 Albatool AlKhazal
Albatool AlKhazal Samiha Chohan
Samiha Chohan Destani J. Ross1
Destani J. Ross1 Jinhwan Kim
Jinhwan Kim
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 04 March 2025
Sec. Pediatric Oncology
Volume 15 - 2025 | https://doi.org/10.3389/fonc.2025.1553511
This article is part of the Research Topic Novel Molecular Targets and Therapies for Pediatric Extracranial Solid Tumors View all 7 articles
Neuroblastoma is a pediatric cancer that originates from neural crest cells and is the most common extracranial solid tumor in children under five years of age. While low-risk neuroblastoma often regresses spontaneously, high-risk neuroblastoma poses a significant clinical challenge. Recent advances in understanding neuroblastoma’s molecular mechanisms have led to the development of targeted therapies that aim to selectively inhibit specific pathways involved in tumor growth and progression, improving patient outcomes while minimizing side effects. This review provides a comprehensive review of neuroblastoma biology and emerging therapeutic strategies. Key topics include (a) immunotherapies and immunotargets, (b) non-coding RNAs (long non-coding RNA, microRNA, and circular RNA), (c) molecular biomarkers and pathways, and (d) limitations and future directions.
Neuroblastoma is the most common extracranial solid tumor in children under the age of five, accounting for approximately 15% of childhood cancer-related deaths (1). It originates from immature neural crest cells within the sympathetic nervous system (2). More specifically, neuroblastoma is thought to arise from sympathoadrenal progenitor cells, which first emerge in the sympathetic ganglia and adrenal glands during development. These progenitor cells co-express markers indicative of both sympathetic neurons and chromaffin cell differentiation (3). During normal development, sympathoadrenal (SA) cells migrate to their designated locations, where extrinsic signals guide their differentiation into either sympathetic neurons or chromaffin cells (4). However, when this differentiation process is disrupted, tumors can form along the sympathetic nervous system, including in the adrenal gland and paraspinal ganglia from the head and neck down to the pelvis (5, 6).
Neuroblastoma is often referred to as a “tumor of extremes” due to its biological heterogeneity, manifested in the presence of two primary cell types: undifferentiated mesenchymal cells and more differentiated adrenergic cells (7). These cell types can switch back and forth (interconvert), resembling cells at different stages of normal neural crest development (7). This cellular diversity likely underlies the disease’s highly variable clinical course, ranging from spontaneous regression in some cases to aggressive, fatal progression in others. This variability has intrigued researchers since its discovery (8, 9). Neuroblastoma’s Neuroblastoma was first identified as a pediatric cancer in the mid-nineteenth century, and its neuronal origin was established by Wright in 1910 (10). Following the emergence of pediatric surgery after World War I, Robert Gross perfected surgical techniques for children and reported on over 200 neuroblastoma cases (11). In collaboration with radiologist Martin Wittenborg, Gross used orthovoltage X-ray therapy to achieve local control of the disease (12). They discovered that older patients with advanced disease had poorer outcomes compared to younger patients, who had more favorable survival rates. By the mid-20th century, chemotherapy, radiotherapy, and surgery became the standard of care, and in 1971, a formal staging system for the neuroblastoma was established (13).
Neuroblastoma patients are categorized into low-, intermediate-, and high-risk groups based on clinical, pathologic, and genetic factors, including tumor stage, age at diagnosis, and molecular markers (14). High-risk neuroblastoma (HRNB) constitutes approximately 50-60% of all neuroblastoma cases and is associated with poor prognostic indicators such as MYCN amplification, metastatic disease, and unfavorable histology. Despite significant advancements in treatment, outcomes for children with HRNB remain poor, with a 5-year overall survival (OS) rate ranging from 51-62% (14). Tumor heterogeneity and development of treatment resistance in HRNB present significant therapeutic challenges (15). However, with advancements in biotechnology and sequencing technologies, precision medicine has emerged as a promising approach for improving outcomes, particularly through targeted therapies aimed at the molecular drivers of the disease.
The current treatment approach for HRNB includes intensive multimodal and systemic regimens such as chemotherapy, immunotherapy, and stem cell transplantation. These systemic therapies damage both cancerous and healthy cells and notoriously result in significant short and long-term adverse effects, negatively impacting the quality of life for patients. This underscores the need for more effective and targeted therapies that address the underlying molecular mechanisms of the disease (16). Targeted therapies may target proteins expressed on cancer cells or address mutations in DNA or protein expression patterns. Mechanisms of action for targeted therapies include blocking chemical signaling pathways or delivering toxins directly to cancer cells, all of which work to suppress tumor cell proliferation. As genome-wide sequencing continues to uncover more molecular biomarkers specific to these heterogeneous tumors, targeted and precision medicine strategies can be further refined, ultimately improving outcomes and reducing the cytotoxicity of current treatments. This review focuses on understanding the biology of HRNB and explores ongoing research and clinical strategies aimed at developing and integrating targeted therapies (Figure 1).
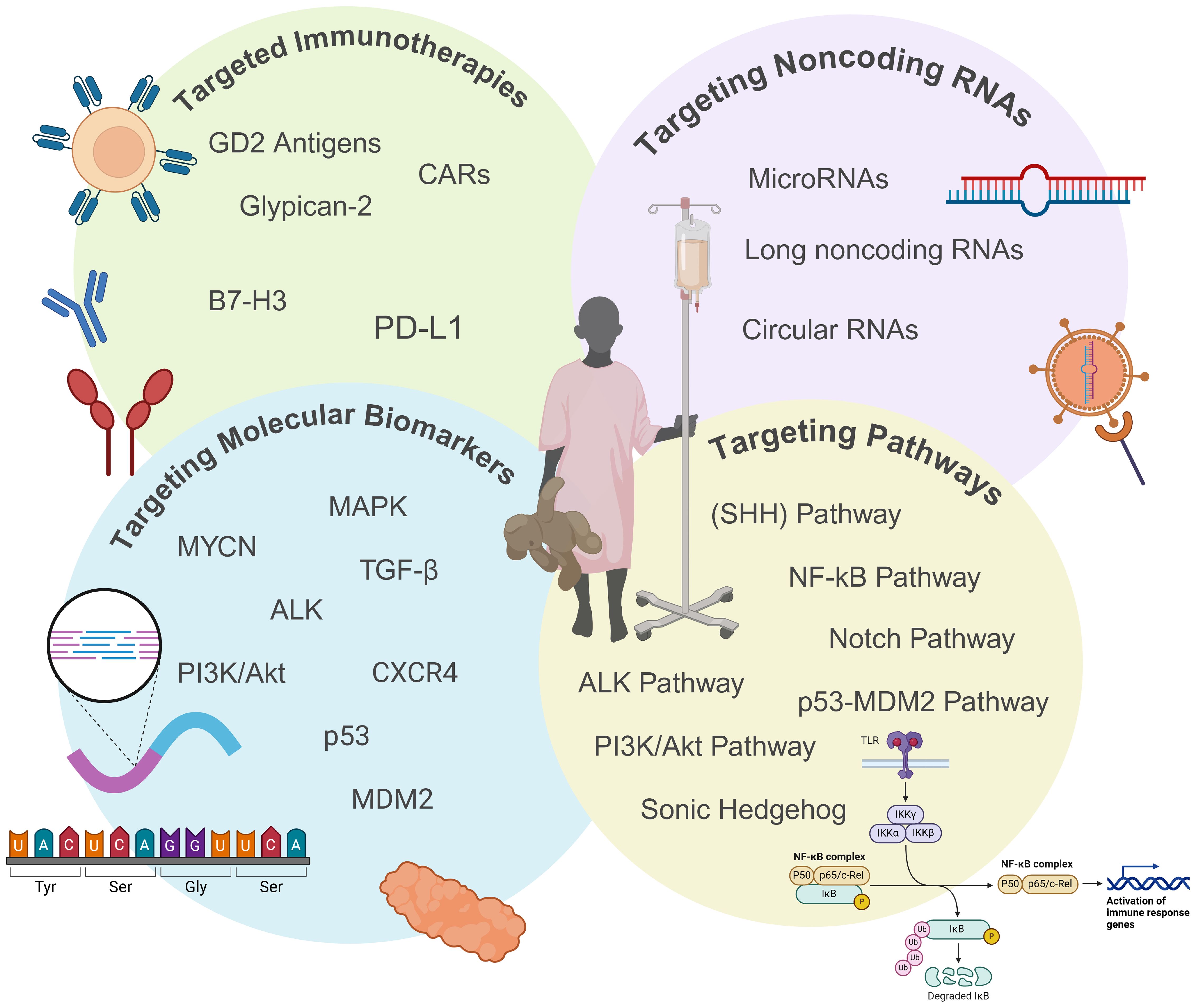
Figure 1. Overview of emerging targeted therapeutic approaches for high-risk neuroblastoma (HRNB). This figure highlights emerging targeted therapies for HRNB, including immunotherapies, noncoding RNAs, biomarkers, and dysregulated pathways. These approaches aim to overcome the limitations and toxicities of traditional treatments by offering more precise and effective therapeutic options to improve outcomes for HRNB patients. Created with BioRender.
Due to neuroblastoma’s variable clinical spectrum, risk stratification plays a crucial role in determining treatment plans and predicting outcomes. Historical staging systems have been replaced by the International Neuroblastoma Risk Group (INRG) Staging System introduced in 2005, which allows for preoperative risk stratification and incorporates the assessment of image-defined risk factors (IDRFs) to distinguish non-metastatic tumors into stages L1 and L2 (17). L1 tumors represent tumors without evidence of encasement or compression of critical nerves, blood vessels, or organs; while the presence of these imaging features indicate an L2 tumor. Tumors with IDRFs have a higher risk of disease recurrence, more challenging surgical resection and overall poorer outcomes. Children with metastatic disease are classified as stage M; while children under 18 months of age with metastasis limited to the skin, liver, or bone marrow are a unique subset of patients classified as stage MS. In addition to the INRG stage, factors such as the patient’s age at diagnosis, tumor histology and cytogenetic features are used to further refine risk classification. In 2021, the Children’s Oncology Group (COG) added segmental chromosomal aberrations (SCAs) to the risk assessment, as these aberrations are associated with poor outcomes (14).
Revisions to the neuroblastoma risk classification by COG reflect the impact of ongoing research efforts to better understand neuroblastoma tumor biology and advancements in predicting prognosis. However, despite these efforts, HRNB remains a major treatment challenge, with a 5-year event-free survival (EFS) rate of just 51% for HRNB patients, compared to over 85% for those in low- and intermediate-risk groups (14).
The treatment of HRNB typically involves three phases: induction, consolidation, and post-consolidation. The induction phase aims to eliminate visible disease and achieve remission, using five to eight cycles of intensive chemotherapy with drugs such as platinum agents, topoisomerase inhibitors, and alkylating agents. Ongoing COG trials utilize a combination of topotecan, vincristine, doxorubicin, cisplatin, cyclophosphamide, and etoposide chemotherapeutics (18). Autologous stem cell collection occurs during induction, as stem cells are purged of malignant cells before being stored for later use in consolidation therapy. Surgery is also a key component of induction, typically performed after several cycles of chemotherapy (19). The goal is a complete resection of the primary tumor, however, a 90% resection is acceptable in order to minimize surgical complications such as major vascular injury or end-organ damage (20). Despite aggressive induction therapy, approximately 10% of patients experience disease progression and only 20% achieve a complete response. Improving induction therapy outcomes are a major focus of ongoing clinical trials in HRNB (21).
In the consolidation phase, the primary aim is to eradicate any residual disease. Patients undergo autologous stem cell transplantation (ASCT) and radiation therapy. During ASCT, patients receive high doses of myelosuppressive chemotherapy, followed by stem cell reinfusion to restore bone marrow function. Studies show improved outcomes for patients who undergo ASCT compared to those who receive continued chemotherapy (22, 23). After ASCT, 21.6 Gy of external beam radiation is administered to the primary tumor and any sites of persistent disease (24). While neuroblastoma is radiosensitive, there is ongoing research to reduce radiation exposure due to its associated long-term toxicity.
The post-consolidation phase focuses on preventing relapse and includes multimodal therapies such as immunotherapy and isotretinoin, which will be discussed in detail in later sections. However, despite the intensive multimodal approach, outcomes for relapsed and refractory neuroblastoma remain poor. Surgery, radiotherapy, and chemotherapy, while effective in some cases, carry significant side effects and chemotherapy is limited by the development of treatment resistance (25). Additionally, HRNB treatments are associated with long-term toxicities that can severely affect patients’ quality of life, leading to increased morbidity and mortality. Nevertheless, development of targeted therapies, such as anti-GD2 immunotherapy’s success, highlights the potential of such an approach to not only improve survival rates but also reduce toxic side effects, offering a better quality of life for HRNB patients (25).
The molecular biology of neuroblastoma plays a crucial role in both clinical risk stratification and the development of targeted therapies (26). Neuroblastoma is characterized by somatically acquired genetic alterations that lead to changes in gene expression and correlate with tumor aggressiveness. Key genetic drivers of neuroblastoma include MYCN amplification, anaplastic lymphoma kinase (ALK) mutations or amplifications, telomerase reverse transcriptase (TERT) rearrangements, alpha-thalassemia/mental retardation X-linked (ATRX) deletions or mutations, and segmental chromosomal aberrations (SCAs).
MYCN amplification, the predominant genetic alteration associated with HRNB, occurs in approximately 20% cases and marks aggressive disease (27). MYCN amplification is linked to tumor metastasis, poor prognosis, and treatment challenges (28, 29). Other critical genetic changes include 1p loss of heterozygosity, commonly associated with unresectable metastatic disease and MYCN amplification, and 11q deletion, which correlates with advanced disease stages and poor survival (30–32). ALK mutations are also frequently found in HRNB, and ALK amplification is independently associated with aggressive tumor features such as tumor progression and therapy resistance (33).
ATRX is a frequently mutated tumor-suppressor gene, including in neuroblastoma, where it plays a critical role in chromatin remodeling by regulating chromatin state, gene expression, and DNA damage repair (34). ATRX mutations are commonly found in high-risk neuroblastoma patients and are associated with a poor prognosis (35). Genomic aberrations such as recurrent ATRX deletions and inactivation lead to alternative lengthening of telomeres (ALT), a mechanism that cancer cells use to bypass telomere shortening and maintain telomere length. Another critical mechanism for telomere maintenance is the activation of telomerase via TERT expression. TERT plays a major role in maintaining chromosomal stability by regulating telomere length, thus enabling cancer cells to evade senescence and continue proliferating (36). In HRNB, whole-genome sequencing revealed structural rearrangements of TERT in 17 of 75 cases, which were linked to increased TERT expression and associated with a very poor prognosis (37).
SCAs have emerged as an important risk factor in neuroblastoma. These structural genetic alterations are associated with aggressive tumor behavior, poor prognosis, and a higher likelihood of relapse independent of the poor prognosis associated with MYCN amplification. As part of the revised neuroblastoma risk classification system, SCAs help refine prognostic stratification, identifying patients who may require more intensive therapeutic strategies (14). Additionally, epigenetic modifications in high-risk neuroblastoma, such as DNA methylation, histone modification, and non-coding RNAs, significantly contribute to tumor progression. MicroRNAs (miRs), which regulate gene expression by targeting mRNA transcripts, are often dysregulated in HRNB and their roles in tumorigenesis and as biomarkers are an area of active research (38, 39).
Several signaling pathways play significant roles in HRNB. The most common of these pathways are the ALK and Ras-Mitogen activated protein kinase (RAS-MAPK) pathways (40, 41). Mutations in ALK, found in 12-14% of HRNB cases, activate downstream pathways PI3K/Akt and MAPK, promoting tumor cell survival and proliferation (42, 43). PI3K/Akt and Wnt/β-catenin pathways are crucial for tumor growth, while the Wnt pathway is involved in the maintenance and proliferation of neural crest cells (44, 45). Mutations in key signaling pathways, such as PI3K/Akt and Wnt/β-catenin, are common in relapsed neuroblastomas, suggesting their clinical significance (44, 45). Sonic Hedgehog (SHH) pathway is notable for maintaining neuroblastoma tumorigenicity and promoting cell proliferation (46). NF-kB activation contributes to tumor growth, metastasis, and chemokine receptor (CXCR4) upregulation, enhancing invasion (47, 48). TGF-β and Notch pathway is also important where TGF-β suppresses cell growth while Notch signaling inhibits tumor cell proliferation (49, 50). Finally, dysregulation of the p53-MDM2 pathway contributes as MDM2 overexpression leads to the degradation of p53, inhibiting tumor suppression and increasing MYCN activity, thus contributing to poor prognosis and resistance to therapy (51, 52). These collectively disrupted pathways highlight the complexity of HRNB pathobiology and suggest potential targets for novel therapies (Figure 2).
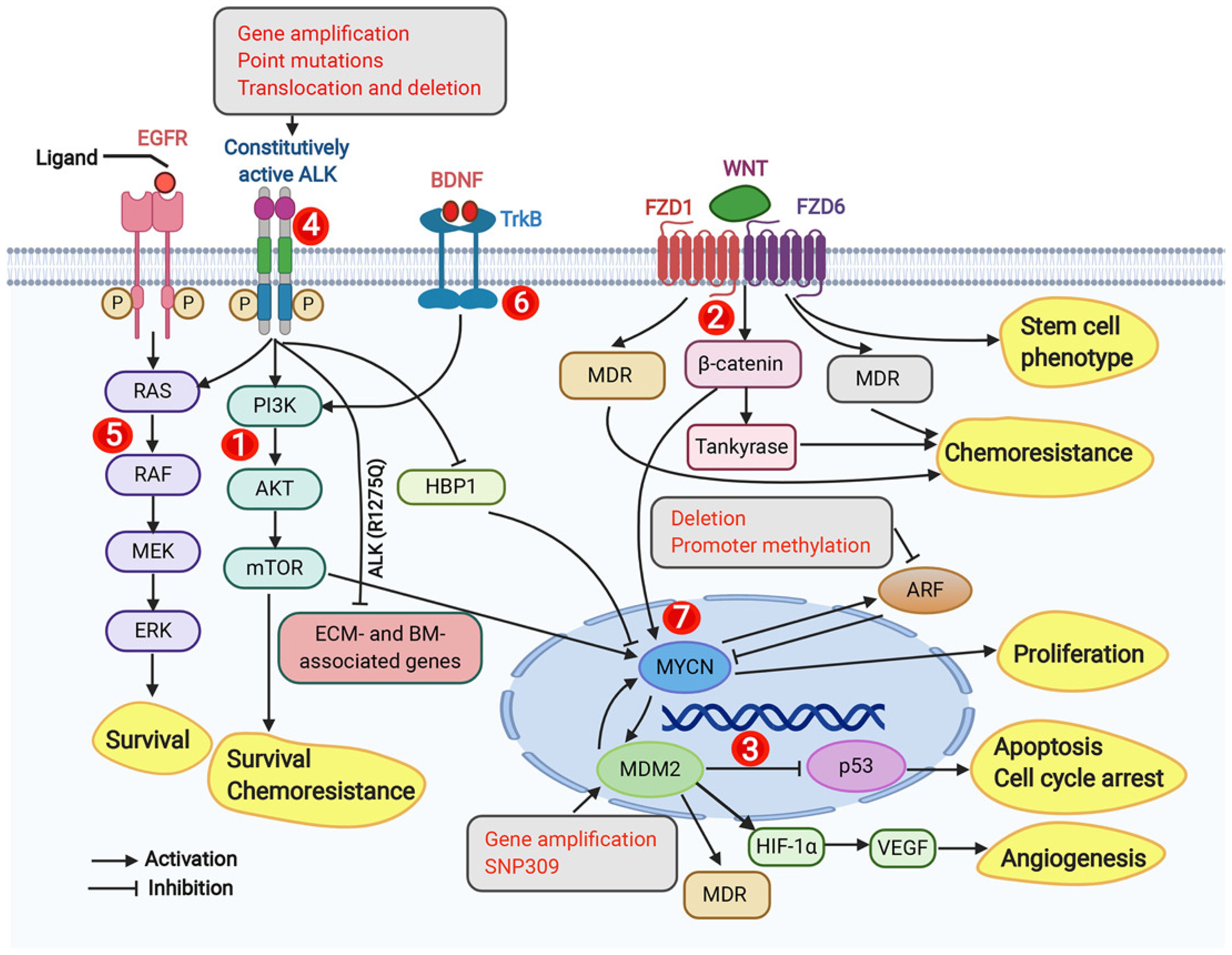
Figure 2. Dysregulated signaling pathways, genomic, and molecular targets in neuroblastoma. This figure from Zafar et al. (2020) highlights key molecular signaling pathways, genomic targets, and molecular targets implicated in neuroblastoma, emphasizing the complex network of dysregulated pathways, including PI3K/AKT/mTOR, Wnt, p53-MDM2, ALK, RAS-MAPK, TrkB, and MYCN, that drive tumor survival, chemoresistance, and progression. These targets represent potential therapeutic interventions for neuroblastoma treatment. Adapted from Zafar et al., 2021, Medicinal Research Reviews, 41(2):961–1021 with permission from Wiley Periodicals LLC.
A major challenge in treating neuroblastoma is its ability to evade the immune system. This resistance is due to neuroblastoma’s low immunogenicity profile, resulting in fewer neoantigens for immune cells to target (53). Furthermore, neuroblastoma’s tumor microenvironment (TME) is characterized by low levels of tumor-infiltrating lymphocytes (TILs) and the presence of immune-suppressing cells (54, 55). These factors hinder immune responses, and research efforts to enhance HRNB’s immunogenicity and improve immune-based therapies are ongoing.
GD2 is a ganglioside found on the outer cell membrane of peripheral neurons and the central nervous system, and skin melanocytes (25). It is highly expressed on the surface of neuroblastoma cells compared to normal cells, making it an attractive target for HRNB immunotherapy (25). GD2-targeting monoclonal antibodies (mAbs), such as Dinutuximab (ch14.18; Unituxin), were the first immunotherapy approved for use in HRNB. Dinutuximab a murine-chimeric mAb, binds to GD2, triggering the binding of C1q and initiating complement-dependent cytotoxicity (CDC) (56). This process engages the body’s immune system, facilitating antibody-dependent cell-mediated cytotoxicity (ADCC) by NK cells and granulocytes, leading to tumor cell destruction. Additionally, the downregulation of the PI3K/Akt/mTOR signaling network has been observed upon the binding of mAbs to GD2 (57). Combining Dinutuximab with IL-2, granulocyte-macrophage colony-stimulating factor (GM-CSF), and isotretinoin was a major breakthrough in HRNB treatment, increasing event-free survival (66 ± 5% vs. 46 ± 5%) and overall survival (86 ± 4% vs. 75 ± 5%) at two years (57). However, Dinutuximab therapy comes with significant side effects, primarily capillary leak syndrome, hypersensitivity reactions, and severe pain, due to GD2 expression on normal nerve cells. An alternative, Dinutuximab beta (ch14.18/CHO), produced by re-cloning in Chinese hamster ovary (CHO) cells, exhibits a more favorable glycosylation pattern, reducing immune responses and improving tolerability (58). While similar to ch14.18 in efficacy, Dinutuximab beta has been associated with reduced pain and was approved by the European medicines agency in 2017 for HRNB treatment (55). However, pain reduction was insufficient for escalating doses or exploring alternative administration methods (59).
Murine antibodies, including Dinutuximab, can also induce severe infusion reactions due to immunogenicity, specifically human anti-mouse antibodies (HAMA). To address this, humanized monoclonal antibodies targeting GD2 have been developed. Naxitamab (Hu3F8-IgG1), a humanized version of the monoclonal antibody murine 3F8 that targets GD2, was granted FDA approval in 2020 for treating relapsed or refractory HRNB in children aged one and older and adults with bone or bone marrow involvement (60). Naxitamab demonstrated cytolytic activity against neuroblastoma cells in vitro and led to objective responses in 34% to 45% of patients with refractory advanced neuroblastoma (60). Another humanized antibody, hu14.18K322A (hu14), modified from another murine monoclonal antibody (14G2a) that targets GD2, shows reduced complement-dependent cytotoxicity while retaining ADCC activity and synergizes with common chemotherapies in vitro (61, 62). In a Phase II study, combining hu14 with chemotherapy, IL-2, and GM-CSF resulted in improved early objective responses (66.7% vs. 39.1%) and a 73.7% 3-year event-free survival rate, while similar adverse effects were observed (62).
Due to the severe adverse effects of GD2 monoclonal antibodies, researchers are exploring other methods to target GD2, enhance its expression on HRNB cells, or increase antibody sensitivity. A study by Galassi et al. (63), found that increasing GD2 expression using Nanofenretinide, a nanoformulation of fenretinide, alongside Naxitamab, enhanced cytotoxicity in vitro. However, increasing GD2 expression alone did not always improve efficacy, as shown when combining Nanospermidine with Naxitamab, which did not increase cytotoxicity despite elevated GD2 levels. Another promising approach is radioimmunotherapy, where GD2-targeting antibodies deliver radioactivity to HRNB tumor sites (64). One recent example is the use of radio-labeled Dinutuximab, that can bind to GD2 and offer high tumor-to-organ dose ratios. However, it is important to consider that prolonged blood residence leads to absorption in blood-rich organs such as the liver, spleen, and kidneys (64).
Furthermore, GD2’s high expression on neuroblastoma cell surfaces has made it a target for chimeric antigen receptor T-cells (CAR-T). CAR-T cell therapy directs engineered T cells to a specific antigen, such as GD2, via chimeric antigen receptors (65). Although CAR-T therapy targeting GD2 in neuroblastoma shows promise, it faces limitations such as T cell exhaustion, loss of functionality, and poor tumor infiltration. A Phase I trial (NCT02107963) demonstrated the feasibility and safety of GD2-CAR T-cell therapy, with no dose-dependent cytotoxicity (66). While GD2-CAR T-cells expanded in all patients, their persistence was limited, and all patients eventually progressed (67). Research suggests that expansion is influenced by factors like CXCR3+/– expression, with better expansion associated with naive T cells and CXCR3+ monocytes (68). In response to the challenges faced by CAR-T therapy arising from the intrinsic limitations of T-cells, CAR-NK T cells are being explored as an alternative to CAR-T cells alone. This leverages the unique antitumor activity of Natural Killer-T cells. These cells have been shown to expand and localize to tumors in relapsed neuroblastoma patients (Figure 3) (69). In a Phase I trial (NCT03294954), three patients underwent lymphodepleting conditioning followed by CAR-NK T cells infusion, with no dose-limiting toxicities (69, 70). One patient exhibited an objective response with regression of bone metastatic lesions. These promising results suggest that CAR-NKT cells can be expanded to clinical scale and safely used for HRNB treatment and beyond (70).
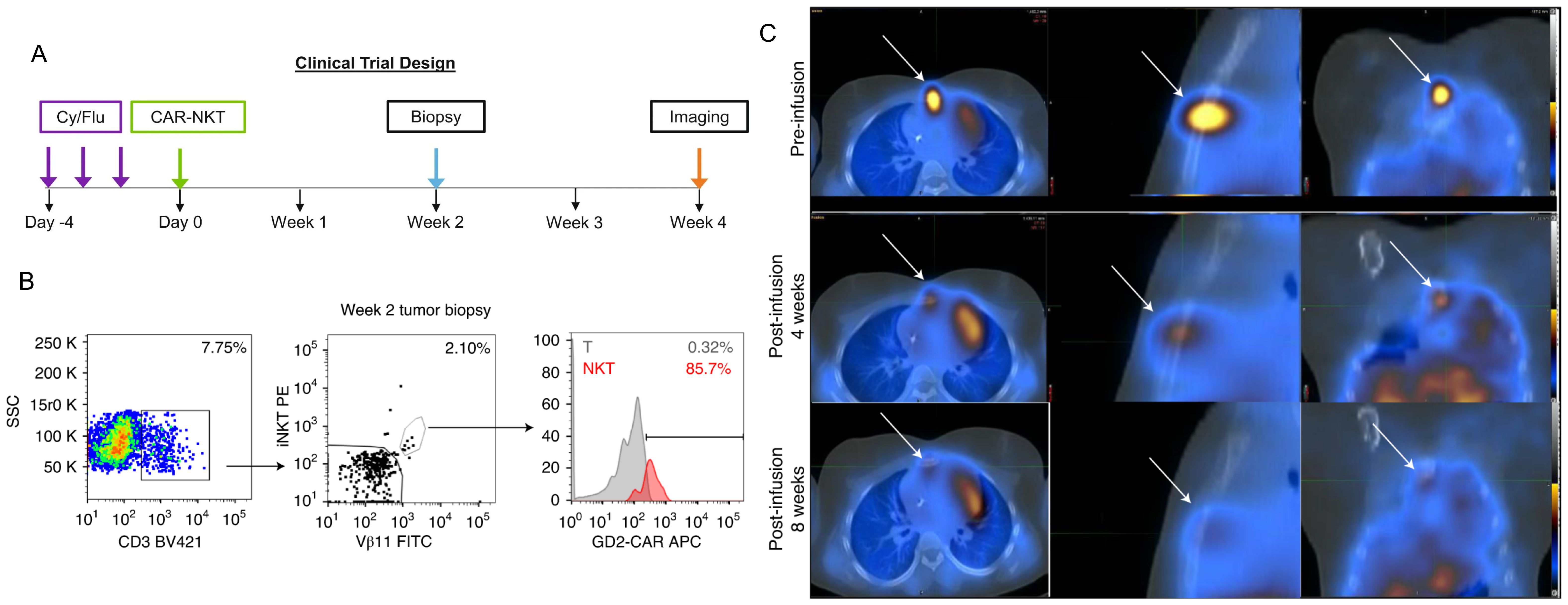
Figure 3. CAR-NKT cells infiltrate tumor sites and mediate tumor regression in refractory neuroblastoma patients. Figures 2 and Extended Data Figure 1 from Heczey et al. demonstrate that NKT cells are present at higher frequencies within tumor-infiltrating lymphocytes compared to peripheral blood lymphocytes, with 85.7% of tumor-infiltrating NKT cells expressing CAR-GD2 (B). Panel (C) illustrates the reduction in size of neuroblastoma bone metastasis. Panel (A) depicts the clinical trial design for this study. These findings highlight the therapeutic potential of CAR-NKT cells in treating neuroblastoma tumors and promoting their regression. Adapted from Heczey, A., Courtney, A.N., Montalbano, A., et al. Anti-GD2 CAR-NKT cells in patients with relapsed or refractory neuroblastoma: an interim analysis. Nat Med 26, 1686–1690 (2020).
GD2 remains a critical therapeutic target for HRNB, with multiple strategies, including monoclonal antibodies, radioimmunotherapy, and CAR therapies, and the introduction of anti-GD2 immunotherapy notably improved survival for HRNB. However, the toxicities associated with currently approved immunotherapies are significant and limit the current clinical treatment strategies so further research to optimize GD2-targeted therapies for HRNB.
Glypican-2 (GPC2) is a heparan sulfate proteoglycan oncoprotein belonging to the glypican family that is anchored to the cell membrane via a glycosylphosphatidylinositol (GPI) anchor (71). GPC2 is expressed on the surface of neuroblastoma cells, activating the WNT signaling pathway and stimulating growth. This is correlated with worse outcomes with HRNB patients with higher GPC2 expression (71). Recent studies have highlighted differences in GPC2 transcript expression between neuroblastoma cells and normal tissues, suggesting the existence of a tumor-selective GPC2 sequence that can be targeted specifically to the tumor (72). GPC2’s selective expression to neuroblastoma makes it an attractive target for immunotherapy, particularly for CAR-T therapies, as a means to enhance neuroblastoma cell specificity and limit cytotoxicity to normal cells. Repeated testing of CAR constructs has shown that a CAR composed of a single chain variable fragment (scFv) derived from an antibody that targets GPC2 (CT3) exhibits superior anti-tumor activity against HRNB (Figure 4) (73). Additionally, combining GPC2 targeted CAR-T cell therapy with immunomodulatory agents like long-acting interleukin-7 (rhIL-7-hyFc) has demonstrated enhanced T-cell expansion, reduced exhaustion markers, and improved efficacy against solid tumors with low antigen density (74). While GPC2 therapy is promising there is significant heterogeneity in GPC2 expression in neuroblastoma, meaning the therapy is only effective in patients who express GPC2 (72–74). For sustained anti-tumor activity, the persistent presence of CAR-T cells is crucial. However, CAR-T cells can have a limited lifespan in vivo, which can impact their long-term effectiveness (72–74).
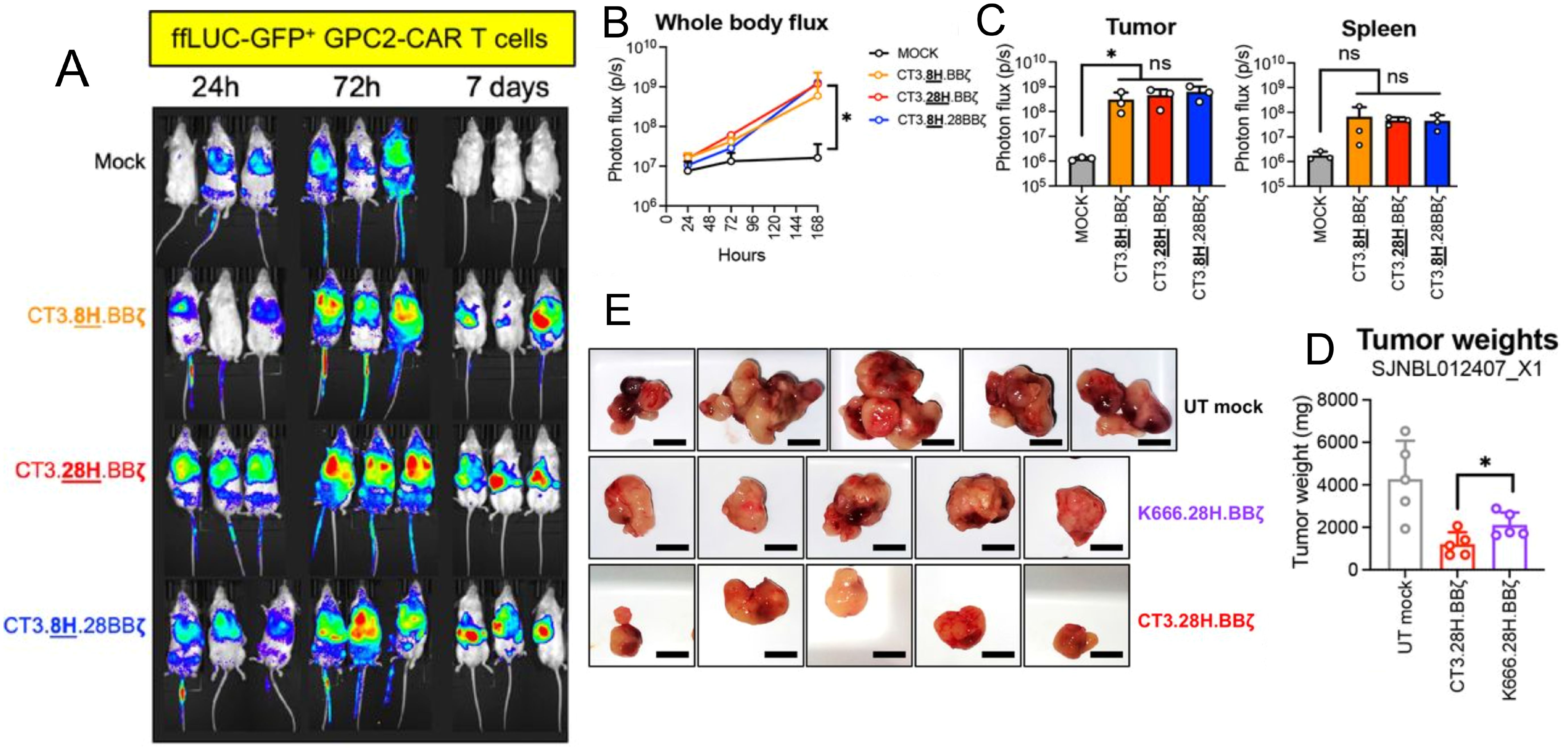
Figure 4. Preclinical evaluation of GPC2-targeting CAR T cells for neuroblastoma treatment. (A-C) show that GPC2-CAR T cells efficiently home to the tumor microenvironment (TME), expand, and enrich as a cytotoxic effector population, highlighting their potential for targeted tumor eradication. (D, E) compare the performance of GPC2-CAR T cells with GD2-CAR T cells (K666.28H.BBζ), showing superior in vitro cytotoxicity, tumor rechallenge response, and reduced tumor burden in vivo. Adapted from Sun et al., 2023, Journal for ImmunoTherapy of Cancer, 11(1):e005881 with permission from BMJ Specialist Journals.
B7-H3 is a checkpoint inhibitor ligand that is overexpressed in neuroblastoma cells. Studies have shown that B7-H3 contributes to tumorigenesis, angiogenesis, invasion, and metastasis through various mechanisms (75, 76). Blocking B7-H3 with B7-H3 mAb (5B14) in vitro has enhanced the ability of NK cells to kill neuroblastoma cells (77). Recently, pre-clinical studies are exploring CAR targeting B7-H3 as a way to deliver immune cells directly to the tumor. Engineered CAR-NK92 cells targeting B7-H3 have successfully eliminated neuroblastoma cell lines in monolayer cultures and 3D models in vitro (78). However, genetic modification of primary NK cells are challenging due to low transfection efficiency and poor CAR function (78). Similarly, engineered CAR-T cells targeting B7-H3 show strong activity against neuroblastoma cells in vivo (79). For instance, SynNotch GD2-B7-H3 CAR T-cells exhibited high stability, high specificity, and enhanced cytotoxicity against neuroblastoma cells in vitro and in xenograft mouse models compared to conventional CAR-T cells (80). The SynNotch gating strategy, which uses GD2 as the gate and B7-H3 as the inducible target, is being leveraged to improve the precision of CAR T-cell therapies and enhancing the safety profile of using GD2 therapy alone (80). However, GD2-B7-H3 CAR-T cells showed lower cytotoxic efficiency with longer times required for gated CAR to show similar levels of cytotoxicity compared to conventional CAR-T cells (80). This can be resolved with more rounds of therapy; however other issues might arise like tumor escape due to loss of either needed antigen (80).
The Nectin cell adhesion molecule-2 and T-cell immunoreceptor with Ig and ITIM domains (NECTIN2-TIGIT) axis is an immune checkpoint involved in immune evasion. NECTIN2, a cell adhesion molecule expressed on the surface of HRNB cells, acts as a binding partner for TIGIT, an immune checkpoint receptor found on immune cells such as T cells and NK cells (81). This axis is strongly associated with the dysfunction of both NK cells and T cells. Research has shown that blocking TIGIT and programmed death-ligand 1 (PD-L1) can induce a complete response in vivo, including in chemotherapy-resistant neuroblastoma cells (81). Another pre-clinical study showed that blocking both PD-L1 and TIGIT increased the efficacy of Dinutuximab beta (GD2 antibody) in HRNB tumors in vivo causing complete removal of tumor compared to blocking either TIGIT and PD-L1 separately (82). These findings highlight the potential of targeting the NECTIN2-TIGIT axis to improve the TME and enhance the immune response in HRNB, but further research is needed to explore immunotherapy efficacy while managing the risk of immunotoxicity, prior to implementing this strategy in clinical practice (81).
Delta-like homologue 1 (DLK1) is a surface protein that belongs to the epidermal growth factor (EGF)-like protein family and is believed to suppress the Notch signaling pathway (83). Recent research has identified DLK1 as a promising target for immunotherapy, particularly in neuroblastoma. DLK1 is overexpressed in neuroblastoma cells, and its expression is largely restricted to the adrenal medulla and pituitary gland (84). According to Hamilton et al. DLK1 may play a role in maintaining the undifferentiated state of neuroblastoma cells, preventing their maturation (84). Further studies have shown that depletion of DLK1 in neuroblastoma cells promotes cell differentiation. Additionally, ADCT-701, an antibody-drug conjugate currently in an ongoing phase I clinical trial (NCT06041516) for adult patients with neuroendocrine tumors, has demonstrated effectiveness in targeting and killing DLK1-positive neuroblastoma cells in vivo (84, 85). Further research needs to done be in order to detect off target toxicity since ADCT-701 does not bind to mouse DLK-1 (84).
Immune checkpoints, such as programmed cell death protein 1 (PD-1) and cytotoxic T lymphocyte-associated protein 4 (CTLA-4), are critical regulators of the immune system, controlling its ability to recognize and eliminate tumor cells (86). Under normal conditions, these checkpoints prevent excessive immune activation, maintaining self-tolerance and preventing autoimmunity. However, many tumors exploit these regulatory mechanisms to evade immune detection, leading to immune suppression (87). One such immune checkpoint, PD-L1, is a ligand expressed on the surface of neuroblastoma cells, where it binds to PD-1 receptors on activated T cells, inhibiting T cell activation and proliferation (88). PD-L1 inhibitors such as nivolumab are currently being investigated in early-phase clinical trials. These agents work by blocking the inhibitory signals from PD-1 engagement, thus enhancing T cell activity against tumors. Early-phase clinical trials are exploring the combination of nivolumab with dinutuximab (GD2 antibody), and preliminary results suggest this combination may enhance immune-mediated tumor responses and improve overall survival in patients with relapsed or refractory neuroblastoma (89). While PD-1 signaling blockade can improve antitumor responses and lead to long-lasting clinical benefits, 30%–60% of patients do not respond to PD-1/PD-L1 inhibitors and many formed resistance to them (88).
Another checkpoint inhibitor, Cytotoxic T-Lymphocyte Antigen-4 (CTLA-4), acts as an inhibitory receptor on T cells by competing with CD28 for binding to B7-1 and B7-2 molecules on antigen-presenting cells (90). This competition negatively regulates T cell activation, contributing to immune evasion by tumors. A recent study in vivo demonstrated that combining CpG-PBNP-PTT (a form of photothermal therapy) with anti-CTLA-4 antibodies resulted in complete tumor regression in treated tumors and slower progression in untreated ones. This combination also led to long-term survival and the generation of protective immunity against tumor rechallenge (91). However, HRNB heterogeneity and poor immunogenicity limit the efficacy of CTLA-4 therapy (86).
MicroRNAs (miRs) are small, non-coding RNA molecules, typically 19-23 nucleotides in length, that regulate gene expression by binding to target miRs, leading to their degradation or translational inhibition (92). Dysregulation of miRs has been implicated in neuroblastoma tumorigenesis, with known roles in cell differentiation, tumor promotion, and tumor inhibition (93). Studies have shown that manipulating miR levels—either by increasing levels with miR mimics or decreasing levels with miR inhibitors—holds therapeutic potential for treating cancers like neuroblastoma (93).
Recently it was found that higher levels of the miR-29 family (miR-29a, miR-29b, and miR-29c) were associated with better survival in neuroblastoma patients (Figure 5) (94). The mechanism of action of miR-29 is believed to involve the activation of NK cells through targeting B7-H3 in neuroblastoma (94). It was shown that neuroblastoma cells exhibited higher B7-H3 expression and lower levels of miR-29, when neuroblastoma cells were transfected with miR-29 mimics for 48 hours it resulted in a significant depletion of B7-H3 mRNA resulting in enhanced NK cell activation and cytotoxicity (94, 95). Furthermore, when activated immune cells were combined with miR-29-expressing neuroblastoma cells, there was a marked increase in CD107 and perforin expression, indicating NK cell activation, enhanced tumor lysis and higher IL-2 secretion (94, 95).
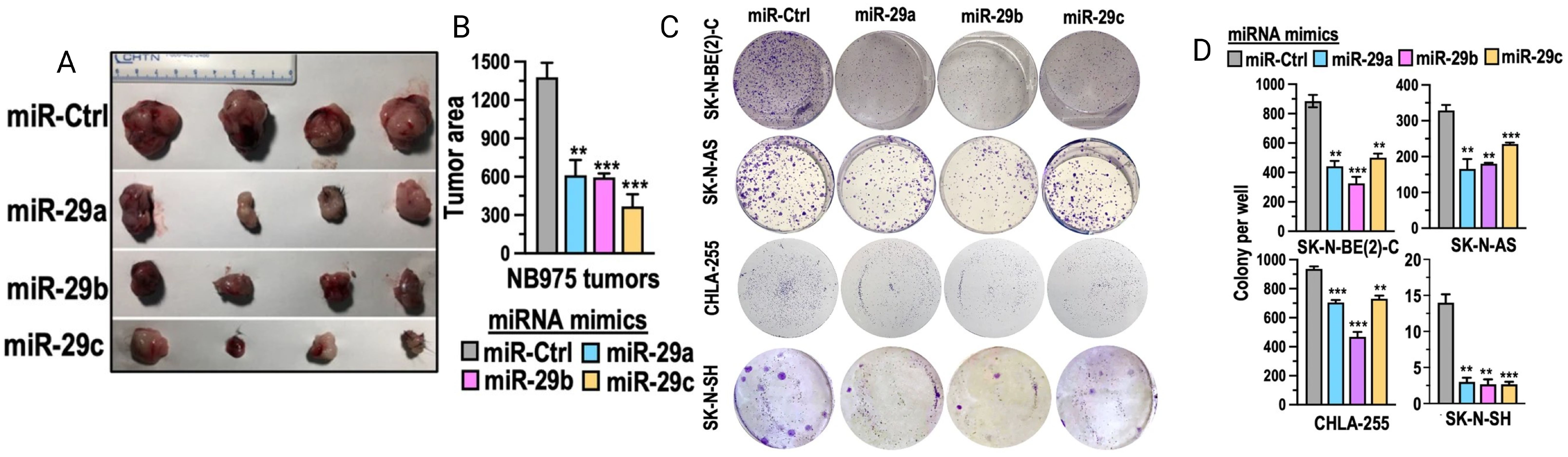
Figure 5. miRNA-29 demonstrates anti-tumor activity by inhibiting cell growth, colony formation, and reducing tumor size and area. (A, B) show tumor area reduction following miR-29 transfection in vivo. (C) depicts the inhibition of various neuroblastoma cell lines in vitro after treatment with miR-29. Adapted from Pathania et al., 2024, Cell Death & Disease, 15 (6):428 with permission from Springer Nature.
The miR-124 family is another miR group with potential implications for neuroblastoma treatment. miR-124, a conserved miR predominantly expressed in neurons, plays a key role in regulating the development and function of the nervous system (96). However, its role in neuroblastoma remains controversial, likely due to the heterogeneity of the disease (97). Nolan et al. showed that miR-124 directly targets key cytoskeletal genes, leading to a shift from a mesenchymal (invasive) phenotype to a more epithelial (less invasive) phenotype, which enhances neuroblastoma cells’ sensitivity to chemotherapy (98). This is consistent with findings by Sharif et al. and Zhao et al. which displayed that miR-124 can arrest the cell cycle at the G1 phase, preventing cell proliferation in HRNB cell lines (99, 100). In contrast, Zhang et al. found that miR-124 directly downregulates VAMP3, which was shown to promote cell death, which correlates with poorer patient outcomes in neuroblastoma (97). This is due to the interplay between miR-124 and VAMP3, which influences the expression of genes involved in various cellular processes. These findings align with Huang et al. who showed that inhibition of miR-124 led to increased AHR expression and resulted in cell differentiation in HRNB cell lines (101).
The miR-34 family is another widely studied miR family with important implications targeting MYCN. Overexpression of miR-34a has been shown to significantly reduce proliferation, induce cell cycle arrest, and activate apoptosis and levels are lower in HRNB (102). Possible targets include key oncogenes MYCN, ALK and LIN28B along with HNF4α and the autophagy-related gene 5 (ATG5) (102–104). These miRs can also enhance tumor sensitivity to chemotherapy by targeting multiple signaling pathways (103).
The let-7 miR family plays a crucial role in suppressing neuroblastoma tumors by targeting several oncogenes (DICER1, ARID3B, MYC, HMGA2) (105, 106). However, let-7 is frequently downregulated in neuroblastoma through multiple mechanisms. One mechanism involves the LIN28B protein, which is often overexpressed in high-risk neuroblastoma (HRNB) and associated with poor patient outcomes (107). LIN28B suppresses let-7 by increasing MYCN protein levels, which in turn targets and inhibits Let-7 (107). This LIN28B-MYCN pathway is associated with disrupting neuroblast differentiation (107). Furthermore, MYCN has been shown to suppress let-7 independently of LIN28B. In high-risk neuroblastoma (HRNB) cells with LIN28B knocked down, where MYCN levels were elevated, let-7 miRNA expression was still suppressed (106). This is because excess MYCN mRNA can directly bind to and degrade let-7 (106). Intriguingly, the frequency of let-7 gene loss differs between MYCN-amplified and non-amplified tumors (106). Loss of let-7 genes is common in non-MYCN-amplified tumors (63.4%), but much less frequent in MYCN-amplified tumors (16.7%) (106). This suggests that in MYCN-amplified tumors, the excess MYCN protein functionally sequesters and inactivates let-7, thus reducing the selective pressure for the tumor cells to physically delete the let-7 genes (106).
While miR-29, miR-124, miR-34, and miR Let-7 are commonly recognized as neuroblastoma-related miRs, there are many other miRs currently under investigation and our understanding of the role of miRs in cell proliferation and differentiation in neuroblastoma is rapidly evolving. miRs have demonstrated successful reduction of tumor cell proliferation in pre-clinical studies of neuroblastoma and other cancers, but clinical studies have been limited by cytotoxicity.
Furthermore, miRs are actively being explored as potential biomarkers for disease surveillance and prognostic indicators. Further research is needed to explore how miRs can be combined with other therapeutic strategies for improved treatment outcomes.
Long noncoding RNAs (lncRNAs) are a class of RNA molecules that range in length from 200 nucleotides to 100 kilobases and do not encode proteins (108). Despite this, they play crucial roles in transcriptional and epigenetic regulation of gene expression (109). LncRNAs have been implicated in various cancers, where they regulate tumor-associated genes and other cellular processes (110). For example, the metastasis-associated lung adenocarcinoma transcript (MALAT1) is upregulated in neuroblastoma cells under hypoxic conditions compared to normal conditions (111). MALAT1 promotes the upregulation of Fibroblast Growth Factor 2 (FGF2) mRNA and protein expression in neuroblastoma cells and enhances FGF2 secretion into the extracellular fluid. This leads to endothelial cell migration, invasion, and vasculature formation. Additionally, MALAT1 has been shown to have a significant positive relationship with Axl, a transmembrane receptor tyrosine kinase (RTK) that acts as an oncogene and is overexpressed in neuroblastoma (112). Bi et al. demonstrated that targeting Axl with its inhibitor has been shown to suppress neuroblastoma cell metastasis by regulating tumor-driven angiogenesis, which highlights its potential as a promising therapeutic target for neuroblastoma treatment (112).
Small nucleolar RNA host gene-7 (SNHG7) is a lncRNA that is upregulated in neuroblastoma, where it plays a significant role in cell proliferation and is associated with poor prognosis. SNHG7 is suggested to promote cell proliferation, migration, and invasion by modulating miR-653-5p, a miR that inhibits STAT2 (113). STAT2 is involved in promoting the expression of oncogenic gene STAT3 (114). Thus lncRNA SNHG7 induces STAT2 overexpression which contributes to neuroblastoma progression (113). In vivo studies show that silencing SNHG7 significantly inhibits tumor growth in xenograft models, leading to an increase in miR-323a-5p and miR-342-5p expression (115). Additionally, silencing SNHG7 was shown to enhance Cisplatin sensitivity (116). SNHG7 acts as a sponge for miR-329-3p, which plays a role in reducing Cisplatin resistance and autophagy inhibition, resulting in the upregulation of MYO10 which plays a role in reducing Cisplatin sensitivity (116). Further research is essential to better understand how lncRNAs interact with other molecular targets and how they can be integrated with complementary therapeutic strategies to enhance treatment outcomes.
Circular RNAs (circRNAs) are mainly non-coding RNAs, typically 19–23 nucleotides in length, that play important roles in cell regulation (117). Their name is derived from the unique structure they form by binding the ends of the RNA together to create a circular polynucleotide, which enhances their stability and makes them resistant to exonucleases (117). Pre-clinical in vivo studies have shown that circRNAs can influence both tumor survival and suppression in HRNB, depending on their target (118–122). Table 1 provides examples of different circRNAs and their roles in HRNB. Further research is needed to explore their potential as a promising therapeutic target for neuroblastoma treatment.

Table 1. Examples of circular RNAs and their effects in HRNB.
Genomic aberrations of the ALK gene are found in approximately 14% of patients with HRNB (123); therefore it is a critical potential target for therapeutic development. Crizotinib, the first ALK inhibitor to undergo clinical assessment, was evaluated in two phase III trials which confirmed its efficacy over standard first-line chemotherapy, leading to its FDA approval in 2011 (124–126). In 2013, a phase I trial was initiated in which crizotinib was administered with chemotherapy (127) and subsequently expanded to phase III trial due to promising results (128).
However, early clinical trials of first-generation ALK inhibitors, like crizotinib, demonstrated limited efficacy (129). More potent second- and third- generation ALK inhibitors, such as ceritinib, lorlatinib, brigatinib, alectinib, and repotrectinib, have shown improved therapeutic activity in neuroblastomas with ALK mutations, though they achieve complete responses in only a small number of patients (130–135).
Among these, lorlatinib, a third-generation ALK inhibitor, was designed to overcome resistance and shows superior potency compared to crizotinib, particularly against three of the most common ALK mutations (132, 136–138). Despite these promising results, treatment resistance remains a significant clinical challenge with prolonged use of these targeted therapies.
In addition to ALK inhibitors, focal adhesion kinase (FAK) inhibitors are of interest. A preclinical study using patient-derived xenografts of neuroblastoma evaluated the effects of FAK inhibitors PF-573, 228 and Y15. The inhibitors significantly reduced cell survival, proliferation, and motility, while inducing cell cycle arrest. FAK inhibition also decreased tumor sphere formation and reduced the expression of stem cell markers, suggesting a reduction in cancer stem cell-like properties. These results position FAK as a promising therapeutic target for neuroblastoma (139).
Chugh et al. evaluated the dual ALK and FAK inhibitor ESK440 in preclinical HRNB models (140). ESK440 significantly inhibited cell proliferation in vitro with ALK aberrations, and reduced ALK, FAK, and downstream target activation, impairing migration and invasion. ESK440 significantly inhibited cell proliferation in vitro with ALK aberrations, and reduced ALK, FAK, and downstream target activation, impairing migration and invasion (140). Importantly, ESK440 also reduced MYCN levels. In neuroblastoma cell lines and patient-derived xenografts, ESK440 treatment reduced tumor growth with no observed toxicity. ESK440 showed comparable or enhanced efficacy over lorlatinib and maintained effectiveness in lorlatinib-resistant models (140). These findings suggest that ESK440 is a promising targeted therapy for ALK-driven neuroblastoma and warrant further clinical investigation.
MYCN is the hallmark oncogene of neuroblastoma and is a prognostic factor that is indicative of unfavorable outcomes in patients with HRNB. MYCN has been described as a transcription factor that controls several cellular processes. Many strategies have been proposed to directly target MYCN; however, translation to clinical therapeutics have not yet been successful.
An approach to target MYCN is through the use of ornithine decarboxylase 1 (ODC1) inhibitors because the ODC1 gene encodes the rate-limiting enzyme ODC involved in the biosynthesis of polyamines (141). Polyamines are well-studied cationic molecules integral to normal and cancer cell growth and their biosynthesis pathway is a well-characterized direct Myc transcription target (142). A study by Rounbehler et al. demonstrated the upregulated polyamine biosynthesis in MYCN-amplified neuroblastoma contributes to aggressive tumor growth (143). The study further demonstrated the significance of the MYCN and polyamine pathway in neuroblastoma tumorigenesis by using difluoromethylornithine, an ODC inhibitor, which decreased polyamine levels. The decrease of polyamine levels disabled Myc’s ability to drive cell growth and slow the cell cycle.
Inhibition of Aurora A (AURKA) and Aurora B kinases (AURKB) has been widely researched and is currently a potential way to target MYCN. The family of aurora kinases are associated with centrosome separation in the cell cycle (144). AURKA complexes with MYCN during the S-phase of cell division leading to MYCN stabilization and decreased susceptibility to proteasomal degradation (145). The same study by Roeschart et al. showed that combinatorial inhibition of Aurora-A and ataxia telangiectasia and Rad3 related (ATR) kinase which suppresses double-strand break accumulation, results in regression of MYCN-amplified tumors. AURKB is a direct transcriptional target of MYCN and both aurora kinases have been correlated with a poor prognosis in neuroblastoma (146, 147). Neuroblastoma cells have been treated with alisertib, a specific AURKA inhibitor, and were shown to inhibit cell growth, G2/M arrest, degradation of MYC and tumor growth inhibition (148).
One of the hallmarks of cancer is a dysregulation in cell division which leads to aberrant cell proliferation; inhibiting cell division is a common approach for cancer therapy. Cell division is controlled by a complex of cyclin and cyclin-dependent kinases (CDKs) (149), and in neuroblastoma, CDK4/6 inhibitors are of particular interest. Ferguson et al. demonstrated that CDK4/6 inhibitor palbociclib was able to inhibit proliferation and promote extensive neuronal differentiation of adrenergic neuroblastoma cells in vivo (150). The first clinical study exploring a CDK4/6 inhibitor in a pediatric population utilized ribociclib, an orally bioavailable and highly specific CDK4/6 inhibitor (151). The study demonstrated that ribociclib was well-tolerated in pediatric patients, but its efficacy in HRNB specifically remains unclear due to the complex molecular heterogeneity of neuroblastoma tumors. Further studies must be conducted to determine the specific efficacy, optimal patient population, and long-term benefits of ribociclib in HRNB patients.
The PHOX2B (paired like homeobox 2B) gene is essential in the development of the autonomic nervous system, promoting neural crest cell differentiation, and PHOX2B mutations are associated with peripheral neuroblastic tumors such as neuroblastoma (4, 152). In fact, it was the first gene for which germline mutations were discovered in neuroblastoma (153) and low expression of PHOX2B is associated with a higher tumor stage, poor outcome and poor survival (154). PHOX2B binds to the ALK gene promoter (155) and activates the Delta-Notch pathway to induce neuroblastoma cell proliferation (156, 157). Mycophenolate mofetil and XAV939 are two molecules that have demonstrated potential to inhibit PHOX2B in neuroblastoma cells in preclinical studies. Di Zanni et al. showed that mycophenolate mofetil decreases PHOX2B mRNA and protein expression in neuroblastoma cells in vitro (157), while Suebsoonthron et al. shows PHOX2B downregulation with XAV939 treatment. Additionally, XAV939 increases chemosensitivity of neuroblastoma cell lines to doxorubicin, a common chemotherapeutic associated with significant treatment toxicity (158).
Some limitations that must be addressed before advancing the targeting of PHOX2B in neuroblastoma treatment include the need for more selective inhibitors to avoid off-target effects (157), the potential for resistance mechanisms to develop (158), the variability in tumor responses (157), and the requirement for more comprehensive in vivo studies to assess long-term safety and efficacy (158). Additionally, the complex interaction between PHOX2B and other signaling pathways in neuroblastoma suggests that combination therapies may be necessary, which could introduce challenges in terms of optimizing treatment regimens and minimizing toxicity (158).
MDM2 inhibitors have demonstrated in vitro and in vivo anticancer activities (159, 160). Nutlins are small molecule MDM2 inhibitors that can induce cell cycle arrest or apoptosis in p53 wildtype cancer cells (161). Peirce and Findley demonstrated Nutlin-3’s ability to sensitize p53-null neuroblastoma cells to doxorubicin (162), but such first-generation MDM2 inhibitors were limited by poor bioavailability and pharmacokinetics, which hindered their in vivo efficacy (163).
RG7388, or idasanutlin, is a second-generation Nutlin developed to improve efficacy and toxicity of the Nutlins. Treatment with RG7388 induces functional activation of the p53 pathway and apoptosis of neuroblastoma cell lines in vitro (164), and combination therapy with RG7388 treatment and chemotherapeutics demonstrated increased apoptosis in p53 wild-type neuroblastoma cells (165). An active phase I/II clinical study (NCT04029688) is investigating the safety and efficacy of RG7388 for solid tumors, including neuroblastoma (166).
The aberrant activation of the phosphatidylinositol 3’-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) pathway serves as a pro-survival signaling cascade (167). Pathologic activation of Akt has been shown to frequently occur in neuroblastoma and is associated with a poor prognosis and is possibly related to MYCN amplification (168). Strong phosphorylation of the S6 ribosomal protein (a target of mTOR) was also observed in neuroblastoma samples; therefore, the PI3/Akt/mTOR pathway affects several pathways or proteins promoting a more aggressive cancer cell phenotype. Notably, the PI3/Akt/mTOR pathway contributes to MYCN oncogene stabilization which is associated with cell proliferation (169).
A study by Chesler et al. (170) has shown that the PI3/Akt/mTOR pathway inhibition leads to a decrease in the levels of N-Myc protein in NB. Furthermore, a clinical study (NCT00776867) evaluating the efficacy of perifosine (Akt inhibitor) was conducted in pediatric patients with solid tumors including HRNB (NCT00776867). In this phase I/Ib trial, 27 HRNB patients were treated with the AKT inhibitor perifosine, with nine MYCN-non-amplified patients remaining progression-free for up to 74 months, including one complete response. Toxicity was minimal even with extended treatment (11-62 months) (171). The study supports perifosine as a potential therapy for MYCN-non-amplified HRNB, either as monotherapy or in combination with other treatments. However, limitations such as the small sample size, lack of a control group, and the inclusion of only one patient with MYCN-amplified HRNB need to be overcome to improve the generalizability of the results.
Eukaryotic initiation factors (eIFs) are essential proteins that regulate the initiation phase of translation, the process by which ribosomes synthesize proteins from mRNA templates (172). These factors, including eIF1, eIF2, eIF3, eIF4, and eIF5, facilitate the assembly of the translation machinery and ensure accurate protein synthesis (173). As mentioned in previous sections, MYCN gene is amplified and a marker of HRNB. The 5’ untranslated region (UTR) of MYCN mRNA plays a crucial role in its translation by interacting with eIFs, thereby influencing MYCN protein levels.
Additionally, rocaglates, a class of compounds that stabilize eIF4A, have been shown to inhibit translation initiation by forming steric barriers that block initiation ribosomes (174). Amidino-rocaglates, synthetic derivatives of rocaglates, are among the most potent inhibitors in this class (175). CR-1-31-B, a small molecular rocaglate compound, functions as an inhibitor of eukaryotic translation initiation by targeting eIF4A, an RNA helicase that unwinds RNA secondary structures for translation initiation. A study by Skofler et al. is a preclinical in vitro investigation that explores the potential of targeting eIF4A1 as a novel therapeutic approach for neuroblastoma (176). They found that eIF4A1 is overexpressed in neuroblastoma tissues, contributing to tumorigenesis through its role in translation initiation. By using the synthetic rocaglate CR-1-31-B, which clamps eIF4A1 to mRNA and inhibits translation initiation, they observed decreased cell viability, increased apoptosis, and alterations in cell cycle distribution in neuroblastoma cell lines. Notably, CR-1-31-B showed effectiveness at low nanomolar doses, without affecting non-malignant cells, highlighting the therapeutic potential of targeting translation initiation in neuroblastoma (176). A recent preclinical in vitro study by Volegova et al. also demonstrated that inhibiting eIF4A1, a key initiation factor, using the compound CMLD012824, led to growth inhibition in MYCN-amplified neuroblastoma models without generalized toxicity (177).
Targeting eukaryotic initiation factors (eIFs) has shown therapeutic potential in HRNB but several limitations must be addressed before moving into clinical studies. Challenges such as off-target effects, the need for more selective inhibitors, potential toxicity, and the complexity of targeting translation initiation in vivo must be overcome to ensure the specificity, safety, and efficacy of eIF-targeted therapies. These factors are critical to consider for advancing eIF-targeting strategies from preclinical research to clinical application.
HRNB is a devastating pediatric cancer prone to aggressive tumors, metastatic disease, and development of treatment resistance. Despite multimodal therapy and decades of research, the overall survival remains poor, highlighting the need for more effective therapies. A more targeted approach to therapy could potentially increase treatment efficacy while simultaneously reducing treatment side effects, which can be significant and life-limiting. Current approaches for targeted therapies for neuroblastoma include targeting genetic aberrations, disrupted signaling pathways, epigenetic regulators, and Bcl-2 family proteins. While immunotherapies such as GD2 antibody-based treatments have become a standard treatment option for neuroblastoma, their optimization and alternative immunotherapy technologies such as CAR T-cell therapy are areas of ongoing research.
To bridge the gap and improve translational research, more physiologically relevant and technically reproducible model systems are needed. Most in vivo neuroblastoma model systems consist of mice, zebrafish, or less commonly, chick chorioallantoic membrane (CAM) (178). Each of these models may answer specific scientific questions but cannot completely reproduce the human TME, creating a significant gap in research efforts and a lack of translation from pre-clinical studies into successful clinical developments (178). Novel methods such a patient-derived xenograft models, humanized mouse models, and 3D tissue-engineered system may be more relevant models in which to test neuroblastoma therapeutics (178).
In parallel with the development of better models, personalized medicine based on patient-specific genetic profiles holds promise for enhancing treatment efficacy. Studies identifying genomic variations at the time of relapse to determine optimal combination of small molecule inhibitors treatments are underway and strongly supported by preclinical studies. Additionally, miRs represent ways to study individual tumor biology and leverage functional miRs as monotherapy, combination therapy, or to establish synergism with existing therapies such as chemotherapy, immunotherapy, and radiation therapy. Furthermore, liquid biopsies utilizing cell-free DNA can potentially detect genetic alterations not captured by a single-site biopsy, providing insights into tumor heterogeneity and expanding actionable targets for therapy (179).
Additionally, strategies to enhance delivery of therapeutics directly to the tumor are another area of active research. One approach is liposomal drug delivery, where drugs are encapsulated in liposomes decorated with molecules or ions that specifically target neuroblastoma cells (180). Other methods, such as carbon nanotubes, drug-loaded silk films, amphiphilic diblock polymers, and nanoparticles, can also be functionalized with targeting ligands to improve drug delivery efficiency (180). Another promising delivery method involves mesenchymal stem cells (MSCs), which can migrate to tumors and release growth factors to modulate the tumor microenvironment. MSC-derived extracellular vesicles are being explored as drug delivery vehicles due to their ability to efficiently load anticancer agents and target tumor cells, offering a potential avenue for targeted therapy in solid tumors (181).
Ultimately, understanding neuroblastoma’s genetic complexities can create the ability to personalize medicine based on unique tumor characteristics. As mutants arise and new signaling pathways are identified, novel therapies targeting these pathways can be explored. Research determining the mechanism of treatment resistance and strategies to overcome treatment resistance will be critical areas for neuroblastoma research to continue progress improving the dismal outcomes associated with HRNB. Striving to improve overall survival while also reducing treatment associated toxicity and side effects is the driving force behind ongoing research efforts, with the hope of significantly improving both survival rates and quality of life for children diagnosed with HRNB.
AA: Writing – original draft, Writing – review & editing. SC: Writing – original draft, Writing – review & editing. DR: Writing – review & editing. JK: Funding acquisition, Supervision, Writing – original draft, Writing – review & editing. EB: Funding acquisition, Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Institute of Health (NIH) under grant R00CA263016, the UC Davis Comprehensive Cancer Center Support Grant P30CA093373, and the Hartwell Foundation Award A24-2412.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Cheung NKV, Dyer MA. Neuroblastoma: developmental biology, cancer genomics, and immunotherapy. Nat Rev Cancer. (2013) 13:397. doi: 10.1038/nrc3526
2. Newman EA, Abdessalam S, Aldrink JH, Austin M, Heaton TE, Bruny J, et al. Update on neuroblastoma. J Pediatr Surg. (2019) 54:383–9. doi: 10.1016/j.jpedsurg.2018.09.004
3. Anderson D, Carnahan J, Michelsohn A, Patterson P. Antibody markers identify a common progenitor to sympathetic neurons and chromaffin cells in vivo and reveal the timing of commitment to neuronal differentiation in the sympathoadrenal lineage. J Neurosci. (1991) 11:3507–19. doi: 10.1523/JNEUROSCI.11-11-03507.1991
4. Huber K, Franke A, Brühl B, Krispin S, Ernsberger U, Schober A, et al. Persistent expression of BMP-4 in embryonic chick adrenal cortical cells and its role in chromaffin cell development. Neural Dev. (2008) 3:28. doi: 10.1186/1749-8104-3-28
5. Cohen MA, Zhang S, Sengupta S, Ma H, Bell GW, Horton B, et al. Formation of human neuroblastoma in mouse-human neural crest chimeras. Cell Stem Cell. (2020) 26:579–592.e6. doi: 10.1016/j.stem.2020.02.001
6. Mahapatra S, Challagundla KB. Neuroblastoma. In: StatPearls [Internet]. Treasure Island (FL: StatPearls Publishing (2023). Available at: https://www.ncbi.nlm.nih.gov/sites/books/NBK448111/.
7. van Groningen T, Koster J, Valentijn LJ, Zwijnenburg DA, Akogul N, Hasselt NE, et al. Neuroblastoma is composed of two super-enhancer-associated differentiation states. Nat Genet. (2017) 49:1261–6. doi: 10.1038/ng.3899
8. Capasso M, Brignole C, Lasorsa VA, Bensa V, Cantalupo S, Sebastiani E, et al. From the identification of actionable molecular targets to the generation of faithful neuroblastoma patient-derived preclinical models. J Trans Med. (2024) 22:151. doi: 10.1186/s12967-024-04954-w
9. Park JR, Eggert A, Caron H. Neuroblastoma: biology, prognosis, and treatment. Pediatr Clin North Am. (2008) 55:97–120. doi: 10.1016/j.pcl.2007.10.014
10. Wright JH. Neurocytoma or neuroblastoma, a kind of tumor not generally recognized. J Exp Med. (1910) 12:556–61. doi: 10.1084/jem.12.4.556
11. Gross RE, Martin LW. Neuroblastoma sympatheticum; a study and report of 217 cases. Pediatrics. (1959) 23:1179–91. doi: 10.1542/peds.23.6.1179
12. Wittenborg MH. Roentgen therapy in neuroblastoma; a review of seventy three cases. Radiology. (1950) 54:679–88. doi: 10.1148/54.5.679
13. Evans AE, D’Angio GJ, Randolph J. A proposed staging for children with neuroblastoma. children’s cancer study group a. Cancer. (1971) 27:374–8. doi: 10.1002/1097-0142(197102)27:2<374::AID-CNCR2820270221>3.0.CO;2-G
14. Irwin MS, Naranjo A, Zhang FF, Cohn SL, London WB, Gastier-Foster JM, et al. Revised neuroblastoma risk classification system: A report from the children’s oncology group. JCO. (2021) 39:3229–41. doi: 10.1200/JCO.21.00278
15. Goto S, Umehara S, Gerbing RB, Stram DO, Brodeur GM, Seeger RC, et al. Histopathology (International neuroblastoma pathology classification) and MYCN status in patients with peripheral neuroblastic tumors: a report from the children’s cancer group. Cancer. (2001) 92:2699–708. doi: 10.1002/1097-0142(20011115)92:10<2699::AID-CNCR1624>3.0.CO;2-A
16. Zafar A, Wang W, Liu G, Wang X, Xian W, McKeon F, et al. Molecular targeting therapies for neuroblastoma: Progress and challenges. Med Res Rev. (2021) 41:961–1021. doi: 10.1002/med.21750
17. Monclair T, Brodeur GM, Ambros PF, Brisse HJ, Cecchetto G, Holmes K, et al. The international neuroblastoma risk group (INRG) staging system: an INRG task force report. J Clin Oncol. (2009) 27:298–303. doi: 10.1200/JCO.2008.16.6876
18. Testing the addition of 131I-MIBG or lorlatinib to intensive therapy in people Q18 with high-risk neuroblastoma (NBL) – NCI. (2016). Available at: https://www.cancer.gov/about-cancer/treatment/clinical-trials/search/v?id=NCT03126916 (Accessed December 15, 2024).
19. Krystal J, Foster JH. Treatment of high-risk neuroblastoma. Children. (2023) 10:1302. doi: 10.3390/children10081302
20. Ryan AL, Akinkuotu A, Pierro A, Morgenstern DA, Irwin MS. The role of surgery in high-risk neuroblastoma. J Pediatr Hematology/Oncol. (2020) 42:1. doi: 10.1097/MPH.0000000000001607
21. ANBL1531: A phase 3 study of 131I-metaiodobenzylguanidine (131I-MIBG) or crizotinib added to intensive therapy for children with newly diagnosed high-risk neuroblastoma (NBL) . Available at: https://www.cincinnatichildrens.org/service/c/clinical-trials/studies/ANBL1531 (Accessed December 15, 2024).
22. Matthay KK, Reynolds CP, Seeger RC, Shimada H, Adkins ES, Haas-Kogan D, et al. Long-term results for children with high-risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13-cis-retinoic acid: A children’s oncology group study. J Clin Oncol. (2009) 27:1007–13. doi: 10.1200/JCO.2007.13.8925
23. Matthay KK, Villablanca JG, Seeger RC, Stram DO, Harris RE, Ramsay NK, et al. Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. New Engl J Med. (1999) 341:1165–73. doi: 10.1056/NEJM199910143411601
24. Braunstein SE, London WB, Kreissman SG, Villablanca JG, Davidoff AM, DeSantes K, et al. Role of the extent of prophylactic regional lymph node radiotherapy on survival in high-risk neuroblastoma: A report from the COG A3973 study. Pediatr Blood Cancer. (2019) 66:e27736. doi: 10.1002/pbc.27736
25. Sait S, Modak SI. Anti-GD2 immunotherapy for neuroblastomas. Expert Rev Anticancer Ther. (2017) 17:889–904. doi: 10.1080/14737140.2017.1364995
26. DuBois SG, Macy ME, Henderson TO. High-risk and relapsed neuroblastoma: toward more cures and better outcomes. Am Soc Clin Oncol Educ Book. (2022) 42:1–13. doi: 10.1200/EDBK_349783
27. Maris JM, Matthay KK. Molecular biology of neuroblastoma. JCO. (1999) 17:2264–4. doi: 10.1200/JCO.1999.17.7.2264
28. Schwab M, Alitalo K, Klempnauer KH, Varmus HE, Bishop JM, Gilbert F, et al. Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature. (1983) 305:245–8. doi: 10.1038/305245a0
29. Kohl NE, Legouy E, DePinho RA, Nisen PD, Smith RK, Gee CE, et al. Human nmyc is closely related in organization and nucleotide sequence to c-myc. Nature. (1986) 319:73–7. doi: 10.1038/319073a0
30. Franke F, Rudolph B, Christiansen H, Harbott J, Lampert F. Tumour karyotype may be important in the prognosis of human neuroblastoma. J Cancer Res Clin Oncol. (1986) 111:266–72. doi: 10.1007/BF00389243
31. Maris JM, Guo C, White PS, Hogarty MD, Thompson PM, Stram DO, et al. Allelic deletion at chromosome bands 11q14-23 is common in neuroblastoma. Med Pediatr Oncol. (2001) 36:24–7. doi: 10.1002/1096-911X(20010101)36:1<24::AIDMPO1007>3.0.CO;2-7
32. Bellini A, Pötschger U, Bernard V, Lapouble E, Baulande S, Ambros PF, et al. Frequency and prognostic impact of ALK amplifications and mutations in the european neuroblastoma study group (SIOPEN) high-risk neuroblastoma trial (HRNBL1). JCO. (2021) 39:3377–90. doi: 10.1200/JCO.21.00086
33. Aguilera P, López-Contreras AJ. ATRX, a guardian of chromatin. Trends Genet. (2023) 39:505–19. doi: 10.1016/j.tig.2023.02.009
34. van Gerven MR, Bozsaky E, Matser YAH, Vosseberg J, Taschner-Mandl S, Koster J, et al. Mutational spectrum of ATRX aberrations in neuroblastoma and associated patient and tumor characteristics. Cancer Sci. (2022) 113:2167–78. doi: 10.1111/cas.v113.6
35. Dratwa M, Wysoczańska B, Łacina P, Kubik T, Bogunia-Kubik K. TERT–regulation and roles in cancer formation. Front Immunol. (2020) 11:589929. doi: 10.3389/fimmu.2020.589929
36. Valentijn LJ, Koster J, Zwijnenburg DA, Hasselt NE, van Sluis P, Volckmann R, et al. TERT rearrangements are frequent in neuroblastoma and identify aggressive tumors. Nat Genet. (2015) 47:1411–4. doi: 10.1038/ng.3438
37. Peng Y, Croce CM. The role of MicroRNAs in human cancer. Sig Transduct Target Ther. (2016) 1:1–9. doi: 10.1038/sigtrans.2015.4
38. De Preter K, Mestdagh P, Vermeulen J, Zeka F, Naranjo A, Bray I, et al. miRNA expression profiling enables risk stratification in archived and fresh neuroblastoma tumor samples. Clin Cancer Res. (2011) 17:7684–92. doi: 10.1158/1078-0432.CCR-11-0610
39. Mossë YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, et al. Identification of ALK as the major familial neuroblastoma predisposition gene. Nature. (2008) 455:930–5. doi: 10.1038/nature07261
40. Eleveld TF, Schild L, Koster J, Zwijnenburg DA, Alles LK, Ebus ME, et al. RASMAPK pathway-driven tumor progression is associated with loss of CIC and other genomic aberrations in neuroblastoma. Cancer Res. (2018) 78:6297–307. doi: 10.1158/0008-5472.CAN-18-1045
41. Iwahara T, Fujimoto J, Wen D, Cupples R, Bucay N, Arakawa T, et al. Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene. (1997) 14:439–49. doi: 10.1038/sj.onc.1200849
42. Yao S, Cheng M, Zhang Q, Wasik M, Kelsh R, Winkler C. Anaplastic lymphoma kinase is required for neurogenesis in the developing central nervous system of zebrafish. PloS One. (2013) 8:e63757. doi: 10.1371/journal.pone.0063757
43. Fransson S, Abel F, Kogner P, Martinsson T, Ejeskär K. Stage-dependent expression of PI3K/Akt–pathway genes in neuroblastoma. Int J Oncol. (2013) 42:609–16. doi: 10.3892/ijo.2012.1732
44. Ahmad MH, Ghosh B, Rizvi MA, Ali M, Kaur L, Mondal AC. Neural crest cells development and neuroblastoma progression: Role of wnt signaling. J Cell Physiol. (2023) 238:306–28. doi: 10.1002/jcp.v238.2
45. Xu L, Wang X, Wan J, Li T, Gong X, Zhang K, et al. Sonic hedgehog pathway is essential for neuroblastoma cell proliferation and tumor growth. Mol Cell Biochem. (2012) 364:235–41. doi: 10.1007/s11010-011-1222-6
46. Bianchi ME, Mezzapelle R. The chemokine receptor CXCR4 in cell proliferation and tissue regeneration. Front Immunol. (2020) 11:2109/full. doi: 10.3389/fimmu.2020.02109/full
47. Zhi Y, Duan Y, Zhou X, Yin X, Guan G, Zhang H, et al. NF-kB signaling pathway confers neuroblastoma cells migration and invasion ability via the regulation of CXCR4. Med Sci Monit. (2014) 20:2746–52. doi: 10.12659/MSM.892597
48. Iolascon A, Giordani L, Borriello A, Carbone R, Izzo A, Tonini GP, et al. Reduced expression of transforming growth factor-beta receptor type III in high stage neuroblastomas. Br J Cancer. (2000) 82:1171–6. doi: 10.1054/bjoc.1999.1058
49. Zage PE, Nolo R, Fang W, Stewart J, Garcia-Manero G, Zweidler-McKay PA. Notch pathway activation induces neuroblastoma tumor cell growth arrest. Pediatr Blood Cancer. (2012) 58:682–9. doi: 10.1002/pbc.23202
50. Nag S, Qin J, Srivenugopal KS, Wang M, Zhang R. The MDM2-p53 pathway revisited. J BioMed Res. (2013) 27:254–71. doi: 10.7555/JBR.27.20130030
51. He J, Gu L, Zhang H, Zhou M. Crosstalk between MYCN and MDM2-p53 signal pathways regulates tumor cell growth and apoptosis in neuroblastoma. Cell Cycle. (2011) 10:2994–3002. doi: 10.4161/cc.10.17.17118
52. Persaud NV, Park JA, Cheung NKV. High-risk neuroblastoma challenges and opportunities for antibody-based cellular immunotherapy. J Clin Med. (2024) 13:4765. doi: 10.3390/jcm13164765
53. Wienke J, Dierselhuis MP, Tytgat GAM, Künkele A, Nierkens S, Molenaar JJ. The immune landscape of neuroblastoma: Challenges and opportunities for novel therapeutic strategies in pediatric oncology. Eur J Cancer. (2021) 144:123–50. doi: 10.1016/j.ejca.2020.11.014
54. Pieniażek B, Cencelewicz K, Bździuch P, Młynarczyk Ł, Lejman M, Zawitkowska J, et al. Neuroblastoma–a review of combination immunotherapy. Int J Mol Sci. (2024) 25:7730. doi: 10.3390/ijms25147730
55. Pastor ER, Mousa SA. Current management of neuroblastoma and future direction. Crit Rev Oncology/Hematol. (2019) 138:38–43. doi: 10.1016/j.critrevonc.2019.03.013
56. Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, et al. Interleukin-2, and isotretinoin for neuroblastoma. New Engl J Med. (2010) 363:1324–34. doi: 10.1056/NEJMoa0911123
57. Zeng Y, Fest S, Kunert R, Katinger H, Pistoia V, Michon J, et al. Antineuroblastoma effect of ch14.18 antibody produced in CHO cells is mediated by NK-cells in mice. Mol Immunol. (2005) 42:1311–9. doi: 10.1016/j.molimm.2004.12.018
58. Barone G, Barry A, Bautista F, Brichard B, Defachelles AS, Herd F, et al. Managing adverse events associated with dinutuximab beta treatment in patients with high-risk neuroblastoma: practical guidance. Paediatr Drugs. (2021) 23:537–48. doi: 10.1007/s40272-021-00469-9
59. LiverTox: Clinical and research information on drug-induced liver injury [Internet]. Bethesda (MD: National Institute of Diabetes and Digestive and Kidney Diseases; 2012-. Naxitamab: Short Chapter (2024). Available at: https://www.ncbi.nlm.nih.gov/books/NBK605692/.
60. Sorkin LS, Otto M, Baldwin WM, Vail E, Gillies SD, Handgretinger R, et al. Anti- GD(2) with an FC point mutation reduces complement fixation and decreases antibody-induced allodynia. Pain. (2010) 149:135–42. doi: 10.1016/j.pain.2010.01.024
61. Furman WL, McCarville B, Shulkin BL, Davidoff A, Krasin M, Hsu CW, et al. Improved outcome in children with newly diagnosed high-risk neuroblastoma treated with chemoimmunotherapy: updated results of a phase II study using hu14.18K322A. JCO. (2022) 40:335–44. doi: 10.1200/JCO.21.01375
62. Galassi L, Rossi M, Lodeserto P, Lenzi M, Borsetti F, Voltattorni M, et al. Naxitamab activity in neuroblastoma cells is enhanced by nanofenretinide and nanospermidine. Pharmaceutics. (2023) 15:648. doi: 10.3390/pharmaceutics15020648
63. Zhang Y, Kupferschlaeger J, Lang P, Reischl G, Handgretinger RJ, la Fougère C, et al. 131I-GD2-ch14.18 scintigraphy to evaluate option for radioimmunotherapy in patients with advanced tumors. J Nucl Med. (2022) 63:205–11. doi: 10.2967/jnumed.120.261854
64. June CH, Sadelain M. Chimeric antigen receptor therapy. New Engl J Med. (2018) 379:64–73. doi: 10.1056/NEJMra1706169
65. National Cancer Institute (NCI). A phase I trial of T cells expressing an anti- GD2 chimeric antigen receptor in children and young adults with GD2+ solid tumors. (2024). Available at: https://clinicaltrials.gov/study/NCT02107963 (Accessed December 15, 2024).
66. Ramakrishna S, Kaczanowska S, Murty T, Contreras CF, Merchant M, Glod J, et al. Abstract CT142: GD2.Ox40.CD28.z CAR T cell trial in neuroblastoma and osteosarcoma. Cancer Res. (2022) 82:CT142. doi: 10.1158/1538-7445.am2022-ct142
67. Kaczanowska S, Murty T, Alimadadi A, Contreras CF, Duault C, Subrahmanyam PB, et al. Immune determinants of CAR-t cell expansion in solid tumor patients receiving GD2 CAR-t cell therapy. Cancer Cell. (2024) 42:35–51.e8. doi: 10.1016/j.ccell.2023.11.011
68. Heczey A, Courtney AN, Montalbano A, Robinson S, Liu K, Li M, et al. Anti- GD2 CAR-NKT cells in patients with relapsed or refractory neuroblastoma: an interim analysis. Nat Med. (2020) 26:1686–90. doi: 10.1038/s41591-020-1074-2
69. Heczey A. GD2 specific chimeric antigen receptor (CAR) and interleukin-15 expressing autologous natural killer T-cells to treat children with neuroblastoma. (2024). Available at: https://clinicaltrials.gov/study/NCT03294954 (Accessed December 15, 2024).
70. Bosse KR, Raman P, Zhu Z, Lane M, Martinez D, Heitzeneder S, et al. Identification of GPC2 as an oncoprotein and candidate immunotherapeutic target in high-risk neuroblastoma. Cancer Cell. (2017) 32:295–309. doi: 10.1016/j.ccell.2017.08.003
71. Li N, Torres MB, Spetz MR, Wang R, Peng L, Tian M, et al. CAR T cells targeting tumor-associated exons of glypican 2 regress neuroblastoma in mice. Cell Rep Med. (2021) 2:100297. doi: 10.1016/j.xcrm.2021.100297
72. Sun M, Cao Y, Okada R, Reyes-González JM, Stack HG, Qin H, et al. Preclinical optimization of a GPC2-targeting CAR T-cell therapy for neuroblastoma. J Immunother Cancer. (2023) 11:e005881. doi: 10.1136/jitc-2022-005881
73. Li D, Liang T, Hutchins LE, Wolfarth AA, Ferrando-Martinez S, Lee BH, et al. rhIL-7-hyFc, a long-acting interleukin-7, improves efficacy of CAR-t cell therapy in solid tumors. J Immunother Cancer. (2024) 12:e008989. doi: 10.1136/jitc-2024-008989
74. Liu S, Liang J, Liu Z, Zhang C, Wang Y, Watson AH, et al. The role of CD276 in cancers. Front Oncol. (2021) 11:654684/full. doi: 10.3389/fonc.2021.654684/full
75. Kontos F, Michelakos T, Kurokawa T, Sadagopan A, Schwab JH, Ferrone CR, et al. B7-H3: an attractive target for antibody-based immunotherapy. Clin Cancer Res. (2021) 27:1227–35. doi: 10.1158/1078-0432.CCR-20-2584
76. Castriconi R, Dondero A, Augugliaro R, Cantoni C, Carnemolla B, Sementa AR, et al. Identification of 4Ig-B7-H3 as a neuroblastoma-associated molecule that exerts a protective role from an NK cell-mediated lysis. Proc Natl Acad Sci USA. (2004) 101:12640–5. doi: 10.1073/pnas.0405025101
77. Grote S, Chan KCH, Baden C, Bösmüller H, Sulyok M, Frauenfeld L, et al. CD276 as novel CAR NK-92 therapeutic target for neuroblastoma. Adv IN Cell AND Gene Ther. (2021) 4:e105. doi: 10.1002/acg2.105
78. Majzner RG, Theruvath JL, Nellan A, Heitzeneder S, Cui Y, Mount CW, et al. CAR T cells targeting B7-H3, a pan-cancer antigen, demonstrate potent preclinical activity against pediatric solid tumors and brain tumors. Clin Cancer Res. (2019) 25:2560–74. doi: 10.1158/1078-0432.CCR-18-0432
79. Moghimi B, Muthugounder S, Jambon S, Tibbetts R, Hung L, Bassiri H, et al. Preclinical assessment of the efficacy and specificity of GD2-B7H3 SynNotch CAR-t in metastatic neuroblastoma. Nat Commun. (2021) 12:511. doi: 10.1038/s41467-020-20785-x
80. Wienke J, Visser LL, Kholosy WM, Keller KM, Barisa M, Poon E, et al. Integrative analysis of neuroblastoma by single-cell RNA sequencing identifies the NECTIN2-TIGIT axis as a target for immunotherapy. Cancer Cell. (2024) 42:283–300.e8. doi: 10.1016/j.ccell.2023.12.008
81. Siebert N, Zumpe M, Schwencke CH, Biskupski S, Troschke-Meurer S, Leopold J, et al. Combined blockade of TIGIT and PD-L1 enhances anti-neuroblastoma efficacy of GD2-directed immunotherapy with dinutuximab beta. Cancers. (2023) 15:3317. doi: 10.3390/cancers15133317
82. van Limpt VAE, Chan AJ, van Sluis PG, Caron HN, van Noesel CJM, Versteeg R. High delta-like 1 expression in a subset of neuroblastoma cell lines corresponds to differentiated chromaffin cell type. Int J Cancer. (2003) 105:61–9. doi: 10.1002/ijc.v105:1
83. Hamilton AK, Radaoui AB, Tsang M, Martinez D, Conkrite KL, Patel K, et al. A proteogenomic surfaceome study identifies DLK1 as an immunotherapeutic target in neuroblastoma. Cancer Cell. (2024) 42:1970–1982.e7. doi: 10.1016/j.ccell.2024.10.003
84. National cancer institute (NCI). a first-in-human phase I trial with antibody drug conjugate ADCT-701 in neuroendocrine tumors and carcinomas. (2024). Available at: https://clinicaltrials.gov/study/NCT06041516 (Accessed December 15, 2024).
85. Pathania AS, Prathipati P, Murakonda SP, Murakonda AB, Srivastava A, Avadhesh, et al. Immune checkpoint molecules in neuroblastoma: A clinical perspective. Semin Cancer Biol. (2022) 86:247–58. doi: 10.1016/j.semcancer.2022.06.013
86. Zeng L, Xu H, Li SH, Xu SY, Chen K, Qin LJ, et al. Cross-cohort analysis identified an immune checkpoint-based signature to predict the clinical outcomes of neuroblastoma. J Immunother Cancer. (2023) 11:e005980. doi: 10.1136/jitc-2022-005980
87. Makuku R, Khalili N, Razi S, Keshavarz-Fathi M, Rezaei N. Current and future perspectives of PD-1/PDL-1 blockade in cancer immunotherapy. J Immunol Res. (2021) 2021:6661406. doi: 10.1155/2021/6661406
88. Ehlert K, Hansjuergens I, Zinke A, Otto S, Siebert N, Henze G, et al. Nivolumab dinutuximab beta in two patients with refractory neuroblastoma. J Immunother Cancer. (2020) 8:e000540. doi: 10.1136/jitc-2020-000540
89. Sansom DM. CD28, CTLA-4 and their ligands: who does what and to whom? Immunology. (2000) 101:169–77. doi: 10.1046/j.1365-2567.2000.00121.x
90. Cano-Mejia J, Shukla A, Ledezma DK, Palmer E, Villagra A, Fernandes R. CpGcoated pRussian blue nanoparticles-based photothermal therapy combined with anti-CTLA-4 immune checkpoint blockade triggers a robust abscopal effect against neuroblastoma. Trans Oncol. (2020) 13:100823. doi: 10.1016/j.tranon.2020.100823
91. O’Brien J, Hayder H, Zayed Y, Peng C. Overview of microRNA biogenesis, mechanisms of actions, and circulation. Front Endocrinol (Lausanne). (2018) 9:402. doi: 10.3389/fendo.2018.00402
92. Aravindan N, Subramanian K, Somasundaram DB, Herman TS, Aravindan S. MicroRNAs in neuroblastoma tumorigenesis, therapy resistance, and disease evolution. Cancer Drug Resist. (2019) 2:1086–105. doi: 10.20517/cdr.2019.68
93. Pathania AS, Chava H, Chaturvedi NK, Chava S, Byrareddy SN, Coulter DW, et al. The miR-29 family facilitates the activation of NK-cell immune responses by targeting the B7-H3 immune checkpoint in neuroblastoma. Cell Death Dis. (2024) 15:428. doi: 10.1038/s41419-024-06791-7
94. Cheung IY, Farazi TA, Ostrovnaya I, Xu H, Tran H, Mihailovic A, et al. Deep MicroRNA sequencing reveals downregulation of miR-29a in neuroblastoma central nervous system metastasis. Genes Chromosomes Cancer. (2014) 53:803–14. doi: 10.1002/gcc.v53.10
95. Ko HY, Lee J, Lee YS, Choi Y, Ali BA, Al-Khedhairy AA, et al. Bioimaging of transcriptional activity of microRNA124a during neurogenesis. Biotechnol Lett. (2015) 37:2333–40. doi: 10.1007/s10529-015-1912-3
96. Zhang X, Yang C, Meng Z, Zhong H, Hou X, Wang F, et al. miR-124 and VAMP3 act antagonistically in human neuroblastoma. Int J Mol Sci. (2023) 24:14877. doi: 10.3390/ijms241914877
97. Nolan JC, Salvucci M, Carberry S, Barat A, Segura MF, Fenn J, et al. A contextdependent role for miR-124-3p on cell phenotype, viability and chemosensitivity in neuroblastoma in vitro. Front Cell Dev Biol. (2020) 8:559553. doi: 10.3389/fcell.2020.559553
98. Sharif S, Ghahremani MH, Soleimani M. Induction of morphological and functional differentiation of human neuroblastoma cells by miR-124. J Biosci. (2017) 42:555–63. doi: 10.1007/s12038-017-9714-5
99. Zhao Z, Ma X, Hsiao TH, Lin G, Kosti A, Yu X, et al. A high-content morphological screen identifies novel microRNAs that regulate neuroblastoma cell differentiation. Oncotarget. (2014) 5:2499–512. doi: 10.18632/oncotarget.1703
100. Huang TC, Chang HY, Chen CY, Wu PY, Lee H, Liao YF, et al. Silencing of miR-124 induces neuroblastoma SK-N-SH cell differentiation, cell cycle arrest and apoptosis through promoting AHR. FEBS Lett. (2011) 585:3582–6. doi: 10.1016/j.febslet.2011.10.025
101. Cheng X, Xu Q, Zhang Y, Shen M, Zhang S, Mao F, et al. miR-34a inhibits progression of neuroblastoma by targeting autophagy-related gene 5. Eur J Pharmacol. (2019) 850:53–63. doi: 10.1016/j.ejphar.2019.01.071
102. Di Paolo D, Pastorino F, Brignole C, Corrias MV, Emionite L, Cilli M, et al. Combined replenishment of miR-34a and let-7b by targeted nanoparticles inhibits tumor growth in neuroblastoma preclinical models. Small. (2020) 16:1906426. doi: 10.1002/smll.201906426
103. Li Z, Chen H. miR-34a inhibits proliferation, migration and invasion of pediatric neuroblastoma cells via targeting HNF4a. Artif Cells Nanomed Biotechnol. (2019) 47:3072–8. doi: 10.1080/21691401.2019.1637886
104. Liu T, Gu L, Wu Z, Albadari N, Li W, Zhou M. MYCN mRNA degradation and cancer suppression by a selective small-molecule inhibitor in MYCN-amplified neuroblastoma. Front Oncol. (2022) 12:1058726. doi: 10.3389/fonc.2022.1058726
105. Powers JT, Tsanov KM, Pearson DS, Roels F, Spina CS, Ebright R, et al. Multiple mechanisms disrupt the let-7 microRNA family in neuroblastoma. Nature. (2016) 535:246–51. doi: 10.1038/nature18632
106. Molenaar JJ, Domingo-Fernández R, Ebus ME, Lindner S, Koster J, Drabek K, et al. LIN28B induces neuroblastoma and enhances MYCN levels via let-7 suppression. Nat Genet. (2012) 44:1199–206. doi: 10.1038/ng.2436
107. Mattick JS, Amaral PP, Carninci P, Carpenter S, Chang HY, Chen LL, et al. Long non-coding RNAs: definitions, functions, challenges and recommendations. Nat Rev Mol Cell Biol. (2023) 24:430–47. doi: 10.1038/s41580-022-00566-8
108. Statello L, Guo CJ, Chen LL, Huarte M. Gene regulation by long non-coding RNAs and its biological functions. Nat Rev Mol Cell Biol. (2021) 22:96–118. doi: 10.1038/s41580-020-00315-9
109. Nandwani A, Rathore S, Datta M. LncRNAs in cancer: Regulatory and therapeutic implications. Cancer Lett. (2021) 501:162–71. doi: 10.1016/j.canlet.2020.11.048
110. Tee AE, Liu B, Song R, Li J, Pasquier E, Cheung BB, et al. The long noncoding RNA MALAT1 promotes tumor-driven angiogenesis by up-regulating pro-angiogenic gene expression. Oncotarget. (2016) 7:8663–75. doi: 10.18632/oncotarget.6675
111. Bi S, Wang C, Li Y, Zhang W, Zhang J, Lv Z, et al. LncRNA-MALAT1-mediated promotes cell invasion and migration in human neuroblastoma. Tumour Biol. (2017) 39:1010428317699796. doi: 10.1177/1010428317699796
112. Chi R, Chen X, Liu M, Zhang H, Li F, Fan X, et al. Role of SNHG7-miR-653-5p- STAT2 feedback loop in regulating neuroblastoma progression. J Cell Physiol. (2019) 234:13403–12. doi: 10.1002/jcp.v234.8
113. Gamero AM, Young MR, Mentor-Marcel R, Bobe G, Scarzello AJ, Wise J, et al. STAT2 contributes to promotion of colorectal and skin carcinogenesis. Cancer Prev Res (Phila). (2010) 3:495–504. doi: 10.1158/1940-6207.CAPR-09-0105
114. Jia J, Zhang D, Zhang J, Yang L, Zhao G, Yang H, et al. Long non-coding RNA SNHG7 promotes neuroblastoma progression through sponging miR-323a-5p and miR-342-5p. BioMed Pharmacother. (2020) 128:110293. doi: 10.1016/j.biopha.2020.110293
115. Wang SY, Wang X, Zhang CY. LncRNA SNHG7 enhances chemoresistance in neuroblastoma through cisplatin-induced autophagy by regulating miR-329-3p/ MYO10 axis. Eur Rev Med Pharmacol Sci. (2020) 24:3805–17. doi: 10.26355/eurrev_202004_20847
116. Karami Fath M, Pourbagher Benam S, Salmani K, Naderi S, Fahham Z, Ghiabi S, et al. Circular RNAs in neuroblastoma: Pathogenesis, potential biomarker, and therapeutic target. Pathol - Res Pract. (2022) 238:154094. doi: 10.1016/j.prp.2022.154094
117. Yang J, Liu B, Xu Z, Feng M. Silencing of circ_0135889 restrains proliferation and tumorigenicity of human neuroblastoma cells. J Surg Res. (2022) 279:135–47. doi: 10.1016/j.jss.2022.05.025
118. Chang S, Ren D, Zhang L, Liu S, Yang W, Cheng H, et al. Therapeutic SHPRH- 146aa encoded by circ-SHPRH dynamically upregulates P21 to inhibit CDKs in neuroblastoma. Cancer Lett. (2024) 598:217120. doi: 10.1016/j.canlet.2024.217120
119. Du J, Zhuo Y, Sun X, Nie M, Yang J, Luo X, et al. hsa_circ_0000285 sponging miR-582-3p promotes neuroblastoma progression by regulating the wnt/b-catenin signaling pathway. Open Med (Wars). (2023) 18:20230726. doi: 10.1515/med-2023-0726
120. Chen Y, Lin L, Hu X, Li Q, Wu M. Silencing of circular RNA circPDE5A suppresses neuroblastoma progression by targeting the miR-362-5p/NOL4L axis. Int J Neurosci. (2023) 133:141–51. doi: 10.1080/00207454.2021.1896505
121. Bian J, Ding H, Hu A, Wang J. Circ_0001361/miR-490-5p/IGF2 axis regulates the viability and apoptosis of neuroblastoma cells. Neurochem Res. (2024) 49:3060–8. doi: 10.1007/s11064-024-04225-6
122. Rosswog C, Fassunke J, Ernst A, Schömig-Markiefka B, Merkelbach-Bruse S, Bartenhagen C, et al. Genomic ALK alterations in primary and relapsed neuroblastoma. Br J Cancer. (2023) 128:1559–71. doi: 10.1038/s41416-023-02208-y
123. Pfizer. Phase 3, randomized, open-label study of the efficacy and safety of crizotinib versus pemetrexed&x2F;Cisplatin or pemetrexed&x2F;Carboplatin in previously untreated patients with non-squamous carcinoma of the lung harboring a translocation or inversion event involving the anaplastic lymphoma kinase (Alk) gene locus. (2017). Available at: https://clinicaltrials.gov/study/NCT01154140 (Accessed December 15, 2024).
124. Pfizer. Phase 3, randomized, open-label study of the efficacy and safety of pf- 02341066 versus standard of care chemotherapy (Pemetrexed or docetaxel) in patients with advanced non-small cell lung cancer (Nsclc) harboring a translocation or inversion event involving the anaplastic lymphoma kinase (Alk) gene locus. (2016). Available at: https://clinicaltrials.gov/study/NCT00932893 (Accessed December 15, 2024).
125. Solomon BJ, Mok T, Kim DW, Wu YL, Nakagawa K, Mekhail T, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. (2014) 371:2167–77. doi: 10.1056/NEJMoa1408440
126. Children’s Oncology Group. A phase 1 study of crizotinib in combination with conventional chemotherapy for relapsed or refractory solid tumors or anaplastic large cell lymphoma. (2023). Available at: https://clinicaltrials.gov/study/NCT01606878 (Accessed December 15, 2024).
127. Children’s oncology group. a phase 3 study of 131I-metaiodobenzylguanidine (131I-MIBG) or ALK inhibitor therapy added to intensive therapy for children with newly diagnosed high-risk neuroblastoma (NBL). (2024). Available at: https://clinicaltrials.gov/study/NCT03126916 (Accessed December 15, 2024).
128. Carpenter EL, Mossé YP. Targeting ALK in neuroblastoma–preclinical and clinical advancements. Nat Rev Clin Oncol. (2012) 9:391–9. doi: 10.1038/nrclinonc.2012.72
129. Iyer R, Wehrmann L, Golden RL, Naraparaju K, Croucher JL, MacFarland SP, et al. Entrectinib is a potent inhibitor of trk-driven neuroblastomas in a xenograft mouse model. Cancer Lett. (2016) 372:179–86. doi: 10.1016/j.canlet.2016.01.018
130. Liu T, Merguerian MD, Rowe SP, Pratilas CA, Chen AR, Ladle BH. Exceptional response to the ALK and ROS1 inhibitor lorlatinib and subsequent mechanism of resistance in relapsed ALK F1174L-mutated neuroblastoma. Cold Spring Harb Mol Case Stud. (2021) 7:a006064. doi: 10.1101/mcs.a006064
131. Infarinato NR, Park JH, Krytska K, Ryles HT, Sano R, Szigety KM, et al. The ALK/ROS1 inhibitor PF-06463922 overcomes primary resistance to crizotinib in ALKdriven neuroblastoma. Cancer Discovery. (2016) 6:96–107. doi: 10.1158/2159-8290.CD-15-1056
132. Siaw JT, Wan H, Pfeifer K, Rivera VM, Guan J, Palmer RH, et al. Brigatinib, an anaplastic lymphoma kinase inhibitor, abrogates activity and growth in ALK-positive neuroblastoma cells, drosophila and mice. Oncotarget. (2016) 7:29011–22. doi: 10.18632/oncotarget.v7i20
133. Alam MW, Borenäs M, Lind DE, Cervantes-Madrid D, Umapathy G, Palmer RH. And hallberg b alectinib, an anaplastic lymphoma kinase inhibitor, abolishes ALK activity and growth in ALK-positive neuroblastoma cells. Front Oncol. (2019) 9:579. doi: 10.3389/fonc.2019.00579
134. Cervantes-Madrid D, Szydzik J, Lind DE, Borenäs M, Bemark M, Cui J, et al. Repotrectinib (TPX-0005), effectively reduces growth of ALK driven neuroblastoma cells. Sci Rep. (2019) 9:19353. doi: 10.1038/s41598-019-55060-7
135. Zou HY, Friboulet L, Kodack DP, Engstrom LD, Li Q, West M, et al. PF-06463922, an ALK/ROS1 inhibitor, overcomes resistance to first and second generation ALK inhibitors in preclinical models. Cancer Cell. (2015) 28:70–81. doi: 10.1016/j.ccell.2015.05.010
136. Guan J, Tucker ER, Wan H, Chand D, Danielson LS, Ruuth K, et al. The ALK inhibitor PF-06463922 is effective as a single agent in neuroblastoma driven by expression of ALK and MYCN. Dis Models Mechan. (2016) 9:941–52. doi: 10.1242/dmm.024448
137. Shaw AT, Friboulet L, Leshchiner I, Gainor JF, Bergqvist S, Brooun A, et al. Resensitization to crizotinib by the lorlatinib ALK resistance mutation L1198F. New Engl J Med. (2016) 374:54–61. doi: 10.1056/NEJMoa1508887
138. Stafman LL, Williams AP, Marayati R, Aye JM, Markert HR, Garner EF, et al. Focal adhesion kinase inhibition contributes to tumor cell survival and motility in neuroblastoma patient-derived xenografts. Sci Rep. (2019) 9:13259. doi: 10.1038/s41598-019-49853-z
139. Chugh S, Tien JC, Hon J, Kenum C, Mannan R, Cheng Y, et al. Therapeutic benefit of the dual ALK/FAK inhibitor ESK440 in ALK-driven neuroblastoma. Neoplasia. (2024) 6:100964. doi: 10.1016/j.neo.2024.100964
140. Bachmann AS, Geerts D. Polyamine synthesis as a target of MYC oncogenes. J Biol Chem. (2018) 293:18757–69. doi: 10.1074/jbc.TM118.003336
141. Wagner AJ, Meyers C, Laimins LA, Hay N. C-myc induces the expression and activity of ornithine decarboxylase. Cell Growth Differ. (1993) 4:879–83.
142. Rounbehler RJ, Li W, Hall MA, Yang C, Fallahi M, Cleveland JL. Targeting ornithine decarboxylase impairs development of MYCN-amplified neuroblastoma. Cancer Res. (2009) 69:547–53. doi: 10.1158/0008-5472.CAN-08-2968
143. Nigg EA. Mitotic kinases as regulators of cell division and its checkpoints. Nat Rev Mol Cell Biol. (2001) 2:21–32. doi: 10.1038/35048096
144. Roeschert I, Poon E, Henssen AG, Garcia HD, Gatti M, Giansanti C, et al. Combined inhibition of aurora-a and ATR kinase results in regression of MYCNamplified neuroblastoma. Nat Cancer. (2021) 2:312–26. doi: 10.1038/s43018-020-00171-8
145. Bogen D, Wei JS, Azorsa DO, Ormanoglu P, Buehler E, Guha R, et al. Aurora b kinase is a potent and selective target in MYCN-driven neuroblastoma. Oncotarget. (2015) 6:35247–62. doi: 10.18632/oncotarget.v6i34
146. Richards MW, Burgess SG, Poon E, Carstensen A, Eilers M, Chesler L, et al. Structural basis of n-myc binding by aurora-a and its destabilization by kinase inhibitors. Proc Natl Acad Sci USA. (2016) 113:13726–31. doi: 10.1073/pnas.1610626113
147. Yang Y, Ding L, Zhou Q, Fen L, Cao Y, Sun J, et al. Silencing of AURKA augments the antitumor efficacy of the AURKA inhibitor MLN8237 on neuroblastoma cells. Cancer Cell Int. (2020) 20:9. doi: 10.1186/s12935-019-1072-y
148. Zhang M, Zhang L, Hei R, Li X, Cai H, Wu X, et al. CDK inhibitors in cancer therapy, an overview of recent development. Am J Cancer Res. (2021) 11:1913–35.
149. Ferguson KM, Gillen SL, Chaytor L, Poon E, Marcos D, Gomez RL, et al. Palbociclib releases the latent differentiation capacity of neuroblastoma cells. Dev Cell. (2023) 58:1967–1982.e8. doi: 10.1016/j.devcel.2023.08.028
150. Geoerger B, Bourdeaut F, DuBois SG, Fischer M, Geller JI, Gottardo NG, et al. A phase I study of the CDK4/6 inhibitor ribociclib (LEE011) in pediatric patients with malignant rhabdoid tumors, neuroblastoma, and other solid tumors. Clin Cancer Res. (2017) 23:2433–41. doi: 10.1158/1078-0432.CCR-16-2898
151. Lee JP, Hung YP, O’Dorisio TM, Howe JR, Hornick JL, Bellizzi AM. Examination of PHOX2B in adult neuroendocrine neoplasms reveals relatively frequent expression in phaeochromocytomas and paragangliomas. Histopathology. (2017) 71:503–10. doi: 10.1111/his.2017.71.issue-4
152. Mosse YP, Laudenslager M, Khazi D, Carlisle AJ, Winter CL, Rappaport E, et al. Germline PHOX2B mutation in hereditary neuroblastoma. Am J Hum Genet. (2004) 75:727–30. doi: 10.1086/424530
153. Abel F, Dalevi D, Nethander M, Jörnsten R, De Preter K, Vermeulen J, et al. A 6-gene signature identifies four molecular subgroups of neuroblastoma. Cancer Cell Int. (2011) 11:9. doi: 10.1186/1475-2867-11-9
154. Bachetti T, Di Paolo D, Di Lascio S, Mirisola V, Brignole C, Bellotti M, et al. PHOX2B-mediated regulation of ALK expression: in vitro identification of a functional relationship between two genes involved in neuroblastoma. PloS One. (2010) 5:e13108. doi: 10.1371/journal.pone.0013108
155. Revet I, Huizenga G, Chan A, Koster J, Volckmann R, van Sluis P, et al. The MSX1 homeobox transcription factor is a downstream target of PHOX2B and activates the delta–notch pathway in neuroblastoma. Exp Cell Res. (2008) 314:707–19. doi: 10.1016/j.yexcr.2007.12.008
156. Di Zanni E, Fornasari D, Ravazzolo R, Ceccherini I, Bachetti T. Identification of novel pathways and molecules able to down-regulate PHOX2B gene expression by in vitro drug screening approaches in neuroblastoma cells. Exp Cell Res. (2015) 336:43–57. doi: 10.1016/j.yexcr.2015.03.025
157. Suebsoonthron J, Jaroonwitchawan T, Yamabhai M, Noisa P. Inhibition of WNT signaling reduces differentiation and induces sensitivity to doxorubicin in human malignant neuroblastoma SH-SY5Y cells. Anticancer Drugs. (2017) 28:469–79. doi: 10.1097/CAD.0000000000000478
158. Burgess A, Chia KM, Haupt S, Thomas D, Haupt Y, Lim E. Clinical overview of MDM2/X-targeted therapies. Front Oncol. (2016) 6:7. doi: 10.3389/fonc.2016.00007
159. Tisato V, Voltan R, Gonelli A, Secchiero P, Zauli G. MDM2/X inhibitors under clinical evaluation: perspectives for the management of hematological malignancies and pediatric cancer. J Hematol Oncol. (2017) 10:133. doi: 10.1186/s13045-017-0500-5
160. Arya AK, El-Fert A, Devling T, Eccles RM, Aslam MA, Rubbi CP, et al. Nutlin- 3, the small-molecule inhibitor of MDM2, promotes senescence and radiosensitises laryngeal carcinoma cells harbouring wild-type p53. Br J Cancer. (2010) 103:186–95. doi: 10.1038/sj.bjc.6605739
161. Peirce SK, Findley HW. The MDM2 antagonist nutlin-3 sensitizes p53-null neuroblastoma cells to doxorubicin via E2F1 and TAp73. Int J Oncol. (2009) 34:1395–402. doi: 10.3892/ijo_00000267
162. Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. (2004) 303:844–8. doi: 10.1126/science.1092472
163. Lakoma A, Barbieri E, Agarwal S, Jackson J, Chen Z, Kim Y, et al. The MDM2 small-molecule inhibitor RG7388 leads to potent tumor inhibition in p53 wild-type neuroblastoma. Cell Death Discovery. (2015) 1:1–9. doi: 10.1038/cddiscovery.2015.26
164. Chen L, Pastorino F, Berry P, Bonner J, Kirk C, Wood KM, et al. Preclinical evaluation of the first intravenous small molecule MDM2 antagonist alone and in combination with temozolomide in neuroblastoma. Int J Cancer. (2019) 144:3146–59. doi: 10.1002/ijc.v144.12
165. Roche H-L. A phase I&x2F;II, multicenter, open-label, multi-arm study evaluating the safety, tolerability, pharmacokinetics, and preliminary activity of idasanutlin in combination with either chemotherapy or venetoclax in the treatment of pediatric and young adult patients with relapsed/refractory acute leukemias or solid tumors. (2024). Available at: https://clinicaltrials.gov/study/NCT04029688 (Accessed December 15, 2024).
166. Fulda S. The PI3K/akt/mTOR pathway as therapeutic target in neuroblastoma . Available online at: https://www.eurekaselect.com/article/15113 (Accessed December 15, 2024).
167. Opel D, Poremba C, Simon T, Debatin KM, Fulda S. Activation of akt predicts poor outcome in neuroblastoma. Cancer Res. (2007) 67:735–45. doi: 10.1158/0008-5472.CAN-06-2201
168. Hogarty MD, Maris JM. PI3King on MYCN to improve neuroblastoma therapeutics. Cancer Cell. (2012) 21:145–7. doi: 10.1016/j.ccr.2012.01.018
169. Chesler L, Schlieve C, Goldenberg DD, Kenney A, Kim G, McMillan A, et al. Inhibition of phosphatidylinositol 3-kinase destabilizes mycn protein and blocks malignant progression in neuroblastoma. Cancer Res. (2006) 66:8139–46. doi: 10.1158/0008-5472.CAN-05-2769
170. Kushner BH, Cheung NKV, Modak S, Becher OJ, Basu EM, Roberts SS, et al. A phase I/Ib trial targeting the Pi3k/Akt pathway using perifosine: Long-term progression-free survival of patients with resistant neuroblastoma. Int J Cancer. (2017) 140:480–4. doi: 10.1002/ijc.v140.2
171. Hinnebusch AG, Lorsch JR. The mechanism of eukaryotic translation initiation: new insights and challenges. Cold Spring Harb Perspect Biol. (2012) 4:a011544. doi: 10.1101/cshperspect.a011544
172. Hao P, Yu J, Ward R, Liu Y, Hao Q, An S, et al. Eukaryotic translation initiation factors as promising targets in cancer therapy. Cell Commun Signaling. (2020) 18:175. doi: 10.1186/s12964-020-00607-9
173. Chu J, Zhang W, Cencic R, O’Connor PBF, Robert F, Devine WG, et al. Rocaglates induce gain-of-Function alterations to eIF4A and eIF4F. Cell Rep. (2020) 30:2481–2488.e5. doi: 10.1016/j.celrep.2020.02.002
174. Chu J, Zhang W, Cencic R, Devine WG, Beglov D, Henkel T, et al. Amidinorocaglates: A potent class of eIF4A inhibitors. Cell Chem Biol. (2019) 26:1586–1593.e3. doi: 10.1016/j.chembiol.2019.08.008
175. Skofler C, Kleinegger F, Krassnig S, Birkl-Toeglhofer AM, Singer G, Till H, et al. Eukaryotic translation initiation factor 4AI: A potential novel target in neuroblastoma. Cells. (2021) 10:301. doi: 10.3390/cells10020301
176. Volegova MP, Brown LE, Banerjee U, Dries R, Sharma B, Kennedy A, et al. The MYCN 5’ UTR as a therapeutic target in neuroblastoma. Cell Rep. (2024) 43:114134. doi: 10.1016/j.celrep.2024.114134
177. Nolan JC, Frawley T, Tighe J, Soh H, Curtin C, Piskareva O. Preclinical models for neuroblastoma: Advances and challenges. Cancer Lett. (2020) 474:53–62. doi: 10.1016/j.canlet.2020.01.015
178. Chicard M, Boyault S, Colmet Daage L, Richer W, Gentien D, Pierron G, et al. Genomic copy number profiling using circulating free tumor DNA highlights heterogeneity in neuroblastoma. Clin Cancer Res. (2016) 22:5564–73. doi: 10.1158/1078-0432.CCR-16-0500
179. Mobasheri T, Rayzan E, Shabani M, Hosseini M, Mahmoodi Chalbatani G, Rezaei N. Neuroblastoma-targeted nanoparticles and novel nanotechnology-based treatment methods. J Cell Physiol. (2021) 236:1751–75. doi: 10.1002/jcp.v236.3
Keywords: high-risk neuroblastoma, neuroblastoma biology, targeted therapy, pediatric cancer, clinical and preclinical research
Citation: AlKhazal A, Chohan S, Ross DJ, Kim J and Brown EG (2025) Emerging clinical and research approaches in targeted therapies for high-risk neuroblastoma. Front. Oncol. 15:1553511. doi: 10.3389/fonc.2025.1553511
Received: 30 December 2024; Accepted: 06 February 2025;
Published: 04 March 2025.
Edited by:
Aditi Vedi, Cambridge University Hospitals NHS Foundation Trust, United KingdomReviewed by:
Thomas Eleveld, Princess Maxima Center for Pediatric Oncology, NetherlandsCopyright © 2025 AlKhazal, Chohan, Ross, Kim and Brown. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinhwan Kim, ampua2ltQHVjZGF2aXMuZWR1; Erin G. Brown, ZWdicm93bkB1Y2RhdmlzLmVkdQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.