 Jia-Xing Sun
Jia-Xing Sun Hong-Xiang Yan
Hong-Xiang Yan Dan Hu
Dan Hu Jian Zhou
Jian Zhou Yu-Sheng Wang
Yu-Sheng Wang Xu Hou
Xu Hou
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Ophthalmol., 03 June 2022
Sec. Glaucoma
Volume 2 - 2022 | https://doi.org/10.3389/fopht.2022.902898
This article is part of the Research TopicRising Stars in Glaucoma: 2022View all 6 articles
Background: Mutations in the Crumbs homolog-1 (CRB1) gene are associated with a variety of retinal degenerations including Leber congenital amaurosis (LCA) and retinitis pigmentosa (RP). It is also important to highlight atypical features to make proper diagnosis and treatment.
Case Presentation: We present the case of a 7-year-old girl with biallelic heterozygous CRB1 mutations. The clinical features include macular retinoschisis, Coats-like vasculopathy, short axial length, and angle-closure glaucoma (ACG). We also briefly review the current opinion on CRB1 mutation-related diseases.
Conclusion: CRB1 mutations could result in a combined manifestation in anterior and posterior segments. This case emphasizes the importance of genetic diagnosis for those young patients with complicated rare clinical features to call for a specific treatment and follow-up plan. It also highlights the crucial role of CRB1 in eyeball development.
The Crumbs homolog-1 (CRB1) gene is one of the human homologs of the Crumbs gene in Drosophila. Mapped to chromosome 1q31.3, CRB1 mutations have been associated with a variety of retinal degenerations, including Leber congenital amaurosis (LCA), early-onset rod–cone dystrophy (EORCD), retinitis pigmentosa (RP), and cone–rod dystrophy (CRD) (1). Some atypical clinical features were also reported, such as isolated macular dystrophy and foveal retinoschisis (2–4), hyperopia and nanophthalmos (5, 6), optic disk drusen (6), and Coats-like vasculopathy (7). Thus, it is important to highlight those less common clinical features in order to improve both diagnosis and treatment.
In this paper, we report the case of a 7-year-old girl with biallelic heterozygous CRB1 mutations, whose clinical features included macular retinoschisis, Coats-like retinal capillary leakage, short axial length, and angle-closure glaucoma (ACG). We also briefly review the current opinion on genotype and phenotype analyses as well as treatment approaches for CRB1 mutation-related diseases. This case report was prepared following the CARE guidelines.
A 7-year-old girl with healthy, non-consanguineous parents was admitted to our hospital in 2016 with a complaint of poor visual acuity of both eyes, discovered by her mother occasionally. A history of premature birth, trauma, or systemic diseases was not reported by her parents. Her brother had normal vision.
No abnormality was found by general physical examination. Her best-corrected visual acuity (BCVA) was 20/400 in the right eye and counting fingers (CF) at 30 cm in the left eye. The intraocular pressure (IOP) was 38 mmHg in the right eye and 35 mmHg in the left eye measured by Goldmann applanation tonometry. Slit-lamp examination showed mild conjunctival hyperemia. The pupil was around 4 mm in diameter, with delayed light reflex in both eyes. A shallow but clear anterior chamber was observed, combined with a lucent lens and vitreous opacities with cells (Figure 1A). Fundus evaluation showed a cup–disk ratio (C/D) of 0.8 and dull macular reflex in the posterior pole, with no pigmentary changes in the peripheral retina, in both eyes (Figure 1B). Gonioscopy further revealed closed anterior chamber angles in the nasal quadrant and narrow angles in other quadrants, with no signs of hyperpigmentation or neovascularization. The central cornea thickness was 606 μm in the right eye and 612 μm in the left.
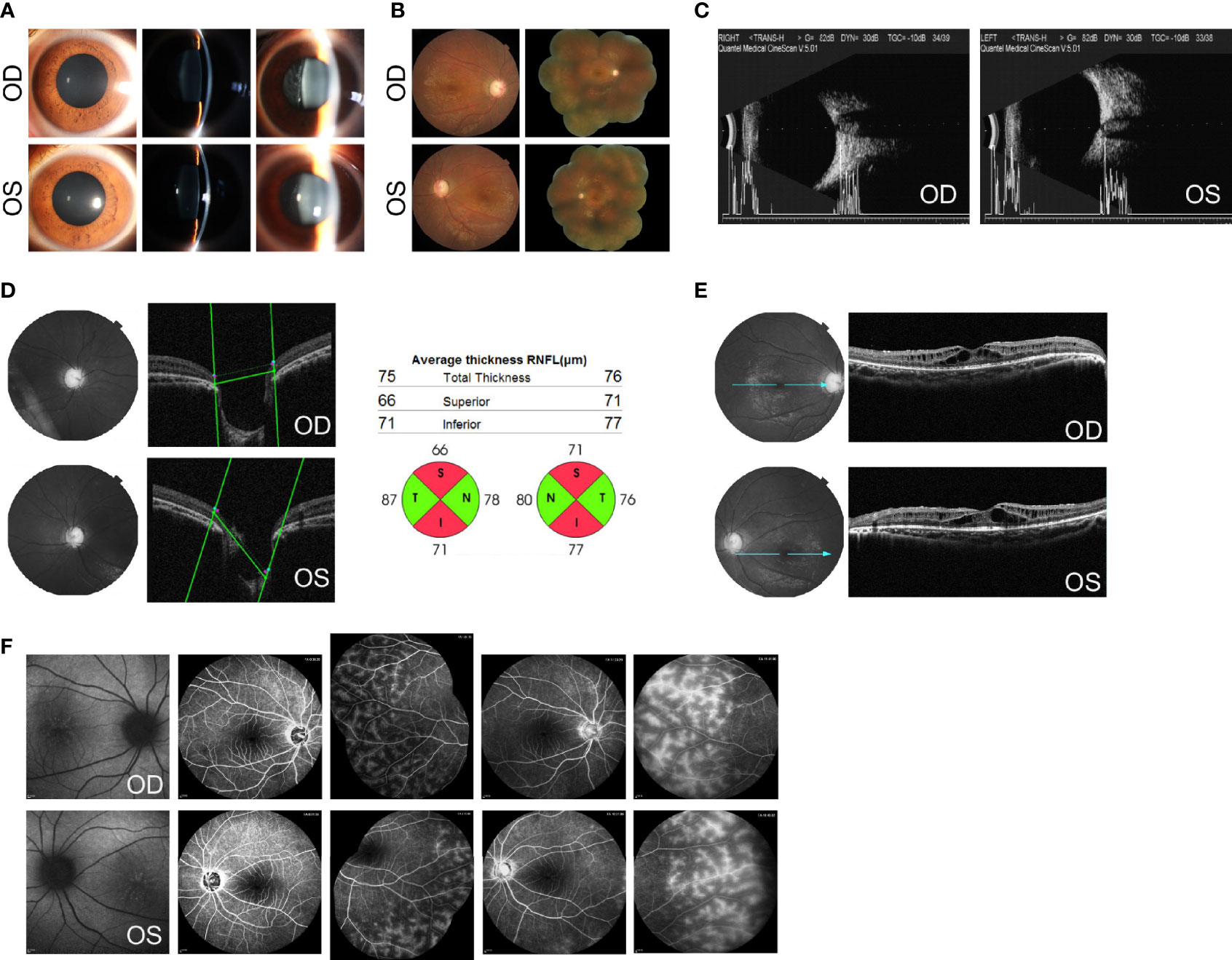
Figure 1 Ocular findings at the initial examination. (A) Slit-lamp aspects of the ocular anterior segment show moderate mydriasis, shallow anterior chamber, lucent lens, and vitreous opacities with cells in both eyes. (B) Fundus photographs show a C/D of 0.8 and dull macular reflex in the posterior pole, without pigmentary changes in both eyes. (C) B-mode ultrasound scanning shows a short axial length (20.0 mm for the right and 20.2 mm for the left) with optic disk excavation in both eyes. (D) Optical coherence tomography (OCT) shows an enlarged and depressed optic disk with RNFL thickness in both eyes. (E) OCT shows macular retinoschisis in the inner and outer nuclear layers, with the disruption of the ellipsoid zone and external limiting membrane within the fovea. (F) Fundus autofluorescence (AF) shows foveal hyperfluorescent specks in the right eye and streaks of radial hyper-AF originating from the central fovea in the left eye. The early and late phases of FA show spoke-wheel-like dye accumulation without leakage within the fovea and peripheral retinal telangiectasia with mild leakage in both eyes. OCT images of the optic disk and values of RNFL thickness were acquired using Topcon 3D OCT-2000 (Topcon Corporation, Tokyo, Japan). OCT images of the macula were acquired using Heidelberg SPECTRALIS OCT (Heidelberg Engineering, Heidelberg, Germany). AF images were acquired using Heidelberg SPECTRALIS HRA (Heidelberg Engineering, Heidelberg, Germany).
Imageological examinations were followed to figure out the possible reasons for high IOP, enlarged C/D, and vitreous opacities. Her axial length, measured by a B-mode ultrasound scanner, was 20.0 mm for the right eye and 20.2 mm for the left eye, respectively, with optic disk excavation in both eyes (Figure 1C). The thickness of the retinal nerve fiber layer (RNFL), tested by optical coherence tomography (OCT), was significantly thinner on the superior and inferior side around the optic papilla bilaterally, compared with the normal value databank (Figure 1D). The macular region showed marked cystoid spaces in the inner and outer nuclear layers. Within the regions of the fovea, the disruption of the ellipsoid zone and external limiting membrane was also noted (Figure 1E). On fundus autofluorescence (AF), there were several hyperfluorescent specks within the fovea in the right eye and streaks of radial hyper-AF originating from the central fovea in the left eye (Figure 1F). Fluorescein angiography (FA) revealed no signs of optic disk or macular leakage apart from a little spoke-wheel-like dye accumulation within the fovea, as well as retinal telangiectasia with mild leakage of the peripheral retina in both eyes (Figure 1F).
The abnormal ocular phenotype included bilateral anterior angle closure, short axial length, optic atrophy, macular retinoschisis, and Coats-like response in the peripheral retina. In consideration of the later age of onset, normal size of the cornea without Haab’s striae, and closed anterior chamber angles, the diagnosis of primary congenital glaucoma were excluded. Further laboratory investigations referring to rheumatism, ankylosing spondylitis, tuberculosis, or other systemic autoimmune or infectious diseases were all negative, indicating no systemic abnormality. Therefore, the proposed diagnosis was childhood glaucoma associated with non-acquired ocular anomalies.
To further investigate the pathology, DNA samples were screened to detect mutations in the most frequently involved genes in retinal diseases (Supplementary Table S1). The genetic testing showed combined heterozygous missense mutation (paternal p.C469G and maternal p.R905Q) in CRB1 in the affected girl (Figures 2A–C), which might account for her clinical features including macular retinoschisis, short axial length, and Coats-like vasculopathy.
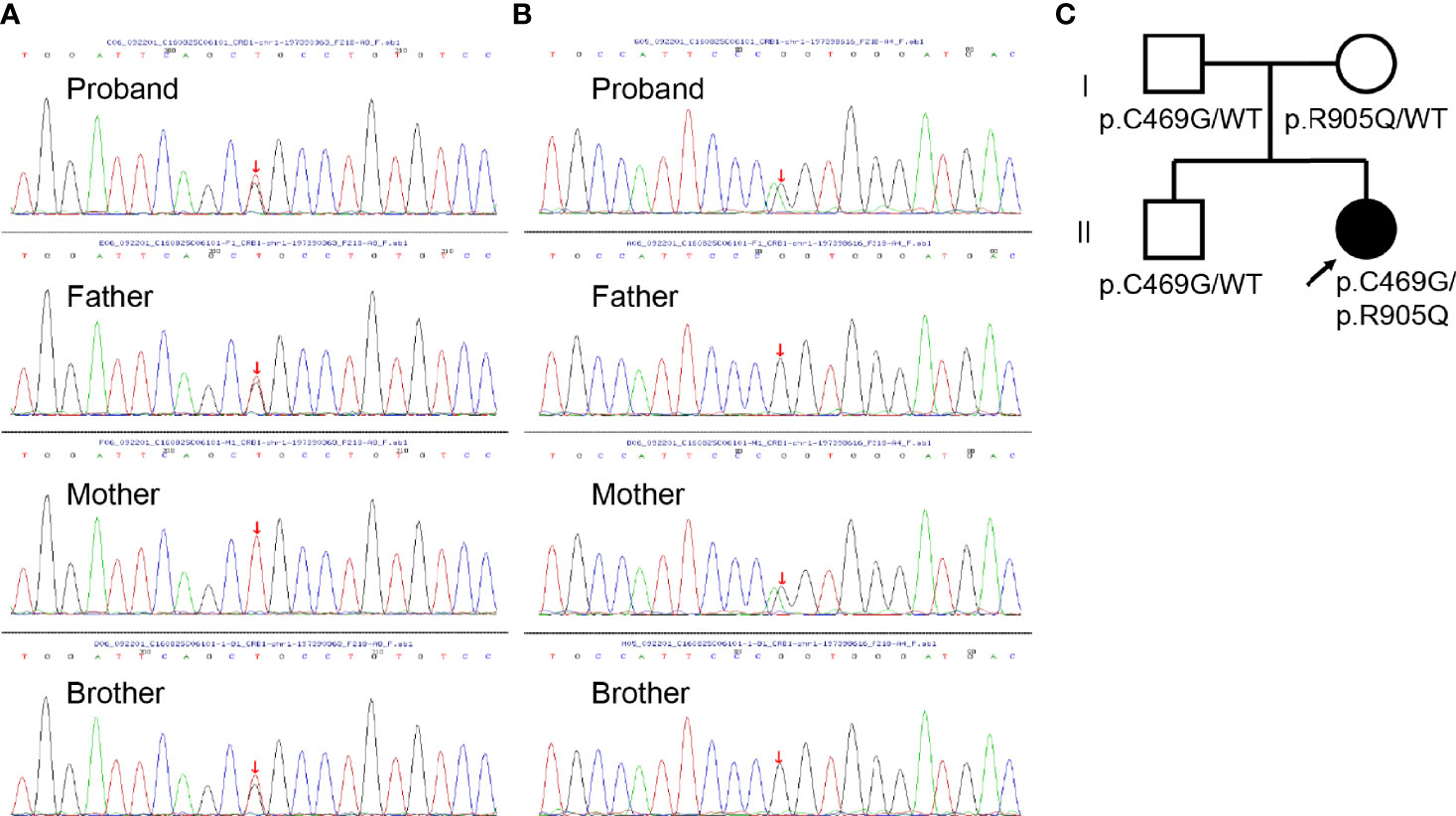
Figure 2 Genomic sequencing and pedigree of the present family. (A) Sanger sequencing revealing the heterozygous c.1405T>G mutation (red arrow) in CRB1 detected in the proband, her father, and brother. (B) Sanger sequencing revealing the heterozygous c.2714G>A mutation (red arrow) in CRB1 detected in the proband and her mother. (C) Pedigree of the present family with the proband indicated (black arrow). Squares indicate men, circles women, shaded symbols individuals with symptoms, and unshaded ones individuals without any symptoms.
Initial treatment involved the following combined medications: beta-blockers, carbonic anhydrase inhibitors, alpha agonists, and finally prostaglandin analogs. Laser peripheral iridectomy (LPI) was performed to achieve a lower IOP fluctuation in both eyes. However, the IOP was beyond control with the maximum tolerated medications 1 year later, and the girl was treated with trabeculectomy combined with mitomycin C in both eyes one after another. In 2018, decreased bleb function was noted, and secondary surgery including phacoemulsification, intraocular lens (IOL) implantation, anterior vitrectomy, surgical peripheral iridotomy, and visco-goniosynechialysis was performed in both eyes. Then, routine follow-up based on the IOP and visual function was advised.
In August 2020, the BCVA was 20/400 in both eyes, and the IOP was 18 mmHg in the right and 17 mmHg in the left (combined with 0.5% timolol BID and 1% brinzolamide TID). Slit-lamp examination revealed a limited functioning filtering bleb in both eyes. The anterior chamber was deep and IOL was in the capsular bag (Figure 3A). A pale optic disk was found, with a C/D of 0.9–1.0 (Figure 3B). An ultrasound biomicroscope (UBM) test further revealed closed anterior chamber angles in all quadrants in the right eye and in the nasal quadrant in the left eye (Figure 3C). OCT revealed more serious cystoid changes and disrupted structures of the outer retina within the macula, as well as excavation of the optic disk (Figure 3D). Blue AF indicated retinal pigment epithelium (RPE) atrophy in the macula (Figure 3E). For electrophysiological testing, the pattern visual evoked potential (PVEP) P100 component was undetectable, while delayed and reduced waves were found in each recording on full-field electroretinography (ERG) (Figure 3F). Decreased central amplitude density in multifocal ERG (mfERG) in both eyes was found as well (Figure 3F).
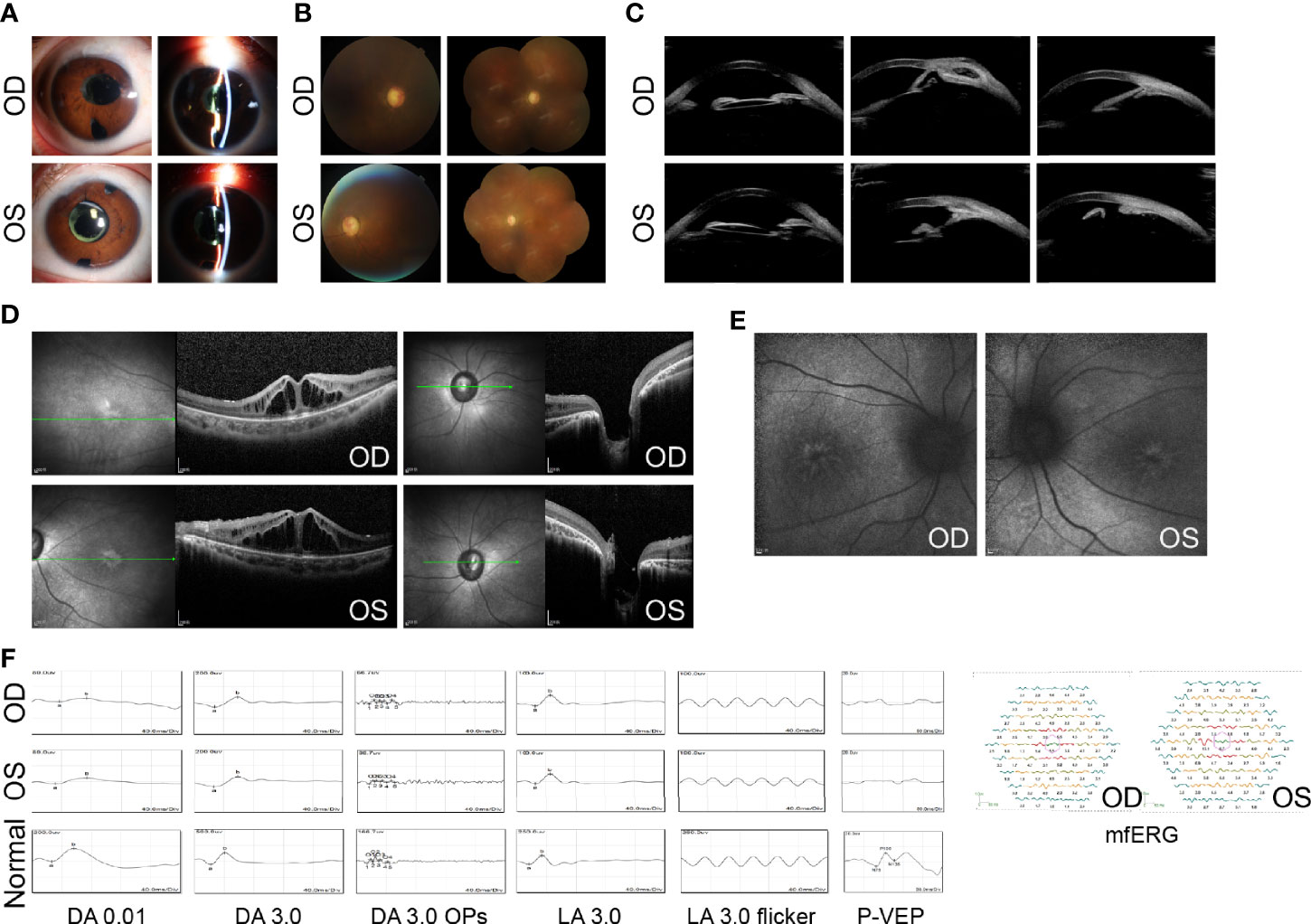
Figure 3 Ocular manifestations after treatment. (A) Slit-lamp examination shows limited functioning filtering bleb, deep anterior chamber, and IOLs in both eyes. (B) Fundus photographs show a pale optic disk with C/D of 0.9–1.0 in both eyes. (C) The UBM test shows closed anterior chamber angles in both eyes. (D) OCT shows more serious cystoid changes within the macula and optic disk excavation in both eyes. (E) AF reveals hyper-AF streaks and mass within the central fovea in both eyes. (F) ERGs show delayed and reduced waves for each recording, while PVEP potentials are undetectable in both eyes. mfERG shows decreased central amplitude density in both eyes. Age-matched normal ERGs and PVEP are shown for comparison. DA, dark-adapted; OPs, oscillatory potentials; LA, light-adapted. UBM images were acquired using Aviso (Quantel Medical, Cournon d’Auvergne, France). All OCT images were acquired using Heidelberg SPECTRALIS OCT (Heidelberg Engineering, Heidelberg, Germany).
The Crumbs protein was first identified in Drosophila. In mammals, the CRB family consists of four members, namely, CRB1, CRB2, CRB3A, and CRB3B (8). The prototype CRB protein is a single transmembrane protein with a large extracellular domain including a signal peptide sequence, 19 epidermal growth factor (EGF)-like domains, and 3 laminin A globular (AG)-like domains. The cytoplasmic domain includes conserved FERM- and PDZ-binding motifs (9) (Figure 4A). They are localized primarily at the subapical region above the adherens junctions (AJ) between photoreceptors, Müller glia cells, and between photoreceptors and Müller glial cells (12) and form the core of the CRB complex which maintains the correct apical–basal polarity and adhesion of retinal neuro-epithelium (8). Therefore, any possible disruption of CRB1 might break the AJ structure and cell polarity, induce photoreceptor injury and finally lead to retinal degeneration resulting in vision impairment.
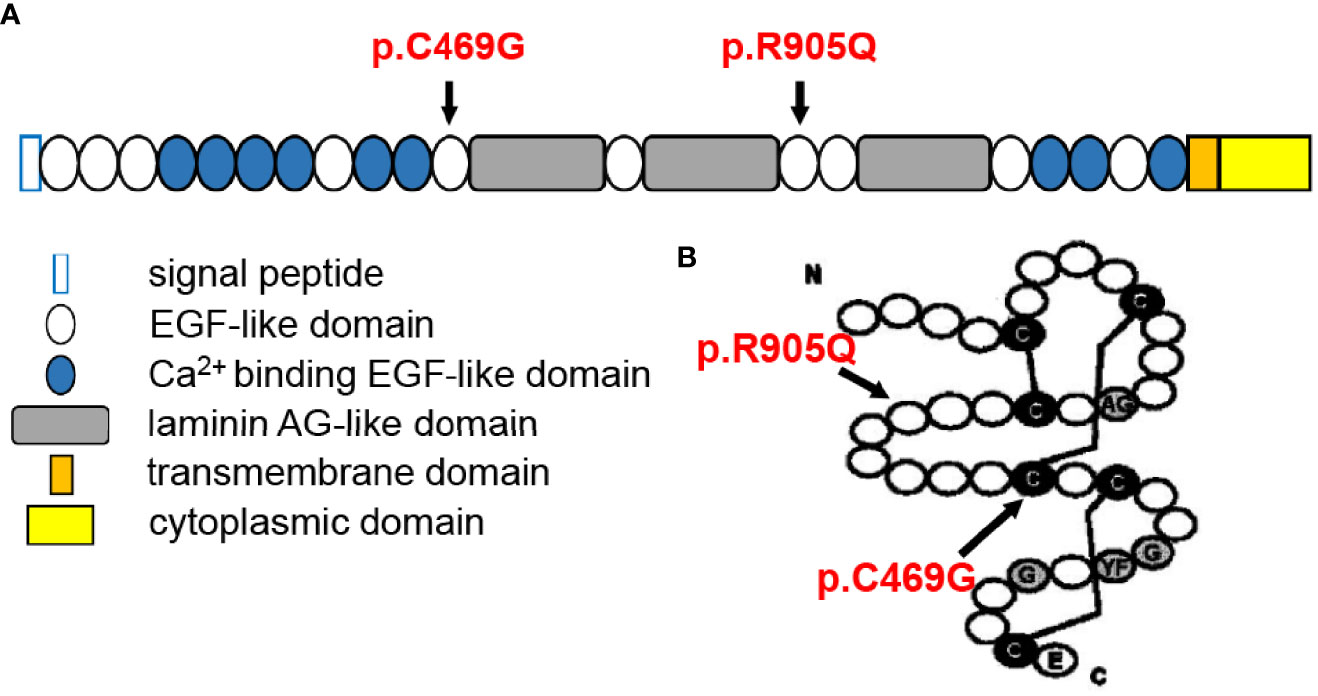
Figure 4 Structural illustration of canonical CRB1-A protein showing the location of the two mutations [diagrammatic drawings are modified and cited from den Hollander et al. (10) and Bujakowska et al. (11)]. (A) Position of the two mutations in relation to CRB1-A protein domains, showing that both mutations are located at the EGF-like domain. (B) Structure of the EGF-like domain with indications of the present mutations, showing that p.C469G mutation might disturb the formation of a disulfide bond between cysteine molecules.
To date, 553 variants in CRB1 have been described [based on the Leiden Open Variation Database (LOVD), https://databases.lovd.nl/shared/genes/CRB1, updated April 13, 2022], of which 450 were labeled pathogenic/pathogenic-like, resulting in a range of retinal disease phenotypes. Ehrenberg et al. (1) reviewed that CRB1 mutation was detected in around 0%–17% of LCA patients, 7% of EORCD patients, and 0%–6.5% of RP patients. CRB1-associated RP, designated “RP12”, appears severe, autosomal recessive and is characterized by a preserved para-arteriolar retinal pigment epithelium (PPRPE) in the early-to-middle stages of disease (13). For LCA, CRB1 mutations cause LCA8, characterized by the following retinal hallmarks: nummular pigment clumps, yellow-white retinal dots, and salt-and-pepper retinal pigmentation (1, 14). Notably, besides those typical phenotypes, a series of rare consequences, including isolated maculopathy, keratoconus, hyperopia or nanophthalmos, and Coats-like vasculopathy, have recently been observed. Firstly, based on previous reports, CRB1-associated maculopathy is characterized by the presence of intraretinal cysts in the inner and outer retinal layers, which could be detected as spoke wheel appearance at the fovea with marked schitic changes at OCT (4, 15–21). In some cases, these changes were described as “macular edema” (18). However, as what was shown in our case, there might be no macular leakage on FFA. Khan et al. (17) described that intraretinal cysts tended to appear in younger patients than older ones, as the latter presented outer retinal degeneration and macular atrophy instead. Mucciolo et al. (20) reported a long-term follow-up of a patient with CRB1-associated maculopathy and found a spontaneous passage from a foveal schitic shape to a cystic profile and finally atrophic maculopathy, without signs of leakage on FFA, which might be regarded as the natural evolution in the course of the disease. Secondly, relative short axial length was another atypical appearance reported due to CRB1 mutation. RP12 and LCA8 have been frequently associated with hyperopia (1, 5, 7, 21). Nanophthalmos has also been reported in CRB1 mutants (6, 22). In a retrospective cohort study, 7 out of 55 patients with CRB1 mutations had associated glaucoma (2). More recently, Liu et al. (23) and Abe et al. (24) have described cases with CRB1-associated RP combined with primary ACG. Therefore, it is indicated that CRB1 mutation may account for the girl’s short axial length, crowded anterior segment, and ACG. Another point in our case is retinal peripheral telangiectasia, which might refer to an early stage of Coats-like response or inflammatory features. On the one hand, it is reported that CRB1 plays a role in neurovascular interaction that regulates layered vascular network development and integrity (25). den Hollander et al. (7) suggested that CRB1 mutations were strongly associated with the development of Coats-like exudative vasculopathy in patients with RP. On the other hand, the vasculopathy observed in CRB1 mutations could also be affected by microenvironmental factors. Murro et al. (26) reported a case of CRB1-associated retinal dystrophy characterized by vitritis, retinal capillaritis, and cystoid change of macular. Bujakowska et al. (9) analyzed published phenotypes of CRB1 mutants and showed that the presence/absence of Coats-like vasculopathy did not reveal a particular mutation pattern, which suggests the involvement of additional genetic and/or environmental modifying factors. Notably, similar signatures, including retinoschisis, mild peripheral angiographic leakage, and ACG, could also be found in autosomal recessive bestrophinopathy (ARB) caused by mutations in both alleles of the BEST1 gene (27, 28). Therefore, genetic testing is essential for differential diagnosis.
Though scientists attempted to understand the phenotype–genotype correlation of CRB1 mutations, current meta-analyses failed to conclude any clear correlation, apart from the agreement that null mutations were more likely to cause more severe phenotypes, indicating additional modifying factors (1, 2, 9, 10, 16, 17). Moreover, it is reasonable to consider that mutations that occurred in key locations relating to protein special structure are more dangerous. Recently, it has been found that there are three main CRB1 isoforms in the human retina: the canonical CRB1-A localized in Müller glial cells (12 exons), the potentially secreted CRB1-C, and a new isoform CRB1-B predominant in photoreceptors (7 exons) (29). Therefore, the positions of mutation may also affect distinct isoforms and lead to different phenotypes. Indeed, Mairot et al. (30) reported that the phenotype in CRB1 patients might be dependent on the severity of Müller cell impairment related to isoform diversity. In addition, experiments based on gene knockout mouse models indicated a modifying role of CRB2 (31–34). In our case, each of the two mutations was inherited paternally and maternally, respectively, and both were located in EGF-like domains (Figure 4A). These two mutations have been previously reported to be associated with LCA (11, 35). The p.C469G mutation affects exon 6 of CRB1-A and, therefore, affects all three isoforms and might disturb the formation of a disulfide bond between cysteine molecules leading to structural alteration. The p.R905Q affects exon 8 of CRB1-A and, therefore, only affects CRB1-A and CRB1-B but does not affect CRB1-C(Figure 4B).
The key points in the management of this case are to stabilize the IOP, monitor macular atrophy, and prevent peripheral capillary leakage. It is still controversial whether the non-edema macular cysts in retinal dystrophy need to be treated or not. Some studies showed that oral uptake of carbonic anhydrase inhibitors (i.e., acetazolamide) was effective in promoting resolution, as they could regulate fluid movement from the retina across the RPE to the choroid and strengthen retinal adhesiveness (4, 36, 37). However, studies also revealed a spontaneous resolution of the cystic abnormalities toward atrophic evolution, which indicated the minimal necessity for drug treatment (20). In this context, long-term follow-up is required to monitor disease progression and adjust therapeutic strategies. Recent studies on mouse models take new insights into gene therapy for CRB1 using adeno-associated viral (AAV) vector-based gene augmentation (38–41). Moreover, the use of CRISPR/Cas9 to correct specific point mutations in patients is also discussed as a potentially viable method (12). In addition, CRB1 patient-induced pluripotent stem cell (iPSC)-derived human retinal organoids could mimic the retinal phenotype and might serve as good models for testing gene therapeutic approaches including both gene augmentation and gene editing (42). Recently, a French biotech company called Horama has signed an exclusive license agreement with Leiden University Medical Center targeting CRB1 gene mutations to treat inherited retinal dystrophies, which is expected to initiate a phase I/II clinical study with the drug candidate in 2023 (41). Those efforts may bring new approaches to a better outcome for patients suffering from CRB1 mutation.
In this paper, we report a rare phenotype of biallelic heterozygous CRB1 mutations which are characterized by macular retinoschisis, Coats-like vasculopathy, short axial length, and ACG. This case emphasizes the importance of securing a genetic diagnosis for patients with combined manifestation in anterior and posterior segments, especially when it appears as cryptogenic retinal atrophy, schisis, or cystic profile. Moreover, it highlights the vital role of CRB1 in maintaining normal macular structure and eyeball development.
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
J-XS, DH, JZ, Y-SW, JW, X-JS, and XH examined and treated the patient. H-XY and X-JS helped with the genome analysis. J-XS and H-XY drafted the manuscript. XH initiated the concept, supervised the writing of the manuscript, and was responsible for the clinical data. All authors contributed to the writing of the final manuscript.
This study was supported by the National Natural Science Foundation of China (81900870 and 81371034).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fopht.2022.902898/full#supplementary-material
1. Ehrenberg M, Pierce EA, Cox GF, Fulton AB. Crb1: One Gene, Many Phenotypes. Semin Ophthalmol (2013) 28(5-6):397–405. doi: 10.3109/08820538.2013.825277
2. Talib M, van Schooneveld MJ, van Genderen MM, Wijnholds J, Florijn RJ, Ten Brink JB, et al. Genotypic and Phenotypic Characteristics of Crb1-Associated Retinal Dystrophies: A Long-Term Follow-Up Study. Ophthalmology (2017) 124(6):884–95. doi: 10.1016/j.ophtha.2017.01.047
3. Morarji J, Lenassi E, Black GC, Ashworth JL. Atypical Presentation of Crb1 Retinopathy. Acta Ophthalmologica (2016) 94(6):e513–4. doi: 10.1111/aos.12997
4. Vincent A, Ng J, Gerth-Kahlert C, Tavares E, Maynes JT, Wright T, et al. Biallelic Mutations in Crb1 Underlie Autosomal Recessive Familial Foveal Retinoschisis. Invest Ophthalmol Visual Sci (2016) 57(6):2637–46. doi: 10.1167/iovs.15-18281
5. Abouzeid H, Li Y, Maumenee IH, Dharmaraj S, Sundin O. A G1103r Mutation in Crb1 Is Co-Inherited With High Hyperopia and Leber Congenital Amaurosis. Ophthalmic Genet (2006) 27(1):15–20. doi: 10.1080/13816810500481840
6. Paun CC, Pijl BJ, Siemiatkowska AM, Collin RW, Cremers FP, Hoyng CB, et al. A Novel Crumbs Homolog 1 Mutation in a Family With Retinitis Pigmentosa, Nanophthalmos, and Optic Disc Drusen. Mol Vision (2012) 18:2447–53.
7. den Hollander AI, Heckenlively JR, van den Born LI, de Kok YJ, van der Velde-Visser SD, Kellner U, et al. Leber Congenital Amaurosis and Retinitis Pigmentosa With Coats-Like Exudative Vasculopathy Are Associated With Mutations in the Crumbs Homologue 1 (Crb1) Gene. Am J Hum Genet (2001) 69(1):198–203. doi: 10.1086/321263
8. Alves CH, Pellissier LP, Wijnholds J. The Crb1 and Adherens Junction Complex Proteins in Retinal Development and Maintenance. Prog Retinal Eye Res (2014) 40:35–52. doi: 10.1016/j.preteyeres.2014.01.001
9. Bujakowska K, Audo I, Mohand-Said S, Lancelot ME, Antonio A, Germain A, et al. Crb1 Mutations in Inherited Retinal Dystrophies. Hum Mutat (2012) 33(2):306–15. doi: 10.1002/humu.21653
10. den Hollander AI, Davis J, van der Velde-Visser SD, Zonneveld MN, Pierrottet CO, Koenekoop RK, et al. Crb1 Mutation Spectrum in Inherited Retinal Dystrophies. Hum Mutat (2004) 24(5):355–69. doi: 10.1002/humu.20093
11. Xu K, Xie Y, Sun T, Zhang X, Chen C, Li Y. Genetic and Clinical Findings in a Chinese Cohort With Leber Congenital Amaurosis and Early Onset Severe Retinal Dystrophy. Br J Ophthalmol (2020) 104(7):932–7. doi: 10.1136/bjophthalmol-2019-314281
12. Quinn PM, Pellissier LP, Wijnholds J. The Crb1 Complex: Following the Trail of Crumbs to a Feasible Gene Therapy Strategy. Front Neurosci (2017) 11:175. doi: 10.3389/fnins.2017.00175
13. den Hollander AI, ten Brink JB, de Kok YJ, van Soest S, van den Born LI, van Driel MA, et al. Mutations in a Human Homologue of Drosophila Crumbs Cause Retinitis Pigmentosa (Rp12). Nat Genet (1999) 23(2):217–21. doi: 10.1038/13848
14. den Hollander AI, Roepman R, Koenekoop RK, Cremers FP. Leber Congenital Amaurosis: Genes, Proteins and Disease Mechanisms. Prog Retinal Eye Res (2008) 27(4):391–419. doi: 10.1016/j.preteyeres.2008.05.003
15. Tsang SH, Burke T, Oll M, Yzer S, Lee W, Xie YA, et al. Whole Exome Sequencing Identifies Crb1 Defect in an Unusual Maculopathy Phenotype. Ophthalmology (2014) 121(9):1773–82. doi: 10.1016/j.ophtha.2014.03.010
16. Khan AO, Aldahmesh MA, Abu-Safieh L, Alkuraya FS. Childhood Cone-Rod Dystrophy With Macular Cystic Degeneration From Recessive Crb1 Mutation. Ophthalmic Genet (2014) 35(3):130–7. doi: 10.3109/13816810.2013.804097
17. Khan KN, Robson A, Mahroo OAR, Arno G, Inglehearn CF, Armengol M, et al. A Clinical and Molecular Characterisation of Crb1-Associated Maculopathy. Eur J Hum Genet EJHG (2018) 26(5):687–94. doi: 10.1038/s41431-017-0082-2
18. Wolfson Y, Applegate CD, Strauss RW, Han IC, Scholl HP. Crb1-Related Maculopathy With Cystoid Macular Edema. JAMA Ophthalmol (2015) 133(11):1357–60. doi: 10.1001/jamaophthalmol.2015.2814
19. Shah N, Damani MR, Zhu XS, Bedoukian EC, Bennett J, Maguire AM, et al. Isolated Maculopathy Associated With Biallelic Crb1 Mutations. Ophthalmic Genet (2017) 38(2):190–3. doi: 10.3109/13816810.2016.1155225
20. Mucciolo DP, Murro V, Giorgio D, Passerini I, Sodi A, Virgili G, et al. Long-Term Follow-Up of a Crb1-Associated Maculopathy. Ophthalmic Genet (2018) 39(4):522–5. doi: 10.1080/13816810.2018.1479431
21. Henderson RH, Mackay DS, Li Z, Moradi P, Sergouniotis P, Russell-Eggitt I, et al. Phenotypic Variability in Patients With Retinal Dystrophies Due to Mutations in Crb1. Br J Ophthalmol (2011) 95(6):811–7. doi: 10.1136/bjo.2010.186882
22. Zenteno JC, Buentello-Volante B, Ayala-Ramirez R, Villanueva-Mendoza C. Homozygosity Mapping Identifies the Crumbs Homologue 1 (Crb1) Gene as Responsible for a Recessive Syndrome of Retinitis Pigmentosa and Nanophthalmos. Am J Med Genet Part A (2011) 155A(5):1001–6. doi: 10.1002/ajmg.a.33862
23. Liu X, Li J, Lin S, Xiao X, Luo J, Wei W, et al. Evaluation of the Genetic Association Between Early-Onset Primary Angle-Closure Glaucoma and Retinitis Pigmentosa. Exp Eye Res (2020) 197:108118. doi: 10.1016/j.exer.2020.108118
24. Abe RY, Makarczyk LSQ, Avila MP, Sallum JMF. Early Occurrence of Primary Angle-Closure Glaucoma in a Patient With Retinitis Pigmentosa and Crb1 Gene Variations. Arq Bras Oftalmol (2022). doi: 10.5935/0004-2749.20220078
25. Son S, Cho M, Lee J. Crumbs Proteins Regulate Layered Retinal Vascular Development Required for Vision. Biochem Biophys Res Commun (2020) 521(4):939–46. doi: 10.1016/j.bbrc.2019.11.013
26. Murro V, Mucciolo DP, Sodi A, Vannozzi L, De Libero C, Simonini G, et al. Retinal Capillaritis in a Crb1-Associated Retinal Dystrophy. Ophthalmic Genet (2017) 38(6):555–8. doi: 10.1080/13816810.2017.1281966
27. Khojasteh H, Azarmina M, Ebrahimiadib N, Daftarian N, Riazi-Esfahani H, Naraghi H, et al. Autosomal Recessive Bestrophinopathy: Clinical and Genetic Characteristics of Twenty-Four Cases. J Ophthalmol (2021) 2021:6674290. doi: 10.1155/2021/6674290
28. Boon CJ, van den Born LI, Visser L, Keunen JE, Bergen AA, Booij JC, et al. Autosomal Recessive Bestrophinopathy: Differential Diagnosis and Treatment Options. Ophthalmology (2013) 120(4):809–20. doi: 10.1016/j.ophtha.2012.09.057
29. Ray TA, Cochran K, Kozlowski C, Wang J, Alexander G, Cady MA, et al. Comprehensive Identification of Mrna Isoforms Reveals the Diversity of Neural Cell-Surface Molecules With Roles in Retinal Development and Disease. Nat Commun (2020) 11(1):3328. doi: 10.1038/s41467-020-17009-7
30. Mairot K, Smirnov V, Bocquet B, Labesse G, Arndt C, Defoort-Dhellemmes S, et al. Crb1-Related Retinal Dystrophies in a Cohort of 50 Patients: A Reappraisal in the Light of Specific Muller Cell and Photoreceptor Crb1 Isoforms. Int J Mol Sci (2021) 22(23). doi: 10.3390/ijms222312642
31. Pellissier LP, Lundvig DM, Tanimoto N, Klooster J, Vos RM, Richard F, et al. Crb2 Acts as a Modifying Factor of Crb1-Related Retinal Dystrophies in Mice. Hum Mol Genet (2014) 23(14):3759–71. doi: 10.1093/hmg/ddu089
32. Quinn PM, Mulder AA, Henrique Alves C, Desrosiers M, de Vries SI, Klooster J, et al. Loss of Crb2 in Muller Glial Cells Modifies a Crb1-Associated Retinitis Pigmentosa Phenotype Into a Leber Congenital Amaurosis Phenotype. Hum Mol Genet (2019) 28(1):105–23. doi: 10.1093/hmg/ddy337
33. Quinn PM, Alves CH, Klooster J, Wijnholds J. Crb2 in Immature Photoreceptors Determines the Superior-Inferior Symmetry of the Developing Retina to Maintain Retinal Structure and Function. Hum Mol Genet (2018) 27(18):3137–53. doi: 10.1093/hmg/ddy194
34. Pellissier LP, Alves CH, Quinn PM, Vos RM, Tanimoto N, Lundvig DM, et al. Targeted Ablation of Crb1 and Crb2 in Retinal Progenitor Cells Mimics Leber Congenital Amaurosis. PloS Genet (2013) 9(12):e1003976. doi: 10.1371/journal.pgen.1003976
35. Zernant J, Kulm M, Dharmaraj S, den Hollander AI, Perrault I, Preising MN, et al. Genotyping Microarray (Disease Chip) for Leber Congenital Amaurosis: Detection of Modifier Alleles. Invest Ophthalmol Visual Sci (2005) 46(9):3052–9. doi: 10.1167/iovs.05-0111
36. Ganesh A, Stroh E, Manayath GJ, Al-Zuhaibi S, Levin AV. Macular Cysts in Retinal Dystrophy. Curr Opin Ophthalmol (2011) 22(5):332–9. doi: 10.1097/ICU.0b013e328349229e
37. Cordovez JA, Traboulsi EI, Capasso JE, Sadagopan KA, Ganesh A, Rychwalski PJ, et al. Retinal Dystrophy With Intraretinal Cystoid Spaces Associated With Mutations in the Crumbs Homologue (Crb1) Gene. Ophthalmic Genet (2015) 36(3):257–64. doi: 10.3109/13816810.2014.881505
38. Buck TM, Vos RM, Alves CH, Wijnholds J. Aav-Crb2 Protects Against Vision Loss in an Inducible Crb1 Retinitis Pigmentosa Mouse Model. Mol Ther Methods Clin Dev (2021) 20:423–41. doi: 10.1016/j.omtm.2020.12.012
39. Boon N, Alves CH, Mulder AA, Andriessen CA, Buck TM, Quinn PMJ, et al. Defining Phenotype, Tropism, and Retinal Gene Therapy Using Adeno-Associated Viral Vectors (Aavs) in New-Born Brown Norway Rats With a Spontaneous Mutation in Crb1. Int J Mol Sci (2021) 22(7). doi: 10.3390/ijms22073563
40. Pellissier LP, Quinn PM, Alves CH, Vos RM, Klooster J, Flannery JG, et al. Gene Therapy Into Photoreceptors and Muller Glial Cells Restores Retinal Structure and Function in Crb1 Retinitis Pigmentosa Mouse Models. Hum Mol Genet (2015) 24(11):3104–18. doi: 10.1093/hmg/ddv062
41. Boon N, Wijnholds J, Pellissier LP. Research Models and Gene Augmentation Therapy for Crb1 Retinal Dystrophies. Front Neurosci (2020) 14:860. doi: 10.3389/fnins.2020.00860
Keywords: Crumbs homolog-1 (CRB1), macular retinoschisis, Coats-like vasculopathy, angle-closure glaucoma, mutation
Citation: Sun J-X, Yan H-X, Hu D, Zhou J, Wang Y-S, Wu J, Song X-J and Hou X (2022) Biallelic Heterozygous Mutations in Crumbs Homolog-1 Gene Associated With Macular Retinoschisis and Angle-Closure Glaucoma: A Case Report and Literature Review. Front. Ophthalmol. 2:902898. doi: 10.3389/fopht.2022.902898
Received: 23 March 2022; Accepted: 03 May 2022;
Published: 03 June 2022.
Edited by:
Yiqin Du, University of Pittsburgh, United StatesReviewed by:
ShiYing Li, Xiamen University, ChinaCopyright © 2022 Sun, Yan, Hu, Zhou, Wang, Wu, Song and Hou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xu Hou, aHhmbW11QDE2My5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.