
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Ophthalmol. , 04 July 2024
Sec. Retina
Volume 4 - 2024 | https://doi.org/10.3389/fopht.2024.1410874
This article is part of the Research Topic Retinal metabolism in health and disease View all 7 articles
 Sudhat Ashok1
Sudhat Ashok1 Sriganesh Ramachandra Rao1,2,3*
Sriganesh Ramachandra Rao1,2,3*Membrane-anchored proteins play critical roles in cell signaling, cellular architecture, and membrane biology. Hydrophilic proteins are post-translationally modified by a diverse range of lipid molecules such as phospholipids, glycosylphosphatidylinositol, and isoprenes, which allows their partition and anchorage to the cell membrane. In this review article, we discuss the biochemical basis of isoprenoid synthesis, the mechanisms of isoprene conjugation to proteins, and the functions of prenylated proteins in the neural retina. Recent discovery of novel prenyltransferases, prenylated protein chaperones, non-canonical prenylation-target motifs, and reversible prenylation is expected to increase the number of inherited systemic and blinding diseases with aberrant protein prenylation. Recent important investigations have also demonstrated the role of several unexpected regulators (such as protein charge, sequence/protein-chaperone interaction, light exposure history) in the photoreceptor trafficking of prenylated proteins. Technical advances in the investigation of the prenylated proteome and its application in vision research are discussed. Clinical updates and technical insights into known and putative prenylation-associated retinopathies are provided herein. Characterization of non-canonical prenylation mechanisms in the retina and retina-specific prenylated proteome is fundamental to the understanding of the pathogenesis of protein prenylation-associated inherited blinding disorders.
Post-translational modifications (PTMs) of proteins are covalent chemical modifications of target peptide motifs which influences protein folding, structure, stability, localization, protein-protein interactions, and function (1, 2). Protein lipidation is a class of PTMs involving enzymatic conjugation with various lipid molecules such as cholesterol (3), phospholipids (4), glycosylphophatidylionositine (GPI), and isoprenes (5). Protein lipidation facilitates protein anchorage to the plasma membrane, protein-protein interactions, and membrane-cytoskeleton interaction (6, 7). The focus of this review article includes isoprene-modification of proteins, and inherited retinal dystrophies with underlying protein prenylation defects.
Protein prenylation involves thioether linkage of an isoprenoid molecule, either a 15-carbon farnesyl pyrophosphate (FPP) or a 20-carbon geranylgeranyl pyrophosphate (GGPP), to one or more cysteine residues of a carboxy terminal target peptide motif (8–10). The biological role of prenylation is best known in the context of cancer biology. Ras GTPases, well-known prenylated protein targets, play critical roles in signal transduction, cell growth, and are implicated in various forms of cancer and cancer progression. For review of Ras prenylation, the reader is referred to the following review articles (11, 12).
In this review, we aim to provide a thorough background of protein prenylation mechanisms, recent advances in approaches to study protein prenylation, and to discuss the role of prenylated proteins in retinal physiology and pathology. We also discuss putative approaches for determining prenylated proteome in the retina.
The lipid moieties required for protein prenylation, FPP and GGPP, are key isoprenoid intermediates of the mevalonate/cholesterol synthesis pathway (Figure 1). Briefly, acetyl-CoA is converted to HMG-CoA in a sequential manner catalyzed by ACAT1, ACAT2, and HMG-CoA synthase 1 (HMGCS1), which is then converted to mevalonate by HMG-CoA reductase (HMGCR) (13). HMGCR is the rate-limiting step in the mevalonate pathway and is the mechanistic target of statins, a commonly used drug used in the treatment of hypertension and hypercholesteremia (14). Mevalonate kinase (MVK) catalyzes the phosphorylation of mevalonic acid to phosphomevalonate and is subject to feedback inhibition by downstream isoprene intermediates like geranylpyrophosphate (GPP), FPP, and GGPP (15). Phosphomevalonate is converted to isopentenyl pyrophosphate (IPP) by phosphorylation and decarboxylation, catalyzed by phosphomevalonate kinase (PMK) and mevalonate pyrophosphate decarboxylase (MPDC), respectively (16). IPP undergoes reversible isomerization to dimethylallyl pyrophosphate (DMAPP). The condensation of IPP and DMAPP results in the 10-carbon isoprenoid geranyl pyrophosphate (GPP). Subsequent condensation of 1 or 2 more IPP units to GPP yields the 15-carbon and the 20-carbon isoprenoids FPP and GGPP, respectively (17). Key mevalonate pathway intermediates IPP, FPP, and GGPP feed into the synthesis of squalene, dolichols, ubiquinone, sterols and derivative steroid hormones, as well as protein prenylation (18). A schematic representation of isoprene synthesis through the mevalonate pathway is provided in Figure 1.
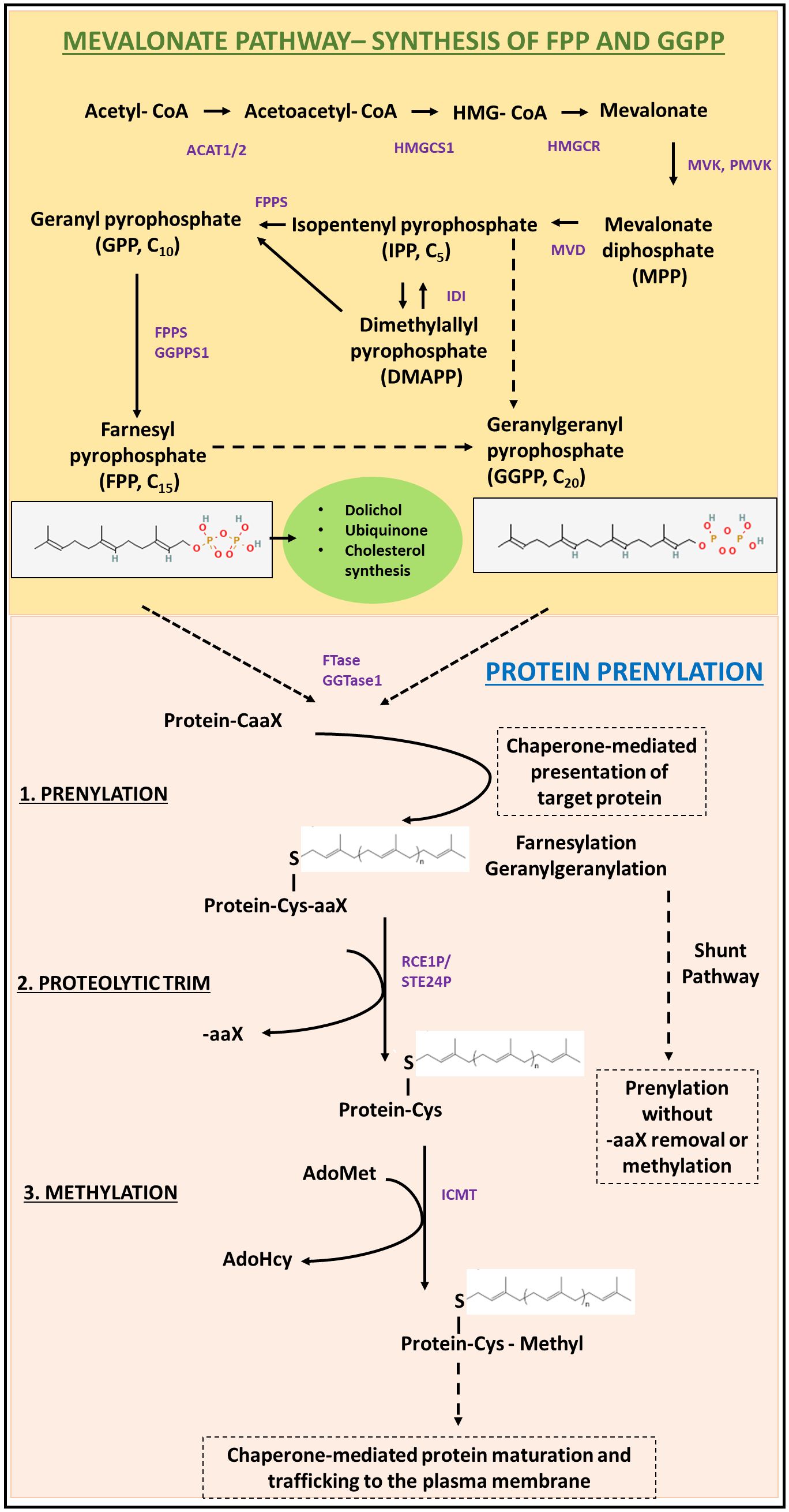
Figure 1 Schematic representation of the biochemical requirements and processes involved in protein prenylation. The mevalonate/cholesterol synthesis pathway is required for the generation of the isoprene moieties (FPP and GGPP) required for protein prenylation. A detailed description of the pre-squalene pathway essential for de novo synthesis of isoprenes has been described in Section 1.1. The prenyl groups are then conjugated to target motifs by the action of prenyltransferases (FTase or GGTase). Following prenylation, the target protein undergoes sequential proteolysis (by RCE1), and subsequently methylated before protein maturation and chaperone-mediated membrane trafficking. Alternatively, some prenylated proteins remain cytosolic, through the shunt pathway; without further trafficking to the plasma membrane.
After isoprenoid generation, irreversible prenylation is catalyzed by protein farnesyltransferase (FTase) or protein geranylgeranyltransferase type I (GGTase-I), which differ in target carboxy-terminal signal peptide recognition and the isoprenoid substrate (9). FTase catalyzes the addition of farnesyl pyrophosphate (FPP), while GGTase-I catalyzes the addition of geranylgeranyl pyrophosphate (GGPP, Figure 1) (8–10, 19, 20). The C-terminal recognition motif for prenylation is the CAAX (also referred to as Ca1a2X) domain, in which ‘C’ is the cysteine to be prenylated, ‘A’ is any aliphatic amino acid, and ‘X’ is an amino acid which determines enzyme specificity (8, 21, 22). The target motifs for FTase typically have serine, methionine, alanine, or glutamine at the X residue position, while GGTase-I usually recognizes leucine or phenylalanine. Some substrates may also be recognized by both enzymes as targets for prenylation (23–26). Recent studies have shown that proteins terminating in C(X)3X and CXX may also potentially be substrates of FTase, where X represents any amino acid (27–29).
Following prenylation at the cysteine residue in the target motif, most prenylated proteins then undergo two additional modifications before transport to the cell membrane. First, the terminal -AAX residues are cleaved by the CAAX proteases Ras converting enzyme 1 (RCE1P) or zinc metallopeptidase (STE24). The resulting carboxylate then undergoes methylation by the S-adenosylmethionine-dependent isoprenylcysteine methyltransferase (ICMT) to form a post-translationally modified prenylcysteine methyl ester C-terminus on the protein (30, 31) (Figure 1). The three steps (prenylation, proteolytic cleavage, and methylation) described above are necessary for function of most prenylated proteins but recently it has been found that heat shock protein chaperone Ydj1p in yeast undergoes prenylation without subsequent proteolysis or methylation in what has been deemed the “shunt pathway”, with evidence of these processing steps actually being deleterious to the protein’s function (32).
The other enzyme responsible for the transfer of 20-carbon geranylgeranyl-group from GGPP to the target motif is termed geranylgeranyltransferase type II (GGTase-II). GGTase-II is unique in that it can modify diverse C-terminal motifs from the Rab family of proteins including CCXX, CXC, or XXCC, where cysteine residues undergo geranylgeranylation and X represents any amino acid (33–36). GGTase-II also requires Rab escort protein (REP) for substrate recognition. REP binds to the substrate motif to be prenylated and presents the catalytic site of GGTase-II for prenylation (37). Recently, a fourth non-canonical prenyltransferase, GGTase-III, was identified which adds a second isoprenoid geranylgeranyl group onto a pre-farnesylated protein substrate with a -CCFarAIM C-terminal motif (38, 39). Interestingly, a chemical proteomic study described the discovery of non-canonical prenylated protein, ALDH9A1, which lacks any classic prenylation motif (40). Moreover, this modification was found to be reversible and not inhibited by known prenyltransferase inhibitors suggesting the possibility of a yet-to-be identified prenyltransferase (40). Single cell RNA seq data analysis of the developing mouse retina (41, 42) suggests the expression of this non-canonical prenylated protein in majority of cell types in the developing retina. Taken together, the discoveries of non-canonical C-terminal sequences for FTase recognition, GGTase-III, and the possibility of other non-canonical prenyltransferases have challenged our understanding of this modification and have greatly expanded the potential pool of prenylated proteins.
The mechanistic understanding of the isoprene substrate (FPP vs. GGPP) specificity of prenyltransferases stems from structural studies, structure-function studies, peptide library studies, computational approaches, radiolabeling, and affinity tagging approaches (22, 43–45). Both FTase and GGTase-I are heterodimeric metalloenzymes consisting of an identical α subunit and distinct, homologous β subunits (46–48). The isoprene-binding site of both enzymes is constituted by the interface of the α and β subunits, and predominantly consists of amino acid residues of the β subunit. The isoprene binding pocket of both GGTase-I and FTase is lined with conserved aromatic residues. The selectivity of FTase for the shorter FPP (5 carbon) substrate is explained by the presence of bulky tryptophan and tyrosine residues of the β subunit (W102 and Y365), within the FPP binding pocket. This blocks the binding of the larger 20-carbon GGPP prenyl moiety (47).
To identify the prenyltransferase enzyme and target protein interactions responsible for substrate selectivity, a series of peptide substrates were co-crystallized with each prenyltransferase (26). This study revealed that the various CAAX sequences bind and interact along one face of the funnel-shaped peptide-binding site in both prenyltransferases except for the C-terminal X residue. FTase utilizes two complementary binding pockets for the X residue depending on the identity of that residue. For example, an X residue of S, Q and M interacts with a binding pocket comprised of Y131α, A98β, S99β, W102β, H149β, A151β and P152β, while F, L, H and N residues interact with a separate binding pocket comprised of residues L96, S99, W102, W106, A151 of the β subunit (26). GGTase-I differs greatly in its X residue specificity in that it is composed of one binding pocket which contains the residues T49, H121, A123, F174 of its β subunit. GGTase-I appears to prefer hydrophobic residues at the X position in its target substrates (26). Taken together, differential binding of prenyltransferases for the X residue in the CAAX domain, and the presence or absence of bulky residues in the isoprene-binding site of FTase and GGTase-I underlie the difference between protein farnesylation and geranylgeranylation.
The discovery of farnesylation of rod transducin gamma subunit in the early 1990s (49) spurred a series of protein biochemistry and pharmacological studies investigating protein prenylation in the retina, especially photoreceptor (PR) cells. Investigations of protein prenylation-associated retinopathies (discussed in sections below) have also provided critical insights into the retinal functions of prenylated proteins. We first discuss some important pharmacological and protein engineering approaches that have provided general insights into the role and requirement of protein prenylation in the neural retina.
Several upstream inhibitors of mevalonate pathway are known to suppress protein prenylation by depleting the cellular isoprene pool. The best-known class of drugs that impact protein prenylation are statins. Statins block HMG-CoA reductase activity, the rate limiting step thus decreasing the pathway flux, and thereby decreasing the levels of intermediate isoprenes. Statins mimic the natural substrate molecule, HMG-CoA, and competitively inhibit HMG-CoA reductase (50). Statins therefore inhibit the synthesis of a broad range of important isoprenoid metabolites. Intravitreal injection of lovastatin led to early changes in the structural organization of the neural retina characterized by formation of rosette-like arrangements of PRs and eventually necrosis of the retina by 4 days (51, 52). Surprisingly, pharmacological inhibition of downstream squalene epoxidase in the neural retina, by intravitreal injection of NB-598 did not lead to acute retinal dystrophy despite cholesterol synthesis inhibition (51, 52). Retinal sensitivity to intravitreal injection of lovastatin was found to be due to defective protein prenylation in the retina, suggesting an indispensable role of protein prenylation in retinal development (51, 52).
Other small molecule inhibitors of mevalonate pathway targeting farnesyl diphosphate synthase (FPPS) and geranylgeranyl diphosphate synthase (GGPPS) like the nitrogen-containing bisphosphonates (N-BPs) and their analogues have been studied in the setting of lytic bone disease and cancer (53, 54). Prenyltransferases have been extensively targeted for developing cancer therapeutic compounds such as tipifarnib (FTase inhibitor) and GGTI-2418 (GGTase-I inhibitor) (55–57). The effect of such inhibitors in the developing and mature retina has not been studied, and may provide key insights into the function of prenylated proteins in retinal development and/or retinal pathologies. Post-prenylation enzymes RCE1P, STE24, and ICMT inhibitors have also been of interest as cancer therapeutic strategies (58), but their effects on the retinal structure and function remains to be investigated. Conditional gene deletion strategies or small molecule inhibitor studies are necessary to further elucidate the role and requirement of FTase, GGTase-I, FPPS, GGPPS, and post-prenylation processing enzymes in the maintenance of normal retinal structure and function. Given that protein prenylation is essential in all tissues and cell types, including those in the retina, one may expect pan-retinal expression of critical players of protein farnesylation and geranylgeranylation. We used publicly available datasets and transcriptome datamining tools to demonstrate the expression of key players of protein prenylation pathway in essentially all retinal cell types of the developing murine retina (41, 42) (Figure 2).
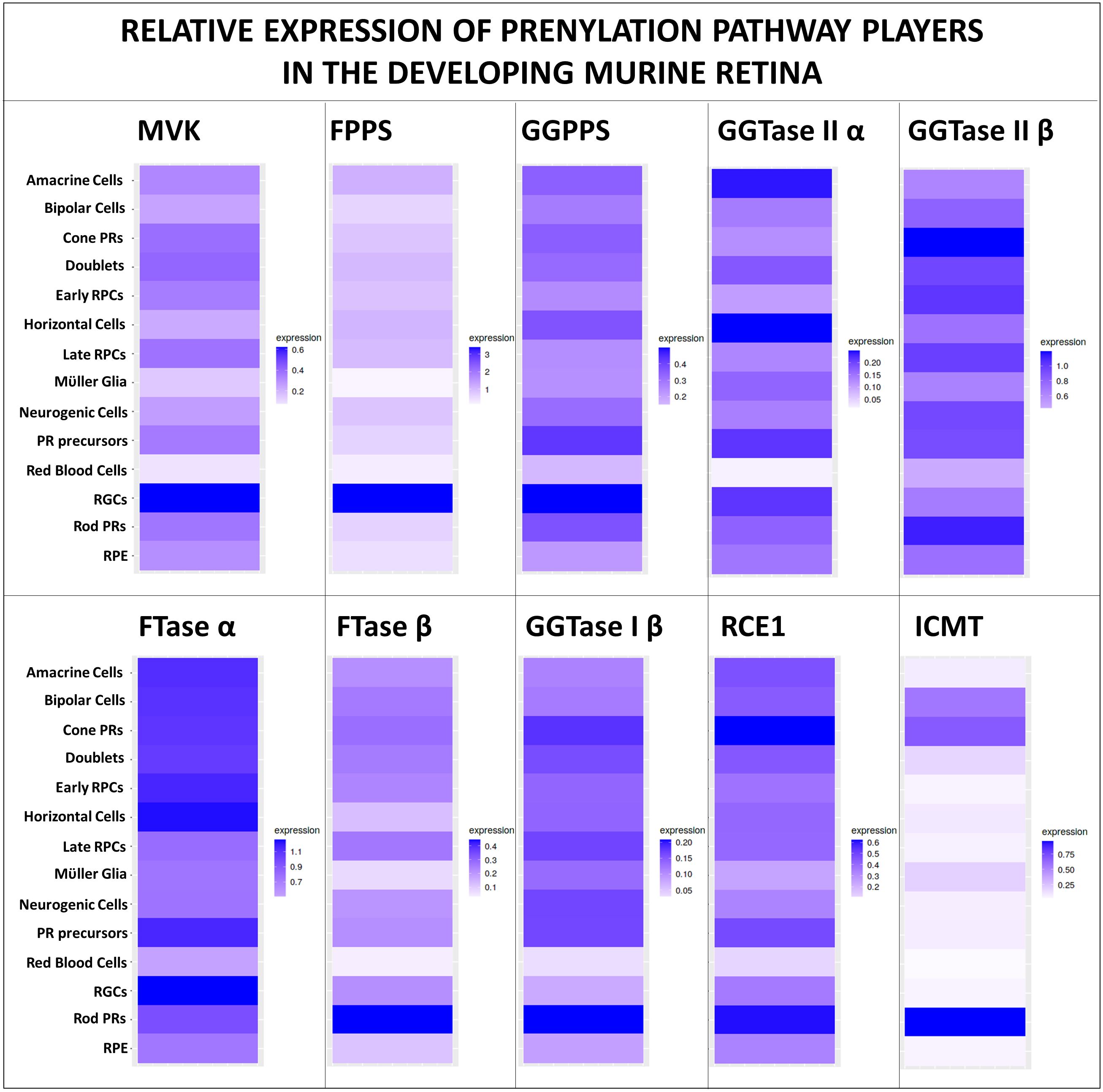
Figure 2 Expression of protein prenylation players in the developing retina. Prenylation is necessary for the maintenance of all retinal cell types, and hence the maintenance of normal retinal structure and function. Query of publicly available mouse retinal single-cell RNAseq dataset (41) using a simple, user-friendly, open source RNA-seq data mining platform (42) demonstrates the expression of key mevalonate pathway players, prenyltransferase subunits, proteases, methyltransferase in essentially all cell types of the developing murine retina (between E11 to PN 8 days).
The mechanism behind the distribution of prenylated targets in rod PRs was explored by Maza and coworkers using chimeric fluorescent probes consisting of different prenyl groups (farnesyl vs. geranylgeranyl), and charged or neutral amino acids upstream of the prenylation site (59). It was shown that prenylated fluorescent protein probes exhibit weak membrane binding and are dynamic in their distribution, suggesting that the membrane compartmentalization mechanisms are not dependent on the nature of the lipid moiety alone (59). Endogenously prenylated proteins may be targeted more efficiently to the cell membrane by interacting with dedicated chaperones. The connecting cilium is suggested to act as an active sorting platform, facilitating the interaction of the target prenylated protein with prenyl-binding chaperones like PrBPδ, for efficient trafficking to the rod outer segment (59). Positively charged prenylated proteins were enriched in the synapse and inner segment (IS) compared to their negatively charged counterparts (59). The absence of positively charged prenylated probes from the outer segment (OS) was initially attributed to the stronger binding at nascent and basal discs at the OS-IS junction. However, this behavior was found to be rather dynamic as photobleaching resulted in the decline of signal strength at the OS-IS junction suggesting a nuanced mechanism of compartmentalization that balances specific interactions with dynamic redistribution (59). Lastly, it was shown that PrBPδ, a chaperone for trafficking prenylated proteins to the outer segment, is dependent on specific structural elements beyond just the prenylated site (59). In summary, trafficking of prenylated proteins in PRs is dependent on prenyl-lipid moiety, charge on the upstream amino acids, prenylated cargo interaction with appropriate chaperones for efficient membrane targeting and may be even influenced by light exposure.
We have discussed the biochemical basis of protein prenylation, the cellular requirement of de novo synthesis of isoprenes, and provided some insights into protein prenylation in the retina. An excellent, comprehensive review by Roosing et. al., covered this topic of interest nearly a decade ago (5). We herein provide the background and updates on protein prenylation-associated inherited retinal diseases, and insights from transgenic mouse models of retinal prenylopathies.
The first-described example of prenylation defect-induced visual system dysfunction was X-linked choroideremia, wherein point deletion mutation in the gene CHM/REP1 (Chromosome X, location: Xq21.2), encoding Rab escort protein 1 (REP-1) was identified. REP-1 serves as a chaperone for several Rab proteins and is a crucial accessory protein for GGTase-II. Mutations in REP1 causes progressive mid-peripheral atrophy of the retina, RPE and the choroid, resulting in progressive vision loss, ultimately leading to blindness (60). More specifically, male patients initially present with night blindness in childhood, which then advances to peripheral visual field impairment and ultimately to total blindness later in life (61). Whereas female carriers are usually asymptomatic but may exhibit a unique speckling pattern on fundus autofluorescence imaging, and sometimes night blindness as well (62).
REP-1 chaperones unprenylated Rabs and presents the cargo to GGTase-II for geranylgeranylation (63, 64). REP-1 deficiency leads to the accumulation of unprenylated Rab proteins in the retina (65). Immunolabeling studies showed that REP-1 is expressed in both rods and cones, predominantly in the inner segment and the perinuclear cytoplasm (66). Interestingly, while CHM gene expression is found in extraocular tissues as well, the gene mutation predominantly affects the retina and choroid in patients. This is due to shared functional redundancies between REP-2, which may partially compensate for the geranylgeranylation of Rab GTPases in extraocular tissues (67). For example, it was shown that while Rab27a exhibits a similar affinity for REP-1 and REP-2, REP-1-Rab27a complex (Figure 3) has a greater preference for GGTase-II compared to the REP-2-Rab27a complex in the retina (69).
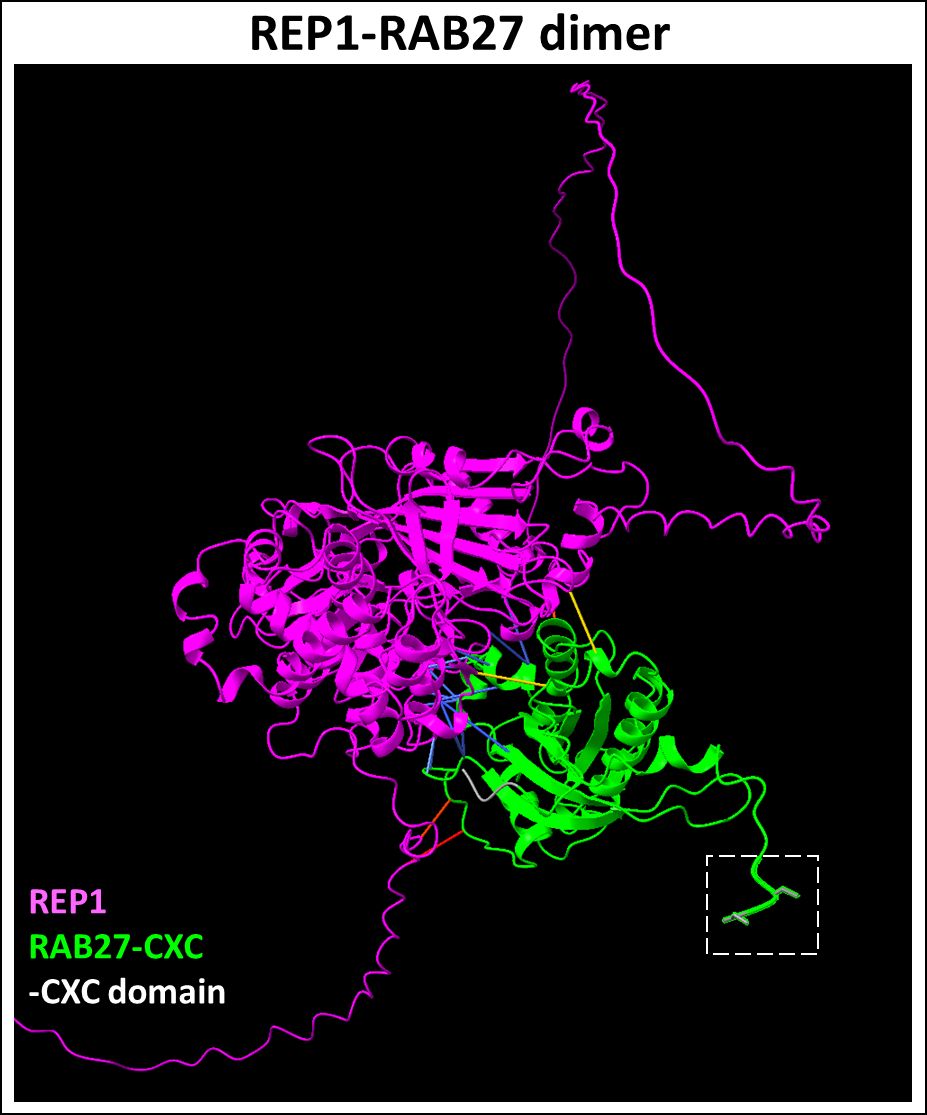
Figure 3 Predicted REP1-RAB27 dimer complex. REP1 (Uniprot ID: P24386) is a chaperone required for presenting Rab27 (Uniprot ID: P51159) to GGTase-II for prenylation at the CXC domain. REP1-Rab27 protein-protein interactions (highlighted in blue) represent predicted binding contacts. Loss of this interaction inhibits the prenylation of the target cargo. Figure generated using ChimeraX (68).
The notion of CHM manifesting as a non-syndromic retinopathy has been challenged recently (70). Whole metabolomic analysis of plasma samples from 25 CHM patients versus age- and sex-matched controls showed plasma alterations in oxidative stress-related metabolites, in addition to alterations in tryptophan metabolism, leading to significantly elevated serotonin levels (70). Lipid metabolism was found to be disrupted in CHM patients with evidence of dysfunctional lipid oxidation, and dyshomeostasis in several sphingolipids and glycerophospholipid levels (70). Aberrations in sphingolipid metabolism has been implicated in neurodegenerative diseases, metabolic disorders, immune function, and cancer (71).
Several preclinical and clinical studies targeting CHM have yielded important insights into factors that affect developing effective therapeutic interventions (72–78). In vivo studies on CHM gene transfer utilizing HIV-based lentiviral vectors led to a decrease in the geranylgeranylated Rab protein load. However, this approach also showed limited transfection of PRs by lentiviral vectors (73). Alternatively, adeno-associated virus (AAV) vectors, such as adeno-associated virus serotype 2 (AAV2) mediated delivery of CHM have been shown to achieve efficient transduction of REP1 in PRs and RPE, with an acceptable safety profile (72, 79–81). However, recent results from phase III of the Efficacy and Safety of BIIB111 for the Treatment of Choroideremia (STAR) trial did not meet the primary endpoint of best-corrected visual acuity (BCVA) for FDA approval. The work highlights the need to consider the stage of disease, anatomical differences, surgical variability, and dose administration among participants (76). Gene therapy for choroideremia may still provide significant visual acuity gains if treated early. Episomal scaffold/matrix attachment region (S/MAR)-based plasmid vectors carrying the human CHM gene in CHM patient-derived fibroblasts and a CHM mutant zebrafish model showed some promise. This may provide an alternative delivery strategy for gene augmentation in CHM patients (82).
Proteins of the sixth family of type I cyclic nucleotide phosphodiesterases (PDE6) are key effector enzymes in the phototransduction cascade in rod and cone PRs (83, 84). PDE6 isoforms are regulated by small inhibitory γ-subunits (Pγ); G-protein mediated disinhibition is vital for PDE6 activity and the phototransduction cascade (84). Rod PDE6 complex is composed of a catalytic PDE6A/B heterodimer and two copies of PDE6G (or Pγr) (Figure 4), whereas cone PDE6 is a homodimer of catalytic PDE6C subunits each associated with the cone specific Pγc subunit.
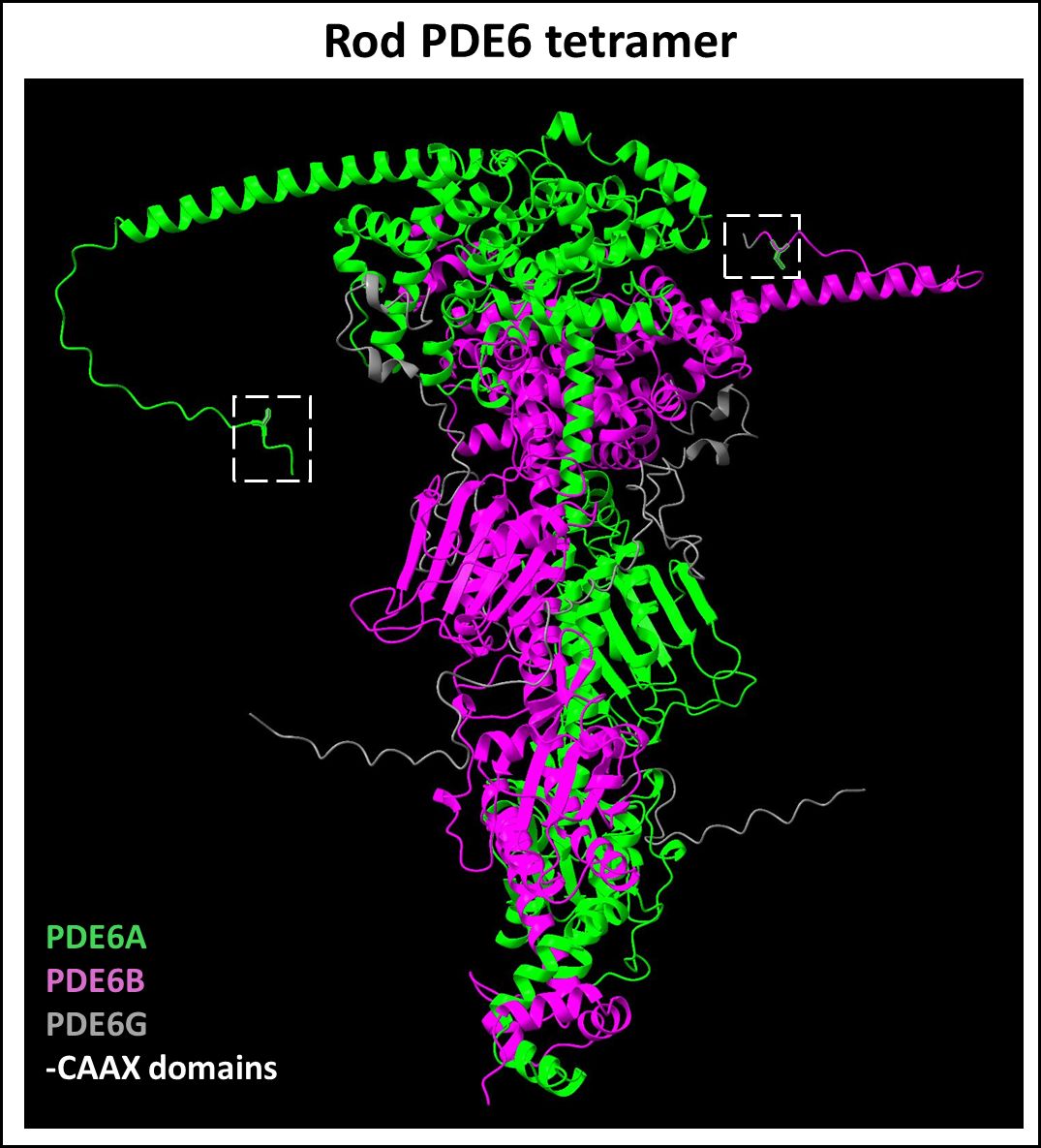
Figure 4 Predicted Rod PDE6 tetramer complex. The rod PDE6 complex is composed of a catalytic PDE6A (Uniprot ID: P16499) and PDE6B (Uniprot ID: P35913) heterodimer with their respective C-terminal prenylation motifs and two copies of small inhibitory γ-subunits (Uniprot ID: P18545). Figure generated using ChimeraX (68).
PDE6 complex assembly occurs in the inner segment of PRs including complex formation with HSP90 and AIPL1 co-chaperones. Moreover, the Pγ subunit of transducin also plays a critical chaperone-like role in PDE6 complex formation and was found to cause rapid retinal degeneration in Pγ-deficient knockout mouse model (85, 86). The prenylated and processed CAAX motif on the γ subunit serves to enhance membrane binding of the transducin- βγ dimer (Figure 5). It was shown that in contrast to the cone PDE6, the rod PDE6A/B requires Pγ for proper folding and maturation (87). While rod PDE6A is farnesylated, the rod PDE6B subunit and cone PDE6 undergo geranylgeranylation (88, 89). Prenylation facilitates membrane attachment of PDE6 complex and enhances its binding with AIPL1 (90, 91). Rod PDE6A/B not only requires prenylation for membrane trafficking to the outer segment of the retina, but it is also dependent on post-processing by RCE1 (92). Loss of carboxymethylation following prenylation and proteolytic processing does not hinder trafficking of PDE6 complex to the outer segments, but still leads to PR degeneration in an Icmt homozygous knockout mouse model (93). Mutations in rod PDE6 are responsible for a small subset of autosomal-recessive RP cases (OMIM #613801) (94, 95). A recent animal model of autosomal recessive achromatopsia (OMIM #613093) highlighted the critical role of prenylation of the catalytic subunit of cone PDE6 (96). Notably, this study showed that cones from homozygous mutants exhibited functional deficits, whereas heterozygous mutant cones were functional. In addition, cones from homozygous mutants displayed structural deficits suggesting a potential role for PDE6 in maintaining cone OS length and morphology (96).
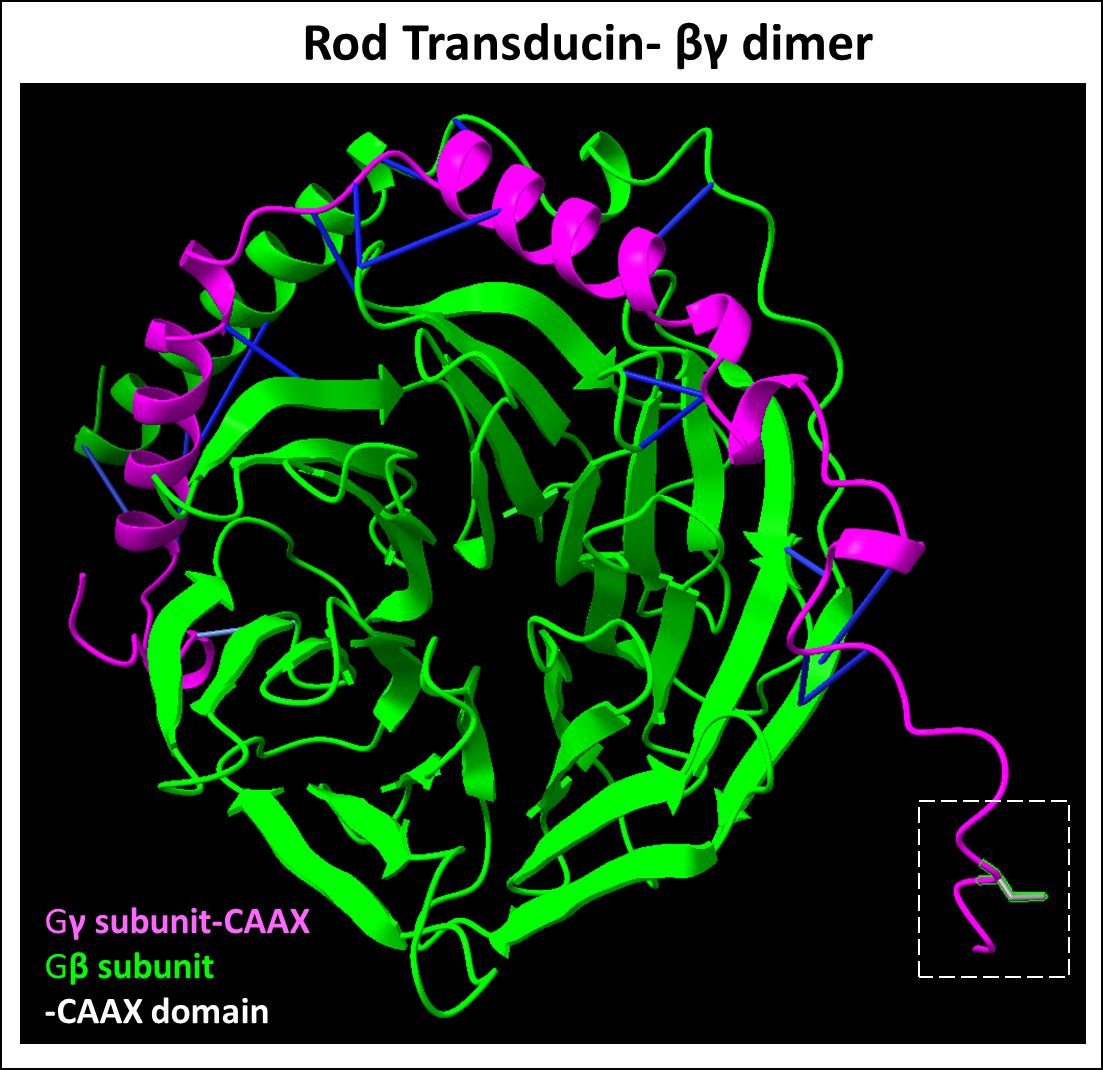
Figure 5 Predicted Rod transducin βγ dimer complex. Transducin βγ dimer is essential for phototransduction. The β (Uniprot ID: P62873) and γ (Uniprot ID: P63211) subunit protein-protein interactions (highlighted in blue) represent predicted binding contacts. The prenylated and processed CAAX motif on the γ subunit serves to enhance membrane binding. Figure generated using ChimeraX (68).
Prenyl-binding protein (PrBP/δ) is a 17 kDa ubiquitous solubilization factor involved in intracellular trafficking of both farnesylated and geranylgeranylated proteins (97–99). It was first discovered in PR cells as the fourth subunit of cGMP phosphodiesterase 6 (PDE6), hence also termed PDE6δ (97). The crystal structure of PrBP/δ unveiled a hydrophobic pocket between two beta sheets, facilitating the insertion of prenyl groups (100). As an illustration, we have provided the predicted dimerization of PrBP/δ and Rab28 highlighting the hydrophobic interactions between the two proteins (Figure 6). In PR cells, PrBP/δ traffics prenylated cargo such as PDE6, Rab28, and rhodopsin kinase (GRK1) from the site of protein synthesis (inner segment) to the retinal outer segment, and its homozygous deletion causes slow, progressive rod-cone dystrophy (101, 102). AlphaFold2/ChimeraX simulation of PrBP/δ-GRK1 complex is provided in Figure 7. PrBP/δ was also reported to interact with the prenylated RPGR (retinitis pigmentosa GTPase regulator) isoform, RPGR1-19 (103, 104). Mutations in PDE6D are associated with Joubert Syndrome (JBTS) (OMIM #615665), a complex neuronal ciliopathy characterized by developmental cerebellar malformations and accompanying retinal degeneration (105, 106).

Figure 6 Predicted PrBP/δ-RAB28 dimer complex. PrBP/δ (Uniprot ID: O43924) was recently identified as a Rab28 (Uniprot ID: P51157) protein interactor, which is farnesylation dependent. This interaction allows for the trafficking of Rab28 protein. Figure generated using ChimeraX (68).
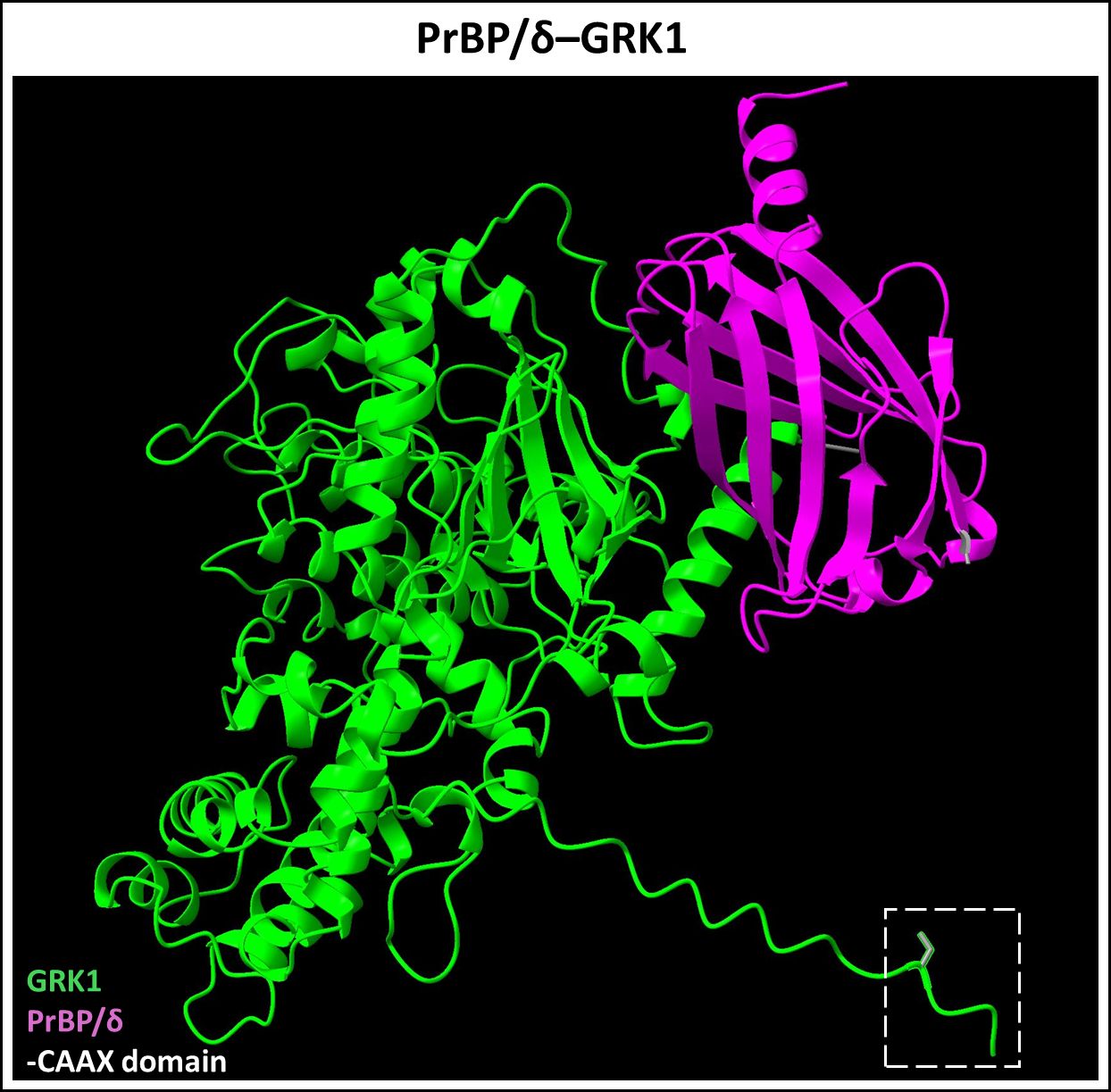
Figure 7 Predicted PrBP/δ-GRK1 dimer complex. PrBP/δ (Uniprot ID: O43924) binds prenylated rhodopsin kinase (GRK1, Uniprot ID: Q15835). This interaction allows for the trafficking of prenylated GRK1 from the inner segment to the outer segment of neural retina. Figure generated using ChimeraX (68).
Several groups showed that PrBP/δ function enhances ciliary targeting of INPP5E, a farnesylated protein implicated in JBTS (OMIM #213300) and Intellectual disability-truncal obesity-retinal dystrophy-micropenis syndrome (OMIM #610156) (105, 107, 108). It is important to note, however, that while the interaction between PrBP/δ and INPP5E is farnesylation dependent, the ciliary targeting of INPP5E is complex and relies on the interplay of four other ciliary localization signals (109). Recently, an affinity proteomics study identified a novel set of PrBP/δ-interacting prenylated proteins that are involved in PR integrity, GTPase activity, nuclear import, or ubiquitination (110). The prenylated protein interactors identified include ciliogenesis factors such as FAM219A, serine/threonine-protein kinase NEM1 (NIM1K), and ubiquitin-like protein 3 (UBL3) (110).
Several frameshift, missense, and nonsense mutations in the gene coding for Aryl hydrocarbon-interacting protein-like 1 (AIPL1) are reportedly associated with autosomal recessive Leber congenital amaurosis type IV (LCA4) (OMIM #604393) (111, 112). Patients with LCA4 typically present with early-onset severe visual impairment, nystagmus, and poor pupillary light responses compared to other LCA types (113, 114). Both scotopic and photopic Electroretinography (ERG) responses were severely diminished in rods and cones, respectively. Disc drusen, optic disc edema, and macular atrophy have also been noted (113, 114). Optical coherence tomography (OCT) imaging in older patients showed severe cone dystrophy (114).
AIPL1 is a specialized PR-specific co-chaperone required for PDE6 maturation and stabilization (115–117). Retinal degeneration caused by AIPL1 defects were described in two mouse models of LCA4 (115, 116). In Aipl1 knockout mouse model, rapid rod-cone dystrophy was observed by postnatal 3 weeks of age (115). While the AIPL1 h/h mouse model, carrying homozygous, hypomorphic mutant allele for AIPL1, causes diminished expression of AIPL1, resulting in relatively slower PR dystrophy beginning after 12 weeks of age with almost complete loss of PRs by 5 months of age (116). In both models, PDE6 expression was found to be drastically perturbed, suggesting an indispensable role of AIPL1 in PDE6 maturation.
The AIPL1 protein contains an N-terminal FK506-binding protein (FKBP) domain and a C-terminal tetratricopeptide repeat (TPR) domain with three tetratricopeptide repeats (111). Structural studies confirmed that the PDE6 prenylation is required for its binding to the FKBP domain of AIPL1 (91, 118). The TPR domain serves as a binding site for cytosolic HSP90 and the regulatory Pγ subunit of the PDE6 complex (119, 120). Interaction of PDE6 with the TPR domain of AIP1L also plays a key role in the chaperone-dependent folding and maturation of the PDE6 complex (117, 121). Intriguingly, AIPL1 appears to be colocalized predominantly from the synapse to the inner segment of PR cells, with an enrichment in the connecting cilium, while the PDE6 complex is eventually trafficked to the outer segment (122). This may suggest that AIPL1 chaperone function is limited to PDE6 maturation and does not extend to the trafficking of PDE6 to the outer segments. A recent transgenic mouse study showed that targeted mutation of PDE6A at the prenylation consensus cysteine target, does not affect its trafficking to the outer segment (123). Prenylation of PDE6C was found to be important for the formation of a stable ternary chaperone complex comprising of AIPL1, HSP90, and PDE6 subunits in HEK293 cells (123). Future work is required to determine if AIPL1 plays a role, if any, in a relay-type system where it offloads mature PDE6 complex to another trafficking chaperone like PrBP/δ.
Heterotrimeric G-protein transducin, Gt, is an essential signal transducer and amplifier in retinal PR cells for phototransduction (124). It is a membrane-bound G-protein comprised of α, β, and γ subunits, and is bound to a guanine diphosphate (GDP) molecule (124). Both α and γ subunits are lipid-modified, wherein the α subunit is acylated and the γ subunit is farnesylated, despite both subunits possessing a CAAX motif (125, 126). These modifications allow for outer segment membrane localization and subsequent protein interaction (127). Prenylation also enhances the binding of Gβγ to G-protein α subunits (128). The importance of Gγ subunit farnesylation was shown by Kassai and co-workers, who demonstrated that replacing the farnesyl group of Gγ with the more hydrophobic geranylgeranyl attachment rendered the Gβγ dimer incapable of undergoing light-driven translocation to rod outer segments (129).
Homozygous deletion of Gγ subunit (coded by the gene Gngt1) in mice showed rod atrophy starting at PN 1 month, with almost complete rod degeneration by 6 months of age and a significant decrease in the protein levels of Gα and Gβ, despite mRNA levels comparable to wild-type controls (130). In contrast, another study found only poor phototransduction signal amplification in intact rods and showed a decline in visual sensitivity of rods in a Gγ-deficient mouse model, without prompt retinal degeneration (131). The pivotal role of farnesylation of Gγ in mediating phototransduction was further confirmed in point mutant models where the Gγ farnesylation site was removed (132). This study showed that farnesylation was not essential for Gβγ dimerization (Figure 5), but essential for localization to the rod outer segments and participation in phototransduction (132). Surprisingly, despite the clear functional role of farnesylated Gγ in the phototransduction cascade, there are no known mutations of Gngt1 linked with inherited retinopathies. This is perhaps attributable to the low frequency of mutation reported in this relatively small gene (225 bp open-reading frame) (5).
Rab small GTPases, which belong to the Ras superfamily, are key regulators of membrane trafficking and cell growth (133, 134). Rab28 carries a C-terminal CAAX motif that is farnesylated in contrast to the common Rab geranylgeranylation and was the first prenylated small Rab GTPase identified to be directly involved in an inherited retinal disease (135–137). Immunohistochemical analysis suggested that Rab28 colocalized at the PR basal body and the ciliary rootlet (137). Rab28 mutations cause cone-rod dystrophy 18 (CRD18) (OMIM #615374), presenting with central retinal atrophy and hyperpigmented appearance of the fovea. Both scotopic and photopic ERG responses were significantly diminished, suggesting decreased rod and cone response to light, respectively (136–138). A transgenic knockout mouse model of Rab28 showed Rab28 requirement for disc shedding by cone PRs and its subsequent phagocytosis. Loss of Rab28 function in knockout mice faithfully mimics the human CRD18 phenotype (139). Moreover, this study identified Rab28 protein interactors such as KCNJ13 and PrBP/δ (PRBP/δ and Rab28 complex, simulated in Figure 6). Autosomal recessive mutations of KCNJ13 leads to Leber congenital amaurosis type 16 (LCA16) (OMIM #614186), wherein KCNJ13 regulates phagocytic uptake of shed outer segments by the RPE (139, 140).
Autosomal dominant mutations in RPGR gene (Chromosome X, Xp11.4) account for over 70% of X-linked retinitis pigmentosa cases (XLRP) (OMIM #300455) worldwide (141). Two major spliced isoforms of RPGR have been identified in the mammalian retina: RPGR1−19 and RPGRORF15 (142). Both protein isoforms share an identical N-terminal tandem repeat structure named RCC1-like domain (RLD), while the C-terminal domains are not conserved due to altered mRNA splicing. The C-terminus of RPGR1−19 constitutes a target prenylation motif which is geranylgeranylated, whereas RPGRORF15 contains a Glu-Gly-rich low complexity region in the C-terminus (142, 143). The shared N-terminal RLD region may confer some functional redundancy between the two RPGR isoforms, while the differential prenylation of RPGR1−19 may govern its differential subcellular colocalization and function. This has been demonstrated in vitro using transformed human RPE1 cells wherein the prenylated RPGR1−19 isoform localized to the RPE primary cilia, while RPGRORF15 does not traffic to cilia (144, 145). Interestingly, rescue experiments in RPGR knockout mice showed that the RPGRORF15 isoform sufficiently rescued PR functioning (146, 147).
A report showed a decrease in RPGR1−19 isoform, with concurrent upregulation in the RPGRORF15 isoform throughout eye development (147). Thus, suggesting a duality in the role of the two RPGR isoforms during retinal development vs.. functioning of the mature retina. However, the exact role of prenylated RPGR1−19 in early retinal development has not yet been directly established. Recently, Zhang et.al., showed the effects of common missense mutations of the shared RLD on the RPGR protein interaction network, and demonstrated that the RLD domain is indispensable for interactions with PrBP/δ and INPP5E in RPE1 cells (148). Prenylation of RPGR1−19 isoform is necessary for its ciliary targeting via PrBP/δ trafficking and that prenylation also enhances the RLD binding region for protein interaction (148). Future work should aim to gain the mechanistic understanding underpinning protein interaction disruption on retinal health and whether prenylation represents a dispensable modification in the setting of XLRP pathogenesis.
Mevalonate kinase (MVK) catalyzes the phosphorylation of mevalonic acid to phosphomevalonate. Autosomal recessive mutations in MVK causes metabolic disorder with poor genotype-phenotype correlation (149). It encompasses a severe form of the disease, mevalonic aciduria (MEVA) (OMIM #610377), and a milder periodic fever syndrome, hyperimmunoglobulinemia D syndrome (HIDS) (OMIM #260920) (150). HIDS is characterized by regular episodes of high fever and systemic inflammation in the affected pediatric patient population. In contrast, mevalonic aciduria patients present with chronic systemic inflammation, along with neurodevelopmental issues such as cerebellar atrophy, dystonia, and ataxia. MKD patients also present with gastrointestinal complications and retinal dystrophy (151–153). A study in 2013 showed that mutations in MVK causes late-onset non-syndromic RP, accompanied by mild MKD symptoms (154). It was thought that the accumulation of mevalonic acid may be responsible for the neuronal degeneration including, PR deterioration, observed in MKD patients (153). Two recent studies have shown that patients with MKD exhibit significant accumulation of unprenylated Rab proteins (155, 156). Therefore, the retinal dystrophy phenotype associated with MKD may arise from decreased de novo synthesis of isoprenoid metabolites (dolichol, cholesterol, FPP, and GGPP), as well as cellular insufficiency in prenylation of the target proteome.
Recent discoveries of novel prenyltansferases, targets motifs, and non-canonical mechanisms of prenylation, has increased the probability of discovering yet unidentified prenylopathies. Here, we briefly discuss the case of a rare autosomal recessive mutation that primarily affects the cis-prenyltransferase (CPT) complex involved in dolichol synthesis. The cis-prenyltransferase complex is a heterotetrameric complex comprised of the catalytic subunit dehydrodolichyldiphosphate synthase (DHDDS), and its interacting partner NUS1 or Nogo-B receptor (NgBR). The CPT complex catalyzes the serial cis-condensation of multiple IPP molecules to FPP, to ultimately generate dolichol, an isoprenoid metabolite which serves as the obligate glycan carrier, and is indispensable for protein glycosylation. We recently discussed the visual disorders pertaining to the dolichol synthesis pathway (157).
Recessive mutations in NUS1 leads to a systemic disorder including neurodevelopmental issues, retinal dystrophy, and optic neuropathy, unsurprisingly, given the requirement of dolichol synthesis and protein glycosylation in all cells (158). NUS1 performs several other critical roles in maintaining lysosomal homeostasis (159). Interestingly, a recent study showed that NgBR independently binds to farnesylated Ras protein, outside of its CPT function, and plays a key role in membrane localization of H-Ras (Figure 8) (160). Moreover, NgBR binds not only to farnesylated H-Ras with its canonical CAAX (CVLS) motif, but also a recombinant geranylgeranylated H-Ras protein with GGTase-I target CAAX sequence (CLVL) (160). We have independently demonstrated that conditional ablation of Nus1 in the developing PRs, using a CRX-Cre mouse line, causes total PR and bipolar cell loss by PN 2–3 weeks (Ramachandra Rao et.al., unpublished results). However, the underlying degenerative mechanism remains to be understood. These findings highlight the need for investigating the additional chaperone-like roles of NgBR in binding and trafficking prenylated proteins in the retina.
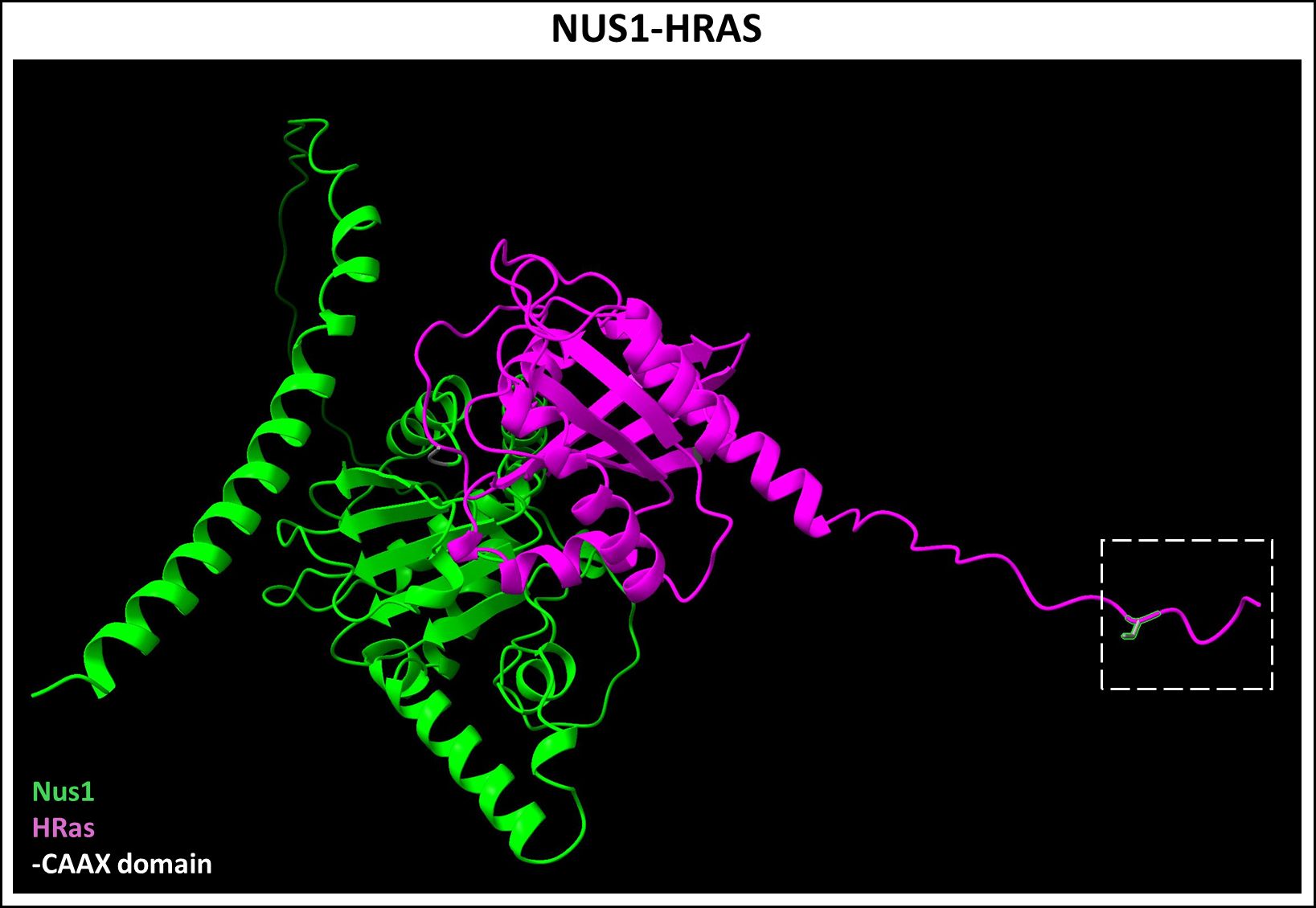
Figure 8 Predicted NUS1-HRAS dimer complex. Nogo B-Receptor (NUS1, Uniprot ID: Q96E22), a part of the cis-prenyltransferase complex, was recently shown to bind farnesylated HRAS (Uniprot ID: P01112) and plays a chaperone-like role in its membrane localization. Figure generated using ChimeraX (68).
We have provided a brief, tabular summary of the clinical features and molecular basis of known, above discussed prenylation-associated retinopathies in Table 1. Defects in prenylation or the chaperone-mediated maturation and trafficking of key membrane-bound prenylated proteins essentially manifest as early rod-cone or cone-rod dystrophies, depending on the predominant affected cell type.

Table 1 Overview of ocular outcomes in protein prenylation-associated inherited retinopathies.
We have thus far discussed the biochemical mechanisms underlying protein prenylation, and inherited blinding diseases characterized by defective prenylation of proteins expressed in the neural retina. With the discovery of novel prenyltransferases and non-canonical target motifs, it would be unsurprising if other prenylation-associated retinopathies and/or systemic disorders are discovered. It is therefore essential to develop approaches to investigate the retinal prenylome. We now turn our attention to technical advances and available methodologies in predicting and/or directly determining the cellular prenylome. We also discuss possible applications of computational, proteomic, and in vitro approaches to determine the total retinal prenylated proteome.
Computational approaches have been developed to enable prediction of prenylated proteome. Maurer-Stroh and Eisenhaber developed the algorithm PrePS to predict prenyltransferase substrates, based on data from experimentally derived prenylated motifs and prediction of new motifs acting as a substrate for farnesylation or geranylgeranylation (161). The training dataset consisted of 692 FTase and 486 GGTase-I substrates curated by literature survey as well as BLASTP analysis with known prenylated substrates against an NCBI database. In addition, an 11 amino acid sequence upstream of the prenyl cysteine was added to refine the algorithm. With this refinement of PrePS, the algorithm expands the rules which predict the prenylation of a substrate to a 15-amino acid sequence (162, 163). However, experimental determination of the target peptide prenylation is still necessary to determine true false positive rate of this predictive methodology. FlexPepBind is a structure-based computational modeling approach for predicting FTase substrates (162). This algorithm predicts peptide binding through a structure-based modeling approach by aligning different peptide sequences onto a template peptide-receptor complex. This structure-based predictive method was further experimentally validated using in vitro assays, wherein 26 of the 29 tested novel peptide were FTase targets suggesting a very low false negative prediction (162).
Computational approaches dependent on training datasets are expected to be biased against non-canonical, unidentified novel prenylation target motifs. To address this discrepancy, a recent study used yeast Hsp40 Ydj1p chaperone as a genetic reporter (164). Ydj1p is prenylated but is subject to the shunt pathway in which the prenylated protein does not undergo the downstream proteolytic processing and remains cytosolic. Prenylated Ydj1p produces a thermotolerant phenotype in yeast which serves as a rapid proxy to monitor protein prenylation status. Using this mutation-phenotype screen, the authors evaluated over 67,000 recombination events, correlating to about 93.5% of the total possible amino acid combinations which may code for CAAX motif. The hits identified from the Ydj1p-based genetic screen did not greatly overlap with those found in other screenings that utilized common targets such as Ras, suggesting that a much larger set of motifs may be serving as prenylation motifs than was previously thought (164). Only a small subset of prenylated motifs identified using the Ydj1p-based screening were identified as having a high probability of prenylation by FlexPepBind and PrePS with results of 27% and 7%, respectively. However, it is important to note that this discrepancy may also reflect different target peptide-specificities for the mammalian and yeast prenyltransferases enzymes.
Data mining methods using experimentally determined retinal proteome and transcriptome data may be highly effective in predicting the retinal prenylome, including non-canonical prenylation motifs. Applying targeted data mining to the transcriptome or proteome specific to the RPE and neural retina—separated into macular and peripheral regions—could be particularly powerful (165). These methods may help distinguish the prenylome specific to the human macula and peripheral retina and allow for better understanding of protein prenylation-associated vision disorders. Furthermore, pathway analysis tools like Ingenuity Pathway Analysis may reveal the potential cellular and retina-specific functions of these predicted prenylated proteins (166). The latest advancements in AI-driven prediction of protein structures, protein-protein interactions, and protein-ligand interactions such as AlphaFold2 and AlphaFold3, could provide preliminary computational validation of these predicted retinal prenylomes (167). It is important to acknowledge that novel hits arising from predictive, computational methodologies need to be experimentally validated using in vivo and in vitro approaches, as discussed below.
Fluorescence tagging of proteins has been extensively utilized to study prenylation at endogenous levels, providing insights into prenylated protein maturation and trafficking, and has also facilitated studies of prenyltransferase inhibitors since membrane localization is an easily measurable experimental endpoint. Diffuse fluorescence throughout the cell indicates cytoplasmic localization, which implies a protein either not undergoing prenylation or becoming prenylated without further processing as noted in the shunt pathway. For instance, the effect of proteolysis on membrane colocalization of K-Ras protein was determined using the above discussed chimeric approach (168). Using Rce1+/+ and Rce1-/- fibroblast cells transfected with a chimeric GFP-K-Ras construct, it was shown that fluorescence was localized to the plasma membrane in Rce1+/+ cells but diffuse in Rce1-/- cells, suggesting that proteolysis was necessary for Ras protein membrane localization (168).
An alternative approach for detecting protein prenylation inside mammalian systems that is not reliant on membrane localization as proxy endpoint, is called Protein Lipidation Quantification (PLQ). This method is derived from an established method of Micellar Electro-Kinetic Chromatography (MEKC) that allows selective partition of prenylated proteins into detergent micelles (using SDS) during capillary electrophoresis (169, 170). Detergent micelles serve as pseudo-stationary phase and exhibit differential electrophoretic migration compared to unprenylated free analyte. PLQ has been successfully used to separate prenylated full length fluorescent protein with a C-terminus CAAX motif in in-cell studies (171).
Chemical proteomic approaches facilitate the direct detection of prenylation in vivo and are based on the development of analogues of FPP and GGPP that are functionalized with biochemical affinity tags, immunogenic tags for detection by antibodies, and chemo-selective tags for bio-orthogonal labeling. Spielmann and coworkers showed the application of an immunogenic tag by using anilinogeraniol (AGOH) to detect FTase substrates (172). Anilinogeraniol is an analogue of an upstream precursor of FPP, which undergoes cellular kinase-dependent diphosphorylation to 8-anilinogeranyl diphosphate (AGPP), and where the third isoprene unit of FPP is replaced with an aniline moiety that then serves as an epitope for detection by Western blot (172, 173). Biotin-geranyl diphosphate (BGPP) has been generated for use as an affinity tag (174). BGPP only allowed for the efficient identification of GGTase-II substrates as the bulky nature of the biotin group was found to interfere with the protein substrate binding for FTase and GGTase-I. Engineered FTase and GGTase-I variants can utilize BGPP as a donor for protein modification (174). Lysates from cells expressing wild-type or the engineered prenyltransferase variants were incubated with BGPP, followed by avidin pull down and proteomic analysis, which allowed the identification of GGTase-II target proteins. Several Rab proteins such as Rab7 and Rab27, which were differentially prenylated in WT prenyltransferase expressing cells, were identified as GGTase-II substrates (174).
Distefano and coworkers designed alkynylated FPP analogue C15AlkOPP, which has been used to study protein prenylation inhibitors, labeling of proteins sensitive to human pathogens, and the delineation of the prenylome of Plasmodium falciparum, the causative agent of malaria (175–180). More recently, new alkyne-tagged isoprenoid analogues which closely mimic FPP (YnF) and GGPP (YnGG) allowed click-chemistry based detection of prenylated proteins via mass spectrometry in a human endothelial cell line (181). These analogues allow quantitative analysis of endogenous prenylated proteome since they do not alter the specific activity of prenyltransferases. Upon addition of the alkyne tagged analogue, proteins are captured via click CuAAC ligation to azide-containing fluor/biotin-tagged reagents. This facilitates the enrichment of prenylated proteins and subsequent analysis via LC-MS/MS. These analogues also proved useful in investigating protein prenylation in vivo. Intracerebroventricular (ICV) injection of the C15AlkOPP analogue in brains of a transgenic mouse model (APP/PSI) of Alzheimer’s Disease found the upregulation of several prenylated proteins in diseased mice (182). The development of these protein prenylation probes has greatly enhanced our understanding of protein prenylation biochemistry. Appropriate application of similar approaches in in vitro and in vivo models may prove fruitful in delineating the underlying degenerative mechanisms of prenylation-associated retinopathies.
Experimental determination of retinal prenylome in vivo is challenging since current prenylation screening assays utilize in vitro approaches. While proteomic and lipidomic strategies have proved to be powerful in refining our understanding of the prenylome, these approaches are expensive and labor-intensive. However, both the in vitro as well as the proteomic/lipidomic approaches described above may be highly relevant in defining the retinal cell type-specific prenylome. For instance, RPE prenylome and target-specific prenylation mechanisms may be easily investigated by applying such approaches in induced pluripotent stem cell (iPSC)-derived RPE cells, as well as primary RPE cells (183). Similarly, primary Müller glia and ganglion cell cultures may also be extremely useful in determining retinal cell type-specific prenylome. Identification of novel retinal cell type-specific prenylated hits may then be further validated in vivo using careful conditional gene ablation approaches.
Defining the total retinal prenylated proteome, as well as cell type-specific expression, processing, and functions of prenylated proteins will provide a wholesome understanding of the role and requirement of protein prenylation on retinal functioning. Achieving this goal also requires identification and validation of potential novel prenyltransferases, chaperones, and target proteins expressed in the neural retina. As discussed above, careful application of computational approaches will not only facilitate the identification of retinal prenylated proteome, but also help determine differential expression of prenylated proteins in the human macula vs. peripheral retina. These frontiers promise a comprehensive understanding of prenylation-associated retinopathies and their respective mechanisms. The challenges in determining the retinal prenylated proteome, retinal outcomes in inherited prenylopathies, and putative approaches to achieve those ends are summarized in Figure 9.
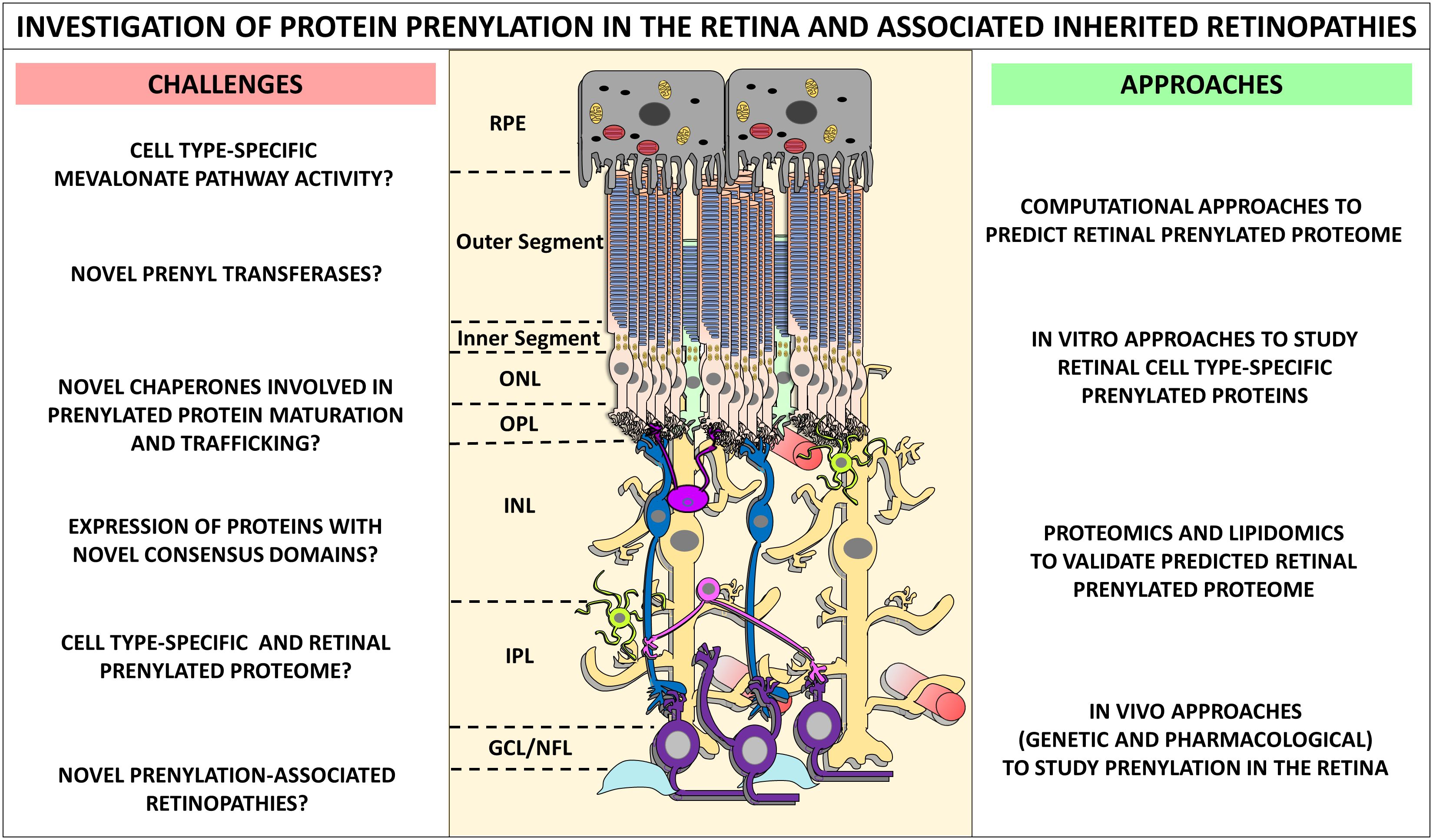
Figure 9 The challenges, approaches and future directions in the investigation of protein prenylation in the retina and associated inherited retinopathies. Determination of retinal prenylated proteome, and the retinal function of such prenylated proteins is challenging. We identify some of the immediate questions and challenges, as well as state-of-the-art approaches in better understanding prenylation mechanisms in the retina. A comprehensive approach linking computational, in vitro, proteomics and lipidomics, and in vivo strategies can help fully define the retinal prenylome. Such approaches may vastly improve our understanding of prenylation-associated retinopathies, and fast-track the development of therapeutics for these devastating blinding disorders.
Protein prenylation in PR plays a key role in protein subcellular localization and trafficking. A recent study by Maza et.al., highlights several important considerations and dynamics involved in the subcellular colocalization of prenylated proteins in the PR cell (59). As described, several prenylated retinal proteins are implicated in various retinal pathologies. The underlying role of aberrant prenylated proteins in retinal degeneration may be garnered from their known roles in retinal development and functioning. For example, G protein transducin, PDE6A, and PDE6B are involved in phototransduction cascade, while RAB28 and RPGR are involved in transport regulation. Moreover, mutations affecting AIPL1 and PrBP/δ results in a more generalized prenylation defect as both chaperones are involved in the appropriate trafficking of their cargo prenylated retinal proteins. The reported inherited retinopathies involve the complex interplay of more than just the implicated prenylated protein including protein chaperones and other protein interactors. In the case of MKD, one might hypothesize that supplementing the retina with the deficient isoprenoid groups may rescue the retinal dystrophy phenotype and reverse the accumulation of unprenylated Rabs (5). However, stimulating the synthesis of prenyl groups in the retina may cause undesirable side effects, such as neoplastic growth as is the case with overexpression of prenylated Ras oncogenes products. It is also difficult to predict the impact on isoprene synthesis with this approach given there are multiple feedback mechanisms that regulates mevalonate pathway (18, 184). So far, gene augmentation via lentiviral vectors and more recently, episomal scaffold/matrix attachment region (S/MAR)-based plasmid vectors seem to offer promising avenues in the treatment of prenylation-associated retinopathy such as CHM. However, recent results from phase III STAR trial underscore the need to set realistic expectations for clinical outcomes and emphasize the need for early therapeutic intervention to attain better clinical outcomes.
Finally, to fully capture the role of prenylation in the retina and retinal disorders and thus design therapeutic strategies, it is important to define the complete prenylome of the retina. More than 150 proteins are thought to be involved in the molecular pathways responsible for producing the prenyl lipid moieties or serve as potential substrates for prenylation as determined by PRENbase (http://mendel.imp.ac.at/PrePS/PRENbase/) (44). In addition, it is estimated that over 1166 proteins in the human proteome contain the CAAX motif suggesting that the number of prenylated proteins might be underrepresented in PRENbase. To add to the growing number of potentially prenylated proteins, studies have identified prenylation substrates in both yeast and mammalian systems that diverge from the canonical CAAX box and calls for the expansion of the classically recognized prenylation motif (27, 28). There are estimated to be more than 1000 proteins each with either a C(x)3X or CXX motif at their respective C-terminus. The discovery of heat shock protein chaperone Ydj1p in yeast which undergoes prenylation without subsequent proteolysis or methylation further adds to the complexity of this lipid modification (32). These studies highlight the need to identify potentially “shunted” proteins and proteins containing non-canonical C-terminal sequences, especially in the retina. Application of chemical proteomic approaches, genetic modeling, and targeted mining of retinal proteome to identify putative retinal prenylome will provide a fulsome mechanistic understanding of prenylation-associated blinding disorders.
SA: Conceptualization, Software, Visualization, Writing – original draft, Writing – review & editing. SR: Conceptualization, Funding acquisition, Software, Supervision, Visualization, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The original research by the authors that served as some of basis for this Review Article was supported, in part: R01 EY033298 (SJF: PI, SRR: Co-I, SUNY Buffalo, UB) from the National Eye Institute/N.I.H.; by two Career Starter Grants from the Knights Templar Eye Foundation (SR); startup funds from the Department of Ophthalmology, Jacobs School of Medicine and Biomedical Sciences, SUNY-University at Buffalo (SR); and by facilities and resources provided by the Jacobs School of Medicine and Biomedical Sciences, SUNY-University at Buffalo.
The authors would like to acknowledge the resources and services made available by the Ross Eye Institute, Department of Ophthalmology, Jacobs School of Medicine and Biomedical Sciences, SUNY Buffalo. The authors would like to thank Dr. Steven J. Fliesler (Dept. of Ophthalmology, SUNY Buffalo) for multiple fruitful discussions on the topic of interest presented here. Molecular graphics and analyses performed with UCSF ChimeraX, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from National Institutes of Health R01-GM129325 and the Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases. The authors acknowledge the resources and facilities provided by the VA Western New York Healthcare System. The opinions expressed herein do not reflect those of the Department of Veteran Affairs, the National Institutes of Health, or any other agency of the U.S. Government.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Mann M, Jensen ON. Proteomic analysis of post-translational modifications. Nat Biotechnol. (2003) 21:255–61. doi: 10.1038/nbt0303-255
2. Walsh C. Posttranslational modification of proteins: expanding nature’s inventory. Greenwood Village, CO, United States: Roberts and Company Publishers (2006).
3. Porter JA, Young KE, Beachy PA. Cholesterol modification of hedgehog signaling proteins in animal development. Science. (1996) 274:255–9. doi: 10.1126/science.274.5285.255
4. Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci. (2004) 117:2805–12. doi: 10.1242/jcs.01131
5. Roosing S, Collin RW, den Hollander AI, Cremers FP, Siemiatkowska AM. Prenylation defects in inherited retinal diseases. J Med Genet. (2014) 51:143–51. doi: 10.1136/jmedgenet-2013-102138
6. Resh MD. Covalent lipid modifications of proteins. Curr Biol. (2013) 23:R431–R5. doi: 10.1016/j.cub.2013.04.024
7. Ganesan L, Levental I. Pharmacological inhibition of protein lipidation. J membrane Biol. (2015) 248:929–41. doi: 10.1007/s00232-015-9835-4
8. Benetka W, Koranda M, Eisenhaber F. Protein prenylation: An (almost) comprehensive overview on discovery history, enzymology, and significance in physiology and disease. Monatshefte für Chemie/Chemical Month. (2006) 137:1241–81. doi: 10.1007/s00706-006-0534-9
9. Casey PJ, Seabra MC. Protein prenyltransferases. J Biol Chem. (1996) 271:5289–92. doi: 10.1074/jbc.271.10.5289
10. Zhang FL, Casey PJ. Protein prenylation: molecular mechanisms and functional consequences. Annu Rev Biochem. (1996) 65:241–69. doi: 10.1146/annurev.bi.65.070196.001325
11. Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. (2003) 3:459–65. doi: 10.1038/nrc1097
12. Campbell PM, Der CJ. Oncogenic Ras and its role in tumor cell invasion and metastasis. Semin Cancer Biol. (2004) 14:105–14. doi: 10.1016/j.semcancer.2003.09.015
13. Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. (1990) 343:425–30. doi: 10.1038/343425a0
15. Hinson DD, Chambliss KL, Toth MJ, Tanaka RD, Gibson KM. Post-translational regulation of mevalonate kinase by intermediates of the cholesterol and nonsterol isoprene biosynthetic pathways. J Lipid Res. (1997) 38:2216–23. doi: 10.1016/S0022-2275(20)34935-X
16. Manaswiyoungkul P, de Araujo ED, Gunning PT. Targeting prenylation inhibition through the mevalonate pathway. RSC Med Chem. (2020) 11:51–71. doi: 10.1039/C9MD00442D
17. Blanden MJ, Ashok S, Hougland JL. Mechanisms of caaX protein processing: protein prenylation by FTase and GGTase-I. In: Liu HW, Begley TP. editors. Comprehensive Natural Products III. Amsterdam, NL: Elsevier (2020) p. 497–527. doi: 10.1016/B978-0-12-409547-2.14837-1
18. Rao SR, Fliesler SJ. Cholesterol homeostasis in the vertebrate retina: biology and pathobiology. J Lipid Res. (2021) 62:100057. doi: 10.1194/jlr.TR120000979
19. Casey PJ. Lipid modifications of G proteins. Curr Opin Cell Biol. (1994) 6:219–25. doi: 10.1016/0955-0674(94)90139-2
20. Marshall CJ. Protein prenylation: a mediator of protein-protein interactions. Science. (1993) 259:1865–6. doi: 10.1126/science.8456312
21. Caplin BE, Ohya Y, Marshall MS. Amino acid residues that define both the isoprenoid and CAAX preferences of the Saccharomyces cerevisiae protein farnesyltransferase. Creating the perfect farnesyltransferase. J Biol Chem. (1998) 273:9472–9. doi: 10.1074/jbc.273.16.9472
22. Lane KT, Beese LS. Thematic review series: lipid posttranslational modifications. Struct Biol Protein farnesyltransferase geranylgeranyltransferase type I. J Lipid Res. (2006) 47:681–99. doi: 10.1194/jlr.R600002-JLR200
23. Hartman HL, Hicks KA, Fierke CA. Peptide specificity of protein prenyltransferases is determined mainly by reactivity rather than binding affinity. Biochemistry. (2005) 44:15314–24. doi: 10.1021/bi0509503
24. Hicks KA, Hartman HL, Fierke CA. Upstream polybasic region in peptides enhances dual specificity for prenylation by both farnesyltransferase and geranylgeranyltransferase type I. Biochemistry. (2005) 44:15325–33. doi: 10.1021/bi050951v
25. Krzysiak AJ, Aditya AV, Hougland JL, Fierke CA, Gibbs RA. Synthesis and screening of a CaaL peptide library versus FTase reveals a surprising number of substrates. Bioorg Med Chem Lett. (2010) 20:767–70. doi: 10.1016/j.bmcl.2009.11.011
26. Reid TS, Terry KL, Casey PJ, Beese LS. Crystallographic analysis of CaaX prenyltransferases complexed with substrates defines rules of protein substrate selectivity. J Mol Biol. (2004) 343:417–33. doi: 10.1016/j.jmb.2004.08.056
27. Blanden MJ, Suazo KF, Hildebrandt ER, Hardgrove DS, Patel M, Saunders WP, et al. Efficient farnesylation of an extended C-terminal C(x)(3)X sequence motif expands the scope of the prenylated proteome. J Biol Chem. (2018) 293:2770–85. doi: 10.1074/jbc.M117.805770
28. Ashok S, Hildebrandt ER, Ruiz CS, Hardgrove DS, Coreno DW, Schmidt WK, et al. Protein farnesyltransferase catalyzes unanticipated farnesylation and geranylgeranylation of shortened target sequences. Biochemistry. (2020) 59:1149–62. doi: 10.1021/acs.biochem.0c00081
29. Schey GL, Buttery PH, Hildebrandt ER, Novak SX, Schmidt WK, Hougland JL, et al. MALDI-MS analysis of peptide libraries expands the scope of substrates for farnesyltransferase. Int J Mol Sci. (2021) 22:12042. doi: 10.3390/ijms222112042
30. Boyartchuk VL, Ashby MN, Rine J. Modulation of Ras and a-factor function by carboxyl-terminal proteolysis. Science. (1997) 275:1796–800. doi: 10.1126/science.275.5307.1796
31. Winter-Vann AM, Casey PJ. Post-prenylation-processing enzymes as new targets in oncogenesis. Nat Rev Cancer. (2005) 5:405–12. doi: 10.1038/nrc1612
32. Hildebrandt ER, Cheng M, Zhao P, Kim JH, Wells L, Schmidt WK. A shunt pathway limits the CaaX processing of Hsp40 Ydj1p and regulates Ydj1p-dependent phenotypes. Elife. (2016) 5:e15899. doi: 10.7554/eLife.15899
33. Leung KF, Baron R, Seabra MC. Thematic review series: lipid posttranslational modifications. geranylgeranylation of Rab GTPases. J Lipid Res. (2006) 47:467–75. doi: 10.1194/jlr.R500017-JLR200
34. Guo Z, Wu YW, Das D, Delon C, Cramer J, Yu S, et al. Structures of RabGGTase-substrate/product complexes provide insights into the evolution of protein prenylation. EMBO J. (2008) 27:2444–56. doi: 10.1038/emboj.2008.164
35. Khosravi-Far R, Clark GJ, Abe K, Cox AD, McLain T, Lutz RJ, et al. Ras (CXXX) and Rab (CC/CXC) prenylation signal sequences are unique and functionally distinct. J Biol Chem. (1992) 267:24363–8. doi: 10.1016/S0021-9258(18)35774-0
36. Stenmark H, Olkkonen VM. The rab GTPase family. Genome Biol. (2001) 2:REVIEWS3007. doi: 10.1186/gb-2001-2-5-reviews3007
37. Shen F, Seabra MC. Mechanism of digeranylgeranylation of Rab proteins. Formation of a complex between monogeranylgeranyl-Rab and Rab escort protein. J Biol Chem. (1996) 271:3692–8. doi: 10.1074/jbc.271.7.3692
38. Kuchay S, Wang H, Marzio A, Jain K, Homer H, Fehrenbacher N, et al. GGTase3 is a newly identified geranylgeranyltransferase targeting a ubiquitin ligase. Nat Struct Mol Biol. (2019) 26:628–36. doi: 10.1038/s41594-019-0249-3
39. Shirakawa R, Goto-Ito S, Goto K, Wakayama S, Kubo H, Sakata N, et al. A SNARE geranylgeranyltransferase essential for the organization of the Golgi apparatus. EMBO J. (2020) 39:e104120. doi: 10.15252/embj.2019104120
40. Suazo KF, Belicek J, Schey GL, Auger SA, Petre AM, Li L, et al. Thinking outside the CaaX-box: an unusual reversible prenylation on ALDH9A1. RSC Chem Biol. (2023) 4:913–25. doi: 10.1039/D3CB00089C
41. Clark BS, Stein-O’Brien GL, Shiau F, Cannon GH, Davis-Marcisak E, Sherman T, et al. Single-cell RNA-seq analysis of retinal development identifies NFI factors as regulating mitotic exit and late-born cell specification. Neuron. (2019) 102:1111–26 e5. doi: 10.1016/j.neuron.2019.04.010
42. Voigt AP, Whitmore SS, Lessing ND, DeLuca AP, Tucker BA, Stone EM, et al. Spectacle: An interactive resource for ocular single-cell RNA sequencing data analysis. Exp Eye Res. (2020) 200:108204. doi: 10.1016/j.exer.2020.108204
43. Zverina EA, Lamphear CL, Wright EN, Fierke CA. Recent advances in protein prenyltransferases: substrate identification, regulation, and disease interventions. Curr Opin Chem Biol. (2012) 16:544–52. doi: 10.1016/j.cbpa.2012.10.015
44. Maurer-Stroh S, Koranda M, Benetka W, Schneider G, Sirota FL, Eisenhaber F. Towards complete sets of farnesylated and geranylgeranylated proteins. PloS Comput Biol. (2007) 3:e66. doi: 10.1371/journal.pcbi.0030066
45. Wang YC, Distefano MD. Synthetic isoprenoid analogues for the study of prenylated proteins: Fluorescent imaging and proteomic applications. Bioorg Chem. (2016) 64:59–65. doi: 10.1016/j.bioorg.2015.12.003
46. Seabra MC, Reiss Y, Casey PJ, Brown MS, Goldstein JL. Protein farnesyltransferase and geranylgeranyltransferase share a common alpha subunit. Cell. (1991) 65:429–34. doi: 10.1016/0092-8674(91)90460-G
47. Taylor JS, Reid TS, Terry KL, Casey PJ, Beese LS. Structure of mammalian protein geranylgeranyltransferase type-I. EMBO J. (2003) 22:5963–74. doi: 10.1093/emboj/cdg571
48. Strickland CL, Windsor WT, Syto R, Wang L, Bond R, Wu Z, et al. Crystal structure of farnesyl protein transferase complexed with a CaaX peptide and farnesyl diphosphate analogue. Biochemistry. (1998) 37:16601–11. doi: 10.1021/bi981197z
49. Lai RK, Perez-Sala D, Canada FJ, Rando RR. The gamma subunit of transducin is farnesylated. Proc Natl Acad Sci U S A. (1990) 87:7673–7. doi: 10.1073/pnas.87.19.7673
50. Al-Janabi A, Lightman S, Tomkins-Netzer O. ‘Statins in retinal disease’. Eye (Lond). (2018) 32:981–91. doi: 10.1038/s41433-018-0066-7
51. Pittler SJ, Fliesler SJ, Fisher PL, Keller PK, Rapp LM. In vivo requirement of protein prenylation for maintenance of retinal cytoarchitecture and photoreceptor structure. J Cell Biol. (1995) 130:431–9. doi: 10.1083/jcb.130.2.431
52. Pittler SJ, Fliesler SJ, Rapp LM. Novel morphological changes in rat retina induced by intravitreal injection of lovastatin. Exp Eye Res. (1992) 54:149–52. doi: 10.1016/0014-4835(92)90080-C
53. Kavanagh KL, Guo K, Dunford JE, Wu X, Knapp S, Ebetino FH, et al. The molecular mechanism of nitrogen-containing bisphosphonates as antiosteoporosis drugs. Proc Natl Acad Sci U S A. (2006) 103:7829–34. doi: 10.1073/pnas.0601643103
54. Waller DD, Park J, Tsantrizos YS. Inhibition of farnesyl pyrophosphate (FPP) and/or geranylgeranyl pyrophosphate (GGPP) biosynthesis and its implication in the treatment of cancers. Crit Rev Biochem Mol Biol. (2019) 54:41–60. doi: 10.1080/10409238.2019.1568964
55. Caponigro F, Casale M, Bryce J. Farnesyl transferase inhibitors in clinical development. Expert Opin Investig Drugs. (2003) 12:943–54. doi: 10.1517/13543784.12.6.943
56. Dinsmore CJ, Bell IM. Inhibitors of farnesyltransferase and geranylgeranyltransferase-I for antitumor therapy: substrate-based design, conformational constraint and biological activity. Curr Top Med Chem. (2003) 3:1075–93. doi: 10.2174/1568026033452113
57. Karasic TB, Chiorean EG, Sebti SM, O’Dwyer PJ. A phase I study of GGTI-2418 (Geranylgeranyl transferase I inhibitor) in patients with advanced solid tumors. Target Oncol. (2019) 14:613–8. doi: 10.1007/s11523-019-00661-5
58. Sabt A, Tawfik HO, Khaleel EF, Badi RM, Ibrahim HAA, Elkaeed EB, et al. An overview of recent advancements in small molecules suppression of oncogenic signaling of K-RAS: an updated review. Mol Divers. (2024) 1–28. doi: 10.1007/s11030-023-10777-6
59. Maza NA, Schiesser WE, Calvert PD. An intrinsic compartmentalization code for peripheral membrane proteins in photoreceptor neurons. J Cell Biol. (2019) 218:3753–72. doi: 10.1083/jcb.201906024
60. Sarkar H, Moosajee M. Choroideremia: molecular mechanisms and therapies. Trends Mol Med. (2022) 28:378–87. doi: 10.1016/j.molmed.2022.02.011
61. Mitsios A, Dubis AM, Moosajee M. Choroideremia: from genetic and clinical phenotyping to gene therapy and future treatments. Ther Adv ophthalmol. (2018) 10:2515841418817490. doi: 10.1177/2515841418817490
62. Preising MN, Wegscheider E, Friedburg C, Poloschek CM, Wabbels BK, Lorenz B. Fundus autofluorescence in carriers of choroideremia and correlation with electrophysiologic and psychophysical data. Ophthalmology. (2009) 116:1201–9. e2. doi: 10.1016/j.ophtha.2009.01.016
63. Seabra MC, Brown MS, Goldstein JL. Retinal degeneration in choroideremia: deficiency of rab geranylgeranyl transferase. Science. (1993) 259:377–81. doi: 10.1126/science.8380507
64. Andres DA, Seabra MC, Brown MS, Armstrong SA, Smeland TE, Cremers FP, et al. cDNA cloning of component A of Rab geranylgeranyl transferase and demonstration of its role as a Rab escort protein. Cell. (1993) 73:1091–9. doi: 10.1016/0092-8674(93)90639-8
65. Preising M, Ayuso C. Rab escort protein 1 (REP1) in intracellular traffic: a functional and pathophysiological overview. Ophthal Genet. (2004) 25:101–10. doi: 10.1080/13816810490514333
66. MacDonald I, Chan CC, Hiriyanna K, Shen D, Fariss R. REP–1 localization in the eye. Invest Ophthalmol Visual Sci. (2005) 46:540.
67. Cremers FP, Armstrong SA, Seabra MC, Brown MS, Goldstein JL. REP-2, a Rab escort protein encoded by the choroideremia-like gene. J Biol Chem. (1994) 269:2111–7. doi: 10.1016/S0021-9258(17)42142-9
68. Meng EC, Goddard TD, Pettersen EF, Couch GS, Pearson ZJ, Morris JH, et al. UCSF ChimeraX: Tools for structure building and analysis. Protein Sci. (2023) 32:e4792. doi: 10.1002/pro.4792
69. Larijani B, Hume AN, Tarafder AK, Seabra MC. Multiple factors contribute to inefficient prenylation of Rab27a in Rab prenylation diseases. J Biol Chem. (2003) 278:46798–804. doi: 10.1074/jbc.M307799200
70. Cunha DL, Richardson R, Tracey-White D, Abbouda A, Mitsios A, Horneffer-van der Sluis V, et al. REP1 deficiency causes systemic dysfunction of lipid metabolism and oxidative stress in choroideremia. JCI Insight. (2021) 6:e146934. doi: 10.1172/jci.insight.146934
71. Hannun YA, Obeid LM. Sphingolipids and their metabolism in physiology and disease. Nat Rev Mol Cell Biol. (2018) 19:175–91. doi: 10.1038/nrm.2017.107
72. Tolmachova T, Tolmachov OE, Barnard AR, de Silva SR, Lipinski DM, Walker NJ, et al. Functional expression of Rab escort protein 1 following AAV2-mediated gene delivery in the retina of choroideremia mice and human cells ex vivo. J Mol Med (Berl). (2013) 91:825–37. doi: 10.1007/s00109-013-1006-4
73. Tolmachova T, Tolmachov OE, Wavre-Shapton ST, Tracey-White D, Futter CE, Seabra MC. CHM/REP1 cDNA delivery by lentiviral vectors provides functional expression of the transgene in the retinal pigment epithelium of choroideremia mice. J Gene Med. (2012) 14:158–68. doi: 10.1002/jgm.1652
74. Miyoshi H, Takahashi M, Gage FH, Verma IM. Stable and efficient gene transfer into the retina using an HIV-based lentiviral vector. Proc Natl Acad Sci U S A. (1997) 94:10319–23. doi: 10.1073/pnas.94.19.10319
75. Dimopoulos IS, Chan S, MacLaren RE, MacDonald IM. Pathogenic mechanisms and the prospect of gene therapy for choroideremia. Expert Opin Orphan Drugs. (2015) 3:787–98. doi: 10.1517/21678707.2015.1046434
76. MacLaren RE, Fischer MD, Gow JA, Lam BL, Sankila EK, Girach A, et al. Subretinal timrepigene emparvovec in adult men with choroideremia: a randomized phase 3 trial. Nat Med. (2023) 29:2464–72. doi: 10.1038/s41591-023-02520-3
77. MacLaren RE, Groppe M, Barnard AR, Cottriall CL, Tolmachova T, Seymour L, et al. Retinal gene therapy in patients with choroideremia: initial findings from a phase 1/2 clinical trial. Lancet. (2014) 383:1129–37. doi: 10.1016/S0140-6736(13)62117-0
78. Trapani I, Auricchio A. Has retinal gene therapy come of age? From bench to bedside and back to bench. Hum Mol Genet. (2019) 28:R108–R18. doi: 10.1093/hmg/ddz130
79. Rodrigues GA, Shalaev E, Karami TK, Cunningham J, Slater NKH, Rivers HM. Pharmaceutical development of AAV-based gene therapy products for the eye. Pharm Res. (2018) 36:29. doi: 10.1007/s11095-018-2554-7
80. Anand V, Barral DC, Zeng Y, Brunsmann F, Maguire AM, Seabra MC, et al. Gene therapy for choroideremia: in vitro rescue mediated by recombinant adenovirus. Vision Res. (2003) 43:919–26. doi: 10.1016/S0042-6989(02)00389-9
81. Vasireddy V, Mills JA, Gaddameedi R, Basner-Tschakarjan E, Kohnke M, Black AD, et al. AAV-mediated gene therapy for choroideremia: preclinical studies in personalized models. PloS One. (2013) 8:e61396. doi: 10.1371/journal.pone.0061396
82. Toualbi L, Toms M, Almeida PV, Harbottle R, Moosajee M. Gene augmentation of CHM using non-viral episomal vectors in models of choroideremia. Int J Mol Sci. (2023) 24:15225. doi: 10.3390/ijms242015225
83. Conti M, Beavo J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem. (2007) 76:481–511. doi: 10.1146/annurev.biochem.76.060305.150444
84. Cote RH. Photoreceptor phosphodiesterase (PDE6): activation and inactivation mechanisms during visual transduction in rods and cones. Pflugers Arch. (2021) 473:1377–91. doi: 10.1007/s00424-021-02562-x
85. Tsang SH, Gouras P, Yamashita CK, Kjeldbye H, Fisher J, Farber DB, et al. Retinal degeneration in mice lacking the gamma subunit of the rod cGMP phosphodiesterase. Science. (1996) 272:1026–9. doi: 10.1126/science.272.5264.1026
86. Gopalakrishna KN, Boyd K, Artemyev NO. Mechanisms of mutant PDE6 proteins underlying retinal diseases. Cell Signal. (2017) 37:74–80. doi: 10.1016/j.cellsig.2017.06.002
87. Singh S, Srivastava D, Boyd K, Artemyev NO. Reconstitution of the phosphodiesterase 6 maturation process important for photoreceptor cell function. J Biol Chem. (2024) 300:105576. doi: 10.1016/j.jbc.2023.105576
88. Anant JS, Ong OC, Xie HY, Clarke S, O’Brien PJ, Fung BK. In vivo differential prenylation of retinal cyclic GMP phosphodiesterase catalytic subunits. J Biol Chem. (1992) 267:687–90. doi: 10.1016/S0021-9258(18)48336-6
89. Christiansen JR, Ramamurthy V. Greasing the protein biosynthesis machinery of photoreceptor neurons: role for postprenylation processing of proteins. Cell logist. (2012) 2:15–9. doi: 10.4161/cl.19804
90. Cook TA, Ghomashchi F, Gelb MH, Florio SK, Beavo JA. Binding of the delta subunit to rod phosphodiesterase catalytic subunits requires methylated, prenylated C-termini of the catalytic subunits. Biochemistry. (2000) 39:13516–23. doi: 10.1021/bi001070l
91. Majumder A, Gopalakrishna KN, Cheguru P, Gakhar L, Artemyev NO. Interaction of aryl hydrocarbon receptor-interacting protein-like 1 with the farnesyl moiety. J Biol Chem. (2013) 288:21320–8. doi: 10.1074/jbc.M113.476242
92. Christiansen JR, Kolandaivelu S, Bergo MO, Ramamurthy V. RAS-converting enzyme 1-mediated endoproteolysis is required for trafficking of rod phosphodiesterase 6 to photoreceptor outer segments. Proc Natl Acad Sci U S A. (2011) 108:8862–6. doi: 10.1073/pnas.1103627108
93. Christiansen JR, Pendse ND, Kolandaivelu S, Bergo MO, Young SG, Ramamurthy V. Deficiency of isoprenylcysteine carboxyl methyltransferase (ICMT) leads to progressive loss of photoreceptor function. J Neurosci. (2016) 36:5107–14. doi: 10.1523/JNEUROSCI.0176-16.2016
94. McLaughlin ME, Sandberg MA, Berson EL, Dryja TP. Recessive mutations in the gene encoding the beta-subunit of rod phosphodiesterase in patients with retinitis pigmentosa. Nat Genet. (1993) 4:130–4. doi: 10.1038/ng0693-130
95. Dryja TP, Li T. Molecular genetics of retinitis pigmentosa. Hum Mol Genet. (1995) 4:1739–43. doi: 10.1093/hmg/4.suppl_1.1739
96. Moakedi F, Aljammal R, Poria D, Saravanan T, Rhodes SB, Reid C, et al. Prenylation is essential for the enrichment of cone phosphodiesterase-6 (PDE6) in outer segments and efficient cone phototransduction. Hum Mol Genet. (2023) 32:2735–50. doi: 10.1093/hmg/ddad108
97. Gillespie PG, Prusti RK, Apel ED, Beavo JA. A soluble form of bovine rod photoreceptor phosphodiesterase has a novel 15-kDa subunit. J Biol Chem. (1989) 264:12187–93. doi: 10.1016/S0021-9258(18)63839-6
98. Florio SK, Prusti RK, Beavo JA. Solubilization of membrane-bound rod phosphodiesterase by the rod phosphodiesterase recombinant delta subunit. J Biol Chem. (1996) 271:24036–47. doi: 10.1074/jbc.271.39.24036
99. Baehr W. Membrane protein transport in photoreceptors: the function of PDEdelta: the Proctor lecture. Invest Ophthalmol Vis Sci. (2014) 55:8653–66. doi: 10.1167/iovs.14-16066
100. Hanzal-Bayer M, Renault L, Roversi P, Wittinghofer A, Hillig RC. The complex of Arl2-GTP and PDEδ: from structure to function. EMBO J. (2002) 21:2095–106. doi: 10.1093/emboj/21.9.2095
101. Zhang H, Li S, Doan T, Rieke F, Detwiler P, Frederick J, et al. Deletion of PrBP/δ impedes transport of GRK1 and PDE6 catalytic subunits to photoreceptor outer segments. Proc Natl Acad Sci. (2007) 104:8857–62. doi: 10.1073/pnas.0701681104
102. Zhang H, Constantine R, Frederick JM, Baehr W. The prenyl-binding protein PrBP/delta: a chaperone participating in intracellular trafficking. Vision Res. (2012) 75:19–25. doi: 10.1016/j.visres.2012.08.013
103. Linari M, Ueffing M, Manson F, Wright A, Meitinger T, Becker J. The retinitis pigmentosa GTPase regulator, RPGR, interacts with the delta subunit of rod cyclic GMP phosphodiesterase. Proc Natl Acad Sci U S A. (1999) 96:1315–20. doi: 10.1073/pnas.96.4.1315
104. Lee JJ, Seo S. PDE6D binds to the C-terminus of RPGR in a prenylation-dependent manner. EMBO Rep. (2015) 16:1581–2. doi: 10.15252/embr.201541220
105. Thomas S, Wright KJ, Le Corre S, Micalizzi A, Romani M, Abhyankar A, et al. A homozygous PDE6D mutation in Joubert syndrome impairs targeting of farnesylated INPP5E protein to the primary cilium. Hum Mutat. (2014) 35:137–46. doi: 10.1002/humu.2014.35.issue-1
106. Wang SF, Kowal TJ, Ning K, Koo EB, Wu AY, Mahajan VB, et al. Review of ocular manifestations of Joubert syndrome. Genes (Basel). (2018) 9:605. doi: 10.3390/genes9120605
107. Jacoby M, Cox JJ, Gayral S, Hampshire DJ, Ayub M, Blockmans M, et al. INPP5E mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse. Nat Genet. (2009) 41:1027–31. doi: 10.1038/ng.427
108. Kösling SK, Fansa EK, Maffini S, Wittinghofer A. Mechanism and dynamics of INPP5E transport into and inside the ciliary compartment. Biol Chem. (2018) 399:277–92. doi: 10.1515/hsz-2017-0226
109. Cilleros-Rodriguez D, Martin-Morales R, Barbeito P, Deb Roy A, Loukil A, Sierra-Rodero B, et al. Multiple ciliary localization signals control INPP5E ciliary targeting. Elife. (2022) 11:e78383. doi: 10.7554/eLife.78383.sa2
110. Faber S, Letteboer SJF, Junger K, Butcher R, Tammana TVS, van Beersum SEC, et al. PDE6D mediates trafficking of prenylated proteins NIM1K and UBL3 to primary cilia. Cells. (2023) 12:312. doi: 10.3390/cells12020312
111. Sohocki MM, Bowne SJ, Sullivan LS, Blackshaw S, Cepko CL, Payne AM, et al. Mutations in a new photoreceptor-pineal gene on 17p cause Leber congenital amaurosis. Nat Genet. (2000) 24:79–83. doi: 10.1038/71732
112. Sacristan-Reviriego A, Le HM, Georgiou M, Meunier I, Bocquet B, Roux AF, et al. Clinical and functional analyses of AIPL1 variants reveal mechanisms of pathogenicity linked to different forms of retinal degeneration. Sci Rep. (2020) 10:17520. doi: 10.1038/s41598-020-74516-9
113. Heher KL, Traboulsi EI, Maumenee IH. The natural history of Leber’s congenital amaurosis. Age-related findings in 35 patients. Ophthalmology. (1992) 99:241–5. doi: 10.1016/S0161-6420(92)31985-2
114. Aboshiha J, Dubis AM, van der Spuy J, Nishiguchi KM, Cheeseman EW, Ayuso C, et al. Preserved outer retina in AIPL1 Leber’s congenital amaurosis: implications for gene therapy. Ophthalmology. (2015) 122:862–4. doi: 10.1016/j.ophtha.2014.11.019
115. Ramamurthy V, Niemi GA, Reh TA, Hurley JB. Leber congenital amaurosis linked to AIPL1: a mouse model reveals destabilization of cGMP phosphodiesterase. Proc Natl Acad Sci U S A. (2004) 101:13897–902. doi: 10.1073/pnas.0404197101
116. Liu X, Bulgakov OV, Wen XH, Woodruff ML, Pawlyk B, Yang J, et al. AIPL1, the protein that is defective in Leber congenital amaurosis, is essential for the biosynthesis of retinal rod cGMP phosphodiesterase. Proc Natl Acad Sci U S A. (2004) 101:13903–8. doi: 10.1073/pnas.0405160101
117. Gopalakrishna KN, Boyd K, Yadav RP, Artemyev NO. Aryl hydrocarbon receptor-interacting protein-like 1 is an obligate chaperone of phosphodiesterase 6 and is assisted by the γ-subunit of its client. J Biol Chem. (2016) 291:16282–91. doi: 10.1074/jbc.M116.737593
118. Yadav RP, Gakhar L, Yu L, Artemyev NO. Unique structural features of the AIPL1-FKBP domain that support prenyl lipid binding and underlie protein malfunction in blindness. Proc Natl Acad Sci U S A. (2017) 114:E6536–e45. doi: 10.1073/pnas.1704782114
119. Hidalgo-de-Quintana J, Evans RJ, Cheetham ME, van der Spuy J. The Leber congenital amaurosis protein AIPL1 functions as part of a chaperone heterocomplex. Invest Ophthalmol Vis Sci. (2008) 49:2878–87. doi: 10.1167/iovs.07-1576
120. Yadav RP, Majumder A, Gakhar L, Artemyev NO. Extended conformation of the proline-rich domain of human aryl hydrocarbon receptor-interacting protein-like 1: implications for retina disease. J Neurochem. (2015) 135:165–75. doi: 10.1111/jnc.13223
121. Aguilà M, Bevilacqua D, McCulley C, Schwarz N, Athanasiou D, Kanuga N, et al. Hsp90 inhibition protects against inherited retinal degeneration. Hum Mol Genet. (2014) 23:2164–75. doi: 10.1093/hmg/ddt613
122. van der Spuy J, Chapple JP, Clark BJ, Luthert PJ, Sethi CS, Cheetham ME. The Leber congenital amaurosis gene product AIPL1 is localized exclusively in rod photoreceptors of the adult human retina. Hum Mol Genet. (2002) 11:823–31. doi: 10.1093/hmg/11.7.823
123. Yadav RP, Boyd K, Artemyev NO. Molecular insights into the maturation of phosphodiesterase 6 by the specialized chaperone complex of HSP90 with AIPL1. J Biol Chem. (2022) 298:101620. doi: 10.1016/j.jbc.2022.101620
124. Arshavsky VY, Burns ME. Current understanding of signal amplification in phototransduction. Cell Logist. (2014) 4:e29390. doi: 10.4161/cl.29390
125. Neubert TA, Johnson RS, Hurley JB, Walsh KA. The rod transducin alpha subunit amino terminus is heterogeneously fatty acylated. J Biol Chem. (1992) 267:18274–7. doi: 10.1016/S0021-9258(19)36955-8
126. Fukada Y, Takao T, Ohguro H, Yoshizawa T, Akino T, Shimonishi Y. Farnesylated gamma-subunit of photoreceptor G protein indispensable for GTP-binding. Nature. (1990) 346:658–60. doi: 10.1038/346658a0
127. Wedegaertner PB, Wilson PT, Bourne HR. Lipid modifications of trimeric G proteins. J Biol Chem. (1995) 270:503–6. doi: 10.1074/jbc.270.2.503
128. Iniguez-Lluhi JA, Simon MI, Robishaw JD, Gilman AG. G protein beta gamma subunits synthesized in Sf9 cells. Functional characterization and the significance of prenylation of gamma. J Biol Chem. (1992) 267:23409–17. doi: 10.1016/S0021-9258(18)50106-X
129. Kassai H, Aiba A, Nakao K, Nakamura K, Katsuki M, Xiong WH, et al. Farnesylation of retinal transducin underlies its translocation during light adaptation. Neuron. (2005) 47:529–39. doi: 10.1016/j.neuron.2005.07.025
130. Lobanova ES, Finkelstein S, Herrmann R, Chen YM, Kessler C, Michaud NA, et al. Transducin gamma-subunit sets expression levels of alpha- and beta-subunits and is crucial for rod viability. J Neurosci. (2008) 28:3510–20. doi: 10.1523/JNEUROSCI.0338-08.2008
131. Kolesnikov AV, Rikimaru L, Hennig AK, Lukasiewicz PD, Fliesler SJ, Govardovskii VI, et al. G-protein betagamma-complex is crucial for efficient signal amplification in vision. J Neurosci. (2011) 31:8067–77. doi: 10.1523/JNEUROSCI.0174-11.2011
132. Brooks C, Murphy J, Belcastro M, Heller D, Kolandaivelu S, Kisselev O, et al. Farnesylation of the transducin G protein gamma subunit is a prerequisite for its ciliary targeting in rod photoreceptors. Front Mol Neurosci. (2018) 11. doi: 10.3389/fnmol.2018.00016
133. Hutagalung AH, Novick PJ. Role of Rab GTPases in membrane traffic and cell physiology. Physiol Rev. (2011) 91:119–49. doi: 10.1152/physrev.00059.2009
134. Pfeffer SR. Rab GTPase regulation of membrane identity. Curr Opin Cell Biol. (2013) 25:414–9. doi: 10.1016/j.ceb.2013.04.002
135. Brauers A, Schurmann A, Massmann S, Muhl-Zurbes P, Becker W, Kainulainen H, et al. Alternative mRNA splicing of the novel GTPase Rab28 generates isoforms with different C-termini. Eur J Biochem. (1996) 237:833–40. doi: 10.1111/j.1432-1033.1996.0833p.x
136. Lee G-I, Lee C, Subramanian S, Kim NK, Ki C-S, Park W-Y, et al. A novel likely pathogenic variant in the RAB28 gene in a Korean patient with cone–rod dystrophy. Ophthal Genet. (2017) 38:587–9. doi: 10.1080/13816810.2017.1301965
137. Roosing S, Rohrschneider K, Beryozkin A, Sharon D, Weisschuh N, Staller J, et al. Mutations in RAB28, encoding a farnesylated small GTPase, are associated with autosomal-recessive cone-rod dystrophy. Am J Hum Genet. (2013) 93:110–7. doi: 10.1016/j.ajhg.2013.05.005
138. Riveiro-Alvarez R, Xie YA, Lopez-Martinez MA, Gambin T, Perez-Carro R, Avila-Fernandez A, et al. New mutations in the RAB28 gene in 2 Spanish families with cone-rod dystrophy. JAMA Ophthalmol. (2015) 133:133–9. doi: 10.1001/jamaophthalmol.2014.4266
139. Ying G, Boldt K, Ueffing M, Gerstner CD, Frederick JM, Baehr W. The small GTPase RAB28 is required for phagocytosis of cone outer segments by the murine retinal pigmented epithelium. J Biol Chem. (2018) 293:17546–58. doi: 10.1074/jbc.RA118.005484
140. Kanzaki Y, Fujita H, Sato K, Hosokawa M, Matsumae H, Shiraga F, et al. KCNJ13 gene deletion impairs cell alignment and phagocytosis in retinal pigment epithelium derived from human-induced pluripotent stem cells. Invest Ophthalmol Vis Sci. (2020) 61:38. doi: 10.1167/iovs.61.5.38
141. Stone EM, Andorf JL, Whitmore SS, DeLuca AP, Giacalone JC, Streb LM, et al. Clinically focused molecular investigation of 1000 consecutive families with inherited retinal disease. Ophthalmology. (2017) 124:1314–31. doi: 10.1016/j.ophtha.2017.04.008
142. Kirschner R, Rosenberg T, Schultz-Heienbrok R, Lenzner S, Feil S, Roepman R, et al. RPGR transcription studies in mouse and human tissues reveal a retina-specific isoform that is disrupted in a patient with X-linked retinitis pigmentosa. Hum Mol Genet. (1999) 8:1571–8. doi: 10.1093/hmg/8.8.1571
143. He S, Parapuram SK, Hurd TW, Behnam B, Margolis B, Swaroop A, et al. Retinitis Pigmentosa GTPase Regulator (RPGR) protein isoforms in mammalian retina: insights into X-linked Retinitis Pigmentosa and associated ciliopathies. Vision Res. (2008) 48:366–76. doi: 10.1016/j.visres.2007.08.005
144. Rao KN, Zhang W, Li L, Anand M, Khanna H. Prenylated retinal ciliopathy protein RPGR interacts with PDE6δ and regulates ciliary localization of Joubert syndrome-associated protein INPP5E. Hum Mol Genet. (2016) 25:4533–45. doi: 10.1093/hmg/ddw281
145. Fansa EK, O’Reilly NJ, Ismail S, Wittinghofer A. The N- and C-terminal ends of RPGR can bind to PDE6δ. EMBO Rep. (2015) 16:1583–5. doi: 10.15252/embr.201541404
146. Hong DH, Pawlyk BS, Adamian M, Sandberg MA, Li T. A single, abbreviated RPGR-ORF15 variant reconstitutes RPGR function in vivo. Invest Ophthalmol Vis Sci. (2005) 46:435–41. doi: 10.1167/iovs.04-1065
147. Wright RN, Hong DH, Perkins B. Misexpression of the constitutive Rpgr(ex1–19) variant leads to severe photoreceptor degeneration. Invest Ophthalmol Vis Sci. (2011) 52:5189–201. doi: 10.1167/iovs.11-7470
148. Zhang Q, Giacalone JC, Searby C, Stone EM, Tucker BA, Sheffield VC. Disruption of RPGR protein interaction network is the common feature of RPGR missense variations that cause XLRP. Proc Natl Acad Sci U S A. (2019) 116:1353–60. doi: 10.1073/pnas.1817639116
149. Drenth JP, Cuisset L, Grateau G, Vasseur C, van de Velde-Visser SD, de Jong JG, et al. Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. International Hyper-IgD Study Group. Nat Genet. (1999) 22:178–81. doi: 10.1038/9696
150. van der Burgh R, Ter Haar NM, Boes ML, Frenkel J. Mevalonate kinase deficiency, a metabolic autoinflammatory disease. Clin Immunol. (2013) 147:197–206. doi: 10.1016/j.clim.2012.09.011
151. Balgobind B, Wittebol-Post D, Frenkel J. Retinitis pigmentosa in mevalonate kinase deficiency. J Inherit Metab Dis. (2005) 28:1143–5. doi: 10.1007/s10545-005-0178-7
152. Prietsch V, Mayatepek E, Krastel H, Haas D, Zundel D, Waterham HR, et al. Mevalonate kinase deficiency: enlarging the clinical and biochemical spectrum. Pediatrics. (2003) 111:258–61. doi: 10.1542/peds.111.2.258
153. Simon A, Kremer HP, Wevers RA, Scheffer H, De Jong JG, van der Meer JW, et al. Mevalonate kinase deficiency: Evidence for a phenotypic continuum. Neurology. (2004) 62:994–7. doi: 10.1212/01.WNL.0000115390.33405.F7
154. Siemiatkowska AM, van den Born LI, van Hagen PM, Stoffels M, Neveling K, Henkes A, et al. Mutations in the mevalonate kinase (MVK) gene cause nonsyndromic retinitis pigmentosa. Ophthalmology. (2013) 120:2697–705. doi: 10.1016/j.ophtha.2013.07.052
155. Munoz MA, Jurczyluk J, Simon A, Hissaria P, Arts RJW, Coman D, et al. Defective protein prenylation in a spectrum of patients with mevalonate kinase deficiency. Front Immunol. (2019) 10:1900. doi: 10.3389/fimmu.2019.01900
156. Munoz MA, Skinner OP, Masle-Farquhar E, Jurczyluk J, Xiao Y, Fletcher EK, et al. Increased core body temperature exacerbates defective protein prenylation in mouse models of mevalonate kinase deficiency. J Clin Invest. (2022) 132:e160929. doi: 10.1172/JCI160929
157. Rao SR, Pittler SJ, Fliesler SJ. Perspectives on retinal dolichol metabolism, and visual deficits in dolichol metabolism-associated inherited disorders. Adv Exp Med Biol. (2023) 1415:449–56. doi: 10.1007/978-3-031-27681-1_66
158. Park EJ, Grabinska KA, Guan Z, Stranecky V, Hartmannova H, Hodanova K, et al. Mutation of Nogo-B receptor, a subunit of cis-prenyltransferase, causes a congenital disorder of glycosylation. Cell Metab. (2014) 20:448–57. doi: 10.1016/j.cmet.2014.06.016
159. Harrison KD, Miao RQ, Fernandez-Hernando C, Suarez Y, Davalos A, Sessa WC. Nogo-B receptor stabilizes Niemann-Pick type C2 protein and regulates intracellular cholesterol trafficking. Cell Metab. (2009) 10:208–18. doi: 10.1016/j.cmet.2009.07.003
160. Zhao B, Hu W, Kumar S, Gonyo P, Rana U, Liu Z, et al. The Nogo-B receptor promotes Ras plasma membrane localization and activation. Oncogene. (2017) 36:3406–16. doi: 10.1038/onc.2016.484
161. Maurer-Stroh S, Eisenhaber F. Refinement and prediction of protein prenylation motifs. Genome Biol. (2005) 6:R55. doi: 10.1186/gb-2005-6-6-r55
162. London N, Lamphear CL, Hougland JL, Fierke CA, Schueler-Furman O. Identification of a novel class of farnesylation targets by structure-based modeling of binding specificity. PloS Comput Biol. (2011) 7:e1002170. doi: 10.1371/journal.pcbi.1002170
163. Hougland JL, Hicks KA, Hartman HL, Kelly RA, Watt TJ, Fierke CA. Identification of novel peptide substrates for protein farnesyltransferase reveals two substrate classes with distinct sequence selectivities. J Mol Biol. (2010) 395:176–90. doi: 10.1016/j.jmb.2009.10.038
164. Berger BM, Kim JH, Hildebrandt ER, Davis IC, Morgan MC, Hougland JL, et al. Protein isoprenylation in yeast targets COOH-terminal sequences not adhering to the CaaX consensus. Genetics. (2018) 210:1301–16. doi: 10.1534/genetics.118.301454
165. Orozco LD, Owen LA, Hofmann J, Stockwell AD, Tao J, Haller S, et al. A systems biology approach uncovers novel disease mechanisms in age-related macular degeneration. Cell Genom. (2023) 3:100302. doi: 10.1016/j.xgen.2023.100302
166. Kramer A, Green J, Pollard J Jr., Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics. (2014) 30:523–30. doi: 10.1093/bioinformatics/btt703
167. Abramson J, Adler J, Dunger J, Evans R, Green T, Pritzel A, et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature. (2024) 630:493–500. doi: 10.1038/s41586-024-07487-w
168. Kim E, Ambroziak P, Otto JC, Taylor B, Ashby M, Shannon K, et al. Disruption of the mouse Rce1 gene results in defective Ras processing and mislocalization of Ras within cells. J Biol Chem. (1999) 274:8383–90. doi: 10.1074/jbc.274.13.8383
169. Hancu G, Simon B, Rusu A, Mircia E, Gyéresi Á. Principles of micellar electrokinetic capillary chromatography applied in pharmaceutical analysis. Adv Pharm Bullet. (2013) 3:1. doi: 10.5681/apb.2013.001
170. Gong F, Yang H, Sun W, Cao J, Liu W. Development and validation of a micellar electrokinetic capillary chromatography method for the determination of goserelin and related substances. Electrophoresis. (2016) 37:623–9. doi: 10.1002/elps.201500328
171. Shala-Lawrence A, Blanden MJ, Krylova SM, Gangopadhyay SA, Beloborodov SS, Hougland JL, et al. Simultaneous analysis of a non-lipidated protein and its lipidated counterpart: enabling quantitative investigation of protein lipidation’s impact on cellular regulation. Anal Chem. (2017) 89:13502–7. doi: 10.1021/acs.analchem.7b03846
172. Troutman JM, Roberts MJ, Andres DA, Spielmann HP. Tools to analyze protein farnesylation in cells. Bioconjug Chem. (2005) 16:1209–17. doi: 10.1021/bc050068+
173. Troutman JM, Subramanian T, Andres DA, Spielmann HP. Selective modification of CaaX peptides with ortho-substituted anilinogeranyl lipids by protein farnesyl transferase: competitive substrates and potent inhibitors from a library of farnesyl diphosphate analogues. Biochemistry. (2007) 46:11310–21. doi: 10.1021/bi700516m
174. Nguyen UT, Guo Z, Delon C, Wu Y, Deraeve C, Franzel B, et al. Analysis of the eukaryotic prenylome by isoprenoid affinity tagging. Nat Chem Biol. (2009) 5:227–35. doi: 10.1038/nchembio.149
175. Hosokawa A, Wollack JW, Zhang Z, Chen L, Barany G, Distefano MD. Evaluation of an alkyne-containing analogue of farnesyl diphosphate as a dual substrate for protein-prenyltransferases. Int J Pept Res Ther. (2007) 13:345–54. doi: 10.1007/s10989-007-9090-3
176. DeGraw AJ, Palsuledesai C, Ochocki JD, Dozier JK, Lenevich S, Rashidian M, et al. Evaluation of alkyne-modified isoprenoids as chemical reporters of protein prenylation. Chem Biol Drug Des. (2010) 76:460–71. doi: 10.1111/j.1747-0285.2010.01037.x
177. Palsuledesai CC, Ochocki JD, Markowski TW, Distefano MD. A combination of metabolic labeling and 2D-DIGE analysis in response to a farnesyltransferase inhibitor facilitates the discovery of new prenylated proteins. Mol Biosyst. (2014) 10:1094–103. doi: 10.1039/C3MB70593E
178. Charron G, Li MM, MacDonald MR, Hang HC. Prenylome profiling reveals S-farnesylation is crucial for membrane targeting and antiviral activity of ZAP long-isoform. Proc Natl Acad Sci U S A. (2013) 110:11085–90. doi: 10.1073/pnas.1302564110
179. Gisselberg JE, Zhang L, Elias JE, Yeh E. The prenylated proteome of plasmodium falciparum reveals pathogen-specific prenylation activity and drug mechanism-of-action. Mol Cell Proteom. (2017) 16:S54–64. doi: 10.1074/mcp.M116.064550
180. Suazo KF, Schaber C, Palsuledesai CC, Odom John AR, Distefano MD. Global proteomic analysis of prenylated proteins in Plasmodium falciparum using an alkyne-modified isoprenoid analogue. Sci Rep. (2016) 6:38615. doi: 10.1038/srep38615
181. Storck EM, Morales-Sanfrutos J, Serwa RA, Panyain N, Lanyon-Hogg T, Tolmachova T, et al. Dual chemical probes enable quantitative system-wide analysis of protein prenylation and prenylation dynamics. Nat Chem. (2019) 11:552–61. doi: 10.1038/s41557-019-0237-6
182. Jeong A, Auger SA, Maity S, Fredriksen K, Zhong R, Li L, et al. In vivo prenylomic profiling in the brain of a transgenic mouse model of Alzheimer’s disease reveals increased prenylation of a key set of proteins. ACS Chem Biol. (2022) 17:2863–76. doi: 10.1021/acschembio.2c00486
183. Lakkaraju A, Umapathy A, Tan LX, Daniele L, Philp NJ, Boesze-Battaglia K, et al. The cell biology of the retinal pigment epithelium. Prog Retin Eye Res. (2020) 78:100846. doi: 10.1016/j.preteyeres.2020.100846
Keywords: post-translational modifications, mevalonate pathway, retina, prenylation, farnesylation, geranylgeranylation, inherited retinopathies
Citation: Ashok S and Ramachandra Rao S (2024) Updates on protein-prenylation and associated inherited retinopathies. Front. Ophthalmol. 4:1410874. doi: 10.3389/fopht.2024.1410874
Received: 01 April 2024; Accepted: 13 June 2024;
Published: 04 July 2024.
Edited by:
Daisy Y. Shu, University of New South Wales, AustraliaReviewed by:
Glenn Prazere Lobo, University of Minnesota Twin Cities, United StatesCopyright © 2024 Ashok and Ramachandra Rao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sriganesh Ramachandra Rao, c3JhbWFjaGFAYnVmZmFsby5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.