 Madhumita S. Manivannan
Madhumita S. Manivannan Anthea Peters
Anthea Peters Spencer B. Gibson
Spencer B. Gibson
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 24 February 2025
Sec. Molecular and Cellular Oncology
Volume 15 - 2025 | https://doi.org/10.3389/fonc.2025.1549792
Lysosomes are dynamic organelles integral to cellular homeostasis, secretory pathways, immune responses, and cell death regulation. In cancers, lysosomes become dysregulated to sustain proliferative signaling, metabolism, and invasion. In hematological malignancies such as acute myeloid leukemia (AML), acute lymphoblastic leukemia (ALL), chronic myeloid leukemia (CML), and chronic lymphocytic leukemia (CLL), leukemia cells demonstrate lysosome dysregulation with increased lysosomal activity, mTORC1 signaling, catabolic reactions, and autophagy. This supports the survival, metabolism, and proliferation of the leukemia cells. Lysosomes also play a critical role in treatment resistance by promoting cell survival and sequestration of drugs. This has led to the development of lysosome-targeted therapies such as cationic amphiphilic drugs (CAD), ATPase inhibitors or autophagy inhibitors to treat hematological malignancies. Lysosome-targeted treatments have shown effectiveness at inducing cell death by inhibiting cell survival mechanisms and inducing apoptosis. In addition, the combination of lysosome-targeted therapies with standard treatments gives synergistic apoptotic responses in leukemia cells. In this review, we will describe the lysosomal functions, their dysregulation in hematological malignancies and the development of lysosomal targeted therapies for leukemia treatment. By understanding lysosome dysregulation and developing lysosome-targeted agents, innovative treatment strategies could be effective in overcoming drug resistance in hematological malignancies.
Lysosomes, membrane-bound organelles rich in hydrolytic enzymes, are essential for degrading and recycling cellular waste. Discovered by Christian de Duve in the 1950s, lysosomes were initially characterized as the cell’s digestive system (1). However, their roles extend far beyond degradation, encompassing processes such as autophagy, antigen presentation, secretion, and regulation of cell death (2–4). These multifaceted functions are crucial for maintaining cellular homeostasis and responding to various stressors (5).
Lysosomes have garnered significant attention in hematological malignancies such as leukemia due to their involvement in cancer cell metabolism, survival, and resistance to therapy (6). Leukemias are cancers of the blood, bone marrow, and lymphatic systems characterized by the uncontrolled proliferation of malignant cells. Traditional treatments, including chemotherapy and radiation, often face challenges such as drug resistance and adverse side effects. This has driven the search for novel therapeutic targets to improve treatment efficacy and patient outcomes.
Lysosomes play a pivotal role in the pathology of these malignancies through their involvement in autophagy, a cellular process that cancer cells exploit for survival under stress conditions, including nutrient deprivation and chemotherapy (7). Dysregulation of lysosomal pathways can lead to altered autophagy, contributing to cancer progression and resistance to treatment (8). Additionally, lysosomal enzymes and their role in apoptosis regulation are critical in determining the sensitivity of cancer cells to therapy-induced cell death.
Targeting lysosomal functions and pathways offers a promising therapeutic strategy in hematological malignancies. Inhibitors of autophagy, lysosomal membrane permeabilization, and enzyme activity are being explored to enhance the efficacy of existing treatments and overcome drug resistance (9). By disrupting the lysosomal machinery that cancer cells rely on, these therapies selectively induce cell death in malignant cells while sparing normal cells.
Understanding the role of lysosomes in hematological malignancies’ pathogenesis and therapy resistance is crucial for developing targeted treatments. Ongoing research is focused on elucidating the molecular mechanisms governing lysosomal functions and identifying potential therapeutic targets within these pathways. This approach holds promise for improving the prognosis and quality of life for patients with hematological malignancies, highlighting the lysosome’s potential as a key player in cancer therapy. This review will summarize the current understanding of lysosomal functions in general and in hematological malignancies, specifically leukemia, and explore the potential of targeting lysosomal pathways as a novel therapeutic strategy for these cancers.
Lysosomes are membrane-bound organelles characterized by their acidic interior, which is maintained by the vacuolar H+-ATPase (V-ATPase) that actively pumps protons into the lysosome (10). This acidic environment is essential for the optimal activity of lysosomal enzymes and for the degradation of macromolecules. The lysosomal membrane is composed of a lipid bilayer enriched with cholesterol and specific lysosomal membrane proteins such as lysosome-associated membrane proteins (LAMP-1 and LAMP-2). These proteins play crucial roles in maintaining lysosomal integrity, facilitating the transport of materials, and protecting the lysosome from autolysis (11). The membrane also contains transporters and ion channels, including those for calcium and protons, which regulate lysosomal function and signalling.
Lysosomes house over 60 hydrolases, including proteases, lipases, nucleases, and glycosidases (2). Key proteolytic enzymes include cathepsins (e.g., cathepsin B, D, and L), which degrade proteins into amino acids. These enzymes are synthesized as inactive precursors and activated in the acidic lysosomal environment. Their coordinated activity enables the breakdown of diverse substrates, from damaged organelles to extracellular material.
Lysosomes are central to cellular signalling pathways, including those regulating autophagy, metabolism, and immune responses. The mechanistic target of rapamycin complex 1 (mTORC1) is a critical lysosome-associated signalling hub. mTORC1 senses nutrient availability and regulates cell growth and autophagy (12). Additionally, lysosomes interact with transcription factors such as TFEB (transcription factor EB), which governs lysosomal biogenesis and autophagy by responding to cellular stress and nutrient signals (13) (Figure 1).
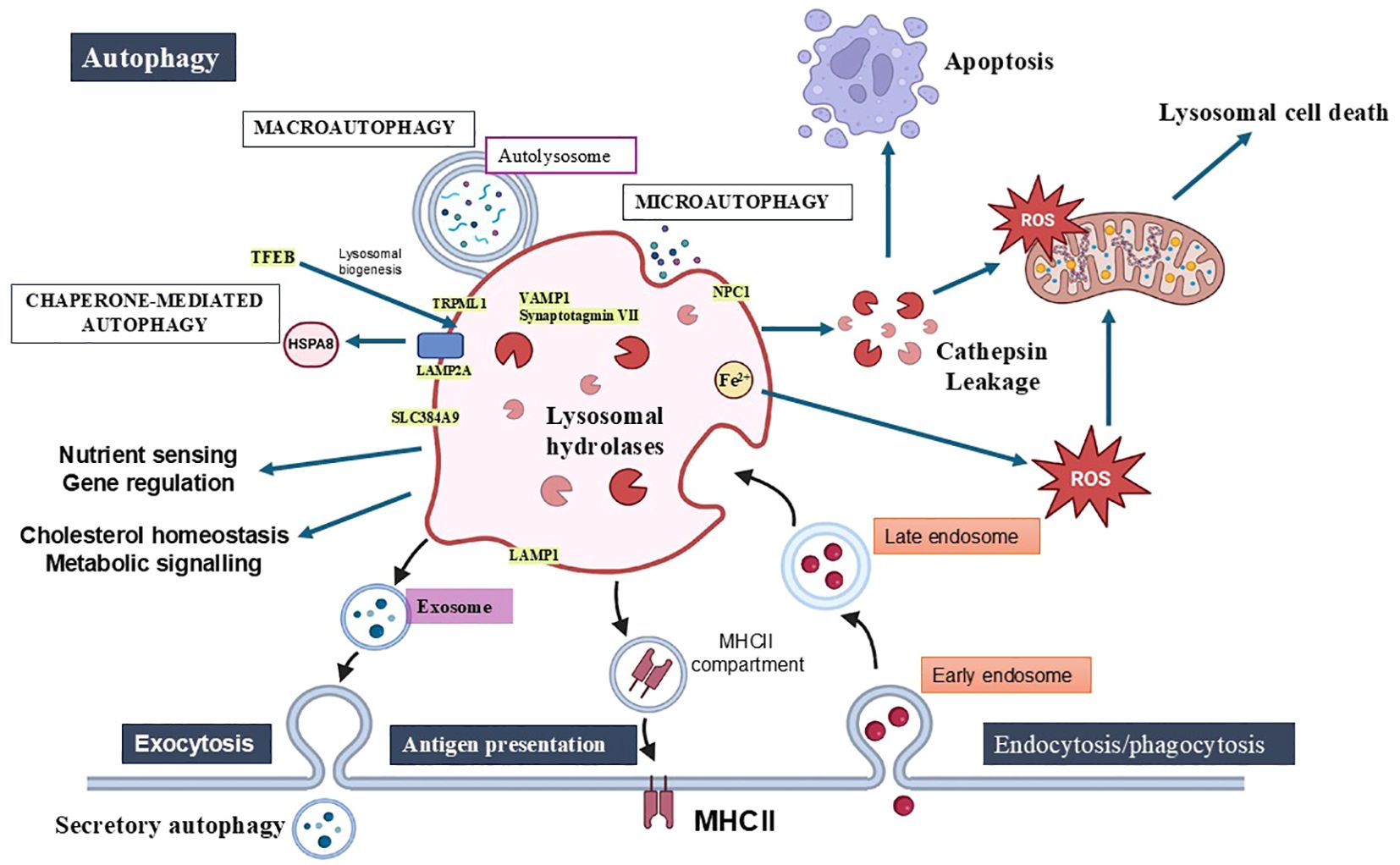
Figure 1. Lysosomal functions in cellular homeostasis, autophagy, immunity, and cell death. Lysosomes play key roles in maintaining homeostasis by degrading and recycling macromolecules, supporting nutrient sensing, cholesterol regulation, metabolic signalling, and energy homeostasis. The three types of autophagy—macroautophagy, microautophagy, and chaperone-mediated autophagy—are essential for removing damaged organelles and intracellular pathogens. Lysosomes also contribute to immune functions through phagocytosis, antigen processing via MHC-II and lysosomal exocytosis. Lysosomal membrane permeabilization (LMP) is a pivotal event that triggers various forms of cell death, including apoptosis, necrosis, pyroptosis, and ferroptosis, through the release of hydrolases, cathepsins, and reactive oxygen species (ROS). This figure highlights lysosomes’ dual role in promoting both cellular survival through autophagy and inducing cell death when lysosomal integrity is disrupted. Key components of the lysosomal membrane include integral proteins (LAMP1, LAMP2A), ion channels (TRPMLs), trafficking and fusion proteins (Synaptotagmin VII, VAMP7), lipid and amino acid transporters (NPC1, SLC38A9).
Calcium ions play a vital role in lysosomal dynamics and signalling. Lysosomes act as calcium stores, releasing calcium through channels like TRPML1 (transient receptor potential mucolipin 1) in response to stimuli. This calcium release is critical for processes such as vesicle trafficking, fusion with other organelles, and activation of signalling cascades (14). Dysregulation of lysosomal calcium signalling is implicated in various diseases, including neurodegenerative disorders and cancer (15).
By integrating these characteristics, lysosomes emerge as multifunctional organelles pivotal to cellular health and disease, making them promising targets for therapeutic intervention in conditions like leukemia and lymphoma.
Cellular homeostasis involves the synthesis, folding, trafficking, and degradation of proteins, lipids, nucleic acids, and sugars crucial for cellular function and survival (16). Lysosomes play a critical role in degrading and recycling these cellular macromolecules or redundant/damaged organelles (17). For example, lysosomes provide amino acids and glucose, which promote the translocation of mTORC1 to the lysosomal surface, mediating cellular and organismal growth (18, 19). In addition, lysosomes also digest extracellular material supplied by endocytosis or phagocytosis. In leukemia and other cancers, lysosomal dysfunction can disrupt cellular homeostasis, impairing the degradation and recycling of essential macromolecules and damaging organelles (20). This dysfunction can lead to metabolic adaptations that support cancer cell growth, survival, and resistance to treatments, as lysosomes provide key nutrients like amino acids and glucose, which fuel processes such as mTORC1 activation (21).
Lysosomes play a direct role in cellular homeostasis through the degradation of macromolecules. This is accomplished through lysosomes containing many acid hydrolases such as proteases, nucleases, glycosidases, lipases, phospholipases, phosphatases, peptidases and sulfatases responsible for this degradation. Lysosomes have membrane proton pump proteins of the H+-ATPase family that ensure an acidic pH in the lysosome (10). These proteins are highly glycosylated protecting them from lysosome protease degradation. This provides an acidic pH in lysosomes to maintain acidic hydrolase enzymatic activity. The lysosome membrane also contains transport proteins that transfer the degraded products to the cytosol for secretion or reuse by the cell.
Lysosomal positioning within the cell is also important for coordinating catabolic and anabolic processes in response to nutrient availability. Under nutrient-rich conditions, lysosomes are found at the cell periphery, associated with activated mTORC1. In contrast, during starvation, lysosomes cluster around the perinuclear region, facilitating the fusion of autophagosomes with lysosomes and the subsequent release of nutrients (22). This strategic positioning allows lysosomes to effectively manage nutrient sensing and energy homeostasis.
Lysosomal hydrolases play a direct role in the mobilization of energy stores, digesting and mobilizing nutrients under growth-promoting conditions. However, when undigested lipids or glycogen accumulate inside lysosomes, they become toxic, leading to pathological states ranging from mild disease to death (23, 24). By degrading macromolecules and mobilizing nutrients like amino acids and glucose, lysosomes support cellular energy homeostasis, fueling cancer cell proliferation and survival (25). In nutrient-rich conditions, lysosomes’ interaction with activated mTORC1 promotes cell growth and metabolic reprogramming, a common feature in cancer cells. Conversely, during nutrient deprivation or stress, lysosomes facilitate autophagy, allowing cancer cells to adapt by recycling cellular components for energy (26). However, dysfunction in lysosomal hydrolases, transport proteins, or membrane acidification can lead to the accumulation of undigested materials, contributing to cellular toxicity and promoting tumour progression (27). Furthermore, altered lysosomal positioning and dysfunction in nutrient sensing can exacerbate metabolic imbalances in cancer cells, enhancing their resistance to therapies such as chemotherapy and radiation (8). The central role of lysosomes in maintaining nutrient homeostasis and regulating cellular metabolism underscores their potential as therapeutic targets in cancer.
Autophagy is a process that maintains cellular homeostasis. There are three main types of autophagy: macroautophagy, microautophagy, and chaperone-dependent autophagy. Microautophagy is a non-selective process in that fragments of the cytoplasm are absorbed by lysosomes by invagination of the lysosome membrane. This is important to eliminate intracellular pathogens when they enter the cells via phagocytosis. This is degraded upon phagosome fusion with a lysosome to form a phagolysosome (28). However, the degradation pathway may fail, and pathogens can escape the phagosome and enter the cytoplasm. In this condition, the autophagic machinery is activated, and the autophagosome engulfs the pathogen and fuses with the lysosome for degradation (29). Chaperone-dependent autophagy is a highly selective process where proteins are marked and transported to autophagosomes for degradation. The protein chaperones including p62 and HSP70, deliver these proteins to the autolysosome through translocation complexes in the membrane (30). Macroautophagy is the most common type of autophagy. It is a multi-step process controlled by proteins from the ATG family. A double membrane (phagophore) is formed around cytoplasmic material and then through recruitment of ATG family members including LC3 for an autophagosome. The generation of the autophagosome then fuses with lysosomes to form an autolysosome that degrades the cytoplasmic material (31). The cytoplasmic material could be macromolecules such as proteins and lipids but could also contain organelles. One organelle example is damaged mitochondria by being engulfed in autophagosomes and degraded in autolysosomes. This process is termed mitophagy (32). This process is repeated, forming an autophagy flux to maintain cellular homeostasis to ensure cell survival under stressful conditions such as starvation and oxidative stress. If autophagy is prolonged or the number of autolysosomes increases too fast, cells will undergo cell death, termed autophagy-mediated cell death. The balance will determine whether cellular homeostasis is beneficial to the organism. Cancer cells exploit autophagy to degrade and recycle cellular components, including damaged organelles like mitochondria (mitophagy), to maintain energy production and promote survival (33). This adaptation is particularly important in hematological malignancies such as leukemia, where rapidly proliferating cells face metabolic stress.
The final step in phagocytosis requires fusion between the phagosome and the lysosomes. Phagocytes such as neutrophils, release the lysosomal enzyme elastase, which has a proteolytic activity that can degrade bacterial components and endogenous elements such as matrix proteins. Elastase is released in response to inflammatory signals such as cytokine TNF and lipidic mediators such as leukotriene B4. The endosomal receptor TLR9 can also be activated by mitochondrial DNA following mitophagy, leading to SNARE protein recycling and supporting autophagic flux. Antigen-presenting cells (APC), endocytosis and phagocytosis are linked to antigen processing on major histocompatibility class II molecules (MHC-II) (34). Antigens are taken up by phagocytosis or, in the case of B cells, through surface receptor-triggered endocytosis and can be processed on MHC-II molecules and MHC-1 (35). Lysosomal exocytosis is also important in immune cells. Lysosomal exocytosis also occurs at the B cell synapse formed with APCs, such as follicular dendritic cells that present native particulate antigens at their surface (36). Autophagy regulates inflammation by limiting the production of ROS and the activity of inflammasome. Dysregulated lysosomal function in leukemia cells can impair phagocytic activity and antigen processing, weakening immune surveillance and promoting chronic inflammation (37).
mTOR (mechanistic target of rapamycin) signaling pathway is critical for maintaining the balance between Hematopoietic Stem and Progenitor Cells (HSPC) quiescence and activation (38). In their quiescent state, HSCs predominantly rely on glycolysis for energy production rather than mitochondrial oxidative phosphorylation (OXPHOS). Lysosomal activity influences this metabolic preference, thereby preserving HSC quiescence. Lysosomes participate in the degradation of ferritin, a process known as ferritinophagy, to release iron (39). Iron is essential for erythropoiesis (red blood cell production). Autophagy modulates the differentiation of HSPCs into various blood cell lineages, such as erythrocytes, granulocytes, and lymphocytes. For example: In erythropoiesis, autophagy helps degrade mitochondria (mitophagy) during the maturation of red blood cells. In lymphopoiesis, autophagy supports T and B cell development by regulating organelle turnover and metabolic reprogramming. During hypoxia or nutrient deprivation, autophagy protects HSPCs by reducing oxidative stress and maintaining cellular homeostasis (40).
Autophagy-related genes, such as ATG5, ATG7, and Fip200, are essential regulators of hematopoiesis. ATG5 and ATG7 are crucial for autophagy initiation and support HSPC quiescence and survival, particularly under stress conditions (41–43).Fip200, involved in autophagosome formation, is key to HSPC differentiation into various blood cell lineages, including erythrocytes, granulocytes, and lymphocytes (44). Disruption of these genes impairs hematopoietic differentiation and increases susceptibility to hematologic diseases, underscoring their vital role in maintaining hematopoietic homeostasis. For instance, mice lacking functional autophagy genes, including FIP200, Atg7, and Atg12, experience a severe loss of hematopoietic stem cells (HSCs), leading to conditions like anemia and leukemia (45). These findings underscore the vital role of autophagy in maintaining hematopoietic homeostasis.
Cells have a strategic alternative to lysosomal degradation to dispose of the waste through either an autophagic machinery-dependent manner, the process is defined as secretory autophagy, lysosomal-exocytosis-or lysosome derived ectosomes. Autophagy-related secretory pathways called LC3-dependent EV loading and secretion (LDELS) capture proteins at late endosomes. It facilitates their secretion outside the cell (46). Lysosomal exocytosis is the fusion of mature lysosomes to the plasma membrane allowing lysosomal contents to be released into the extracellular space. Lysosomal ectosomes are formed when lysosomes fuse with the plasma membrane forming an outward budding microvesicle. The lysosome ectosomes are then released into the extracellular space. These lysosomes’ secretory pathways release their enzymatic contents into the extracellular space and influence the cellular microenvironment. For example, lysosomal enzymes degrade extracellular matrix components, facilitating cell migration and dissemination. Furthermore, the pro-inflammatory cytokine lacking signal peptide IL1β is delivered to the extracellular space by autophagy-based UPS mechanism and stimulation of autophagy leads to inflammasome-dependent IL1β secretion (47, 48). In leukemia, secretory autophagy and lysosomal exocytosis contribute to the dissemination of malignant cells by releasing enzymes that degrade the extracellular matrix, facilitating tumor invasion and metastasis. Additionally, the secretion of pro-inflammatory cytokines like IL1β through these lysosomal pathways can promote chronic inflammation in the tumor microenvironment, supporting leukemia progression and immune evasion (49, 50).
When lysosomes are ruptured, lytic content is released into the cytoplasm such as hydrolases and cathepsins through a mechanism called lysosomal membrane permeabilization (LMP). This triggers cell death often coined lysosome-mediated cell death (51). Lysosomal membrane damage can be induced by free radicals, lysosomotropic agents, endogenous pore-forming proteins, and accumulation of sphingomyelin or protein aggregates (52). LMP induces apoptosis mediated by cathepsins. Cathepsins cleave pro-apoptotic member Bid into an active truncated form (tBid) that translocates to the mitochondria leading to pro-apoptotic Bcl-2 family members BAX and BAK oligomerization (53). This leads to cytochrome c release, activation of caspases and cell death. In addition, cathepsins degrade anti-apoptotic proteins Bcl-2, Bcl-xl and Mcl-1 promoting apoptosis. Besides cathepsins, reactive oxygen species (ROS) are generated following LMP due to the release of acidic contents in lysosomes (54). This leads to mitochondrial membrane permeabilization, caspase activation, and apoptosis. Besides apoptosis, LMP leads to necrosis-like cell death through the proteolytic disintegration of cellular organelles (55). Pyroptosis is an inflammatory regulated form of cell death where the inflammatory protein complex (inflammasome) activates pro-caspase 1 and induces an inflammatory response through the IL1β and IL18 cleavage. LMP induced by ROS or protein aggregates releases cathepsins and induces the inflammasome leading to pyroptosis (56). Lysosomes contain reactive iron that upon LMP induces lipid peroxidation leading to iron-mediated cell death (ferroptosis) (57). Autophagy-induced cell death can be activated when the effector mechanisms of apoptosis are inhibited, either due to the presence of caspase inhibitors or the double knockout of BAX and BAK (58, 59). Excessive engulfment of cytoplasmic material, including mitochondria, through autophagy, leads to cell death. LMP participates through further induction of autophagy flux through ROS, and accumulation of cholesterol and ceramide in lysosomes causing membrane disruption and release of lysosomal contents (60). When lysosome rupture exceeds lysosome biogenesis, it can also inhibit autophagy flux and disrupt autophagy survival function (61). The regulation between the survival and cell death function of autophagy still needs to be elucidated. In the context of leukemia, LMP-induced cell death could be integrated into existing treatment strategies by using agents that promote lysosomal destabilization, thereby sensitizing leukemia cells to chemotherapy or other therapies. These agents, such as lysosomotropic drugs or compounds that induce ROS production, could enhance the cytotoxic effects of standard treatments by amplifying LMP, accelerating tumor cell death, and overcoming treatment resistance (62).
A cancer cell contains more active lysosomes than a healthy cell and demonstrates high levels of mTORC1 signaling, catabolic reactions and autophagy which aid in cancer cell survival, metabolism and proliferation (63). Lysosomal neuraminidase (NEU1) enhances lysosomal exocytosis and lysosomal hydrolase activity, remodeling the extracellular matrix within the tumor and invading the neighboring tissue thus promoting cancer metastasis (64). Metastatic cells are more vulnerable to lysosome-targeting drugs because lysosomes are highly diverse in size, content, location and activity compared to normal cells (65). However, this lysosomal heterogeneity also poses a challenge in treatment, as chemotherapeutic drugs often become sequestered within lysosomes, contributing to chemoresistance (66).
Autophagy plays a critical role in maintaining cellular homeostasis by degrading damaged proteins and organelles. In the context of myeloproliferative neoplasms, specific oncogenes such as JAK2V617F, a mutation commonly found in polycythemia vera, essential thrombocythemia, and myelofibrosis, may interact with autophagic pathways (67). In addition to JAK2 (Janus kinase 2), MPL plays a significant role in the pathogenesis of myeloproliferative neoplasms (MPN), particularly in essential thrombocythemia (ET) and myelofibrosis (MF) (68). MPL is a crucial oncogene signalling protein that functions as a receptor for thrombopoietin (TPO), a key regulator of platelet production (69). Mutations in MPL, particularly MPLW515L/K, have been identified in a subset of MPN cases, contributing to the dysregulation of hematopoiesis and abnormal megakaryocyte proliferation (70). These mutations lead to constitutive activation of MPL signalling pathways, promoting excessive megakaryocyte production and platelet overproduction, hallmarks of ET and MF. MPL also undergoes autophagic degradation, particularly in the context of JAK2 and CALR mutations, which are common in MPN (71). In these mutated forms, MPL interacts with the autophagic machinery, contributing to the regulation of protein homeostasis within the cell. Autophagic degradation of MPL serves as a mechanism to modulate its signalling activity, preventing excessive signalling that could lead to uncontrolled cell proliferation. Dysregulation of this autophagic process may contribute to the pathogenesis of MPN by allowing for sustained MPL signalling, leading to the expansion of abnormal hematopoietic progenitors (72). Thus, the interplay between MPL, JAK2 and autophagy highlights the complex role of autophagic pathways in regulating oncogene signaling in MPNs. Mutations or balance, dysfunction that disrupt this balance may promote hematopoietic malignancy. Understanding the role of autophagic degradation involving lysosomes in these neoplasms could provide new insights into potential treatment strategies (Figure 2).
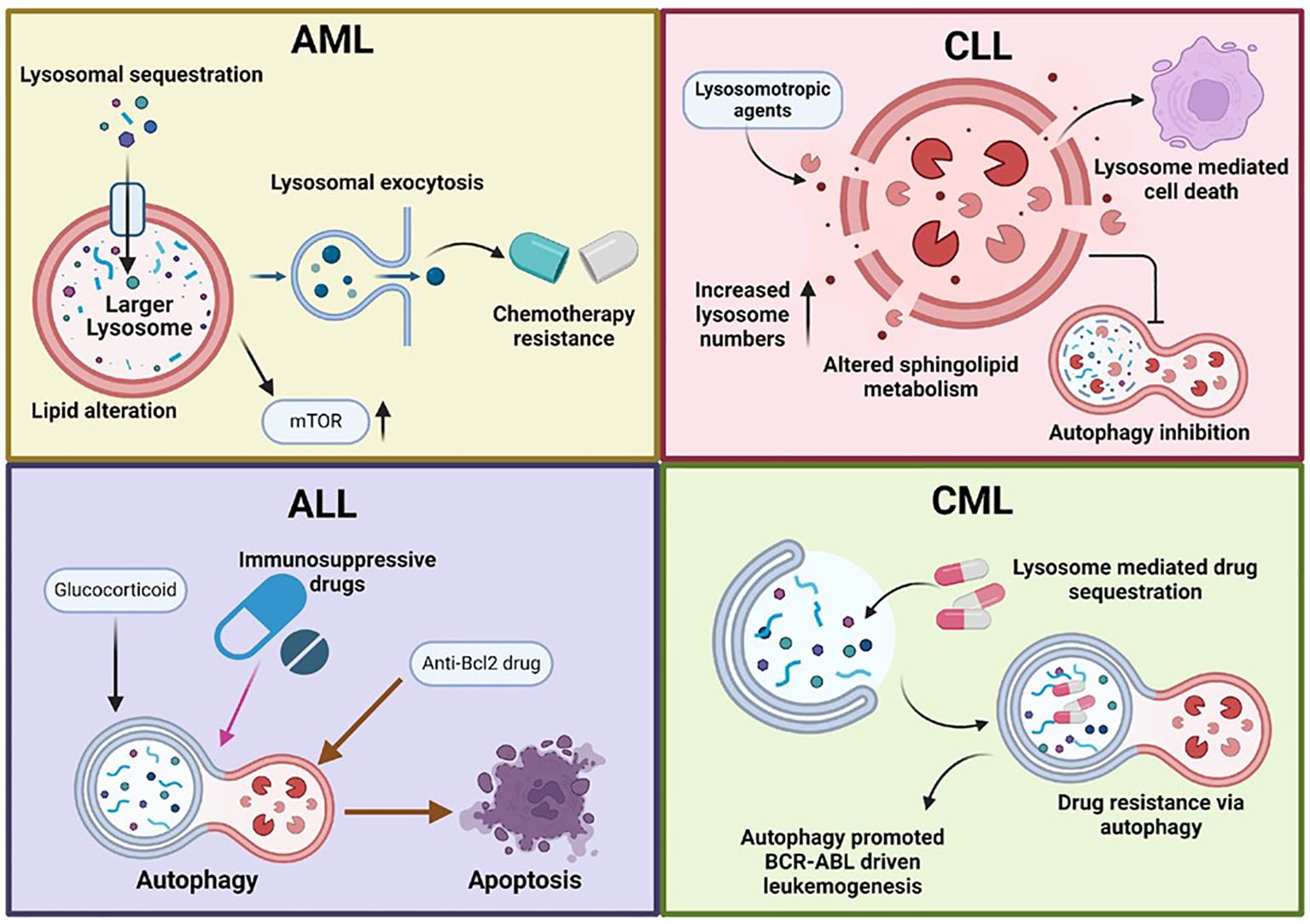
Figure 2. Lysosomal dysfunction in hematological malignancies. This figure illustrates the role of dysfunctional lysosomes in various hematological malignancies, including acute myeloid leukemia (AML), acute lymphocytic leukemia (ALL), chronic myeloid leukemia (CML), and chronic lymphocytic leukemia (CLL). Lysosomal dysfunction in cancer cells is characterized by increased lysosomal biogenesis, larger lysosomes, enhanced lysosomal activity, and altered autophagy pathways. These abnormalities promote cancer cell survival, proliferation, and drug resistance. AML cells often have larger, more active lysosomes that enhance autophagy, promoting survival and proliferation. These include drug sequestration, altered acidification, mTORC1 signalling, and changes in lipid profiles. Lysosomal exocytosis also aids in extracellular remodelling, supporting tumour invasion and metastasis. In ALL, autophagy is reduced in BCR-ABL+ cells. Immunosuppressive and glucocorticoid drugs induce autophagy, while mTORC1 inhibition can trigger autophagy-mediated cell death. Anti-Bcl-2 drugs also cause autophagy-dependent cell death through the ATG5 pathway. In chronic lymphocytic leukemia (CLL), lysosomes play a key role in BCR signaling and cell death. There is altered sphingolipid metabolism, increased lysosome numbers, and selective disruption by lysosomotropic agents. Additionally, autophagy is inhibited in CLL. In CML, autophagy contributes to drug resistance and promotes BCR-ABL-driven leukemogenesis. Additionally, lysosomes mediate drug sequestration, further aiding in resistance mechanisms.
Most studies have focused on solid tumors, but lysosomes contribute to the development of various hallmarks of hematological malignancies, such as sustained proliferative signaling (mTORC1 signaling), metabolism (catabolic reactions, autophagy) and invasion (lysosomal exocytosis) (73). Furthermore, lysosome-targeted therapies have shown promise in hematological malignancies, particularly leukemia, where they induce cytotoxic effects on leukemia cells (Table 1).
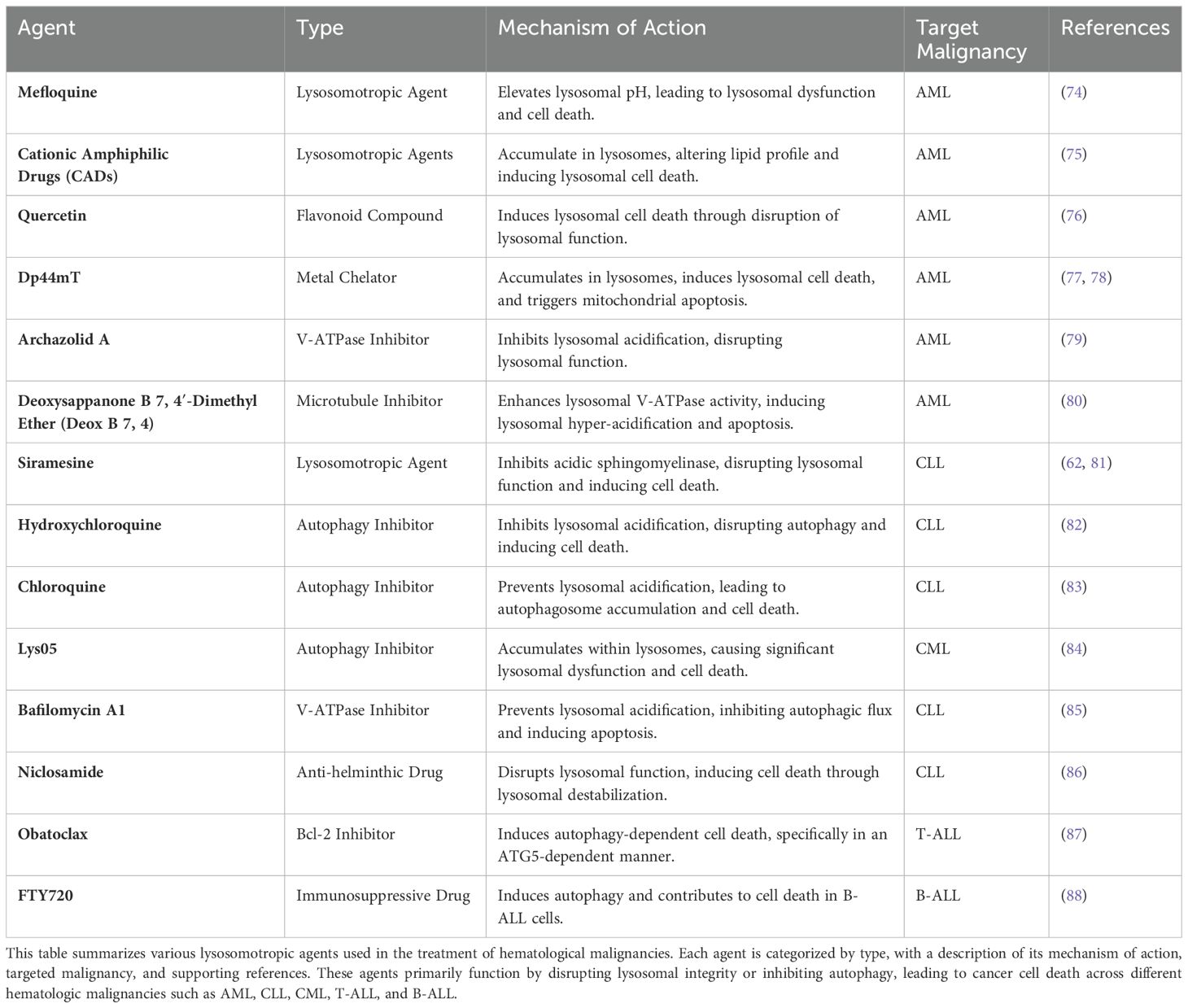
Table 1. Lysosomotropic agents and their mechanisms of action in hematological malignancies.
Acute Myeloid Leukemia (AML) is a hematological malignancy characterized by an excessive accumulation of immature blood-forming cells in the bone marrow, impairing the formation of normal blood cells (89). Treatment is tailored to cytogenetic risk, consisting of induction chemotherapy with cytarabine and anthracycline plus gemtuzumab-ozogamicin (IgG4-kappa monoclonal antibody linked to a cytotoxic drug) or FLT3-inhibitor midostaurin in young fit adult patients and HDAC inhibitor azacitidine with or without BCL2-inhibitor venetoclax in older or unfit adults (90). Consolidation therapy occurs after initial remission and usually involves additional rounds of treatment. Finally, allogeneic stem cell transplantations are performed if needed for certain subtypes when there is a suitable donor. A subtype of AML is acute promyelocytic leukemia (APL) and is treated with all-trans retinoic acid (ATRA) and either arsenic trioxide or anthracyclines (91).
Using gene clusters, it was demonstrated that AML cells with higher lysosome-related genes predict the prognosis of AML cells and response to chemotherapy. AML cells also show larger lysosomes and increased expression of lysosome biogenesis genes indicating higher lysosome activity (92). Indeed, increased lysosome activity supports the survival of senescent AML cells. Furthermore, the expression of transient receptor potential mucolipin (TRPML1) found in lysosomes is upregulated in AML cells (93). In contrast, lysosome biogenesis transcription factor TFEB acts as a tumor suppressor that induces differentiation and cell death in normal and malignant myeloid progenitor cells, thus controlling myelopoiesis (94). This has been linked to its role in MYC signaling and epigenetic controls and not to its function in lysosomal biosynthesis. This indicates that AML cells have altered lysosomes to elevated biomass and bioenergetic demands.
Besides altered lysosomal biosynthesis, autophagy plays an important role in AML progression. Several common chromosomal deletions in AML include regions of autophagy genes. Depletion of autophagy gene ATG5 led to increased proliferation and more aggressive leukemia in the MLL-ENL-induced murine AML model. Deletion of ATG7 in OCI-AML3 cells also increased chemotherapy resistance in xenograft mice (95). In addition, inhibition of autophagy, followed by deletion of ATG5 or ATG7, reduces leukemia-initiating cells (LICs) and increases mitochondrial ROS (96). Inhibition of autophagy also promotes the survival and proliferation of AML cells (97). Mutations in SQSTM1/p62 are believed to impact mitophagy and myeloid leukemia development (98). In FLT3-ITD-driven AML, mTORC1 activation suppresses autophagy and prevents the autolysosome degradation of the FLT3 protein. In contrast, FLT3-ITD upregulates transcription factor ATF4 increasing basal autophagy in AML cells and inhibiting autophagy increasing survival of the FLT3-ITD-driven AML mice (99). This illustrates the multiple role autophagy plays in AML progression.
Targeting lysosomes is one of many strategies to combat chemoresistance. Lysosomotropic agents are chemicals that accumulate in the lysosomal lumen and elevate lysosomal pH, leading to lysosomal dysfunction and membrane permeabilization (LMP). Mefloquine, a lysosome-damaging agent, releases lysosomal CTSB and CTSL into the cytosol, inducing cell death in AML cells (74). Similarly, cationic-amphiphilic antihistamines target leukemia cells in patients (100). Archazolid A, a V-ATPase inhibitor, has shown anti-leukemic effects by suppressing lysosomal acidification (79, 80). Conversely, the microtubule inhibitor deoxysappanone B 7, 4′-dimethyl ether (Deox B 7, 4) demonstrates anti-leukemic activity by enhancing lysosomal V-ATPase activity, resulting in hyper-acidification of lysosomes and inducing apoptosis in AML cells (80). Cationic amphiphilic drugs (CAD) are another category of small molecules that accumulate in lysosomes, altering the lipid profile in the lysosomal lumen and inducing lysosomal cell death in multiple AML cell lines (75). The flavonoid quercetin, a polyphenol compound, induces lysosomal cell death in leukemia cells (76). The metal chelator Dp44mT, which accumulates in lysosomes, induces lysosomal cell death (77). Dp44mT also triggers the release of CTSD from lysosomes into the cytosol, initiating mitochondrial cytochrome-c-dependent apoptosis (78, 101). In summary, lysosomal acidification plays a significant role in AML and targeted therapeutic approaches can be selected based on the status of lysosomal acidification (102).
Acute lymphocytic leukemia (ALL) belongs to a highly heterogeneous group of hematological malignancies from the lymphoid lineage. ALL is characterized by uncontrolled proliferation of clonal neoplastic cells in the bone marrow leading to bone marrow failure and death if untreated. B cell-derived ALL (B-ALL) represents 80% of all ALL cases (103). It affects mostly children but can occur in adults. T cell-derived ALL (T-ALL) accounts for 10-15% of all ALL cases and is characterized by uncontrolled proliferation of lymphoblast arising from the thymus (104). Intensive combination chemotherapy has improved survival, particularly in children, but drug resistance still occurs (105). Newer agents include bispecific T cell engager (CD19-CD3) blinatumomab and antibody (anti-CD22) - drug conjugate inotuzumab. Allogeneic stem cell transplantation or CAR-T cell therapy is used in high-risk and refractory cases.
The role of lysosomes in ALL has focused on autophagy in the survival and death of leukemia cells. In B-ALL, cells with the p185 form of BCR-ABL show reduced basal levels of autophagy, but when autophagy was blocked by the knockdown of ATG3, cells underwent apoptosis. The immunosuppressive drug FTY720 induced cell death in B-ALL cells in a caspase-independent manner but also induced autophagy, contributing to cell survival (88). In contrast, Glucocorticoids (GC) have been shown to induce cell death through the activation of the autophagic machinery in B-ALL cell lines and primary cells (106, 107). Using a mTORC1 inhibitor everolimus, autophagy was increased in B-ALL cell lines and primary samples. Knocking down Beclin-1 resulted in less everolimus cytotoxicity suggesting autophagy provided a cell death response. Nevertheless, using an autophagy inhibitor, 3-methyladenine failed to block everolimus-induced cell death in B-ALL cells. Obatoclax induces autophagy-dependent cell death in ALL, specifically in an ATG5-dependent but Beclin-1-independent manner (87). Taken together, the context of autophagy activation in B-ALL cells will determine whether autophagy contributes to cell survival or death.
In T-ALL cells, inhibition of either PI3K/mTOR or Akt pathways has been shown to induce autophagy. Treatment of T-ALL cells with AKT inhibitors demonstrated that autophagy was protective as shown by knockdown of Beclin or using autophagy inhibitor chloroquine. Autophagy may also play an active role in the cell death of T-ALL cells, as evidenced by a study where Jurkat cells were treated with selenite, a drug known for its anti-tumor efficacy, exerting both pro-apoptotic and pro-autophagic effects (102). Metformin (LKB/AMPK inhibitor) induced autophagy contributed to cell death in T-ALL cells. Using the anti-Bcl-2 drug obatoclax in GC-resistant T-ALL cell lines showed increased autophagy mediated by dissociation of Beclin-1 from Bcl-2 family member Mcl-1 and reduced mTORC1 activity (108). This led to an autophagy-dependent necroptosis in these cells. Similar to B-ALL, the context of autophagy activation will determine whether it contributes to cell survival or death.
Chronic myeloid leukemia (CML) is a myeloid-derived leukemia characterized by the expression of the BCR: ABL fusion oncoprotein. This constitutively active tyrosine kinase leads to uncontrolled proliferation and growth. Using ABL tyrosine kinase inhibitors (TKI) such as imatinib and newer generation TKIs has proven to be an effective therapy however drug resistance remains a problem. The lysosome sequestration of hydrophobic weak base drugs such as imatinib could be the mechanism of drug resistance but no imatinib-resistant CML cell line has shown lysosomal mediated resistance (109). Autophagy is another lysosome-regulated mechanism that could contribute to drug resistance. Autophagy is induced in both CML cell lines and Leukemic Stem Cells (LSCs) following in vitro treatment with imatinib, suggesting that BCR-ABL acts as a negative regulator of autophagy (110). However, other studies indicate that the expression of this oncogene promotes autophagosome formation and that autophagy is essential for BCR-ABL-dependent leukemogenesis. Combining mTOR and autophagy inhibition has proven effective in targeting TKI-resistant CML cells. In a CML Patient-Derived Xenograft (PDX) model, treatment with talazoparib in combination with autophagy inhibitor chloroquine significantly enhanced the anti-tumour effect of talazoparib (111). Talazoparib markedly triggered autophagy in CML cells, as confirmed by the accumulation of autophagosomes, decreased SQSTM1 levels, and upregulation of LC3-II (111, 112). This suggests autophagy inhibition might be an effective treatment to overcome TKI drug resistance in CML.
Chronic lymphocytic leukemia (CLL) is the most common type of leukemia in the Western world. It is characterized by the accumulation of CD5+ and CD19+ monoclonal B cells. Standard therapies currently utilize a second-generation BTK tyrosine kinase inhibitor such as acalabrutinib or an anti-Bcl-2 mimetic drug, venetoclax alone or in combination with anti-CD20 antibodies. Unfortunately, drug resistance remains a problem. Lysosome-mediated cell death presents a compelling therapeutic avenue for CLL. In CLL cells, lysosomes are involved in regulating the BCR signaling pathway by targeting the TOSO: IgM complex, which is critical for cell survival and proliferation (113). Lysosome numbers are also increased in CLL cells compared to normal B cells. In addition, CLL cells have altered sphingolipid metabolism, leading to increased levels of sphingosine. Sphingosine leads to lysosomal membrane instability. CLL cells also have higher basal levels of autophagy, contributing to cell survival.
Lysosomes are involved in the effectiveness of standard treatment for CLL. We have shown that Venetoclax can act as an autophagy inhibitor by suppressing the expression of ATG12 and in combination with starvation or ibrutinib treatment increased cell death. Rituximab, a type I anti-CD20 monoclonal antibody, targets lysosomes for FcγRIIB- promoted internalization of CD20: anti-CD20 complexes, reducing CD20 expression on the cell surface and contributing to rituximab resistance (114). Conversely, GA101, a type II anti-CD20 monoclonal antibody induces lysosome-dependent cell death through lysosome disruption (115). This indicates that lysosomes play a role in CLL treatments.
Due to the alteration in lysosomes, several lysosomotropic agents have been used to treat CLL cells. Siramesine is a lysosomotropic agent shown to induce cell death in several different types of cancer including breast and brain cancers through its inhibition of the acidic sphingomyelinase (ASMase). In CLL cells, siramesine was shown to selectively target lysosome disruption compared to normal B cells and was effective in combination with venetoclax. Besides siramesine, other lysosomotropic agents such as antihistamines and antimalarial drugs induce lysosome-mediated cell death in CLL cells. Furthermore, these drugs synergize with ibrutinib to increase apoptosis. This indicates that lysosomotropic agents could be effective treatments for CLL.
Several drugs targeting the autophagy pathway through inhibiting autolysosomes induce cell death in CLL cells. Hydroxychloroquine, traditionally used for malaria and autoimmune diseases, inhibits lysosomal acidification, disrupting autophagy and promoting CLL cells (116). Similarly, chloroquine prevents lysosomal acidification, leading to the accumulation of autophagosomes and cell death in CLL cells (83). Lys05, a more potent derivative of chloroquine, accumulates within lysosomes, causing significant lysosomal dysfunction and resulting in the death of CLL cells (84). Bafilomycin A1, a V-ATPase inhibitor, prevents lysosomal acidification, leading to the inhibition of autophagic flux and apoptosis in CLL cells (85). Niclosamide, an anti-helminthic drug, disrupts lysosomal function and autophagy, inducing cell death in leukemia cells through mechanisms involving lysosomal destabilization (86). These studies suggest that targeting lysosome function in autophagy might render CLL cells sensitive to cell death.
Dysfunctional lysosomes play a critical role in the pathogenesis and treatment resistance of various leukemias, including AML, ALL, CML, and CLL (Figure 2). Understanding these mechanisms offers valuable insights into developing targeted therapies that can enhance treatment efficacy and improve patient outcomes in these hematological malignancies. Ongoing research into lysosomal functions and their regulation holds promise for identifying novel therapeutic strategies to combat leukemia and lymphoma (Table 1).
Several lysosomal drugs are currently in clinical trials for the treatment of leukemia, targeting lysosomal pathways to induce apoptosis or enhance the efficacy of existing therapies. Venetoclax, a BCL-2 inhibitor, disrupts mitochondrial and lysosomal integrity and is approved for chronic lymphocytic leukemia (CLL) and acute myeloid leukemia (AML), with ongoing trials exploring its use in combination therapies. Hydroxychloroquine, a lysosomal autophagy inhibitor, is in Phase 1/2 trials for various leukemia types, often combined with Venetoclax or chemotherapy to overcome drug resistance (117). Similarly, chloroquine, another autophagy inhibitor, is in early-phase trials and shows potential to enhance the efficacy of standard therapies in AML and CLL (118). Dactinomycin, known for inducing lysosomal stress, is approved for acute lymphoblastic leukemia (ALL) and remains a part of combination regimens for pediatric cases (119). Lysosome-targeted therapies are emerging as a promising approach in the treatment of leukemia, particularly due to the critical roles lysosomes play in cell survival, apoptosis, and immune evasion. Research is ongoing to identify optimal combinations and treatment schedules to maximize the benefits of lysosome-targeted therapies. Biomarker-driven approaches may help tailor these therapies to individual patients, improving efficacy and minimizing adverse effects. By understanding the roles of lysosomal dysfunction in leukemia pathogenesis, therapeutic strategies can potentially enhance treatment outcomes, overcome resistance mechanisms, and provide a more effective means of targeting this heterogeneous group of malignancies. Clinical trials of lysosome-targeted agents alone and in combination with standard therapies will be crucial for advancing these innovative approaches and improving patient care in leukemia.
Lysosomes play important functions within cells beyond just maintaining cellular homeostasis. In hematological malignancies, lysosomes are altered to maintain increased demand for biomass and bioenergetics leading to cell survival and growth. Disrupting the lysosomal membrane integrity or modulating lysosomal enzyme activity has shown a potential to selectively induce cell death in leukemia cells. The efficacy of lysosome-mediated cell death in pre-clinical studies suggests that it could be a valuable addition to the existing therapies, to overcoming resistance and improving patient outcomes. Further clinical trials are necessary to fully understand the safety and effectiveness of these interventions in the context of hematological malignancies.
MM: Writing – original draft. AP: Writing – review & editing. SG: Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. de Duve C, Pressman BC, Gianetto R, Wattiaux R, Appelmans F. Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem J. (1955) 60:604–17. doi: 10.1042/bj0600604
2. Ballabio A, Bonifacino JS. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat Rev Mol Cell Biol. (2020) 21:101–18. doi: 10.1038/s41580-019-0185-4
3. Meng Y, Heybrock S, Neculai D, Saftig P. Cholesterol handling in lysosomes and beyond. Trends Cell Biol. (2020) 30:452–66. doi: 10.1016/j.tcb.2020.02.007
4. Lum JJ, DeBerardinis RJ, Thompson CB. Autophagy in metazoans: cell survival in the land of plenty. Nat Rev Mol Cell Biol. (2005) 6:439–48. doi: 10.1038/nrm1660
5. Cao M, Luo X, Wu K, He X. Targeting lysosomes in human disease: from basic research to clinical applications. Signal Transduct Target Ther. (2021) 6:379. doi: 10.1038/s41392-021-00778-y
6. You L, Jin S, Zhu L, Qian W. Autophagy, autophagy-associated adaptive immune responses and its role in hematologic Malignancies. Oncotarget. (2017) 8:12374–88. doi: 10.18632/oncotarget.13583
7. Kumar S, Sánchez-Álvarez M, Lolo FN, Trionfetti F, Strippoli R, Cordani M. Autophagy and the lysosomal system in cancer. Cells. (2021) 10:2752. doi: 10.3390/cells10102752
8. MaChado ER, Annunziata I, van de Vlekkert D, Grosveld GC, d’Azzo A. Lysosomes and cancer progression: A Malignant liaison. Front Cell Dev Biol. (2021) 9. doi: 10.3389/fcell.2021.642494
9. Lee MJ, Park JS, Bin J, Joe YA. Enhancing anti-cancer therapy with selective autophagy inhibitors by targeting protective autophagy. Biomol Ther (Seoul). (2023) 31:1–15. doi: 10.4062/biomolther.2022.153
10. Mindell JA. Lysosomal acidification mechanisms. Annu Rev Physiol. (2012) 74:69–86. doi: 10.1146/annurev-physiol-012110-142317
11. Li J, Pfeffer SR. Lysosomal membrane glycoproteins bind cholesterol and contribute to lysosomal cholesterol export. Elife. (2016) 5:1–15. doi: 10.7554/eLife.21635
12. Wang G, Chen L, Qin S, Zhang T, Yao J, Yi Y, et al. Mechanistic target of rapamycin complex 1: from a nutrient sensor to a key regulator of metabolism and health. Adv Nutr. (2022) 13:1882–900. doi: 10.1093/advances/nmac055
13. Napolitano G, Ballabio A. TFEB at a glance. J Cell Sci. (2016) 129:2475–81. doi: 10.1242/jcs.146365
14. Cao Q, Yang Y, Zhong XZ, Dong XP. The lysosomal Ca2+ release channel TRPML1 regulates lysosome size by activating calmodulin. J Biol Chem. (2017) 292:8424–35. doi: 10.1074/jbc.M116.772160
15. Piao S, Amaravadi RK. Targeting the lysosome in cancer. Ann N Y Acad Sci. (2016) 1371:45–54. doi: 10.1111/nyas.2016.1371.issue-1
16. Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. (2012) 31:1095–108. doi: 10.1038/emboj.2012.32
17. Ferrari V, Tedesco B, Cozzi M, Chierichetti M, Casarotto E, Pramaggiore P, et al. Lysosome quality control in health and neurodegenerative diseases. Cell Mol Biol Lett. (2024) 29:116. doi: 10.1186/s11658-024-00633-2
18. Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. (2010) 141:290–303. doi: 10.1016/j.cell.2010.02.024
19. Efeyan A, Sabatini DM. Nutrients and growth factors in mTORC1 activation. Biochem Soc Trans. (2013) 41:902–5. doi: 10.1042/BST20130063
20. Liu J, Wu Y, Meng S, Xu P, Li S, Li Y, et al. Selective autophagy in cancer: mechanisms, therapeutic implications, and future perspectives. Mol Cancer. (2024) 23:22. doi: 10.1186/s12943-024-01934-y
21. Takahara T, Amemiya Y, Sugiyama R, Maki M, Shibata H. Amino acid-dependent control of mTORC1 signaling: a variety of regulatory modes. J BioMed Sci. (2020) 27:87. doi: 10.1186/s12929-020-00679-2
22. Korolchuk VI, Saiki S, Lichtenberg M, Siddiqi FH, Roberts EA, Imarisio S, et al. Lysosomal positioning coordinates cellular nutrient responses. Nat Cell Biol. (2011) 13:453–60. doi: 10.1038/ncb2204
23. Anderson RA, Rao N, Byrum RS, Rothschild CB, Bowden DW, Hayworth R, et al. In situ localization of the genetic locus encoding the lysosomal acid lipase/cholesteryl esterase (LIPA) deficient in Wolman disease to chromosome 10q23.2-q23.3. Genomics. (1993) 15:245–7. doi: 10.1006/geno.1993.1052
24. van der Ploeg AT, Reuser AJJ. Pompe’s disease. Lancet. (2008) 372:1342–53. doi: 10.1016/S0140-6736(08)61555-X
25. Lim SM, Mohamad Hanif EA, Chin SF. Is targeting autophagy mechanism in cancer a good approach? The possible double-edge sword effect. Cell Biosci. (2021) 11:56. doi: 10.1186/s13578-021-00570-z
26. Rabanal-Ruiz Y, Otten EG, Korolchuk VI. mTORC1 as the main gateway to autophagy. Essays Biochem. (2017) 61:565–84. doi: 10.1042/EBC20170027
27. Tang T, Yang ZY, Wang D, Yang XY, Wang J, Li L, et al. The role of lysosomes in cancer development and progression. Cell Biosci. (2020) 10:131. doi: 10.1186/s13578-020-00489-x
28. Wong CO, Gregory S, Hu H, Chao Y, Sepúlveda VE, He Y, et al. Lysosomal degradation is required for sustained phagocytosis of bacteria by macrophages. Cell Host Microbe. (2017) 21:719–730.e6. doi: 10.1016/j.chom.2017.05.002
29. Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. (2013) 13:722–37. doi: 10.1038/nri3532
30. Tedesco B, Vendredy L, Timmerman V, Poletti A. The chaperone-assisted selective autophagy complex dynamics and dysfunctions. Autophagy. (2023) 19:1619–41. doi: 10.1080/15548627.2022.2160564
31. Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res. (2014) 24:24–41. doi: 10.1038/cr.2013.168
32. Saito T, Sadoshima J. Molecular mechanisms of mitochondrial autophagy/mitophagy in the heart. Circ Res. (2015) 116:1477–90. doi: 10.1161/CIRCRESAHA.116.303790
33. White E, Mehnert JM, Chan CS. Autophagy, metabolism, and cancer. Clin Cancer Res. (2015) 21:5037–46. doi: 10.1158/1078-0432.CCR-15-0490
34. Stern LJ, Potolicchio I, Santambrogio L. MHC class II compartment subtypes: structure and function. Curr Opin Immunol. (2006) 18:64–9. doi: 10.1016/j.coi.2005.11.005
35. Colbert JD, Cruz FM, Rock KL. Cross-presentation of exogenous antigens on MHC I molecules. Curr Opin Immunol. (2020) 64:1–8. doi: 10.1016/j.coi.2019.12.005
36. Yuseff MI, Reversat A, Lankar D, Diaz J, Fanget I, Pierobon P, et al. Polarized secretion of lysosomes at the B cell synapse couples antigen extraction to processing and presentation. Immunity. (2011) 35:361–74. doi: 10.1016/j.immuni.2011.07.008
37. Xu Y, Shao B, Zhang Y. The significance of targeting lysosomes in cancer immunotherapy. Front Immunol. (2024) 15. doi: 10.3389/fimmu.2024.1308070
38. Gan B, DePinho R. mTORC1 signaling governs hematopoietic stem cell quiescence. Cell Cycle. (2009) 8:1003–6. doi: 10.4161/cc.8.7.8045
39. Wang J, Wu N, Peng M, Oyang L, Jiang X, Peng Q, et al. Ferritinophagy: research advance and clinical significance in cancers. Cell Death Discovery. (2023) 9:463. doi: 10.1038/s41420-023-01753-y
40. Arif T. Lysosomes and their role in regulating the metabolism of hematopoietic stem cells. Biol (Basel). (2022) 11:1410. doi: 10.3390/biology11101410
41. Gomez-Puerto MC, Folkerts H, Wierenga ATJ, Schepers K, Schuringa JJ, Coffer PJ, et al. Autophagy proteins ATG5 and ATG7 are essential for the maintenance of human CD34+ Hematopoietic stem-progenitor cells. Stem Cells. (2016) 34:1651–63. doi: 10.1002/stem.2347
42. Mortensen M, Soilleux EJ, Djordjevic G, Tripp R, Lutteropp M, Sadighi-Akha E, et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J Exp Med. (2011) 208:455–67. doi: 10.1084/jem.20101145
43. Liu Y, Shi L, Chen Y, Luo S, Chen Y, Chen H, et al. Autophagy regulates the maturation of hematopoietic precursors in the embryo. Nat Commun. (2024) 15:2255. doi: 10.1038/s41467-024-46453-y
44. Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, et al. ULK-atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. (2009) 20:1992–2003. doi: 10.1091/mbc.e08-12-1249
45. Montazersaheb S, Ehsani A, Fathi E, Farahzadi R, Vietor I. An overview of autophagy in hematopoietic stem cell transplantation. Front Bioeng Biotechnol. (2022) 10. doi: 10.3389/fbioe.2022.849768
46. Gardner JO, Leidal AM, Nguyen TA, Debnath J. LC3-dependent EV loading and secretion (LDELS) promotes TFRC (transferrin receptor) secretion via extracellular vesicles. Autophagy. (2023) 19:1551–61. doi: 10.1080/15548627.2022.2140557
47. Claude-Taupin A, Bissa B, Jia J, Gu Y, Deretic V. Role of autophagy in IL-1β export and release from cells. Semin Cell Dev Biol. (2018) 83:36–41. doi: 10.1016/j.semcdb.2018.03.012
48. Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J. (2011) 30:4701–11. doi: 10.1038/emboj.2011.398
49. Zhang Z, Yue P, Lu T, Wang Y, Wei Y, Wei X. Role of lysosomes in physiological activities, diseases, and therapy. J Hematol Oncol. (2021) 14:79. doi: 10.1186/s13045-021-01087-1
50. Rafiq S, McKenna SL, Muller S, Tschan MP, Humbert M. Lysosomes in acute myeloid leukemia: potential therapeutic targets? Leukemia. (2021) 35:2759–70. doi: 10.1038/s41375-021-01388-x
51. Repnik U, Hafner Česen M, Turk B. Lysosomal membrane permeabilization in cell death: Concepts and challenges. Mitochondrion. (2014) 19:49–57. doi: 10.1016/j.mito.2014.06.006
52. Wang F, Gómez-Sintes R, Boya P. Lysosomal membrane permeabilization and cell death. Traffic. (2018) 19:918–31. doi: 10.1111/tra.2018.19.issue-12
53. Żołnierczyk JD, Kiliańska ZM. Main pro-apoptotic member of Bcl-2 family proteins – Bax. Folia Biologica Oecologica. (2010) 6:5–32. doi: 10.2478/v10107-009-0004-3
54. Denamur S, Tyteca D, Marchand-Brynaert J, Van Bambeke F, Tulkens PM, Courtoy PJ, et al. Role of oxidative stress in lysosomal membrane permeabilization and apoptosis induced by gentamicin, an aminoglycoside antibiotic. Free Radic Biol Med. (2011) 51:1656–65. doi: 10.1016/j.freeradbiomed.2011.07.015
55. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. (2018) 25:486–541. doi: 10.1038/s41418-017-0012-4
56. Campden RI, Zhang Y. The role of lysosomal cysteine cathepsins in NLRP3 inflammasome activation. Arch Biochem Biophys. (2019) 670:32–42. doi: 10.1016/j.abb.2019.02.015
57. Villalpando-Rodriguez GE, Blankstein AR, Konzelman C, Gibson SB. Lysosomal destabilizing drug siramesine and the dual tyrosine kinase inhibitor lapatinib induce a synergistic ferroptosis through reduced heme oxygenase-1 (HO-1) levels. Oxid Med Cell Longev. (2019) 2019:1–14. doi: 10.1155/2019/9561281
58. Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, et al. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science. (2004) 304:1500–2. doi: 10.1126/science.1096645
59. Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, et al. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol. (2004) 6:1221–8. doi: 10.1038/ncb1192
60. Liu S, Yao S, Yang H, Liu S, Wang Y. Autophagy: Regulator of cell death. Cell Death Dis. (2023) 14:648. doi: 10.1038/s41419-023-06154-8
61. Das G, Shravage BV, Baehrecke EH. Regulation and function of autophagy during cell survival and cell death. Cold Spring Harb Perspect Biol. (2012) 4:a008813–a008813. doi: 10.1101/cshperspect.a008813
62. Manivannan MS, Yang X, Patel N, Peters A, Johnston JB, Gibson SB. Lysosome-disrupting agents in combination with venetoclax increase apoptotic response in primary chronic lymphocytic leukemia (CLL) cells mediated by lysosomal cathepsin D release and inhibition of autophagy. Cells. (2024) 13:1041. doi: 10.3390/cells13121041
63. Davidson SM, Vander Heiden MG. Critical functions of the lysosome in cancer biology. Annu Rev Pharmacol Toxicol. (2017) 57:481–507. doi: 10.1146/annurev-pharmtox-010715-103101
64. MaChado E, White-Gilbertson S, van de Vlekkert D, Janke L, Moshiach S, Campos Y, et al. Regulated lysosomal exocytosis mediates cancer progression. Sci Adv. (2015) 1:1–16. doi: 10.1126/sciadv.1500603
65. Morgan MJ, Fitzwalter BE, Owens CR, Powers RK, Sottnik JL, Gamez G, et al. Metastatic cells are preferentially vulnerable to lysosomal inhibition. Proc Natl Acad Sci. (2018) 115:E8476–88. doi: 10.1073/pnas.1706526115
66. Zhitomirsky B, Yunaev A, Kreiserman R, Kaplan A, Stark M, Assaraf YG. Lysosomotropic drugs activate TFEB via lysosomal membrane fluidization and consequent inhibition of mTORC1 activity. Cell Death Dis. (2018) 9:1191. doi: 10.1038/s41419-018-1227-0
67. Courdy C, Platteeuw L, Ducau C, De Araujo I, Boet E, Sahal A, et al. Targeting PP2A-dependent autophagy enhances sensitivity to ruxolitinib in JAK2V617F myeloproliferative neoplasms. Blood Cancer J. (2023) 13:106. doi: 10.1038/s41408-023-00875-x
68. Kang MG, Choi HW, Lee JH, Choi YJ, Choi HJ, Shin JH, et al. Coexistence of JAK2 and CALR mutations and their clinical implications in patients with essential thrombocythemia. Oncotarget. (2016) 7:57036–49. doi: 10.18632/oncotarget.10958
69. Hitchcock IS, Hafer M, Sangkhae V, Tucker JA. The thrombopoietin receptor: revisiting the master regulator of platelet production. Platelets. (2021) 32:770–8. doi: 10.1080/09537104.2021.1925102
70. Chaligné R, James C, Tonetti C, Besancenot R, Le Couédic JP, Fava F, et al. Evidence for MPL W515L/K mutations in hematopoietic stem cells in primitive myelofibrosis. Blood. (2007) 110:3735–43. doi: 10.1182/blood-2007-05-089003
71. Han L, Schubert C, Köhler J, Schemionek M, Isfort S, Brümmendorf TH, et al. Calreticulin-mutant proteins induce megakaryocytic signaling to transform hematopoietic cells and undergo accelerated degradation and Golgi-mediated secretion. J Hematol Oncol. (2016) 9:45. doi: 10.1186/s13045-016-0275-0
72. Yung Y, Lee E, Chu HT, Yip PK, Gill H. Targeting abnormal hematopoietic stem cells in chronic myeloid leukemia and philadelphia chromosome-negative classical myeloproliferative neoplasms. Int J Mol Sci. (2021) 22:659. doi: 10.3390/ijms22020659
73. Jeger JL. Endosomes, lysosomes, and the role of endosomal and lysosomal biogenesis in cancer development. Mol Biol Rep. (2020) 47:9801–10. doi: 10.1007/s11033-020-05993-4
74. Sukhai MA, Prabha S, Hurren R, Rutledge AC, Lee AY, Sriskanthadevan S, et al. Lysosomal disruption preferentially targets acute myeloid leukemia cells and progenitors. J Clin Invest. (2013) 123:315–28. doi: 10.1172/JCI64180
75. Nielsen IØ, Groth-Pedersen L, Dicroce-Giacobini J, Jonassen ASH, Mortensen M, Bilgin M, et al. Cationic amphiphilic drugs induce elevation in lysoglycerophospholipid levels and cell death in leukemia cells. Metabolomics. (2020) 16:91. doi: 10.1007/s11306-020-01710-1
76. Maruszewska A, Tarasiuk J. Quercetin triggers induction of apoptotic and lysosomal death of sensitive and multidrug resistant leukaemia HL60 cells. Nutr Cancer. (2021) 73:484–501. doi: 10.1080/01635581.2020.1752745
77. Lovejoy DB, Jansson PJ, Brunk UT, Wong J, Ponka P, Richardson DR. Antitumor activity of metal-chelating compound Dp44mT is mediated by formation of a redox-active copper complex that accumulates in lysosomes. Cancer Res. (2011) 71:5871–80. doi: 10.1158/0008-5472.CAN-11-1218
78. Seebacher NA, Richardson DR, Jansson PJ. A mechanism for overcoming P-glycoprotein-mediated drug resistance: novel combination therapy that releases stored doxorubicin from lysosomes via lysosomal permeabilization using Dp44mT or DpC. Cell Death Dis. (2016) 7:e2510–0. doi: 10.1038/cddis.2016.381
79. Zhang S, Schneider LS, Vick B, Grunert M, Jeremias I, Menche D, et al. Anti-leukemic effects of the V-ATPase inhibitor Archazolid A. Oncotarget. (2015) 6:43508–28. doi: 10.18632/oncotarget.v6i41
80. Bernard D, Gebbia M, Prabha S, Gronda M, MacLean N, Wang X, et al. Select microtubule inhibitors increase lysosome acidity and promote lysosomal disruption in acute myeloid leukemia (AML) cells. Apoptosis. (2015) 20:948–59. doi: 10.1007/s10495-015-1123-3
81. Dielschneider RF, Eisenstat H, Mi S, Curtis JM, Xiao W, Johnston JB, et al. Lysosomotropic agents selectively target chronic lymphocytic leukemia cells due to altered sphingolipid metabolism. Leukemia. (2016) 30:1290–300. doi: 10.1038/leu.2016.4
82. Mansilla E, Marin GH, Nuñez L, Drago H, Sturla F, Mertz C, et al. The lysosomotropic agent, hydroxychloroquine, delivered in a biodegradable nanoparticle system, overcomes drug resistance of B-chronic lymphocytic leukemia cells in vitro. Cancer Biother Radiopharm. (2010) 25:97–103. doi: 10.1089/cbr.2009.0655
83. Mauthe M, Orhon I, Rocchi C, Zhou X, Luhr M, Hijlkema KJ, et al. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy. (2018) 14:1435–55. doi: 10.1080/15548627.2018.1474314
84. McAfee Q, Zhang Z, Samanta A, Levi SM, Ma XH, Piao S, et al. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc Natl Acad Sci. (2012) 109:8253–8. doi: 10.1073/pnas.1118193109
85. Mauvezin C, Neufeld TP. Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy. (2015) 11:1437–8. doi: 10.1080/15548627.2015.1066957
86. Huang FL, Yu SJ, Liao EC, Li LY, Shen PW, Li CL. Niclosamide suppresses T-cell acute lymphoblastic leukemia growth through activation of apoptosis and autophagy. Oncol Rep. (2021) 47:30. doi: 10.3892/or.2021.8241
87. Heidari N, Hicks MA, Harada H. GX15-070 (obatoclax) overcomes glucocorticoid resistance in acute lymphoblastic leukemia through induction of apoptosis and autophagy. Cell Death Dis. (2010) 1:e76–6. doi: 10.1038/cddis.2010.53
88. Wallington-Beddoe CT, Hewson J, Bradstock KF, Bendall LJ. FTY720 produces caspase-independent cell death of acute lymphoblastic leukemia cells. Autophagy. (2011) 7:707–15. doi: 10.4161/auto.7.7.15154
89. Chen Y, Li J, Xu L, Găman MA, Zou Z. The genesis and evolution of acute myeloid leukemia stem cells in the microenvironment: From biology to therapeutic targeting. Cell Death Discovery. (2022) 8:397. doi: 10.1038/s41420-022-01193-0
90. Stubbins RJ, Francis A, Kuchenbauer F, Sanford D. Management of acute myeloid leukemia: A review for general practitioners in oncology. Curr Oncol. (2022) 29:6245–59. doi: 10.3390/curroncol29090491
91. Sanz J, Montesinos P, Sanz MA. Role of hematopoietic stem cell transplantation in acute promyelocytic leukemia. Front Oncol. (2021) 11. doi: 10.3389/fonc.2021.614215
92. Wan P, Zhong L, Yu L, Shen C, Shao X, Chen S, et al. Lysosome-related genes predict acute myeloid leukemia prognosis and response to immunotherapy. Front Immunol. (2024) 15. doi: 10.3389/fimmu.2024.1384633
93. Dai M, Lin B, Li H, Wang Y, Wu M, Wei Y, et al. Lysosomal cation channel TRPML1 suppression sensitizes acute myeloid leukemia cells to chemotherapeutics by inhibiting autophagy. Mol Cell Biochem. (2024), 1–15. doi: 10.1007/s11010-024-05054-5
94. Yun S, Vincelette ND, Yu X, Watson GW, Fernandez MR, Yang C, et al. TFEB links MYC signaling to epigenetic control of myeloid differentiation and acute myeloid leukemia. Blood Cancer Discovery. (2021) 2:162–85. doi: 10.1158/2643-3230.BCD-20-0029
95. Liu Q, Chen L, Atkinson JM, Claxton DF, Wang HG. Atg5-dependent autophagy contributes to the development of acute myeloid leukemia in an MLL-AF9-driven mouse model. Cell Death Dis. (2016) 7:e2361. doi: 10.1038/cddis.2016.264
96. Chen Y, Chen J, Zou Z, Xu L, Li J. Crosstalk between autophagy and metabolism: implications for cell survival in acute myeloid leukemia. Cell Death Discovery. (2024) 10:46. doi: 10.1038/s41420-024-01823-9
97. Watson A, Riffelmacher T, Stranks A, Williams O, De Boer J, Cain K, et al. Autophagy limits proliferation and glycolytic metabolism in acute myeloid leukemia. Cell Death Discovery. (2015) 1:15008. doi: 10.1038/cddiscovery.2015.8
98. Nguyen TD, Shaid S, Vakhrusheva O, KosChade SE, Klann K, Thölken M, et al. Loss of the selective autophagy receptor p62 impairs murine myeloid leukemia progression and mitophagy. Blood. (2019) 133:168–79. doi: 10.1182/blood-2018-02-833475
99. Heydt Q, Larrue C, Saland E, Bertoli S, Sarry JE, Besson A, et al. Oncogenic FLT3-ITD supports autophagy via ATF4 in acute myeloid leukemia. Oncogene. (2018) 37:787–97. doi: 10.1038/onc.2017.376
100. Cornet-Masana JM, Banús-Mulet A, Carbó JM, Torrente MÁ, Guijarro F, Cuesta-Casanovas L, et al. Dual lysosomal-mitochondrial targeting by antihistamines to eradicate leukaemic cells. EBioMedicine. (2019) 47:221–34. doi: 10.1016/j.ebiom.2019.08.021
101. Yuan J, Lovejoy DB, Richardson DR. Novel di-2-pyridyl–derived iron chelators with marked and selective antitumor activity: in vitro and in vivo assessment. Blood. (2004) 104:1450–8. doi: 10.1182/blood-2004-03-0868
102. Jiang Q, Li F, Shi K, Wu P, An J, Yang Y, et al. ATF4 activation by the p38MAPK–eIF4E axis mediates apoptosis and autophagy induced by selenite in Jurkat cells. FEBS Lett. (2013) 587:2420–9. doi: 10.1016/j.febslet.2013.06.011
103. Szymańska K, Park S. Acute lymphocytic leukemia: diagnosis and treatment ☆. In: Reference Module in Biomedical Sciences. United States: Elsevier (2018).
104. Raetz EA, Teachey DT. T-cell acute lymphoblastic leukemia. Hematology. (2016) 2016:580–8. doi: 10.1182/asheducation-2016.1.580
105. Malard F, Mohty M. Acute lymphoblastic leukaemia. Lancet. (2020) 395:1146–62. doi: 10.1016/S0140-6736(19)33018-1
106. Laane E, Tamm KP, Buentke E, Ito K, Khahariza P, Oscarsson J, et al. Cell death induced by dexamethasone in lymphoid leukemia is mediated through initiation of autophagy. Cell Death Differ. (2009) 16:1018–29. doi: 10.1038/cdd.2009.46
107. Teuffel O, Kuster SP, Hunger SP, Conter V, Hitzler J, Ethier MC, et al. Dexamethasone versus prednisone for induction therapy in childhood acute lymphoblastic leukemia: a systematic review and meta-analysis. Leukemia. (2011) 25:1232–8. doi: 10.1038/leu.2011.84
108. Huang FL, Yu SJ, Li CL. Role of autophagy and apoptosis in acute lymphoblastic leukemia. Cancer Control. (2021) 28:107327482110191. doi: 10.1177/10732748211019138
109. Ruzickova E, Skoupa N, Dolezel P, Smith DA, Mlejnek P. The lysosomal sequestration of tyrosine kinase inhibitors and drug resistance. Biomolecules. (2019) 9:675. doi: 10.3390/biom9110675
110. Sheng Z, Ma L, Sun JE, Zhu LJ, Green MR. BCR-ABL suppresses autophagy through ATF5-mediated regulation of mTOR transcription. Blood. (2011) 118:2840–8. doi: 10.1182/blood-2010-12-322537
111. Mitchell R, Hopcroft LEM, Baquero P, Allan EK, Hewit K, James D, et al. Targeting BCR-ABL-independent TKI resistance in chronic myeloid leukemia by mTOR and autophagy inhibition. JNCI: J Natl Cancer Institute. (2018) 110:467–78. doi: 10.1093/jnci/djx236
112. Liu Y, Song H, Song H, Feng X, Zhou C, Huo Z. Targeting autophagy potentiates the anti-tumor effect of PARP inhibitor in pediatric chronic myeloid leukemia. AMB Express. (2019) 9:108. doi: 10.1186/s13568-019-0836-z
113. Vire B, David A, Wiestner A. TOSO, the fcμ Receptor, is highly expressed on chronic lymphocytic leukemia B cells, internalizes upon IgM binding, shuttles to the lysosome, and is downregulated in response to TLR activation. J Immunol. (2011) 187:4040–50. doi: 10.4049/jimmunol.1100532
114. Lim SH, Vaughan AT, Ashton-Key M, Williams EL, Dixon SV, Chan HTC, et al. Fc gamma receptor IIb on target B cells promotes rituximab internalization and reduces clinical efficacy. Blood. (2011) 118:2530–40. doi: 10.1182/blood-2011-01-330357
115. Jak M, van Bochove GGW, Reits EA, Kallemeijn WW, Tromp JM, Umana P, et al. CD40 stimulation sensitizes CLL cells to lysosomal cell death induction by type II anti-CD20 mAb GA101. Blood. (2011) 118:5178–88. doi: 10.1182/blood-2011-01-331702
116. Cook KL, Wärri A, Soto-Pantoja DR, Clarke PAG, Cruz MI, Zwart A, et al. Chloroquine inhibits autophagy to potentiate antiestrogen responsiveness in ER+ Breast cancer. Clin Cancer Res. (2014) 20:3222–32. doi: 10.1158/1078-0432.CCR-13-3227
117. Grønningsæter IS, Reikvam H, Aasebø E, Bartaula-Brevik S, Hernandez-Valladares M, Selheim F, et al. Effects of the autophagy-inhibiting agent chloroquine on acute myeloid leukemia cells; characterization of patient heterogeneity. J Pers Med. (2021) 11:1–23. doi: 10.3390/jpm11080779
118. Wang HL, Li JN, Kan WJ, Xu GY, Luo GH, Song N, et al. Chloroquine enhances the efficacy of chemotherapy drugs against acute myeloid leukemia by inactivating the autophagy pathway. Acta Pharmacol Sin. (2023) 44:2296–306. doi: 10.1038/s41401-023-01112-8
Keywords: lysosomes, autophagy, hematological malignancies, lysosomal dysfunction, chemotherapy resistance, targeted treatment, drug resistance, leukemia
Citation: Manivannan MS, Peters A and Gibson SB (2025) Lysosome targeted therapies in hematological malignancies. Front. Oncol. 15:1549792. doi: 10.3389/fonc.2025.1549792
Received: 22 December 2024; Accepted: 06 February 2025;
Published: 24 February 2025.
Edited by:
Tamer Saad Kaoud, The University of Texas at Austin, United StatesReviewed by:
Arunkumar Venkatesan, Upstate Medical University, United StatesCopyright © 2025 Manivannan, Peters and Gibson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Spencer B. Gibson, c2dpYnNvbjJAdWFsYmVydGEuY2E=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.