 Mary Anne Garner
Mary Anne Garner Ryan G. Strickland
Ryan G. Strickland Christopher A. Girkin1,2
Christopher A. Girkin1,2 Alecia K. Gross
Alecia K. Gross
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Ophthalmol., 09 December 2022
Sec. Retina
Volume 2 - 2022 | https://doi.org/10.3389/fopht.2022.1007103
This article is part of the Research TopicWomen in Retina: 2022View all 7 articles
The maintenance of intraocular pressure (IOP) is critical to preserving the pristine optics required for vision. Disturbances in IOP can directly impact the optic nerve and retina, and inner retinal injury can occur following acute and chronic IOP elevation. There are a variety of animal models that have been developed to study the effects of acute and chronic elevation of IOP on the retina, retinal ganglion cell (RGC) morphology, intracellular signaling, gene expression changes, and survival. Acute IOP models induce injury that allows for the study of RGC response to well characterized injury and potential recovery. This review will focus on the initial impact of acute IOP elevation on RGC injury and recovery as these early responses may be the best targets for potential therapeutic interventions to promote RGC survival in glaucoma.
The loss of retinal ganglion cells (RGCs) in the retina and degeneration of the optic nerve is a hallmark of glaucomatous optic neuropathy, a leading cause for irreversible blindness worldwide (1, 2). Lowering of intraocular pressure (IOP) is currently the primary therapeutic intervention for glaucoma, though there are other possible causative factors that are responsible for RGC death in the disease (3, 4). In the last decade, several reviews have focused on the molecular mechanisms of (5–10) and neuroprotective strategies for (11–13) chronic glaucoma. While glaucoma is a heterogenous disease encompassing several different pathological phenotypes, here we review the morphological and molecular changes that RGCs and retinal glial cells undergo in models of acute increases in IOP.
Acute models of ocular hypertension vary in their execution, duration, and severity but generally produce a reversible loss of inner retinal function without inducing immediate RGC death. While the mechanism of RGC injury from either acute subischemic or ischemic injury may differ from the changes seen with chronic elevated IOP exposure, the acute IOP model can be performed with precisely controlled pressures and durations and provides a window into the mechanisms of RGC recovery. Additionally, electroretinography (ERG), used to measure retinal function, can be used as a metric for ensuring that the elevated IOP-induced injury remains subischemic since very high pressures can affect inner retinal function resulting in diminished b-waves (14).
There are numerous animal models of acute hypertension (15–34), and although nearly every model is different in mechanism, duration, and pressure elevation, we have included those that were evaluated within hours or days of pressure elevation unless otherwise explicitly stated. These models have resulted in an extensive body of knowledge regarding genes expressed and proteins up- or down-regulated. Throughout, we state whether the model discussed is acute or transient in nature and have added relevant findings from genetic and chronic models.
While the optic nerve and retinal structure vary across species, models of elevated IOP exhibit consistent patterns of RGC damage following injury. Though glaucoma is a chronic degenerative optic neuropathy, early disease stage signaling pathways and cell morphological changes are uncovered by studying the response of RGCs and their milieu to acute injury and the mechanisms by which they recover from induced damage (35).
The neural retina is an organized tissue with distinct layers of neuronal and glial cells. The neuronal cell types include: 1) rod and cone photoreceptors, which transduce the energy of a photon of light into an electrical signal; 2) horizontal cells in the inner nuclear layer which synapse with photoreceptors; 3) bipolar cells, which receive input from photoreceptors and form synapses with RGCs; 4) amacrine cells in the inner plexiform layer form synapses with RGCs; and 5) RGC somas, dendrites, and axons. RGC axons form the optic nerve that projects to the lateral geniculate nucleus and suprachiasmatic nucleus of the thalamus (in primates) as well as to the superior colliculus of the brainstem and primary visual cortex within the occipital lobe (in primates and rodent models). The retina is also home to glial cells including astrocytes, Müller glia, and microglia. Each of these glial cell types play a role in RGC support, and each can subsequently become deleterious to RGC survival when activated for extended periods of time (e.g., following ocular hypertension).
Molecular profiling has indicated that there are approximately 40 subtypes of RGCs described in mice (36–39) and at least 18 different subtypes of RGCs in the primate retina (40, 41). Of the four major types studied in glaucomatous animal models (ON-sustained or transient and OFF-sustained or transient), the αOFF-RGCs, whose dendritic arbors form synapses with bipolar cells in the OFF sublamina of the inner retina, are the most vulnerable to morphological changes in dendritic arborization, synapse formation, and cell death following IOP elevation by laser (42) and microbead injection (43, 44). However, this effect may be due to age-related changes in dendritic morphology with OFF-RGCs being more adaptable and therefore able to recover more quickly following injury in younger animals (45). Additionally, the reported susceptibility of the larger αOFF-RGCs could be due to quantification of RGC subtypes based on soma size and an unintentional bias towards the loss of cells with larger somas if RGC cell bodies have shrunk in response to injury. ON- and OFF-sustained RGCs show evidence of normal dendritic arbors but loss of excitatory synapses within those arbors following microbead-induced IOP elevation (43), implying that changes in synapse formation may precede dendritic arbor morphological changes.
The mechanism of injury from acute IOP elevation is incompletely understood as elevation of IOP can impact the RCG axons directly and indirectly within the optic nerve head (ONH), and can alter inner retinal and ONH perfusion to the RGC soma and axons. In humans and higher primates, the axons of the RGCs pass through the lamina cribrosa before becoming myelinated and forming the optic nerve. Though rodent models lack the connective tissue found in human lamina cribrosae, they do possess a glial lamina containing astrocytes, and axoplasmic stasis can occur in the ONH of these models as well (46). There is an extensive body of work analyzing the effects of axonal damage on retrograde and anterograde transport and synapse degeneration, discussed below. The lamina cribrosa has been studied extensively as it is particularly susceptible to the damage induced by elevated IOP (47), resulting in axoplasmic stasis within the RGC axons. Remodeling of the lamina cribrosa in response to elevated IOP has been recently reviewed (48).
In addition to the axonal injury within the ONH region, there are also effects on the retinal vasculature and neural retina, especially in the inner retina, which can affect receptors in the dendritic membranes and calcium concentrations within the cells (49). Early gene expression and cell signaling changes that occur because of elevated IOP on the retina and on axonal transport are essential to understanding the early stages of glaucomatous damage (26).
RGC function, measured via ERG, is lost as a result of increases in IOP in animal models of experimental glaucoma while the outer retinal function is preserved (14, 16, 26, 50, 51). With acute models of very high IOP produced either transiently (17) or for a prolonged acute period (1 hour) (52), the functional recovery time for RGCs can be longer than that of the outer retina and may result in a lack of recovery even 4 weeks following insult (53). Transient increases in IOP are achieved through manometrically elevating IOP using a saline solution and are often used to produce pressures above that of the mean arterial pressure, producing an ischemic insult and preventing ocular perfusion. These high pressures not only affect RGC function but also result in loss of function of the outer retinal photoreceptors and bipolar cells. These studies often focus on functional recovery over time following insults of varying degrees. Studies have also indicated a relationship between blood pressure (BP), and therefore ocular perfusion pressure, and functional losses due to IOP (54).
Acute increases in IOP lead to nerve fiber layer thinning with a decrease in RGC soma number in rats (14, 52, 55) and mice (16), and degeneration of the optic nerve (18). Interestingly, there have also been reports of retinal nerve fiber layer thickening that is resolved by 3 weeks in rats exposed to 8 hours of IOP at 50 mmHg (56).
How retinal cells initially respond to changes in IOP may be due to detection of mechanical stimuli via transient receptor potential (TRP) channels found throughout the retina (57) including in RGCs (58) and glia (59). This family of 28 cationic channels respond to mechanical stimuli such as strain or shear, inflammatory signals, changes in metabolic energy and oxidation (among other stimuli). An excellent recent review of TRP channels in all retinal cell types (60) discusses their role in retinal homeostasis and in response to injury, including their ability to induce astrocyte cytoskeletal reorganization and migration in response to elevated IOP (59, 61). Because TRP channels play a role in cellular homeostasis, their overactivation and inhibition can both result in gliosis. TRP channels remain a focus for investigation because their early sensing of mechanical changes following IOP may be what initially induce subcellular changes in retinal glia and RGCs.
One of the earliest morphological responses to elevated IOP is the interruption of axonal transport (Figure 1) (29, 30, 32, 62). Since neurotrophins can be target-derived and retrogradely transported along with their receptors in RGCs, the increase in IOP and subsequent axoplasmic stasis is detrimental to this vital process. The neurotrophin brain-derived neurotrophic factor (BDNF) and its receptor, TrkB, are normally trafficked retrogradely via dynein along axonal microtubules in RGCs. Retrograde transport of BDNF (22, 63) and TrkB (but not TrkA) are affected following acutely increased IOP in rats, similar to a chronic model of glaucoma in non-human primates (22). It is important to note that these studies detect effects in axonal transport hours following acute IOP elevation. When the analysis occurs one to two weeks following acute IOP elevation, there are no changes in anterograde or retrograde transport (56). This difference highlights the ability of RGCs to recover from acute models of hypertension. BDNF levels are notably low in glaucoma patients (5, 64, 65), and treatment with BDNF can be protective in glaucoma models (13, 66–68). BDNF protects RGCs exposed to increased IOP (69, 70) and is sufficient to increase proximal and distal axon density (71). However, the protections offered by BDNF are transient (72) and treatment with any neurotrophic factor would need to overcome the physical blood-retinal barrier as well as the limitation of neurotrophin half-life (73).
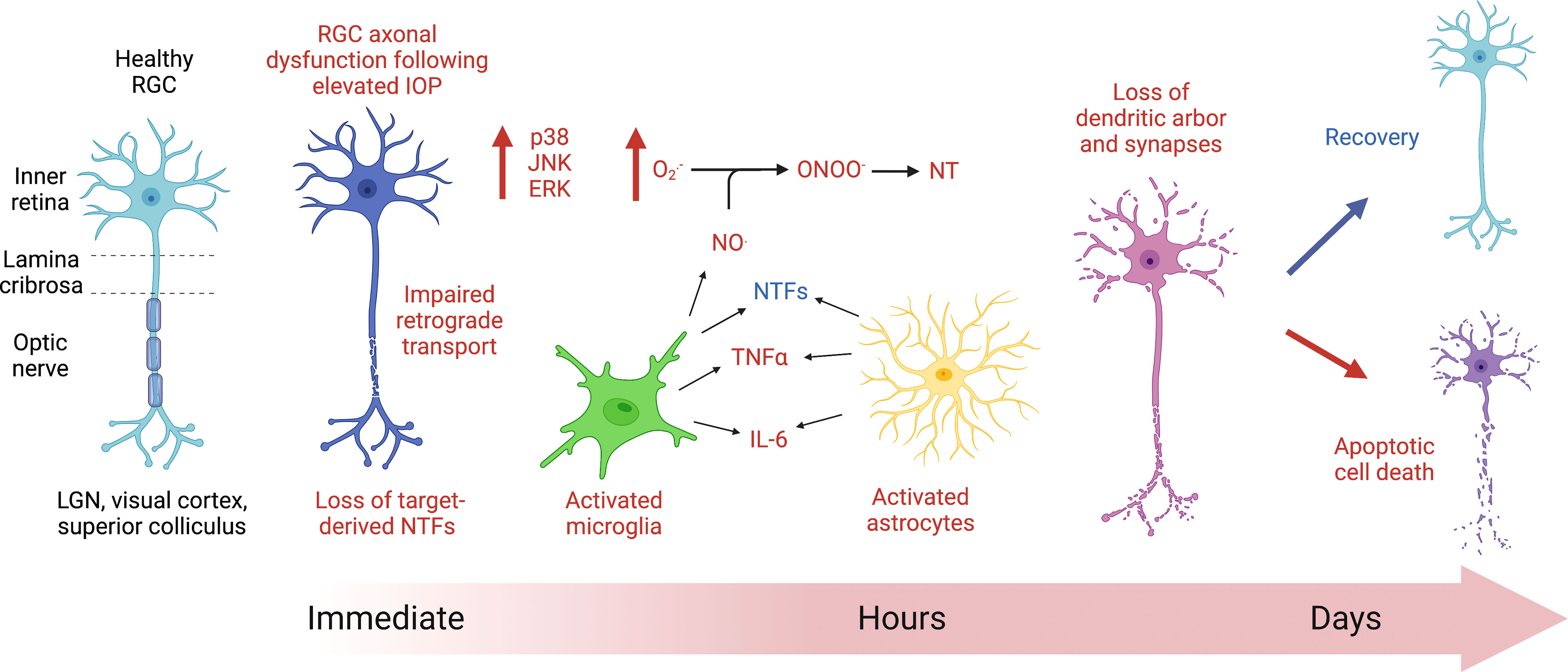
Figure 1 Cartoon diagram depicting the initial response of retinal ganglion cells (RGCs) to acute ocular hypertension. Axoplasmic stasis occurs immediately following RGC injury because of acute increases in intraocular pressure (IOP), affecting axonal transport. Within hours of injury, cell signaling changes occur, notably with the expression of mitogen activated protein kinases (MAPK, such as p38, JNK, and ERK). Glial cells, including microglia (green) and astrocytes (yellow), show evidence of increased reactivity within hours of the insult. Activated glial cells release nitric oxide (NO.), which, when combined with superoxide (), can produce the highly reactive intermediate compound, peroxynitrite (ONOO-), which can then nitrate proteins on tyrosine residues (NT), interfering with protein function. There are noted morphological changes in RGC dendritic arborization within hours and throughout days following injury. Ultimately, the RGCs affected by increases in ocular hypertension will either recover or die via apoptosis.
Mitochondria are also transported along axons in RGCs, and mitochondrial health is implicated in glaucoma since aged animals exhibit lower rates of transport and RGC dysfunction, resulting in the upregulation of Parkin, LC3-I and II, LAMP, and Optineurin (74). Effects on mitochondrial transport have been noted at 3 days following elevated IOP in a rodent model (75), though these trafficking defects may occur earlier as seen with BDNF and TrkB (22, 63). These early changes in axonal transport can precede RGC damage in acute and chronic disease models as axoplasmic statis has been shown to occur prior to RGC soma loss or presynaptic losses (Figure 1) (76). In an acute rat model of laser-induced ocular hypertension, axon transport deficits preceded RGC soma loss (77). In the chronic DBA/2J mouse model of ocular hypertension, anterograde transport is also affected early, even without exposure to increased IOP, and RGCs die via apoptosis (78).
Studies of IOP elevation in rats indicate initial increases in cell proliferation markers (79) and down-regulated pathways involved in axon extension, dendrite morphogenesis and metabolism of RGCs prior to the onset of apoptotic cell death (80). RGC degeneration involves dendritic changes including a loss of arborization and a decrease in synapse number. Dendritic branching and complexity can be assessed using a Sholl analysis where concentric circles are placed at regular intervals around the soma of the RGC and intersections are counted (81). Additionally, synapse number can be assessed using pre- and post-synaptic markers (e.g., synapsin, PSD95), and individual RGCs can be assessed using Thy-1 YFP transgenic mice where less than 1% of RGCs express the YFP, allowing for visualization of the entire dendritic arbor (81). Alterations in dendritic morphology occur prior to soma loss following acute IOP elevation in mice (82) as they do after optic nerve transection and chronic IOP elevation (81). In fact, dendritic morphological changes have been noted as soon as 90 minutes following acute elevation in IOP (82) (Figure 1). Various studies have investigated the early changes in RGC compartments after ocular hypertension (5, 6, 83–85). These changes include alterations in soma and dendrites as well as failures in axonal transport distally (83). How these changes relate to axonal injury at the nerve head or alterations in retinal perfusion remain unclear.
Microarray studies in retina indicate an upregulation in inflammatory and immune pathways in the early stages of elevated IOP-induced damage. While these studies are retina-wide and do not distinguish among the cell types in which the changes occur, they provide evidence for changes in the early stages of glaucomatous damage. Early immune responses are detectable prior to RGC neurodegeneration, but whether these responses are protective or detrimental is unclear.
When neuronal damage occurs, glial inflammatory responses may initially produce a protective response, releasing neurotrophic factors and antioxidants, but prolonged reactivity of astrocytes and microglia can become excitotoxic to RGCs. Some acute models provide evidence for the upregulation of genes indicating activated microglia (Aif1) (79, 80) and reactive astrocytes and Müller glial (Gfap) (80), though others did not find changes in Gfap gene expression (79). There is also evidence for increased GFAP protein expression in rats (14, 86) and mice (16, 87) as early as 1 day following acute ocular hypertension (Figure 1). Glial reactivity and immune upregulation may therefore occur in early-to-mid states of glaucomatous disease (88), may not be upregulated uniformly across the retina or ONH, and may depend on the extent and duration of ocular hypertensive injury. GFAP protein expression is certainly increased in chronic models of ocular hypertension and in glaucomatous eyes (89).
Microglia are resident immune cells within the nervous system that respond to neuronal damage by proliferating and producing inflammatory cytokines such as tumor necrosis factor alpha (TNFα) (90). While initially neuroprotective (91), prolonged microglial activation can become detrimental. In glaucoma, microglial activity occurs during the early stages of the disease, prior to RGC apoptosis (91, 92), and treating DBA/2J mice with an inhibitor of microglial activation, minocycline, prevents some RGC apoptosis (93). Recent work has shown, however, that ablation of microglia causes significantly more RGC loss in a chronic microbead mouse model (94). The upregulation of heat shock proteins (HSP) by RGCs in response to elevated IOP can trigger microglial activation and inflammatory responses (95). HSPs are also elevated in human patients with glaucoma.
Acute elevation in IOP is sufficient to induce morphological changes in microglia in as little as one hour (Figure 1) (96), and activated microglia release pro-inflammatory molecules (e.g., complement proteins, nitric oxide (NO.), TNFα, interleukin-6 (IL-6), and FasL) which contribute to RGC damage and apoptotic cell death in glaucoma (97, 98). Cytokines such as IL6 and Lif have also been shown to be immediately upregulated in a rat model of acute IOP (79). Macrophages respond after acute IOP in rats (99) and mice (100) with increases in MCP-1, a macrophage chemokine, as early as 6 hours following acute IOP (101).
Superoxide () is upregulated in the optic nerve and in the RGC somas within 24 hours (102), and free radical scavengers offer protection (103) in acute models. Superoxide and nitric oxide (NO.) can combine to form peroxynitrite (ONOO-) (Figure 1), which is a highly reactive negatively charged molecule that can nitrate tyrosine residues in proteins, altering their function. Nitrotyrosine levels have long been linked with inflammation and neurodegeneration and are elevated in primary open angle glaucoma (POAG) patient serum and in aqueous humor of animal models of glaucoma (104). The antioxidant ubiquinol (the reduced form of CoQ10) has been shown to be neuroprotective in one model of acute hypertension for at least 2 weeks following injury (105) as well as in the DBA/2J mouse model of ocular hypertension (106) by decreasing Bax expression and caspase-3 release, thereby preventing RGC apoptosis (105).
Increases in mitogen activated protein kinases (MAPKs) such as phosphorylated p38, JNK, and ERK are elevated within one hour following acute increases in IOP in the rat (106) as they are in glaucomatous eyes (98) (Figure 1). Inhibition of p38 MAPK has been shown to prevent degeneration of RGC axons in a microbead model of elevated IOP in rats while also protecting anterograde axonal transport (107–109). While JNK3 (110) and JNK2 (111) deficiencies are not protective, inhibition of JNK1/2/3 did spare RGCs exposed to acute high IOP (112).
Acute models of elevated IOP are used to elucidate the retinal response to this acute multifactorial insult. While chronic models of elevated IOP are able to analyze the fate of RGCs and their environment, acute models of IOP elevation provide a window into the initial subcellular and morphological changes that occur in RGCs and their neighboring retinal cells. The wide variety of models used to examine the effects of acute ocular hypertension on RGC morphology and cell signaling as well as glial reactivity have resulted in an enormous body of work across multiple animal species. Overall, they reveal the sensitivity of the inner retina to IOP related injury and the extent of the autoregulatory capacity of the inner retina, illuminating several critical pathways that mediate damage to the inner retina. While the mechanisms of acute IOP injury likely involve differing degrees of primary retinal ischemia when compared to chronic glaucoma, the use of acute models of ocular hypertension across several species is further strengthened by the fact that many of the pathways upregulated are also found in more prolonged models of ocular hypertension and in the later stages of chronic disease models as well as in human postmortem glaucomatous eyes. Uncovering the early responses of RGCs and their milieu to initial insults will be important in the search for therapeutic interventions in glaucoma since it is these initial changes that will likely be the optimal targets for pharmacological and/or genetic therapeutic interventions early in the disease, protecting RGCs from injury prior to irreversible loss. Expansion of acute models to include the human eye would further complement the work accomplished in animal models, providing translational validation for mechanisms of and treatments for the human disease.
MG: Conceptualization, writing – original draft preparation, writing – review and editing, investigation. RS: writing – review and editing. CG: Conceptualization, review and editing, funding acquisition, supervision. AG: Conceptualization, writing – review and editing, funding acquisition, supervision. All authors contributed to the article and approved the submitted version.
National Eye Institute: R01 EY0300096 (AG), National Eye Institute: R01 EY028284 (CG), EyeSight Foundation of Alabama (CG), Research to Prevent Blindness (CG).
We would like to thank Dr. Lynn Dobrunz for invaluable insights for this work.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Quigley HA, Broman AT. The number of people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol (2006) 90(3):262–7. doi: 10.1136/bjo.2005.081224
2. Tham Y-C, Li X, Wong TY, Quigley HA, Aung T, Cheng C-Y. Global prevalence of glaucoma and projections of glaucoma burden through 2040: A systematic review and meta-analysis. Ophthalmology (2014) 121(11):2081–90. doi: 10.1016/j.ophtha.2014.05.013
3. Leske MC, Heijl A, Hussein M, Bengtsson B, Hyman L, Komaroff E, et al. Factors for glaucoma progression and the effect of treatment: The early manifest glaucoma trial. Arch Ophthalmol (2003) 121(1):48–56. doi: 10.1001/archopht.121.1.48
4. Heijl A, Leske C, Bengtsson B, Hyman L, Bengtsson B, Hussein M, et al. Reduction of intraocular pressure and glaucoma progression. Arch Ophthalmol (2002) 120(10):1268. doi: 10.1001/archopht.120.10.1268
5. Almasieh M, Wilson AM, Morquette B, Cueva Vargas JL, Di Polo A. The molecular basis of retinal ganglion cell death in glaucoma. Prog Retin Eye Res (2012) 31(2):152–81. doi: 10.1016/j.preteyeres.2011.11.002
6. Agostinone J, Di Polo A. Retinal ganglion cell dendrite pathology and synapse loss: Implications for glaucoma. Prog Brain Res (2015) 220:199–216. doi: 10.1016/bs.pbr.2015.04.012
7. Williams PA, Marsh-Armstrong N, Howell GR. Lasker/IRRF initiative on astrocytes and glaucomatous neurodegeneration participants. neuroinflammation in glaucoma: A new opportunity. Exp Eye Res (2017) 157:20–7. doi: 10.1016/j.exer.2017.02.014
8. Ito YA, Di Polo A. Mitochondrial dynamics, transport, and quality control: A bottleneck for retinal ganglion cell viability in optic neuropathies. Mitochondrion (2017) 36:186–92. doi: 10.1016/j.mito.2017.08.014
9. Quaranta L, Bruttini C, Micheletti E, Konstas AGP, Michelessi M, Oddone F, et al. Glaucoma and neuroinflammation: An overview. Surv Ophthalmol (2021) 66(5):693–713. doi: 10.1016/j.survophthal.2021.02.003
10. Boccuni I, Fairless R. Retinal glutamate neurotransmission: From physiology to pathophysiological mechanisms of retinal ganglion cell degeneration. Life (Basel) (2022) 12(5): 638–671. doi: 10.3390/life12050638
11. Lambuk L, Mohd Lazaldin MA, Ahmad S, Iezhitsa I, Agarwal R, Uskoković V, et al. Brain-derived neurotrophic factor-mediated neuroprotection in glaucoma: A review of current state of the art. Front Pharmacol (2022) 13:875662. doi: 10.3389/fphar.2022.875662
12. Yuan Y, Xiong R, Wu Y, Ha J, Wang W, Han X, et al. Associations of statin use with the onset and progression of open-angle glaucoma: A systematic review and meta-analysis. E Clin Med (2022) 46:101364. doi: 10.1016/j.eclinm.2022.101364
13. Fudalej E, Justyniarska M, Kasarełło K, Dziedziak J, Szaflik JP, Cudnoch-Jędrzejewska A. Neuroprotective factors of the retina and their role in promoting survival of retinal ganglion cells: A review. Ophthalmic Res (2021) 64(3):345–55. doi: 10.1159/000514441
14. Hannon BG, Feola AJ, Gerberich BG, Read AT, Prausnitz MR, Ethier CR, et al. Using retinal function to define ischemic exclusion criteria for animal models of glaucoma. Exp Eye Res (2021) 202:108354. doi: 10.1016/j.exer.2020.108354
15. Kong YX, Crowston JG, Vingrys AJ, Trounce IA, Bui VB. Functional changes in the retina during and after acute intraocular pressure elevation in mice. Invest Ophthalmol Vis Sci (2009) 50(12):5732–40. doi: 10.1167/iovs.09-3814
16. Kong YXG, van Bergen N, Bui BV, Chrysostomou V, Vingrys AJ, Trounce IA, et al. Impact of aging and diet restriction on retinal function during and after acute intraocular pressure injury. Neurobiol Aging (2012) 33(6):1126.e15–25. doi: 10.1016/j.neurobiolaging.2011.11.026
17. Chong RS, Busoy JMF, Tan B, Yeo SW, Lee YS, Barathi AV, et al. A minimally invasive experimental model of acute ocular hypertension with acute angle closure characteristics. Transl Vis Sci Technol (2020) 9(7):24. doi: 10.1167/tvst.9.7.24
18. He Z, Bui BV, Vingrys AJ. The rate of functional recovery from acute IOP elevation. Invest Ophthalmol Vis Sci (2006) 47(11):4872–80. doi: 10.1167/iovs.06-0590
19. Joos KM, Li C, Sappington RM. Morphometric changes in the rat optic nerve following short-term intermittent elevations in intraocular pressure. Invest Ophthalmol Vis Sci (2010) 51(12):6431–40. doi: 10.1167/iovs.10-5212
20. Morrison JC, Johnson E, Cepurna WO. Rat models for glaucoma research. Prog Brain Res (2008) 173:285–301. doi: 10.1016/S0079-6123(08)01121-7
21. Johnson TV, Tomarev SI. Rodent models of glaucoma. Brain Res Bull (2010) 81(2–3):349–58. doi: 10.1016/j.brainresbull.2009.04.004
22. Bouhenni RA, Dunmire J, Sewell A, Edward DP. Animal models of glaucoma. J BioMed Biotechnol (2012) 2012:692609. doi: 10.1155/2012/692609
23. Pease ME, McKinnon SJ, Quigley HA, Kerrigan–Baumrind LA, Zack DJ. Obstructed axonal transport of BDNF and its receptor TrkB in experimental glaucoma | IOVS | ARVO journals. Invest Ophthalmol Visual Sci (2000) 41(3):764–74.
24. Lagreze WA, Knörl R. Memantine is neuroprotective in a rat model of pressure induced retinal ischemia. Invest Ophthalmol Vis Sci (1998) 39(6):4.
25. Selles-Navarro I, Villegas-Pérez M. Retinal ganglion cell death after different transient periods of pressure-induced ischemia and survival intervals. Invest Ophthalmol Vis Sci (1996) 37(10):13.
26. Johnson NF, Foulds WS. The effect of total acute ischemia on the structure of the rabbit retina. Exp Eye Res (1978) 27(1): 45–59. doi: 10.1016/0014-4835(78)90052-0.
27. Siliprandi R, Bucci MG, Canella R, Carmignoto G. Flash and pattern electroretinograms during and after acute intraocular pressure elevation in cats. Invest Ophthalmol Vis Sci (1988) 29(4):558–65.
28. Grehn F, Prost M. Function of retinal nerve fibers depends on perfusion pressure: neurophysiologic investigations during acute intraocular pressure elevation. Invest Ophthalmol Vis Sci (1983) 24:7.
29. Hamor RE GJP. Evaluation of short-term increased intraocular pressure on flash and pattern-generated electroretinograms of dogs. Am J Vet Res (2000) 61(9): 1087–91. doi: 10.2460/ajvr.2000.61.1087
30. Anderson DR, Hendrickson A. Effect of intraocular pressure on rapid axoplasmic transport in monkey optic nerve. Invest Ophthalmol (1974) 13(10):771–83.
31. Minckler DS, Bunt AH, Johanson GW. Orthograde and retrograde axoplasmic transport during acute ocular hypertension in the monkey. Invest Ophthalmol Vis Sci (1977) 16(5):426–41.
32. Coleman AL, Quigley HA, Vitale S, Dunkelberger G. Displacement of the optic nerve head by acute changes in intraocular pressure in monkey eyes. Ophthalmology (1991) 98(1):35–40. doi: 10.1016/S0161-6420(91)32345-5
33. Quigley HA, Anderson DR. Distribution of axonal transport blockade by acute intraocular pressure elevation in the primate optic nerve head. Invest Ophthalmol Vis Sci (1977) 16(7):640–4.
34. Strouthidis NG, Fortune B, Yang H, Sigal IA, Burgoyne CF. Effect of acute intraocular pressure elevation on the monkey optic nerve head as detected by spectral domain optical coherence tomography. Invest Ophthalmol Vis Sci (2011) 52(13):9431–7. doi: 10.1167/iovs.11-7922
35. Crowston JG, Kong YXG, Trounce IA, Dang TM, Fahy ET, Bui BV, et al. An acute intraocular pressure challenge to assess retinal ganglion cell injury and recovery in the mouse. Exp Eye Res (2015) 141:3–8. doi: 10.1016/j.exer.2015.03.006
36. Laboissonniere LA, Goetz JJ, Martin GM, Bi R, Lund TJS, Ellson L, et al. Molecular signatures of retinal ganglion cells revealed through single cell profiling. Sci Rep (2019) 9(1):15778. doi: 10.1038/s41598-019-52215-4
37. Laboissonniere LA, Sonoda T, Lee SK, Trimarchi JM, Schmidt TM. Single-cell RNA-seq of defined subsets of retinal ganglion cells. J Vis Exp (2017) 123: 55229–39. doi: 10.3791/55229
38. Rheaume BA, Jereen A, Bolisetty M, Sajid MS, Yang Y, Renna K, et al. Single cell transcriptome profiling of retinal ganglion cells identifies cellular subtypes. Nat Commun (2018) 9(1):2759. doi: 10.1038/s41467-018-05134-3
39. Baden T, Berens P, Franke K, Román Rosón M, Bethge M, Euler T. The functional diversity of retinal ganglion cells in the mouse. Nature (2016) 529(7586):345–50. doi: 10.1038/nature16468
40. Yamada ES, Bordt AS, Marshak DW. Wide-field ganglion cells in macaque retinas. Vis Neurosci (2005) 22(4):383–93. doi: 10.1017/S095252380522401X
41. Masri RA, Percival KA, Koizumi A, Martin PR, Grünert U. Survey of retinal ganglion cell morphology in marmoset. J Comp Neurol (2019) 527(1):236–58. doi: 10.1002/cne.24157
42. Ou Y, Jo RE, Ullian EM, Wong ROL, Della Santina L. Selective vulnerability of specific retinal ganglion cell types and synapses after transient ocular hypertension. J Neurosci (2016) 36(35):9240–52. doi: 10.1523/JNEUROSCI.0940-16.2016
43. Della Santina L, Inman DM, Lupien CB, Horner PJ, Wong ROL. Differential progression of structural and functional alterations in distinct retinal ganglion cell types in a mouse model of glaucoma. J Neurosci (2013) 33(44):17444–57. doi: 10.1523/JNEUROSCI.5461-12.2013
44. El-Danaf RN, Huberman AD. Characteristic patterns of dendritic remodeling in early-stage glaucoma: Evidence from genetically identified retinal ganglion cell types. J Neurosci (2015) 35(6):2329–43. doi: 10.1523/JNEUROSCI.1419-14.2015
45. Lee PY, Zhao D, Wong VHY, Chrysostomou V, Crowston JG, Bui BV. The effect of aging on retinal function and retinal ganglion cell morphology following intraocular pressure elevation. Front Aging Neurosci (2022) 14:859265. doi: 10.3389/fnagi.2022.859265
46. Zhou L, Chen W, Lin D, Hu W, Tang Z. Neuronal apoptosis, axon damage and synapse loss occur synchronously in acute ocular hypertension. Exp Eye Res (2019) 180:77–85. doi: 10.1016/j.exer.2018.12.006
47. Sigal IA, Flanagan JG, Tertinegg I, Ethier CR. Modeling individual-specific human optic nerve head biomechanics. part I: IOP-induced deformations and influence of geometry. Biomech Model Mechanobiol (2009) 8(2):85–98.
48. Strickland RG, Garner MA, Gross AK, Girkin CA. Remodeling of the lamina cribrosa: mechanisms and potential therapeutic approaches for glaucoma. Int J Mol Sci (2022) 23(15):8068–87. doi: 10.3390/ijms23158068
49. Wang AY, Lee PY, Bui BV, Jobling AI, Greferath U, Brandli A, et al. Potential mechanisms of retinal ganglion cell type-specific vulnerability in glaucoma. Clin Exp Optom (2020) 103(5):562–71. doi: 10.1111/cxo.13031
50. Choh V, Gurdita A, Tan B, Prasad RC, Bizheva K, Joos KM. Short-term moderately elevated intraocular pressure is associated with elevated scotopic electroretinogram responses. Invest Ophthalmol Vis Sci (2016) 57(4):2140–51. doi: 10.1167/iovs.15-18770
51. Choh V, Gurdita A, Tan B, Feng Y, Bizheva K, McCulloch DL, et al. Isoflurane and ketamine:xylazine differentially affect intraocular pressure-associated scotopic threshold responses in sprague-dawley rats. Doc Ophthalmol (2017) 135(2):121–32. doi: 10.1007/s10633-017-9597-7
52. Oka T, Tamada Y, Nakajima E, Shearer TR, Azuma M. Presence of calpain-induced proteolysis in retinal degeneration and dysfunction in a rat model of acute ocular hypertension. J Neurosci Res (2006) 83(7):1342–51. doi: 10.1002/jnr.20827
53. Bui BV, He Z, Vingrys AJ, Nguyen CTO, Wong VHY, Fortune B. Using the electroretinogram to understand how intraocular pressure elevation affects the rat retina. J Ophthalmol (2013) 2013:262467. doi: 10.1155/2013/262467
54. He Z, Nguyen CTO, Armitage JA, Vingrys AJ, Bui BV. Blood pressure modifies retinal susceptibility to intraocular pressure elevation. PloS One (2012) 7(2):e31104. doi: 10.1371/journal.pone.0031104
55. Tan C, Hu T, Peng M, Liu S, Tong J, Ouyang W, et al. Age of rats seriously affects the degree of retinal damage induced by acute high intraocular pressure. Curr Eye Res (2015) 40(3):300–6. doi: 10.3109/02713683.2014.922194
56. Abbott CJ, Choe TE, Lusardi TA, Burgoyne CF, Wang L, Fortune B. Evaluation of retinal nerve fiber layer thickness and axonal transport 1 and 2 weeks after 8 hours of acute intraocular pressure elevation in rats. Invest Ophthalmol Vis Sci (2014) 55(2):674–87. doi: 10.1167/iovs.13-12811
57. Gilliam JC, Wensel TG. TRP channel gene expression in the mouse retina. Vision Res (2011) 51(23-24):2440–52. doi: 10.1016/j.visres.2011.10.009
58. Koser DE, Thompson AJ, Foster SK, Dwivedy A, Pillai EK, Sheridan GK, et al. Mechanisensing is critical for axon growth in the developing brain. Nat Neurosci (2016) 19(12):1592–8. doi: 10.1038/nn.4394
59. Morozumi W, Inagaki S, Iwata Y, Nakamura S, Hara H, Shimazawa M. Piezo channel plays a part in retinal ganglion cell damage. Exp Eye Res (2020) 191:107900. doi: 10.1016/j.exer.2019.107900
60. Križaj D, Cordeiro S, Strauß O. Retinal TRP channels: Cell-type-specific regulators of retinal homeostasis and multimodal integration. Prog Retin Eye Res (2022), 101114. doi: 10.1016/j.preteyeres.2022.101114
61. Choi HJ, Sun D, Jakobs TC. Astrocytes in the optic nerve head express putative mechanisensitive channels. Mol Vision (2015) 21:749–66.
62. Johansson JO. Inhibition and recovery of retrograde axoplasmic transport in rat optic nerve during and after elevated IOP in vivo. Exp Eye Res (1988) 46(2):223–7. doi: 10.1016/S0014-4835(88)80079-4
63. Quigley HA, McKinnon. Retrograde axonal transport of BDNF in retinal ganglion cells is blocked by acute IOP elevation in rats. IOVS (2000) 41(11):7.
64. Ghaffariyeh A, Honarpisheh N, Heidari MH, Puyan S, Abasov F. Brain-derived neurotrophic factor as a biomarker in primary open-angle glaucoma. Optom Vis Sci (2011) 88(1):80–5. doi: 10.1097/OPX.0b013e3181fc329f
65. Oddone F, Roberti G, Micera A, Busanello A, Bonini S, Quaranta L, et al. Exploring serum levels of brain derived neurotrophic factor and nerve growth factor across glaucoma stages. PloS One (2017) 12(1):e0168565. doi: 10.1371/journal.pone.0168565
66. Ren R, Li Y, Liu Z, Liu K, He S. Long-term rescue of rat retinal ganglion cells and visual function by AAV-mediated BDNF expression after acute elevation of intraocular pressure. Invest Ophthalmol Vis Sci (2012) 53(2):1003–11. doi: 10.1167/iovs.11-8484
67. Gao H, Qiao X, Cantor LB, WuDunn D. Up-regulation of brain-derived neurotrophic factor expression by brimonidine in rat retinal ganglion cells. Arch Ophthalmol (2002) 120(6):797–803. doi: 10.1001/archopht.120.6.797
68. Mohd Lazaldin MA, Iezhitsa I, Agarwal R, Bakar NS, Agarwal P, Mohd Ismail N. Neuroprotective effects of brain-derived neurotrophic factor against amyloid beta 1-40-induced retinal and optic nerve damage. Eur J Neurosci (2020) 51(12):2394–411. doi: 10.1111/ejn.14662
69. Cheng L, Sapieha P, Kittlerova P, Hauswirth WW, Di Polo A. TrkB gene transfer protects retinal ganglion cells from axotomy-induced death in vivo. J Neurosci (2002) 22(10):3977–86. doi: 10.1523/JNEUROSCI.22-10-03977.2002
70. Di Polo A, Aigner LJ, Dunn RJ, Bray GM, Aguayo AJ. Prolonged delivery of brain-derived neurotrophic factor by adenovirus-infected müller cells temporarily rescues injured retinal ganglion cells. Proc Natl Acad Sci USA (1998) 95(7):3978–83. doi: 10.1073/pnas.95.7.3978
71. Du R, Wang X, He S. BDNF improves axon transportation and rescues visual function in a rodent model of acute elevation of intraocular pressure. Sci China Life Sci (2020) 63(9):1337–46. doi: 10.1007/s11427-019-1567-0
72. Johnson EC, Morrison JC. Friend or foe? resolving the impact of glial responses in glaucoma. J Glaucoma (2009) 18(5):341–53. doi: 10.1097/IJG.0b013e31818c6ef6
73. Mallone F, Sacchetti M, Bruscolini A, Scuderi L, Marenco M, Lambiase A. Neurotrophic factors in glaucoma and innovative delivery systems. Appl Sci (2020) 10(24):9015. doi: 10.3390/app10249015
74. Dai Y, Hu X, Sun X. Overexpression of parkin protects retinal ganglion cells in experimental glaucoma. Cell Death Dis (2018) 9(2):88. doi: 10.1038/s41419-017-0146-9
75. Takihara Y, Inatani M, Eto K, Inoue T, Kreymerman A, Miyake S, et al. In vivo imaging of axonal transport of mitochondria in the diseased and aged mammalian CNS. Proc Natl Acad Sci USA (2015) 112(33):10515–20. doi: 10.1073/pnas.1509879112
76. Buckingham BP, Inman DM, Lambert W, Oglesby E, Calkins DJ, Steele MR, et al. Progressive ganglion cell degeneration precedes neuronal loss in a mouse model of glaucoma. J Neurosci (2008) 28(11):2735–44. doi: 10.1523/JNEUROSCI.4443-07.2008
77. Lambert WS, Ruiz L, Crish SD, Wheeler LA, Calkins DJ. Brimonidine prevents axonal and somatic degeneration of retinal ganglion cell neurons. Mol Neurodegener (2011) 6(1):4. doi: 10.1186/1750-1326-6-4
78. Libby RT, Li Y, Savinova OV, Barter J, Smith RS, Nickells RW, et al. Susceptibility to neurodegeneration in a glaucoma is modified by bax gene dosage. PloS Genet (2005) 1(1):17–26. doi: 10.1371/journal.pgen.0010004
79. Morrison JC, Cepurna WO, Tehrani S, Choe TE, Jayaram H, Lozano DC, et al. A period of controlled elevation of IOP (CEI) produces the specific gene expression responses and focal injury pattern of experimental rat glaucoma. Invest Ophthalmol Vis Sci (2016) 57(15):6700–11. doi: 10.1167/iovs.16-20573
80. Guo Y, Cepurna WO, Dyck JA, Doser TA, Johnson EC, Morrison JC. Retinal cell responses to elevated intraocular pressure: A gene array comparison between the whole retina and retinal ganglion cell layer. Invest Ophthalmol Vis Sci (2010) 51(6):3003–18. doi: 10.1167/iovs.09-4663
81. Henderson DCM, Vianna JR, Gobran J, Di Pierdomenico J, Hooper ML, Farrell SRM, et al. Longitudinal in vivo changes in retinal ganglion cell dendritic morphology after acute and chronic optic nerve injury. Invest Ophthalmol Vis Sci (2021) 62(9):5. doi: 10.1167/iovs.62.9.5
82. Li Z, Liu S, Weinreb RN, Lindsey JD, Yu M, Liu L, et al. Tracking dendritic shrinkage of retinal ganglion cells after acute elevation of intraocular pressure. Invest Ophthalmol Vis Sci (2011) 52(10):7205–12. doi: 10.1167/iovs.10-6868
83. Crish SD, Sappington RM, Inman DM, Horner PJ, Calkins DJ. Distal axonopathy with structural persistence in glaucomatous neurodegeneration. Proc Natl Acad Sci USA (2010) 107(11):5196–201. doi: 10.1073/pnas.0913141107
84. Whitmore AV, Libby RT, John SWM. Glaucoma: Thinking in new ways-a role for autonomous axonal self-destruction and other compartmentalised processes? Prog Retin Eye Res (2005) 24(6):639–62. doi: 10.1016/j.preteyeres.2005.04.004
85. Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. The classical complement cascade mediates CNS synapse elimination. Cell (2007) 131(6):1164–78. doi: 10.1016/j.cell.2007.10.036
86. Cho KJ, Kim JH, Park H-YL, Park CK. Glial cell response and iNOS expression in the optic nerve head and retina of the rat following acute high IOP ischemia-reperfusion. Brain Res (2011) 1403:67–77. doi: 10.1016/j.brainres.2011.06.005
87. Chrysostomou V, Crowston JG. The photopic negative response of the mouse electroretinogram: reduction by acute elevation of intraocular pressure. Invest Ophthalmol Vis Sci (2013) 54(7):4691–7. doi: 10.1167/iovs.13-12415
88. Guo Y, Johnson EC, Cepurna WO, Dyck JA, Doser T, Morrison JC. Early gene expression changes in the retinal ganglion cell layer of a rat glaucoma model. Invest Ophthalmol Vis Sci (2011) 52(3):1460–73. doi: 10.1167/iovs.10-5930
89. Tezel G, Chauhan BC, LeBlanc RP, Wax MB. Immunohistochemical assessment of the glial mitogen-activated protein kinase activation in glaucoma. Invest Ophthalmol Vis Sci (2003) 44(7):3025–33. doi: 10.1167/iovs.02-1136
90. Wei X, Cho K-S, Thee EF, Jager MJ, Chen DF. Neuroinflammation and microglia in glaucoma: time for a paradigm shift. J Neurosci Res (2019) 97(1):70–6. doi: 10.1002/jnr.24256
91. Ramirez AI, de Hoz R, Salobrar-Garcia E, Salazar JJ, Rojas B, Ajoy D, et al. The role of microglia in retinal neurodegeneration: Alzheimer’s disease, Parkinson, and glaucoma. Front Aging Neurosci (2017) 9:214. doi: 10.3389/fnagi.2017.00214
92. Bosco A, Romero CO, Breen KT, Chagovetz AA, Steele MR, Ambati BK, et al. Neurodegeneration severity can be predicted from early microglia alterations monitored in vivo in a mouse model of chronic glaucoma. Dis Model Mech (2015) 8(5):443–55. doi: 10.1242/dmm.018788
93. Bosco A, Inman DM, Steele MR, Wu G, Soto I, Marsh-Armstrong N, et al. Reduced retina microglial activation and improved optic nerve integrity with minocycline treatment in the DBA/2J mouse model of glaucoma. Invest Ophthalmol Vis Sci (2008) 49(4):1437–46. doi: 10.1167/iovs.07-1337
94. Tan Z, Guo Y, Shrestha M, Sun D, Gregory-Ksander M, Jakobs TC. Microglia depletion exacerbates retinal ganglion cell loss in a mouse model of glaucoma. Exp Eye Res (2022) 225:109273. doi: 10.1016/j.exer.2022.109273
95. Luo C, Yang X, Kain AD, Powell DW, Kuehn MH, Tezel G. Glaucomatous tissue stress and the regulation of immune response through glial toll-like receptor signaling. Invest Ophthalmol Vis Sci (2010) 51(11):5697–707. doi: 10.1167/iovs.10-5407
96. Sun D, Qu J, Jakobs TC. Reversible reactivity by optic nerve astrocytes. Glia (2013) 61(8):1218–35. doi: 10.1002/glia.22507
97. Hanisch U-K, Kettenmann H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci (2007) 10(11):1387–94. doi: 10.1038/nn1997
98. Tezel G, Yang X, Luo C, Cai J, Powell DW. An astrocyte-specific proteomic approach to inflammatory responses in experimental rat glaucoma. Invest Ophthalmol Vis Sci (2012) 53(7):4220–33. doi: 10.1167/iovs.11-9101
99. Huang Y, Li Z, van Rooijen N, Wang N, Pang CP, Cui Q. Different responses of macrophages in retinal ganglion cell survival after acute ocular hypertension in rats with different autoimmune backgrounds. Exp Eye Res (2007) 85(5):659–66. doi: 10.1016/j.exer.2007.07.020
100. Kezic JM, Chrysostomou V, Trounce IA, McMenamin PG, Crowston JG. Effect of anterior chamber cannulation and acute IOP elevation on retinal macrophages in the adult mouse. Invest Ophthalmol Vis Sci (2013) 54(4):3028–36. doi: 10.1167/iovs.13-11865
101. Sun M-H, Pang J-HS, Chen S-L, Han W-H, Ho T-C, Chen K-J, et al. Retinal protection from acute glaucoma-induced ischemia-reperfusion injury through pharmacologic induction of heme oxygenase-1. Invest Ophthalmol Vis Sci (2010) 51(9):4798–808. doi: 10.1167/iovs.09-4086
102. Kanamori A, Catrinescu M-M, Kanamori N, Mears KA, Beaubien R, Levin LA. Superoxide is an associated signal for apoptosis in axonal injury. Brain (2010) 133(9):2612–25. doi: 10.1093/brain/awq105
103. Kanamori A, Catrinescu M-M, Mahammed A, Gross Z, Levin LA. Neuroprotection against superoxide anion radical by metallocorroles in cellular and murine models of optic neuropathy. J Neurochem (2010) 114(2):488–98. doi: 10.1111/j.1471-4159.2010.06781.x
104. Lei Y, Gao Y, Song M, Cao W, Sun X. Peroxynitrite is a novel risk factor and treatment target of glaucoma. Nitric Oxide (2020) 99:17–24. doi: 10.1016/j.niox.2020.03.006
105. Ju W-K, Shim MS, Kim K-Y, Bu JH, Park TL, Ahn S, et al. Ubiquinol promotes retinal ganglion cell survival and blocks the apoptotic pathway in ischemic retinal degeneration. Biochem Biophys Res Commun (2018) 503(4):2639–45. doi: 10.1016/j.bbrc.2018.08.016
106. Edwards G, Lee Y, Kim M, Bhanvadia S, Kim K-Y, Ju W-K. Effect of ubiquinol on glaucomatous neurodegeneration and oxidative stress: Studies for retinal ganglion cell survival and/or visual function. Antioxid (Basel) (2020) 9(10):952–971. doi: 10.3390/antiox9100952
107. Roth S, Shaikh AR, Hennelly MM, Li Q, Bindokas V, Graham CE. Mitogen-activated protein kinases and retinal ischemia. Invest Ophthalmol Vis Sci (2003) 44(12):5383–95. doi: 10.1167/iovs.03-0451
108. Lambert WS, Pasini S, Collyer JW, Formichella CR, Ghose P, Carlson BJ, et al. Of mice and monkeys: neuroprotective efficacy of the p38 inhibitor BIRB 796 depends on model duration in experimental glaucoma. Sci Rep (2020) 10(1):8535. doi: 10.1038/s41598-020-65374-6
109. Dapper JD, Crish SD, Pang I-H, Calkins DJ. Proximal inhibition of p38 MAPK stress signaling prevents distal axonopathy. Neurobiol Dis (2013) 59:26–37. doi: 10.1016/j.nbd.2013.07.001
110. Quigley HA, Cone FE, Gelman SE, Yang Z, Son JL, Oglesby EN, et al. Lack of neuroprotection against experimental glaucoma in c-jun n-terminal kinase 3 knockout mice. Exp Eye Res (2011) 92(4):299–305. doi: 10.1016/j.exer.2011.01.006
111. Nickells RW, Howell GR, Soto I, John SWM. Under pressure: cellular and molecular responses during glaucoma, a common neurodegeneration with axonopathy. Annu Rev Neurosci (2012) 35:153–79. doi: 10.1146/annurev.neuro.051508.135728
Keywords: retina, glaucoma, ocular hypertension, acute intraocular pressure (IOP), retinal ganglion cell
Citation: Garner MA, Strickland RG, Girkin CA and Gross AK (2022) Mechanisms of retinal ganglion cell injury following acute increases in intraocular pressure. Front. Ophthalmol. 2:1007103. doi: 10.3389/fopht.2022.1007103
Received: 29 July 2022; Accepted: 26 October 2022;
Published: 09 December 2022.
Edited by:
Stephanie Louise Grillo, Penn State Milton S. Hershey Medical Center, United StatesReviewed by:
Najam Sharif, Santen Inc., United StatesCopyright © 2022 Garner, Strickland, Girkin and Gross. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alecia K. Gross, YWdyb3NzQHVhYi5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.