cell-specific Scn3a and Scn9a expression. J Physiol. (2014) 592:4677–96. doi: 10.1113/jphysiol.2014.274209
153. Kretschmer T, Happel LT, England JD, Nguyen DH, Tiel RL, Beuerman RW, et al. Accumulation of PN1 and PN3 sodium channels in painful human neuroma-evidence from immunocytochemistry. Acta Neurochir. (2002) 144:803–10. doi: 10.1007/s00701-002-0970-1
154. Akin EJ, Alsaloum M, Higerd GP, Liu S, Zhao P, Dib-Hajj FB, et al. Paclitaxel increases axonal localization and vesicular trafficking of Nav1.7. Brain. (2021) 144:1727–37. doi: 10.1093/brain/awab113
155. Kim KJ, Yoon YW, Chung JM. Comparison of three rodent models of neuropathic pain. Exp Brain Res. (1997) 113:200–6. doi: 10.1007/BF02450318
156. McLachlan EM, Janig W, Michalis M. Peripheral nerve injury triggers noradrenergic sprouting within dorsal root ganglia. Nature. (1993) 363:543–6. doi: 10.1038/363543a0
157. Abdulla FA, Smith PA. Ectopic αsb2/s-adrenoceptors couple to N-type Ca2+ channels in axotomized rat sensory neurons. J Neurosci. (1997) 17:1633–41. doi: 10.1523/JNEUROSCI.17-05-01633.1997
158. Ramer MS, Bisby MA. Sympathetic axons surround neuropeptide-negative axotomized sensory neurons. Neuroreport. (1998) 9:3109–13. doi: 10.1097/00001756-199809140-00035
159. Ramer MS, Bisby MA. Differences in sympathetic innervation of mouse DRG following proximal or distal nerve lesions. Exp Neurol. (1998) 152:197–207. doi: 10.1006/exnr.1998.6855
160. Yang Y, Wang Y, Li S, Xu Z, Li H, Ma L, et al. Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J Med Genet. (2004) 41:171–4. doi: 10.1136/jmg.2003.012153
161. Cummins TR, Dib-Hajj SD, Waxman SG. Electrophysiological properties of mutant Nav1.7 sodium channels in a painful inherited neuropathy. J Neurosci. (2004) 24:8232–6. doi: 10.1523/JNEUROSCI.2695-04.2004
162. Suter MR, Bhuiyan ZA, Laedermann CJ, Kuntzer T, Schaller M, Stauffacher MW, et al. p.L1612P, a novel voltage-gated sodium channel Nav1.7 mutation inducing a cold sensitive paroxysmal extreme pain disorder. Anesthesiology. (2015) 122:414–23. doi: 10.1097/ALN.0000000000000476
163. Fertleman CR, Baker MD, Parker KA, Moffatt S, Elmslie FV, Abrahamsen B, et al. SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron. (2006) 52:767–74. doi: 10.1016/j.neuron.2006.10.006
164. Kerth CM, Hautvast P, Korner J, Lampert A, Meents JE. Phosphorylation of a chronic pain mutation in the voltage-gated sodium channel Nav1.7 increases voltage sensitivity. J Biol Chem. (2021) 296:100227. doi: 10.1074/jbc.RA120.014288
165. Meents JE, Bressan E, Sontag S, Foerster A, Hautvast P, Rosseler C, et al. The role of Nasbv/s1.7 in human nociceptors: insights from human induced pluripotent stem cell-derived sensory neurons of erythromelalgia patients. Pain. (2019) 160:1327–41. doi: 10.1097/j.pain.0000000000001511
166. Blesneac I, Themistocleous AC, Fratter C, Conrad LJ, Ramirez JD, Cox JJ, et al. Rare NaV1.7 variants associated with painful diabetic peripheral neuropathy. Pain. (2018) 159:469–80. doi: 10.1097/j.pain.0000000000001116
167. Estacion M, Han C, Choi JS, Hoeijmakers JG, Lauria G, Drenth JP, et al. Intra- and interfamily phenotypic diversity in pain syndromes associated with a gain-of-function variant of NaV1.7. Mol Pain. (2011) 7:92. doi: 10.1186/1744-8069-7-92
168. Chen L, Wimalasena NK, Shim J, Han C, Lee SI, Gonzalez-Cano R, et al. Two independent mouse lines carrying the Nav1.7 I228M gain-of-function variant display dorsal root ganglion neuron hyperexcitability but a minimal pain phenotype. Pain. (2021) 162:1758–70. doi: 10.1097/j.pain.0000000000002171
169. Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K, et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature. (2006) 444:894–8. doi: 10.1038/nature05413
170. Gingras J, Smith S, Matson DJ, Johnson D, Nye K, Couture L, et al. Global Nav1.7 knockout mice recapitulate the phenotype of human congenital indifference to pain. PLoS ONE. (2014) 9:e105895. doi: 10.1371/journal.pone.0105895
171. Sexton JE, Cox JJ, Zhao J, Wood JN. The genetics of pain: implications for therapeutics. Annu Rev Pharmacol Toxicol. (2017) 58:123–42. doi: 10.1146/annurev-pharmtox-010617-052554
172. Deuis JR, Dekan Z, Wingerd JS, Smith JJ, Munasinghe NR, Bhola RF, et al. Pharmacological characterisation of the highly Nasbv/s17 selective spider venom peptide Pn3a. Sci Rep. (2017) 7:40883. doi: 10.1038/srep40883
173. Alles SRA, Nascimento F, Lujan R, Luiz AP, Millet Q, Bangash MA, et al. Sensory neuron-derived Na(V)17 contributes to dorsal horn neuron excitability. Sci Adv. (2020) 6:eaax4568. doi: 10.1126/sciadv.aax4568
174. Emery EC, Luiz AP, Wood JN. Nav1.7 and other voltage-gated sodium channels as drug targets for pain relief. Expert Opin Ther Targets. (2016) 20:975–83. doi: 10.1517/14728222.2016.1162295
175. Rogawski MA, Tofighy A, White HS, Matagne A, Wolff C. Current understanding of the mechanism of action of the antiepileptic drug lacosamide. Epilepsy Res. (2015) 110:189–205. doi: 10.1016/j.eplepsyres.2014.11.021
176. Jo S, Bean BP. Lacosamide inhibition of Nav1.7 voltage-gated sodium channels: slow binding to fast-inactivated states. Mol Pharmacol. (2017) 91:277–86. doi: 10.1124/mol.116.106401
177. Yang Y, Dib-Hajj SD, Zhang J, Zhang Y, Tyrrell L, Estacion M, et al. Structural modelling and mutant cycle analysis predict pharmacoresponsiveness of a Na(V)17 mutant channel. Nat Commun. (2012) 3:1186. doi: 10.1038/ncomms2184
178. Roecker AJ, Egbertson M, Jones KLG, Gomez R, Kraus RL, Li Y, et al. Discovery of selective, orally bioavailable, N-linked arylsulfonamide Nasbv/s1.7 inhibitors with pain efficacy in mice. Bioorg Med Chem Lett. (2017) 27:2087–93. doi: 10.1016/j.bmcl.2017.03.085
179. Pero JE, Rossi MA, Lehman HDGF, Kelly MJ III, Mulhearn JJ, Wolkenberg SE, et al. Benzoxazolinone aryl sulfonamides as potent, selective Nav1.7 inhibitors with in vivo efficacy in a preclinical pain model. Bioorg Med Chem Lett. (2017) 27:2683–8. doi: 10.1016/j.bmcl.2017.04.040
180. La DS, Kang D, Park JY, Hong SG, Han J. The discovery of benzoxazine sulfonamide inhibitors of NaV1.7: tools that bridge efficacy and target engagement. Bioorg Med Chem Lett. (2017) 27:3477–85. doi: 10.1016/j.bmcl.2017.05.070
181. Bankar G, Howard S, Nelkenbrecher K, Waldbrook M, Dourado M, Shuart NG, et al. Selective NaV1.7 antagonists with long residence time show improved efficacy against inflammatory and neuropathic pain. Cell Rep. (2018) 24:3133–45. doi: 10.1016/j.celrep.2018.08.063
182. Shields SD, Deng L, Reese RM, Dourado M, Tao J, Foreman O, et al. Insensitivity to pain upon adult-onset deletion of Nav1.7 or its blockade with selective inhibitors. J Neurosci. (2018) 38:10180–201. doi: 10.1523/JNEUROSCI.1049-18.2018
183. Golubovic M, Kostic T, Djordjevic M, Peric V, Lazarevic M, Milic DJ, et al. In silico development of potential therapeutic for the pain treatment by inhibiting voltage-gated sodium channel 1.7. Comput Biol Med. (2021) 132:104346. doi: 10.1016/j.compbiomed.2021.104346
184. Hinckley CA, Kuryshev Y, Sers A, Barre A, Buisson B, Naik H, et al. Characterization of vixotrigine, a broad-spectrum voltage-gated sodium channel blocker. Mol Pharmacol. (2021) 99:49–59. doi: 10.1124/molpharm.120.000079
185. Zakrzewska JM, Palmer J, Morisset V, Giblin GM, Obermann M, Ettlin DA, et al. Safety and efficacy of a Nav1.7 selective sodium channel blocker in patients with trigeminal neuralgia: a double-blind, placebo-controlled, randomised withdrawal phase 2a trial. Lancet Neurol. (2017) 16:291–300. doi: 10.1016/S1474-4422(17)30005-4
186. Klint JK, Smith JJ, Vetter I, Rupasinghe DB, Er SY, Senff S, et al. Seven novel modulators of the analgesic target NaV1.7 uncovered using a high-throughput venom-based discovery approach. Br J Pharmacol. (2015) 172:2445–58. doi: 10.1111/bph.13081
187. Green BR, Catlin P, Zhang MM, Fiedler B, Bayudan W, Morrison A, et al. Conotoxins containing nonnatural backbone spacers: cladistic-based design, chemical synthesis, and improved analgesic activity. Chem Biol. (2007) 14:399–407. doi: 10.1016/j.chembiol.2007.02.009
188. Shcherbatko A, Rossi A, Foletti D, Zhu G, Bogin O, Galindo CM, et al. Engineering highly potent and selective microproteins against Nav1.7 sodium channel for treatment of pain. J Biol Chem. (2016) 291:13974–86. doi: 10.1074/jbc.M116.725978
189. Cardoso FC, Lewis RJ. Structure-function and therapeutic potential of spider venom-derived cysteine knot peptides targeting sodium channels. Front Pharmacol. (2019) 10:366. doi: 10.3389/fphar.2019.00366
190. Cardoso FC. Multi-targeting sodium and calcium channels using venom peptides for the treatment of complex ion channels-related diseases. Biochem Pharmacol. (2020) 181:114107. doi: 10.1016/j.bcp.2020.114107
191. Motin L, Durek T, Adams DJ. Modulation of human Nav1.7 channel gating by synthetic alpha-scorpion toxin OD1 and its analogs. Channels.(2016) 10:139–47. doi: 10.1080/19336950.2015.1120392
192. Murray JK, Ligutti J, Liu D, Zou A, Poppe L, Li H, et al. Engineering potent and selective analogues of GpTx-1, a tarantula venom peptide antagonist of the Na(V)1.7 sodium channel. J Med Chem. (2015) 58:2299–314. doi: 10.1021/jm501765v
193. Murray JK, Wu B, Tegley CM, Nixey TE, Falsey JR, Herberich B, et al. Engineering NaV1.7 inhibitory JzTx-V peptides with a potency and basicity profile suitable for antibody conjugation to enhance pharmacokinetics. ACS Chem Biol. (2019) 14:806–18. doi: 10.1021/acschembio.9b00183
194. Cardoso FC, Dekan Z, Rosengren KJ, Erickson A, Vetter I, Deuis JR, et al. Identification and characterization of ProTx-III [μ-TRTX-Tp1a], a new voltage-gated sodium channel inhibitor from venom of the tarantula thrixopelma pruriens. Mol Pharmacol. (2015) 88:291–303. doi: 10.1124/mol.115.098178
195. Cardoso FC, Dekan Z, Smith JJ, Deuis JR, Vetter I, Herzig V, et al. Modulatory features of the novel spider toxin μ-TRTX-Df1a isolated from the venom of the spider Davus fasciatus. Br J Pharmacol. (2017) 174:2528–44. doi: 10.1111/bph.13865
196. Flinspach M, Xu Q, Piekarz AD, Fellows R, Hagan R, Gibbs A, et al. Insensitivity to pain induced by a potent selective closed-state Nav17 inhibitor. Sci Rep. (2017) 7:39662. doi: 10.1038/srep39662
197. Neff RA, Flinspach M, Gibbs A, Shih AY, Minassian NA, Liu Y, et al. Comprehensive engineering of the tarantula venom peptide huwentoxin-IV to inhibit the human voltage-gated sodium channel hNav1.7. J Biol Chem. (2020) 295:1315–27. doi: 10.1016/S0021-9258(17)49888-7
198. Zhang Y, Wang L, Peng D, Zhang Q, Yang Q, Li J, et al. Engineering of highly potent and selective HNTX-III mutant against hNav1.7 sodium channel for treatment of pain. J Biol Chem. (2021) 296:100326. doi: 10.1016/j.jbc.2021.100326
199. Zhang Y, Yang Q, Zhang Q, Peng D, Chen M, Liang S, et al. Engineering gain-of-function analogues of the spider venom peptide HNTX-I, a potent blocker of the hNaV1.7 sodium channel. Toxins. (2018) 10:358. doi: 10.3390/toxins10090358
200. Xie MX, Yang J, Pang RP, Zeng WA, Ouyang HD, Liu YQ, et al. Bulleyaconitine A attenuates hyperexcitability of dorsal root ganglion neurons induced by spared nerve injury: the role of preferably blocking Nav1.7 and Nav1.3 channels. Mol Pain. (2018) 14:1744806918778491. doi: 10.1177/1744806918778491
201. Nakatani Y, Negoro K, Yamauchi M, Katasho M, Ishikura KI, Iwaki A, et al. Neoline, an active ingredient of the processed aconite root in Goshajinkigan formulation, targets Nav1.7 to ameliorate mechanical hyperalgesia in diabetic mice. J Ethnopharmacol. (2020) 259:112963. doi: 10.1016/j.jep.2020.112963
202. Lee JH, Park CK, Chen G, Han Q, Xie RG, Liu T, et al. A monoclonal antibody that targets a NaV1.7 channel voltage sensor for pain and itch relief. Cell. (2014) 157:1393–404. doi: 10.1016/j.cell.2014.03.064
203. Cai W, Zhao Q, Shao J, Zhang J, Li L, Ren X, et al. MicroRNA-182 alleviates neuropathic pain by regulating Nav17 following spared nerve injury in rats. Sci Rep. (2018) 8:16750. doi: 10.1038/s41598-018-34755-3
204. Hull JM, Isom LL. Voltage-gated sodium channel beta subunits: the power outside the pore in brain development and disease. Neuropharmacology. (2018) 132:43–57. doi: 10.1016/j.neuropharm.2017.09.018
205. Ho C, Zhao J, Malinowski S, Chahine M, O'Leary ME. Differential expression of sodium channel beta subunits in dorsal root ganglion sensory neurons. J Biol Chem. (2012) 287:15044–53. doi: 10.1074/jbc.M111.333740
206. Zhao J, O'Leary ME, Chahine M. Regulation of Nav1.6 and Nav1.8 peripheral nerve Na+ channels by auxiliary beta-subunits. J Neurophysiol. (2011) 106:608–19. doi: 10.1152/jn.00107.2011
207. Caffrey JM, Eng DL, Black JA, Waxman SG, Kocsis JD. Three types of sodium channels in adult rat dorsal root ganglion neurons. Brain Res. (1992) 592:283–97. doi: 10.1016/0006-8993(92)91687-A
208. Renganathan M, Cummins TR, Hormuzdiar WN, Waxman SG. alpha -SNS produces the slow TTX-resistant sodium current in large cutaneous afferent DRG neurons. J Neurophysiol. (2000) 84:710–8. doi: 10.1152/jn.2000.84.2.710
209. Djouhri L, Fang X, Okuse K, Wood JN, Berry CM, Lawson SN. The TTX-resistant sodium channel Nav1.8 (SNS/PN3): expression and correlation with membrane properties in rat nociceptive primary afferent neurons. J Physiol. (2003) 550:739–52. doi: 10.1113/jphysiol.2003.042127
210. Choi JS, Dib-Hajj SD, Waxman SG. Differential slow inactivation and use-dependent inhibition of Nav1.8 channels contribute to distinct firing properties in IB4+ and IB4ΓêÆ DRG neurons. J Neurophysiol. (2007) 97:1258–65. doi: 10.1152/jn.01033.2006
211. Shields SD, Ahn HS, Yang Y, Han C, Seal RP, Wood JN, et al. Nav1.8 expression is not restricted to nociceptors in mouse peripheral nervous system. Pain. (2012) 153:2017–30. doi: 10.1016/j.pain.2012.04.022
212. Tan ZY, Piekarz AD, Priest BT, Knopp KL, Krajewski JL, McDermott JS, et al. Tetrodotoxin-resistant sodium channels in sensory neurons generate slow resurgent currents that are enhanced by inflammatory mediators. J Neurosci. (2014) 34:7190–7. doi: 10.1523/JNEUROSCI.5011-13.2014
213. Uchida H, Ma L, Ueda H. Epigenetic gene silencing underlies C-fiber dysfunctions in neuropathic pain. J Neurosci. (2010) 30:4806–14. doi: 10.1523/JNEUROSCI.5541-09.2010
214. Dib-Hajj S, Black JA, Felts P, Waxman SG. Down-regulation of transcripts for Na channel alpha-SNS in spinal sensory neurons following axotomy. Proc Natl Acad Sci USA. (1996) 93:14950–4. doi: 10.1073/pnas.93.25.14950
215. Dib-Hajj SD, Black JA, Cummins TR, Kenney AM, Kocsis JD, Waxman SG. Rescue of alpha-SNS sodium channel expression in small dorsal root ganglion neurons after axotomy by nerve growth factor in vivo. J Neurophysiol. (1998) 79:2668–76. doi: 10.1152/jn.1998.79.5.2668
216. Gold MS, Weinreich D, Kim CS, Wang R, Treanor J, Porreca F, et al. Redistribution of NaV1.8 in uninjured axons enables neuropathic pain. J Neurosci. (2003) 23:158–66. doi: 10.1523/JNEUROSCI.23-01-00158.2003
217. Coward K, Plumpton C, Facer P, Birch R, Carlstedt T, Tate S, et al. Immunolocalization of SNS/PN3 and NaN/SNS2 sodium channels in human pain states. Pain. (2000) 85:41–50. doi: 10.1016/S0304-3959(99)00251-1
218. Faber CG, Lauria G, Merkies IS, Cheng X, Han C, Ahn HS, et al. Gain-of-function Nav1.8 mutations in painful neuropathy. Proc Natl Acad Sci USA. (2012) 109:19444–9. doi: 10.1073/pnas.1216080109
219. Daou I, Beaudry H, Ase AR, Wieskopf JS, Ribeiro-da-Silva A, Mogil JS, et al. Optogenetic silencing of Nav1.8-positive afferents alleviates inflammatory and neuropathic pain. eNeuro. (2016) 3:1. doi: 10.1523/ENEURO.0140-15.2016
220. Pabel S, Ahmad S, Tirilomis P, Stehle T, Mustroph J, Knierim M, et al. Inhibition of NaV1.8 prevents atrial arrhythmogenesis in human and mice. Basic Res Cardiol. (2020) 115:20. doi: 10.1007/s00395-020-0780-8
221. Jarvis MF, Honore P, Shieh CC, Chapman M, Joshi S, Zhang XF, et al. A-803467, a potent and selective Nav1.8 sodium channel blocker, attenuates neuropathic and inflammatory pain in the rat. Proc Natl Acad Sci USA. (2007) 104:8520–5. doi: 10.1073/pnas.0611364104
222. Payne CE, Brown AR, Theile JW, Loucif AJ, Alexandrou AJ, Fuller MD, et al. A novel selective and orally bioavailable Nasbv/s1.8 channel blocker, PF-01247324, attenuates nociception and sensory neuron excitability. Br J Pharmacol. (2015) 172:2654–70. doi: 10.1111/bph.13092
223. Alsaloum M, Higerd GP, Effraim PR, Waxman SG. Status of peripheral sodium channel blockers for non-addictive pain treatment. Nat Rev Neurol. (2020) 16:689–705. doi: 10.1038/s41582-020-00415-2
224. Deuis JR, Dekan Z, Inserra MC, Lee TH, Aguilar MI, Craik DJ, et al. Development of a muO-conotoxin analogue with improved lipid membrane interactions and potency for the analgesic sodium channel Nasbv/s1.8. J Biol Chem. (2016) 291:11829–42. doi: 10.1074/jbc.M116.721662
225. McGaraughty S, Chu KL, Scanio MJ, Kort ME, Faltynek CR, Jarvis MF. A selective Nav1.8 sodium channel blocker, A-803467 [5-(4-chlorophenyl-N-(3,5-dimethoxyphenyl)furan-2-carboxamide], attenuates spinal neuronal activity in neuropathic rats. J Pharmacol Exp Ther. (2008) 324:1204–11. doi: 10.1124/jpet.107.134148
226. Cummins TR, Black JA, Dib-Hajj SD, Waxman SG. Glial-derived neurotrophic factor upregulates expression of functional SNS and NaN sodium channels and their currents in axotomized dorsal root ganglion neurons. J Neurosci. (2000) 20:8754–61. doi: 10.1523/JNEUROSCI.20-23-08754.2000
227. Dib-Hajj SD, Tyrrell L, Cummins TR, Black JA, Wood PM, Waxman SG. Two tetrodotoxin-resistant sodium channels in human dorsal root ganglion neurons. FEBS Lett. (1999) 462:117–20. doi: 10.1016/S0014-5793(99)01519-7
228. Cummins TR, Dib-Hajj SD, Black JA, Akopian AN, Wood JN, Waxman SG. A novel persistent tetrodotoxin-resistant sodium current in SNS-null and wild-type small primary sensory neurons. J Neurosci. (1999) 19:RC43. doi: 10.1523/JNEUROSCI.19-24-j0001.1999
229. Dib-Hajj S, Black JA, Cummins TR, Waxman SG. NaN/Nav1.9: a sodium channel with unique properties. Trends Neurosci. (2002) 25:253–9. doi: 10.1016/S0166-2236(02)02150-1
230. Dib-Hajj SD, Black JA, Waxman SG. NaV1.9: a sodium channel linked to human pain. Nat Rev Neurosci. (2015) 16:511–9. doi: 10.1038/nrn3977
231. Dib-Hajj SD, Tyrrell L, Black JA, Waxman SG. NaN, a novel voltage-gated Na channel, is expressed preferentially in peripheral sensory neurons and down-regulated after axotomy. Proc Natl Acad Sci USA. (1998) 95:8963–8. doi: 10.1073/pnas.95.15.8963
232. Priest BT, Murphy BA, Lindia JA, Diaz C, Abbadie C, Ritter AM, et al. Contribution of the tetrodotoxin-resistant voltage-gated sodium channel NaV1.9 to sensory transmission and nociceptive behavior. Proc Natl Acad Sci USA. (2005) 102:9382–7. doi: 10.1073/pnas.0501549102
233. Binshtok AM, Bean BP, Woolf CJ. Inhibition of nociceptors by TRPV1-mediated entry of impermeant sodium channel blockers. Nature. (2007) 449:607–10. doi: 10.1038/nature06191
234. Stueber T, Eberhardt MJ, Hadamitzky C, Jangra A, Schenk S, Dick F, et al. Quaternary lidocaine derivative QX-314 activates and permeates human TRPV1 and TRPA1 to produce inhibition of sodium channels and cytotoxicity. Anesthesiology. (2016) 124:1153–65. doi: 10.1097/ALN.0000000000001050
235. Roberson DP, Binshtok AM, Blasl F, Bean BP, Woolf CJ. Targeting of sodium channel blockers into nociceptors to produce long-duration analgesia: a systematic study and review. Br J Pharmacol. (2011) 164:48–58. doi: 10.1111/j.1476-5381.2011.01391.x
236. Abdulla FA, Smith PA. Axotomy- and autotomy-induced changes in Ca2+and K+ channel currents of rat dorsal root ganglion neurons. J Neurophysiol. (2001) 85:644–58. doi: 10.1152/jn.2001.85.2.644
237. Everill B, Kocsis JD. Reduction of potassium currents in identified cutaneous afferent dorsal root ganglion neurons after axotomy. J Neurophysiol. (1999) 82:700–8. doi: 10.1152/jn.1999.82.2.700
238. Yang EK, Takimoto K, Hayashi Y, de Groat WC, Yoshimura N. Altered expression of potassium channel subunit mRNA and alpha-dendrotoxin sensitivity of potassium currents in rat dorsal root ganglion neurons after axotomy. Neuroscience. (2004) 123:867–74. doi: 10.1016/j.neuroscience.2003.11.014
239. Rose K, Ooi L, Dalle C, Robertson B, Wood IC, Gamper N. Transcriptional repression of the M channel subunit Kv7.2 in chronic nerve injury. Pain. (2011) 152:742–54. doi: 10.1016/j.pain.2010.12.028
240. Cao XH, Chen SR, Li L, Pan HL. Nerve injury increases brain-derived neurotrophic factor levels to suppress BK channel activity in primary sensory neurons. J Neurochem. (2012) 121:944–53. doi: 10.1111/j.1471-4159.2012.07736.x
241. Tsantoulas C, McMahon SB. Opening paths to novel analgesics: the role of potassium channels in chronic pain. Trends Neurosci. (2014) 37:146–58. doi: 10.1016/j.tins.2013.12.002
242. Takeda M, Tsuboi Y, Kitagawa J, Nakagawa K, Iwata K, Matsumoto S. Potassium channels as a potential therapeutic target for trigeminal neuropathic and inflammatory pain. Mol Pain. (2011) 7:5. doi: 10.1186/1744-8069-7-5
243. Ocana M, Cendan CM, Cobos EJ, Entrena JM, Baeyens JM. Potassium channels and pain: present realities and future opportunities. Eur J Pharmacol. (2004) 500:203–19. doi: 10.1016/j.ejphar.2004.07.026
244. Zemel BM, Ritter DM, Covarrubias M, Muqeem T. A-type KV channels in dorsal root ganglion neurons: diversity, function, and dysfunction. Front Mol Neurosci. (2018) 11:253. doi: 10.3389/fnmol.2018.00253
245. Calvo M, Richards N, Schmid AB, Barroso A, Zhu L, Ivulic D, et al. Altered potassium channel distribution and composition in myelinated axons suppresses hyperexcitability following injury. Elife. (2016) 5:e12661. doi: 10.7554/eLife.12661
246. Wei AD, Gutman GA, Aldrich R, Chandy KG, Grissmer S, Wulff H. International Union of Pharmacology. LII Nomenclature and molecular relationships of calcium-activated potassium channels. Pharmacol Rev. (2005) 57:463–72. doi: 10.1124/pr.57.4.9
247. Gutman GA, Chandy KG, Grissmer S, Lazdunski M, McKinnon D, Pardo LA, et al. International Union of Pharmacology. LIII Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol Rev. (2005) 57:473–508. doi: 10.1124/pr.57.4.10
248. Kubo Y, Adelman JP, Clapham DE, Jan LY, Karschin A, Kurachi Y, et al. International Union of Pharmacology. LIV Nomenclature and molecular relationships of inwardly rectifying potassium channels. Pharmacol Rev. (2005) 57:509–26. doi: 10.1124/pr.57.4.11
249. Goldstein SA, Bayliss DA, Kim D, Lesage F, Plant LD, Rajan S. International Union of Pharmacology. LV Nomenclature and molecular relationships of two-P potassium channels. Pharmacol Rev. (2005) 57:527–40. doi: 10.1124/pr.57.4.12
250. Laumet G, Garriga J, Chen SR, Zhang Y, Li DP, Smith TM, et al. G9a is essential for epigenetic silencing of K+ channel genes in acute-to-chronic pain transition. Nat Neurosci. (2015) 18:1746–55. doi: 10.1038/nn.4165
251. Everill B, Kocsis JD. Nerve growth factor maintains potassium conductance after nerve injury in adult cutaneous afferent dorsal root ganglion neurons [In Process Citation]. Neuroscience. (2000) 100:417–22. doi: 10.1016/S0306-4522(00)00263-3
252. Yang JW, Vacher H, Park KS, Clark E, Trimmer JS. Trafficking-dependent phosphorylation of Kv1.2 regulates voltage-gated potassium channel cell surface expression. Proc Natl Acad Sci USA. (2007) 104:20055–60. doi: 10.1073/pnas.0708574104
253. Nesti E, Everill B, Morielli AD. Endocytosis as a mechanism for tyrosine kinase-dependent suppression of a voltage-gated potassium channel. Mol Biol Cell. (2004) 15:4073–88. doi: 10.1091/mbc.e03-11-0788
254. Cao XH, Byun HS, Chen SR, Cai YQ, Pan HL. Reduction in voltage-gated K+ channel activity in primary sensory neurons in painful diabetic neuropathy: role of brain-derived neurotrophic factor. J Neurochem. (2010) 114:1460–75. doi: 10.1111/j.1471-4159.2010.06863.x
255. Kim DS, Choi JO, Rim HD, Cho HJ. Downregulation of voltage-gated potassium channel alpha gene expression in dorsal root ganglia following chronic constriction injury of the rat sciatic nerve. Brain Res Mol Brain Res. (2002) 105:146–52. doi: 10.1016/S0169-328X(02)00388-1
256. Hao J, Padilla F, Dandonneau M, Lavebratt C, Lesage F, Noel J, et al. Kv1.1 channels act as mechanical brake in the senses of touch and pain. Neuron. (2013) 77:899–914. doi: 10.1016/j.neuron.2012.12.035
257. Lu Q, Peevey J, Jow F, Monaghan MM, Mendoza G, Zhang H, et al. Disruption of Kv1.1 N-type inactivation by novel small molecule inhibitors (disinactivators). Bioorg Med Chem. (2008) 16:3067–75. doi: 10.1016/j.bmc.2007.12.031
258. Zhang J, Rong L, Shao J, Zhang Y, Liu Y, Zhao S, et al. Epigenetic restoration of voltage-gated potassium channel Kv1.2 alleviates nerve injury-induced neuropathic pain. J Neurochem. (2021) 156:367–78. doi: 10.1111/jnc.15117
259. Zhao X, Tang Z, Zhang H, Atianjoh FE, Zhao JY, Liang L, et al. A long noncoding RNA contributes to neuropathic pain by silencing Kcna2 in primary afferent neurons. Nat Neurosci. (2013) 16:1024–31. doi: 10.1038/nn.3438
260. Sun L, Gu X, Pan Z, Guo X, Liu J, Atianjoh FE, et al. Contribution of DNMT1 to neuropathic pain genesis partially through epigenetically repressing Kcna2 in primary afferent neurons. J Neurosci. (2019) 39:6595–607. doi: 10.1523/JNEUROSCI.0695-19.2019
261. Fan L, Guan X, Wang W, Zhao JY, Zhang H, Tiwari V, et al. Impaired neuropathic pain and preserved acute pain in rats overexpressing voltage-gated potassium channel subunit Kv1.2 in primary afferent neurons. Mol Pain. (2014) 10:8. doi: 10.1186/1744-8069-10-8
262. Ishikawa K, Tanaka M, Black JA, Waxman SG. Changes in expression of voltage-gated potassium channels in dorsal root ganglion neurons following axotomy. Muscle Nerve. (1999) 22:502–7. doi: 10.1002/(SICI)1097-4598(199904)22:4<502::AID-MUS12>3.0.CO;2-K
263. Li Z, Guo Y, Ren X, Rong L, Huang M, Cao J, et al. HDAC2, but not HDAC1, regulates Kv1.2 expression to mediate neuropathic pain in CCI rats. Neuroscience. (2019) 408:339–48. doi: 10.1016/j.neuroscience.2019.03.033
264. Miao J, Zhou X, Ji T, Chen G. NF-kappaB p65-dependent transcriptional regulation of histone deacetylase 2 contributes to the chronic constriction injury-induced neuropathic pain via the microRNA-183/TXNIP/NLRP3 axis. J Neuroinflamm. (2020) 17:225. doi: 10.1186/s12974-020-01901-6
265. Pollema-Mays SL, Centeno MV, Apkarian AV, Martina M. Expression of DNA methyltransferases in adult dorsal root ganglia is cell-type specific and up regulated in a rodent model of neuropathic pain. Front Cell Neurosci. (2014) 8:217. doi: 10.3389/fncel.2014.00217
266. Mo K, Wu S, Gu X, Xiong M, Cai W, Atianjoh FE, et al. MBD1 contributes to the genesis of acute pain and neuropathic pain by epigenetic silencing of Oprm1 and Kcna2 genes in primary sensory neurons. J Neurosci. (2018) 38:9883. doi: 10.1523/JNEUROSCI.0880-18.2018
267. Zhao JY, Liang L, Gu X, Li Z, Wu S, Sun L, et al. DNA methyltransferase DNMT3a contributes to neuropathic pain by repressing Kcna2 in primary afferent neurons. Nat Commun. (2017) 8:14712. doi: 10.1038/ncomms14712
268. Wu Q, Wei G, Ji F, Jia S, Wu S, Guo X, et al. TET1 overexpression mitigates neuropathic pain through rescuing the expression of ++-opioid receptor and Kv1.2 in the primary sensory neurons. Neurotherapeutics. (2019) 16:491–504. doi: 10.1007/s13311-018-00689-x
269. Tsantoulas C, Zhu L, Yip P, Grist J, Michael GJ, McMahon SB. Kv2 dysfunction after peripheral axotomy enhances sensory neuron responsiveness to sustained input. Exp Neurol. (2014) 251:115–26. doi: 10.1016/j.expneurol.2013.11.011
270. Tsantoulas C, Zhu L, Shaifta Y, Grist J, Ward JPT, Raouf R, et al. Sensory neuron downregulation of the Kv9.1 potassium channel subunit mediates neuropathic pain following nerve injury. J Neurosci. (2012) 32:17502–13. doi: 10.1523/JNEUROSCI.3561-12.2012
271. Kerschensteiner D, Soto F, Stocker M. Fluorescence measurements reveal stoichiometry of K+ channels formed by modulatory and delayed rectifier alpha-subunits. Proc Natl Acad Sci USA. (2005) 102:6160–5. doi: 10.1073/pnas.0500468102
272. Bocksteins E, Van d V, Van Bogaert PP, Snyders DJ. Kv3 channels contribute to the delayed rectifier current in small cultured mouse dorsal root ganglion neurons. Am J Physiol Cell Physiol. (2012) 303:C406–15. doi: 10.1152/ajpcell.00343.2011
273. Brown DA, Passmore GM. Neural KCNQ (Kv7) channels. Br J Pharmacol. (2009) 156:1185–95. doi: 10.1111/j.1476-5381.2009.00111.x
274. Adams PR, Brown DA, Constanti A. M-currents and other potassium currents in bullfrog sympathetic neurones. J Physiol. (1982) 330:537–72. doi: 10.1113/jphysiol.1982.sp014357
275. Barkai O, Goldstein RH, Caspi Y, Katz B, Lev S, Binshtok AM. The role of Kv7/M potassium channels in controlling ectopic firing in nociceptors. Front Mol Neurosci. (2017) 10:181. doi: 10.3389/fnmol.2017.00181
276. Zheng Q, Fang D, Liu M, Cai J, Wan Y, Han JS, et al. Suppression of KCNQ/M (Kv7) potassium channels in dorsal root ganglion neurons contributes to the development of bone cancer pain in a rat model. Pain. (2013) 154:434–48. doi: 10.1016/j.pain.2012.12.005
277. King CH, Lancaster E, Salomon D, Peles E, Scherer SS. Kv7.2 regulates the function of peripheral sensory neurons. J Comp Neurol. (2014) 522:3262–80. doi: 10.1002/cne.23595
278. Blackburn-Munro G, Jensen BS. The anticonvulsant retigabine attenuates nociceptive behaviours in rat models of persistent and neuropathic pain. Eur J Pharmacol. (2003) 460:109–16. doi: 10.1016/S0014-2999(02)02924-2
279. Djouhri L, Malki MI, Zeidan A, Nagi K, Smith T. Activation of Kv7 channels with the anticonvulsant retigabine alleviates neuropathic pain behaviour in the streptozotocin rat model of diabetic neuropathy. J Drug Target. (2019) 27:1118–26. doi: 10.1080/1061186X.2019.1608552
280. Jones F, Gamper N, Gao H. Kv7 Channels and Excitability Disorders. Berlin: Springer Berlin Heidelberg (2021). p 1–46.
281. Abd-Elsayed A, Jackson M, Gu SL, Fiala K, Gu J. Neuropathic pain and Ksbv/s7 voltage-gated potassium channels: the potential role of Ksbv/s7 activators in the treatment of neuropathic pain. Mol Pain. (2019) 15:1744806919864256. doi: 10.1177/1744806919864256
282. Barrese V, Stott JB, Greenwood IA. KCNQ-encoded potassium channels as therapeutic targets. Annu Rev Pharmacol Toxicol. (2018) 58:625–48. doi: 10.1146/annurev-pharmtox-010617-052912
283. Rivera-Arconada I, Roza C, Lopez-Garcia JA. Enhancing m currents: a way out for neuropathic pain? Front Mol Neurosci. (2009) 2:10. doi: 10.3389/neuro.02.010.2009
284. Du X, Gamper N. Potassium channels in peripheral pain pathways: expression, function and therapeutic potential. Curr Neuropharmacol. (2013) 11:621–40. doi: 10.2174/1570159X113119990042
285. Du X, Gao H, Jaffe D, Zhang H, Gamper N. M-type K(+) channels in peripheral nociceptive pathways. Br J Pharmacol. (2018) 175:2158–72. doi: 10.1111/bph.13978
286. Barkai O, Puig S, Lev S, Title B, Katz B, Eli-Berchoer L, et al. Platelet-derived growth factor activates nociceptive neurons by inhibiting M-current and contributes to inflammatory pain. Pain. (2019) 160:1281–96. doi: 10.1097/j.pain.0000000000001523
287. Mucha M, Ooi L, Linley JE, Mordaka P, Dalle C, Robertson B, et al. Transcriptional control of KCNQ channel genes and the regulation of neuronal excitability. J Neurosci. (2010) 30:13235–45. doi: 10.1523/JNEUROSCI.1981-10.2010
288. Zhang F, Gigout S, Liu Y, Wang Y, Hao H, Buckley NJ, et al. Repressor element 1-silencing transcription factor drives the development of chronic pain states. Pain. (2019) 160:2398–408. doi: 10.1097/j.pain.0000000000001633
289. Ooi L, Wood IC. Chromatin crosstalk in development and disease: lessons from REST. Nat Rev Genet. (2007) 8:544–54. doi: 10.1038/nrg2100
290. Willis DE, Wang M, Brown E, Fones L, Cave JW. Selective repression of gene expression in neuropathic pain by the neuron-restrictive silencing factor/repressor element-1 silencing transcription (NRSF/REST). Neurosci Lett. (2016) 625:20–5. doi: 10.1016/j.neulet.2015.12.003
291. Rugo HS, Jacobs I, Sharma S, Scappaticci F, Paul TA, Jensen-Pergakes K, et al. The promise for histone methyltransferase inhibitors for epigenetic therapy in clinical oncology: a narrative review. Adv Ther. (2020) 37:3059–82. doi: 10.1007/s12325-020-01379-x
292. Ritter DM, Zemel BM, Lepore AC, Covarrubias M. Kv3.4 channel function and dysfunction in nociceptors. Channels. (2015) 9:209–17. doi: 10.1080/19336950.2015.1056949
293. Grabauskas G, Heldsinger A, Wu X, Xu D, Zhou S, Owyang C. Diabetic visceral hypersensitivity is associated with activation of mitogen-activated kinase in rat dorsal root ganglia. Diabetes. (2011) 60:1743–51. doi: 10.2337/db10-1507
294. Shinoda M, Fukuoka T, Takeda M, Iwata K, Noguchi K. Spinal glial cell line-derived neurotrophic factor infusion reverses reduction of Kv4.1-mediated A-type potassium currents of injured myelinated primary afferent neurons in a neuropathic pain model. Mol Pain. (2019) 15:1744806919841196. doi: 10.1177/1744806919841196
295. Duan KZ, Xu Q, Zhang XM, Zhao ZQ, Mei YA, Zhang YQ. Targeting A-type K(+) channels in primary sensory neurons for bone cancer pain in a rat model. Pain. (2012) 153:562–74. doi: 10.1016/j.pain.2011.11.020
296. Sakai A, Saitow F, Maruyama M, Miyake N, Miyake K, Shimada T, et al. MicroRNA cluster miR-17-92 regulates multiple functionally related voltage-gated potassium channels in chronic neuropathic pain. Nat Commun. (2017) 8:16079. doi: 10.1038/ncomms16079
297. Chien LY, Cheng JK, Chu D, Cheng CF, Tsaur ML. Reduced expression of A-type potassium channels in primary sensory neurons induces mechanical hypersensitivity. J Neurosci. (2007) 27:9855–65. doi: 10.1523/JNEUROSCI.0604-07.2007
298. Ritter DM, Zemel BM, Hala TJ, O'Leary ME, Lepore AC, Covarrubias M. Dysregulation of Kv3.4 channels in dorsal root ganglia following spinal cord injury. J Neurosci. (2015) 35:1260–73. doi: 10.1523/JNEUROSCI.1594-14.2015
299. Kuo YL, Cheng JK, Hou WH, Chang YC, Du PH, Jian JJ, et al. K(+) channel modulatory subunits KChIP and DPP participate in Kv4-mediated mechanical pain control. J Neurosci. (2017) 37:4391–404. doi: 10.1523/JNEUROSCI.1619-16.2017
300. Yunoki T, Takimoto K, Kita K, Funahashi Y, Takahashi R, Matsuyoshi H, et al. Differential contribution of Kv4-containing channels to A-type, voltage-gated potassium currents in somatic and visceral dorsal root ganglion neurons. J Neurophysiol. (2014) 112:2492–504. doi: 10.1152/jn.00054.2014
301. Matsuyoshi H, Takimoto K, Yunoki T, Erickson VL, Tyagi P, Hirao Y, et al. Distinct cellular distributions of Kv4 pore-forming and auxiliary subunits in rat dorsal root ganglion neurons. Life Sci. (2012) 91:258–63. doi: 10.1016/j.lfs.2012.07.007
302. Uchida H, Sasaki K, Ma L, Ueda H. Neuron-restrictive silencer factor causes epigenetic silencing of Kv4.3 gene after peripheral nerve injury. Neuroscience. (2016) 166:1–4. doi: 10.1016/j.neuroscience.2009.12.021
303. Amarillo Y, De Santiago-Castillo JA, Dougherty K, Maffie J, Kwon E, Covarrubias M, et al. Ternary Kv4.2 channels recapitulate voltage-dependent inactivation kinetics of A-type K+ channels in cerebellar granule neurons. J Physiol. (2008) 586:2093–106. doi: 10.1113/jphysiol.2007.150540
304. Pongs O, Schwarz JR. Ancillary subunits associated with voltage-dependent K+ channels. Physiol Rev. (2010) 90:755–96. doi: 10.1152/physrev.00020.2009
305. Jerng HH, Dougherty K, Covarrubias M, Pfaffinger PJ. A novel N-terminal motif of dipeptidyl peptidase-like proteins produces rapid inactivation of KV4.2 channels by a pore-blocking mechanism. Channels. (2009) 3:448–61. doi: 10.4161/chan.3.6.10216
306. Abdulla FA, Smith PA. Nerve injury increases an excitatory action of neuropeptide Y and Y2- agonists on dorsal root ganglion neurons. Neuroscience. (1999) 89:43–60. doi: 10.1016/S0306-4522(98)00443-6
307. Li W, Gao SB, Lv CX, Wu Y, Guo ZH, Ding JP, et al. Characterization of voltage-and Ca2+-activated K+ channels in rat dorsal root ganglion neurons. J Cell Physiol. (2007) 212:348–57. doi: 10.1002/jcp.21007
308. Zhang XL, Mok LP, Katz EJ, Gold MS. BKCa currents are enriched in a subpopulation of adult rat cutaneous nociceptive dorsal root ganglion neurons. Eur J Neurosci. (2010) 31:450–62. doi: 10.1111/j.1460-9568.2009.07060.x
309. Sarantopoulos CD, McCallum JB, Rigaud M, Fuchs A, Kwok WM, Hogan QH. Opposing effects of spinal nerve ligation on calcium-activated potassium currents in axotomized and adjacent mammalian primary afferent neurons. Brain Res. (2007) 1132:84–99. doi: 10.1016/j.brainres.2006.11.055
310. Chen SR, Cai YQ, Pan HL. Plasticity and emerging role of BKCa channels in nociceptive control in neuropathic pain. J Neurochem. (2009) 110:352–62. doi: 10.1111/j.1471-4159.2009.06138.x
311. Zhang FX, Gadotti VM, Souza IA, Chen L, Zamponi GW. BK potassium channels suppress Cavalpha2delta subunit function to reduce inflammatory and neuropathic pain. Cell Rep. (2018) 22:1956–64. doi: 10.1016/j.celrep.2018.01.073
312. Roy S, Morayo AA, Large RJ, Webb TI, Camarasu C, Sergeant GP, et al. Structure-activity relationships of a novel group of large-conductance Ca(2+)-activated K(+) (BK) channel modulators: the GoSlo-SR family. ChemMedChem. (2012) 7:1763–9. doi: 10.1002/cmdc.201200321
313. Soder RP, Petkov GV. Large conductance Ca2+ -activated K+ channel activation with NS1619 decreases myogenic and neurogenic contractions of rat detrusor smooth muscle. Eur J Pharmacol. (2011) 670:252–9. doi: 10.1016/j.ejphar.2011.08.013
314. Kraft R, Krause P, Jung S, Basrai D, Liebmann L, Bolz J, et al. BK channel openers inhibit migration of human glioma cells. Pflugers Arch. (2003) 446:248–55. doi: 10.1007/s00424-003-1012-4
315. Layne JJ, Nausch B, Olesen SP, Nelson MT. BK channel activation by NS11021 decreases excitability and contractility of urinary bladder smooth muscle. Am J Physiol Regul Integr Comp Physiol. (2010) 298:R378–84. doi: 10.1152/ajpregu.00458.2009
316. Bentzen BH, Nardi A, Calloe K, Madsen LS, Olesen SP, Grunnet M. The small molecule NS11021 is a potent and specific activator of Ca2+-activated big-conductance K+ channels. Mol Pharmacol. (2007) 72:1033–44. doi: 10.1124/mol.107.038331
317. Bentzen BH, Andersen RW, Olesen SP, Grunnet M, Nardi A. Synthesis and characterisation of NS13558: a new important tool for addressing KCa1.1 channel function ex vivo. Naunyn Schmiedebergs Arch Pharmacol. (2010) 381:271–83. doi: 10.1007/s00210-009-0456-2
318. Sakamoto K, Nonomura T, Ohya S, Muraki K, Ohwada T, Imaizumi Y. Molecular mechanisms for large conductance Ca2+-activated K+ channel activation by a novel opener, 12,14-dichlorodehydroabietic acid. J Pharmacol Exp Ther. (2006) 316:144–53. doi: 10.1124/jpet.105.093856
319. Lu R, Flauaus C, Kennel L, Petersen J, Drees O, Kallenborn-Gerhardt W, et al. KCa3.1 channels modulate the processing of noxious chemical stimuli in mice. Neuropharmacology. (2017) 125:386–95. doi: 10.1016/j.neuropharm.2017.08.021
320. Takeshita N, Oe T, Kiso T, Kakimoto S. A KCa3.1 channel opener, ASP0819, modulates nociceptive signal processing from peripheral nerves in fibromyalgia-like pain in rats. J Pain Res. (2021) 14:23–34. doi: 10.2147/JPR.S274563
321. Arnold LM, Blauwet MB, Tracy K, Cai N, Walzer M, Blahunka P, et al. Efficacy and safety of ASP0819 in patients with fibromyalgia: results of a proof-of-concept, randomized, double-blind, placebo-controlled trial. J Pain Res. (2020) 13:3355–69. doi: 10.2147/JPR.S274562
322. Zoga V, Kawano T, Liang MY, Bienengraeber M, Weihrauch D, McCallum B, et al. KATP channel subunits in rat dorsal root ganglia: alterations by painful axotomy. Mol Pain. (2010) 6:6. doi: 10.1186/1744-8069-6-6
323. Kawano T, Zoga V, McCallum JB, Wu HE, Gemes G, Liang MY, et al. ATP-sensitive potassium currents in rat primary afferent neurons: biophysical, pharmacological properties, and alterations by painful nerve injury. Neuroscience. (2009) 162:431–43. doi: 10.1016/j.neuroscience.2009.04.076
324. Campbell JD, Sansom MS, Ashcroft FM. Potassium channel regulation. EMBO Rep. (2003) 4:1038–42. doi: 10.1038/sj.embor.7400003
325. Welch SP, Dunlow LD. Antinociceptive activity of intrathecally administered potassium channel openers and opioid agonists: a common mechanism of action? J Pharmacol Exp Ther. (1993) 267:390–9.
326. Kawano T, Zoga V, Gemes G, McCallum JB, Wu HE, Pravdic D, et al. Suppressed Ca2+/CaM/CaMKII-dependent K(ATP) channel activity in primary afferent neurons mediates hyperalgesia after axotomy. Proc Natl Acad Sci USA. (2009) 106:8725–30. doi: 10.1073/pnas.0901815106
327. Gada K, Plant LD. Two-pore domain potassium channels: emerging targets for novel analgesic drugs: IUPHAR review 26. Br J Pharmacol. (2019) 176:256–66. doi: 10.1111/bph.14518
328. Dobler T, Springauf A, Tovornik S, Weber M, Schmitt A, Sedlmeier R, et al. TRESK two-pore-domain K+ channels constitute a significant component of background potassium currents in murine dorsal root ganglion neurones. J Physiol. (2007) 585:867–79. doi: 10.1113/jphysiol.2007.145649
329. Tulleuda A, Cokic B, Callejo G, Saiani B, Serra J, Gasull X. TRESK channel contribution to nociceptive sensory neurons excitability: modulation by nerve injury. Mol Pain. (2011) 7:30. doi: 10.1186/1744-8069-7-30
330. Castellanos A, Pujol-Coma A, Andres-Bilbe A, Negm A, Callejo G, Soto D, et al. TRESK background K+ channel deletion selectively uncovers enhanced mechanical and cold sensitivity. J Physiol. (2020) 598:1017–38. doi: 10.1113/JP279203
331. Pollema-Mays SL, Centeno MV, Ashford CJ, Apkarian AV, Martina M. Expression of background potassium channels in rat DRG is cell-specific and down-regulated in a neuropathic pain model. Mol Cell Neurosci. (2013) 57:1–9. doi: 10.1016/j.mcn.2013.08.002
332. Singh S, Agarwal P, Ravichandiran V. Two-pore domain potassium channel in neurological disorders. J Membr Biol. (2021) 254:367–80. doi: 10.1007/s00232-021-00189-8
333. Loucif AJC, Saintot PP, Liu J, Antonio BM, Zellmer SG, Yoger K, et al. GI-530159, a novel, selective, mechanosensitive two-pore-domain potassium (K2P) channel opener, reduces rat dorsal root ganglion neuron excitability. Br J Pharmacol. (2018) 175:2272–83. doi: 10.1111/bph.14098
334. Snutch TP, Zamponi GW. Recent advances in the development of T-type calcium channel blockers for pain intervention. Br J Pharmacol. (2017) 175:2375–83. doi: 10.1111/bph.13906
335. Tibbs GR, Posson DJ, Goldstein PA. Voltage-gated ion channels in the PNS: novel therapies for neuropathic pain? Trends Pharmacol Sci. (2016) 37:522–42. doi: 10.1016/j.tips.2016.05.002
336. Catterall WA, Perez-Reyes E, Snutch TP, Striessnig J. International Union of Pharmacology. XLVIII Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol Rev. (2005) 57:411–25. doi: 10.1124/pr.57.4.5
337. Dolphin AC. Voltage-gated calcium channels and their auxiliary subunits: physiology and pathophysiology and pharmacology. J Physiol. (2016) 594:5369–90. doi: 10.1113/JP272262
338. Ramachandra R, Hassan B, McGrew SG, Dompor J, Farrag M, Ruiz-Velasco V, et al. Identification of CaV channel types expressed in muscle afferent neurons. J Neurophysiol. (2013) 110:1535–43. doi: 10.1152/jn.00069.2013
339. Zamponi GW. Targeting voltage-gated calcium channels in neurological and psychiatric diseases. Nat Rev Drug Discov. (2015). doi: 10.1038/nrd.2015.5
340. Bourinet E, Altier C, Hildebrand ME, Trang T, Salter MW, Zamponi GW. Calcium-permeable ion channels in pain signaling. Physiol Rev. (2014) 94:81–140. doi: 10.1152/physrev.00023.2013
341. Pan B, Guo Y, Wu HE, Park J, Trinh VN, Luo ZD, et al. Thrombospondin-4 divergently regulates voltage-gated Ca2+ channel subtypes in sensory neurons after nerve injury. Pain. (2016) 157:2068–80. doi: 10.1097/j.pain.0000000000000612
342. Baccei ML, Kocsis JD. Voltage-gated calcium currents in axotomized adult rat cutaneous afferent neurons. J Neurophysiol. (2000) 83:2227–38. doi: 10.1152/jn.2000.83.4.2227
343. Chaplan SR, Pogrel JW, Yaksh TL. Role of voltage-dependent calcium channel subtypes in experimental tactile allodynia. J Pharmacol Exp Ther. (1994) 269:1117–23.
344. Patel R, Montagut-Bordas C, Dickenson AH. Calcium channel modulation as a target in chronic pain control. Br J Pharmacol. (2018) 175:2173–84. doi: 10.1111/bph.13789
345. Field MJ, Cox PJ, Stott E, Melrose H, Offord J, Su TZ, et al. Identification of the {alpha}2-{delta}-1 subunit of voltage-dependent calcium channels as a molecular target for pain mediating the analgesic actions of pregabalin. PNAS. (2006) 103:17537–42. doi: 10.1073/pnas.0409066103
346. Dolphin AC. Calcium channel auxiliary alpha2delta and beta subunits: trafficking and one step beyond. Nat Rev Neurosci. (2012) 13:542–55. doi: 10.1038/nrn3311
347. Saegusa H, Kurihara T, Zong S, Kazuno A, Matsuda Y, Nonaka T, et al. Suppression of inflammatory and neuropathic pain symptoms in mice lacking the N-type Ca2+ channel. EMBO J. (2001) 20:2349–56. doi: 10.1093/emboj/20.10.2349
348. Alles SR, Garcia E, Balasubramanyan S, Jones K, Tyson JR, Joy T, et al. Peripheral nerve injury increases contribution of L-type calcium channels to synaptic transmission in spinal lamina II: role of alpha2delta-1 subunits. Mol Pain. (2018) 14:1744806918765806. doi: 10.1177/1744806918765806
349. Patel R, Bauer CS, Nieto-Rostro M, Margas W, Ferron L, Chaggar K, et al. α2δ-1 gene deletion affects somatosensory neuron function and delays mechanical hypersensitivity in response to peripheral nerve damage. J Neurosci. (2013) 33:16412–26. doi: 10.1523/JNEUROSCI.1026-13.2013
350. Yu C, Lin PX, Fitzgerald S, Nelson P. Heterogeneous calcium currents and transmitter release in cultured mouse spinal cord and dorsal root ganglion neurons. J Neurophysiol. (1992) 67:561–75. doi: 10.1152/jn.1992.67.3.561
351. Hogan QH, McCallum JB, Sarantopoulos C, Aason M, Mynlieff M, Kwok WM, et al. Painful neuropathy decreases membrane calcium current in mammalian primary afferent neurons. Pain. (2000) 86:43–53. doi: 10.1016/S0304-3959(99)00313-9
352. Fossat P, Dobremez E, Bouali-Benazzouz R, Favereaux A, Bertrand SS, Kilk K, et al. Knockdown of L calcium channel subtypes: differential effects in neuropathic pain. J Neurosci. (2010) 30:1073–85. doi: 10.1523/JNEUROSCI.3145-09.2010
353. Radwani H, Lopez-Gonzalez MJ, Cattaert D, Roca-Lapirot O, Dobremez E, Bouali-Benazzouz R, et al. Cav1.2 and Cav1.3 L-type calcium channels independently control short- and long-term sensitization to pain. J Physiol. (2016) 594:6607–26. doi: 10.1113/JP272725
354. Savalli N, Pantazis A, Sigg D, Weiss JN, Neely A, Olcese R. The alpha2delta-1 subunit remodels CaV1.2 voltage sensors and allows Ca2+ influx at physiological membrane potentials. J Gen Physiol. (2016) 148:147–59. doi: 10.1085/jgp.201611586
355. Muizelaar JP, Kleyer M, Hertogs IA, DeLange DC. Complex regional pain syndrome (reflex sympathetic dystrophy and causalgia): management with the calcium channel blocker nifedipine and/or the alpha-sympathetic blocker phenoxybenzamine in 59 patients. Clin Neurol Neurosurg. (1997) 99:26–30. doi: 10.1016/S0303-8467(96)00594-X
356. Hartung JE, Moy JK, Loeza-Alcocer E, Nagarajan V, Jostock R, Christoph T, et al. Voltage gated calcium channels in human dorsal root ganglion neurons. Pain. (2021). doi: 10.1097/j.pain.0000000000002465. [Epub ahead of print].
357. Bladen C, Gadotti VM, Gunduz MG, Berger ND, Simsek R, Safak C, et al. 1,4-Dihydropyridine derivatives with T-type calcium channel blocking activity attenuate inflammatory and neuropathic pain. Pflugers Arch. (2015) 467:1237–47. doi: 10.1007/s00424-014-1566-3
358. Gadotti VM, Bladen C, Zhang FX, Chen L, Gunduz MG, Simsek R, et al. Analgesic effect of a broad-spectrum dihydropyridine inhibitor of voltage-gated calcium channels. Pflugers Arch. (2015) 467:2485–93. doi: 10.1007/s00424-015-1725-1
359. Westenbroek RE, Hoskins L, Catterall WA. Localization of Ca2+ channel subtypes on rat spinal motor neurons, interneurons, and nerve terminals. J Neurosci. (1998) 18:6319–30. doi: 10.1523/JNEUROSCI.18-16-06319.1998
360. Rettig J, Sheng ZH, Kim DK, Hodson CD, Snutch TP, Catterall WA. Isoform-specific interaction of the alpha1A subunits of brain Ca2+ channels with the presynaptic proteins syntaxin and SNAP-25. Proc Natl Acad Sci USA. (1996) 93:7363–8. doi: 10.1073/pnas.93.14.7363
361. Sheng ZH, Rettig J, Cook T, Catterall WA. Calcium-dependent interaction of N-type calcium channels with the synaptic core complex. Nature. (1996) 379:451–4. doi: 10.1038/379451a0
362. Miljanich GP. Ziconotide: neuronal calcium channel blocker for treating severe chronic pain. Curr Med Chem. (2004) 11:3029–40. doi: 10.2174/0929867043363884
363. Matis G, De NP, Dupoiron D, Likar R, Zuidema X, Rasche D. Intrathecal pain management with ziconotide: time for consensus? Brain Behav. (2021) 11(Suppl. 1):e02055. doi: 10.1002/brb3.2055
364. Zhu CZ, Vortherms TA, Zhang M, Xu J, Swensen AM, Niforatos W, et al. Mechanistic insights into the analgesic efficacy of A-1264087, a novel neuronal Ca(2+) channel blocker that reduces nociception in rat preclinical pain models. J Pain. (2014) 15:387–14. doi: 10.1016/j.jpain.2013.12.002
365. Swensen AM, Herrington J, Bugianesi RM, Dai G, Haedo RJ, Ratliff KS, et al. Characterization of the substituted N-triazole oxindole TROX-1, a small-molecule, state-dependent inhibitor of Ca(V)2 calcium channels. Mol Pharmacol. (2012) 81:488–97. doi: 10.1124/mol.111.075226
366. Ogiyama T, Yonezawa K, Inoue M, Katayama N, Watanabe T, Yoshimura S, et al. Discovery of an 8-methoxytetrahydroisoquinoline derivative as an orally active N-type calcium channel blocker for neuropathic pain without CYP inhibition liability. Bioorg Med Chem. (2015) 23:4638–48. doi: 10.1016/j.bmc.2015.05.053
367. Ogiyama T, Yonezawa K, Inoue M, Watanabe T, Sugano Y, Gotoh T, et al. Discovery of a 1-isopropyltetrahydroisoquinoline derivative as an orally active N-type calcium channel blocker for neuropathic pain. Bioorg Med Chem. (2015) 23:4624–37. doi: 10.1016/j.bmc.2015.05.030
368. Brittain JM, Piekarz AD, Wang Y, Kondo T, Cummins TR, Khanna R. An atypical role for collapsin response mediator protein 2 (CRMP-2) in neurotransmitter release via interaction with presynaptic voltage-gated calcium channels. J Biol Chem. (2009) 284:31375–90. doi: 10.1074/jbc.M109.009951
369. Xie JY, Chew LA, Yang X, Wang Y, Qu C, Wang Y, et al. Sustained relief of ongoing experimental neuropathic pain by a CRMP2 peptide aptamer with low abuse potential. Pain. (2016) 157:2124–40. doi: 10.1097/j.pain.0000000000000628
370. Dolphin AC. G protein modulation of voltage-gated calcium channels. Pharmacol Rev. (2003) 55:607–27. doi: 10.1124/pr.55.4.3
371. Puke MJ, Xu XJ, Wiesenfeld-Hallin Z. Intrathecal administration of clonidine suppresses autotomy, a behavioral sign of chronic pain in rats after sciatic nerve section. Neurosci Lett. (1991) 133:199–202. doi: 10.1016/0304-3940(91)90569-F
372. Giovannitti JAJr, Thoms SM, Crawford JJ. Alpha-2 adrenergic receptor agonists: a review of current clinical applications. Anesth Prog. (2015) 62:31–9. doi: 10.2344/0003-3006-62.1.31
373. Yen LD, Bennett GJ, Ribeiro-da-Silva A. Sympathetic sprouting and changes in nociceptive sensory innervation in the glabrous skin of the rat hind paw following partial peripheral nerve injury. J Comp Neurol. (2006) 495:679–90. doi: 10.1002/cne.20899
374. Bauer CS, Rahman W, Tran-Van-Minh A, Lujan R, Dickenson AH, Dolphin AC. The anti-allodynic alpha(2)delta ligand pregabalin inhibits the trafficking of the calcium channel alpha(2)delta-1 subunit to presynaptic terminals in vivo. Biochem Soc Trans. (2010) 38:525–8. doi: 10.1042/BST0380525
375. Dworkin RH, Kirkpatrick P. Pregabalin. Nat Rev Drug Discov. (2005) 4:455–6. doi: 10.1038/nrd1756
376. Cundy KC, Branch R, Chernov-Rogan T, Dias T, Estrada T, Hold K, et al. XP13512 [(+/-)-1-([(alpha-isobutanoyloxyethoxy)carbonyl] aminomethyl)-1-cyclohexane acetic acid], a novel gabapentin prodrug: I. Design, synthesis, enzymatic conversion to gabapentin, and transport by intestinal solute transporters. J Pharmacol Exp Ther. (2004) 311:315–23. doi: 10.1124/jpet.104.067934
377. Biggs JE, Stemkowski PL, Knaus EE, Chowdhury MA, Ballanyi K, Smith PA. Suppression of network activity in dorsal horn by gabapentin permeation of TRPV1 channels; implications for drug access to cytoplasmic targets. Neurosci Lett. (2015) 584:397–402. doi: 10.1016/j.neulet.2014.07.033
378. Yang SN, Berggren PO. The role of voltage-gated calcium channels in pancreatic beta-cell physiology and pathophysiology. Endocr Rev. (2006) 27:621–76. doi: 10.1210/er.2005-0888
379. Saegusa H, Tanabe T. N-type voltage-dependent Ca2+ channel in non-excitable microglial cells in mice is involved in the pathophysiology of neuropathic pain. Biochem Biophys Res Commun. (2014) 450:142–7. doi: 10.1016/j.bbrc.2014.05.103
380. Huntula S, Saegusa H, Wang X, Zong S, Tanabe T. Involvement of N-type Ca(2+) channel in microglial activation and its implications to aging-induced exaggerated cytokine response. Cell Calcium. (2019) 82:102059. doi: 10.1016/j.ceca.2019.102059
381. Fernandez FR, Iftinca MC, Zamponi GW, Turner RW. Modeling temperature- and Cav3 subtype-dependent alterations in T-type calcium channel mediated burst firing. Mol Brain. (2021) 14:115. doi: 10.1186/s13041-021-00813-7
382. Jacus MO, Uebele VN, Renger JJ, Todorovic SM. Presynaptic CaV3.2 channels regulate excitatory neurotransmission in nociceptive dorsal horn neurons. J Neurosci. (2012) 32:9374–82. doi: 10.1523/JNEUROSCI.0068-12.2012
383. Garcia-Caballero A, Gadotti VM, Stemkowski P, Weiss N, Souza IA, Hodgkinson V, et al. The deubiquitinating enzyme USP5 modulates neuropathic and inflammatory pain by enhancing Cav3.2 channel activity. Neuron. (2014) 83:1144–58. doi: 10.1016/j.neuron.2014.07.036
384. Weiss N, Hameed S, Fernandez-Fernandez JM, Fablet K, Karmazinova M, Poillot C, et al. A Ca(v)3.2/syntaxin-1A signaling complex controls T-type channel activity and low-threshold exocytosis. J Biol Chem. (2012) 287:2810–8. doi: 10.1074/jbc.M111.290882
385. Rose KE, Lunardi N, Boscolo A, Dong X, Erisir A, Jevtovic-Todorovic V, et al. Immunohistological demonstration of CaV3.2 T-type voltage-gated calcium channel expression in soma of dorsal root ganglion neurons and peripheral axons of rat and mouse. Neuroscience. (2013) 250:263–74. doi: 10.1016/j.neuroscience.2013.07.005
386. Talley EM, Cribbs LL, Lee JH, Daud A, Perez-Reyes E, Bayliss DA. Differential distribution of three members of a gene family encoding low voltage-activated (T-type) calcium channels. J Neurosci. (1999) 19:1895–911. doi: 10.1523/JNEUROSCI.19-06-01895.1999
387. Altier C, Zamponi GW. Targeting Ca2+ channels to treat pain: T-type versus N-type. Trends Pharmacol Sci. (2004) 25:465–70. doi: 10.1016/j.tips.2004.07.004
388. Jagodic MM, Pathirathna S, Nelson MT, Mancuso S, Joksovic PM, Rosenberg ER, et al. Cell-specific alterations of t-type calcium current in painful diabetic neuropathy enhance excitability of sensory neurons. J Neurosci. (2007) 27:3305–16. doi: 10.1523/JNEUROSCI.4866-06.2007
389. Jagodic MM, Pathirathna S, Joksovic PM, Lee W, Nelson MT, Naik AK, et al. Upregulation of the T-type calcium current in small rat sensory neurons after chronic constrictive injury of the sciatic nerve. J Neurophysiol. (2008) 99:3151–6. doi: 10.1152/jn.01031.2007
390. Lauzadis J, Liu H, Lu Y, Rebecchi MJ, Kaczocha M, Puopolo M. Contribution of T-type calcium channels to spinal cord injury-induced hyperexcitability of nociceptors. J Neurosci. (2020) 40:7229–40. doi: 10.1523/JNEUROSCI.0517-20.2020
391. Francois A, Laffray S, Pizzoccaro A, Eschalier A, Bourinet E. T-type calcium channels in chronic pain: mouse models and specific blockers. Pflugers Arch. (2014) 466:707–17. doi: 10.1007/s00424-014-1484-4
392. Todorovic SM, Jevtovic-Todorovic V. Neuropathic pain: role for presynaptic T-type channels in nociceptive signaling. Pflugers Arch. (2013) 465:921–7. doi: 10.1007/s00424-012-1211-y
393. Todorovic SM, Jevtovic-Todorovic V. Targeting of CaV3.2 T-type calcium channels in peripheral sensory neurons for the treatment of painful diabetic neuropathy. Pflugers Arch. (2014) 466:701–6. doi: 10.1007/s00424-014-1452-z
394. Francois A, Schuetter N, Laffray S, Sanguesa J, Pizzoccaro A, Dubel S, et al. The low-threshold calcium channel Cav3.2 determines low-threshold mechanoreceptor function. Cell Rep. (2015) 10:370–82. doi: 10.1016/j.celrep.2014.12.042
395. Stemkowski PL, Garcia-Caballero A, Gadotti VM, M'Dahoma S, Chen L, Souza IA, et al. Identification of interleukin-1 beta as a key mediator in the upregulation of Cav3. 2–USP5 interactions in the pain pathway. Mol Pain. (2017) 13:1744806917724698. doi: 10.1177/1744806917724698
396. Tomita S, Sekiguchi F, Kasanami Y, Naoe K, Tsubota M, Wake H, et al. Cav3.2 overexpression in L4 dorsal root ganglion neurons after L5 spinal nerve cutting involves Egr-1, USP5 and HMGB1 in rats: an emerging signaling pathway for neuropathic pain. Eur J Pharmacol. (2020) 888:173587. doi: 10.1016/j.ejphar.2020.173587
397. Weiss N, Black SA, Bladen C, Chen L, Zamponi GW. Surface expression and function of Cav3.2 T-type calcium channels are controlled by asparagine-linked glycosylation. Pflugers Arch. (2013) 465:1159–70. doi: 10.1007/s00424-013-1259-3
398. Orestes P, Osuru HP, McIntire WE, Jacus MO, Salajegheh R, Jagodic MM, et al. Reversal of neuropathic pain in diabetes by targeting glycosylation of Ca(V)3.2 T-type calcium channels. Diabetes. (2013) 62:3828–38. doi: 10.2337/db13-0813
399. Wang H, Wei Y, Pu Y, Jiang D, Jiang X, Zhang Y, et al. Brain-derived neurotrophic factor stimulation of T-type Ca(2+) channels in sensory neurons contributes to increased peripheral pain sensitivity. Sci Signal. (2019) 12:600. doi: 10.1126/scisignal.aaw2300
400. Boakye PA, Rancic V, Whitlock KH, Simmons D, Longo FM, Ballanyi K, et al. Receptor dependence of BDNF actions in superficial dorsal horn: relation to central sensitization and actions of macrophage colony stimulating factor 1. J Neurophysiol. (2019) 121:2308–22. doi: 10.1152/jn.00839.2018
401. Smith PA. BDNF: no gain without pain? Neuroscience. (2014) 283:107−23. doi: 10.1016/j.neuroscience.2014.05.044
402. Gandini MA, Souza IA, Kullar A, Gambeta E, Zamponi GW. Regulation of CaV3.2 channels by the receptor for activated C kinase 1 (Rack-1). Pflugers Archiv Euro J Physiol. (2021). doi: 10.1007/s00424-021-02631-1. [Epub ahead of print].
403. Lu R, Fan B, Yin D, Li Y, Wang B, Zhu S, et al. Receptor for activated C kinase 1 mediates the chronic constriction injury-induced neuropathic pain in the rats' peripheral and central nervous system. Neurosci Lett. (2019) 712:134477. doi: 10.1016/j.neulet.2019.134477
404. Dogrul A, Gardell LR, Ossipov MH, Tulunay FC, Lai J, Porreca F. Reversal of experimental neuropathic pain by T-type calcium channel blockers. Pain. (2003) 105:159–68. doi: 10.1016/S0304-3959(03)00177-5
405. M'Dahoma S, Gadotti VM, Zhang FX, Park B, Nam JH, Onnis V, et al. Effect of the T-type channel blocker KYS-05090S in mouse models of acute and neuropathic pain. Pflugers Arch. (2015) 468:193–9. doi: 10.1007/s00424-015-1733-1
406. Zhang Q, Xia Z, Joshi S, Scott VE, Jarvis MF. Optimization of ADME properties for sulfonamides leading to the discovery of a T-type calcium channel blocker, ABT-639. ACS Med Chem Lett. (2015) 6:641–4. doi: 10.1021/acsmedchemlett.5b00023
407. Jarvis MF, Scott VE, McGaraughty S, Chu KL, Xu J, Niforatos W, et al. A peripherally acting, selective T-type calcium channel blocker, ABT-639, effectively reduces nociceptive and neuropathic pain in rats. Biochem Pharmacol. (2014) 89:536–44. doi: 10.1016/j.bcp.2014.03.015
408. Ziegler D, Duan WR, An G, Thomas JW, Nothaft W. A randomized double-blind, placebo-, and active-controlled study of T-type calcium channel blocker ABT-639 in patients with diabetic peripheral neuropathic pain. Pain. (2015) 156:2013–20. doi: 10.1097/j.pain.0000000000000263
409. Choe W, Messinger RB, Leach E, Eckle VS, Obradovic A, Salajegheh R, et al. TTA-P2 is a potent and selective blocker of T-type calcium channels in rat sensory neurons and a novel antinociceptive agent. Mol Pharmacol. (2011) 80:900–10. doi: 10.1124/mol.111.073205
410. Francois A, Kerckhove N, Meleine M, Alloui A, Barrere C, Gelot A, et al. State-dependent properties of a new T-type calcium channel blocker enhance Ca(V)3.2 selectivity and support analgesic effects. Pain. (2013) 154:283–93. doi: 10.1016/j.pain.2012.10.023
411. Tringham E, Powell KL, Cain SM, Kuplast K, Mezeyova J, Weerapura M, et al. T-type calcium channel blockers that attenuate thalamic burst firing and suppress absence seizures. Sci Transl Med. (2012) 4:121ra19. doi: 10.1126/scitranslmed.3003120
412. Harding EK, Dedek A, Bonin RP, Salter MW, Snutch TP, Hildebrand ME. The T-type calcium channel antagonist, Z944, reduces spinal excitability and pain hypersensitivity. Br J Pharmacol. (2021) 178:3517–32. doi: 10.1111/bph.15498
413. Zamponi GW, Lewis RJ, Todorovic SM, Arneric SP, Snutch TP. Role of voltage-gated calcium channels in ascending pain pathways. Brain Res Rev. (2009) 60:84–9. doi: 10.1016/j.brainresrev.2008.12.021
414. Bezencon O, Heidmann B, Siegrist R, Stamm S, Richard S, Pozzi D, et al. Discovery of a potent, selective T-type calcium channel blocker as a drug candidate for the treatment of generalized epilepsies. J Med Chem. (2017) 60:9769–89. doi: 10.1021/acs.jmedchem.7b01236
415. Berger ND, Gadotti VM, Petrov RR, Chapman K, Diaz P, Zamponi GW. NMP-7 inhibits chronic inflammatory and neuropathic pain via block of Cav3.2 T-type calcium channels and activation of CB2 receptors. Mol Pain. (2014) 10:77. doi: 10.1186/1744-8069-10-77
416. Hofmann F, Biel M, Kaupp UB. International Union of Pharmacology. LI Nomenclature and structure-function relationships of cyclic nucleotide-regulated channels. Pharmacol Rev. (2005) 57:455–62. doi: 10.1124/pr.57.4.8
417. Smith T, Al OM, Sathish J, Djouhri L. Increased expression of HCN2 channel protein in L4 dorsal root ganglion neurons following axotomy of L5- and inflammation of L4-spinal nerves in rats. Neuroscience. (2015) 295:90–102. doi: 10.1016/j.neuroscience.2015.03.041
418. Young GT, Emery EC, Mooney ER, Tsantoulas C, McNaughton PA. Inflammatory and neuropathic pain are rapidly suppressed by peripheral block of hyperpolarisation-activated cyclic nucleotide-gated ion channels. Pain. (2014) 155:1708–19. doi: 10.1016/j.pain.2014.05.021
419. Emery EC, Young GT, McNaughton PA. HCN2 ion channels: an emerging role as the pacemakers of pain. Trends Pharmacol Sci. (2012) 33:456–63. doi: 10.1016/j.tips.2012.04.004
420. Emery EC, Young GT, Berrocoso EM, Chen L, McNaughton PA. HCN2 ion channels play a central role in inflammatory and neuropathic pain. Science. (2011) 333:1462–6. doi: 10.1126/science.1206243
421. Luo L, Chang L, Brown SM, Ao H, Lee DH, Higuera ES, et al. Role of peripheral hyperpolarization-activated cyclic nucleotide-modulated channel pacemaker channels in acute and chronic pain models in the rat. Neuroscience. (2007) 144:1477–85. doi: 10.1016/j.neuroscience.2006.10.048
422. Antal M, Papp I, Bahaerguli N, Veress G, Vereb G. Expression of hyperpolarization-activated and cyclic nucleotide-gated cation channel subunit 2 in axon terminals of peptidergic nociceptive primary sensory neurons in the superficial spinal dorsal horn of rats. Eur J Neurosci. (2004) 19:1336–42. doi: 10.1111/j.1460-9568.2004.03235.x
423. Papp I, Szucs P, Hollo K, Erdelyi F, Szabo G, Antal M. Hyperpolarization-activated and cyclic nucleotide-gated cation channel subunit 2 ion channels modulate synaptic transmission from nociceptive primary afferents containing substance P to secondary sensory neurons in laminae I-IIo of the rodent spinal dorsal horn. Eur J Neurosci. (2006) 24:1341–52. doi: 10.1111/j.1460-9568.2006.05013.x
424. Chaplan SR, Guo HQ, Lee DH, Luo L, Liu C, Kuei C, et al. Neuronal hyperpolarization-activated pacemaker channels drive neuropathic pain. J Neurosci. (2003) 23:1169–78. doi: 10.1523/JNEUROSCI.23-04-01169.2003
425. Noh S, Kumar N, Bukhanova N, Chen Y, Stemkowsi PL, Smith PA. The heart-rate-reducing agent, ivabradine, reduces mechanical allodynia in a rodent model of neuropathic pain. Eur J Pain. (2014) 18:1139–47. doi: 10.1002/j.1532-2149.2014.00460.x
426. Dini L, Del LM, Resta F, Melchiorre M, Spinelli V, Di Cesare ML, et al. Selective blockade of HCN1/HCN2 channels as a potential pharmacological strategy against pain. Front Pharmacol. (2018) 9:1252. doi: 10.3389/fphar.2018.01252
427. Lainez S, Tsantoulas C, Biel M, McNaughton PA. HCN3 ion channels: roles in sensory neuronal excitability and pain. J Physiol. (2019) 597:4661–75. doi: 10.1113/JP278211
428. Hsiao HT, Liu YC, Liu PY, Wu SN. Concerted suppression of Ih and activation of IK(M) by ivabradine, an HCN-channel inhibitor, in pituitary cells and hippocampal neurons. Brain Res Bull. (2019) 149:11–20. doi: 10.1016/j.brainresbull.2019.03.016
429. Ding W, You Z, Shen S, Chen L, Zhu S, Mao J. Inhibition of HCN channel activity in the thalamus attenuates chronic pain in rats. Neurosci Lett. (2016) 631:97–103. doi: 10.1016/j.neulet.2016.08.021
430. Lee MC, Bond S, Wheeler D, Scholtes I, Armstrong G, McNaughton P, et al. A randomised, double-blind, placebo-controlled crossover trial of the influence of the HCN channel blocker ivabradine in a healthy volunteer pain model: an enriched population trial. Pain. (2019) 160:2554–65. doi: 10.1097/j.pain.0000000000001638
431. Sartiani L, Mannaioni G, Masi A, Novella Romanelli M, Cerbai E. The hyperpolarization-activated cyclic nucleotide-gated channels: from biophysics to pharmacology of a unique family of ion channels. Pharmacol Rev. (2017) 69:354. doi: 10.1124/pr.117.014035
432. Bagal SK, Omoto K, Blakemore DC, Bungay PJ, Bilsland JG, Clarke PJ, et al. Discovery of allosteric, potent, subtype selective, and peripherally restricted TrkA kinase inhibitors. J Med Chem. (2019) 62:247–65. doi: 10.1021/acs.jmedchem.8b00280
433. Bagal SK, Andrews M, Bechle BM, Bian J, Bilsland J, Blakemore DC, et al. Discovery of potent, selective, and peripherally restricted pan-trk kinase inhibitors for the treatment of pain. J Med Chem. (2018) 61:6779–800. doi: 10.1021/acs.jmedchem.8b00633
434. Surur AS, Beirow K, Bock C, Schulig L, Kindermann MK, Bodtke A, et al. Flupirtine analogues: explorative synthesis and influence of chemical structure on KV7.2/KV7.3 channel opening activity. Chem Open. (2019) 8:41–4. doi: 10.1002/open.201800244
435. Messinger RB, Naik AK, Jagodic MM, Nelson MT, Lee WY, Choe WJ, et al. In vivo silencing of the Ca(V)3.2 T-type calcium channels in sensory neurons alleviates hyperalgesia in rats with streptozocin-induced diabetic neuropathy. Pain. (2009) 145:184–95. doi: 10.1016/j.pain.2009.06.012
436. Stemkowski PL, Smith PA. An overview of animal models of neuropathic pain. In: Toth C, Moulin DE, editors. Neuropathic Pain, Causes, Management and Understanding. Cambridge: Cambridge University Press (2013). p. 33−50.
437. Bouali-Benazzouz R, Landry M, Benazzouz A, Fossat P. Neuropathic pain modeling: focus on synaptic and ion channel mechanisms. Prog Neurobiol. (2021) 201:102030. doi: 10.1016/j.pneurobio.2021.102030
439. Mogil JS. Laboratory environmental factors and pain behavior: the relevance of unknown unknowns to reproducibility and translation. Lab Anim. (2017) 46:136–41.
440. Harte SE, Meyers JB, Donahue RR, Taylor BK, Morrow TJ. Mechanical Conflict System: A Novel Operant Method for the Assessment of Nociceptive Behavior. PLoS One. (2016) 11:e0150164. doi: 10.1371/journal.pone.0150164 eCollection 2016.
441. Mauderli AP, Acosta-Rua A, Vierck CJ. An operant assay of thermal pain in conscious, unrestrained rats. J Neurosci Methods. (2000) 97:19–29.
442. Negus SS, Vanderah TW, Brandt MR, Bilsky EJ, Becerra L, Borsook D. Preclinical assessment of candidate analgesic drugs: recent advances and future challenges. J Pharmacol Exp Ther. (2006) 319:507–14.
443. Mogil JS. Sex differences in pain and pain inhibition: multiple explanations of a controversial phenomenon. Nat Rev Neurosci. (2012) 13:859–66. doi: 10.1038/nrn3360
444. Mogil JS. Sources of individual differences in pain. Ann Rev Neurosci. (2021) 44:1–25. doi: 10.1146/annurev-neuro-092820-105941
445. Mapplebeck JC, Beggs S, Salter MW. Molecules in pain and sex: a developing story. Mol Brain. (2017) 10:9. doi: 10.1186/s13041-017-0289-8
446. Fillingim RB, King CD, Ribeiro-Dasilva MC, Rahim-Williams B, Riley JL III. Sex, gender, and pain: a review of recent clinical and experimental findings. J Pain. (2009) 10:447–85. doi: 10.1016/j.jpain.2008.12.001
447. Johnston KJA, Ward J, Ray PR, Adams MJ, McIntosh AM, Smith BH, et al. Sex-stratified genome-wide association study of multisite chronic pain in UK Biobank. PLoS Genet. (2021) 17:e1009428. doi: 10.1371/journal.pgen.1009428
448. Mogil JS. Qualitative sex differences in pain processing: emerging evidence of a biased literature. Nat Rev Neurosci. (2020) 21:353–65. doi: 10.1038/s41583-020-0310-6
449. Martin LJ, Acland EL, Cho C, Gandhi W, Chen D, Corley E, et al. Male-specific conditioned pain hypersensitivity in mice and humans. Curr Biol. (2019) 29:192–201. doi: 10.1016/j.cub.2018.11.030
450. Hendrich J, Alvarez P, Joseph EK, Ferrari LF, Chen X, Levine JD. In vivo and in vitro comparison of female and male nociceptors. J Pain. (2012) 13:1224–31. doi: 10.1016/j.jpain.2012.09.009
451. Sorge RE, Mapplebeck JC, Rosen S, Beggs S, Taves S, Alexander JK, et al. Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat Neurosci. (2015) 18:1081–3. doi: 10.1038/nn.4053
452. Sorge RE, Totsch SK. Sex differences in pain. J Neurosci Res. (2016) 95:1271–81. doi: 10.1002/jnr.23841
453. Szabo-Pardi TA, Syed UM, Castillo ZW, Burton MD. Use of integrated optical clearing and 2-photon imaging to investigate sex differences in neuroimmune interactions after peripheral nerve injury. Front Cell Dev Biol. (2021) 9:119. doi: 10.3389/fcell.2021.624201
454. Mifflin KA, Benson C, Thorburn KC, Baker GB, Kerr BJ. Manipulation of neurotransmitter levels has differential effects on formalin-evoked nociceptive behavior in male and female mice. J Pain. (2016) 17:483–98. doi: 10.1016/j.jpain.2015.12.013
455. Dedek A, Xu J, Lorenzo L-E, Godin AG, Kandegedara CM, Glavina G, et al. Sexual dimorphism in a neuronal mechanism of spinal hyperexcitability across rodent and human models of pathological pain. BioRxiv [Preprint]. (2021). doi: 10.1101/2021.06.15.447407
456. Luo X, Chen O, Wang Z, Bang S, Ji J, Lee SH, et al. IL-23/IL-17A/TRPV1 axis produces mechanical pain via macrophage-sensory neuron crosstalk in female mice. Neuron. (2021) 109:2691–706. doi: 10.1016/j.neuron.2021.06.015
457. O'Brien MS, Philpott HTA, McDougall JJ. Targeting the Nav1.8 ion channel engenders sex-specific responses in lysophosphatidic acid-induced joint neuropathy. Pain. (2019) 160:269–78. doi: 10.1097/j.pain.0000000000001399
458. Ferreira MA, Luckemeyer DD, Macedo-Junior SJ, Schran RG, Silva AM, Prudente AS, et al. Sex-dependent Cav2.3 channel contribution to the secondary hyperalgesia in a mice model of central sensitization. Brain Res. (2021) 1764:147438. doi: 10.1016/j.brainres.2021.147438
459. Shansky RM, Murphy AZ. Considering sex as a biological variable will require a global shift in science culture. Nat Neurosci. (2021) 24:457–64. doi: 10.1038/s41593-021-00806-8
460. Baron R, Maier C, Attal N, Binder A, Bouhassira D, Cruccu G, et al. Peripheral neuropathic pain: a mechanism-related organizing principle based on sensory profiles. Pain. (2017) 158:261–72. doi: 10.1097/j.pain.0000000000000753
461. Brewer CL, Li J, O'Conor K, Serafin EK, Baccei ML. Neonatal injury evokes persistent deficits in dynorphin inhibitory circuits within the adult mouse superficial dorsal horn. J Neurosci. (2020) 40:3882–95. doi: 10.1523/JNEUROSCI.0029-20.2020
462. Moriarty O, Tu Y, Sengar AS, Salter MW, Beggs S, Walker SM. Priming of adult incision response by early-life injury: neonatal microglial inhibition has persistent but sexually dimorphic effects in adult rats. J Neurosci. (2019) 39:3081–93. doi: 10.1523/JNEUROSCI.1786-18.2019
463. Beggs S, Currie G, Salter MW, Fitzgerald M, Walker SM. Priming of adult pain responses by neonatal pain experience: maintenance by central neuroimmune activity. Brain. (2012) 135:404–17. doi: 10.1093/brain/awr288
464. Luo JL, Qin HY, Wong CK, Tsang SY, Huang Y, Bian ZX. Enhanced excitability and down-regulated voltage-gated potassium channels in colonic drg neurons from neonatal maternal separation rats. J Pain. (2011) 12:600–9. doi: 10.1016/j.jpain.2010.11.005
465. Dworsky-Fried Z, Kerr BJ, Taylor AMW. Microbes, microglia, and pain. Neurobiol Pain. (2020) 7:100045. doi: 10.1016/j.ynpai.2020.100045
466. Fitzgerald M, McKelvey R. Nerve injury and neuropathic pain - a question of age. Exp Neurol. (2016) 275(Pt 2):296–302. doi: 10.1016/j.expneurol.2015.07.013
467. Gaudet AD, Fonken LK, Ayala MT, Maier SF, Watkins LR. Aging and miR-155 in mice influence survival and neuropathic pain after spinal cord injury. Brain Behav Immun. (2021) 97:365–70. doi: 10.1016/j.bbi.2021.07.003
468. Peirs C, Williams SG, Zhao X, Arokiaraj CM, Ferreira DW, Noh MC, et al. Mechanical allodynia circuitry in the dorsal horn is defined by the nature of the injury. Neuron. (2021) 109:73–90. doi: 10.1016/j.neuron.2020.10.027
469. Decosterd I, Woolf CJ. Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain. (2000) 87:149–58. doi: 10.1016/S0304-3959(00)00276-1
470. Noh MC, Mikler B, Joy T, Smith PA. Time course of inflammation in dorsal root ganglia correlates with differential reversibility of mechanical allodynia. Neuroscience. (2020) 428:199–216. doi: 10.1016/j.neuroscience.2019.12.040
471. Rolke R, Baron R, Maier C, Tolle TR, Treede -DR, Beyer A, et al. Quantitative sensory testing in the German Research Network on Neuropathic Pain (DFNS): standardized protocol and reference values. Pain. (2006) 123:231–43. doi: 10.1016/j.pain.2006.01.041
472. Vollert J, Maier C, Attal N, Bennett DLH, Bouhassira D, Enax-Krumova EK, et al. Stratifying patients with peripheral neuropathic pain based on sensory profiles: algorithm and sample size recommendations. Pain. (2017) 158:1446–55. doi: 10.1097/j.pain.0000000000000935
473. Backonja MM, Attal N, Baron R, Bouhassira D, Drangholt M, Dyck PJ, et al. Value of quantitative sensory testing in neurological and pain disorders: NeuPSIG consensus. Pain. (2013) 154:1807–19. doi: 10.1016/j.pain.2013.05.047
474. Bannister K, Sachau J, Baron R, Dickenson AH. Neuropathic pain: mechanism-based therapeutics. Annu Rev Pharmacol Toxicol. (2020) 60:257–74. doi: 10.1146/annurev-pharmtox-010818-021524
475. Renthal W, Chamessian A, Curatolo M, Davidson S, Burton M, Dib-Hajj S, et al. Human cells and networks of pain: transforming pain target identification and therapeutic development. Neuron. (2021) 109:1426–9. doi: 10.1016/j.neuron.2021.04.005
476. Murphy PG, Ramer MS, Borthwick L, Gauldie J, Richardson PM, Bisby MA. Endogenous interleukin-6 contributes to hypersensitivity to cutaneous stimuli and changes in neuropeptides associated with chronic nerve constriction in mice. Eur J Neurosci. (1999) 11:2243–53. doi: 10.1046/j.1460-9568.1999.00641.x
477. Taylor PC, Lee YC, Fleischmann R, Takeuchi T, Perkins EL, Fautrel B, et al. Achieving pain control in rheumatoid arthritis with baricitinib or adalimumab plus methotrexate: results from the RA-BEAM trial. J Clin Med. (2019) 8:6. doi: 10.3390/jcm8060831
478. Gilron I, Jensen TS, Dickenson AH. Combination pharmacotherapy for management of chronic pain: from bench to bedside. Lancet Neurol. (2013) 12:1084–95. doi: 10.1016/S1474-4422(13)70193-5
479. Middleton SJ, Barry AM, Comini M, Li Y, Ray PR, Shiers S, et al. Studying human nociceptors: from fundamentals to clinic. Brain. (2021) 144:1312–36. doi: 10.1093/brain/awab048
480. Rostock C, Schrenk-Siemens K, Pohle J, Siemens J. Human vs. mouse nociceptors - similarities and differences. Neuroscience. (2018) 387:13–27. doi: 10.1016/j.neuroscience.2017.11.047
481. Cheng C, Guo GF, Martinez JA, Singh V, Zochodne DW. Dynamic plasticity of axons within a cutaneous milieu. J Neurosci. (2010) 30:14735–44. doi: 10.1523/JNEUROSCI.2919-10.2010
482. Acharjee S, Noorbakhsh F, Stemkowski PL, Olechowski C, Cohen EA, Ballanyi K, et al. HIV-1 viral protein R causes peripheral nervous system injury associated with in vivo neuropathic pain. FASEB J. (2010) 24:4343–53. doi: 10.1096/fj.10-162313
483. Shiers S, Klein RM, Price TJ. Quantitative differences in neuronal subpopulations between mouse and human dorsal root ganglia demonstrated with RNAscope in situ hybridization. Pain. (2020) 161:2410–24. doi: 10.1097/j.pain.0000000000001973
484. Chambers SM, Qi Y, Mica Y, Lee G, Zhang XJ, Niu L, et al. Combined small-molecule inhibition accelerates developmental timing and converts human pluripotent stem cells into nociceptors. Nat Biotechnol. (2012) 30:715–20. doi: 10.1038/nbt.2249
485. Young GT, Gutteridge A, Fox H, Wilbrey AL, Cao L, Cho LT, et al. Characterizing human stem cell-derived sensory neurons at the single-cell level reveals their ion channel expression and utility in pain research. Mol Ther. (2014) 22:1530–43. doi: 10.1038/mt.2014.86
486. Lampert A, Bennett DL, McDermott LA, Neureiter A, Eberhardt E, Winner B, et al. Human sensory neurons derived from pluripotent stem cells for disease modelling and personalized medicine. Neurobiol Pain. (2020) 8:100055. doi: 10.1016/j.ynpai.2020.100055
487. Haile Y, Nakhaei-Nejad M, Boakye PA, Baker G, Smith PA, Murray AG, et al. Reprogramming of HUVECs into induced pluripotent stem cells (HiPSCs), generation and characterization of HiPSC-derived neurons and astrocytes. PLoS One. (2015) 10:e0119617. doi: 10.1371/journal.pone.0119617
488. Vojnits K, Mahammad S, Collins TJ, Bhatia M. Chemotherapy-induced neuropathy and drug discovery platform using human sensory neurons converted directly from adult peripheral blood. Stem Cells Transl Med. (2019) 8:1180–91. doi: 10.1002/sctm.19-0054
489. Vallbo AB. Microneurography: how it started and how it works. J Neurophysiol. (2018) 120:1415–27. doi: 10.1152/jn.00933.2017
490. Serra J, Bostock H, Sola R, Aleu J, Garcia E, Cokic B, et al. Microneurographic identification of spontaneous activity in C-nociceptors in neuropathic pain states in humans and rats. Pain. (2012) 153:42–55. doi: 10.1016/j.pain.2011.08.015
491. Paschon V, Correia FF, Morena BC, da Silva VA, Dos Santos GB, da Silva MCC, et al. CRISPR, prime editing, optogenetics, and DREADDs: new therapeutic approaches provided by emerging technologies in the treatment of spinal cord injury. Mol Neurobiol. (2020) 57:2085–100. doi: 10.1007/s12035-019-01861-w
492. Calvo M, Davies AJ, Hebert HL, Weir GA, Chesler EJ, Finnerup NB, et al. The genetics of neuropathic pain from model organisms to clinical application. Neuron. (2019) 104:637–53. doi: 10.1016/j.neuron.2019.09.018
493. Alles SRA, Walsh P, Truong V. Development of Novel Human Induced Pluripotent Stem Cell Derived (hiPSC) Sensory Neuron Lines From Pain Patient Blood Samples. U.S. Provisional Patent Application No. 63/132,168 (2020).
Keywords: Nav1.3, Nav1.7, Nav1.8, Cav3.2, Kv7.2/7.3, dorsal root ganglia (DRG), primary afferent, allodynia
Citation: Alles SRA and Smith PA (2021) Peripheral Voltage-Gated Cation Channels in Neuropathic Pain and Their Potential as Therapeutic Targets. Front. Pain Res. 2:750583. doi: 10.3389/fpain.2021.750583
Received: 30 July 2021; Accepted: 10 November 2021;
Published: 13 December 2021.
Edited by:
Susanne Katharina Sauer, University of Erlangen Nuremberg, GermanyReviewed by:
Pradipta R. Ray, The University of Texas at Dallas, United StatesFernanda C. Cardoso, The University of Queensland, Australia
Copyright © 2021 Alles and Smith. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter A. Smith, cGFzM0B1YWxiZXJ0YQ==