 Benjamin I. Siegel
Benjamin I. Siegel Prabhumallikarjun Patil3,4
Prabhumallikarjun Patil3,4 Akul Prakash
Akul Prakash Darren M. Klawinski
Darren M. Klawinski Eugene I. Hwang
Eugene I. Hwang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 28 February 2025
Sec. Pediatric Oncology
Volume 15 - 2025 | https://doi.org/10.3389/fonc.2025.1504803
This article is part of the Research Topic Recent Biological Insights into Pediatric Brain Tumors View all 9 articles
Central nervous system tumors represent the leading cause of cancer-related mortality in children. Conventional therapies of surgery, radiation, and cytotoxic chemotherapy have insufficient efficacy for some pediatric CNS tumors and are associated with significant morbidity, prompting an ongoing need for novel treatment approaches. Identification of molecular alterations driving tumorigenesis has led to a rising interest in developing targeted therapies for these tumors. The present narrative review focuses on recent progress in targeted therapies for pediatric CNS tumors. We outline the key implicated cellular pathways, discuss candidate molecular therapies for targeting each pathway, and present an overview of the clinical trial landscape for targeted therapies in pediatric CNS tumors. We then discuss challenges and future directions for targeted therapy, including combinatorial approaches and real-time drug screening for personalized treatment planning.
The core treatment modalities for pediatric central nervous system (CNS) tumors are surgery, radiation, and cytotoxic chemotherapy. While effective for some patients, these modalities are inadequate for many CNS tumor types and can cause significant morbidity. Recently, with better understanding of the underlying molecular drivers of pediatric cancer, targeted therapy has emerged as a promising alternative, or adjunct, to traditional cancer treatment. Targeted therapy aims to disrupt specific molecular pathways that drive tumor growth and progression. The underlying principle is that by targeting specific molecules involved in the growth and spread of cancer cells, on-target effects will increase and damage to healthy tissues will be attenuated.
In this narrative review, we outline the key pathways implicated in pediatric CNS tumors and evaluate specific targets for therapeutic intervention. Using these molecular pathways as a framework, we present a primer on the clinical trial landscape for targeted therapies in pediatric CNS tumors by surveying key completed (Table 1) and ongoing (Table 2) trials. Finally, we discuss innovative approaches to employing targeted therapy, including combinatorial regimens and real-time drug screening for personalized treatment planning.
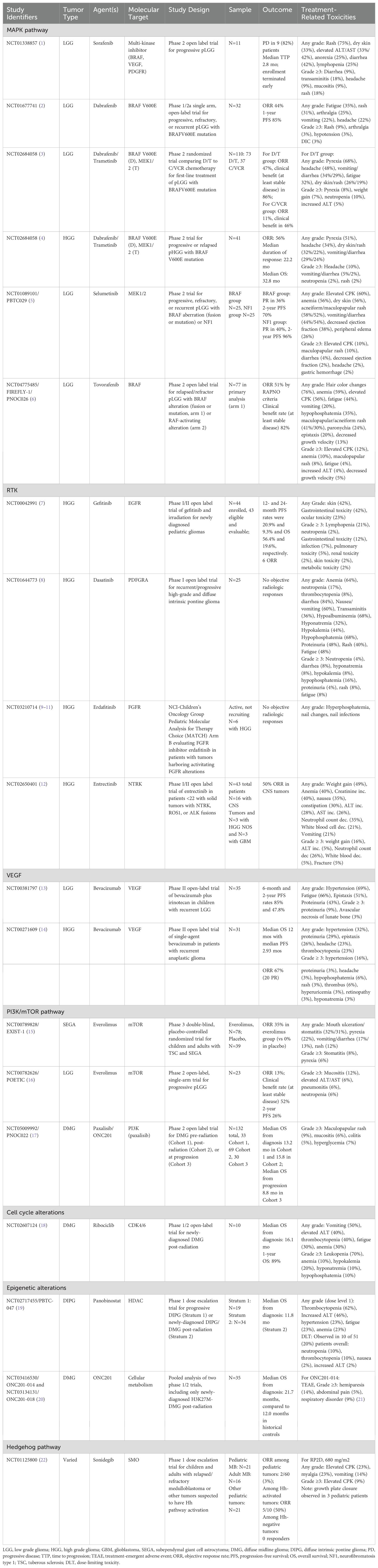
Table 1. Summary of key completed trials using molecular targeted therapy in pediatric CNS tumors.
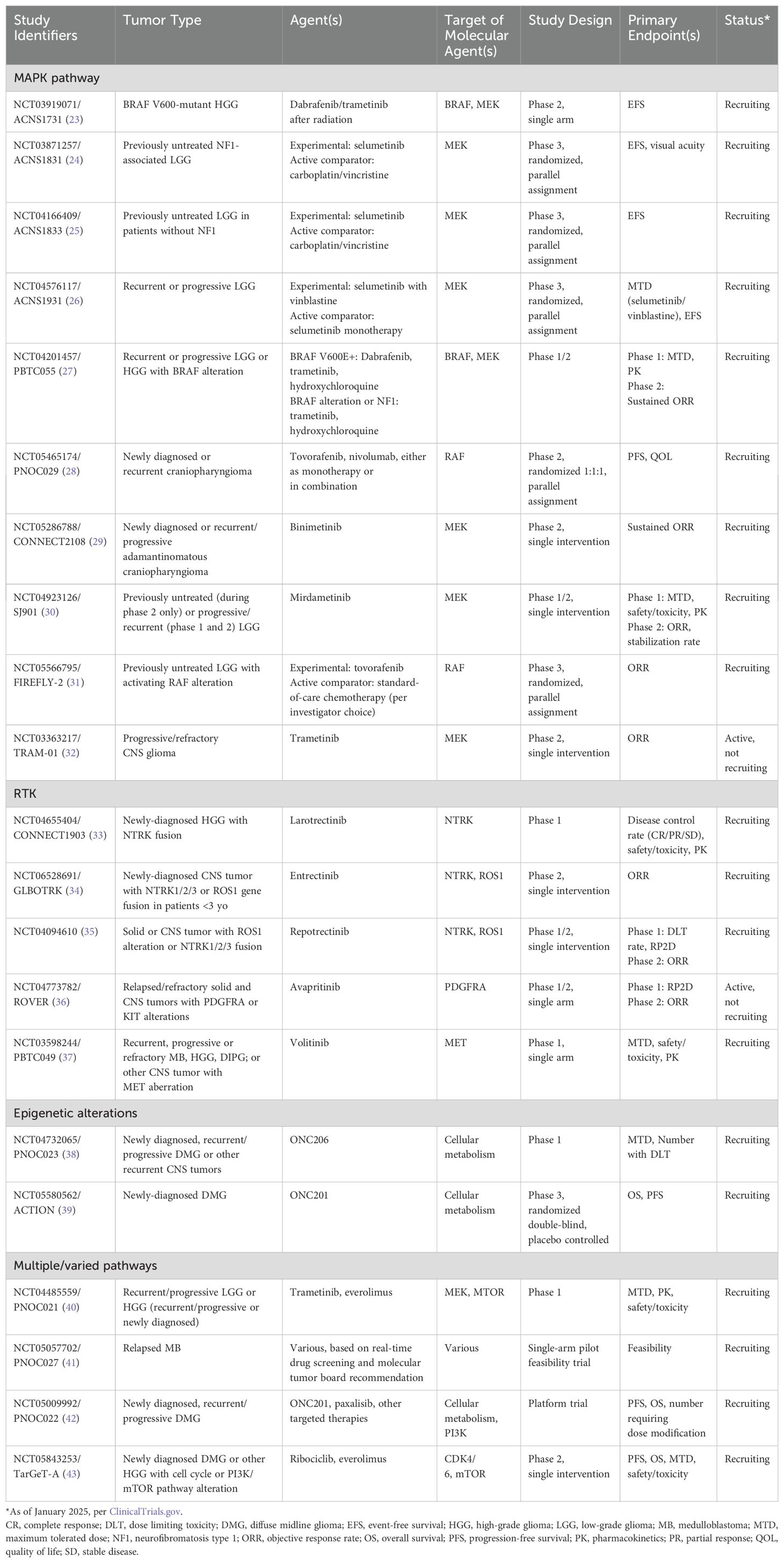
Table 2. Ongoing clinical trials using molecular targeted therapy in pediatric CNS tumors.
The mitogen-activated protein kinase (MAPK) signaling cascade is one of the first described and most extensively studied pathways in cell biology. MAPK signaling is complex with multiple upstream and downstream interactions with other major pathways involved in cell proliferation, differentiation and other metabolic signals supporting tumor sustenance and growth (Figure 1). Direct downstream RAS signaling is composed of RAS–RAF–MAPK kinase (MEK) – ERK. In physiologic conditions, this cascade is activated by receptor tyrosine kinase (RTK), which triggers the GTPase-dependent RAS. Neurofibromin-1 (NF1) is a negative regulator of RAS, which in turn activates RAF kinase. RAF is a primary mediator of the MAPK pathway and is responsible for the sequential activation of downstream targets MEK1/2 and the transcription factor ERK 1/2. ERK1/2 transcriptionally regulates genes involved in proliferation and cell survival, including cAMP response element–binding protein, as well as the transcriptional regulator Myc-like (c-Myc) and nuclear factor kappa B (NFKB) (44).
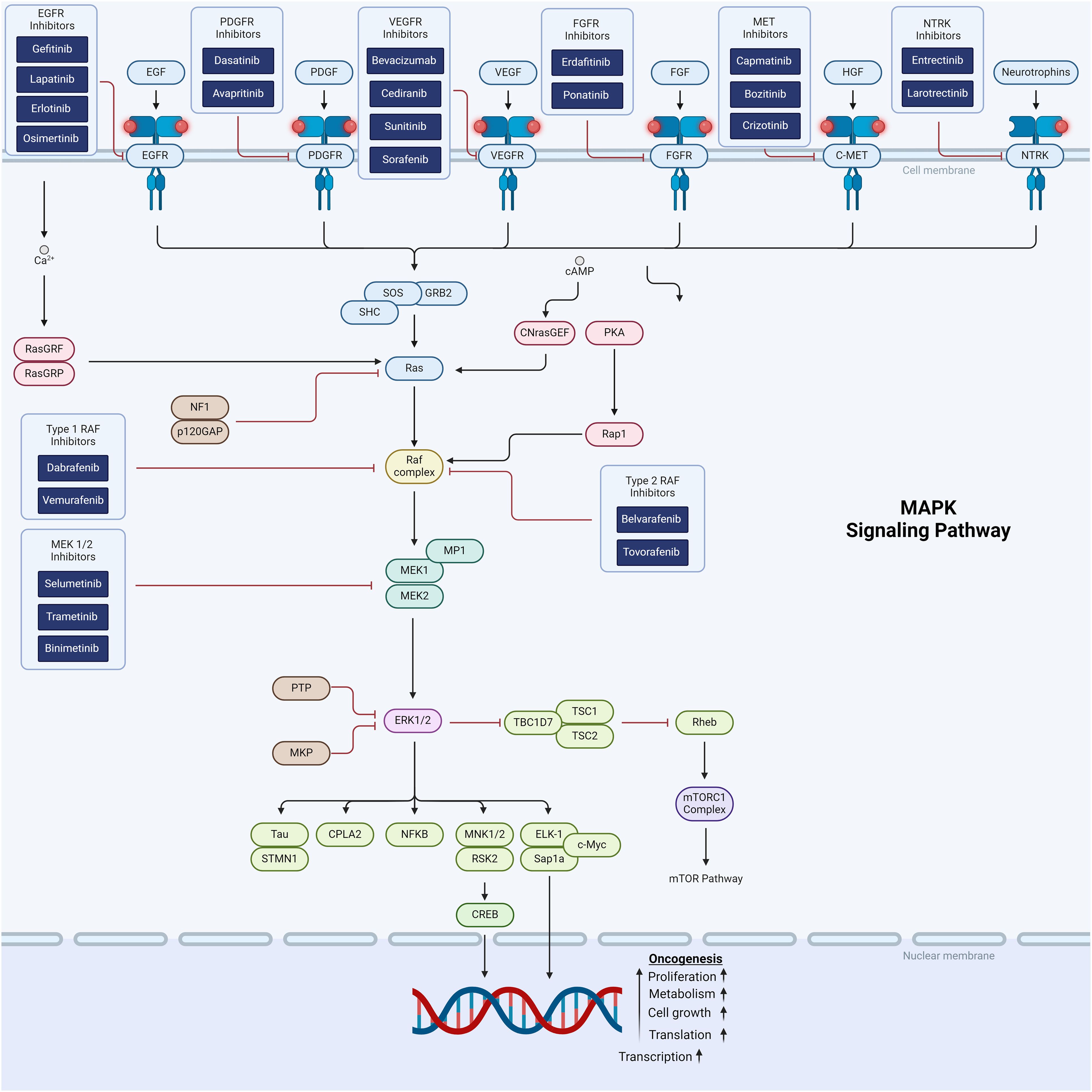
Figure 1. Schematic representation of the MAPK and RTK cellular signaling showing targets for therapeutic intervention.
The MAPK pathway is implicated in the vast majority of pediatric low grade gliomas (pLGGs), to the point that pLGG is thought to act as single pathway disease (45). Over 85% of pLGGs exhibit molecular aberrations of RAF, which ultimately upregulate the MAPK pathway (46, 47). Activating alterations of BRAF can occur as point mutations, in-frame deletions, or fusions with other kinases. The two most common alterations are the BRAFV600E mutation, caused by nucleotide transversion resulting in the substitution of valine (V) with glutamic acid (E) at position 600 (i.e., V600E point mutation) and tandem duplication resulting in BRAF-KIAA 1549 fusion (48–50). Amongst pLGGs, pleomorphic xanthoastrocytomas and gangliogliomas histologies are more commonly are associated with BRAFV600E, whereas pilocytic astrocytoma mostly harbor fusion of BRAF: KIAA1549 (51, 52). Although most data is in pLGG, 5-10% of pediatric high-grade gliomas also have MAPK pathway alterations (53). MAPK pathway activation has also been implicated in adamantinomatous craniopharyngioma (54), providing a target for multiple ongoing craniopharyngioma trials (Table 2).
The BRAF V600E mutation is seen in 15-20% of pLGG and is associated with higher resistance to chemotherapy and progression to higher grade malignancy (47, 51, 55, 56). Type-1 RAF inhibitors stabilize RAF in its active confirmation and block its catalytic activity. Dabrafenib is a Type-1 RAF inhibitor originally approved for advanced BRAFV600E-positive melanoma. In pediatric oncology, dabrafenib was first applied as a monotherapy in a phase I/IIa multicenter, open-label study in pediatric patients with advanced BRAF V600E mutation–positive solid tumors (NCT01677741). The drug was well-tolerated overall, with the most common adverse events being mild to moderate mucocutaneous and gastrointestinal toxicity. For those with pLGG, meaningful clinical benefit was noted, with an objective response rate of 44% and a 1-year estimated progression-free survival rate of 85% by independent review (2, 57). Similar safety and efficacy signals were noted in an early-phase trial of vemurafenib, another Type-1 RAF inihibitor (58, 59). A phase II study for vemurafenib in pLGG is ongoing (NCT01748149) (58).
The combination of BRAF inhibition with downstream MEK inhibition was first shown to be of clinical value in adults with non-small cell lung cancer (NSCLC) and anaplastic thyroid cancer (60–64). Based on the adult experience, a phase 1/2 trial in children with relapsed or refractory BRAFv600E pLGG were treated with either trametinib monotherapy or dual therapy with trametinib and dabrafenib. The combination group had a PR rate of 25%, compared to 15% in the monotherapy group (65). A subsequent phase 2 trial with combination of dabrafenib and trametinib was conducted for BRAFV600E pLGG in the upfront setting (3). This randomized trial compared the dabrafenib/trametinib combination to traditional chemotherapy with carboplatin/vincristine The targeted therapy group had an overall response rate of 47%, compared to 11% in the chemotherapy group. Additionally, clinical benefit (at least stable disease for >24 weeks) was observed in 86% of patients receiving dabrafenib/trametinib, compared to 46% in those receiving carboplatin/vincristine. These results led to FDA approval of dabrafenib/trametinib for upfront treatment of BRAF V600E-mutant pLGG (66). A rollover trial NCT03975829 aims to study long-term effects of therapy with dabrafenib, trametinib, or a combination of both drugs in pediatric patients.
The BRAF V600E mutation also occurs in 5-10% pHGGs (53). Data from case reports and retrospective reviews indicate that dabrafenib may be effective in relapsed and refractory pHGG with BRAFV600E mutation (4, 67). An ongoing phase 2 COG trial NCT03919071 aims to now study upfront targeted therapy, combining dabrafenib and trametinib after focal radiation for BRAF V600E–mutant pHGG.
The BRAF-KIAA fusion causes dysfunction of the BRAF N-terminal regulatory domain, which normally regulates downstream RAS/MAPK signaling. Approximately one third of pLGG exhibit the BRAF-KIAA fusion (45, 49). MEK1/2 inhibitors inhibit the MAPK pathway downstream of RAS and RAF (Figure 1), and have shown clinical activity in BRAF-altered pLGG.
Selumetinib has shown clinical benefit in phase -1 and phase -2 clinical trials of recurrent and refractory pediatric low-grade gliomas with BRAF aberrations (5, 68, 69). In a key phase 2 trial run by the PBTC consortium, selumetinib showed an objective response rate of 40% for BRAF-altered progressive or recurrent pLGG and a 2-year PFS of 70%. By comparison, the landmark COG trial evaluating carboplatin/vincristine in the upfront setting for pLGG reported an objective response rate of 46% and a 2-year PFS of 87% (5, 70). Two ongoing phase III studies aim to study standard chemotherapy to upfront selumetinib in patients with newly diagnosed pLGG in patients with or without NF1, respectively (NCT03871257 and NCT04166409).
Other MEK inhibitors studied in pediatric CNS tumors include trametinib and binimetinib. In addition to its role in upfront therapy BRAFV600E pLGG, trametinib is currently being examined in other pLGG subtypes. An ongoing clinical trial NCT03363217/TRAM-01 aims to study trametinib as a monotherapy in a basket trial involving four groups of progressive tumors (KIAA1540-BRAF fusion, NF1-associated plexiform neurofibromas, NF1-associated other gliomas, and other MAPK-ERK pathway–activated gliomas) (71). Interim analysis on 53 evaluable patients reported 25 (47%) with at least a minor response and 48 (91%) with at least stable disease (72). In a pre-clinical model, binimietinb demonstrated superior CNS penetration compared to other MEK inhibitors, prompting a phase 2 trial evaluating its efficacy in progressive pLGG (72). Of 28 evaluable patients with BRAF fusion, 12 (43%) had a partial response and 26 (93%) had at least stable disease. However, significant toxicity was observed, with 22% discontinuing due to toxicity and 49% requiring dose-reduction.
Early experience with type -1 RAF inhibitors in LGG with BRAF fusions demonstrated paradoxical pathway activation through RAF dimerization, resulting in rapid tumor progression (1). Subsequently, Type 2 RAF inhibitors have been developed including tovorafenib, which has high CNS penetrance and does not paradoxically activate RAS. The FIREFLY-1 study (NCT04775485), a phase 2 trial of tovorafenib in progressive or recurrent BRAF-altered pLGG, reported an overall response rate of 51% by RAPNO criteria and a clinical benefit rate (defined as at least stable disease) of 82% (6). The FIREFLY-1 study led to FDA approval for tovorafenib for relapsed/refractory pLGG in 2024. FIREFLY-2 (NCT05566795), a follow-up phase 3 trial evaluating tovarafenib in for pLGG in the upfront setting, is underway (9).
Receptor tyrosine kinases (RTKs) are a family of cell surface proteins which act as receptors for growth factors, hormones, cytokines, neurotrophic factors, and other extracellular signaling molecules. This family of receptors is divided into subfamilies including epidermal growth factor receptor (EGFR), platelet-derived growth factor receptor (PDGFR), fibroblast growth factor receptor (FGFR), insulin and insulin-like growth factor receptor (IGFR), vascular endothelial growth factor receptor (VEGFR), and hepatocyte growth factor receptor (HGFR/C-MET) (10, 73–77). Once activated, RTKs initiate a signal cascade primarily through two downstream pathways: RAS/MAPK/ERK and RAS/PI3K/AKT. Ultimately, these pathways result in cell proliferation, invasiveness, survival, and angiogenesis. Aberrations in RTKs are commonly found in both pediatric high- and low-grade gliomas and are therefore promising therapeutic targets for treatment (10, 73–77). The most common genetic RTK alterations in these tumors occur in the EGFR family, followed by altered PDGFR and MET tyrosine kinase pathways. Multiple RTK inhibitors have and are currently being developed and evaluated in clinical trials (10, 73–77).
Mutations in EGFR are the most common RTK aberrations in glioblastoma (GBM) and thus are an important therapeutic target. Both amplification and mutations in EGFR have been detected and are implicated in the pathogenesis and resistance to treatment of GBM cells (10, 73–77). To date, most studies with EGFR-tyrosine kinase inhibitors (TKI) or antibodies have shown limited efficacy likely due to poor CNS penetration of these drugs. Phase I and II clinical trials with first generation EGFR inhibitors gefitinib, lapatinib, and erlotinib have demonstrated marginal therapeutic response in primary and recurrent GBM. While second generation EGFR inhibitors did show response in GBM xenograft models, they showed limited activity in clinical trials of recurrent GBM. Osimertinib, a third generation EGFR inhibitor, is currently approved as first-line treatment for CNS metastatic disease in non-small cell lung cancer with EGFR mutations secondary to its high CNS penetrance and preclinical and clinical activity (74, 78). Preclinical data have demonstrated that osimertinib can reach high concentrations in the CNS and can be effective against EGFR mutated glioblastoma (78). This has prompted the use of osimertinib alone or in addition to conventional chemotherapy in patients with EGFR mutated GBM including the current study NCT03732352 (74, 79, 80). There are various reports of clinical experience using osimertinib in combination with bevacizumab or temozolomide which have not only shown the feasibility of combining EFGR inhibition with other therapy, but also a prognostic benefit in the upfront and recurrent setting following radiation (79, 81).
Dysregulation of PDGFR signaling contributes to oncogenesis in high-grade gliomas and have been associated with worse prognosis. The most common are mutations leading to amplification of PDGFRA in approximately 15% of pediatric high-grade gliomas and lead to significantly higher PDFGRA expression (10, 73–77, 82). Interestingly, an analysis of 290 pHGG reported that the mutation itself, rather than PDGFRA amplification, was of prognostic significance (82). The drug dasatinib has demonstrated high PDGFR inhibition and CNS penetration, but when used as a single agent in recurrent adult GBM it did not show efficacy (77, 82). In preclinical studies, the PDGFRA inhibitor avapritinib demonstrated significant decrease in tumor growth and improved survival in mouse models of pediatric PDGFRA mutated H3K27M DMG. Subsequently, the drug was used in 8 pediatric and young adult patients with PDGFRA-altered diffuse midline glioma (DMG) or other high-grade glioma. There were no significant acute toxicities within the cohort and 50% of patients exhibited a radiographic response (83). These findings have led to the Phase 1/2 study of avapritinib (ROVER) in pediatric patients with relapsed/refractory solid tumors dependent on KIT or PDGFRA signaling (84).
Abnormal expression of fibroblast growth factor receptors (FGFR) is the second-most common molecular aberration in sporadic pLGG (behind BRAF). In particular, FGFR1 has been shown to be disrupted through either point mutations or copy number variations and mutations (10, 73–77). The drug erdafitinib has demonstrated preclinical and clinical activity in pediatric gliomas (85) harboring FGFR mutations and has been investigated in a phase I trial for solid tumors including GBM showing partial responses. Another FGFR inhibitor ponatinib has also demonstrated favorable CNS penetration on pharmacokinetic analysis (75, 77).
The neurotrophic tropomyosin kinase (NTRK) genes (NTRK-1, NTRK-2, and NTRK-3) are located on chromosomes 1 (1q22), 9 (9q22), and 15 (15q25) and code for the receptor tyrosine kinase proteins TRK-A, TRK-B, and TRK-C, respectively. Activation of these receptors leads to downstream signaling cascades including Ras/MAPK, phospholipase C-γ (PLC-γ), and PI3-K and are involved in normal neurodevelopment (86). NTRK gene fusions are frequently reported in both pediatric and adult tumor populations and lead to constitutively activated TRK and tumorigenesis. More than 50 fusions have been described; however, the general structural rearrangement is preserved with the overall result of a chimeric protein keeping the NTRK tyrosine kinase domain ligand-independent (86). Aberrations involving the NTRK genes have been found in both pLGG and pHGG, including infant high-grade glioma (45, 53, 56, 86, 87). The prevalence of NTRK fusions has been reported in as high as 40% in infant high-grade gliomas, 10% in non-brain stem pediatric high-grade gliomas, 4% in diffuse intrinsic pontine gliomas, and < 1% of pediatric low-grade glioma (12, 86–88). Entrectinib was the first drug developed for NTRK fusionsand has good CNS penetrance. Entrectinib is further appealing for use infantile hemispheric high grade glioma because it also targets ALK and ROS1 fusions, which, in addition to NTRK fusions, are commonly seen in this tumor type (87). Entrectinib was tested in the phase I and phase II STARTRK trials and showed promising results in pediatric and adult CNS tumors harboring NTRK fusions with an ORR of 50% (12, 86), and is being further evaluated in a phase 2 trial of children less than 3 years old with CNS tumors harboring NTRK or ROS1 fusion (34). Larotrectinib was developed as a highly specific NTRK inhibitor with good CNS penetrance and antitumor activity in patients with NTRK fused CNS malignancy. Larotrectinib has been evaluated in the pediatric clinical trial SCOUT and the adult and pediatric trial NAVIGATE, which both included patients with primary CNS tumors. A pooled analysis of these trials showed that 82% of patients with measurable disease had tumor shrinkage with a 12-month PFS of 56% and favorable safety profile (86, 89, 90). A recent multicenter retrospective cohort study included 16 pediatric patients with NTRK-fusion gliomas treated with larotectinib and demonstrated an objective response rate in 11 (69%) patients (91). An early-phase clinical trial using larotrectinib in the upfront setting for pediatric HGG with NTRK fusion is underway (NCT04655404) (92).
Mesenchymal-epithelial transition (MET) is an RTK that contributes to growth and angiogenesis of pediatric high-grade glioma as it is expressed and activated in tumor cells and vascular endothelial cells resulting in cellular proliferation and invasion (10, 73–77). Genetic alterations in MET have been associated with poor prognosis in GBM (74). Inhibition of MET with capmatinib was studied in a phase II trial in adult GBM and showed no clear activity while the MET inhibitor bozitinib was tested in 18 pediatric patients with recurrent high-grade glioma with partial response seen in only 2 patients (77). Another inhibitor of MET crizotinib is being studied in combination with temozolomide and radiotherapy for newly diagnosed GBM (NCT02270034) and with dasatinib in pediatric patients with diffuse intrinsic pontine glioma and high-grade glioma (NCT01644773) (74).
High-grade gliomas have structurally and functionally abnormal vasculature. Beginning in the mid-1990s, studies showed that inhibition of vascular endothelial growth factor (VEGF) expression in GBM reduced vasculature formation and suppressed tumor growth (10, 74). The most widely used drug to target VEGF is the humanized monoclonal anti-angiogenic antibody bevacizumab, which is the first FDA-approved targeted treatment for recurrent GBM. Bevacizumab has showed improved progression-free survival (PFS) in GBM, but has not shown benefit to overall survival when used alone (14, 74). In pediatric neuro-oncology, bevacizumab has shown efficacy in combination with irinotecan in progressive or recurrent low-grade glioma (13, 93). Bevacizumab may also have specific application for optic pathway gliomas, where it has demonstrated a favorable association with visual outcomes (94, 95). The VEGFR inhibitor cediranib was investigated in a phase II clinical trial as monotherapy in recurrent GBM showing improved radiographic response at 6 months, but no overall survival benefit. The drug sunitinib, which targets PDGFRA/B in addition to VEGFR, showed preclinical promise, but it did not show improvement of PFS in patients with recurrent GBM in a phase II clinical trial (74, 77).
The PI3K/mTOR pathway is a signal transduction pathway involved in cell growth and proliferation and is another critical target for tumor-directed therapy in pediatric CNS tumors. Once activated at the cell membrane, phosphatidylinositol 3-kinase (PI3K) leads to the accumulation of PIP3, which in turn leads to phosphorylation and activation of Akt (96) (Figure 2). Activated Akt inhibits the GTPase tuberin-hamartin (TSC) complex. Inactivation of the TSC complex disinhibits mTOR, initiating downstream promotion of cell proliferation and survival. Activation of PI3K, a critical entry point to the PI3K/mTOR pathway, can be achieved through activation of transmembrane proteins including RTKs (eg, FGFR) and insulin-like growth factor 1 receptor (IGF-1R). PI3K can also be activated directly by RAS, leading to signficant crosstalk between the PI3K/mTOR and MAPK pathways. Alterations in the PI3K/mTOR pathway are ubiquitous in human cancer and have been specifically implicated in pediatric high- and low-grade glioma (97).
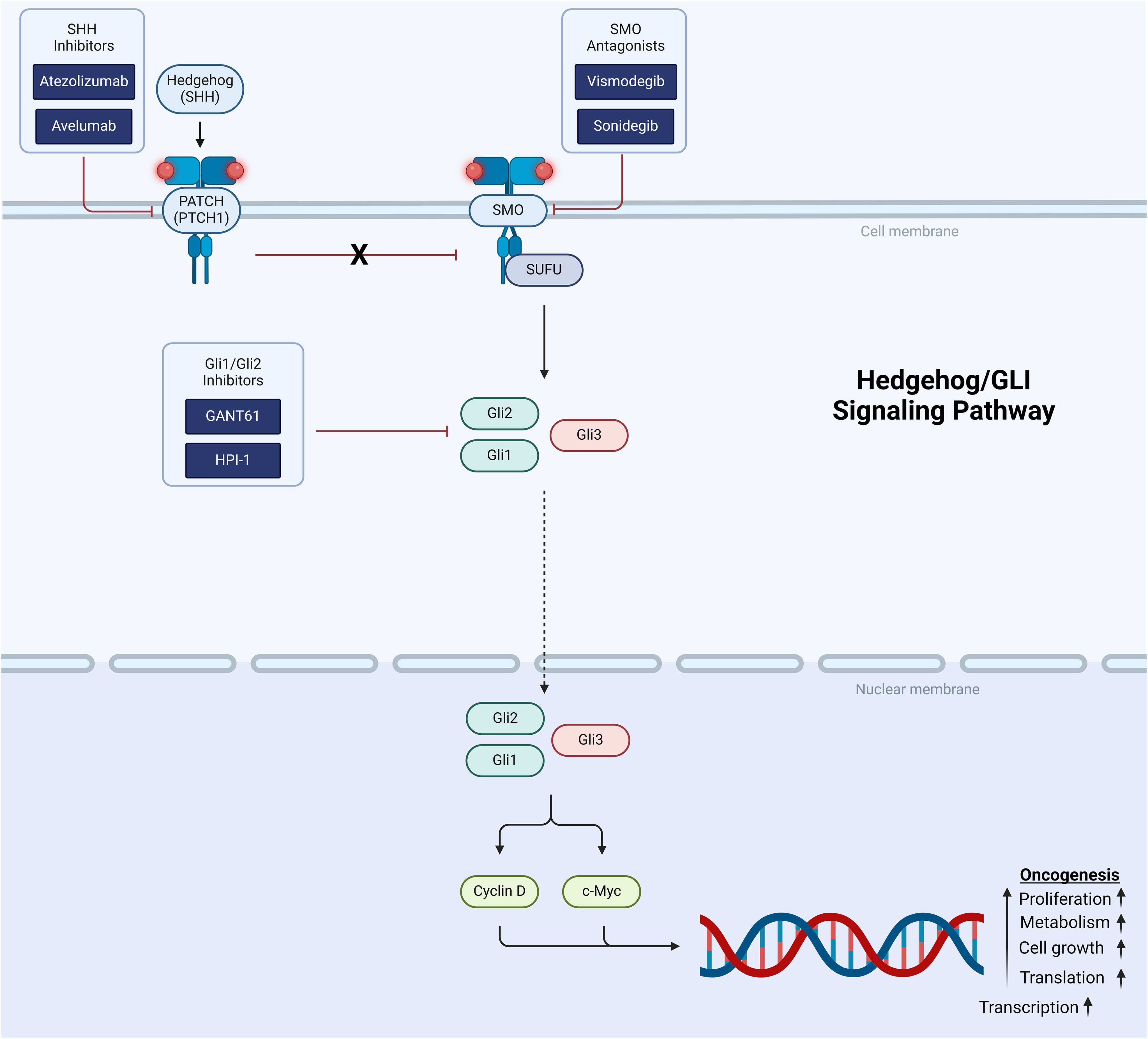
Figure 2. Schematic representation of the PI3K/mTOR signaling pathway showing target points for therapeutic intervention.
Most efforts to target the pathway are focused on inhibiting PI3K or mTOR. For example, for subependymal giant cell astrocytoma (SEGA) associated with the tumor predisposition syndrome tuberous sclerosis, the mTOR inhibitor everolimus is now FDA-approved in the upfront setting based on a landmark phase 3 clinical trial showing its efficacy (15). Attempts to apply mTOR inhibitor monotherapy to other tumor types have been less successful. A phase 2 trial of everolimus monotherapy in recurrent or progressive pediatric LGG showed a partial response rate in only 2 of 23 (13%) patients (16), and a separate study demonstrated that PI3k/mTOR pathway activation did not correlate with response (98). Similarly low response rates were observed in a trial of recurrent NF1-associated LGG treated with everolimus (99).
The crosstalk between the MAPK and PI3K/mTOR pathways creates an appealing opportunity for combinatorial treatment approaches. The combination of everolimus with the MEK inhibitor trametinib is currently being evaluated in the clinical trial NCT04485559 for patients with pediatric gliomas (either low or high grade).
Alterations in the PI3K/Akt/mTOR pathway have also been identified as molecular drivers in H3K27-altered diffuse midline glioma (53). The PI3K inhibitor paxalisib has demonstrated efficacy in pre-clinical DMG models when used in combination with other agents (100, 101), prompting its incorporation into an ongoing clinical trial with using an adaptive platform design (PNOC022/NCT05009992). Preliminary data from this trial indicate that the combination of paxalisib and ONC201 is generally well-tolerated, with the most common treatment-related adverse events being myelosuppression, rash, mucositis, and colitis (17).
Cyclin-dependent kinases (CDKs) are protein complexes involved in cell cycle regulation. CDK4 and CDK6 are of specific relevance in cancer and promote the transition from G1 to S cell cycle phases through the phosphorylation of retinoblastoma protein (Rb) (102). Inhibition of CDK4/6 by the tumor suppressor protein P16 causes G1 cell arrest by inhibiting CDK4/6 activity (103). Homozygous deletions of CDKN2A, the gene that codes for P16, result in unregulated cell division through disinhibition of CDK4/6 and have been implicated in multiple cancer types, including in BRAF V600E gliomas. Indeed, CDKN2A deletion is an independent negative predictor of outcome in V600E-mutant pLGG (51). CDKN2A homozygous deletions are also common in IDH-WT HGGs, can occur independently of BRAF alterations, and are associated with a worse clinical outcome (104).
CDK4/6 inhibitors are a class of small-molecule drugs designed to recapitulate the physiologic function of P16 that is lost with CDKN2A deletion, thereby promoting cell cycle arrest. Ribociclib is a CDK4/6 inhibitor with good CNS penetration (105). In a phase I/II clinical trial for DIPG, ribociclib was well-tolerated and associated with increased necrotic tumor volume but did not provide significant clinical benefit (18). A putative explanation for failure of CDK4/6 inhibitor monotherapy is reversal of cell-cycle arrest when the drug is withdrawn. This has led to efforts for combinatorial therapies as such as the TarGeT-A trial, which combines ribociclib with the mTOR inhibitor everolimus (Table 2).
Epigenetic changes involve modification to gene expression, rather than alteration to genes themselves. A complex interplay between DNA and histone modification results in a dynamic switching of genes “on” and “off”, as well as modulation of the level of gene expression (106). In the nucleus, DNA is packaged as chromatin. The basic structural unit of chromatin is the nucleosome, which is a coil of DNA wrapped around a histone core. The histone core is an octamer made up of 8 proteins, 2 each of H2A, H2B, H3, and H4. Each histone protein has an amino acid tail, which is relevant for gene expression, and are abundant in lysine and arginine. H3 is of specific relevance to pediatric brain tumors.
One of the ways that transcription is regulated is through modulating the wrapping and unwrapping of DNA around histone octamers. This is done through modification of the DNA itself, through methylation, and through modification of the histone amino acid tails - byacetylation and methylation. Histone acetylation results in a relative negative charge. DNA is also negatively charged, so when there is histone acetylation, there is loosening of the DNA coil around the histone, facilitating transcription. In turn, histone methylation creates a docking site for chromatin-associated proteins. Histone methylation can result in activated or repressed chromatin, depending on the site. Methylation of specific lysine residues on the amino acid tails of H3 and H4, including H3K9, H3K27, and H4K20 result in transcriptional suppression. Of particular interest in CNS tumors is trimethylation of H3K27, abbreviated H3K27me3, which results in transcriptional suppression by Polycomb Repressive Complex 2 (PRC2). By contrast, methylation of other lysine residues, including H3K4, H3K36, and H3K79 results in transcriptional activation.
In contrast to adult HGG, histone modifications are seen in about 80% of pHGG. The most common somatic alteration seen in pHGG are variants resulting in the presence of a methionine (M) instead of a lysine (K) at position 27 on the amino acid tail of histone H3, H3K27M. This results in chromatin remodeling and loss of trimethylation of H3K27 (H3K27me3) with subsequent transcriptional activation and tumorigenesis. The H3K27M mutation is a hallmark molecular finding in diffuse midline glioma (DMG) (53). DMG is associated with dismal clinical outcomes and multiple chemotherapeutic regimens have been evaluated without significant improvement on survival. In the past decade, there has been rising interest in the small molecule ONC201/dordaviprone, which showed signs of efficacy in adult patients with progressive H3K27M DMG (107). Recently, evidence has emerged indicating that the anti-tumor effect of ONC201 in DIPG is through disruption of the TCA cycle within mitochondria, leading to an inhibitory effect on histone lysine demethylases and increase in genomic H3K27me3 (20). A pooled analysis of two clinical trials (NCT03416530/ONC201-014 and NCT03134131/ONC201-018) evaluating ONC201 in non-recurrent H3K27M-mutant DMG identified a modest improvement in median overall survival compared to historical controls (OS 21.7 months vs 12.0 months, respectively) (20). ACTION (NCT05580562) is an ongoing randomized trial comparing ONC201 to placebo in newly-diagnosed H3K27M-mutant DMG (39). Another histone alteration involves replacement of glycine (G) with valine (V) or arginine (R) at position 34 on histone H3.3. G34R and G34V mutants result in transcriptional activation and are commonly seen in pediatric-type hemispheric high grade gliomas (53).
Histone deacetylases (HDACs) are enzymes which catalyze the removal of acetyl functional groups from histone proteins, ultimately resulting in gene inactivation. Inhibition of HDACs prevents deacetylation and therefore results in gene activation through chromatin opening. Panobinostat is a non-selective HDAC inhibitor that is FDA-approved for the treatment of multiple myeloma. Panobinostat has demonstrated efficacy in orthotopic xenograft models of DIPG (108). However, its application in humans has been limited by significant dose-limiting toxicity (particularly myelosuppression) (19) and limited CNS penetration (109). To achieve adequate target exposure at tolerable doses of panobinostat, novel approaches using convection-enhanced delivery (CED) are being explored (110). Fimepinostat is another HDAC inhibitor which, when used in combination with gemcitabine, demonstrated a synergistic anti-tumor effect in an orthotopic H3K27M DIPG xenograft model and represents a potential therapeutic strategy for future trials (111).
Epigenetic alterations are also seen in embryonal tumors, including medulloblastoma and atypical teratoid/rhabdoid tumor (ATRT). In both medulloblastoma and ATRT, there is overexpression of EZH2, an enzyme component of PRC2 involved in the methylation of H3K27 (106, 112). Overexpression of EZH2 results in widespread trimethylation of H3K27 and ultimately tumorigenesis through decreased tumor suppressor gene activity (113). Tazemetostat is a selective EZH2 inhibitor FDA-approved in epithelioid sarcoma. Early experience in ATRT has been promising; in a case series of 4 pediatric patients with ATRT treated with tazemetostat in the upfront setting following resection and conventional chemotherapy, a 2 had a PR and 2 had a CR, with 3 of 4 patients alive at last follow-up (OS 30-34 mo) (7).
The Hedgehog/Glioma-associated oncogene homolog (HH/GLI) pathway controls various processes during embryonic development including cerebellar maturation and tissue regeneration. Sonic hedgehog (SHH), a component of the HH/GLI pathway, is critical for normal cerebellar development, but constitutive activation of SHH signaling results in tumorigenesis (8, 11, 114, 115). Hedgehog pathway signaling involves the 12 pass-transmembrane receptor, PATCH (PTCH1) and when bound releases its inhibition of smoothened (SMO), a protein that activates the downstream portion of the pathway by binding to the cell fusion inhibitor called suppressor of fused (SUFU) and induces nuclear translocation of activators Gli1 and Gli2 and a repressor Gli3 (Figure 3). The Gli proteins regulate the expression of downstream targets including Cyclin D and MYC involved in cell survival, proliferation, and differentiation (8, 11, 114). Mutations in this pathway drive the initiation and progression of the SHH subtype of medulloblastoma as well as other solid tumors. Therefore, various agents have been developed targeting SHH, SMO and Gli1 and Gli 2 (8, 11, 114).
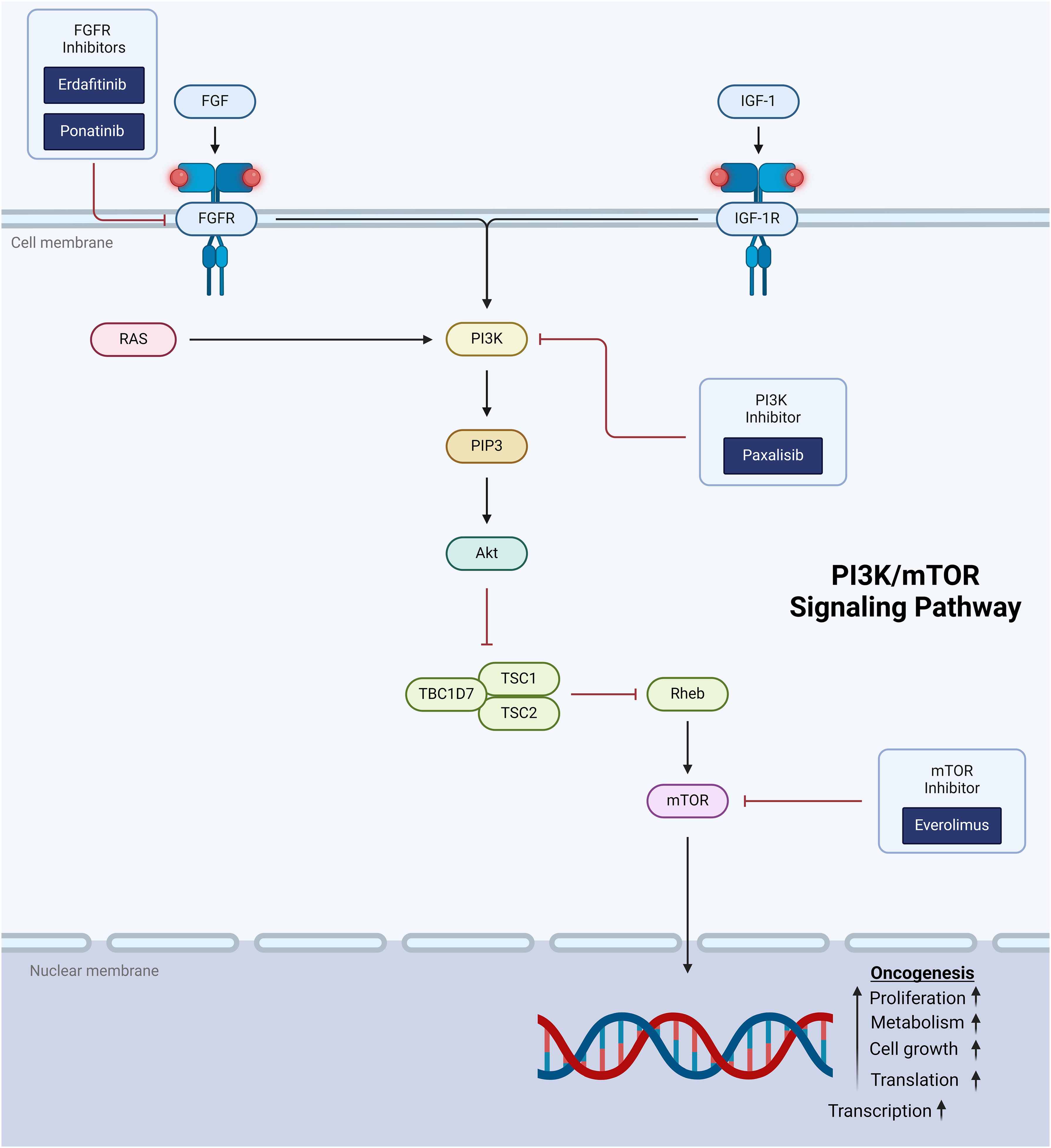
Figure 3. Schematic representation of the hedgehog/GLI signaling pathway showing targets for therapeutic intervention.
SHH inhibitors atezolizumab and avelumab prevent the binding of SHH to PTCH1 and are being studied in the preclinical setting. Vismodegib and sonidegib are SMO antagonists and were approved by the FDA in 2012 and 2015, respectively, for the treatment of advanced or metastatic basal cell carcinoma (8, 11, 114). In initial an early Phase I study with vismodegib only 1 of 3 patients with SHH activated medulloblastoma had antitumor response. This has been corroborated subsequent SMO inhibitor trials (22, 116), and may be explained by intra-group heterogeneity within the SHH subgroup. Vismodegib was also found to induce irreversible growth plate fusion with prolonged exposure (117). The drugs GANT61 and HPI-1 target Gli1 and Gli2 but have not been tested in medulloblastoma (8, 114). Resistance to SMO inhibition was first described in 2009 and the patient with medulloblastoma had mutations in the SMO gene. It has since been described that mutations in SMO lead to both de novo and acquired resistance and neither vismodegib or sonidegib are effective in certain mutations (114). Hyperactivation of GLI has been shown to lead to chemoresistance and radiation resistance in multiple cancers including medulloblastoma. This increase in the pathway ultimately suppresses the anti-tumor response from the immune system (8, 11, 114, 115, 118, 119).
The development of targeted therapies has been driven by a deepening understanding of the molecular underpinnings of pediatric CNS tumors. This expanding knowledge base has enabled the identification of specific molecular targets for therapeutic intervention. Nevertheless, success has been uneven across the pediatric neuro-oncology landscape. The greatest advances have been seen in low grade glioma, a largely single-pathway disease and currently the only pediatric CNS tumor with FDA-approved targeted therapies – dabrafenib/trametinib for upfront treatment of BRAF V600E mutant LGG and tovorafenib for BRAF-altered LGG in the recurrent setting. A common theme in this review is that monotherapy with targeted agents is rarely sufficient for durable treatment response. As illustrated in Table 2, several current clinical trials are using combinatorial approaches to achieve the sweet spot of optimizing efficacy while mitigating toxicity. These include employing multiple targeted agents addressing discrete implicated pathways (eg, everolimus/trametinib for LGG and HGG, NCT04485559), combining conventional treatment modalities with targeted agents, and using molecular therapy along with immunotherapy to create a synergistic anti-tumor effect.
Other trials are using a personalized medicine approach via a “molecular tumor board” that incorporates a patient’s individual tumor molecular profile to determine a treatment plan. In addition to providing rationale treatment recommendations for a given tumor’s molecular profile, this centralized approach has appealing equity implications by improving access to specialized care (120). The PNOC003/NCT02274987 trial used a molecular tumor board to recommend personalized treatment regimens for children with DIPG (121). The multidisciplinary tumor board considered clinical and genomic data before providing a consensus recommendation of up to 4 FDA-approved drugs to be included in the treatment regimen within 21 days of surgery (122). Thirty-eight participants were enrolled, 28 of whom were evaluable by the tumor board (123). Nineteen (68%) patients followed tumor board treatment recommendations, supporting the feasibility of the approach. There was no difference in survival for those who followed tumor board recommendations compared to those who did not. Nevertheless, experience from this trial informed the development of PNOC022, on ongoing platform trial that includes an arm for molecularly-guided combinatorial molecular therapy for DMG.
Another innovative trial design uses real-time drug screening with live tumor tissue to provide the molecular tumor board with more robust information in developing an individualized treatment plan (124). For example, the ongoing trial PNOC027/NCT05057702 conducts high-throughput drug screening on freshly isolated tumor cells of children with relapsed medulloblastoma. The platform evaluates responses to 232 clinically-available compounds. A recent preliminary report of 9 patients enrolled on the study demonstrated the feasibility of the approach: 8 of the 9 patients successfully completed real-time drugs screening, with a median turnaround time of 7 days from sample receipt (125).
While outside the scope of this review, there is also considerable interest in optimizing delivery of targeted therapies beyond traditional oral, intravenous, or intrathecal routes. For example, convection enhanced delivery, low intensity focused ultrasound, and nanoparticle-based therapies have been employed as tools to circumvent the blood-brain barrier and modulate the immune microenvironment (126–129).
An underlying impetus for developing targeted therapy, in addition to increasing treatment efficacy, is avoiding the systemic toxicities seen in traditional cytotoxic chemotherapy. With notable exceptions (eg, bevacizumab), targeted therapies have the additional benefit of availability of oral formulations, sparing patients the need for durable central venous access or hospital admissions for drug administration. Many targeted therapies are also less immunosuppressive compared to cytotoxic chemotherapies, decreasing the risk for serious infections. Nevertheless, as presented in Table 1, these drugs are not without adverse effects. Grade 1/2 “nuisance” toxicities including mucocutaneous and gastrointestinal effects are common, and serious effects on cardiac, liver, and bone marrow function have been observed. Additionally, due to their novelty, long-term effects are not well characterized.
Although promising, molecular targeted therapy remains in its infancy. Questions remain regarding the optimal sequence and duration of therapies. Strategies to address “rebound” phenomena, in which early growth is observed after therapy discontinuation, remain unsettled. Finally, while immediate toxicity profiles are generally favorable compared to traditional cytotoxic chemotherapy, long-term effects of targeted therapies on growth, fertility, and cognitive function are not yet known.
BS: Conceptualization, Supervision, Writing – original draft, Writing – review & editing. PP: Conceptualization, Visualization, Writing – original draft, Writing – review & editing. AP: Visualization, Writing – original draft, Writing – review & editing. DK: Conceptualization, Writing – original draft, Writing – review & editing. EH: Conceptualization, Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Pediatric Cancer Foundation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Karajannis MA, Legault G, Fisher MJ, Milla SS, Cohen KJ, Wisoff JH, et al. Phase II study of sorafenib in children with recurrent or progressive low-grade astrocytomas. Neuro Oncol. (2014) 16:1408–16. doi: 10.1093/neuonc/nou059
2. Hargrave DR, Bouffet E, Tabori U, Broniscer A, Cohen KJ, Hansford JR, et al. Efficacy and safety of dabrafenib in pediatric patients with BRAF V600 mutation-positive relapsed or refractory low-grade glioma: results from a phase I/IIa study. Clin Cancer Res. (2019) 25:7303–11. doi: 10.1158/1078-0432.CCR-19-2177
3. Bouffet E, Hansford JR, Garre ML, Hara J, Plant-Fox A, Aerts I, et al. Dabrafenib plus trametinib in pediatric glioma with BRAF V600 mutations. N Engl J Med. (2023) 389:1108–20. doi: 10.1056/NEJMoa2303815
4. Hargrave DR, Terashima K, Hara J, Kordes UR, Upadhyaya SA, Sahm F, et al. Phase II trial of dabrafenib plus trametinib in relapsed/refractory BRAF V600-mutant pediatric high-grade glioma. J Clin Oncol. (2023) 41:5174–83. doi: 10.1200/JCO.23.00558
5. Fangusaro J, Onar-Thomas A, Young Poussaint T, Wu S, Ligon AH, Lindeman N, et al. Selumetinib in paediatric patients with BRAF-aberrant or neurofibromatosis type 1-associated recurrent, refractory, or progressive low-grade glioma: a multicentre, phase 2 trial. Lancet Oncol. (2019) 20:1011–22. doi: 10.1016/S1470-2045(19)30277-3
6. Kilburn LB, Khuong-Quang DA, Hansford JR, Landi D, van der Lugt J, Leary SES, et al. The type II RAF inhibitor tovorafenib in relapsed/refractory pediatric low-grade glioma: the phase 2 FIREFLY-1 trial. Nat Med. (2024) 30:207–17. doi: 10.1038/s41591-023-02668-y
7. Vejmelkova K, Pokorna P, Noskova K, Faustmannova A, Drabova K, Pavelka Z, et al. Tazemetostat in the therapy of pediatric INI1-negative Malignant rhabdoid tumors. Sci Rep. (2023) 13:21623. doi: 10.1038/s41598-023-48774-2
8. Huang SY, Yang JY. Targeting the hedgehog pathway in pediatric medulloblastoma. Cancers (Basel). (2015) 7:2110–23. doi: 10.3390/cancers7040880
9. van Tilburg CM, Kilburn LB, Perreault S, Schmidt R, Azizi AA, Cruz-Martinez O, et al. LOGGIC/FIREFLY-2: a phase 3, randomized trial of tovorafenib vs. chemotherapy in pediatric and young adult patients with newly diagnosed low-grade glioma harboring an activating RAF alteration. BMC Cancer. (2024) 24:147. doi: 10.1186/s12885-024-11820-x
10. Pearson JRD, Regad T. Targeting cellular pathways in glioblastoma multiforme. Signal Transduct Target Ther. (2017) 2:17040. doi: 10.1038/sigtrans.2017.40
11. Li QR, Zhao H, Zhang XS, Lang H, Yu K. Novel-smoothened inhibitors for therapeutic targeting of naive and drug-resistant hedgehog pathway-driven cancers. Acta Pharmacol Sin. (2019) 40:257–67. doi: 10.1038/s41401-018-0019-5
12. Desai AV, Robinson GW, Gauvain K, Basu EM, Macy ME, Maese L, et al. Entrectinib in children and young adults with solid or primary CNS tumors harboring NTRK, ROS1, or ALK aberrations (STARTRK-NG). Neuro Oncol. (2022) 24:1776–89. doi: 10.1093/neuonc/noac087
13. Gururangan S, Fangusaro J, Poussaint TY, McLendon RE, Onar-Thomas A, Wu S, et al. Efficacy of bevacizumab plus irinotecan in children with recurrent low-grade gliomas - A Pediatric Brain Tumor Consortium study. Neuro-Oncology. (2014) 16:310–7. doi: 10.1093/neuonc/not154
14. Kreisl TN, Zhang W, Odia Y, Shih JH, Butman JA, Hammoud D, et al. A phase II trial of single-agent bevacizumab in patients with recurrent anaplastic glioma. Neuro Oncol. (2011) 13:1143–50. doi: 10.1093/neuonc/nor091
15. Franz DN, Belousova E, Sparagana S, Bebin EM, Frost M, Kuperman R, et al. Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet. (2013) 381:125–32. doi: 10.1016/S0140-6736(12)61134-9
16. Wright KD, Yao X, London WB, Kao PC, Gore L, Hunger S, et al. A POETIC Phase II study of continuous oral everolimus in recurrent, radiographically progressive pediatric low-grade glioma. Pediatr Blood Cancer. (2021) 68:e28787. doi: 10.1002/pbc.28787
17. Kline C, Franson A, Banerjee A, Reddy AT, Raber S, Hoffman C, et al. Trls-14. Pnoc022 report: A combination therapy trial using an adaptive platform design for patients with diffuse midline glioma at initial diagnosis, post-radiation therapy, or progression. Neuro-Oncology. (2024) 26. doi: 10.1093/neuonc/noae064.167
18. DeWire M, Fuller C, Hummel TR, Chow LML, Salloum R, de Blank P, et al. A phase I/II study of ribociclib following radiation therapy in children with newly diagnosed diffuse intrinsic pontine glioma (DIPG). J Neurooncol. (2020) 149:511–22. doi: 10.1007/s11060-020-03641-2
19. Monje M, Cooney T, Glod J, Huang J, Peer CJ, Faury D, et al. Phase I trial of panobinostat in children with diffuse intrinsic pontine glioma: A report from the Pediatric Brain Tumor Consortium (PBTC-047). Neuro Oncol. (2023) 25:2262–72. doi: 10.1093/neuonc/noad141
20. Venneti S, Kawakibi AR, Ji S, Waszak SM, Sweha SR, Mota M, et al. Clinical efficacy of ONC201 in H3K27M-mutant diffuse midline gliomas is driven by disruption of integrated metabolic and epigenetic pathways. Cancer Discovery. (2023) 13:2370–93. doi: 10.1158/2159-8290.CD-23-0131
21. Gardner SL, Tarapore RS, Allen J, McGovern SL, Zaky W, Odia Y, et al. Phase I dose escalation and expansion trial of single agent ONC201 in pediatric diffuse midline gliomas following radiotherapy. Neurooncol Adv. (2022) 4:vdac143. doi: 10.1093/noajnl/vdac143
22. Kieran MW, Chisholm J, Casanova M, Brandes AA, Aerts I, Bouffet E, et al. Phase I study of oral sonidegib (LDE225) in pediatric brain and solid tumors and a phase II study in children and adults with relapsed medulloblastoma. Neuro Oncol. (2017) 19:1542–52. doi: 10.1093/neuonc/nox109
23. A phase 2 Study of Dabrafenib (NSC763760) With Trametinib (NSC763093) After Local Irradiation in Newly-Diagnosed BRAF V600-Mutant High-Grade Glioma (HGG) [Internet] (2019). Available online at: https://clinicaltrials.gov/study/NCT03919071 (Accessed January 6, 2025).
24. A Phase 3 Randomized Study of Selumetinib Versus Carboplatin/Vincristine in Newly Diagnosed or Previously Untreated Neurofibromatosis Type 1 (NF1) Associated Low-Grade Glioma (LGG) [Internet] (2019). Available online at: https://clinicaltrials.gov/study/NCT03871257 (Accessed January 6, 2025).
25. A Phase 3 Randomized Non-Inferiority Study of Carboplatin and Vincristine Versus Selumetinib (NSC748727) in Newly Diagnosed or Previously Untreated Low-Grade Glioma (LGG) Not Associated With BRAFV600E Mutations or Systemic Neurofibromatosis Type 1 (NF1) [Internet] (2019). Available online at: https://clinicaltrials.gov/study/NCT04166409 (Accessed January 6, 2025).
26. A Phase 3 Study of Selumetinib (NSC748727) or Selumetinib in Combination With Vinblastine for Non-NF1, Non-TSC Patients With Recurrent or Progressive Low-Grade Gliomas (LGGs) Lacking BRAFV600E or IDH1 Mutations [Internet] (2020). Available online at: https://clinicaltrials.gov/study/NCT04576117 (Accessed January 6, 2025).
27. Phase I/II Trial of Dabrafenib, Trametinib, and Hydroxychloroquine (HCQ) for BRAF V600E-mutant or Trametinib and HCQ for BRAF Fusion/Duplication Positive or NF1-associated Recurrent or Progressive Gliomas in Children and Young Adults [Internet] (2019). Available online at: https://clinicaltrials.gov/study/NCT04201457 (Accessed January 6, 2025).
28. Nivolumab and Tovorafenib for the Treatment of Newly Diagnosed or Recurrent Craniopharyngioma in Children and Young Adults [Internet] (2022). Available online at: https://clinicaltrials.gov/study/NCT05465174 (Accessed January 6, 2025).
29. Phase 2 Study of the MEK Inhibitor MEKTOVI® (Binimetinib) for the Treatment of Pediatric Adamantinomatous Craniopharyngioma (2022). Available online at: https://clinicaltrials.gov/study/NCT05286788 (Accessed January 6, 2025).
30. SJ901: Phase 1/2 Evaluation of Single Agent Mirdametinib (PD-0325901), a Brain-Penetrant MEK1/2 Inhibitor, for the Treatment of Children, Adolescents, and Young Adults With Low-Grade Glioma [Internet] (2021). Available online at: https://clinicaltrials.gov/study/NCT04923126 (Accessed January 6, 2025).
31. LOGGIC/FIREFLY-2: a Phase 3, Randomized, International Multicenter Trial of DAY101 Monotherapy Versus Standard of Care Chemotherapy in Patients with Pediatric Low-Grade Glioma Harboring an Activating RAF Alteration Requiring First-Line Systemic Therapy [Internet] (2022). Available online at: https://clinicaltrials.gov/study/NCT05566795 (Accessed January 6, 2025).
32. A Phase 2 Study of Trametinib for Patients With Pediatric Glioma or Plexiform Neurofibroma With Refractory Tumor and Activation of the MAPK/ERK Pathway [Internet] (2017). Available online at: https://clinicaltrials.gov/study/NCT03363217 (Accessed January 6, 2025).
33. A Pilot and Surgical Study of Larotrectinib for Treatment of Children With Newly-Diagnosed High-Grade Glioma With NTRK Fusion [Internet] (2020). Available online at: https://clinicaltrials.gov/study/NCT04655404 (Accessed January 6, 2025).
34. PHASE 2 Study of Entrectinib as a Single Agent in Upfront Therapy for Children <3 Years of Age With NTRK1/2/3 or ROS1-FUSED CNS Tumors (GLOBOTRK) [Internet] (2024). Available online at: https://clinicaltrials.gov/study/NCT06528691 (Accessed January 6, 2025).
35. A Phase 1/2, Open-Label, Safety, Tolerability, Pharmacokinetics, and Anti-Tumor Activity Study of Repotrectinib in Pediatric and Young Adult Subjects With Advanced or Metastatic Malignancies Harboring ALK, ROS1, NTRK1-3 Alterations [Internet] (2019). Available online at: https://clinicaltrials.gov/study/NCT04094610 (Accessed January 6, 2025).
36. A Phase 1/2, Single-arm Study to Evaluate the Safety, Pharmacokinetics, and Antitumor Activity of Avapritinib in Pediatric Patients With Solid Tumors Dependent on KIT or PDGFRA Signaling [Internet] (2021). Available online at: https://clinicaltrials.gov/study/NCT04773782 (Accessed January 6, 2025).
37. A Phase I Study of Savolitinib in Recurrent, Progressive or Refractory Medulloblastoma, High-Grade Glioma, Diffuse Intrinsic Pontine Glioma, and CNS Tumors Harboring MET Aberrations [Internet] (2018). Available online at: https://clinicaltrials.gov/study/NCT03598244 (Accessed January 6, 2025).
38. PNOC023: Open Label Phase 1 and Target Validation Study of ONC206 in Children and Young Adults With Newly Diagnosed or Recurrent Diffuse Midline Glioma (DMG), and Other Recurrent Primary Malignant Central Nervous System (CNS) Tumors [Internet] (2021). Available online at: https://clinicaltrials.gov/study/NCT04732065 (Accessed January 6, 2025).
39. Arrillaga-Romany I, Lassman A, McGovern SL, Mueller S, Nabors B, van den Bent M, et al. ACTION: a randomized phase 3 study of ONC201 (dordaviprone) in patients with newly diagnosed H3 K27M-mutant diffuse glioma. Neuro Oncol. (2024) 26:S173–S81. doi: 10.1093/neuonc/noae031
40. PNOC021: A Phase I Trial Evaluating the Combination of Trametinib and Everolimus in Pediatric and Young Adult Patients With Recurrent Low-Grade Gliomas and High Grade Gliomas [Internet] (2020). Available online at: https://clinicaltrials.gov/study/NCT04485559 (Accessed January 6, 2025).
41. A Pilot Trial of Real Time Drug Screening and Genomic Testing to Determine an Individualized Treatment Plan in Children and Young Adults With Relapsed Medulloblastoma [Internet] (2021). Available online at: https://clinicaltrials.gov/study/NCT05057702 (Accessed January 6, 2025).
42. A Combination Therapy Trial Using an Adaptive Platform Design for Children and Young Adults With Diffuse Midline Gliomas (DMGs) Including Diffuse Intrinsic Pontine Gliomas (DIPGs) at Initial Diagnosis, Post-Radiation Therapy and at Time of Progression [Internet] (2021). Available online at: https://clinicaltrials.gov/study/NCT05009992 (Accessed January 6, 2025).
43. PhaseII Study of Ribociclib and Everolimus Following Radiotherapy in Pediatric and Young Adult Patients Newly Diagnosed With HGG Including DIPG, Which Harbor Alterations of the Cell Cycle and/or PI3K/mTOR Pathways [Internet] (2023). Available online at: https://clinicaltrials.gov/study/NCT05843253 (Accessed January 6, 2025).
44. Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. (2003) 3:459–65. doi: 10.1038/nrc1097
45. Ryall S, Tabori U, Hawkins C. Pediatric low-grade glioma in the era of molecular diagnostics. Acta Neuropathol Commun. (2020) 8:30. doi: 10.1186/s40478-020-00902-z
46. Schindler G, Capper D, Meyer J, Janzarik W, Omran H, Herold-Mende C, et al. Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol. (2011) 121:397–405. doi: 10.1007/s00401-011-0802-6
47. Pfister S, Janzarik WG, Remke M, Ernst A, Werft W, Becker N, et al. BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J Clin Invest. (2008) 118:1739–49. doi: 10.1172/JCI33656
48. Dougherty MJ, Santi M, Brose MS, Ma C, Resnick AC, Sievert AJ, et al. Activating mutations in BRAF characterize a spectrum of pediatric low-grade gliomas. Neuro Oncol. (2010) 12:621–30. doi: 10.1093/neuonc/noq007
49. Jones DT, Kocialkowski S, Liu L, Pearson DM, Backlund LM, Ichimura K, et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res. (2008) 68:8673–7. doi: 10.1158/0008-5472.CAN-08-2097
50. Penman CL, Faulkner C, Lowis SP, Kurian KM. Current understanding of BRAF alterations in diagnosis, prognosis, and therapeutic targeting in pediatric low-grade gliomas. Front Oncol. (2015) 5:54. doi: 10.3389/fonc.2015.00054
51. Lassaletta A, Zapotocky M, Mistry M, Ramaswamy V, Honnorat M, Krishnatry R, et al. Therapeutic and prognostic implications of BRAF V600E in pediatric low-grade gliomas. J Clin Oncol. (2017) 35:2934–41. doi: 10.1200/JCO.2016.71.8726
52. Zhang J, Wu G, Miller CP, Tatevossian RG, Dalton JD, Tang B, et al. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet. (2013) 45:602–12. doi: 10.1038/ng.2611
53. Mackay A, Burford A, Carvalho D, Izquierdo E, Fazal-Salom J, Taylor KR, et al. Integrated molecular meta-analysis of 1,000 pediatric high-grade and diffuse intrinsic pontine glioma. Cancer Cell. (2017) 32:520–37 e5. doi: 10.1016/j.ccell.2017.08.017
54. Apps JR, Gonzalez-Meljem JM, Guiho R, Pickles JC, Prince E, Schwalbe E, et al. Recurrent adamantinomatous craniopharyngiomas show MAPK pathway activation, clonal evolution and rare TP53-loss-mediated Malignant progression. Acta Neuropathol Commun. (2024) 12:127. doi: 10.1186/s40478-024-01838-4
55. Dahiya S, Haydon DH, Alvarado D, Gurnett CA, Gutmann DH, Leonard JR. BRAF(V600E) mutation is a negative prognosticator in pediatric ganglioglioma. Acta Neuropathol. (2013) 125:901–10. doi: 10.1007/s00401-013-1120-y
56. Ryall S, Zapotocky M, Fukuoka K, Nobre L, Guerreiro Stucklin A, Bennett J, et al. Integrated molecular and clinical analysis of 1,000 pediatric low-grade gliomas. Cancer Cell. (2020) 37:569–83 e5. doi: 10.1016/j.ccell.2020.03.011
57. Kieran MW, Geoerger B, Dunkel IJ, Broniscer A, Hargrave D, Hingorani P, et al. A phase I and pharmacokinetic study of oral dabrafenib in children and adolescent patients with recurrent or refractory BRAF V600 mutation-positive solid tumors. Clin Cancer Res. (2019) 25:7294–302. doi: 10.1158/1078-0432.CCR-17-3572
58. Nicolaides T, Nazemi KJ, Crawford J, Kilburn L, Minturn J, Gajjar A, et al. Phase I study of vemurafenib in children with recurrent or progressive BRAF(V600E) mutant brain tumors: Pacific Pediatric Neuro-Oncology Consortium study (PNOC-002). Oncotarget. (2020) 11:1942–52. doi: 10.18632/oncotarget.v11i21
59. Nobre L, Zapotocky M, Ramaswamy V, Ryall S, Bennett J, Alderete D, et al. Outcomes of BRAF V600E pediatric gliomas treated with targeted BRAF inhibition. JCO Precis Oncol. (2020) 4:561–71. doi: 10.1200/PO.19.00298.#91
60. Subbiah V, Kreitman RJ, Wainberg ZA, Cho JY, Schellens JHM, Soria JC, et al. Dabrafenib and trametinib treatment in patients with locally advanced or metastatic BRAF V600-mutant anaplastic thyroid cancer. J Clin Oncol. (2018) 36:7–13. doi: 10.1200/JCO.2017.73.6785
61. Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet. (2015) 386:444–51. doi: 10.1016/S0140-6736(15)60898-4
62. Kim KB, Kefford R, Pavlick AC, Infante JR, Ribas A, Sosman JA, et al. Phase II study of the MEK1/MEK2 inhibitor Trametinib in patients with metastatic BRAF-mutant cutaneous melanoma previously treated with or without a BRAF inhibitor. J Clin Oncol. (2013) 31:482–9. doi: 10.1200/JCO.2012.43.5966
63. Wen PY, Stein A, van den Bent M, De Greve J, Wick A, de Vos F, et al. Dabrafenib plus trametinib in patients with BRAF(V600E)-mutant low-grade and high-grade glioma (ROAR): a multicentre, open-label, single-arm, phase 2, basket trial. Lancet Oncol. (2022) 23:53–64. doi: 10.1016/S1470-2045(21)00578-7
64. Planchard D, Smit EF, Groen HJM, Mazieres J, Besse B, Helland A, et al. Dabrafenib plus trametinib in patients with previously untreated BRAF(V600E)-mutant metastatic non-small-cell lung cancer: an open-label, phase 2 trial. Lancet Oncol. (2017) 18:1307–16. doi: 10.1016/S1470-2045(17)30679-4
65. Bouffet E, Geoerger B, Moertel C, Whitlock JA, Aerts I, Hargrave D, et al. Efficacy and safety of trametinib monotherapy or in combination with dabrafenib in pediatric BRAF V600-mutant low-grade glioma. J Clin Oncol. (2023) 41:664–74. doi: 10.1200/JCO.22.01000
66. Barbato MI, Nashed J, Bradford D, Ren Y, Khasar S, Miller CP, et al. FDA approval summary: dabrafenib in combination with trametinib for BRAFV600E mutation-positive low-grade glioma. Clin Cancer Res. (2024) 30:263–8. doi: 10.1158/1078-0432.CCR-23-1503
67. Rosenberg T, Yeo KK, Mauguen A, Alexandrescu S, Prabhu SP, Tsai JW, et al. Upfront molecular targeted therapy for the treatment of BRAF-mutant pediatric high-grade glioma. Neuro Oncol. (2022) 24:1964–75. doi: 10.1093/neuonc/noac096
68. Banerjee A, Jakacki RI, Onar-Thomas A, Wu S, Nicolaides T, Young Poussaint T, et al. A phase i trial of the MEK inhibitor selumetinib (AZD6244) in pediatric patients with recurrent or refractory low-grade glioma: A Pediatric Brain Tumor Consortium (PBTC) study. Neuro-Oncology. (2017) 19:1135–44. doi: 10.1093/neuonc/now282
69. Fangusaro J, Onar-Thomas A, Poussaint TY, Wu S, Ligon AH, Lindeman N, et al. A phase II trial of selumetinib in children with recurrent optic pathway and hypothalamic low-grade glioma without NF1: a Pediatric Brain Tumor Consortium study. Neuro Oncol. (2021) 23:1777–88. doi: 10.1093/neuonc/noab047
70. Packer RJ, Lange B, Ater J, Nicholson HS, Allen J, Walker R, et al. Carboplatin and vincristine for recurrent and newly diagnosed low-grade gliomas of childhood. J Clin Oncol: Off J Am Soc Clin Oncol. (1993) 11:850–6. doi: 10.1200/JCO.1993.11.5.850
71. Perreault S, Larouche V, Tabori U, Hawkin C, Lippe S, Ellezam B, et al. A phase 2 study of trametinib for patients with pediatric glioma or plexiform neurofibroma with refractory tumor and activation of the MAPK/ERK pathway: TRAM-01. BMC Cancer. (2019) 19:1250. doi: 10.1186/s12885-019-6442-2
72. Perreault S, Sadat Kiaei D, Dehaes M, Larouche V, Tabori U, Hawkin C, et al. A phase 2 study of trametinib for patients with pediatric glioma or plexiform neurofibroma with refractory tumor and activation of the MAPK/ERK pathway. J Clin Oncol. (2022) 40:2042. doi: 10.1200/JCO.2022.40.16_suppl.2042
73. Regad T. Targeting RTK signaling pathways in cancer. Cancers (Basel). (2015) 7:1758–84. doi: 10.3390/cancers7030860
74. Qin A, Musket A, Musich PR, Schweitzer JB, Xie Q. Receptor tyrosine kinases as druggable targets in glioblastoma: Do signaling pathways matter? Neurooncol Adv. (2021) 3:vdab133. doi: 10.1093/noajnl/vdab133
75. Liu H, Qiu W, Sun T, Wang L, Du C, Hu Y, et al. Therapeutic strategies of glioblastoma (GBM): The current advances in the molecular targets and bioactive small molecule compounds. Acta Pharm Sin B. (2022) 12:1781–804. doi: 10.1016/j.apsb.2021.12.019
76. Hayden E, Holliday H, Lehmann R, Khan A, Tsoli M, Rayner BS, et al. Therapeutic targets in diffuse midline gliomas—An emerging landscape. Cancers. (2021) 13:6251. doi: 10.3390/cancers13246251
77. Schwark K, Messinger D, Cummings JR, Bradin J, Kawakibi A, Babila CM, et al. Receptor tyrosine kinase (RTK) targeting in pediatric high-grade glioma and diffuse midline glioma: Pre-clinical models and precision medicine. Front Oncol. (2022) 12:922928. doi: 10.3389/fonc.2022.922928
78. Chagoya G, Kwatra SG, Nanni CW, Roberts CM, Phillips SM, Nullmeyergh S, et al. Efficacy of osimertinib against EGFRvIII+ glioblastoma. Oncotarget. (2020) 11:2074–82. doi: 10.18632/oncotarget.v11i22
79. Cardona AF, Jaramillo-Velasquez D, Ruiz-Patino A, Polo C, Jimenez E, Hakim F, et al. Efficacy of osimertinib plus bevacizumab in glioblastoma patients with simultaneous EGFR amplification and EGFRvIII mutation. J Neurooncol. (2021) 154:353–64. doi: 10.1007/s11060-021-03834-3
80. Makhlin I, Salinas RD, Zhang D, Jacob F, Ming GL, Song H, et al. Clinical activity of the EGFR tyrosine kinase inhibitor osimertinib in EGFR-mutant glioblastoma. CNS Oncol. (2019) 8:CNS43. doi: 10.2217/cns-2019-0014
81. Abousaud M, Faroqui NM, Lesser G, Strowd RE, Ramkissoon SH, Kwatra M, et al. Clinical experience using osimertinib in patients with recurrent Malignant gliomas containing EGFR alterations. J Cancer Sci Clin Ther. (2021) 5:210–20. doi: 10.26502/jcsct.5079114
82. Koschmann C, Zamler D, MacKay A, Robinson D, Wu YM, Doherty R, et al. Characterizing and targeting PDGFRA alterations in pediatric high-grade glioma. Oncotarget. (2016) 7:65696–706. doi: 10.18632/oncotarget.11602
83. Trissal M, Mayr L, Schwark K, LaBelle J, Kong S, Furtner J, et al. Hgg-19. Clinical response to the pdgfra/kit inhibitor avapritinib in pediatric and young adult high-grade glioma patients with H3k27m or pdgfra genomic alterations. Neuro-Oncology. (2023) 25:i43–i4. doi: 10.1093/neuonc/noad073.168
84. Chi S, Hsieh A, Foley M, Shi H, Swamy P, Rodstrom J, et al. Ctni-24. A phase 1/2 study of avapritinib in pediatric patients with solid tumors dependent on kit or pdgfra signaling. Neuro-Oncology. (2021) 23:vi64–vi. doi: 10.1093/neuonc/noab196.249
85. Lee A, Chou AJ, Williams PM, Roy-Chowdhuri S, Patton DR, Coffey BD, et al. Erdafitinb in patients with FGFR-altered tumors: Results from the NCI-COG Pediatric MATCH trial arm B (APEC1621B). J Clin Oncol. (2023) 41:10007. doi: 10.1200/JCO.2023.41.16_suppl.10007
86. Gambella A, Senetta R, Collemi G, Vallero SG, Monticelli M, Cofano F, et al. NTRK fusions in central nervous system tumors: A rare, but worthy target. Int J Mol Sci. (2020) 21:753. doi: 10.3390/ijms21030753
87. Guerreiro Stucklin AS, Ryall S, Fukuoka K, Zapotocky M, Lassaletta A, Li C, et al. Alterations in ALK/ROS1/NTRK/MET drive a group of infantile hemispheric gliomas. Nat Commun. (2019) 10:4343. doi: 10.1038/s41467-019-12187-5
88. Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y, et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet. (2014) 46:444–50. doi: 10.1038/ng.2938
89. Laetsch TW, DuBois SG, Mascarenhas L, Turpin B, Federman N, Albert CM, et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol. (2018) 19:705–14. doi: 10.1016/S1470-2045(18)30119-0
90. Doz F, van Tilburg CM, Geoerger B, Hojgaard M, Ora I, Boni V, et al. Efficacy and safety of larotrectinib in TRK fusion-positive primary central nervous system tumors. Neuro Oncol. (2022) 24:997–1007. doi: 10.1093/neuonc/noab274
91. Lamoureux AA, Fisher MJ, Lemelle L, Pfaff E, Amir-Yazdani P, Kramm C, et al. Clinical characteristics and outcome of central nervous system tumors harboring NTRK gene fusions. Clin Cancer Res. (2024) 31(3):561–72. doi: 10.1158/1078-0432.CCR-24-0581
92. Chi SN, Lazow M, Salloum R, Lane A, Hargrave D, Witt O, et al. Trls-09. Connect1903: A pilot and surgical study of larotrectinib for treatment of children with newly diagnosed hgg with ntrk fusion (Nct04655404). Neuro-Oncology. (2024) 26. doi: 10.1093/neuonc/noae064.162
93. Packer RJ, Jakacki R, Horn M, Rood B, Vezina G, MacDonald T, et al. Objective response of multiply recurrent low-grade gliomas to bevacizumab and irinotecan. Pediatr Blood Cancer. (2009) 52:791–5. doi: 10.1002/pbc.21935
94. Bennebroek CAM, van Zwol J, Porro GL, Oostenbrink R, Dittrich ATM, Groot ALW, et al. Impact of bevacizumab on visual function, tumor size, and toxicity in pediatric progressive optic pathway glioma: A retrospective nationwide multicentre study. Cancers (Basel). (2022) 14:6087. doi: 10.3390/cancers14246087
95. Siegel BI, Nelson D, Peragallo JH, MacDonald TJ, Wolf DS. Visual outcomes after bevacizumab-based therapy for optic pathway glioma. Pediatr Blood Cancer. (2023) 70:e30668. doi: 10.1002/pbc.v70.12
96. Populo H, Lopes JM, Soares P. The mTOR signalling pathway in human cancer. Int J Mol Sci. (2012) 13:1886–918. doi: 10.3390/ijms13021886
97. Glaviano A, Foo ASC, Lam HY, Yap KCH, Jacot W, Jones RH, et al. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol Cancer. (2023) 22:138. doi: 10.1186/s12943-023-01827-6
98. Haas-Kogan DA, Aboian MS, Minturn JE, Leary SES, Abdelbaki MS, Goldman S, et al. Everolimus for children with recurrent or progressive low-grade glioma: results from the phase II PNOC001 trial. J Clin Oncol. (2024) 42:441–51. doi: 10.1200/JCO.23.01838
99. Ullrich NJ, Prabhu SP, Reddy AT, Fisher MJ, Packer R, Goldman S, et al. A phase II study of continuous oral mTOR inhibitor everolimus for recurrent, radiographic-progressive neurofibromatosis type 1-associated pediatric low-grade glioma: a Neurofibromatosis Clinical Trials Consortium study. Neuro Oncol. (2020) 22:1527–35. doi: 10.1093/neuonc/noaa071
100. Duchatel RJ, Jackson ER, Parackal SG, Kiltschewskij D, Findlay IJ, Mannan A, et al. PI3K/mTOR is a therapeutically targetable genetic dependency in diffuse intrinsic pontine glioma. J Clin Invest. (2024) 134. doi: 10.1172/JCI170329
101. Jackson ER, Duchatel RJ, Staudt DE, Persson ML, Mannan A, Yadavilli S, et al. ONC201 in combination with paxalisib for the treatment of H3K27-altered diffuse midline glioma. Cancer Res. (2023) 83:2421–37. doi: 10.1158/0008-5472.CAN-23-0186
102. Hamilton E, Infante JR. Targeting CDK4/6 in patients with cancer. Cancer Treat Rev. (2016) 45:129–38. doi: 10.1016/j.ctrv.2016.03.002
103. Lhermitte B, Wolf T, Chenard MP, Coca A, Todeschi J, Proust F, et al. Molecular heterogeneity in BRAF-mutant gliomas: diagnostic, prognostic, and therapeutic implications. Cancers (Basel). (2023) 15:1268. doi: 10.3390/cancers15041268
104. Reinhardt A, Stichel D, Schrimpf D, Sahm F, Korshunov A, Reuss DE, et al. Anaplastic astrocytoma with piloid features, a novel molecular class of IDH wildtype glioma with recurrent MAPK pathway, CDKN2A/B and ATRX alterations. Acta Neuropathol. (2018) 136:273–91. doi: 10.1007/s00401-018-1837-8
105. Patel YT, Davis A, Baker SJ, Campagne O, Stewart CF. CNS penetration of the CDK4/6 inhibitor ribociclib in non-tumor bearing mice and mice bearing pediatric brain tumors. Cancer Chemother Pharmacol. (2019) 84:447–52. doi: 10.1007/s00280-019-03864-9
106. Maury E, Hashizume R. Epigenetic modification in chromatin machinery and its deregulation in pediatric brain tumors: Insight into epigenetic therapies. Epigenetics. (2017) 12:353–69. doi: 10.1080/15592294.2016.1278095
107. Arrillaga-Romany I, Chi AS, Allen JE, Oster W, Wen PY, Batchelor TT. A phase 2 study of the first imipridone ONC201, a selective DRD2 antagonist for oncology, administered every three weeks in recurrent glioblastoma. Oncotarget. (2017) 8:79298–304. doi: 10.18632/oncotarget.17837
108. Grasso CS, Tang Y, Truffaux N, Berlow NE, Liu L, Debily MA, et al. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat Med. (2015) 21:555–9. doi: 10.1038/nm.3855
109. Zhang W, Oh JH, Zhang W, Rathi S, Larson JD, Wechsler-Reya RJ, et al. Central nervous system distribution of panobinostat in preclinical models to guide dosing for pediatric brain tumors. J Pharmacol Exp Ther. (2023) 387:315–27. doi: 10.1124/jpet.123.001826
110. Mueller S, Kline C, Stoller S, Lundy S, Christopher L, Reddy AT, et al. PNOC015: Repeated convection-enhanced delivery of MTX110 (aqueous panobinostat) in children with newly diagnosed diffuse intrinsic pontine glioma. Neuro Oncol. (2023) 25:2074–86. doi: 10.1093/neuonc/noad105
111. Wang D, Yan K, Yu H, Li H, Zhou W, Hong Y, et al. Fimepinostat impairs NF-kappaB and PI3K/AKT signaling and enhances gemcitabine efficacy in H3K27M-mutated diffuse intrinsic pontine glioma. Cancer Res. (2023) 84(4):598–615. doi: 10.1158/0008-5472.CAN-23-0394
112. Gorini F, Miceli M, de Antonellis P, Amente S, Zollo M, Ferrucci V. Epigenetics and immune cells in medulloblastoma. Front Genet. (2023) 14:1135404. doi: 10.3389/fgene.2023.1135404
113. Kim KH, Roberts CW. Mechanisms by which SMARCB1 loss drives rhabdoid tumor growth. Cancer Genet. (2014) 207:365–72. doi: 10.1016/j.cancergen.2014.04.004
114. Gampala S, Yang JY. Hedgehog pathway inhibitors against tumor microenvironment. Cells. (2021) 10:3135. doi: 10.3390/cells10113135
115. Peer E, Tesanovic S, Aberger F. Next-generation hedgehog/GLI pathway inhibitors for cancer therapy. Cancers (Basel). (2019) 11:538. doi: 10.3390/cancers11040538
116. Robinson GW, Orr BA, Wu G, Gururangan S, Lin T, Qaddoumi I, et al. Vismodegib exerts targeted efficacy against recurrent sonic hedgehog-subgroup medulloblastoma: results from phase II pediatric brain tumor consortium studies PBTC-025B and PBTC-032. J Clin Oncol. (2015) 33:2646–54. doi: 10.1200/JCO.2014.60.1591
117. Robinson GW, Kaste SC, Chemaitilly W, Bowers DC, Laughton S, Smith A, et al. Irreversible growth plate fusions in children with medulloblastoma treated with a targeted hedgehog pathway inhibitor. Oncotarget. (2017) 8:69295–302. doi: 10.18632/oncotarget.20619
118. Rudin CM, Hann CL, Laterra J, Yauch RL, Callahan CA, Fu L, et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N Engl J Med. (2009) 361:1173–8. doi: 10.1056/NEJMoa0902903
119. Avery JT, Zhang R, Boohaker RJ. GLI1: A therapeutic target for cancer. Front Oncol. (2021) 11:673154. doi: 10.3389/fonc.2021.673154
120. Kumar A, Owen JR, Sloat NT, Maynard E, Hill VM, Hubbard CB, et al. Expansion of an academic molecular tumor board to enhance access to biomarker-driven trials and therapies in the rural Southeastern United States. Curr Oncol. (2024) 31:7244–57. doi: 10.3390/curroncol31110534
121. Kline C, Jain P, Kilburn L, Bonner ER, Gupta N, Crawford JR, et al. Upfront biology-guided therapy in diffuse intrinsic pontine glioma: therapeutic, molecular, and biomarker outcomes from PNOC003. Clin Cancer Res. (2022) 28:3965–78. doi: 10.1158/1078-0432.CCR-22-0803
122. Mueller S, Jain P, Liang WS, Kilburn L, Kline C, Gupta N, et al. A pilot precision medicine trial for children with diffuse intrinsic pontine glioma-PNOC003: A report from the Pacific Pediatric Neuro-Oncology Consortium. Int J Cancer. (2019) 145:1889–901. doi: 10.1002/ijc.v145.7
123. Roberts HJ, Ravi K, Marini BL, Schepers A, Kline C, Kilburn L, et al. Retrospective comparison of targeted anticancer drugs predicted by the CNS-TAP tool versus those selected by a molecularly driven tumor board in children with DIPG. J Pediatr Hematol Oncol. (2025) 47:19–30. doi: 10.1097/MPH.0000000000002964
124. Cooney T, Lindsay H, Leary S, Wechsler-Reya R. Current studies and future directions for medulloblastoma: A review from the pacific pediatric neuro-oncology consortium (PNOC) disease working group. Neoplasia. (2023) 35:100861. doi: 10.1016/j.neo.2022.100861
125. Shatara M, Paul MR, Long-Boyle JR, Meyer A, Raber S, Yeh-Nayre L, et al. Trls-04. Early insights from pnoc027: personalized treatment approaches based on real-time drug screening and genomic testing in pediatric and adolescent patients with recurrent medulloblastoma. Neuro-Oncology. (2024) 26. doi: 10.1093/neuonc/noae064.157
126. Goodwin CR, Xu R, Iyer R, Sankey EW, Liu A, Abu-Bonsrah N, et al. Local delivery methods of therapeutic agents in the treatment of diffuse intrinsic brainstem gliomas. Clin Neurol Neurosurg. (2016) 142:120–7. doi: 10.1016/j.clineuro.2016.01.007
127. Laine AL, Huynh NT, Clavreul A, Balzeau J, Bejaud J, Vessieres A, et al. Brain tumour targeting strategies via coated ferrociphenol lipid nanocapsules. Eur J Pharm Biopharm. (2012) 81:690–3. doi: 10.1016/j.ejpb.2012.04.012
128. Beccaria K, Canney M, Bouchoux G, Puget S, Grill J, Carpentier A. Blood-brain barrier disruption with low-intensity pulsed ultrasound for the treatment of pediatric brain tumors: a review and perspectives. Neurosurg Focus. (2020) 48:E10. doi: 10.3171/2019.10.FOCUS19726
Keywords: neuro-oncology, targeted therapy, MAPK, mTOR, epigenetics, brain tumors, VEGF, receptor tyrosine kinase
Citation: Siegel BI, Patil P, Prakash A, Klawinski DM and Hwang EI (2025) Targeted therapy in pediatric central nervous system tumors: a review from the National Pediatric Cancer Foundation. Front. Oncol. 15:1504803. doi: 10.3389/fonc.2025.1504803
Received: 01 October 2024; Accepted: 23 January 2025;
Published: 28 February 2025.
Edited by:
Stefano Cairo, Champions Oncology, Inc., United StatesReviewed by:
Joanna Stefanowicz, Medical University of Gdansk, PolandCopyright © 2025 Siegel, Patil, Prakash, Klawinski and Hwang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Benjamin I. Siegel, YnNpZWdlbDFAY2hpbGRyZW5zbmF0aW9uYWwub3Jn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.