 Rita Silva
Rita Silva
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Pain Res. , 27 July 2021
Sec. Pain Mechanisms
Volume 2 - 2021 | https://doi.org/10.3389/fpain.2021.684684
This article is part of the Research Topic Highlights in Pain Mechanisms 2021/22 View all 9 articles
Injuries to the nervous system can result in a debilitating neuropathic pain state that is often resistant to treatment with available analgesics, which are commonly associated with several side-effects. Growing pre-clinical and clinical evidence over the last two decades indicates that immune cell-mediated mechanisms both in the periphery and in the Central Nervous System (CNS) play significant roles in the establishment and maintenance of neuropathic pain. Specifically, following peripheral nerve injury, microglia, which are CNS resident immune cells, respond to the activity of the first pain synapse in the dorsal horn of spinal cord and also to neuronal activity in higher centres in the brain. This microglial response leads to the production and release of several proinflammatory mediators which contribute to neuronal sensitisation under neuropathic pain states. In this review, we collect evidence demonstrating the critical role played by the Fractalkine/CX3CR1 signalling pathway in neuron-to-microglia communication in neuropathic pain states and explore how strategies that include components of this pathway offer opportunities for innovative targets for neuropathic pain.
Neuropathic pain is a devastating condition which affects around 7–10% of the general population globally, predominantly diagnosed in patients above 50 years of age (1). It is a chronic secondary pain condition, either as a result of peripheral (mechanical trauma, metabolic diseases, infection, etc.) or central (spinal cord injury, stroke or multiple sclerosis) nervous (somatosensory) system lesions or diseases (2). After peripheral nerve damage, both activation of sensory neurons and local inflammation occur at the site of injury. The presence of immune cells, such as macrophages (3), which release pro-inflammatory mediators and alter nociceptors' excitability (specialised sensory neurons that respond to noxious stimuli), facilitates ectopic firing and ongoing nociceptive transmission. With persistent and constant neuronal input from the periphery, dorsal horn nociceptive circuits in the spinal cord are activated, sensitised and undergo plastic changes in the CNS. Such maladaptive plasticity in the nociceptive system correlates with altered behavioural responsiveness to innocuous and noxious stimuli (4, 5). In the dorsal horn of the spinal cord, sensitisation of the first sensory synapse is characterised by a complex set of changes in synaptic efficacy, increased receptor expression and an imbalance of descending facilitatory and inhibitory modulation (6–9). Another active component to the generation of neuropathic pain involves immune cells, especially dorsal horn microglia which amplify and actively contribute to mechanisms of chronic pain (10, 11).
Data from our group and others have revealed that, after peripheral nerve damage, spinal cord microglia accumulate and proliferate in the superficial dorsal horn within the termination area of the injured peripheral nerve fibres (12–14). Peripherally injured sensory afferents instigate the change of microglial cells morphology to an activated state (15). Microglial activation is followed by the release of proinflammatory cytokines and chemokines which interact with dorsal horn neurons and modulate neurotransmission (16). As a result, these proinflammatory cytokines and chemokines are believed to contribute to increased nociceptive hypersensitivity and to the development of allodynia (response to innocuous stimuli) and hyperalgesia (enhanced response to noxious stimuli). Animal studies have spawned great interest in using glial inhibitors since blockade of microglial activity reduces nociceptive behaviours in models of neuropathic pain (14, 17, 18). Although significant spinal microgliosis is evident in both sexes of rodents after injury (19, 20), the interruption of spinal microglial activity following neuropathic injury, preferentially attenuates allodynia in male mice (19). Given the critical role of the immune system in the pathophysiology of neuropathic pain, an improved understanding of the pathways which regulate the communication between microglial and neuronal cells will shed light on innovative microglial targets for the treatment of neuropathic pain.
The identification of such bi-directional signalling pathways between CNS immunocompetent cells and neurons is critical to underpin the mechanisms underlying this interaction. Some of the major signalling pathways that mediates neuroimmune interface at the spinal cord dorsal horn level are mediated by chemokine signalling (21–23). Chemokines belong to a large superfamily of small molecules (from 8–15 kDa) and are believed to be key mediators of the interaction between neurons and neighbouring glial cells, and some chemokines exert potent chemotactic and pro-inflammatory functions. The scientific community has uncovered more than 50 chemokine ligands so far, out of which fractalkine (FKN) and its sole receptor (CX3CR1) require and deserve special attention. In this review we will examine pre-clinical evidence and focus on the role of FKN and CX3CR1 in neuron-to-microglial cell communication in neuropathic pain states and reflect on the pharmacological potential of interfering with this signalling pathway.
FKN is a transmembrane chemokine constitutively expressed in the CNS and found in intrinsic neurons of the dorsal horn of spinal cord (24). It belongs to the CX3C subfamily (25) and binds to the CX3CR1 receptor. This receptor is mainly expressed by microglia in the CNS (24, 26). FKN, in its membrane-bound form, consists of an extracellular N-terminal chemokine domain, with a mucin-like stalk connecting with the cell membrane plus a transmembrane hydrophobic region and an intracellular C-terminal domain (27). FKN is also found in soluble forms, which contains the mucin stalk and the N-terminal chemokine domain, and it is released by proteolysis at a membrane-proximal region (28). This enzymatic cleavage is mediated by either the TNF-α converting enzyme (TACE, ADAM17) (28) or the metalloprotease ADAM10, which are transmembrane proteins similarly to FKN (29). FKN cleavage from neuronal membranes can also be mediated by the microglial-derived cysteine protease cathepsin S (CatS). Despite these differences in structure between full-length and soluble forms of FKN, the affinity of the chemokine for the CX3CR1 receptor is suggested to be identical (30).
A wealth of data from ours and other groups show that FKN binding to microglial CX3CR1 induces the activation of several downstream signalling pathways, especially the activation of intracellular p38 MAPK pathway that leads to the release of CatS and IL-1β (26, 31). The activation of this pathway is linked to nociceptive facilitation after nerve injury (18, 32–34). Similar outcome has been reported in bone cancer pain models which have a neuropathic pain component (35). Pain development in this model correlates with an increased dorsal horn microgliosis and increased expression of p-p38 in microglia (36). Indeed, this chemokine pair FKN/CX3CR1 is involved in neuropathic pain development and maintenance via neuron-microglia interaction in the dorsal horn, and upregulation of CX3CR1 expression is observed when microgliosis is present (21, 37–40). Despite differences observed, sexual dimorphism in FKN/CX3CR1 pathway in the spinal cord is yet to be established.
Accumulating evidence over the last fifteen years suggest an important role of microglia in the pathogenesis of neuropathic pain (41, 42). Following peripheral nerve injury, upregulation of CX3CR1 (24) is observed in spinal microglia in association with marked mechanical allodynia (43). Thermal hyperalgesia and mechanical allodynia can also be elicited in naïve animals by an intrathecal injection of FKN (44) and both effects are abrogated in CX3CR1 knockout mice (34, 45). In addition, the administration of a neutralizing antibody against CX3CR1 (46, 47) reduces pain-like behaviours in neuropathic pain models, indicating that microglia-mediated mechanisms contribute to nociceptive hypersensitivity. Injection of FKN, after unique binding to CX3CR1, activates p38 MAPK signalling pathway (34). Selective inhibition of p38-MAPK with skepinone or SB20358 through intrathecal delivery reduced mechanical allodynia in male rodent models of neuropathic pain (48) highlighting the role of phosphorylated p38 MAPK in neuropathic pain. In addition, a study published by Bäckryd and co-workers has found that FKN and CatS levels are higher in the cerebrospinal fluid (CSF) of fibromyalgia patients when compared to healthy individuals (49).
Besides a well-established role in neuropathic pain at spinal cord level and increased FKN in the CSF, some reports have elucidated the FKN/CX3CR1 role at a supraspinal level. In the brain, increased microglial expression has been reported in pain-related areas such as the thalamus (50) or the periaqueductal grey area (PAG) (51). In a recent study, an upregulation of CatS, CX3CR1 and FKN mRNA and CX3CR1 protein expression was observed in the ventral posterolateral thalamic nucleus after spinal nerve ligation (SNL) in rodent models (52). This is further supported by a study examining patients suffering from lumbar chronic pain in which evidence for microglial activation in the thalamus is noticed (53). Evidence also shows that intracerebroventricular administration of FKN causes thermal hyperalgesia in rodents and is accompanied by an increase in p38 MAPK phosphorylation (54).
These data provide a better understanding of the pathophysiological processes in the spinal cord and in the brain highlighting the potential of the FKN/CX3CR1 system as a target for the treatment of neuropathic pain.
CatS is a lysosomal enzyme belonging to the papain family of cysteine proteases (55) preferentially expressed in mononuclear phagocytic cells (56). CatS expression has been observed in dendritic cells, B cells, macrophages and microglia, which act as antigen presenting cells (APCs) (57). The activity of CatS is not restricted to intracellular compartments since the release of enzymatically active protease has been observed in a number of cell types, including macrophages and microglia (26). Due to its expression in immune cells and the direct involvement of CatS in antigen presentation, this enzyme has been linked to several autoimmune conditions such as multiple sclerosis (58) and rheumatoid arthritis (59, 60).
Like most cathepsins, CatS is a small and monomeric endopeptidase (61). It is synthesized as an inactive zymogen in the lysosomal compartment (62). After removal of the pro-peptide by other proteases, CatS becomes enzymatically active (63). This protease plays an important role in adaptive immune responses by regulating MHC class II surface expression and by cleaving the invariant chain p10 (Lip10)—a fragment of the MHC class II-associated invariant chain peptide (64). Mice lacking the Ctss gene display diminished MHC class II (MHCII) antigen presentation (65). In comparison to many other cysteine cathepsin family members, CatS tissue expression is very restricted. Biochemically, this endopeptidase has the ability to retain activity at a neutral pH and this property showcases its increased potential to be involved in extracellular proteolytic activities (63).
In addition to the intracellular function, CatS also shows extracellular activity when it is released by macrophages and microglia. As reported by Clark and co-workers, upon release of CatS, the latter interacts with FKN on neurons, cleaving into its soluble form that further binds to the CX3CR1 receptor located on microglia. The activation of this receptor leads to the phosphorylation of p38 MAPK pathway contributing to the release of proinflammatory cytokines, such as IL-1β to the extracellular environment (66). These can activate neighbouring neurons and contribute to increased neuronal excitability (67, 68). However, the CatS/FKN/CX3CR1 signalling pathway is only fully operational in the presence of high concentrations of adenosine tri-phosphate (ATP) which contributes to the activation of the P2X7 receptor (69), reflecting the critical role of ATP to induce the release of CatS (Figure 1).
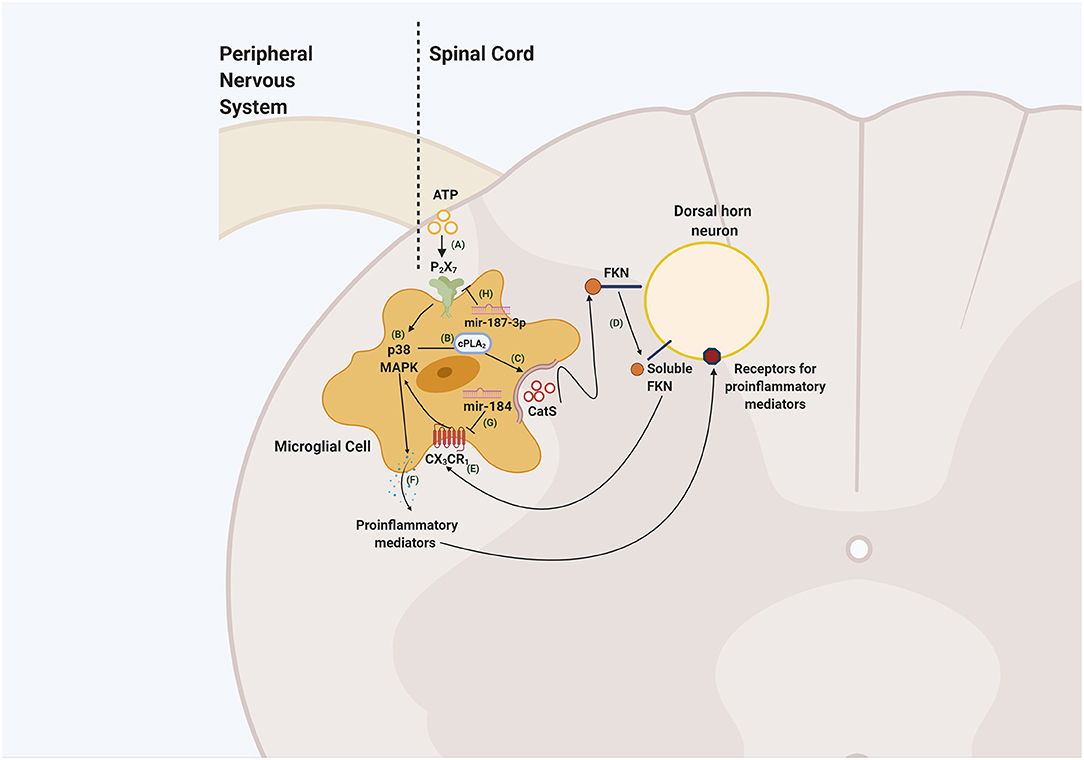
Figure 1. Schematic illustrating neuron-to-glia interaction through the CatS/FKN/CX3CR1 pathway in the spinal cord dorsal horn. ATP released by damaged primary afferents and dorsal horn neurons (A) lead to the activation of P2X7 receptors. The activation of these receptors phosphorylates p38 MAPK signalling pathway and phospholipase A2 (cPLA2) (B) resulting in the release of CatS (C). CatS cleaves FKN present in the membrane of the spinal cord dorsal horn neuron (D). The soluble form of FKN interacts with the CX3CR1 receptor located in microglia (E) that phosphorylates p38 MAPK, induces the release of proinflammatory mediators (F) that may sensitise spinal cord dorsal neurons and contribute to the development of central sensitisation and neuropathic pain. Mediating the expression of mir-184 inhibits the activation of CX3CR1 (G) consequently reducing the release of proinflammatory mediators and microglial activation. On the other hand, upregulating mir-187-3p (H) through the administration of its mimic, downregulates P2X7 expression, potentially disrupting the liberation of CatS. This figure was created with BioRender.com.
CatS expression is upregulated after peripheral nerve injury in the spinal cord dorsal horn and is accompanied by an increase in mechanical sensitivity. This is abrogated by the administration of a neutralizing antibody against FKN suggesting that CatS requires CX3CR1 to exert pro-nociceptive activity (34). This suggestion is further validated by the observation that CatS intrathecally injected in CX3CR1 knockout mice fails to induced and mechanical allodynia (34). In addition, a recent study has shown that one day post peripheral nerve injury CatS mRNA levels are upregulated in the ipsilateral side of the spinal cord and similar observations could be verified after intrathecal administration of colony-stimulating factor 1 (CSF1) (70). This interaction between CSF1 and CatS sheds light into new players that contribute to microglial activation and CatS release. Notably, this further reinforces the idea that CatS is a pro-nociceptive contributor for the central mechanisms underlying neuropathic pain. Despite playing an undeniable pro-nociceptive role centrally, it is important to note that CatS also exerts effects in the periphery by acting on targets such as the protease-activated receptor 2 (PAR2) (71). Activation of this receptor by CatS through enzymatic cleavage has been shown to contribute to preclinical pain (72) and reflects the versatile nature of CatS, emphasizing its powerful potential as a therapeutic target.
Several studies have demonstrated that noncoding RNAs, especially microRNAs, are altered in pain-related regions and these changes are linked with neuropathic pain pathology (73, 74). Several microRNAs have been linked to chemokine signalling. For instance, miR-23a is downregulated in the spinal cord after nerve injury and an increase of the expression of this noncoding RNAs reduces CXCR4 expression and attenuates pain-like behaviours (75). More recently, in a model of bone cancer pain (BCP), the expression of cx3cr1 mRNA expression was upregulated along with increased microglial activation in the spinal cord. Computational analysis revealed that cx3cr1 is a target gene for miR-184 and by activating miR-184, microglial CX3CR1 expression is downregulated (76). Furthermore, in models of ischemia-reperfusion (IR)-induced pain hypersensitivity, downregulation of P2X7 receptor expression by an intrathecal injection of the mimic-187-3p was associated with reduced pain hypersensitivity as well as reduction in cleaved caspase-1 and IL-1β protein levels in the spinal cord (77). By blocking, inactivating, or reducing the expression of P2X7 receptors, our prediction is that CatS release by microglial cells would be halted and, consequently, FKN would not be cleaved into its soluble form (Figure 1). Therefore, miRNAs constitute an innovative and effective strategy to target several players within a pathway involved in neuropathic pain mechanisms.
Based on the emerging appreciation for the role of P2X receptors in mediating nociceptive neurotransmission, several P2X receptors have advanced into clinical trials for inflammation and pain. For instance, intraperitoneal administration of A-438079, a selective competitive P2X7 antagonist and CNS penetrant compound (78), reduces pain-like behaviours in three different rodent animal models of neuropathic pain (79). Regardless of all the promising results obtained in the pre-clinical setting, most of the P2X7 receptor antagonists have not been approved for pain management until today (80).
On the other hand, several pre-clinical studies have reported a successful attenuation of allodynia and hypersensitivity by intrathecal administration of the non-selective CatS inhibitor LHVS as well as following administration of MIV-247, an orally available selective CatS inhibitor that can penetrate the CNS (34, 81). Several other CatS inhibitors (VBY-036 and VBY-891) have gone through Phase I clinical trials and were considered safe for further efficacy studies. Furthermore, the development of a CatS/CatK inhibitor (SAR113137) entered clinical trials for pain management but it was later halted due to initial safety setbacks (82). However successful in preclinical experiments, none of these inhibitors have yet gone through Phase II clinical trials.
At present, molecules targeting P2X7 receptors and CatS have not progressed in clinical trials. However, the CX3CR1 inhibitor AZD8797, which has shown efficacy in models of multiple sclerosis resulting in reduced paralysis, is a good candidate to be considered for treatment of neuropathic pain (83) and its' use for the management of pain may be considered. However, the active involvement of this chemokine pair in other conditions besides chronic pain indicates that pharmacological tools that alter CX3CR1 signalling may result in side effects. For instance, whilst the activation of FKN and/or CX3CR1 signalling may provide novel opportunities for the treatment of Alzheimer's Disease (AD) (84), this does not represent a strategy for neuropathic pain where a blockade of FKN and/or CX3CR1 would be desirable. Even though neurogenerative and chronic pain conditions are both associated with neuroinflammation, including microglial activation, the specific role of the FKN/CX3CR1 signalling pathway in each situation may differ and thus remain to be investigated.
Despite great progress in the study of chemokines and their involvement in the development of pain, especially regarding the FKN/CX3CR1 pair, very few analgesic drugs targeting chemokines have reached later phases of clinical trials. Molecules that target soluble FKN, which is known to mediate nociception, or its respective signalling, may provide reduced side and stronger analgesic effects. Furthermore, targeting upstream regulators of FKN transcription, such as Stat3 (signal transducer and activator of transcription 3) could be explored as a new avenue to regulate FKN expression in neurons.
Considering the paucity of therapies for the treatment of neuropathic pain, we suggest that future studies could investigate the role of CatS/FKN/CX3CR1 in supraspinal areas which may complement research conducted in the spinal cord and in the dorsal root ganglion (DRG). Currently, little information is provided regarding the effect of this signalling pathway in supraspinal areas in a neuropathic pain context. Uncovering the role of FKN and CX3CR1 in the pain-related areas in the brain under neuropathic pain states may aid in the development of innovative therapeutic approaches.
This chemokine system plays an important role in the development of neuropathic pain in preclinical studies. The identification of these neuron-microglia interactions during neuropathic pain states has led to the identification of microglial targets such as the chemokine receptor CX3CR1, the lysosomal protease CatS and the P2X7 receptor. The inhibition or downregulation of these microglial targets, by using different therapeutic tools (inhibitors, miRNAs, etc.) still constitute a powerful tool for addressing whether modulation of this signalling pathway can attenuate neuropathic pain.
All authors contributed to manuscript revision, read, and approved the submitted version.
We acknowledge support by the European Union's Horizon 2020 research and innovation programme TOBeATPAIN under the Marie Skłodowska-Curie grant agreement No 764860.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Colloca L, Ludman T, Bouhassira D, Baron R, Dickenson AH, Yarnitsky D, et al. Neuropathic pain. Nat Rev Dis Primers. (2017) 3:17002. doi: 10.1038/nrdp.2017.2
2. Finnerup NB, Kuner R, Jensen TS. Neuropathic pain: from mechanisms to treatment. Physiol Rev. (2021) 101:259–301. doi: 10.1152/physrev.00045.2019
3. Austin PJ, Moalem-Taylor G. The neuro-immune balance in neuropathic pain: involvement of inflammatory immune cells, immune-like glial cells and cytokines. J Neuroimmunol. (2010) 229:26–50. doi: 10.1016/j.jneuroim.2010.08.013
4. Ji RR, Nackley A, Huh Y, Terrando N, Maixner W. Neuroinflammation and Central sensitization in chronic and widespread pain. Anesthesiology. (2018) 129:343–66. doi: 10.1097/ALN.0000000000002130
5. Costigan M, Scholz J, Woolf CJ. Neuropathic pain: a maladaptive response of the nervous system to damage. Annu Rev Neurosci. (2009) 32:1–32. doi: 10.1146/annurev.neuro.051508.135531
6. Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain. (2009) 10:895–926. doi: 10.1016/j.jpain.2009.06.012
7. Ruscheweyh R, Wilder-Smith O, Drdla R, Liu XG, Sandkühler J. Long-term potentiation in spinal nociceptive pathways as a novel target for pain therapy. Mol Pain. (2011) 7:20. doi: 10.1186/1744-8069-7-20
8. Prescott SA. Synaptic inhibition and disinhibition in the spinal dorsal horn. Prog Mol Biol Transl Sci. (2015) 131:359–83. doi: 10.1016/bs.pmbts.2014.11.008
9. Boadas-Vaello P, Castany S, Homs J, Álvarez-Pérez B, Deulofeu M, Verdú E. Neuroplasticity of ascending and descending pathways after somatosensory system injury: reviewing knowledge to identify neuropathic pain therapeutic targets. Spinal Cord. (2016) 54:330–40. doi: 10.1038/sc.2015.225
10. Watkins LR, Milligan ED, Maier SF. Glial activation: a driving force for pathological pain. Trends Neurosci. (2001) 24:450–5. doi: 10.1016/S0166-2236(00)01854-3
11. McMahon SB, Malcangio M. Current challenges in glia-pain biology. Neuron. (2009) 64:46–54. doi: 10.1016/j.neuron.2009.09.033
12. Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell. (2009) 139:267–84. doi: 10.1016/j.cell.2009.09.028
13. Taves S, Berta T, Chen G, Ji RR. Microglia and spinal cord synaptic plasticity in persistent pain. Neural Plast. (2013) 2013:753656. doi: 10.1155/2013/753656
14. Clark AK, Gentry C, Bradbury EJ, McMahon SB, Malcangio M. Role of spinal microglia in rat models of peripheral nerve injury and inflammation. Eur J Pain. (2007) 11:223–30. doi: 10.1016/j.ejpain.2006.02.003
15. Beggs S, Salter MW. Stereological and somatotopic analysis of the spinal microglial response to peripheral nerve injury. Brain Behav Immun. (2007) 21:624–33. doi: 10.1016/j.bbi.2006.10.017
16. Kawasaki Y, Zhang L, Cheng JK, Ji RR. Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha in regulating synaptic and neuronal activity in the superficial spinal cord. J Neurosci. (2008) 28:5189–94. doi: 10.1523/JNEUROSCI.3338-07.2008
17. Ledeboer A, Sloane EM, Milligan ED, Frank MG, Mahony JH, Maier SF, et al. Minocycline attenuates mechanical allodynia and proinflammatory cytokine expression in rat models of pain facilitation. Pain. (2005) 115:71–83. doi: 10.1016/j.pain.2005.02.009
18. Tsuda M, Mizokoshi A, Shigemoto-Mogami Y, Koizumi S, Inoue K. Activation of p38 mitogen-activated protein kinase in spinal hyperactive microglia contributes to pain hypersensitivity following peripheral nerve injury. Glia. (2004) 45:89–95. doi: 10.1002/glia.10308
19. Sorge RE, Mapplebeck JC, Rosen S, Beggs S, Taves S, Alexander JK, et al. Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat Neurosci. (2015) 18:1081–3. doi: 10.1038/nn.4053
20. Chen G, Luo X, Qadri MY, Berta T, Ji RR. Sex-dependent glial signaling in pathological pain: distinct roles of spinal microglia and astrocytes. Neurosci Bull. (2018) 34:98–108. doi: 10.1007/s12264-017-0145-y
21. Gao YJ, Ji RR. Chemokines, neuronal-glial interactions, and central processing of neuropathic pain. Pharmacol Ther. (2010) 126:56–68. doi: 10.1016/j.pharmthera.2010.01.002
22. Ramesh G, MacLean AG, Philipp MT. Cytokines and chemokines at the crossroads of neuroinflammation, neurodegeneration, and neuropathic pain. Mediators Inflamm. (2013) 2013:480739. doi: 10.1155/2013/480739
23. Montague K, Malcangio M. The therapeutic potential of targeting chemokine signalling in the treatment of chronic pain. J Neurochem. (2017) 141:520–31. doi: 10.1111/jnc.13927
24. Verge GM, Milligan ED, Maier SF, Watkins LR, Naeve GS, Foster AC. Fractalkine (CX3CL1) and fractalkine receptor (CX3CR1) distribution in spinal cord and dorsal root ganglia under basal and neuropathic pain conditions. Eur J Neurosci. (2004) 20:1150–60. doi: 10.1111/j.1460-9568.2004.03593.x
25. Pan Y, Lloyd C, Zhou H, Dolich S, Deeds J, Gonzalo JA, et al. Neurotactin, a membrane-anchored chemokine upregulated in brain inflammation. Nature. (1997) 387:611–7. doi: 10.1038/42491
26. Clark AK, Yip PK, Malcangio M. The liberation of fractalkine in the dorsal horn requires microglial cathepsin S. J Neurosci. (2009) 29:6945–54. doi: 10.1523/JNEUROSCI.0828-09.2009
27. Fong AM, Robinson LA, Steeber DA, Tedder TF, Yoshie O, Imai T, et al. Fractalkine and CX3CR1 mediate a novel mechanism of leukocyte capture, firm adhesion, and activation under physiologic flow. J Exp Med. (1998) 188:1413–9. doi: 10.1084/jem.188.8.1413
28. Garton KJ, Gough PJ, Blobel CP, Murphy G, Greaves DR, Dempsey PJ, et al. Tumor necrosis factor-alpha-converting enzyme (ADAM17) mediates the cleavage and shedding of fractalkine (CX3CL1). J Biol Chem. (2001) 276:37993–8001. doi: 10.1074/jbc.M106434200
29. Hundhausen C, Misztela D, Berkhout TA, Broadway N, Saftig P, Reiss K, et al. The disintegrin-like metalloproteinase ADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1-mediated cell-cell adhesion. Blood. (2003) 102:1186–95. doi: 10.1182/blood-2002-12-3775
30. Harrison JK, Fong AM, Swain PA, Chen S, Yu YR, Salafranca MN, et al. Mutational analysis of the fractalkine chemokine domain. Basic amino acid residues differentially contribute to CX3CR1 binding, signaling, and cell adhesion. J Biol Chem. (2001) 276:21632–41. doi: 10.1074/jbc.M010261200
31. Clark AK, Staniland AA, Malcangio M. Fractalkine/CX3CR1 signalling in chronic pain and inflammation. Curr Pharm Biotechnol. (2011) 12:1707–14. doi: 10.2174/138920111798357465
32. Jin SX, Zhuang ZY, Woolf CJ, Ji RR. p38 mitogen-activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. J Neurosci. (2003) 23:4017–22. doi: 10.1523/JNEUROSCI.23-10-04017.2003
33. Svensson CI, Marsala M, Westerlund A, Calcutt NA, Campana WM, Freshwater JD, et al. Activation of p38 mitogen-activated protein kinase in spinal microglia is a critical link in inflammation-induced spinal pain processing. J Neurochem. (2003) 86:1534–44. doi: 10.1046/j.1471-4159.2003.01969.x
34. Clark AK, Yip PK, Grist J, Gentry C, Staniland AA, Marchand F, et al. Inhibition of spinal microglial cathepsin S for the reversal of neuropathic pain. Proc Natl Acad Sci USA. (2007) 104:10655–60. doi: 10.1073/pnas.0610811104
35. Zajaczkowska R, Kocot-Kepska M, Leppert W, Wordliczek J. Bone pain in cancer patients: mechanisms and current treatment. Int J Mol Sci. (2019) 20:6047. doi: 10.3390/ijms20236047
36. Hu JH, Yang JP, Liu L, Li CF, Wang LN, Ji FH, et al. Involvement of CX3CR1 in bone cancer pain through the activation of microglia p38 MAPK pathway in the spinal cord. Brain Res. (2012) 1465:1–9. doi: 10.1016/j.brainres.2012.05.020
37. Clark AK, Malcangio M. Fractalkine/CX3CR1 signaling during neuropathic pain. Front Cell Neurosci. (2014) 8:121. doi: 10.3389/fncel.2014.00121
38. Abbadie C, Bhangoo S, De Koninck Y, Malcangio M, Melik-Parsadaniantz S, White FA. Chemokines and pain mechanisms. Brain Res Rev. (2009) 60:125–34. doi: 10.1016/j.brainresrev.2008.12.002
39. Old EA, Malcangio M. Chemokine mediated neuron-glia communication and aberrant signalling in neuropathic pain states. Curr Opin Pharmacol. (2012) 12:67–73. doi: 10.1016/j.coph.2011.10.015
40. Milligan ED, Sloane EM, Watkins LR. Glia in pathological pain: a role for fractalkine. J Neuroimmunol. (2008) 198:113–20. doi: 10.1016/j.jneuroim.2008.04.011
41. Inoue K, Tsuda M. Microglia in neuropathic pain: cellular and molecular mechanisms and therapeutic potential. Nat Rev Neurosci. (2018) 19:138–52. doi: 10.1038/nrn.2018.2
42. Zhou LJ, Peng J, Xu YN, Zeng WJ, Zhang J, Wei X, et al. Microglia Are Indispensable for Synaptic Plasticity in the Spinal Dorsal Horn and Chronic Pain. Cell Rep. (2019) 27:3844–59.e6. doi: 10.1016/j.celrep.2019.05.087
43. Zhuang ZY, Kawasaki Y, Tan PH, Wen YR, Huang J, Ji RR. Role of the CX3CR1/p38 MAPK pathway in spinal microglia for the development of neuropathic pain following nerve injury-induced cleavage of fractalkine. Brain Behav Immun. (2007) 21:642–51. doi: 10.1016/j.bbi.2006.11.003
44. Milligan E, Zapata V, Schoeniger D, Chacur M, Green P, Poole S, et al. An initial investigation of spinal mechanisms underlying pain enhancement induced by fractalkine, a neuronally released chemokine. Eur J Neurosci. (2005) 22:2775–82. doi: 10.1111/j.1460-9568.2005.04470.x
45. Staniland AA, Clark AK, Wodarski R, Sasso O, Maione F, D'Acquisto F, et al. Reduced inflammatory and neuropathic pain and decreased spinal microglial response in fractalkine receptor (CX3CR1) knockout mice. J Neurochem. (2010) 114:1143–57. doi: 10.1111/j.1471-4159.2010.06837.x
46. Holmes FE, Arnott N, Vanderplank P, Kerr NC, Longbrake EE, Popovich PG, et al. Intra-neural administration of fractalkine attenuates neuropathic pain-related behaviour. J Neurochem. (2008) 106:640–9. doi: 10.1111/j.1471-4159.2008.05419.x
47. Sessler K, Blechschmidt V, Hoheisel U, Mense S, Schirmer L, Treede RD. Spinal cord fractalkine (CX3CL1) signaling is critical for neuronal sensitization in experimental nonspecific, myofascial low back pain. J Neurophysiol. (2021) 125:1598–611. doi: 10.1152/jn.00348.2020
48. Taves S, Berta T, Liu DL, Gan S, Chen G, Kim YH, et al. Spinal inhibition of p38 MAP kinase reduces inflammatory and neuropathic pain in male but not female mice: Sex-dependent microglial signaling in the spinal cord. Brain Behav Immun. (2016) 55:70–81. doi: 10.1016/j.bbi.2015.10.006
49. Bäckryd E, Tanum L, Lind AL, Larsson A, Gordh T. Evidence of both systemic inflammation and neuroinflammation in fibromyalgia patients, as assessed by a multiplex protein panel applied to the cerebrospinal fluid and to plasma. J Pain Res. (2017) 10:515–25. doi: 10.2147/JPR.S128508
50. LeBlanc BW, Zerah ML, Kadasi LM, Chai N, Saab CY. Minocycline injection in the ventral posterolateral thalamus reverses microglial reactivity and thermal hyperalgesia secondary to sciatic neuropathy. Neurosci Lett. (2011) 498:138–42. doi: 10.1016/j.neulet.2011.04.077
51. Dubový P, Klusáková I, Hradilová-SvíŽenská I, Joukal M, Boadas-Vaello P. Activation of astrocytes and microglial cells and CCL2/CCR2 upregulation in the dorsolateral and ventrolateral nuclei of periaqueductal gray and rostral ventromedial medulla following different types of sciatic nerve injury. Front Cell Neurosci. (2018) 12:40. doi: 10.3389/fncel.2018.00040
52. Blaszczyk L, Maître M, Lesté-Lasserre T, Clark S, Cota D, Oliet SHR, et al. Sequential alteration of microglia and astrocytes in the rat thalamus following spinal nerve ligation. J Neuroinflammation. (2018) 15:349. doi: 10.1186/s12974-018-1378-z
53. Loggia ML, Chonde DB, Akeju O, Arabasz G, Catana C, Edwards RR, et al. Evidence for brain glial activation in chronic pain patients. Brain. (2015) 138 (Pt 3):604–15. doi: 10.1093/brain/awu377
54. Wang A, Yang T, Zhang L, Jia L, Wu Q, Yao S, et al. IP3-Mediated Calcium Signaling Is Involved in the Mechanism of Fractalkine-Induced Hyperalgesia Response. Med Sci Monit. (2018) 24:8804–11. doi: 10.12659/MSM.913787
55. Kirschke H, Schmidt I, Wiederanders B. Cathepsin S. The cysteine proteinase from bovine lymphoid tissue is distinct from cathepsin L (EC 3.4.22.15). Biochem J. (1986) 240:455–9. doi: 10.1042/bj2400455
56. Petanceska S, Canoll P, Devi LA. Expression of rat cathepsin S in phagocytic cells. J Biol Chem. (1996) 271:4403–9. doi: 10.1074/jbc.271.8.4403
57. Wiener JJ, Sun S, Thurmond RL. Recent advances in the design of cathepsin S inhibitors. Curr Top Med Chem. (2010) 10:717–32. doi: 10.2174/156802610791113432
58. Haves-Zburof D, Paperna T, Gour-Lavie A, Mandel I, Glass-Marmor L, Miller A. Cathepsins and their endogenous inhibitors cystatins: expression and modulation in multiple sclerosis. J Cell Mol Med. (2011) 15:2421–9. doi: 10.1111/j.1582-4934.2010.01229.x
59. Schurigt U, Stopfel N, Hückel M, Pfirschke C, Wiederanders B, Bräuer R. Local expression of matrix metalloproteinases, cathepsins, and their inhibitors during the development of murine antigen-induced arthritis. Arthritis Res Ther. (2005) 7:R174–88. doi: 10.1186/ar1466
60. Clark AK, Grist J, Al-Kashi A, Perretti M, Malcangio M. Spinal cathepsin S and fractalkine contribute to chronic pain in the collagen-induced arthritis model. Arthritis Rheum. (2012) 64:2038–47. doi: 10.1002/art.34351
61. Montague-Cardoso K, Malcangio M. Cathepsin S as a potential therapeutic target for chronic pain. Med Drug Discov. (2020) 7:100047. doi: 10.1016/j.medidd.2020.100047
62. Vasiljeva O, Reinheckel T, Peters C, Turk D, Turk V, Turk B. Emerging roles of cysteine cathepsins in disease and their potential as drug targets. Curr Pharm Des. (2007) 13:387–403. doi: 10.2174/138161207780162962
63. Vasiljeva O, Dolinar M, Pungercar JR, Turk V, Turk B. Recombinant human procathepsin S is capable of autocatalytic processing at neutral pH in the presence of glycosaminoglycans. FEBS Lett. (2005) 579:1285–90. doi: 10.1016/j.febslet.2004.12.093
64. Thanei S, Theron M, Silva AP, Reis B, Branco L, Schirmbeck L, et al. Cathepsin S inhibition suppresses autoimmune-triggered inflammatory responses in macrophages. Biochem Pharmacol. (2017) 146:151–64. doi: 10.1016/j.bcp.2017.10.001
65. Nakagawa TY, Brissette WH, Lira PD, Griffiths RJ, Petrushova N, Stock J, et al. Impaired invariant chain degradation and antigen presentation and diminished collagen-induced arthritis in cathepsin S null mice. Immunity. (1999) 10:207–17. doi: 10.1016/S1074-7613(00)80021-7
66. Clark AK, Staniland AA, Marchand F, Kaan TK, McMahon SB, Malcangio M. P2X7-dependent release of interleukin-1beta and nociception in the spinal cord following lipopolysaccharide. J Neurosci. (2010) 30:573–82. doi: 10.1523/JNEUROSCI.3295-09.2010
67. Clark AK, Gruber-Schoffnegger D, Drdla-Schutting R, Gerhold KJ, Malcangio M, Sandkühler J. Selective activation of microglia facilitates synaptic strength. J Neurosci. (2015) 35:4552–70. doi: 10.1523/JNEUROSCI.2061-14.2015
68. Yan X, Weng HR. Endogenous interleukin-1β in neuropathic rats enhances glutamate release from the primary afferents in the spinal dorsal horn through coupling with presynaptic N-methyl-D-aspartic acid receptors. J Biol Chem. (2013) 288:30544–57. doi: 10.1074/jbc.M113.495465
69. Clark AK, Wodarski R, Guida F, Sasso O, Malcangio M. Cathepsin S release from primary cultured microglia is regulated by the P2X7 receptor. Glia. (2010) 58:1710–26. doi: 10.1002/glia.21042
70. Guan Z, Kuhn JA, Wang X, Colquitt B, Solorzano C, Vaman S, et al. Injured sensory neuron-derived CSF1 induces microglial proliferation and DAP12-dependent pain. Nat Neurosci. (2016) 19:94–101. doi: 10.1038/nn.4189
71. Chung K, Pitcher T, Grant AD, Hewitt E, Lindstrom E, Malcangio M. Cathepsin S acts via protease-activated receptor 2 to activate sensory neurons and induce itch-like behaviour. Neurobiol Pain. (2019) 6:100032. doi: 10.1016/j.ynpai.2019.100032
72. Zhao P, Lieu T, Barlow N, Metcalf M, Veldhuis NA, Jensen DD, et al. Cathepsin S causes inflammatory pain via biased agonism of PAR2 and TRPV4. J Biol Chem. (2014) 289:27215–34. doi: 10.1074/jbc.M114.599712
73. Tavares-Ferreira D, Lawless N, Bird EV, Atkins S, Collier D, Sher E, et al. Correlation of miRNA expression with intensity of neuropathic pain in man. Mol Pain. (2019) 15:1744806919860323. doi: 10.1177/1744806919860323
74. Simeoli R, Montague K, Jones HR, Castaldi L, Chambers D, Kelleher JH, et al. Exosomal cargo including microRNA regulates sensory neuron to macrophage communication after nerve trauma. Nat Commun. (2017) 8:1778. doi: 10.1038/s41467-017-01841-5
75. Pan Z, Shan Q, Gu P, Wang XM, Tai LW, Sun M, et al. miRNA-23a/CXCR4 regulates neuropathic pain via directly targeting TXNIP/NLRP3 inflammasome axis. J Neuroinflammation. (2018) 15:29. doi: 10.1186/s12974-018-1073-0
76. Wang A, Tie M, Guo D, Wu N, Yao S, Yan L, et al. A Novel mechanism of BAM8-22 inhibiting microglia activation: represses CX3CR1 expression via upregulating miR-184. J Mol Neurosci. (2020) 70:550–8. doi: 10.1007/s12031-019-01455-0
77. Li XQ, Yu Q, Zhang ZL, Sun XJ, Ma H. MiR-187-3p mimic alleviates ischemia-reperfusion-induced pain hypersensitivity through inhibiting spinal P2X7R and subsequent mature IL-1β release in mice. Brain Behav Immun. (2019) 79:91–101. doi: 10.1016/j.bbi.2019.05.021
78. Nadal-Nicolás FM, Galindo-Romero C, Valiente-Soriano FJ, Barberà-Cremades M, deTorre-Minguela C, Salinas-Navarro M, et al. Involvement of P2X7 receptor in neuronal degeneration triggered by traumatic injury. Sci Rep. (2016) 6:38499. doi: 10.1038/srep38499
79. McGaraughty S, Chu KL, Namovic MT, Donnelly-Roberts DL, Harris RR, Zhang XF, et al. P2X7-related modulation of pathological nociception in rats. Neuroscience. (2007) 146:1817–28. doi: 10.1016/j.neuroscience.2007.03.035
80. Park JH, Kim YC. P2X7 receptor antagonists: a patent review (2010–2015) Expert Opin Ther Pat (2017) 27:257–67. doi: 10.1080/13543776.2017.1246538
81. Hewitt E, Pitcher T, Rizoska B, Tunblad K, Henderson I, Sahlberg BL, et al. Selective cathepsin S inhibition with MIV-247 attenuates mechanical allodynia and enhances the antiallodynic effects of gabapentin and pregabalin in a mouse model of neuropathic pain. J Pharmacol Exp Ther. (2016) 358:387–96. doi: 10.1124/jpet.116.232926
82. Knezevic NN, Cicmil N, Knezevic I, Candido KD. Discontinued neuropathic pain therapy between 2009–2015. Expert Opin Investig Drugs. (2015) 24:1631–46. doi: 10.1517/13543784.2015.1099627
83. Ridderstad Wollberg A, Ericsson-Dahlstrand A, Juréus A, Ekerot P, Simon S, Nilsson M, et al. Pharmacological inhibition of the chemokine receptor CX3CR1 attenuates disease in a chronic-relapsing rat model for multiple sclerosis. Proc Natl Acad Sci USA. (2014) 111:5409–14. doi: 10.1073/pnas.1316510111
Keywords: neuropathic pain, spinal cord, microglia, fractalkine, cathepsin S, CX3CR1
Citation: Silva R and Malcangio M (2021) Fractalkine/CX3CR1 Pathway in Neuropathic Pain: An Update. Front. Pain Res. 2:684684. doi: 10.3389/fpain.2021.684684
Received: 23 March 2021; Accepted: 30 June 2021;
Published: 27 July 2021.
Edited by:
Xuehai Guan, Guangxi Medical University, ChinaReviewed by:
Peter Michael Grace, University of Texas MD Anderson Cancer Center, United StatesCopyright © 2021 Silva and Malcangio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marzia Malcangio, marzia.malcangio@kcl.ac.uk
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.