 Dwarkesh Amin
Dwarkesh Amin Takaaki Kuwajima
Takaaki Kuwajima- Department of Ophthalmology, The Louis J. Fox Center for Vision Restoration, The University of Pittsburgh School of Medicine, Pittsburgh, PA, United States
Retinal ganglion cells (RGCs) are the neurons in the retina which directly project to the brain and transmit visual information along the optic nerve. Glaucoma, one of the leading causes of blindness, is characterized by elevated intraocular pressure (IOP) and degeneration of the optic nerve, which is followed by RGC death. Currently, there are no clinical therapeutic drugs or molecular interventions that prevent RGC death outside of IOP reduction. In order to overcome these major barriers, an increased number of studies have utilized the following combined analytical methods: well-established rodent models of glaucoma including optic nerve injury models and transcriptomic gene expression profiling, resulting in the successful identification of molecules and signaling pathways relevant to RGC protection. In this review, we present a comprehensive overview of pathological features in a variety of animal models of glaucoma and top differentially expressed genes (DEGs) depending on disease progression, RGC subtypes, retinal regions or animal species. By comparing top DEGs among those different transcriptome profiles, we discuss whether commonly listed DEGs could be defined as potential novel therapeutic targets in glaucoma, which will facilitate development of future therapeutic neuroprotective strategies for treatments of human patients in glaucoma.
Introduction
Glaucoma is a retinal neurodegenerative disease that affects 64 million people worldwide. It is estimated that the number of glaucoma patients will increase in the future (1, 2). There are several types of glaucoma: primary glaucoma including open-angle glaucoma and angle-closure glaucoma, congenital glaucoma, and secondary glaucoma. Increased IOP levels in glaucoma patients can lead to optic nerve damage and RGC loss. However, patients with a form of primary open-angle glaucoma, low tension glaucoma, also develop optic nerve damage despite having normal IOP. Importantly, the common pathological change in all types of glaucoma is loss of axonal integrity and RGC death. Unfortunately, there are no FDA-approved drugs that prevent RGC death by acting directly on RGCs themselves. Identifying therapeutic targets that directly prevent RGC death and/or enhance the capacity of RGC regrowth after axonal insult is one of the crucial steps in developing treatments for all types of glaucoma.
Using a variety of rodent models of glaucoma, a number of studies have identified intrinsic and extrinsic molecules and signaling pathways that mediate RGC survival and death (3–17). Therapeutic interventions targeting these identified genes and molecular pathways could be promising therapies. However, whether effective neuroprotection in human glaucoma patients can be achieved by targeting the same molecular pathways studied in animal disease models remains unclear. Thus, comprehensive analyses to identify beneficial molecular targets which can be commonly effective in different animal models could help to establish therapeutic molecular interventions for glaucoma patients.
In this review, first we summarize existing rodent models of glaucoma and optic nerve degeneration. We then explore the powerful impact that transcriptomic technology has made on identifying new therapeutic targets. Several transcriptomic studies with RNA sequencing (RNA-seq) or microarray have uncovered differentially expressed genes (DEGs) in RGCs or whole retina depending on disease progression, retinal regions, animal species or RGC subtypes. Among them, we select nine studies in which the most highly upregulated and downregulated genes have been identified using their own statistical criteria such as FDR P-value <0.05 or P-value <0.05 compared to control conditions and also listed in the main or supplemental figures in each study. We exclude other studies which are redundant to the selected studies due to the use of same disease models. By manually comparing gene names among those top DEGs, we identify which DEGs are commonly listed across different transcriptome profiles. We then provide the summary of previously validated functions of those common DEGs in RGCs or other cells and their molecular mechanisms. Finally, we discuss whether DEGs identified in the rodent models of glaucoma could be applied for treatments of glaucoma patients.
Inherited and IOP-Induced Experimental Rodent Models of Glaucoma and Optic Nerve Degeneration
Genetically or spontaneously generated rodent models of glaucoma are a valuable resource for studying the genetic etiology of glaucoma and identifying future therapeutic treatments in human patients. The DBA/2J mouse strain is one of the most broadly utilized inbred mouse strains in studying glaucoma (18). DBA/2J mice begin displaying pigment dispersion, iris transillumination, iris stromal atrophy, posterior synechiae, and IOP elevation around 6 months of age. By 11 months, optic nerve atrophy, optic nerve cupping, and loss of optic nerve (60 - 90% loss) and RGCs (60 - 70% loss) are evident (16, 18–21). Genetic analysis in DBA/2J mice revealed that the iris pigment dispersion phenotype is caused by a premature stop codon mutation in the Gpnmb (GpnmbR150X) gene which encodes a transmembrane glycoprotein GPNMB. Iris stromal atrophy is caused by a mutation in the b allele of Tyrp gene (Tyrp1b), which encodes a melanosomal enzyme, tyrosinase related protein-1, TRP-1 (22, 23). DBA/2J-Gpnmb+ mice, which are homozygous for a wild-type allele of Gpnmb on a DBA/2J genetic background, do not develop elevated IOP and show no glaucomatous neurodegeneration with age (24). Another inbred mouse strain, YBR/EiJ (YBR) has been characterized as a form of pigmentary glaucoma: high IOP levels are observed at six to seven months of age, and about 60% of eyes display severe axon loss with extensive gliosis and optic nerve damage by 14 months of age (25). A transgenic mouse model of glaucoma, Tg-MYOCY437H, carrying a mutation of human MYOC (Y437H) mimics the pathophysiology of human MYOC-associated glaucoma (26–28). The Tg-MYOCY437H mouse line begins to exhibit elevation of IOP, RGC loss and axon degeneration from three to five months of age, resulting in 17.6% of RGC loss by three to five months of age and 30% of RGC loss by 12 to 14 months of age (29).
Several IOP-induced experimental models have been developed, providing the advantage of acute IOP elevation and faster neurodegeneration compared to inherited rodent models. There are four major IOP-induced experimental models: laser photocoagulation, injection of microbeads, injection of silicon oil into the anterior chamber or injection of hypertonic saline through episcleral veins. (i) Laser photocoagulation to the single or multiple locations in the anterior chamber commonly causes elevation of IOP within 7 – 10 days after injury and sustained for at least a few weeks (30–35). Laser photocoagulation of both limbal and episcleral veins induces 78% of axon loss four - five weeks after injury (33, 34). Similarly, concurrent laser treatments to trabecular meshwork and episcleral veins lead to 60% of axon loss six weeks after injury (30). However, laser treatments to trabecular meshwork alone cause only 20% of axon loss six weeks after injury (30) and only 41% of axon loss as late as 24 weeks after injury (31). Laser photocoagulation of two locations such as both limbal and episcleral veins or both trabecular meshwork and episcleral veins induce around 30% of RGC loss four - five weeks after injury (33, 34) and 60% of RGC loss nine weeks after injury (30). (ii) Injection of polystyrene microbeads to anterior chamber increases IOP levels within a week, but the magnitude of IOP elevation or persistence of high IOP levels varies depending on injection volume (36), size of microbeads (37) and animal strains (38). Extended elevations of IOP for two to four weeks after injections induce 20 - 40% of axon loss and 25 – 38% of RGC loss (36, 37, 39). (iii) Clinical reports that intravitreal injections of silicone oil, which is normally used to treat complex retinal detachments, can lead to high IOP elevation and post-operative secondary glaucoma in humans (40–42). This evidence supports the development of the silicone oil-based rodent model of glaucoma. Direct injections of silicone oil into the anterior chamber leads to rapid IOP elevation within a few days, and the levels remain high for eight weeks. 88% of peripheral RGC loss and 65% of axon loss are appreciated eight weeks after injections (43). (iv) Injections of hypertonic saline through episcleral veins lead to sclerosis of trabecular meshwork and anterior chamber angle, resulting into chronic and constant IOP elevation for a few weeks to months (44–47). Rodents with more than 70% of RGC loss clearly show progressive cupping and abnormal ERG results (47).
The rodent intraorbital optic nerve crush model has been broadly utilized as a model of traumatic optic neuropathy to identify the cellular and molecular mechanisms underlying RGC death and survival, axon degeneration, and axon regeneration (48). Optic nerve crush and ocular hypertension show differing degenerative phenotypes, such as the magnitude of RGC subtype death and topography of RGC loss (49). However, several studies have demonstrated that the optic nerve crush model and inherited or IOP-induced experimental rodent models of glaucoma trigger RGC degeneration through mediating the same downstream molecules, such as Bax (50, 51) and Jun (52, 53). Optic nerve crush causes rapid neurodegeneration, with 20%, 47% and 66% of RGC loss and 38%, 58% and 67% of axon loss at three, five and seven days after injury, respectively (54). The earlier optic nerve transection model (55) shows slightly more severe pan-RGC death than the optic nerve crush model, but the magnitude of death of RGC subtypes is comparable between the two models (56). Establishing multiple rodent models of glaucoma and optic neuropathies enables us to clarify the pathology as observed in glaucoma patients and to find future therapeutic molecular interventions.
Identification of Neuroprotective Genes by Comparative Transcriptome Analysis
I. Disease Progression
RGC loss and axonal degeneration in glaucoma are irreversible, and the magnitude of neurodegeneration worsens over time. In a common hypothesis, the balance between decreasing expression of neuroprotective genes and/or increasing expression of cell death-induced genes could decide the final neurodegenerative outcomes. Several studies have shown RNA-seq profiles at different time points of neurodegeneration in inherited and IOP-induced experimental rodent models of glaucoma (Figure 1A, Supplemental Table 1). [1] The retina of 3 month-old (before disease onset) and 8 month-old DBA/2J mouse (after IOP elevation) (57), [2] RGCs of the eye with mild or moderate IOP elevation (mild IOP increase:1< mmHg <4; moderate IOP increase: ≥4 mmHg) by injections of polystyrene microbeads and RGCs of the normal eye (58), and [3] the retinal ganglion cell layer (RGCL) of the eye with IOP elevation by aqueous outflow obstruction following injecting hypertonic saline unilaterally into episcleral veins and RGCL of the normal eye (59), [4] the retina two days after optic nerve transection and the uninjured retina (60), [5] the retina two days after optic nerve crush and the uninjured retina (61), and [6] the retina one day after optic nerve crush and the uninjured retina (62) have been utilized for RNA-seq or microarray analysis to identify DEGs in each study. First, we compared top DEGs listed in [1] – [6]. We found that crystallin superfamily members (Crygb, Cryba1, Crygs, Cryba2, Cryaa, Crybb1), Lcn2 (lipocalin 2), Lgals3 (galectin-3), Hmox1 (heme oxygenase-1), Ecel1 (endothelin converting enzyme like 1), Atf3 (activating transcription factor 3), Csrp3 (cysteine and glycine rich protein 3), and Serpina3n (serpin family A member 3) are commonly upregulated or downregulated (Figure 1B). Among these common DEGs, we summarize the information of expression and functions of crystallin superfamily members (Crygb, Cryba1, Crygs, Cryba2, Cryaa, Crybb1), Lcn2, Lgals3 Hmox1, Ecel1 and Atf3. We discuss whether these DEGs could be key common therapeutic targets for treatments of glaucoma.
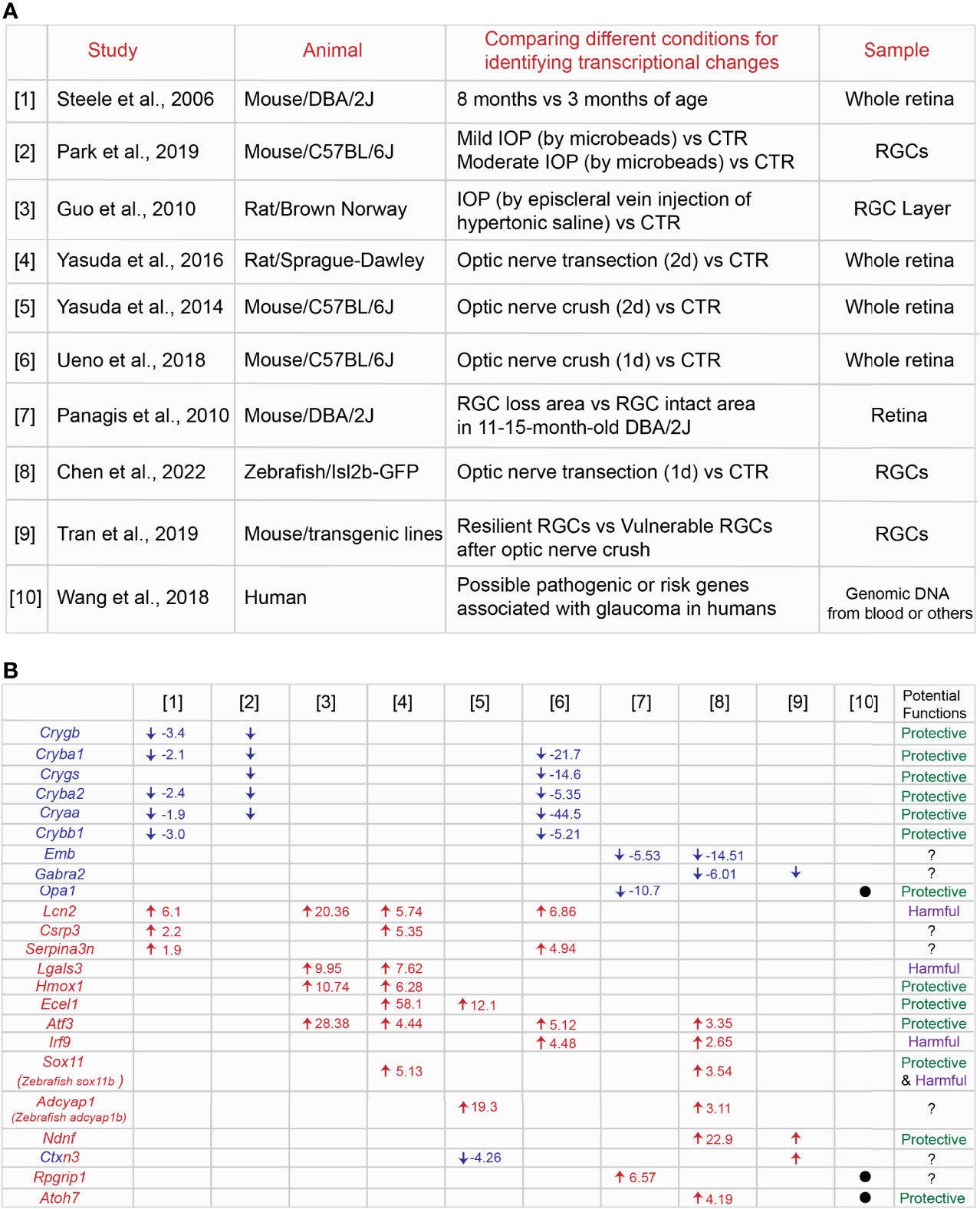
Figure 1 Identification of common genes among different transcriptome profiles at the specific conditions in the animal models of glaucoma. (A) Summary of nine [1] – [9] transcriptome studies in the retina or RGCs in a variety of animal models of glaucoma and [10] possible risk genes associated with glaucoma in humans. (B) Common genes listed across different transcriptome profiles and the fold change of each gene under the specific condition. Potential protective or harmful effects of common DEGs on RGCs in glaucoma have been discussed in the main text.
Crygb, Cryba1, Crygs, Cryba2, Cryaa, Crybb1 - Crystallin Superfamily Members
Crygb, Cryba2, Cryaa, Cryba2 are commonly downregulated in degenerating RGCs or retina as seen in [1] (57), [2] (58) and [6] (62) (Figure 1B). Crystallins include the α, β, and γ subtypes, and the superfamily members are expressed in normal RGCs, but they are downregulated in IOP-induced experimental models of glaucoma (63, 64). Although α-crystallins are downregulated after optic nerve crush, overexpression of αA- and αB-crystallins enhance RGC survival after injury (65). Injection of α-crystallin protein into the vitreous space after optic nerve crush also attenuate optic nerve degeneration (66). Crystallins family members carry out their anti-apoptotic functions through mediating different molecular pathways. They bind to Bax and Bcl-X and repress the translocation from the cytosol into mitochondria during staurosporine-induced cell death (59). They also prevent the activation of caspase 3 and activate PI3K (67) and attenuate endoplasmic reticulum stress (68). Microglial activation and TNF-α and iNOS release are repressed by crystallins (69). Thus, crystallins could be a potential regulator for RGC protection through mediating several molecular pathways in glaucoma.
Lcn2 (Lipocalin 2)
Lcn2 is commonly upregulated in neurodegeneration as shown in [1] (57), [3] (59), [4] (60) and [6] (62) (Figure 1B). Lipocalin-2 (encoded by Lcn2) is a secreted protein that belongs to the Lipocalins, a group of transporters of small lipophilic molecules, and the expression is elevated after administration of lipopolysaccharide (LPS) (70). Several studies have demonstrated that lipocalin-2 has neurotoxic effects. Recombinant lipocalin-2 protein induces apoptosis of neurons in vitro (71). Lipocalin-2 levels are elevated after intracerebral hemorrhage due to the brain injury, and loss of lipocalin-2 reduces microglial activation, brain swelling, brain atrophy, and neurologic deficits compared to control (72). Similarly, thrombin-induced brain swelling, blood-brain barrier disruption, neuronal death and neurologic deficits are markedly reduced by loss of lipocalin-2 (73). After transient middle cerebral artery occlusion, lipocalin-2 expression is upregulated in astrocytes and endothelial cells, and loss of lipocalin-2 attenuates infarct volume, neurologic deficit, and inflammatory response (74). Moreover, the expression is induced in reactive astrocytes in the rodent model of ALS, leading to neuronal death (75). Most recently, in the IOP-induced neurodegeneration in vitro model, recombinant lipocalin-2 protein leads to increased RGC death (76). Thus, lipocalin-2 could be a neurotoxic factor expressed in non-neuronal cells so that suppression of lipocalin-2 expression or blocking the functions could prevent RGCs from death in glaucoma.
Lgals3 (Galectin-3)
Another commonly upregulated gene in neurodegeneration is Lgals3 as seen in [3] (59) and [4] (60) (Figure 1B). Galectin-3 (Gal3) (encoded lgals3) is a member of the lectin family. In the ischemic injury model, Gal3 is expressed in activated microglial cells at the lesion site. Loss of Gal3 exacerbates ischemic damage and increases neuronal apoptosis after cerebral ischemia. This is mediated by an elevation of IL-6 and SOCS3 expression (77). Moreover, treatments with recombinant Gal3 protein reduce neuronal death and brain damage and improve post-ischemic functional recovery (78). This evidence indicates that Gal3 plays critical roles as a neuroprotective factor. However, neurotoxic roles for Gal-3 by immune cells have been also reported. Administration of neutralizing antibodies against galectin-3 attenuates the expression of pro-inflammatory markers such as IL-1β and IL-6 and exerts neuroprotection in the cortex and hippocampus after head injury (79). Loss of Gal3 also enhances RGC protection and reduces the number of degenerated axons after optic nerve crush (80). Taken together, Gal-3-mediated functions may differ depending on the stage of progression of injury and glaucoma.
Hmox1 (Heme Oxygenase-1)
Hmox1 is upregulated in neurodegeneration in [3] (59) and [4] (60) (Figure 1B). Heme oxygenase (HO) enzymes degrade heme to form biliverdin, iron, and carbon monoxide (CO) gas. Heme oxygenase-1 (HO-1, encoded by Hmox1) exerts neuronal and non-neuronal cell protection after visceral organ injury (81) and brain ischemic injury (82). In the retina, overexpression of Hmox1 adenovirus attenuates RGC loss after pressure-induced ischemia (83). Thus, Hmox1 could play a role as a critical neuroprotective factor in glaucoma. On the other hand, light-induced retinal degeneration causes photoreceptor degeneration and increases Hmox1 expression. High expression of Hmox1 induces photoreceptor degeneration even without light stress (84). Taken together, Hmox1 overexpression has a neuroprotective role in only RGCs, but not other retinal cells.
Ecel1 (Endothelin Converting Enzyme Like 1)
Ecel1 is upregulated after optic nerve crush as shown in [4] (60) and [5] (61) (Figure 1B). Endothelin-converting enzyme-like 1 (Ecel1/DINE, encoded by Ecel1) has been identified as an elevated gene response upon nerve injury in the rat brain (85). In the retina, knockdown of Ecel1 by AAV2-CRISPR/CAS9 virus leads to severe RGC loss at four days after optic nerve crush (86). In contrast, loss of Ecel1 has no impacts on RGC survival/death rates at seven days, two and four weeks after optic nerve crush compared to control. Intriguingly, loss of Ecel1 enhances RGC axon regeneration only when Zymosan, a potent monocyte activator and promotes axonal regeneration is present (87). Thus, Ecel1 could exert RGC protection only at the early stages of RGC degeneration in glaucoma.
Atf3 (Activating Transcription Factor 3)
Atf3 is commonly upregulated in [3] (59), [4] (60), [6] (62), and [8] (88) (Figure 1B). ATF3 is a member of the basic leucine zipper (bZip) family of transcription factors, and ATF3 is highly conserved across different species: mouse and zebrafish Atf3 orthologs share 95% and 71% identity with human ATF3 (89). In the optic nerve crush model, Atf3 is preferentially upregulated in αRGCs, one of the RGC subtypes resistant to the injury signals. Overexpression of Atf3 via AAV2 viruses (wherein ~80% of RGCs are infected) enhances RGC survival after optic nerve injury (90). The other study has demonstrated that hippocampal neurons die due to growth factor withdrawal, application of staurosporine or NMDA, or oxygen–glucose deprivation, while ATF3 inhibits neuronal death induced by these toxic factors (91). One of potential molecular pathways for RGC protection by ATF3 is through CREB binding to the Atf3 promoter and subsequent induction of expression (91). CREB is a downstream factor of CaMKII, which shows high neuroprotective effects in NMDA- and IOP-induced RGC degeneration models and after optic nerve crush (92). Thus, CaMKII-CREB-ATF3 molecular axis could enhance RGC survival in glaucoma.
II. Region-Specific Retinal Effects
One of the notable pathological features at the early stages of neurodegeneration in glaucoma is that retinal axon degeneration and loss of RGCs does not occur evenly in the whole retina. Both diffuse and local patterns of RGC loss (93, 94) and specific regions of retinal axon degeneration (95) are observed in glaucoma patients. The rodent models of glaucoma also recapitulate geographically diffuse and focal/regional patterns of RGC loss and axon degeneration (21, 96–102). However, little is known about whether the same molecular mechanisms mediate neurodegeneration in all retinal regions. Most recently, our study has shown that RGCs in the peripheral ventrotemporal (VT) retina are more vulnerable to degenerative signals after optic nerve crush compared to RGCs in other retinal regions. We also demonstrated that neurodegenerative signals conveyed from the optic nerve to VT RGCs after ONC are mediated by SERT-integrin β3 molecular axis, leading to an acute reduction in the expression of GPNMB, which has an RGC protective capacity. In contrast, GPNMB expression in other retinal regions decline slowly. Thus, these molecular interactions confer region-specific RGC vulnerability after optic nerve crush (103). To understand the molecular mechanisms of focal RGC loss in glaucoma, one study has shown through RNA-Seq analysis: [7] the retinal areas where RGCs have already died and the retinal areas where RGCs continue to survive in 11 - 15 month-old DBA/2J mouse (104) (Figure 1A, Supplemental Table 1). We compared top DEGs between [7] and [1] – [6], but we are unable to find any common DEGs. However, we focus on Opa1 (OPA1 mitochondrial dynamin like GTPase) since Opa1 is functionally related to Hmox1 which has been discussed above. Here, we discuss whether Opa1 could be involved in RGC protection through the mitochondrial pathway with other DEGs such as Hmox1 (Figure 1B).
Opa1 (OPA1 Mitochondrial Dynamin Like GTPase)
OPA1 is an inner mitochondrial membrane protein, and loss of OPA1 induces mitochondrial membrane potential reduction and fragmentation of mitochondrial network (105). OPA1 haplo-insufficiency is responsible for the most common form of autosomal dominant optic atrophy (ADOA), a neuropathy leading to degeneration of RGCs and the optic nerve (106). The Opa1 mutant mouse, a mouse carrying a pathogenic mutation in Opa1 shows a significant reduction (35 - 41% decrease) of RGCs compared to WT mice and also axon degeneration with age. Electroretinography (ERG) responses are unaffected, but visually evoked potential (VEP) measurements show significantly reduced amplitudes in the mutant mouse (107, 108). One of the molecular mechanisms underlying Opa1-mediated functions is that BNIP3, a pro-apoptotic member of the Bcl-2 family is induced by stresses such as hypoxia, and it mediates mitochondrial fragmentation and apoptosis by disrupting OPA1 complex (109, 110). Moreover, Opa1 is directly linked to glaucomatous neurodegeneration. In the IOP-induced glaucoma model by translimbal laser photocoagulation of the trabecular meshwork, overexpression of OPA1 via AAV2 viruses attenuates loss of RGCs through reducing expression of BAX and improving mitochondrial health and mitochondrial surface area (111). Thus, Opa1 could maintain mitochondrial quality, leading to RGC protection in glaucoma.
Hmox1 - Opa1 Pathway
As described above, the heme oxygenase-1 (HO-1, encoded by Hmox1) is commonly identified in [3] (59) and [4] (60), and it protects neurons and non-neuronal cells after injury (81, 82). Intriguingly, since the heme oxygenase-1 (HO-1; Hmox1) enzyme system is important for cell protection against oxidative damage in the cardiovascular system, loss of HO-1 shows a reduction in expression of Opa1 and other factors, leading to disruption of mitochondrial quality and then cell death (112). Moreover, loss of HO-1 gene in adipocyte cells reduces thermogenic mitochondrial fusion and fission genes such as Opa1 (113). Thus, vulnerable RGCs locally located in the retina could be determined by dysfunctions of the mitochondrial pathways, whose importance has been also described by the other study (16).
III. Animal Species
Most RGCs die in glaucoma patients and in the rodent models of glaucoma. However, unlike mammals, zebrafish possess high neuroprotective and regenerative capacity in the retina: in the optic nerve crush models, ∼75% of zebrafish RGCs survive after optic nerve injury, even up to 7-weeks post-injury (114). Zebrafish also show robust regenerative responses, regenerate RGC axons and connect to target regions in the brain (115–117). Several transcriptome studies using the retina or RGCs two days (118), three days or later after optic nerve crush (119) have identified DEGs which could potentially regulate axon regeneration. Most recently, to test the hypothesis that RGC protective signals could be acutely evoked after optic nerve damage, we performed RNA-seq analysis using zebrafish RGCs within 24 hours after optic nerve transection [8] (88) (Figure 1A, Supplemental Table 1). Among top DEGs, stat3 and other molecules related to Jak/Stat pathways are highly upregulated after injury, and we found that Jak/Stat pathways mediate RGC protection while microglia/macrophages kill RGCs after optic nerve transection (88). To explore additional neuroprotective genes in RGCs across species, we compared transcriptome profiles between [8] zebrafish RGCs after optic nerve transection (88) and others in the rodent glaucoma and optic nerve injury models [1] – [7]. Intriguingly, zebrafish atf3 (activating transcription factor 3), irf9 (interferon regulatory factor 9), sox11b (mouse Sox11) (SRY-Box transcription factor 11), adcyap1b (mouse adcyap1) (adenylate cyclase activating polypeptide 1) and emb (embigin) are commonly upregulated or downregulated (Figure 1B). Among those common DEGs, since Atf3 has been already discussed above, here we discuss whether other common DEGs such as irf9 and sox11 in mammals could be potential therapeutic targets in glaucoma.
Irf9 (Interferon Regulatory Factor 9)
Irf9 is upregulated in optic nerve crush models [6] (62) and [8] (88) (Figure 1B). Interferon regulatory factor 9 (IRF9 encoded by Irf9) is a transcription factor that regulates innate immune responses. Although there is no direct evidence that IRF9 is involved in RGC protection or death in glaucoma or after optic nerve injury, some studies have shown that IRF9 is connected with neurological pathology (120, 121). IRF9 expression is elevated in neurons after brain ischemia/reperfusion injury, and it potentiates neuronal death via suppression of Sirt1 expression and acetylation of p53 (122). Thus, Irf9 could induce RGC death after nerve injury and in glaucoma in mammals, while in zebrafish, other neuroprotective factors may be more active to overcome IRF9-indcued degenerative signals in RGCs.
Sox11 (Zebrafish Sox11b) (SRY-Box Transcription Factor 11)
Sox11 is upregulated in optic nerve crush models [4] (60) and [8] (88) (Figure 1B). Sox11, a transcription factor is highly expressed in developing RGCs, and it is a critical regulator for RGC differentiation and retinal axon guidance during development (123–126). Although Sox11 expression in RGCs is downregulated after postnatal stages (126), overexpression of Sox11 protects specific RGC types, but not pan-RGCs, and promotes retinal axon regeneration after optic nerve injury (127–129). One of molecular mechanisms is that overexpression of Sox11 increases expression levels of anti-apoptotic factor, Bcl2. However, Sox11 kills αRGCs, one of the resistant RGC subtypes and reduces expression of Spp1, a marker for αRGCs after optic nerve injury (127, 129). Thus, Sox11 could be a unique and selective neuroprotective factor in glaucoma.
IV. RGC Subtypes
46 RGC types in adult mouse retina display differential RGC vulnerability and show distinct molecular signatures, some of which have been identified as novel neuroprotective and/or axon regeneration-related genes using single RNA-seq analysis by [9] (130) (Figure 1A, Supplemental Table 1). For instance, αRGCs are resistant to injury signals after optic nerve crush (131). Moreover, Timp2 and Prph are highly expressed in resilient RGC types, and overexpression of these factors induces RGC protection after optic nerve injury. In contrast, Crhbp and Mmp9 are highly expressed in vulnerable RGCs, and loss of these factors enhances RGC survival (130). Thus, clarifying molecular signatures in resilient RGC subtypes compared to vulnerable RGC subtypes after optic nerve injury enable us to identify them as novel neuroprotective factors in glaucoma. Thus, we compared transcriptome profiles between [9] (130) and [1] – [8]. We found that Ndnf (neuron derived neurotrophic factor), Ctxn3 (cortexin 3) and Gabra2 (gamma-aminobutyric acid type A receptor subunit alpha2) are commonly regulated (Figure 1B). Among them, we focus on Ndnf and discuss potential functions in RGCs.
Ndnf (Neuron Derived Neurotrophic Factor)
Ndnf is upregulated in injured zebrafish RGCs [8] (88) and highly expressed in resilient RGCs [9] (130) after optic nerve injury (Figure 1B). Neuron-derived neurotrophic factor (NDNF encoded by Ndnf) is a glycosylated, disulfide-bonded protein. In the retina, NDNF is more highly expressed in resilient RGCs compared to vulnerable RGCs, and overexpression of Ndnf via AAV2 virus in RGCs promotes RGC survival after optic nerve injury (130). NDNF also has the neuroprotective effects in other CNS neurons. In the hippocampus neuron cultures, NDNF supports neuronal survival and promotes neurite growth and neuron migration (132). Although the molecular mechanisms underlying NDNF-mediated neuronal protection are still unclear, NDNF could be identified as a potential therapeutic gene in glaucoma.
Potential Therapeutic Targets for Glaucoma Patients
A large number of studies on glaucoma inheritance in humans have identified loci that could be associated with specific glaucomatous phenotypes or genetic mutations, suggesting that genetic factors could trigger the initiation of glaucoma. The review article by Wang et al. (133) show that 22 loci of glaucoma are listed and includes the relevant genes such as myocilin (MYOC), optineurin (OPTN), cytochrome P450 subfamily I polypeptide 1 (CYP1B1) and others. [10] These authors also display 74 possible pathogenic or risk genes associated with glaucoma (133) (Figure 1A, Supplemental Table 1). Moreover, single nucleotide polymorphisms (SNPs) and larger variations including copy number variations could also impact the progression and magnitude of glaucomatous phenotypes. Thus, we compare top DEGs identified in [1] – [9] with 74 possible pathogenic or risk genes listed in [10]. We found that OPA1 (OPA1 mitochondrial dynamin like GTPase), RPGRIP1 (RPGR Interacting Protein 1) and ATOH7 (Atonal BHLH Transcription Factor 7) are commonly listed (Figure 1B). Indeed, these genes have been associated with glaucoma in humans: OPA1 (134, 135), RPGRIP1 (136) and ATOH7 (137). OPA1 in rodent RGCs has been discussed above, and it could be a potential neuroprotective gene. Expression and functions of ATOH7 in developing RGCs have been investigated, with ATOH7 mediating RGC survival during development (138). Moreover, most recently, 127 loci associated with open-angle glaucoma have been identified, with specific genes, such as RERE, VCAM1, ZNF638, SMAD6 potentially conferring open-angle glaucoma risk (139). Among those risk genes, MAPT is also listed as one of DEGs in the IOP-induced glaucoma model [3] (59). The microtubule-binding protein tau (tau encoded by MAPT) enhances RGC death after optic nerve crush (140). Alternative splicing of MAPT produces distinct tau isoforms which induce RGC death in the rodent model of Alzheimer’s disease (141). Thus, MAPT could be a mediator of RGC death in glaucoma.
Concluding Marks
Combined analytical studies using well-established animal models of glaucoma and transcriptomic gene expression profiles have provided critical molecular insights into how RGCs can be preserved in glaucoma. In this review, we performed comparative analyses of several transcriptomic profiles using different glaucoma models. We found that some of the DEGs are commonly listed across different species and/or animal disease models, suggesting that they are most likely to be effective therapeutic targets for treatments of RGC degeneration in glaucoma. Because these DEGs have been identified from different cellular sources, such as single RGCs, RGC layer or whole retina, they could affect RGC survival or death through cell autonomous or non-cell autonomous mechanisms. Even if functional studies of these common DEGs unveil no role for them in neurodegeneration, they can still be utilized as molecular markers to inform us about the magnitude of RGC degeneration and/or animal- and cell-type specific degenerative outcomes. There are many other DEGs that we were unable to cover in the scope of this review but may ultimately play important roles in neuroprotection. Several key review articles have previously organized and described relevant genes and molecular mechanisms of neurodegeneration in glaucoma (142–144). Most recently, Wang et al. (145) have also summarized commonly upregulated or downregulated genes by comparing transcriptomic data in several glaucoma mouse models and optic nerve crush models, most of which we have also included in this review. The deep molecular insights into neurodegeneration and neuroprotection, in particular in immune-mediated and other molecular pathways, provided by Wang et al. (145), complement the focus of our review. Together, these primary experimental studies and further cross-analysis through review articles, including ours, will lead to a better understanding of common and differing molecular mechanisms that underly neurodegeneration in disease and injury conditions. Further studies using the existing or novel rodent models of glaucoma with current and new transcriptomic gene expression profiles under a certain condition will be beneficial for gaining more knowledge of targeted molecules for future gene therapy. Moreover, in the comparison of DEGs identified in transcriptomic profiles to disease susceptibility loci identified in epidemiological datasets, only a few DEGs are matched to glaucoma risk alleles in humans. Indeed, many susceptibility loci are related to regulation of IOP since they are expressed in the trabecular meshwork or angle tissues. Thus, further studies aiming at identification of risk genes directly connected to neurodegeneration in glaucoma patients will be needed.
There are still many unanswered questions: how many genes or interventions are required for completely preventing or delaying neurodegeneration? Can we use the same molecular interventions in all types or specific types of glaucoma? What visual functions can be maintained by one or the other gene manipulations in all or specific RGC subtypes? What evaluation system (e.g. 3-D human retinal organoid) is the most reliable to test the neuroprotective efficacy of candidate genes in human glaucoma RGCs? Further research will give answers to these critical questions that are key to development and proper choice of neuroprotective interventions that take into account disease stage and the types of glaucoma.
Author Contributions
All authors contributed to the article and approved the submitted version.
Funding
This work was supported by fund from Research to Prevent Blindness/Ernest & Elizabeth Althouse/Dolly Green Special Scholar Award and Startup funds from the Department of Ophthalmology University of Pittsburgh School of Medicine and the Eye and Ear Foundation of Pittsburgh.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank Jeff Gross, Qing Wang, and the Kuwajima lab for editing and comments on this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fopht.2022.905352/full#supplementary-material
Supplementary Table 1 | Top differentially expressed genes in nine transcriptome studies in the retina or RGCs in different animal models of glaucoma and a list of possible risk genes for glaucoma in humans.
References
1. Quigley HA, Broman AT. The Number of People With Glaucoma Worldwide in 2010 and 2020. Br J Ophthalmol (2006) 90(3):262–7. doi: 10.1136/bjo.2005.081224
2. Tham YC, Li X, Wong TY, Quigley HA, Aung T, Cheng CY. Global Prevalence of Glaucoma and Projections of Glaucoma Burden Through 2040: A Systematic Review and Meta-Analysis. Ophthalmol (2014) 121(11):2081–90. doi: 10.1016/j.ophtha.2014.05.013
3. Bond WS, Hines-Beard J, GoldenMerry YL, Davis M, Farooque A, Sappington RM, et al. Virus-Mediated EpoR76E Therapy Slows Optic Nerve Axonopathy in Experimental Glaucoma. Mol Ther (2016) 24(2):230–9. doi: 10.1038/mt.2015.198
4. Chen H, Cho KS, Vu THK, Shen CH, Kaur M, Chen G, et al. Commensal Microflora-Induced T Cell Responses Mediate Progressive Neurodegeneration in Glaucoma. Nat Commun (2018) 9(1):3209. doi: 10.1038/s41467-018-05681-9
5. Chintalapudi SR, Maria D, Di Wang X, Bailey JNC, consortium N, International Glaucoma Genetics c, et al. Systems Genetics Identifies a Role for Cacna2d1 Regulation in Elevated Intraocular Pressure and Glaucoma Susceptibility. Nat Commun (2017) 8(1):1755. doi: 10.1038/s41467-017-00837-5
6. Cueva Vargas JL, Osswald IK, Unsain N, Aurousseau MR, Barker PA, Bowie D, et al. Soluble Tumor Necrosis Factor Alpha Promotes Retinal Ganglion Cell Death in Glaucoma via Calcium-Permeable AMPA Receptor Activation. J Neurosci (2015) 35(35):12088–102. doi: 10.1523/JNEUROSCI.1273-15.2015
7. Davis BM, Tian K, Pahlitzsch M, Brenton J, Ravindran N, Butt G, et al. Topical Coenzyme Q10 Demonstrates Mitochondrial-Mediated Neuroprotection in a Rodent Model of Ocular Hypertension. Mitochondrion. (2017) 36:114–23. doi: 10.1016/j.mito.2017.05.010
8. Guttenplan KA, Stafford BK, El-Danaf RN, Adler DI, Munch AE, Weigel MK, et al. Neurotoxic Reactive Astrocytes Drive Neuronal Death After Retinal Injury. Cell Rep (2020) 31(12):107776. doi: 10.1016/j.celrep.2020.107776
9. Harder JM, Guymer C, Wood JPM, Daskalaki E, Chidlow G, Zhang C, et al. Disturbed Glucose and Pyruvate Metabolism in Glaucoma With Neuroprotection by Pyruvate or Rapamycin. Proc Natl Acad Sci USA (2020) 117(52):33619–27. doi: 10.1073/pnas.2014213117
10. Harder JM, Williams PA, Soto I, Foxworth NE, Fernandes KA, Freeburg NF, et al. Jnk2 Deficiency Increases the Rate of Glaucomatous Neurodegeneration in Ocular Hypertensive DBA/2J Mice. Cell Death Dis (2018) 9(6):705. doi: 10.1038/s41419-018-0705-8
11. Inman DM, Lambert WS, Calkins DJ, Horner PJ. Alpha-Lipoic Acid Antioxidant Treatment Limits Glaucoma-Related Retinal Ganglion Cell Death and Dysfunction. PloS One (2013) 8(6):e65389. doi: 10.1371/journal.pone.0065389
12. Lee D, Shim MS, Kim KY, Noh YH, Kim H, Kim SY, et al. Coenzyme Q10 Inhibits Glutamate Excitotoxicity and Oxidative Stress-Mediated Mitochondrial Alteration in a Mouse Model of Glaucoma. Invest Ophthalmol Vis Sci (2014) 55(2):993–1005. doi: 10.1167/iovs.13-12564
13. Pease ME, Zack DJ, Berlinicke C, Bloom K, Cone F, Wang Y, et al. Effect of CNTF on Retinal Ganglion Cell Survival in Experimental Glaucoma. Invest Ophthalmol Vis Sci (2009) 50(5):2194–200. doi: 10.1167/iovs.08-3013
14. Sappington RM, Chan M, Calkins DJ. Interleukin-6 Protects Retinal Ganglion Cells From Pressure-Induced Death. Invest Ophthalmol Vis Sci (2006) 47(7):2932–42. doi: 10.1167/iovs.05-1407
15. Tanna AP, Johnson M. Rho Kinase Inhibitors as a Novel Treatment for Glaucoma and Ocular Hypertension. Ophthalmology (2018) 125(11):1741–56. doi: 10.1016/j.ophtha.2018.04.040
16. Williams PA, Harder JM, Foxworth NE, Cochran KE, Philip VM, Porciatti V, et al. Vitamin B3 Modulates Mitochondrial Vulnerability and Prevents Glaucoma in Aged Mice. Science (2017) 355(6326):756–60. doi: 10.1126/science.aal0092
17. Zhang X, Zhang R, Chen J, Wu J. Neuroprotective Effects of DAAO are Mediated via the ERK1/2 Signaling Pathway in a Glaucomatous Animal Model. Exp Eye Res (2020) 190:107892. doi: 10.1016/j.exer.2019.107892
18. John SW, Smith RS, Savinova OV, Hawes NL, Chang B, Turnbull D, et al. Essential Iris Atrophy, Pigment Dispersion, and Glaucoma in DBA/2J Mice. Invest Ophthalmol Vis Sci (1998) 39(6):951–62.
19. Libby RT, Anderson MG, Pang IH, Robinson ZH, Savinova OV, Cosma IM, et al. Inherited Glaucoma in DBA/2J Mice: Pertinent Disease Features for Studying the Neurodegeneration. Vis Neurosci (2005) 22(5):637–48. doi: 10.1017/S0952523805225130
20. John SW. Mechanistic Insights Into Glaucoma Provided by Experimental Genetics the Cogan Lecture. Invest Ophthalmol Vis Sci (2005) 46(8):2649–61. doi: 10.1167/iovs.05-0205
21. Howell GR, Libby RT, Jakobs TC, Smith RS, Phalan FC, Barter JW, et al. Axons of Retinal Ganglion Cells are Insulted in the Optic Nerve Early in DBA/2J Glaucoma. J Cell Biol (2007) 179(7):1523–37. doi: 10.1083/jcb.200706181
22. Anderson MG, Smith RS, Hawes NL, Zabaleta A, Chang B, Wiggs JL, et al. Mutations in Genes Encoding Melanosomal Proteins Cause Pigmentary Glaucoma in DBA/2J Mice. Nat Genet (2002) 30(1):81–5. doi: 10.1038/ng794
23. Chang B, Smith RS, Hawes NL, Anderson MG, Zabaleta A, Savinova O, et al. Interacting Loci Cause Severe Iris Atrophy and Glaucoma in DBA/2J Mice. Nat Genet (1999) 21(4):405–9. doi: 10.1038/7741
24. Howell GR, Libby RT, Marchant JK, Wilson LA, Cosma IM, Smith RS, et al. Absence of Glaucoma in DBA/2J Mice Homozygous for Wild-Type Versions of Gpnmb and Tyrp1. BMC Genet (2007) 8:45. doi: 10.1186/1471-2156-8-45
25. Nair KS, Cosma M, Raghupathy N, Sellarole MA, Tolman NG, de Vries W, et al. YBR/EiJ Mice: A New Model of Glaucoma Caused by Genes on Chromosomes 4 and 17. Dis Model Mech (2016) 9(8):863–71. doi: 10.1242/dmm.024307
26. Fingert JH, Stone EM, Sheffield VC, Alward WL. Myocilin Glaucoma. Surv Ophthalmol (2002) 47(6):547–61. doi: 10.1016/S0039-6257(02)00353-3
27. Xiong S, Kumar A, Tian S, Taher EE, Yang E, Kinchington PR, et al. Stem Cell Transplantation Rescued a Primary Open-Angle Glaucoma Mouse Model. Elife (2021) 10:e63677. doi: 10.7554/eLife.63677
28. Zhou Y, Grinchuk O, Tomarev SI. Transgenic Mice Expressing the Tyr437His Mutant of Human Myocilin Protein Develop Glaucoma. Invest Ophthalmol Vis Sci (2008) 49(5):1932–9. doi: 10.1167/iovs.07-1339
29. Zode GS, Kuehn MH, Nishimura DY, Searby CC, Mohan K, Grozdanic SD, et al. Reduction of ER Stress via a Chemical Chaperone Prevents Disease Phenotypes in a Mouse Model of Primary Open Angle Glaucoma. J Clin Invest. (2011) 121(9):3542–53. doi: 10.1172/JCI58183
30. Levkovitch-Verbin H, Quigley HA, Martin KR, Valenta D, Baumrind LA, Pease ME. Translimbal Laser Photocoagulation to the Trabecular Meshwork as a Model of Glaucoma in Rats. Invest Ophthalmol Vis Sci (2002) 43(2):402–10.
31. Yun H, Lathrop KL, Yang E, Sun M, Kagemann L, Fu V, et al. A Laser-Induced Mouse Model With Long-Term Intraocular Pressure Elevation. PloS One (2014) 9(9):e107446. doi: 10.1371/journal.pone.0107446
32. Aihara M, Lindsey JD, Weinreb RN. Experimental Mouse Ocular Hypertension: Establishment of the Model. Invest Ophthalmol Vis Sci (2003) 44(10):4314–20. doi: 10.1167/iovs.03-0137
33. Fu CT, Sretavan D. Laser-Induced Ocular Hypertension in Albino CD-1 Mice. Invest Ophthalmol Vis Sci (2010) 51(2):980–90. doi: 10.1167/iovs.09-4324
34. Zhang L, Li G, Shi M, Liu HH, Ge S, Ou Y, et al. Establishment and Characterization of an Acute Model of Ocular Hypertension by Laser-Induced Occlusion of Episcleral Veins. Invest Ophthalmol Vis Sci (2017) 58(10):3879–86. doi: 10.1167/iovs.16-20807
35. Grozdanic SD, Betts DM, Sakaguchi DS, Allbaugh RA, Kwon YH, Kardon RH. Laser-Induced Mouse Model of Chronic Ocular Hypertension. Invest Ophthalmol Vis Sci (2003) 44(10):4337–46. doi: 10.1167/iovs.03-0015
36. Sappington RM, Carlson BJ, Crish SD, Calkins DJ. The Microbead Occlusion Model: A Paradigm for Induced Ocular Hypertension in Rats and Mice. Invest Ophthalmol Vis Sci (2010) 51(1):207–16. doi: 10.1167/iovs.09-3947
37. Chen H, Wei X, Cho KS, Chen G, Sappington R, Calkins DJ, et al. Optic Neuropathy Due to Microbead-Induced Elevated Intraocular Pressure in the Mouse. Invest Ophthalmol Vis Sci (2011) 52(1):36–44. doi: 10.1167/iovs.09-5115
38. Eastlake K, Jayaram H, Luis J, Hayes M, Khaw PT, Limb GA. Strain Specific Responses in a Microbead Rat Model of Experimental Glaucoma. Curr Eye Res (2021) 46(3):387–97. doi: 10.1080/02713683.2020.1805472
39. Mukai R, Park DH, Okunuki Y, Hasegawa E, Klokman G, Kim CB, et al. Mouse Model of Ocular Hypertension With Retinal Ganglion Cell Degeneration. PloS One (2019) 14(1):e0208713. doi: 10.1371/journal.pone.0208713
40. Nguyen QH, Lloyd MA, Heuer DK, Baerveldt G, Minckler DS, Lean JS, et al. Incidence and Management of Glaucoma After Intravitreal Silicone Oil Injection for Complicated Retinal Detachments. Ophthalmology (1992) 99(10):1520–6. doi: 10.1016/S0161-6420(92)31771-3
41. Burk LL, Shields MB, Proia AD, McCuen BW 2nd. Intraocular Pressure Following Intravitreal Silicone Oil Injection. Ophthalmic Surg (1988) 19(8):565–9. doi: 10.3928/1542-8877-19880801-11
42. Henderer JD, Budenz DL, Flynn HW Jr., Schiffman JC, Feuer WJ, Murray TG. Elevated Intraocular Pressure and Hypotony Following Silicone Oil Retinal Tamponade for Complex Retinal Detachment: Incidence and Risk Factors. Arch Ophthalmol (1999) 117(2):189–95. doi: 10.1001/archopht.117.2.189
43. Zhang J, Li L, Huang H, Fang F, Webber HC, Zhuang P, et al. Silicone Oil-Induced Ocular Hypertension and Glaucomatous Neurodegeneration in Mouse. Elife (2019) 8:e45881. doi: 10.7554/eLife.45881
44. Morrison JC, Nylander KB, Lauer AK, Cepurna WO, Johnson E. Glaucoma Drops Control Intraocular Pressure and Protect Optic Nerves in a Rat Model of Glaucoma. Invest Ophthalmol Vis Sci (1998) 39(3):526–31.
45. Morrison JC, Cepurna WO, Johnson EC. Modeling Glaucoma in Rats by Sclerosing Aqueous Outflow Pathways to Elevate Intraocular Pressure. Exp Eye Res (2015) 141:23–32. doi: 10.1016/j.exer.2015.05.012
46. He S, Minton AZ, Ma HY, Stankowska DL, Sun X, Krishnamoorthy RR. Involvement of AP-1 and C/EBPbeta in Upregulation of Endothelin B (ETB) Receptor Expression in a Rodent Model of Glaucoma. PloS One (2013) 8(11):e79183. doi: 10.1371/journal.pone.0079183
47. Chauhan BC, Pan J, Archibald ML, LeVatte TL, Kelly ME, Tremblay F. Effect of Intraocular Pressure on Optic Disc Topography, Electroretinography, and Axonal Loss in a Chronic Pressure-Induced Rat Model of Optic Nerve Damage. Invest Ophthalmol Vis Sci (2002) 43(9):2969–76.
48. Chierzi S, Strettoi E, Cenni MC, Maffei L. Optic Nerve Crush: Axonal Responses in Wild-Type and Bcl-2 Transgenic Mice. J Neurosci (1999) 19(19):8367–76. doi: 10.1523/JNEUROSCI.19-19-08367.1999
49. Vidal-Sanz M, Galindo-Romero C, Valiente-Soriano FJ, Nadal-Nicolas FM, Ortin-Martinez A, Rovere G, et al. Shared and Differential Retinal Responses Against Optic Nerve Injury and Ocular Hypertension. Front Neurosci (2017) 11:235. doi: 10.3389/fnins.2017.00235
50. Isenmann S, Wahl C, Krajewski S, Reed JC, Bahr M. Up-Regulation of Bax Protein in Degenerating Retinal Ganglion Cells Precedes Apoptotic Cell Death After Optic Nerve Lesion in the Rat. Eur J Neurosci (1997) 9(8):1763–72. doi: 10.1111/j.1460-9568.1997.tb01534.x
51. Libby RT, Li Y, Savinova OV, Barter J, Smith RS, Nickells RW, et al. Susceptibility to Neurodegeneration in a Glaucoma is Modified by Bax Gene Dosage. PloS Genet (2005) 1(1):17–26. doi: 10.1371/journal.pgen.0010004
52. Syc-Mazurek SB, Fernandes KA, Libby RT. JUN Is Important for Ocular Hypertension-Induced Retinal Ganglion Cell Degeneration. Cell Death Dis (2017) 8(7):e2945. doi: 10.1038/cddis.2017.338
53. Fernandes KA, Harder JM, Kim J, Libby RT. JUN Regulates Early Transcriptional Responses to Axonal Injury in Retinal Ganglion Cells. Exp Eye Res (2013) 112:106–17. doi: 10.1016/j.exer.2013.04.021
54. Li L, Huang H, Fang F, Liu L, Sun Y, Hu Y. Longitudinal Morphological and Functional Assessment of RGC Neurodegeneration After Optic Nerve Crush in Mouse. Front Cell Neurosci (2020) 14:109. doi: 10.3389/fncel.2020.00109
55. Vidal-Sanz M, Bray GM, Villegas-Perez MP, Thanos S, Aguayo AJ. Axonal Regeneration and Synapse Formation in the Superior Colliculus by Retinal Ganglion Cells in the Adult Rat. J Neurosci (1987) 7(9):2894–909. doi: 10.1523/JNEUROSCI.07-09-02894.1987
56. Nadal-Nicolas FM, Sobrado-Calvo P, Jimenez-Lopez M, Vidal-Sanz M, Agudo-Barriuso M. Long-Term Effect of Optic Nerve Axotomy on the Retinal Ganglion Cell Layer. Invest Ophthalmol Vis Sci (2015) 56(10):6095–112. doi: 10.1167/iovs.15-17195
57. Steele MR, Inman DM, Calkins DJ, Horner PJ, Vetter ML. Microarray Analysis of Retinal Gene Expression in the DBA/2J Model of Glaucoma. Invest Ophthalmol Vis Sci (2006) 47(3):977–85. doi: 10.1167/iovs.05-0865
58. Park YH, Snook JD, Ostrin EJ, Kim S, Chen R, Frankfort BJ. Transcriptomic Profiles of Retinal Ganglion Cells Are Defined by the Magnitude of Intraocular Pressure Elevation in Adult Mice. Sci Rep (2019) 9(1):2594. doi: 10.1038/s41598-019-39141-1
59. Guo Y, Cepurna WO, Dyck JA, Doser TA, Johnson EC, Morrison JC. Retinal Cell Responses to Elevated Intraocular Pressure: A Gene Array Comparison Between the Whole Retina and Retinal Ganglion Cell Layer. Invest Ophthalmol Vis Sci (2010) 51(6):3003–18. doi: 10.1167/iovs.09-4663
60. Yasuda M, Tanaka Y, Omodaka K, Nishiguchi KM, Nakamura O, Tsuda S, et al. Transcriptome Profiling of the Rat Retina After Optic Nerve Transection. Sci Rep (2016) 6:28736. doi: 10.1038/srep28736
61. Yasuda M, Tanaka Y, Nishiguchi KM, Ryu M, Tsuda S, Maruyama K, et al. Retinal Transcriptome Profiling at Transcription Start Sites: A Cap Analysis of Gene Expression Early After Axonal Injury. BMC Genomics (2014) 15:982. doi: 10.1186/1471-2164-15-982
62. Ueno S, Yoneshige A, Koriyama Y, Hagiyama M, Shimomura Y, Ito A. Early Gene Expression Profile in Retinal Ganglion Cell Layer After Optic Nerve Crush in Mice. Invest Ophthalmol Vis Sci (2018) 59(1):370–80. doi: 10.1167/iovs.17-22438
63. Piri N, Song M, Kwong JM, Caprioli J. Modulation of Alpha and Beta Crystallin Expression in Rat Retinas With Ocular Hypertension-Induced Ganglion Cell Degeneration. Brain Res (2007) 1141:1–9. doi: 10.1016/j.brainres.2006.11.095
64. Piri N, Kwong JM, Caprioli J. Crystallins in Retinal Ganglion Cell Survival and Regeneration. Mol Neurobiol (2013) 48(3):819–28. doi: 10.1007/s12035-013-8470-2
65. Munemasa Y, Kwong JM, Caprioli J, Piri N. The Role of alphaA- and alphaB-Crystallins in the Survival of Retinal Ganglion Cells After Optic Nerve Axotomy. Invest Ophthalmol Vis Sci (2009) 50(8):3869–75. doi: 10.1167/iovs.08-3138
66. Ying X, Zhang J, Wang Y, Wu N, Wang Y, Yew DT. Alpha-Crystallin Protected Axons From Optic Nerve Degeneration After Crushing in Rats. J Mol Neurosci (2008) 35(3):253–8. doi: 10.1007/s12031-007-9010-1
67. Pasupuleti N, Matsuyama S, Voss O, Doseff AI, Song K, Danielpour D, et al. The Anti-Apoptotic Function of Human alphaA-Crystallin is Directly Related to its Chaperone Activity. Cell Death Dis (2010) 1:e31. doi: 10.1038/cddis.2010.3
68. Ruebsam A, Dulle JE, Myers AM, Sakrikar D, Green KM, Khan NW, et al. A Specific Phosphorylation Regulates the Protective Role of alphaA-Crystallin in Diabetes. JCI Insight (2018) 3(4):e97919. doi: 10.1172/jci.insight.97919
69. Wu N, Yu J, Chen S, Xu J, Ying X, Ye M, et al. Alpha-Crystallin Protects RGC Survival and Inhibits Microglial Activation After Optic Nerve Crush. Life Sci (2014) 94(1):17–23. doi: 10.1016/j.lfs.2013.10.034
70. Kang SS, Ren Y, Liu CC, Kurti A, Baker KE, Bu G, et al. Lipocalin-2 Protects the Brain During Inflammatory Conditions. Mol Psychiatry (2018) 23(2):344–50. doi: 10.1038/mp.2016.243
71. Wang G, Weng YC, Han X, Whaley JD, McCrae KR, Chou WH. Lipocalin-2 Released in Response to Cerebral Ischaemia Mediates Reperfusion Injury in Mice. J Cell Mol Med (2015) 19(7):1637–45. doi: 10.1111/jcmm.12538
72. Ni W, Zheng M, Xi G, Keep RF, Hua Y. Role of Lipocalin-2 in Brain Injury After Intracerebral Hemorrhage. J Cereb Blood Flow Metab (2015) 35(9):1454–61. doi: 10.1038/jcbfm.2015.52
73. Mao S, Xi G, Keep RF, Hua Y. Role of Lipocalin-2 in Thrombin-Induced Brain Injury. Stroke (2016) 47(4):1078–84. doi: 10.1161/STROKEAHA.115.012153
74. Jin M, Kim JH, Jang E, Lee YM, Soo Han H, Woo DK, et al. Lipocalin-2 Deficiency Attenuates Neuroinflammation and Brain Injury After Transient Middle Cerebral Artery Occlusion in Mice. J Cereb Blood Flow Metab (2014) 34(8):1306–14. doi: 10.1038/jcbfm.2014.83
75. Bi F, Huang C, Tong J, Qiu G, Huang B, Wu Q, et al. Reactive Astrocytes Secrete Lcn2 to Promote Neuron Death. Proc Natl Acad Sci USA (2013) 110(10):4069–74. doi: 10.1073/pnas.1218497110
76. Yoneshige A, Hagiyama M, Takashima Y, Ueno S, Inoue T, Kimura R, et al. Elevated Hydrostatic Pressure Causes Retinal Degeneration Through Upregulating Lipocalin-2. Front Cell Dev Biol (2021) 9:664327. doi: 10.3389/fcell.2021.664327
77. Lalancette-Hebert M, Swarup V, Beaulieu JM, Bohacek I, Abdelhamid E, Weng YC, et al. Galectin-3 Is Required for Resident Microglia Activation and Proliferation in Response to Ischemic Injury. J Neurosci (2012) 32(30):10383–95. doi: 10.1523/JNEUROSCI.1498-12.2012
78. Wesley UV, Sutton IC, Cunningham K, Jaeger JW, Phan AQ, Hatcher JF, et al. Galectin-3 Protects Against Ischemic Stroke by Promoting Neuro-Angiogenesis via Apoptosis Inhibition and Akt/Caspase Regulation. J Cereb Blood Flow Metab (2021) 41(4):857–73. doi: 10.1177/0271678X20931137
79. Yip PK, Carrillo-Jimenez A, King P, Vilalta A, Nomura K, Chau CC, et al. Galectin-3 Released in Response to Traumatic Brain Injury Acts as an Alarmin Orchestrating Brain Immune Response and Promoting Neurodegeneration. Sci Rep (2017) 7:41689. doi: 10.1038/srep41689
80. Abreu CA, De Lima SV, Mendonca HR, Goulart CO, Martinez AM. Absence of Galectin-3 Promotes Neuroprotection in Retinal Ganglion Cells After Optic Nerve Injury. Histol Histopathol. (2017) 32(3):253–62. doi: 10.14670/HH-11-788
81. Otterbein LE, Kolls JK, Mantell LL, Cook JL, Alam J, Choi AM. Exogenous Administration of Heme Oxygenase-1 by Gene Transfer Provides Protection Against Hyperoxia-Induced Lung Injury. J Clin Invest (1999) 103(7):1047–54. doi: 10.1172/JCI5342
82. Zhang F, Wang S, Zhang M, Weng Z, Li P, Gan Y, et al. Pharmacological Induction of Heme Oxygenase-1 by a Triterpenoid Protects Neurons Against Ischemic Injury. Stroke (2012) 43(5):1390–7. doi: 10.1161/STROKEAHA.111.647420
83. Hegazy KA, Dunn MW, Sharma SC. Functional Human Heme Oxygenase has a Neuroprotective Effect on Adult Rat Ganglion Cells After Pressure-Induced Ischemia. Neuroreport (2000) 11(6):1185–9. doi: 10.1097/00001756-200004270-00008
84. Li H, Liu B, Lian L, Zhou J, Xiang S, Zhai Y, et al. High Dose Expression of Heme Oxigenase-1 Induces Retinal Degeneration Through ER Stress-Related DDIT3. Mol Neurodegener (2021) 16(1):16. doi: 10.1186/s13024-021-00437-4
85. Kiryu-Seo S, Sasaki M, Yokohama H, Nakagomi S, Hirayama T, Aoki S, et al. Damage-Induced Neuronal Endopeptidase (DINE) Is a Unique Metallopeptidase Expressed in Response to Neuronal Damage and Activates Superoxide Scavengers. Proc Natl Acad Sci USA (2000) 97(8):4345–50. doi: 10.1073/pnas.070509897
86. Sato K, Shiga Y, Nakagawa Y, Fujita K, Nishiguchi KM, Tawarayama H, et al. Ecel1 Knockdown With an AAV2-Mediated CRISPR/Cas9 System Promotes Optic Nerve Damage-Induced RGC Death in the Mouse Retina. Invest Ophthalmol Vis Sci (2018) 59(10):3943–51. doi: 10.1167/iovs.18-23784
87. Kaneko A, Kiryu-Seo S, Matsumoto S, Kiyama H. Damage-Induced Neuronal Endopeptidase (DINE) Enhances Axonal Regeneration Potential of Retinal Ganglion Cells After Optic Nerve Injury. Cell Death Dis (2017) 8(6):e2847. doi: 10.1038/cddis.2017.212
88. Chen S, Lathrop KL, Kuwajima T, Gross JM. Retinal Ganglion Cell Survival After Severe Optic Nerve Injury Is Modulated by Crosstalk Between Jak/Stat Signaling and Innate Immune Responses in the Zebrafish Retina. Development (2022) 149(8):dev199694. doi: 10.1242/dev.199694
89. Katz HR, Arcese AA, Bloom O, Morgan JR. Activating Transcription Factor 3 (ATF3) Is a Highly Conserved Pro-Regenerative Transcription Factor in the Vertebrate Nervous System. Front Cell Dev Biol (2022) 10:824036. doi: 10.3389/fcell.2022.824036
90. Kole C, Brommer B, Nakaya N, Sengupta M, Bonet-Ponce L, Zhao T, et al. Activating Transcription Factor 3 (ATF3) Protects Retinal Ganglion Cells and Promotes Functional Preservation After Optic Nerve Crush. Invest Ophthalmol Vis Sci (2020) 61(2):31. doi: 10.1167/iovs.61.2.31
91. Zhang SJ, Buchthal B, Lau D, Hayer S, Dick O, Schwaninger M, et al. A Signaling Cascade of Nuclear Calcium-CREB-ATF3 Activated by Synaptic NMDA Receptors Defines a Gene Repression Module That Protects Against Extrasynaptic NMDA Receptor-Induced Neuronal Cell Death and Ischemic Brain Damage. J Neurosci (2011) 31(13):4978–90. doi: 10.1523/JNEUROSCI.2672-10.2011
92. Guo X, Zhou J, Starr C, Mohns EJ, Li Y, Chen EP, et al. Preservation of Vision After CaMKII-Mediated Protection of Retinal Ganglion Cells. Cell. (2021) 184(16):4299–314.e12. doi: 10.1016/j.cell.2021.06.031
93. Lei Y, Garrahan N, Hermann B, Fautsch MP, Johnson DH, Hernandez MR, et al. Topography of Neuron Loss in the Retinal Ganglion Cell Layer in Human Glaucoma. Br J Ophthalmol (2009) 93(12):1676–9. doi: 10.1136/bjo.2009.159210
94. Artes PH, Chauhan BC, Keltner JL, Cello KE, Johnson CA, Anderson DR, et al. Longitudinal and Cross-Sectional Analyses of Visual Field Progression in Participants of the Ocular Hypertension Treatment Study. Arch Ophthalmol (2010) 128(12):1528–32. doi: 10.1001/archophthalmol.2010.292
95. Leung CK, Choi N, Weinreb RN, Liu S, Ye C, Liu L, et al. Retinal Nerve Fiber Layer Imaging With Spectral-Domain Optical Coherence Tomography: Pattern of RNFL Defects in Glaucoma. Ophthalmology (2010) 117(12):2337–44. doi: 10.1016/j.ophtha.2010.04.002
96. Vidal-Sanz M, Salinas-Navarro M, Nadal-Nicolas FM, Alarcon-Martinez L, Valiente-Soriano FJ, de Imperial JM, et al. Understanding Glaucomatous Damage: Anatomical and Functional Data From Ocular Hypertensive Rodent Retinas. Prog Retin Eye Res (2012) 31(1):1–27. doi: 10.1016/j.preteyeres.2011.08.001
97. Filippopoulos T, Danias J, Chen B, Podos SM, Mittag TW. Topographic and Morphologic Analyses of Retinal Ganglion Cell Loss in Old DBA/2NNia Mice. Invest Ophthalmol Vis Sci (2006) 47(5):1968–74. doi: 10.1167/iovs.05-0955
98. Jakobs TC, Libby RT, Ben Y, John SW, Masland RH. Retinal Ganglion Cell Degeneration is Topological But Not Cell Type Specific in DBA/2J Mice. J Cell Biol (2005) 171(2):313–25. doi: 10.1083/jcb.200506099
99. Schlamp CL, Li Y, Dietz JA, Janssen KT, Nickells RW. Progressive Ganglion Cell Loss and Optic Nerve Degeneration in DBA/2J Mice is Variable and Asymmetric. BMC Neurosci (2006) 7:66. doi: 10.1186/1471-2202-7-66
100. Guo L, Normando EM, Nizari S, Lara D, Cordeiro MF. Tracking Longitudinal Retinal Changes in Experimental Ocular Hypertension Using the cSLO and Spectral Domain-OCT. Invest Ophthalmol Vis Sci (2010) 51(12):6504–13. doi: 10.1167/iovs.10-5551
101. Davis BM, Guo L, Brenton J, Langley L, Normando EM, Cordeiro MF. Automatic Quantitative Analysis of Experimental Primary and Secondary Retinal Neurodegeneration: Implications for Optic Neuropathies. Cell Death Discov (2016) 2:16031. doi: 10.1038/cddiscovery.2016.31
102. Schaub JA, Kimball EC, Steinhart MR, Nguyen C, Pease ME, Oglesby EN, et al. Regional Retinal Ganglion Cell Axon Loss in a Murine Glaucoma Model. Invest Ophthalmol Vis Sci (2017) 58(5):2765–73. doi: 10.1167/iovs.17-21761
103. Kingston R, Amin D, Misra S, Gross JM, Kuwajima T. Serotonin Transporter-Mediated Molecular Axis Regulates Regional Retinal Ganglion Cell Vulnerability and Axon Regeneration After Nerve Injury. PloS Genet (2021) 17(11):e1009885. doi: 10.1371/journal.pgen.1009885
104. Panagis L, Zhao X, Ge Y, Ren L, Mittag TW, Danias J. Gene Expression Changes in Areas of Focal Loss of Retinal Ganglion Cells in the Retina of DBA/2J Mice. Invest Ophthalmol Vis Sci (2010) 51(4):2024–34. doi: 10.1167/iovs.09-3560
105. Olichon A, Baricault L, Gas N, Guillou E, Valette A, Belenguer P, et al. Loss of OPA1 Perturbates the Mitochondrial Inner Membrane Structure and Integrity, Leading to Cytochrome C Release and Apoptosis. J Biol Chem (2003) 278(10):7743–6. doi: 10.1074/jbc.C200677200
106. Toomes C, Marchbank NJ, Mackey DA, Craig JE, Newbury-Ecob RA, Bennett CP, et al. Spectrum, Frequency and Penetrance of OPA1 Mutations in Dominant Optic Atrophy. Hum Mol Genet (2001) 10(13):1369–78. doi: 10.1093/hmg/10.13.1369
107. Heiduschka P, Schnichels S, Fuhrmann N, Hofmeister S, Schraermeyer U, Wissinger B, et al. Electrophysiological and Histologic Assessment of Retinal Ganglion Cell Fate in a Mouse Model for OPA1-Associated Autosomal Dominant Optic Atrophy. Invest Ophthalmol Vis Sci (2010) 51(3):1424–31. doi: 10.1167/iovs.09-3606
108. Gonzalez-Menendez I, Reinhard K, Tolivia J, Wissinger B, Munch TA. Influence of Opa1 Mutation on Survival and Function of Retinal Ganglion Cells. Invest Ophthalmol Vis Sci (2015) 56(8):4835–45. doi: 10.1167/iovs.15-16743
109. Landes T, Emorine LJ, Courilleau D, Rojo M, Belenguer P, Arnaune-Pelloquin L. The BH3-Only Bnip3 Binds to the Dynamin Opa1 to Promote Mitochondrial Fragmentation and Apoptosis by Distinct Mechanisms. EMBO Rep (2010) 11(6):459–65. doi: 10.1038/embor.2010.50
110. Moulis MF, Millet AM, Daloyau M, Miquel MC, Ronsin B, Wissinger B, et al. OPA1 Haploinsufficiency Induces a BNIP3-Dependent Decrease in Mitophagy in Neurons: Relevance to Dominant Optic Atrophy. J Neurochem (2017) 140(3):485–94. doi: 10.1111/jnc.13894
111. Hu X, Dai Y, Zhang R, Shang K, Sun X. Overexpression of Optic Atrophy Type 1 Protects Retinal Ganglion Cells and Upregulates Parkin Expression in Experimental Glaucoma. Front Mol Neurosci (2018) 11:350. doi: 10.3389/fnmol.2018.00350
112. Suliman HB, Keenan JE, Piantadosi CA. Mitochondrial Quality-Control Dysregulation in Conditional HO-1(-/-) Mice. JCI Insight (2017) 2(3):e89676. doi: 10.1172/jci.insight.89676
113. Singh SP, Grant I, Meissner A, Kappas A, Abraham NG. Ablation of Adipose-HO-1 Expression Increases White Fat Over Beige Fat Through Inhibition of Mitochondrial Fusion and of PGC1alpha in Female Mice. Horm Mol Biol Clin Investig (2017) 31(1). doi: 10.1515/hmbci-2017-0027
114. Zou S, Tian C, Ge S, Hu B. Neurogenesis of Retinal Ganglion Cells is Not Essential to Visual Functional Recovery After Optic Nerve Injury in Adult Zebrafish. PloS One (2013) 8(2):e57280. doi: 10.1371/journal.pone.0057280
115. Diekmann H, Kalbhen P, Fischer D. Active Mechanistic Target of Rapamycin Plays an Ancillary Rather Than Essential Role in Zebrafish CNS Axon Regeneration. Front Cell Neurosci (2015) 9:251. doi: 10.3389/fncel.2015.00251
116. Diekmann H, Kalbhen P, Fischer D. Characterization of Optic Nerve Regeneration Using Transgenic Zebrafish. Front Cell Neurosci (2015) 9:118. doi: 10.3389/fncel.2015.00118
117. Harvey BM, Baxter M, Granato M. Optic Nerve Regeneration in Larval Zebrafish Exhibits Spontaneous Capacity for Retinotopic But Not Tectum Specific Axon Targeting. PloS One (2019) 14(6):e0218667. doi: 10.1371/journal.pone.0218667
118. Dhara SP, Rau A, Flister MJ, Recka NM, Laiosa MD, Auer PL, et al. Cellular Reprogramming for Successful CNS Axon Regeneration is Driven by a Temporally Changing Cast of Transcription Factors. Sci Rep (2019) 9(1):14198. doi: 10.1038/s41598-019-50485-6
119. Veldman MB, Bemben MA, Thompson RC, Goldman D. Gene Expression Analysis of Zebrafish Retinal Ganglion Cells During Optic Nerve Regeneration Identifies KLF6a and KLF7a as Important Regulators of Axon Regeneration. Dev Biol (2007) 312(2):596–612. doi: 10.1016/j.ydbio.2007.09.019
120. Ousman SS, Wang J, Campbell IL. Differential Regulation of Interferon Regulatory Factor (IRF)-7 and IRF-9 Gene Expression in the Central Nervous System During Viral Infection. J Virol (2005) 79(12):7514–27. doi: 10.1128/JVI.79.12.7514-7527.2005
121. Hofer MJ, Li W, Lim SL, Campbell IL. The Type I Interferon-Alpha Mediates a More Severe Neurological Disease in the Absence of the Canonical Signaling Molecule Interferon Regulatory Factor 9. J Neurosci (2010) 30(3):1149–57. doi: 10.1523/JNEUROSCI.3711-09.2010
122. Chen HZ, Guo S, Li ZZ, Lu Y, Jiang DS, Zhang R, et al. A Critical Role for Interferon Regulatory Factor 9 in Cerebral Ischemic Stroke. J Neurosci (2014) 34(36):11897–912. doi: 10.1523/JNEUROSCI.1545-14.2014
123. Jiang Y, Ding Q, Xie X, Libby RT, Lefebvre V, Gan L. Transcription Factors SOX4 and SOX11 Function Redundantly to Regulate the Development of Mouse Retinal Ganglion Cells. J Biol Chem (2013) 288(25):18429–38. doi: 10.1074/jbc.M113.478503
124. Kuwajima T, Soares CA, Sitko AA, Lefebvre V, Mason C. SoxC Transcription Factors Promote Contralateral Retinal Ganglion Cell Differentiation and Axon Guidance in the Mouse Visual System. Neuron. (2017) 93(5):1110–25.e5. doi: 10.1016/j.neuron.2017.01.029
125. Usui A, Mochizuki Y, Iida A, Miyauchi E, Satoh S, Sock E, et al. The Early Retinal Progenitor-Expressed Gene Sox11 Regulates the Timing of the Differentiation of Retinal Cells. Development. (2013) 140(4):740–50. doi: 10.1242/dev.090274
126. Chang KC, Hertz J, Zhang X, Jin XL, Shaw P, Derosa BA, et al. Novel Regulatory Mechanisms for the SoxC Transcriptional Network Required for Visual Pathway Development. J Neurosci (2017) 37(19):4967–81. doi: 10.1523/JNEUROSCI.3430-13.2017
127. Norsworthy MW, Bei F, Kawaguchi R, Wang Q, Tran NM, Li Y, et al. Sox11 Expression Promotes Regeneration of Some Retinal Ganglion Cell Types But Kills Others. Neuron. (2017) 94(6):1112–20.e4. doi: 10.1016/j.neuron.2017.05.035
128. Li Y, Struebing FL, Wang J, King R, Geisert EE. Different Effect of Sox11 in Retinal Ganglion Cells Survival and Axon Regeneration. Front Genet (2018) 9:633. doi: 10.3389/fgene.2018.00633
129. Chang KC, Bian M, Xia X, Madaan A, Sun C, Wang Q, et al. Posttranslational Modification of Sox11 Regulates RGC Survival and Axon Regeneration. eNeuro (2021) 8(1):ENEURO.0358-20.2020.. doi: 10.1523/ENEURO.0358-20.2020
130. Tran NM, Shekhar K, Whitney IE, Jacobi A, Benhar I, Hong G, et al. Single-Cell Profiles of Retinal Ganglion Cells Differing in Resilience to Injury Reveal Neuroprotective Genes. Neuron (2019) 104(6):1039–55.e12. doi: 10.1101/711762
131. Duan X, Qiao M, Bei F, Kim IJ, He Z, Sanes JR. Subtype-Specific Regeneration of Retinal Ganglion Cells Following Axotomy: Effects of Osteopontin and mTOR Signaling. Neuron. (2015) 85(6):1244–56. doi: 10.1016/j.neuron.2015.02.017
132. Kuang XL, Zhao XM, Xu HF, Shi YY, Deng JB, Sun GT. Spatio-Temporal Expression of a Novel Neuron-Derived Neurotrophic Factor (NDNF) in Mouse Brains During Development. BMC Neurosci (2010) 11:137. doi: 10.1186/1471-2202-11-137
133. Wang HW, Sun P, Chen Y, Jiang LP, Wu HP, Zhang W, et al. Research Progress on Human Genes Involved in the Pathogenesis of Glaucoma (Review). Mol Med Rep (2018) 18(1):656–74. doi: 10.3892/mmr.2018.9071
134. Yu-Wai-Man P, Stewart JD, Hudson G, Andrews RM, Griffiths PG, Birch MK, et al. OPA1 Increases the Risk of Normal But Not High Tension Glaucoma. J Med Genet (2010) 47(2):120–5. doi: 10.1136/jmg.2009.067512
135. Bosley TM, Hellani A, Spaeth GL, Myers J, Katz LJ, Moster MR, et al. Down-Regulation of OPA1 in Patients With Primary Open Angle Glaucoma. Mol Vis (2011) 17:1074–9.
136. Fernandez-Martinez L, Letteboer S, Mardin CY, Weisschuh N, Gramer E, Weber BH, et al. Evidence for RPGRIP1 Gene as Risk Factor for Primary Open Angle Glaucoma. Eur J Hum Genet (2011) 19(4):445–51. doi: 10.1038/ejhg.2010.217
137. Mabuchi F, Sakurada Y, Kashiwagi K, Yamagata Z, Iijima H, Tsukahara S. Association Between Genetic Variants Associated With Vertical Cup-to-Disc Ratio and Phenotypic Features of Primary Open-Angle Glaucoma. Ophthalmol (2012) 119(9):1819–25. doi: 10.1016/j.ophtha.2012.02.044
138. Brodie-Kommit J, Clark BS, Shi Q, Shiau F, Kim DW, Langel J, et al. Atoh7-Independent Specification of Retinal Ganglion Cell Identity. Sci Adv (2021) 7(11):eabe4983. doi: 10.1126/sciadv.abe4983
139. Gharahkhani P, Jorgenson E, Hysi P, Khawaja AP, Pendergrass S, Han X, et al. Genome-Wide Meta-Analysis Identifies 127 Open-Angle Glaucoma Loci With Consistent Effect Across Ancestries. Nat Commun (2021) 12(1):1258. doi: 10.1038/s41467-020-20851-4
140. Oku H, Kida T, Horie T, Taki K, Mimura M, Kojima S, et al. Tau Is Involved in Death of Retinal Ganglion Cells of Rats From Optic Nerve Crush. Invest Ophthalmol Vis Sci (2019) 60(6):2380–7. doi: 10.1167/iovs.19-26683
141. Chiasseu M, Alarcon-Martinez L, Belforte N, Quintero H, Dotigny F, Destroismaisons L, et al. Tau Accumulation in the Retina Promotes Early Neuronal Dysfunction and Precedes Brain Pathology in a Mouse Model of Alzheimer's Disease. Mol Neurodegener (2017) 12(1):58. doi: 10.1186/s13024-017-0199-3
142. Jakobs TC. Differential Gene Expression in Glaucoma. Cold Spring Harb Perspect Med (2014) 4(7):a020636. doi: 10.1101/cshperspect.a020636
143. Artero-Castro A, Rodriguez-Jimenez FJ, Jendelova P, VanderWall KB, Meyer JS, Erceg S. Glaucoma as a Neurodegenerative Disease Caused by Intrinsic Vulnerability Factors. Prog Neurobiol (2020) 193:101817. doi: 10.1016/j.pneurobio.2020.101817
144. Enz TJ, Tribble JR, Williams PA. Comparison of Glaucoma-Relevant Transcriptomic Datasets Identifies Novel Drug Targets for Retinal Ganglion Cell Neuroprotection. J Clin Med (2021) 10(17):3938. doi: 10.3390/jcm10173938
Keywords: glaucoma, optic nerve, cell death, vulnerability, transcriptome, retinal ganglion cells
Citation: Amin D and Kuwajima T (2022) Differential Retinal Ganglion Cell Vulnerability, A Critical Clue for the Identification of Neuroprotective Genes in Glaucoma. Front. Ophthalmol. 2:905352. doi: 10.3389/fopht.2022.905352
Received: 27 March 2022; Accepted: 05 May 2022;
Published: 31 May 2022.
Edited by:
Vasantha Rao, Duke University, United StatesReviewed by:
Raghu R. Krishnamoorthy, University of North Texas Health Science Center, United StatesFrancisco M. Nadal-Nicolas, National Eye Institute (NIH), United States
Thomas Johnson, Johns Hopkins University, United States
Copyright © 2022 Amin and Kuwajima. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Takaaki Kuwajima, a3V3YWppbWFAcGl0dC5lZHU=