 Liqin Ruan
Liqin Ruan Lu Wang
Lu Wang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 28 February 2025
Sec. Cancer Molecular Targets and Therapeutics
Volume 15 - 2025 | https://doi.org/10.3389/fonc.2025.1530541
This article is part of the Research Topic Renewed Insight into Cancer Mechanism and Therapy View all 24 articles
Tumors employ a range of strategies to evade detection and eradication by the host’s immune system. These include downregulating antigen expression, altering antigen presentation processes, and inhibiting immune checkpoint pathways. etc. Adoptive Cell Therapy (ACT) represents a strategy that boosts anti-tumor immunity. This is achieved by amplifying or genetically engineering immune cells, which are either sourced from the patient or a donor, in a laboratory setting. Subsequently, these cells are reintroduced into the patient to bolster their immune response against cancer. ACT has successfully restored anti-tumor immune responses by amplifying the activity of T cells from patients or donors. This review focuses on the mechanisms underlying tumor escape, including alterations in tumor cell antigens, the immunosuppressive tumor microenvironment (TME), and modulation of immune checkpoint pathways. It further explores how ACT can avddress these factors to enhance therapeutic efficacy. Additionally, the review discusses the application of gene-editing technologies (such as CRISPR) in ACT, highlighting their potential to strengthen the anti-tumor capabilities of T cells. Looking forward, the personalized design of ACT, combined with immune checkpoint inhibitors and targeted therapies, is expected to significantly improve treatment outcomes, positioning this approach as a key strategy in the field of cancer immunotherapy.
Tumor immune escape refers to the process by which tumor cells evade recognition and elimination by the immune system through various mechanisms, allowing them to survive and proliferate within the body. This phenomenon is recognized as a key driver of tumorigenesis, progression, and recurrence (1). Tumors evade immune surveillance through various mechanisms, including downregulating antigen presentation, inhibiting T cell activity, and altering the function of immune cells in the tumor microenvironment (TME) (2). Notably, the activation of immune checkpoint pathways, such as PD-1/PD-L1 and CTLA-4, enables tumor cells to suppress T cell-mediated anti-tumor responses, thereby supporting their survival (3, 4). In recent years, in-depth research on immune escape mechanisms has provided important basis for the development of immunotherapies targeting these pathways (5, 6).
Adoptive cell therapy(ACT) refers to a treatment method that enhances the anti-tumor activity of a patient’s own or donor’s immune cells through in vitro expansion or genetic modification (7) (Figure 1). As an innovative cancer immunotherapy, ACT can effectively overcome tumor immune evasion and enhance the immune system’s ability to fight against tumors, showing a broad prospect for clinical application (8–10). ACT is currently classified primarily based on different anti-tumor mechanisms, including Tumor-infiltrating lymphocyte (TIL) therapy (11), Chimeric Antigen Receptor T cell (CAR-T) therapy (12), engineered T cell receptor (TCR-T) cell therapy (13) and Cytokine-Induced Killer cells(CIK) therapy (14), etc. (Table 1). In cancer treatment, ACT has become a key component of immunotherapy due to its high targeting and strong anti-tumor effects, especially showing a broad application prospect in the treatment of recurrent and refractory tumors (8, 10). In recent years, the combination of ACT therapy with immune checkpoint inhibitors (ICIs) has also shown significant efficacy (4–6).
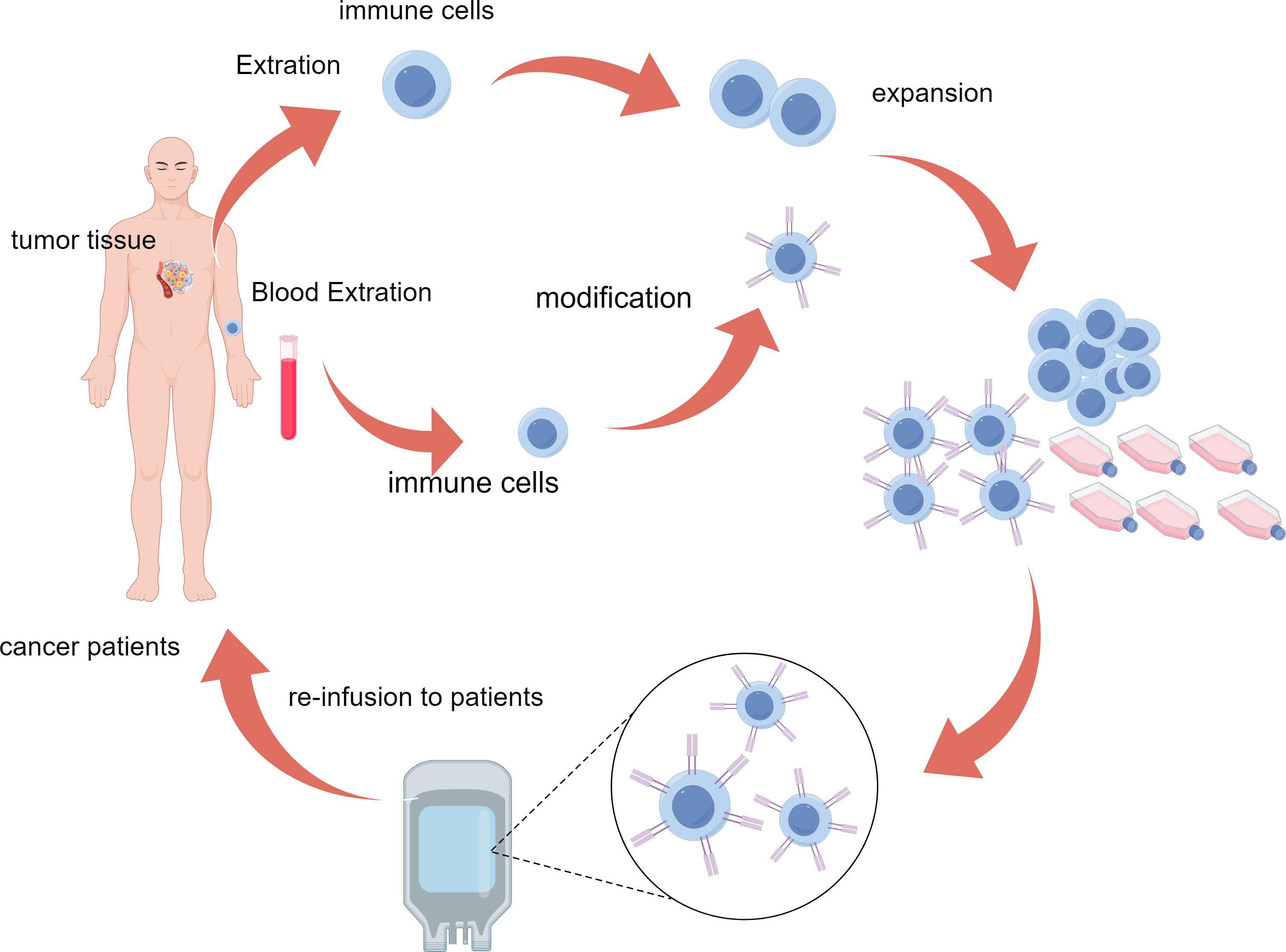
Figure 1. Schematic representation of the ACT process. Immune cells are isolated from patient tumor tissue or blood, genetically modified for enhanced tumor targeting, expanded ex vivo, and reinfused to attack cancer cells in the patient. ACT, adoptive cell therapy.

Table 1. Summary of current adoptive cell therapies in cancer treatment.
ACT effectively overcomes tumor immune evasion through various mechanisms, enhancing the immune system’s ability to recognize and eliminate tumors (56). ACT can enhance the recognition of tumor antigens by in vitro expansion of specific T cells or modification of immune cells (such as CAR-T cells),especially for the downregulation of antigen expression by tumor cells (12, 57, 58). Additionally, ACT can improve the immunosuppressive state in the TME. For instance, CIK cells have MHC-unrestricted killing characteristics and can counteract the negative effects of immunosuppressive cells (e.g., Treg cells, MDSC) in the TME (59–61). By targeting tumor-associated antigens and activating effector T cells, ACT can overcome challenges of immune checkpoint inhibition, such as the PD-1/PD-L1 pathway (4, 10). Furthermore, the combination of ACT with immune checkpoint inhibitors has shown potential to enhance T cell activity and reduce tumor drug resistance (5, 6). Through these innovative mechanisms, ACT not only increases the recognition rate of tumors but also significantly enhances the immune system’s cytotoxic effect on tumor cells (62, 63).
The causes of tumor immune evasion are complex and diverse, making it one of the key factors in tumor growth and metastasis. Firstly, tumor cells evade recognition and attack by T cells by downregulating or altering the expression of major histocompatibility complex (MHC) molecules (61–66). Secondly, the TME is rich in immunosuppressive cells, such as regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs), which weaken the function of effector T cells by secreting inhibitory cytokines (e.g., TGF-β, IL-10) (4, 67, 68). Moreover, tumor cells bind to PD-1 on the surface of T cells by expressing immune checkpoint molecules such as PD-L1, leading to T cell exhaustion and further suppression of anti-tumor immune responses (5, 6, 69). The synergistic effect of these evasion mechanisms enables tumor cells to endure and proliferate within the host (Figure 2).
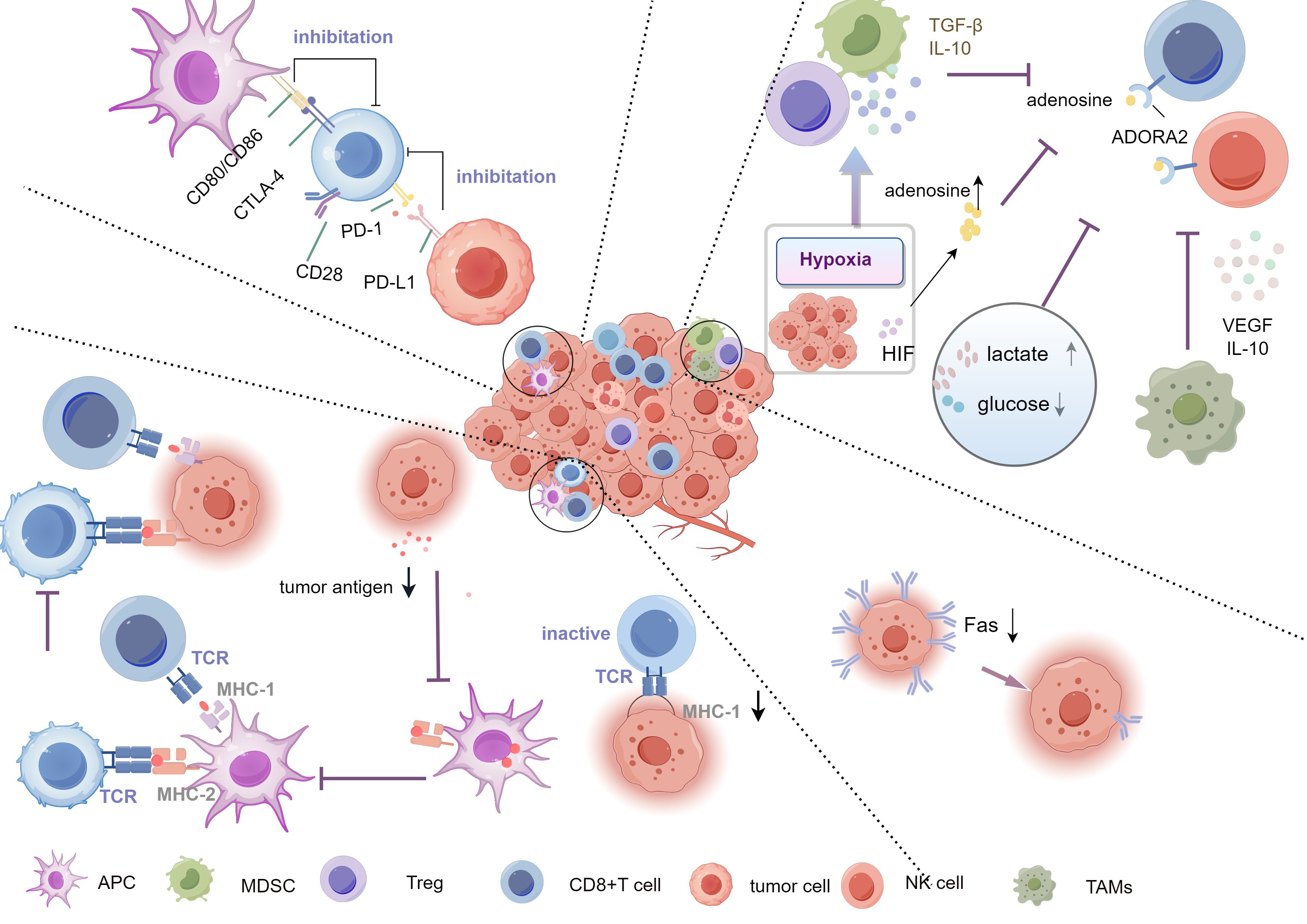
Figure 2. Mechanisms of tumor immune evasion. The Key mechanisms include downregulation of antigen expression or alteration of antigen presentation, preventing effective immune recognition, inhibiting T cell activation via checkpoints (PD-1/PD-L1, CTLA-4), and creating an immunosuppressive microenvironment rich in TGF-β,IL-10, and adenosine. Hypoxia and metabolic changes (high lactate, low glucose) further support evasion, aided by Tregs, TAMs, and MDSCs. Tregs, Regulatory T cells; TAMs, tumor-associated macrophages; MDSCs, myeloid-derived suppressor cells.
Tumor cells evade immune system recognition by reducing antigen expression or altering the antigen presentation process, which is one of the key mechanisms of tumor immune evasion (59, 62, 64). Normally, cells present antigens through MHC molecules, enabling T cells to recognize and kill abnormal cells. The downregulation of tumor neoantigens can be induced through various pathways, such as copy number loss, transcriptional repression, epigenetic silencing, and post-translational mechanisms. These processes affect tumor antigen presentation (70). However, many tumor cells alter this process through various pathways, avoiding immune surveillance (71, 72). Firstly, tumor cells can downregulate the expression of MHC class I (MHC-I) molecules, which are key elements in presenting intracellular protein fragments (including tumor antigens) to CD8+ T cells (73). If the expression of MHC-I molecules is reduced, tumor antigens will not be effectively presented, and CD8+T cells will fail to recognize and attack these tumor cells (3, 62). For instance, downregulation or loss of MHC-I is frequently observed in tumors such as melanoma and lung cancer (74, 75). Moreover, tumor cells can also alter the antigen processing machinery, inhibiting the loading of tumor antigens onto MHC molecules, thereby further reducing the likelihood of immune recognition (76, 77). Additionally, accessory proteins involved in the antigen presentation process may also be modulated. For example, tumor cells can reduce the function of key molecules in the antigen processing machinery, such as the proteasome and TAP transporters, further hindering the presentation of antigens on MHC-I (78). Other proteins in the antigen presentation process, such as β2-microglobulin, are also often downregulated in tumor cells, leading to unstable expression of MHC molecules on the cell surface and further reducing the chances of tumor cells being recognized by the immune system (79).
Furthermore, tumor cells can also produce incompletely or misfolded proteins through antigen mutation, further avoiding T cell recognition (80). In some instances, tumor cells might even cease to express tumor-specific antigens, thereby fundamentally evading immune system assaults (81). Such antigen mutation and loss are common in advanced tumors and are closely related to tumor invasiveness and metastasis (82). This immune evasion mechanism, which reduces antigen expression or alters the antigen presentation process, allows tumor cells to continue growing and expanding under immune surveillance. Strategies to overcome this evasion mechanism have become an important research direction in current cancer immunotherapy. For example, restoring the expression of MHC-I molecules, enhancing antigen presentation capabilities, and ACT that can recognize atypical antigens have shown some efficacy in clinical studies (83, 84).
The immunosuppressive characteristics of the TME play a crucial role in tumor immune evasion (85–87). The TME not only consists of tumor cells, but also includes immune cells, stromal cells, blood vessels, as well as various signaling molecules, which together constitute a complex immunosuppressive network (88). Among them, Tregs and MDSCs in the TME are major participants in immunosuppression. They suppress the activity of effector T cells and natural killer (NK) cells by secreting inhibitory cytokines such as TGF-β and IL-10, weakening the body’s immune response to the tumor (89, 90).
Additionally, tumor-associated macrophages (TAMs) present in the TME also promote immune evasion. TAMs often exhibit an M2 phenotype, which is characterized by their roles in promoting tissue repair and suppressing inflammatory responses. M2-type TAMs facilitate tumor angiogenesis and immunosuppression and inhibit the function of T cells by secreting factors such as VEGF and IL-10 (91, 92). Studies have shown that the density of TAMs at tumor sites is closely associated with tumor progression and poor prognosis (93). Hypoxia is another important characteristic of the TME. Rapid tumor proliferation leads to local hypoxia, activating HIF, which promote the expression of immunosuppressive molecules (94). The hypoxic environment also induces the generation of adenosine, which inhibits the function of T cells and NK cells through the A2A receptor (95). Furthermore, hypoxic conditions can enhance the immunosuppressive effects of Tregs and MDSCs, further weakening the anti-tumor immune response (96).
Metabolic inhibition is also an important mechanism of immune evasion in the TME. Tumor cells in the TME consume large amounts of nutrients such as glucose and glutamine, leading to restricted metabolic activity of effector T cells and preventing them from functioning properly (97). Additionally, the accumulation of lactic acid in the TME also suppresses the proliferation and cytotoxicity of T cells by acidifying the environment (98).
Overall, the TME employs various immunosuppressive mechanisms, including the regulation of immune cell activity, hypoxia, and metabolic changes, to help tumors escape surveillance. The interaction of these mechanisms allows tumors to continue growing and spreading under immune pressure. Therefore, therapies targeting the immunosuppressive TME, such as targeting Tregs, TAMs, or restoring T cell metabolic activity, have become important directions for improving the efficacy of tumor immunotherapy (99).
Immune checkpoint pathways play a crucial role in tumor immune evasion, allowing tumor cells to evade attacks from the host immune system by suppressing T cell functions. Immune checkpoints are important mechanisms that regulate the intensity and duration of immune responses, designed to prevent an overactive immune system from causing autoimmunity (100, 101). By activating these pathways, tumor cells weaken the anti-tumor activity of T cells, thereby helping the survival and spread of tumor cells (100–102). One of the most widely studied immune checkpoint pathways is the PD-1/PD-L1 (programmed death protein-1) pathway. PD-1 is an inhibitory receptor expressed on the surface of T cells, and when it binds to its ligand PD-L1, the PD-1 pathway inhibits T cell proliferation, cytokine secretion, and cytotoxic activity. Many tumor cells highly express PD-L1, which binds to PD-1 on T cells, preventing T cells from attacking tumor cells (62, 103, 104). Studies have shown that overexpression of PD-L1 in various tumors, such as melanoma, lung cancer, and liver cancer, is associated with tumor progression and poor prognosis (105).
In addition to the PD-1/PD-L1 pathway, Cytotoxic T-Lymphocyte Associated Antigen-4 (CTLA-4)is another crucial immune checkpoint. CTLA-4 competitively binds to CD80 and CD86 with the T cell co-stimulatory molecule CD28, and its inhibitory effect on T cell activity is relatively early, primarily occurring in the lymph nodes (6, 106). Beyond directly affecting T cell function, immune checkpoint pathway also further suppresses the anti-tumor immune response by regulating immune suppressive cells in the TME, such as Tregs and MDSCs. Tregs and MDSCs enhance the tumor’s immune evasion capabilities by highly expressing checkpoint molecules like PD-L1 (107, 108). Moreover, tumor cells can also adapt to immunotherapy pressure by inducing the expression of checkpoint molecules, rendering traditional immunotherapy ineffective (109).
In recent years, therapies targeting immune checkpoint pathways, particularly inhibitors of PD-1/PD-L1 and CTLA-4, have become significant breakthroughs in cancer treatment. By blocking these inhibitory pathways, immune checkpoint inhibitors can restore the anti-tumor functions of T cells (10, 110, 111). However, despite the significant clinical efficacy of immune checkpoint inhibitors, some patients still do not respond to treatment or eventually develop resistance, which may be related to the tumor’s ability to evade immune attacks through multiple escape mechanisms (2, 80). Therefore, researching how to overcome the evasion of immune checkpoint pathways is a key direction for improving the effectiveness of cancer immunotherapy in the future.
The mechanisms of tumor immune evasion are diverse. In addition to downregulation of MHC molecules and escape through immune checkpoint pathways, the Fas/FasL pathway blockade is also common mean of immune evasion (112, 113). The Fas/FasL pathway is an important mechanism for regulating apoptosis; under normal conditions, the binding of the Fas receptor to its ligand(FasL) induces apoptosis (114). However, many tumor cells evade apoptosis signals mediated by T cells and B cells by reducing the expression of Fas receptors or altering the function of FasL, thereby achieving immune evasion (3, 62, 115). For instance, research has found that various solid tumors, including melanoma, exhibit dysregulation of the Fas/FasL pathway (116–118). Immune escape mechanisms governed by mutated NOTCH in mature B-cell malignancies, mediated by increased PD-L1 expression and downregulation of MHC class II genes (119). These complex mechanisms allow tumors to evade immune attacks through multiple pathways, suggesting that interventions targeting the Fas/FasL pathway could become new directions for future tumor immunotherapy.
ACT is a treatment method based on T cells from patients or donors, which enhances their anti-tumor activity through in vitro expansion or genetic modification, and then reinfuses the expanded T cells or CIK cells back into the patient’s body to strengthen their anti-tumor activity, avoiding the weakening effects of inhibitory factors in the tumor microenvironment (120). The origin of ACT can be traced back to the 1980s when Rosenberg and colleagues first reported the application of TIL therapy in the treatment of melanoma (121). Since then, ACT has gone through several stages of development. Particularly with the advancement of genetic engineering technology, CAR-T cell therapy and TCR-T cell therapy have become the representatives of modern ACTs (6, 10, 122).
ACT works by overcoming multiple immune evasion mechanisms of tumors. TIL therapy amplifies T cells isolated from tumors, enhancing their ability to recognize and attack tumor cells (99). CAR-T therapy, on the other hand, enables T cells to recognize specific antigens on the surface of tumor cells through genetic modification, thus circumventing immune evasion caused by the downregulation of MHC-I. Moreover, the in vitro modification of CAR-T and TCR-T cells can make them tolerant to immune checkpoint molecules, thereby avoiding inhibition through the PD-1/PD-L1 pathway (2, 5, 10, 122–124).
Specific antigen recognition is one of the core mechanisms by which ACT enhances T cell anti-tumor activity and effectively overcomes the issue of tumor immune evasion (6). Tumor cells often escape immune surveillance by reducing their antigen exposure or downregulating MHC (Figure 3). However, in ACT, specific T cells can recognize tumor-associated antigens and restore their anti-tumor function through genome editing technologies such as CRISPR. Even when MHC expression is reduced, by modifying T cells in vitro, their anti-tumor activity is enhanced, avoiding the interference of tumor evasion mechanisms (10, 12, 63). Particularly, CAR-T therapy uses genetic engineering to equip T cells with specific antigen receptors that can recognize specific antigens on the tumor surface, allowing T cells to bypass these evasion mechanisms and precisely identify tumor cells, avoiding the MHC-dependent recognition process (10, 12). For instance, CAR-T cells can recognize the highly expressed CD19 antigen in hematological tumors, effectively attacking tumor cells regardless of MHC downregulation (125). This design overcomes the tumor’s MHC downregulation evasion strategy, significantly enhancing the anti-tumor effects of T cells.
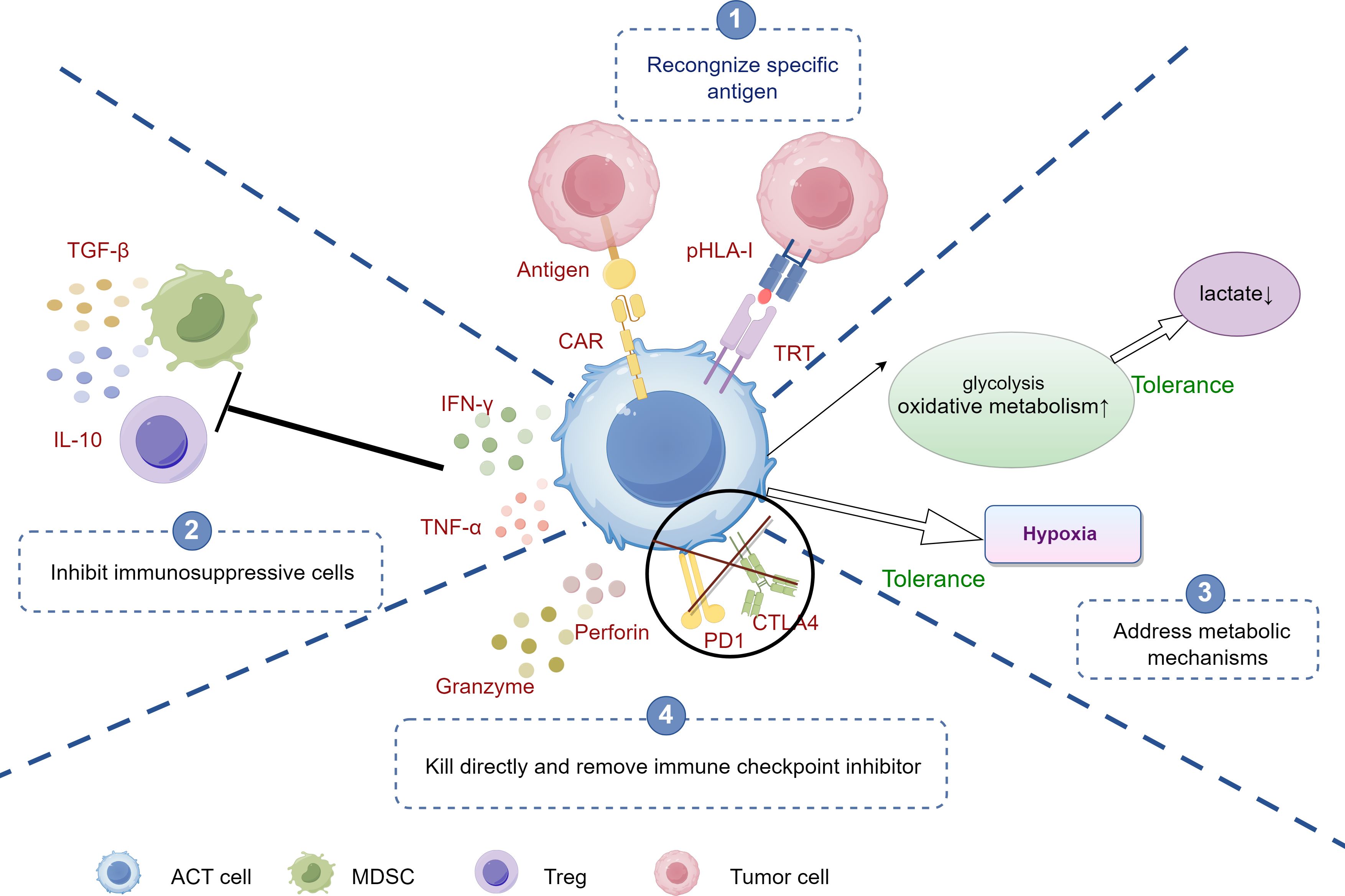
Figure 3. The mechanisms of ACT against tumor immune evasion. ①Antigen Recognition: CAR-T cells recognize specific antigens on tumor cells, initiating targeted attack. ②Inhibition of Immunosuppressive cells: CAR-T cells counteract immunosuppressive cells (e.g., Tregs, MDSCs) by blocking signals, enhancing immune response. ③Addressing Metabolic Barriers: CAR-T cells adapt to the tumor microenvironment by increasing glycolysis and oxidative metabolism, enabling function under hypoxia and low lactate conditions. ④Direct Killing and Immune Checkpoint Inhibition: CAR-T cells release cytotoxic molecules (e.g., perforin, granzyme) to kill tumor cells and target immune checkpoints (e.g., PD-1, CTLA-4) to overcome tumor-induced tolerance. ACT, adoptive cell therapy; CAR-T, Chimeric Antigen Receptor T-Cell; Tregs, Regulatory T cells; TAMs, tumor-associated macrophages; MDSCs, myeloid-derived suppressor cells; Tregs, Regulatory T cells; MDSCs, myeloid-derived suppressor cells.
TCR-T cells are engineered to introduce specific TCRs that enable them to recognize tumor endogenous antigens presented by MHC molecules (13). Although TCR-T cells rely on MHC, they can recognize a variety of tumor endogenous and specific mutational antigens, making them applicable to a broader range of tumor types. For instance, TCR-T cells can recognize the MART-1 antigen in melanoma, effectively activating T cells to kill tumors. Scientists have also genetically modified TCR-T cells to maintain a higher affinity for tumor antigens, further enhancing their recognition and killing capabilities.
To address tumor heterogeneity and antigen escape, bispecific and multispecific T cell technologies are emerging. By designing T cells capable of recognizing two or more tumor antigens, ACT increases the therapeutic coverage and prevents tumors from escaping attack through antigen loss. For example, bispecific CAR-T cells can simultaneously recognize CD19 and CD22, improving anti-tumor efficacy and reducing the risk of relapse (126).
With the advancement of genomics, personalized T cell therapies targeting patient-specific neoantigens have gradually been applied. These neoantigens are generated by mutations and do not exist in normal cells, making them ideal therapeutic targets. By identifying neoantigens through genetic sequencing and designing exclusive TCRs, personalized T cells can be generated that specifically target the patient’s tumor, effectively addressing refractory tumors.
In the TME, tumor cells often suppress the activity of T cells through immune checkpoint pathways, such as PD-1/PD-L1 and CTLA-4, thereby evading immune surveillance. When PD-L1 binds to PD-1 on T cells, it inhibits T cell proliferation and cytotoxicity, ultimately leading to a state of T cell exhaustion, where they cannot recognize and attack tumor cells normally (127). To bypass this inhibitory mechanism, researchers have used gene-editing technologies (such as CRISPR-Cas9) to knock out the PD-1 gene on the surface of T cells, thereby blocking the inhibitory signals of the PD-1/PD-L1 pathway and allowing T cells to remain active in the TME. Studies have shown that PD-1 knockout (KO) T cells exhibit stronger proliferative capacity and anti-tumor activity in both in vitro and in vivo experiments, avoiding negative feedback regulation by immune checkpoints in vivo (2, 10). Particularly, CAR-T cells has been genetically modified to make T cells no longer dependent on MHC molecules recognition of tumors and capable of resisting inhibition by the PD-1 and CTLA-4 pathways (12, 63).
Immune checkpoint inhibitors enhance anti-tumor immune responses by removing inhibitory signals in the immune system, but their clinical application still faces significant bottlenecks, including limited efficacy, uncertainty of predictive biomarkers, immune-related adverse events, and high treatment costs (128). ACT can be used in combination with immune checkpoint inhibitors (such as PD-1, PD-L1, or CTLA-4 inhibitors) to relieve the suppression of T cell functions and further enhance the anti-tumor immune response (5, 99). Combined treatment strategy (chemotherapy, radiotherapy, immune checkpoint blockers) has shown significant efficacy in various types of tumors, especially for patients with advanced and recurrent tumors. The combination of ACT and immune checkpoint inhibitors can significantly improve treatment response rates and patient survival (129, 130). Future research directions aim to further optimize this combined strategy to reduce side effects and improve long-term efficacy.
The TME is rich in immunosuppressive cells, such as Tregs, MDSCs, and TAMs, which weaken the anti-tumor functions of effector T cells by secreting immunosuppressive factors (such as TGF-β, IL-10) (131–135). ACT can reshape the TME and restore immune responses through various pathways.
Initially, adoptive T cell therapy can reverse the immunosuppressive state by secreting cytokines (such as IFN-γ, TNF-α) (10, 136, 137). These cytokines suppress the function of Tregs and MDSCs, reducing their immunosuppressive effects in the TME and restoring the activity of effector T cells and NK cells (63, 138). Likewise, ACT can directly reduce the number of these suppressive cells by clearing Tregs and MDSCs from the TME, promoting anti-tumor immune responses (139, 140).
In addition, the TME is often characterized by hypoxia, nutrient deprivation, and the accumulation of metabolic waste products such as lactate, which significantly suppress the activity of effector T cells (62, 97). ACT modifies T cells genetically to better adapt to these adverse metabolic conditions, thereby maintaining their anti-tumor functions (98). Genetically modified T cells can enhance their glycolytic or oxidative metabolism capabilities, allowing them to remain active in environments lacking glucose or with energy constraints (111, 141).
Tumor cells typically weaken the energy supply of effector T cells through metabolic suppression; however, CAR-T cells can be designed to tolerate nutrient-poor environments, thus preserving their cytotoxic functions (6). ACT can synergize with immune checkpoint blockade therapies to enhance the anti-tumor immune response. Immune checkpoint pathways, such as PD-1/PD-L1 and CTLA-4, are prevalent in the TME and are utilized by tumors to suppress T-cell activity (80). By combining ACT with immune checkpoint inhibitors (e.g., PD-1 and CTLA-4 inhibitors), these inhibitory signals within the TME can be blocked, further activating effector T cells (5, 99). In summary, ACT not only directly enhances the anti-tumor function of T cells but also modulates immunosuppressive mechanisms within the TME, reducing tumor immune evasion and improving the overall efficacy of cancer therapy.
ACT has demonstrated significant potential in overcoming the metabolic suppression within the TME, particularly through genetic modifications that enhance T-cell adaptability. The TME is typically characterized by hypoxia, nutrient deprivation, and the accumulation of metabolic waste products, such as lactate, which severely inhibit the activity of effector T cells (5, 63). ACT leverages genetic modifications to equip T cells to better adapt to these adverse metabolic conditions, thereby sustaining their anti-tumor functionality.
First, hypoxia is a defining feature of the TME. While tumor cells adapt to low oxygen levels by activating the HIF pathway, effector T cells are often inhibited under such conditions (94). Research indicates that genetically modifying adoptive T cells to overexpress HIF-stabilizing proteins or enhance their metabolic activity can improve T-cell survival and function in hypoxic environments (98).
Second, glucose deprivation and lactate accumulation in the TME restrict the energy supply to T cells, suppressing their anti-tumor activity (97). Genetically modified T cells can be engineered to enhance their glycolytic or oxidative metabolic pathways, allowing them to maintain activity even in glucose-deprived or energy-limited environments (63, 99). Moreover, engineered T cells can be made resistant to the high levels of lactate in the TME, thereby minimizing the inhibitory effects of lactate on T-cell function (2, 142).
Lastly, adenosine accumulation in the TME inhibits T-cell function through the A2A receptor pathway (95). Genetically modifying T cells to resist adenosine signaling enables them to retain their cytotoxic activity in adenosine-rich TME (5, 96). These metabolic adaptation strategies not only enhance T-cell survival in the TME but also significantly improve their anti-tumor efficacy (6, 10).
ACT has shown significant therapeutic efficacy in multiple tumor types through enhancing T-cell immune responses to tumors, especially in overcoming tumor immune escape mechanisms. The core of ACT involves extracting T cells from the patient, expanding or genetically modifying them in vitro to enhance their anti-tumor potency, and then re-infusing them into the body to restore or boost the patient’s immune response (12). This treatment has demonstrated potent anti-tumor potential in clinical trials for various tumors, including melanoma, lung cancer, breast cancer, and lymphoma (15, 143–145) (Table 2).
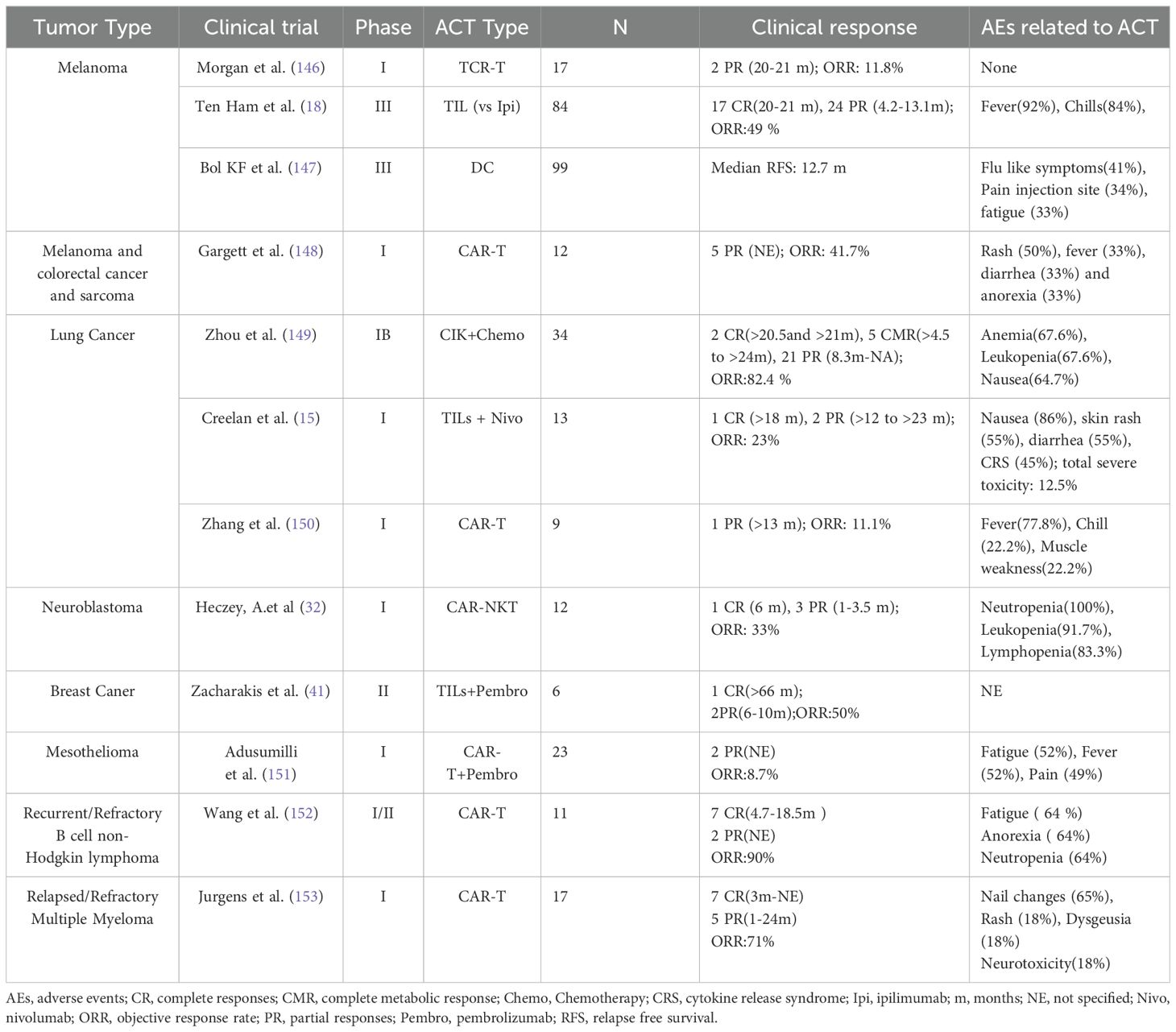
Table 2. Clinical trials of ACT in tumors with published results.
Melanoma is one of the earliest cancers to receive adoptive cell therapy, with research focusing on TIL therapy (154). TIL therapy involves the amplification of T cells isolated from the patient’s tumor tissue, successfully overcoming the tumor’s immune escape through downregulation of antigen expression and immunosuppressive TME (4, 99). A key clinical trial indicated that about 50% of patients with advanced melanoma experienced significant tumor reduction after receiving TIL therapy, with some patients remaining recurrence-free in the long term (18). The antigen specificity of TILs enhances T cell recognition of tumor antigens, while the secretion of cytokines such as IFN-γ suppresses immunosuppressive cells (155). Additionally, studies have shown that the combination of TIL therapy with PD-1 or CTLA-4 inhibitors further enhances the therapeutic effect (156). Immune checkpoint inhibitors release the inhibitory state of T cells, allowing them to exert a stronger cytotoxic effect in the tumor microenvironment (63). This strategy effectively overcomes the immune escape of tumor cells with high PD-L1 expression.
Lung cancer, as a highly heterogeneous tumor, has a variety of immune escape mechanisms. Studies have shown that lung cancer cells often downregulate the expression of MHC-I, weakening antigen presentation capabilities and preventing T cell recognition (157). In a CAR-T therapy trial for lung cancer patients, CAR-T therapy was able to effectively recognize and kill tumor cells with insufficient MHC I expression, compensating for the limitations of traditional T cells that rely on MHC recognition (97, 98). Additionally, hypoxia is another characteristic of the lung cancer microenvironment, leading to immune escape. Genetically modified CAR-T cells, by increasing their adaptability to hypoxic conditions, can remain active even under these adverse conditions (95). This enhanced metabolic adaptability helps T cells function in a microenvironment with nutrient deprivation and accumulation of metabolic waste, overcoming the suppression of T cell function (96).
In the TME of breast cancer patients, the number of Tregs and MDSCs is significantly increased, suppressing the anti-tumor activity of effector T cells (5, 158). In a clinical trial of ACT for advanced breast cancer, researchers used CAR-T cells with enhanced antigen specificity through genetic modification (80). The results showed that CAR-T cells could effectively suppress the immunosuppressive effects of Tregs and MDSCs, restoring the activity of effector T cells (62).
Furthermore, the high expression of the immune checkpoint molecule PD-L1 in breast cancer cells also helps in immune evasion (159). In vitro model studies have shown that the combination of ACT with PD-L1 inhibitors produced a good anti-cancer response in autologous Patient-Derived Xenograft(PDX) models of advanced triple-negative breast cancer (160). This combined strategy not only enhances the recognition of tumors by CAR-T cells but also allows more T cells to function by releasing immune checkpoint inhibition (129, 130, 156, 160).
Lymphoma is another successful application of CAR-T therapy, especially in B-cell lymphoma (161). B-cell lymphoma cells often evade immune surveillance through immune escape mechanisms such as downregulating antigen expression or altering antigen presentation processes (119). In a clinical trial, the use of CD19-targeted CAR-T cell therapy for relapsed B-cell lymphoma achieved a high rate of complete remission (25). CAR-T cells bypass the traditional T cell’s MHC-dependent recognition and directly target the CD19 antigen, effectively addressing the issue of antigen downregulation in lymphoma (162).
However, over time, some patients develop resistance to CAR-T therapy, usually due to tumor cells escaping immune surveillance by losing CD19 antigen expression or increasing the expression of immune checkpoint molecules (163–165). To address this issue, researchers are developing bispecific CAR-T cells that target multiple antigens simultaneously, reducing the risk of immune escape (166, 167).
In addition to melanoma, lung cancer, breast cancer, and lymphoma, the application of adoptive immunotherapy in other solid tumors is also continuously expanding. For instance, in clinical trials for prostate cancer (23), hepatocellular carcinoma (168), and gastric cancer (169), ACT therapy has shown potential to improve immune escape mechanisms. These tumors often evade T cell attacks through metabolic suppression and immune checkpoint escape (97–99). By combining immune checkpoint inhibitors with ACT treatment, the survival and response rates of patients with these solid tumors have been significantly improved (156, 160, 170).
ACT has shown great promise in cancer treatment but faces multiple challenges. First, the high cost and complexity of the treatment limit its widespread application. ACT requires the in vitro expansion and genetic modification of a patient’s T cells, a process that is time-consuming and expensive (10). Second, off-target effects are one of the major safety risks, especially in CAR-T therapy, where T cells may attack healthy tissues, leading to severe side effects (12). Tumor heterogeneity and antigen loss also make some patients unresponsive to treatment, particularly in solid tumors where the immunosuppressive TME weakens the durable action of T cells (63). T cells may become functionally inactivated in the TME, making it difficult to efficiently infiltrate and continuously kill tumors (6). Furthermore, the challenges include the inactivation of immune cells, restricted localization, and diminished efficacy. To tackle these issues, future research must focus on optimizing T cell modification strategies to enhance both efficacy and targeting precision.
The future prospects for ACT are broad, especially driven by new technologies. Gene-editing technologies, such as CRISPR-Cas9, are significantly changing the outlook for ACT applications. With CRISPR technology, scientists can precisely edit the genes of T cells, removing inhibitory signal molecules to enhance their anti-tumor activity (171). For instance, by knocking out PD-1 or other immune checkpoint molecules on the surface of T cells, it is possible to prevent tumors from evading immune surveillance through immune checkpoint pathways (10). Furthermore, CRISPR technology can integrate novel antigen receptors, thereby enhancing T cells’ tumor recognition capabilities (63, 171).
In the future, the combined application of ACT with other treatments will further improve therapeutic effect. The combination of ACT with immune checkpoint inhibitors (such as PD-1/PD-L1 inhibitors) has already shown promising effects in multiple clinical trials (6, 156, 160, 170). Moreover, Combining ACT with targeted therapies (such as BRAF inhibitors) also brings new hope for patients with various solid tumors (12, 172). This multi-pronged treatment strategy not only enhances the efficacy of T cells but also improves the immunosuppressive state of TME, reducing the tumor’s immune escape (80). Future research directions involve continuously optimizing gene-editing technologies and combination therapies to further enhance the anti-tumor capabilities of ACT and reduce side effects (2).
Personalized ACT aims to achieve precision treatment for each patient by detecting tumor-specific antigens and designing personalized treatments. Tumor-specific antigens, such as neoantigens, are proteins expressed in tumor cells due to mutations or abnormal gene expression (70), and can be recognized by T cells. With next-generation sequencing technology, it is possible to quickly screen and identify specific antigens for each patient (10, 171). Using this data, researchers can design customized T cell treatment plans, such as TCR-T or CAR-T therapies, to target tumor cells expressing these antigens (63). Personalized design also involves genetic modification of T cells to enhance their survival and killing efficiency in the TME (173–175). This tailored approach not only improves therapeutic effect but also reduces off-target effects and side effects (12). As precision medicine advances, personalized ACT will become a mainstream direction in cancer treatment.
ACT has demonstrated significant potential in combating tumor immune evasion by enhancing the immune system’s anti-tumor capabilities. This article has detailed how ACT effectively counters the complex strategies of tumor escape through various mechanisms, including enhancing T cell activity, improving the TME, overcoming immune checkpoint inhibition, and metabolic suppression. Furthermore, the article has explored the application of emerging technologies such as CRISPR gene editing, showing the future direction of personalized ACT treatment, especially its broad prospects in combination with immune checkpoint inhibitors and targeted therapies. Although ACT therapy faces challenges such as high costs, off-target effects, and tumor heterogeneity, with the application of new technologies and continuous optimization of treatment plans, ACT is expected to become a key strategy in conquering cancer.
LR: Writing – original draft, Writing – review & editing. LW: Supervision, Validation, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the Jiujiang City Key Research and Development Plan. Grants Awards No.S2024ZDYFN0042.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Senft D. Programming immune escape. Nat Rev Cancer. (2024) 24:294. doi: 10.1038/s41568-024-00688-5
2. Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. (2017) 541:321–30. doi: 10.1038/nature21349
3. Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer. (2019) 19:133–50. doi: 10.1038/s41568-019-0116-x
4. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. (2017) 168:707–23. doi: 10.1016/j.cell.2017.01.017
5. Topalian SL, Forde PM, Emens LA, Yarchoan M, Smith KN, Pardoll DM. Neoadjuvant immune checkpoint blockade: A window of opportunity to advance cancer immunotherapy. Cancer Cell. (2023) 41:1551–66. doi: 10.1016/j.ccell.2023.07.011
6. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. (2018) 359:1350–5. doi: 10.1126/science.aar4060
7. Zhang L, Ding J, Li HY, Wang ZH, Wu J. Immunotherapy for advanced hepatocellular carcinoma, where are we? Biochim Biophys Acta Rev Cancer. (2020) 1874:188441. doi: 10.1016/j.bbcan.2020.188441
8. Gross G, Eshhar Z. Therapeutic potential of T cell chimeric antigen receptors (CARs) in cancer treatment: counteracting off-tumor toxicities for safe CAR T cell therapy. Annu Rev Pharmacol Toxicol. (2016) 56:59–83. doi: 10.1146/annurev-pharmtox-010814-124844
9. Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. (2008) 8:299–308. doi: 10.1038/nrc2355
10. Fesnak AD, June CH, Levine BL. Engineered T cells: the promise and challenges of cancer immunotherapy. Nat Rev Cancer. (2016) 16:566–81. doi: 10.1038/nrc.2016.97
11. Monberg TJ, Borch TH, Svane IM, Donia M. TIL therapy: facts and hopes. Clin Cancer Res. (2023) 29:3275–83. doi: 10.1158/1078-0432.CCR-22-2428
12. June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med. (2018) 379:64–73. doi: 10.1056/NEJMra1706169
13. He J, Yang H, Li D, Liu X, Li S, Liao S, et al. Defined tumor antigen-specific T cells potentiate personalized TCR-T cell therapy and prediction of immunotherapy response. Cell Res. (2022) 32:530–42. doi: 10.1038/s41422-022-00627-9
14. Sharma A, Schmidt-Wolf IGH. 30 years of CIK cell therapy: recapitulating the key breakthroughs and future perspective. J Exp Clin Cancer Res. (2021) 40:388. doi: 10.1186/s13046-021-02184-2
15. Creelan BC, Wang C, Teer JK, Toloza EM, Yao J, et al. Tumor-infiltrating lymphocyte treatment for anti-PD-1-resistant metastatic lung cancer: a phase 1 trial. Nat Med. (2021) 27:1410–8. doi: 10.1038/s41591-021-01462-y
16. Huang H, Nie CP, Liu XF, Song B, Yue JH, Xu JX, et al. Phase I study of adjuvant immunotherapy with autologous tumor-infiltrating lymphocytes in locally advanced cervical cancer. J Clin Invest. (2022) 132(15):e157726. doi: 10.1172/JCI157726
17. Kristensen NP, Heeke C, Tvingsholm SA, Borch A, Draghi A, Crowther MD, et al. Neoantigen-reactive CD8+ T cells affect clinical outcome of adoptive cell therapy with tumor-infiltrating lymphocytes in melanoma. J Clin Invest. (2022) 132(2):e150535. doi: 10.1172/JCI150535
18. Rohaan MW, Borch TH, van den Berg JH, Met Ö, Kessels R, Geukes Foppen MH, et al. Tumor-infiltrating lymphocyte therapy or ipilimumab in advanced melanoma. New Engl J Med. (2022) 387:2113–25. doi: 10.1056/NEJMoa2210233
19. Saberzadeh-Ardestani B, Foster NR, Lee HE, Shi Q, Alberts SR, Smyrk TC, et al. Association of tumor-infiltrating lymphocytes with survival depends on primary tumor sidedness in stage III colon cancers (NCCTG N0147) [Alliance. Ann Oncol. (2022) 33:1159–67. doi: 10.1016/j.annonc.2022.07.1942
20. van den Berg JH, Heemskerk B, van Rooij N, Gomez-Eerland R, Michels S, van Zon M, et al. Tumor infiltrating lymphocytes (TIL) therapy in metastatic melanoma: boosting of neoantigen-specific T cell reactivity and long-term follow-up. J Immunother Cancer. (2020) 8(2):e000848. doi: 10.1136/jitc-2020-000848
21. Mailankody S, Devlin SM, Landa J, Nath K, Diamonte C, Carstens EJ, et al. GPRC5D-targeted CAR T cells for myeloma. N Engl J Med. (2022) 387:1196–206. doi: 10.1056/NEJMoa2209900
22. Majzner RG, Ramakrishna S, Yeom KW, Patel S, Chinnasamy H, Schultz LM, et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature. (2022) 603:934–41. doi: 10.1038/s41586-022-04489-4
23. Narayan V, Barber-Rotenberg JS, Jung IY, Lacey SF, Rech AJ, Davis MM, et al. PSMA-targeting TGFβ-insensitive armored CAR T cells in metastatic castration-resistant prostate cancer: a phase 1 trial. Nat Med. (2022) 28:724–34. doi: 10.1038/s41591-022-01726-1
24. Qi C, Gong J, Li J, Liu D, Qin Y, Ge S, et al. Claudin18.2-specific CAR T cells in gastrointestinal cancers: phase 1 trial interim results. Nat Med. (2022) 28:1189–98. doi: 10.1038/s41591-022-01800-8
25. Zhang J, Hu Y, Yang J, Li W, Zhang M, Wang Q, et al. Non-viral, specifically targeted CAR-T cells achieve high safety and efficacy in B-NHL. Nature. (2022) 609:369–74. doi: 10.1038/s41586-022-05140-y
26. Zhang C, Burger MC, Jennewein L, Genßler S, Schönfeld K, Zeiner P, et al. ErbB2/HER2-specific NK cells for targeted therapy of glioblastoma. J Natl Cancer Inst. (2016) 108(5). doi: 10.1093/jnci/djv375
27. Xie G, Dong H, Liang Y, Ham JD, Rizwan R, Chen J, et al. CAR-NK cells: A promising cellular immunotherapy for cancer. EBioMedicine. (2020) 59:102975. doi: 10.1016/j.ebiom.2020.102975
28. Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. (2020) 382:545–53. doi: 10.1056/NEJMoa1910607
29. Marin D, Basar R, Rafei H, Daher M, Dou J, Mohanty V, et al. Safety, efficacy and determinants of response of allogeneic CD19-specific CAR-NK cells in CD19(+) B cell tumors: a phase 1/2 trial. Nat Med. (2024) 30:772–84. doi: 10.1038/s41591-023-02785-8
30. Zhang W, Liu L, Su H, Liu Q, Shen J, Dai H, et al. Chimeric antigen receptor macrophage therapy for breast tumours mediated by targeting the tumour extracellular matrix. Br J Cancer. (2019) 121:837–45. doi: 10.1038/s41416-019-0578-3
31. Ramos CA, Courtney AN, Robinson SN, Dakhova O, Lulla PD, et al. Allogeneic NKT cells expressing a CD19-specific CAR in patients with relapsed or refractory B-cell Malignancies: an interim analysis. Blood. (2021) 138:2819–9. doi: 10.1182/blood-2021-149712
32. Heczey A, Xu X, Courtney AN, Tian G, Barragan GA, Guo L, et al. Anti-GD2 CAR-NKT cells in relapsed or refractory neuroblastoma: updated phase 1 trial interim results. Nat Med. (2023) 29:1379–88. doi: 10.1038/s41591-023-02363-y
33. Rozenbaum M, Meir A, Aharony Y, Itzhaki O, Schachter J, Bank I, et al. Gamma-delta CAR-T cells show CAR-directed and independent activity against leukemia. Front Immunol. (2020) 11:1347. doi: 10.3389/fimmu.2020.01347
34. Bear AS, Nadler RB, O'Hara MH, Stanton KL, Xu C, Saporito RJ, et al. Natural TCRs targeting KRASG12V display fine specificity and sensitivity to human solid tumors. J Clin Invest. (2024) 134(21):e175790. doi: 10.1172/JCI175790
35. Krakow EF, Brault M, Summers C, Cunningham TM, Biernacki MA, Black RG, et al. HA-1-targeted T-cell receptor T-cell therapy for recurrent leukemia after hematopoietic stem cell transplantation. Blood. (2024) 144:1069–82. doi: 10.1182/blood.2024024105
36. Ma C, Chen P, Du J, Wang L, Lu N, Sun J, et al. Adoptive transfer of CMV-specific TCR-T cells for the treatment of CMV infection after haploidentical hematopoietic stem cell transplantation. J Immunother Cancer. (2024) 12(1):e007735. doi: 10.1136/jitc-2023-007735
37. Wermke M, Holderried TAW, Luke JJ, Morris VK, Alsdorf WH, Wetzko K, et al. First-in-human dose escalation trial to evaluate the clinical safety and efficacy of an anti-MAGEA1 autologous TCR-transgenic T cell therapy in relapsed and refractory solid tumors. J Immunother Cancer. (2024) 12(7):e008668. doi: 10.1136/jitc-2023-008668
38. Yossef R, Krishna S, Sindiri S, Lowery FJ, Copeland AR, Gartner JJ, et al. Phenotypic signatures of circulating neoantigen-reactive CD8(+) T cells in patients with metastatic cancers. Cancer Cell. (2023) 41:2154–2165.e5. doi: 10.1016/j.ccell.2023.11.005
39. Holm JS, Funt SA, Borch A, Munk KK, Bjerregaard AM, Reading JL, et al. Neoantigen-specific CD8 T cell responses in the peripheral blood following PD-L1 blockade might predict therapy outcome in metastatic urothelial carcinoma. Nat Commun. (2022) 13:1935. doi: 10.1038/s41467-022-29342-0
40. Parkhurst M, Goff SL, Lowery FJ, Beyer RK, Halas H, Robbins PF, et al. Adoptive transfer of personalized neoantigen-reactive TCR-transduced T cells in metastatic colorectal cancer: phase 2 trial interim results. Nat Med. (2024) 30:2586–95. doi: 10.1038/s41591-024-03109-0
41. Zacharakis N, Huq LM, Seitter SJ, Kim SP, Gartner JJ, Sindiri S, et al. Breast cancers are immunogenic: immunologic analyses and a phase II pilot clinical trial using mutation-reactive autologous lymphocytes. J Clin Oncol. (2022) 40:1741–54. doi: 10.1200/JCO.21.02170
42. Li N, Tian YW, Xu Y, Meng DD, Gao L, Shen WJ, et al. Combined treatment with autologous CIK cells, radiotherapy and chemotherapy in advanced cervical cancer. Pathol Oncol Res. (2019) 25:691–6. doi: 10.1007/s12253-018-0541-2
43. Ma X, Peng L, Wang J, Gao L, Zhang W, Lu X, et al. Autologous CIK cells combined with chemotherapy as the first-line treatment for locally advanced or metastatic gastric cancer is safe and feasible. Front Immunol. (2023) 14:1267369. doi: 10.3389/fimmu.2023.1267369
44. Wang M, Shi SB, Qi JL, Tang XY, Tian J. S-1 plus CIK as second-line treatment for advanced pancreatic cancer. Med Oncol. (2013) 30:747. doi: 10.1007/s12032-013-0747-9
45. Zhang Y, Wang J, Wang Y, Lu XC, Fan H, Liu Y, et al. Autologous CIK cell immunotherapy in patients with renal cell carcinoma after radical nephrectomy. Clin Dev Immunol 2013. (2013) p:195691. doi: 10.1155/2013/195691
46. Zhang L, Xu Y, Shen J, He F, Zhang D, Chen Z, et al. Feasibility study of DCs/CIKs combined with thoracic radiotherapy for patients with locally advanced or metastatic non-small-cell lung cancer. Radiat Oncol. (2016) 11:60. doi: 10.1186/s13014-016-0635-5
47. Jiang N, Qiao G, Wang X, Morse MA, Gwin WR, Zhou L, et al. Dendritic cell/cytokine-induced killer cell immunotherapy combined with S-1 in patients with advanced pancreatic cancer: A prospective study. Clin Cancer Res. (2017) 23:5066–73. doi: 10.1158/1078-0432.CCR-17-0492
48. Wang X, Ren J, Zhang J, Yan Y, Jiang N, Yu J, et al. Prospective study of cyclophosphamide, thiotepa, carboplatin combined with adoptive DC-CIK followed by metronomic cyclophosphamide therapy as salvage treatment for triple negative metastatic breast cancers patients (aged <45). Clin Transl Oncol. (2016) 18:82–7. doi: 10.1007/s12094-015-1339-2
49. Yang L, Ren B, Li H, Yu J, Cao S, Hao X, et al. Enhanced antitumor effects of DC-activated CIKs to chemotherapy treatment in a single cohort of advanced non-small-cell lung cancer patients. Cancer Immunol Immunother. (2013) 62:65–73. doi: 10.1007/s00262-012-1311-8
50. Zhan HL, Gao X, Pu XY, Li W, Li ZJ, Zhou XF, et al. A randomized controlled trial of postoperative tumor lysate-pulsed dendritic cells and cytokine-induced killer cells immunotherapy in patients with localized and locally advanced renal cell carcinoma. Chin Med J (Engl). (2012) 125:3771–7.
51. Zhao X, Ji CY, Liu GQ, Ma DX, Ding HF, Xu M, et al. Immunomodulatory effect of DC/CIK combined with chemotherapy in multiple myeloma and the clinical efficacy. Int J Clin Exp Pathol. (2015) 8:13146–55.
52. Berrien-Elliott MM, Foltz JA, Russler-Germain DA, Neal CC, Tran J, Gang M, et al. Hematopoietic cell transplantation donor-derived memory-like NK cells functionally persist after transfer into patients with leukemia. Sci Transl Med. (2022) 14:eabm1375. doi: 10.1126/scitranslmed.abm1375
53. Bednarski JJ, Zimmerman C, Berrien-Elliott MM, Foltz JA, Becker-Hapak M, Neal CC, et al. Donor memory-like NK cells persist and induce remissions in pediatric patients with relapsed AML after transplant. Blood. (2022) 139:1670–83. doi: 10.1182/blood.2021013972
54. Ciurea SO, Schafer JR, Bassett R, Denman CJ, Cao K, Willis D, et al. Phase 1 clinical trial using mbIL21 ex vivo-expanded donor-derived NK cells after haploidentical transplantation. Blood. (2017) 130:1857–68. doi: 10.1182/blood-2017-05-785659
55. Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T, et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med. (2016) 8:357ra123. doi: 10.1126/scitranslmed.aaf2341
56. Qin SS, Melucci AD, Chacon AC, Prieto PA. Adoptive T cell therapy for solid tumors: pathway to personalized standard of care. Cells. (2021) 10(4):808. doi: 10.3390/cells10040808
57. Zou F, Lu L, Liu J, Xia B, Zhang W, Hu Q, et al. Engineered triple inhibitory receptor resistance improves anti-tumor CAR-T cell performance via CD56. Nat Commun. (2019) 10:4109. doi: 10.1038/s41467-019-11893-4
58. Fioretti S, Matson CA, Rosenberg KM, Singh NJ. Host B cells escape CAR-T immunotherapy by reversible downregulation of CD19. Cancer Immunol Immunother. (2023) 72:257–64. doi: 10.1007/s00262-022-03231-3
59. Chen Y, Lin J, Guo ZQ, Lin WS, Zhou ZF, Huang CZ, et al. MHC I-related chain a expression in gastric carcinoma and the efficacy of immunotherapy with cytokine-induced killer cells. Am J Cancer Res. (2015) 5:3221–30.
60. Bonanno G, Iudicone P, Mariotti A, Procoli A, Pandolfi A, Fioravanti D, et al. Thymoglobulin, interferon-gamma and interleukin-2 efficiently expand cytokine-induced killer (CIK) cells in clinical-grade cultures. J Transl Med. (2010) 8:129. doi: 10.1186/1479-5876-8-129
61. Yu SJ, Ma C, Heinrich B, Brown ZJ, Sandhu M, Zhang Q, et al. Targeting the crosstalk between cytokine-induced killer cells and myeloid-derived suppressor cells in hepatocellular carcinoma. J Hepatol. (2019) 70:449–57. doi: 10.1016/j.jhep.2018.10.040
62. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. (2011) 331:1565–70. doi: 10.1126/science.1203486
63. Gattinoni L, Powell DJ Jr, Rosenberg SA, Restifo NP. Adoptive immunotherapy for cancer: building on success. Nat Rev Immunol. (2006) 6:383–93. doi: 10.1038/nri1842
64. Yamamoto K, Venida A, Yano J, Biancur DE, Kakiuchi M, Gupta S, et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature. (2020) 581:100–5. doi: 10.1038/s41586-020-2229-5
65. Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer. (2014) 14:135–46. doi: 10.1038/nrc3670
66. Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature. (2012) 482:400–4. doi: 10.1038/nature10755
67. Akdis CA, Akdis M. Mechanisms of immune tolerance to allergens: role of IL-10 and Tregs. J Clin Invest. (2014) 124:4678–80. doi: 10.1172/JCI78891
68. Foubert P, Kaneda MM, Varner JA. PI3Kgamma Activates Integrin alpha(4) and Promotes Immune Suppressive Myeloid Cell Polarization during Tumor Progression. Cancer Immunol Res. (2017) 5:957–68. doi: 10.1158/2326-6066.CIR-17-0143
69. Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, et al. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood. (2009) 114:1537–44. doi: 10.1182/blood-2008-12-195792
70. Xie N, Shen G, Gao W, Huang Z, Huang C, Fu L. Neoantigens: promising targets for cancer therapy. Signal Transduction Targeted Ther. (2023) 8(1):9. doi: 10.1038/s41392-022-01270-x
71. Dersh D, Phelan JD, Gumina ME, Wang B, Arbuckle JH, Holly J, et al. Genome-wide screens identify lineage- and tumor-specific genes modulating MHC-I- and MHC-II-restricted immunosurveillance of human lymphomas. Immunity. (2021) 54:116–131.e10. doi: 10.1016/j.immuni.2020.11.002
72. Chen X, Lu Q, Zhou H, Liu J, Nadorp B, Lasry A, et al. A membrane-associated MHC-I inhibitory axis for cancer immune evasion. Cell. (2023) 186:3903–3920.e21. doi: 10.1016/j.cell.2023.07.016
73. Pishesha N, Harmand TJ, Ploegh HL. A guide to antigen processing and presentation. Nat Rev Immunol. (2022) 22:751–64. doi: 10.1038/s41577-022-00707-2
74. Jaeger AM, Stopfer LE, Ahn R, Sanders EA, Sandel DA, Freed-Pastor WA, et al. Deciphering the immunopeptidome in vivo reveals new tumour antigens. Nature. (2022) 607:149–55. doi: 10.1038/s41586-022-04839-2
75. Xian J, Gao L, Ren Z, Jiang Y, Pan J, Ying Z, et al. Inhibition of autophagy by berbamine hydrochloride mitigates tumor immune escape by elevating MHC-I in melanoma cells. Cells. (2024) 13(18):1537. doi: 10.3390/cells13181537
76. Soler-Agesta R, Anel A, Galluzzi L. Mitochondrial control of antigen presentation in cancer cells. Cancer Cell. (2023) 41:1849–51. doi: 10.1016/j.ccell.2023.10.001
77. Massa C, Wang Y, Marr N, Seliger B. Interferons and resistance mechanisms in tumors and pathogen-driven diseases-focus on the major histocompatibility complex (MHC) antigen processing pathway. Int J Mol Sci. (2023) 24(7):6736. doi: 10.3390/ijms24076736
78. Garrido F, Aptsiauri N. Cancer immune escape: MHC expression in primary tumours versus metastases. Immunology. (2019) 158:255–66. doi: 10.1111/imm.v158.4
79. Seliger B. Strategies of tumor immune evasion. BioDrugs. (2005) 19:347–54. doi: 10.2165/00063030-200519060-00002
80. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. (2016) 375:819–29. doi: 10.1056/NEJMoa1604958
81. Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane JP, et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun. (2017) 8:1136. doi: 10.1038/s41467-017-01062-w
82. Schirrmacher V, Fogel M, Russmann E, Bosslet K, Altevogt P, Beck L. Antigenic variation in cancer metastasis: immune escape versus immune control. Cancer Metastasis Rev. (1982) 1:241–74. doi: 10.1007/BF00046830
83. Kim SP, Vale NR, Zacharakis N, Krishna S, Yu Z, Gasmi B, et al. Adoptive cellular therapy with autologous tumor-infiltrating lymphocytes and T-cell receptor- engineered T cells targeting common p53 neoantigens in human solid tumors. Cancer Immunol Res. (2022) 10:932–46. doi: 10.1158/2326-6066.CIR-22-0040
84. Peng S, Chen S, Hu W, Mei J, Zeng X, Su T, et al. Combination neoantigen-based dendritic cell vaccination and adoptive T-cell transfer induces antitumor responses against recurrence of hepatocellular carcinoma. Cancer Immunol Res. (2022) 10:728–44. doi: 10.1158/2326-6066.CIR-21-0931
85. Zheng Y, Chen Z, Han Y, Han L, Zou X, Zhou B, et al. Immune suppressive landscape in the human esophageal squamous cell carcinoma microenvironment. Nat Commun. (2020) 11:6268. doi: 10.1038/s41467-020-20019-0
86. Deepak KGK, Vempati R, Nagaraju GP, Dasari VR, Nagini S, Rao DN, et al. Tumor microenvironment: Challenges and opportunities in targeting metastasis of triple negative breast cancer. Pharmacol Res. (2020) 153:104683. doi: 10.1016/j.phrs.2020.104683
87. Quail DF, Joyce JA. The microenvironmental landscape of brain tumors. Cancer Cell. (2017) 31:326–41. doi: 10.1016/j.ccell.2017.02.009
88. Liu Y, He S, Wang X-L, Peng W, Chen Q-Y, Chi D-M, et al. Tumour heterogeneity and intercellular networks of nasopharyngeal carcinoma at single cell resolution. Nat Commun. (2021) 12:741. doi: 10.1038/s41467-021-21043-4
89. Xu L, Tanaka S, Bonno M, Ido M, Kawai M, Yamamoto H, et al. Cord blood CD4(+)CD25(+) regulatory T cells fail to inhibit cord blood NK cell functions due to insufficient production and expression of TGF-beta1. Cell Immunol. (2014) 290:89–95. doi: 10.1016/j.cellimm.2014.05.007
90. Gneo L, Rizkalla N, Hejmadi R, Mussai F, de Santo C, Middleton G. TGF-β orchestrates the phenotype and function of monocytic myeloid-derived suppressor cells in colorectal cancer. Cancer Immunol Immunotherapy. (2022) 71:1583–96. doi: 10.1007/s00262-021-03081-5
91. Wang H, Yung MMH, Ngan HYS, Chan KKL, Chan DW. The impact of the tumor microenvironment on macrophage polarization in cancer metastatic progression. Int J Mol Sci. (2021) 22(12):6560. doi: 10.3390/ijms22126560
92. Cheng Y, Zhong X, Nie X, Gu H, Wu X, Li R, et al. Glycyrrhetinic acid suppresses breast cancer metastasis by inhibiting M2-like macrophage polarization via activating JNK1/2 signaling. Phytomedicine. (2023) 114:154757. doi: 10.1016/j.phymed.2023.154757
93. Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. (2014) 41:49–61. doi: 10.1016/j.immuni.2014.06.010
94. Hockel M, Vaupel P. Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J Natl Cancer Inst. (2001) 93:266–76. doi: 10.1093/jnci/93.4.266
95. Allard B, Allard D, Buisseret L, Stagg J. The adenosine pathway in immuno-oncology. Nat Rev Clin Oncol. (2020) 17:611–29. doi: 10.1038/s41571-020-0382-2
96. Sitkovsky M, Lukashev D, Deaglio S, Dwyer K, Robson SC, Ohta A, et al. Adenosine A2A receptor antagonists: blockade of adenosinergic effects and T regulatory cells. Br J Pharmacol. (2008) 153 Suppl 1:S457–64. doi: 10.1038/bjp.2008.23
97. O’Sullivan D, Pearce EL. Targeting T cell metabolism for therapy. Trends Immunol. (2015) 36:71–80. doi: 10.1016/j.it.2014.12.004
98. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. (2007) 109:3812–9. doi: 10.1182/blood-2006-07-035972
99. Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. (2018) 24:541–50. doi: 10.1038/s41591-018-0014-x
100. Guo D, Tong Y, Jiang X, Meng Y, Jiang H, Du L, et al. Aerobic glycolysis promotes tumor immune evasion by hexokinase2-mediated phosphorylation of IkappaBalpha. Cell Metab. (2022) 34:1312–1324.e6. doi: 10.1016/j.cmet.2022.08.002
101. Klement JD, Paschall AV, Redd PS, Ibrahim ML, Lu C, Yang D, et al. An osteopontin/CD44 immune checkpoint controls CD8+ T cell activation and tumor immune evasion. J Clin Invest. (2018) 128:5549–60. doi: 10.1172/JCI123360
102. Ruiz de Galarreta M, Bresnahan E, Molina-Sánchez P, Lindblad KE, Maier B, Sia D, et al. beta-catenin activation promotes immune escape and resistance to anti-PD-1 therapy in hepatocellular carcinoma. Cancer Discovery. (2019) 9:1124–41. doi: 10.1158/2159-8290.CD-19-0074
103. Jiang X, Wang J, Deng X, Xiong F, Ge J, Xiang B, et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol Cancer. (2019) 18:10. doi: 10.1186/s12943-018-0928-4
104. Wang X, Yang X, Zhang C, Wang Y, Cheng T, Duan L, et al. Tumor cell-intrinsic PD-1 receptor is a tumor suppressor and mediates resistance to PD-1 blockade therapy. Proc Natl Acad Sci U.S.A. (2020) 117:6640–50. doi: 10.1073/pnas.1921445117
105. Phung CD, Pham TT, Nguyen HT, Nguyen TT, Ou W, Jeong JH, et al. Anti-CTLA-4 antibody-functionalized dendritic cell-derived exosomes targeting tumor-draining lymph nodes for effective induction of antitumor T-cell responses. Acta Biomater. (2020) 115:371–82. doi: 10.1016/j.actbio.2020.08.008
106. Kennedy PT, Saulters EL, Duckworth AD, Lim YJ, Woolley JF, Slupsky JR, et al. Soluble CTLA-4 attenuates T cell activation and modulates anti-tumor immunity. Mol Ther. (2024) 32:457–68. doi: 10.1016/j.ymthe.2023.11.028
107. Platt JL, Silva I, Balin SJ, Lefferts AR, Farkash E, Ross TM, et al. C3d regulates immune checkpoint blockade and enhances antitumor immunity. JCI Insight. (2017) 2(9):e90201. doi: 10.1172/jci.insight.90201
108. Loeuillard E, Yang J, Buckarma E, Wang J, Liu Y, Conboy C, et al. Targeting tumor-associated macrophages and granulocytic myeloid-derived suppressor cells augments PD-1 blockade in cholangiocarcinoma. J Clin Invest. (2020) 130:5380–96. doi: 10.1172/JCI137110
109. Ferrari V, Lo Cascio A, Melacarne A, Tanasković N, Mozzarelli AM, Tiraboschi L, et al. Sensitizing cancer cells to immune checkpoint inhibitors by microbiota-mediated upregulation of HLA class I. Cancer Cell. (2023) 41:1717–1730.e4. doi: 10.1016/j.ccell.2023.08.014
110. Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U.S.A. (2010) 107:4275–80. doi: 10.1073/pnas.0915174107
111. Agarwal S, et al. Deletion of the inhibitory co-receptor CTLA-4 enhances and invigorates chimeric antigen receptor T cells. Immunity. (2023) 56:2388–2407.e9. doi: 10.1016/j.immuni.2023.09.001
112. Shimonishi T, Aznar MA, Rech AJ, Good CR, Kuramitsu S, Da T, et al. Up-regulation of fas ligand at early stages and down-regulation of Fas at progressed stages of intrahepatic cholangiocarcinoma reflect evasion from immune surveillance. Hepatology. (2000) 32:761–9. doi: 10.1053/jhep.2000.18192
113. Cedeno-Laurent F, Opperman MJ, Barthel SR, Hays D, Schatton T, Zhan Q, et al. Metabolic inhibition of galectin-1-binding carbohydrates accentuates antitumor immunity. J Invest Dermatol. (2012) 132:410–20. doi: 10.1038/jid.2011.335
114. Mahadevan KK, LeBleu VS, Ramirez EV, Chen Y, Li B, Sockwell AM, et al. Elimination of oncogenic KRAS in genetic mouse models eradicates pancreatic cancer by inducing FAS-dependent apoptosis by CD8(+) T cells. Dev Cell. (2023) 58:1562–1577.e8. doi: 10.1016/j.devcel.2023.07.025
115. Xiao W, Ibrahim ML, Redd PS, Klement JD, Lu C, Yang D, et al. Loss of fas expression and function is coupled with colon cancer resistance to immune checkpoint inhibitor immunotherapy. Mol Cancer Res. (2019) 17:420–30. doi: 10.1158/1541-7786.MCR-18-0455
116. Al Subeh ZY, Poschel DB, Redd PS, Klement JD, Merting AD, Yang D, et al. Lipid nanoparticle delivery of fas plasmid restores fas expression to suppress melanoma growth in vivo. ACS Nano. (2022) 16:12695–710. doi: 10.1021/acsnano.2c04420
117. Poulaki V, Mitsiades CS, Mitsiades N. The role of Fas and FasL as mediators of anticancer chemotherapy. Drug Resist Update. (2001) 4:233–42. doi: 10.1054/drup.2001.0210
118. Nakamura M, Nagano H, Sakon M, Yamamoto T, Ota H, Wada H, et al. Role of the Fas/FasL pathway in combination therapy with interferon-alpha and fluorouracil against hepatocellular carcinoma in vitro. J Hepatol. (2007) 46:77–88. doi: 10.1016/j.jhep.2006.07.032
119. Mangolini M, Maiques-Diaz A, Charalampopoulou S, Gerhard-Hartmann E, Bloehdorn J, Moore A, et al. Viral transduction of primary human lymphoma B cells reveals mechanisms of NOTCH-mediated immune escape. Nat Commun. (2022) 13(1):6220. doi: 10.1038/s41467-022-33739-2
120. Chan JD, Lai J, Slaney CY, Kallies A, Beavis PA, Darcy PK, et al. Cellular networks controlling T cell persistence in adoptive cell therapy. Nat Rev Immunol. (2021) 21:769–84. doi: 10.1038/s41577-021-00539-6
121. Rosenberg SA, Spiess P, Lafreniere R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science. (1986) 233:1318–21. doi: 10.1126/science.3489291
122. Zheng N, Fang J, Xue G, Wang Z, Li X, Zhou M, et al. Induction of tumor cell autosis by myxoma virus-infected CAR-T and TCR-T cells to overcome primary and acquired resistance. Cancer Cell. (2022) 40:973–985.e7. doi: 10.1016/j.ccell.2022.08.001
123. Yang XB, Nishimiya D, Löchte S, Jude KM, Borowska M, Savvides CS, et al. Facile repurposing of peptide-MHC-restricted antibodies for cancer immunotherapy. Nat Biotechnol. (2023) 41:932. doi: 10.1038/s41587-022-01567-w
124. Maalej KM, Merhi M, Inchakalody VP, Mestiri S, Alam M, Maccalli C, et al. CAR-cell therapy in the era of solid tumor treatment: current challenges and emerging therapeutic advances. Mol Cancer. (2023) 22(1):20. doi: 10.1186/s12943-023-01723-z
125. Lainscek D, Golob-Urbanc A, Mikolič V, Pantović-Žalig J, Malenšek Š, Jerala R, et al. Regulation of CD19 CAR-T cell activation based on an engineered downstream transcription factor. Mol Ther Oncolytics. (2023) 29:77–90. doi: 10.1016/j.omto.2023.04.005
126. Liu S, Deng B, Yin Z, Lin Y, An L, Liu D, et al. Combination of CD19 and CD22 CAR-T cell therapy in relapsed B-cell acute lymphoblastic leukemia after allogeneic transplantation. Am J Hematol. (2021) 96:671–9. doi: 10.1002/ajh.26160
127. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. (2008) 26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331
128. Dobosz P, Stępień M, Golke A, Dzieciątkowski T. Challenges of the immunotherapy: perspectives and limitations of the immune checkpoint inhibitor treatment. Int J Mol Sci. (2022) 23(5):2847. doi: 10.3390/ijms23052847
129. Zhang A, Wang S, Sun Y, Zhang Y, Zhao L, Yang Y, et al. Targeting and cytotoxicity of chimeric antigen receptor T cells grafted with PD1 extramembrane domain. Exp Hematol Oncol. (2023) 12(1):85. doi: 10.1186/s40164-023-00438-7
130. Ren JT, Liu X, Fang C, Jiang S, June CH, Zhao Y, et al. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin Cancer Res. (2017) 23:2255–66. doi: 10.1158/1078-0432.CCR-16-1300
131. Courau T, Nehar-Belaid D, Florez L, Levacher B, Vazquez T, Brimaud F, et al. TGF-β and VEGF cooperatively control the immunotolerant tumor environment and the efficacy of cancer immunotherapies. JCI Insight. (2016) 1(9):e85974. doi: 10.1172/jci.insight.85974
132. Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity. (2008) 28:546–58. doi: 10.1016/j.immuni.2008.02.017
133. Dhar S, Chakravarti M, Ganguly N, Saha A, Dasgupta S, Bera S, et al. High monocytic MDSC signature predicts multi-drug resistance and cancer relapse in non-Hodgkin lymphoma patients treated with R-CHOP. Front Immunol. (2024) 14. doi: 10.3389/fimmu.2023.1303959
134. Hou Y, Wang H, Zhu M, Ni X, Sun L, Wang W, et al. Platelet-derived TGF-β1 induces functional reprogramming of myeloid-derived suppressor cells in immune thrombocytopenia. Blood. (2023) 144(1):99–112. doi: 10.1182/blood-2023-182619
135. Ichiyama K, Long J, Kobayashi Y, Horita Y, Kinoshita T, Nakamura Y, et al. Article Transcription factor Ikzf1 associates with Foxp3 to repress gene expression in Treg cells and limit autoimmunity and anti-tumor immunity. Immunity. (2024) 57(9):2043–60.e10. doi: 10.1016/j.immuni.2024.07.010
136. Ichiyama K, Long J, Kobayashi Y, Horita Y, Kinoshita T, Nakamura Y, et al. Transcription factor Ikzf1 associates with Foxp3 to repress gene expression in Treg cells and limit autoimmunity and anti-tumor immunity. Immunity. (2024) 57:2043–2060.e10. doi: 10.1016/j.immuni.2024.07.010
137. Qu Y, Wang X, Bai S, Niu L, Zhao G, Yao Y, et al. The effects of TNF-alpha/TNFR2 in regulatory T cells on the microenvironment and progression of gastric cancer. Int J Cancer. (2022) 150:1373–91. doi: 10.1002/ijc.v150.8
138. Potzl J, Roser D, Bankel L, Hömberg N, Geishauser A, Brenner CD, et al. Reversal of tumor acidosis by systemic buffering reactivates NK cells to express IFN-gamma and induces NK cell-dependent lymphoma control without other immunotherapies. Int J Cancer. (2017) 140:2125–33. doi: 10.1002/ijc.30646
139. Villadangos JA. Antigen-specific impairment of adoptive T-cell therapy against cancer: players, mechanisms, solutions and a hypothesis. Immunol Rev. (2016) 272:169–82. doi: 10.1111/imr.2016.272.issue-1
140. Tang N, Cheng C, Zhang X, Qiao M, Li N, Mu W, et al. TGF-beta inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight. (2020) 5(4):e133977. doi: 10.1172/jci.insight.133977
141. Dou BT, Ren S, Qiu L, Zhang X, Zhang N, Cai J, et al. Prophylactic use of interleukin 6 monoclonal antibody can reduce CRS response of CAR-T cell therapy. Front Med. (2024) 10. doi: 10.3389/fmed.2023.1265835
142. Gao TA, Chen YY. Engineering next-generation CAR-T cells: overcoming tumor hypoxia and metabolism. Annu Rev Chem Biomolecular Eng. (2022) 13:193–216. doi: 10.1146/annurev-chembioeng-092120-092914
143. Saberian C, Amaria RN, Najjar AM, Radvanyi LG, Haymaker CL, Forget MA, et al. Randomized phase II trial of lymphodepletion plus adoptive cell transfer of tumor-infiltrating lymphocytes, with or without dendritic cell vaccination, in patients with metastatic melanoma. J Immunotherapy Cancer. (2021) 9(5):e002449. doi: 10.1136/jitc-2021-002449
144. Tchou J, Zhao Y, Levine BL, Zhang PJ, Davis MM, Melenhorst JJ, et al. Safety and efficacy of intratumoral injections of chimeric antigen receptor (CAR) T cells in metastatic breast cancer. Cancer Immunol Res. (2017) 5:1152–61. doi: 10.1158/2326-6066.CIR-17-0189
145. Thieblemont C, Phillips T, Ghesquieres H, Cheah CY, Clausen MR, Cunningham D, et al. Epcoritamab, a novel, subcutaneous CD3xCD20 bispecific T-cell-engaging antibody, in relapsed or refractory large B-cell lymphoma: dose expansion in a phase I/II trial. J Clin Oncol. (2023) 41:2238. doi: 10.1200/JCO.22.01725
146. Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. (2006) 314:126–9. doi: 10.1126/science.1129003
147. Bol KF, Schreibelt G, Bloemendal M, van Willigen WW, Hins-de Bree S, de Goede AL, et al. Adjuvant dendritic cell therapy in stage IIIB/C melanoma: the MIND-DC randomized phase III trial. Nat Commun. (2024) 15(1):1632. doi: 10.1038/s41467-024-45358-0
148. Gargett T, Truong NTH, Gardam B, Yu W, Ebert LM, Johnson A, et al. Safety and biological outcomes following a phase 1 trial of GD2-specific CAR-T cells in patients with GD2-positive metastatic melanoma and other solid cancers. J ImmunoTherapy Cancer. (2024) 12(5):e008659. doi: 10.1136/jitc-2023-008659
149. Zhou L, Xiong Y, Wang Y, Meng Y, Zhang W, Shen M, et al. A phase IB trial of autologous cytokine-induced killer cells in combination with sintilimab, monoclonal antibody against programmed cell death-1, plus chemotherapy in patients with advanced non-small-cell lung cancer. Clin Lung Cancer. (2022) 23:709–19. doi: 10.1016/j.cllc.2022.07.009
150. Zhang Y, Zhang Z, Ding Y, Fang Y, Wang P, Chu W, et al. Phase I clinical trial of EGFR-specific CAR-T cells generated by the piggyBac transposon system in advanced relapsed/refractory non-small cell lung cancer patients. J Cancer Res Clin Oncol. (2021) 147:3725–34. doi: 10.1007/s00432-021-03613-7
151. Adusumilli PS, Zauderer MG, Rivière I, Solomon SB, Rusch VW, O'Cearbhaill RE, et al. A phase I trial of regional mesothelin-targeted CAR T-cell therapy in patients with Malignant pleural disease, in combination with the anti–PD-1 agent pembrolizumab. Cancer Discovery. (2021) 11:2748–63. doi: 10.1158/2159-8290.CD-21-0407
152. Wang L, Fang C, Kang Q, Huang W, Chen Z, Zhao W, et al. Bispecific CAR-T cells targeting CD19/20 in patients with relapsed or refractory B cell non-Hodgkin lymphoma: a phase I/II trial. Blood Cancer J. (2024) 14(1):130. doi: 10.1038/s41408-024-01105-8
153. Jurgens EM, Firestone RS, Chaudhari J, Hosszu K, Devlin SM, Shah UA, et al. Phase I trial of MCARH109, a G protein–coupled receptor class C group 5 member D (GPRC5D)-targeted chimeric antigen receptor T-cell therapy for multiple myeloma: an updated analysis. J Clin Oncol. (2024) 43(5):498–504. doi: 10.1016/S2152-2650(24)01941-4
154. Itoh K, Tilden AB, Balch CM. Interleukin 2 activation of cytotoxic T-lymphocytes infiltrating into human metastatic melanomas. Cancer Res. (1986) 46:3011–7.
155. Draghi A, Chamberlain CA, Khan S, Papp K, Lauss M, Soraggi S, et al. Rapid identification of the tumor-specific reactive TIL repertoire combined detection of CD137, TNF, and IFNγ, following recognition of autologous tumor-antigens. Front Immunol. (2021) 12. doi: 10.3389/fimmu.2021.705422
156. Hall MS, Mullinax JE, Cox CA, Hall AM, Beatty MS, Blauvelt J, et al. Combination nivolumab, CD137 agonism, and adoptive cell therapy with tumor-infiltrating lymphocytes for patients with metastatic melanoma. Clin Cancer Res. (2022) 28:5317–29. doi: 10.1158/1078-0432.CCR-22-2103
157. Morrison BJ, Steel JC, Morris JC. Reduction of MHC-I expression limits T-lymphocyte-mediated killing of Cancer-initiating cells. BMC Cancer. (2018) 18(1):469. doi: 10.1186/s12885-018-4389-3
158. Verma C, Eremin JM, Robins A, Bennett AJ, Cowley GP, El-Sheemy MA, et al. Abnormal T regulatory cells (Tregs: FOXP3, CTLA-4), myeloid-derived suppressor cells (MDSCs: monocytic, granulocytic) and polarised T helper cell profiles (Th1, Th2, Th17) in women with large and locally advanced breast cancers undergoing neoadjuvant chemotherapy (NAC) and surgery: failure of abolition of abnormal treg profile with treatment and correlation of treg levels with pathological response to NAC. J Trans Med. (2013) 11:16. doi: 10.1186/1479-5876-11-16
159. Fang WL, Zhou T, Shi H, Yao M, Zhang D, Qian H, et al. Progranulin induces immune escape in breast cancer via up-regulating PD-L1 expression on tumor-associated macrophages (TAMs) and promoting CD8+T cell exclusion (vol 40, 4, 2021). J Exp Clin Cancer Res. (2022) 41(1):93. doi: 10.1186/s13046-022-02292-7
160. Gammelgaard OL, Terp MG, Kirkin AF, Johansen S, Traynor S, Vever H, et al. Adoptive cell transfer therapy with ex vivo primed peripheral lymphocytes in combination with anti-PDL1 therapy effectively inhibits triple-negative breast cancer growth and metastasis. Mol Cancer. (2024) 23(1):6. doi: 10.1186/s12943-023-01914-8
161. Brudno JN, Lam N, Vanasse D, Shen YW, Rose JJ, Rossi J, et al. Safety and feasibility of anti-CD19 CAR T cells with fully human binding domains in patients with B-cell lymphoma (vol 26, pg 270, 2020). Nat Med. (2020) 26:803–3. doi: 10.1038/s41591-020-0864-x
162. Zah E, Lin MY, Silva-Benedict A, Jensen MC, Chen YY. T cells expressing CD19/CD20 bispecific chimeric antigen receptors prevent antigen escape by Malignant B cells (vol 4, pg 498, 2016). Cancer Immunol Res. (2016) 4:639–41. doi: 10.1158/2326-6066.CIR-16-0108
163. Shah NN, Fry TJ. Mechanisms of resistance to CAR T cell therapy. Nat Rev Clin Oncol. (2019) 16:372–85. doi: 10.1038/s41571-019-0184-6
164. Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, et al. Convergence of acquired mutations and alternative splicing of enables resistance to CART-19 immunotherapy. Cancer Discovery. (2015) 5:1282–95. doi: 10.1158/2159-8290.CD-15-1020
165. Fischer J, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, et al. Isoforms enabling resistance to CART-19 immunotherapy are expressed in B-ALL patients at initial diagnosis. J Immunotherapy. (2017) 40:187–95. doi: 10.1097/CJI.0000000000000169
166. Sun FM, Cheng Y, Wanchai V, Guo W, Mery D, Xu H, et al. Bispecific BCMA/CD24 CAR-T cells control multiple myeloma growth. Nat Commun. (2024) 15(1):615. doi: 10.1038/s41467-024-44873-4
167. Trabolsi A, Arumov A, Schatz JH. Bispecific antibodies and CAR-T cells: dueling immunotherapies for large B-cell lymphomas. Blood Cancer J. (2024) 14(1):27. doi: 10.1038/s41408-024-00997-w
168. Dai HR, Tong C, Shi D, Chen M, Guo Y, Chen D, et al. Efficacy and biomarker analysis of CD133-directed CAR T cells in advanced hepatocellular carcinoma: a single-arm, open-label, phase II trial. Oncoimmunology. (2020) 9(1):1846926. doi: 10.1080/2162402X.2020.1846926
169. Qi CS, Liu C, Gong J, Liu D, Wang X, Zhang P, et al. Claudin18.2-specific CAR T cells in gastrointestinal cancers: phase 1 trial final results. Nat Med. (2024) 30(8):2224–34. doi: 10.1038/s41591-024-03037-z
170. Saji AS, Yang B, Hou WT, Liu X, Ren QP, Wei YF, et al. Combined NK-CIK and PD-1 inhibitor (nivolumab), an effective immunotherapy for treating intrahepatic lymphoepithelioma-like cholangiocarcinoma unassociated with EBV infection: Two case reports and a literature review. Front Oncol. (2023) 13. doi: 10.3389/fonc.2023.1090580
171. Dimitri A, Herbst F, Fraietta JA. Engineering the next-generation of CAR T-cells with CRISPR-Cas9 gene editing. Mol Cancer. (2022) 21(1):78. doi: 10.1186/s12943-022-01559-z
172. Gargett T, Fraser CK, Dotti G, Yvon ES, Brown MP. BRAF and MEK inhibition variably affect GD2-specific chimeric antigen receptor (CAR) T-cell function in vitro. J Immunotherapy. (2015) 38:12–23. doi: 10.1097/CJI.0000000000000061
173. Zhang XY, Zhang C, Qiao M, Cheng C, Tang N, Lu S, et al. Depletion of BATF in CAR-T cells enhances antitumor activity by inducing resistance against exhaustion and formation of central memory cells. Cancer Cell. (2022) 40:1407. doi: 10.1016/j.ccell.2022.09.013
174. Wellhausen N, O'Connell RP, Lesch S, Engel NW, Rennels AK, et al. Epitope base editing CD45 in hematopoietic cells enables universal blood cancer immune therapy. Sci Trans Med. (2023) 15(714):eadi1145. doi: 10.1126/scitranslmed.adi1145
Keywords: adoptive cell therapy, tumor microenvironment, immune evasion, cancer mechanisms, personalized treatment
Citation: Ruan L and Wang L (2025) Adoptive cell therapy against tumor immune evasion: mechanisms, innovations, and future directions. Front. Oncol. 15:1530541. doi: 10.3389/fonc.2025.1530541
Received: 19 November 2024; Accepted: 06 February 2025;
Published: 28 February 2025.
Edited by:
Ahmed Lasfar, Rutgers, The State University of New Jersey, United StatesReviewed by:
Jun Deng, The First Affiliated Hospital of Nanchang University, ChinaCopyright © 2025 Ruan and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lu Wang, eHVlemhpeGkxOTg4QC5vdXRsb29rLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.