 Yuanyuan Zhang1†
Yuanyuan Zhang1† Haiming Gao
Haiming Gao Xiaoliang Liu
Xiaoliang Liu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Oncol. , 19 February 2025
Sec. Cancer Genetics
Volume 15 - 2025 | https://doi.org/10.3389/fonc.2025.1437093
Chromosome 9p21.3 is a locus associated with a rare autosomal dominant cancer predisposition syndrome characterized by early-onset melanoma and a broad spectrum of neural system tumors. Two major tumor-suppressor genes, cyclin-dependent kinase inhibitor 2A and 2B (CDKN2A and CDKN2B), as well as a large non-coding RNA ANRIL, are often co-deleted in the core region. Herein, we report a pregnant woman who had developed more than 20 plexiform neurofibromas since the age of 13 and experienced 11 times of surgical resections. No melanoma or other tumors were found. A germline 9p21.3 deletion involving CDKN2A and the first exon of ANRIL, but not CDKN2B, was identified by whole exome sequencing (WES) and confirmed by quantitative PCR. Prenatal diagnosis was performed through copy number variation-sequencing (CNV-seq), and the pregnancy was terminated with informed choice for an affected fetus. All the eight cases carrying germline 9p21.3 deletions were reviewed for genotype–phenotype correlation, showing that our case with the smallest deletion had plexiform neurofibroma only, and the two cases of Eastern Asian origin had no melanoma. Our data highlight 9p21.3 deletion as a potential differential diagnosis for neurofibroma and emphasize the importance of CNV analysis on the WES data wherein small deletions might be easily overlooked.
Chromosome 9p21.3 is a locus associated with a rare autosomal dominant cancer predisposition syndrome characterized by early-onset cutaneous melanoma and a broad spectrum of neural system tumors. Two major genes cyclin-dependent kinase inhibitor 2A and 2B (CDKN2A and CDKN2B) locate contiguously at 9p21.3, encoding p16INK4A and p14ARF by CDKN2A in an alternative reading frame, and p15INK4B by CDKN2B (1). These encoding proteins are tumor suppressors that prevent cell-cycle progression synergistically through retinoblastoma (Rb) and p53 pathways (2). In addition, a large ANRIL (CDKN2B-AS1) with sequences overlapping CDKN2B and the upstream of CDKN2A is transcribed at the antisense direction into non-coding RNA to modulate the susceptibility to various tumors through transcriptional repression of CDKN2A and CDKN2B (3, 4). Other adjacent genes including the methylthioadenosine phosphorylase (MTAP) and a small antisense non-coding RNA CDKN2A-DT are frequently co-deleted with undetermined effects in tumorigenesis.
Up to now, only seven cases carrying germline 9p21.3 deletions have been reported, with variable deletion sizes and a tumor spectrum (3, 5–10). Here, we report a pregnant woman with germline 9p21.3 heterozygous deletion involving CDKN2A, CDKN2A-DT, and the first exon of ANRIL, who presented with only multiple plexiform neurofibromas (PNFs). This is thus far the smallest 9p21.3 deletion without affecting CDKN2B, and the first prenatal diagnosis and reproductive counseling for such affected individuals.
A 31-year-old gravida 1, para 0 Chinese woman (body weight: 62 kg, height: 162 cm) was referred to genetic counseling at 14 weeks of gestation because of multiple biopsy-proven neurofibromas. She was adopted with unknown familial history (Figure 1A). The first two neurofibromas were found on her left calf at the age of 13, with diameters of 2.5 cm–3.0 cm. Subsequently, the number of neurofibromas increased by 1 or 2 each year and was up to more than 20 by the time of referral. Most of the tumors were subcutaneous centered at the left lower limb, and only one tumor was deep in the pelvic cavity. She had undergone 11 times of surgical resections of these neurofibromas (representative photos shown in Figures 1B-D). The pathological examination supported typical features of PNF (Figure 1E). No other clinical indications of neurofibromatosis type 1 (NF1), including cafe-au-lait spots, axillary freckling, or inguinal freckling, were found by physical examination or cutaneous Woods lamp examination, and no Lisch nodule was identified by ophthalmological examination.
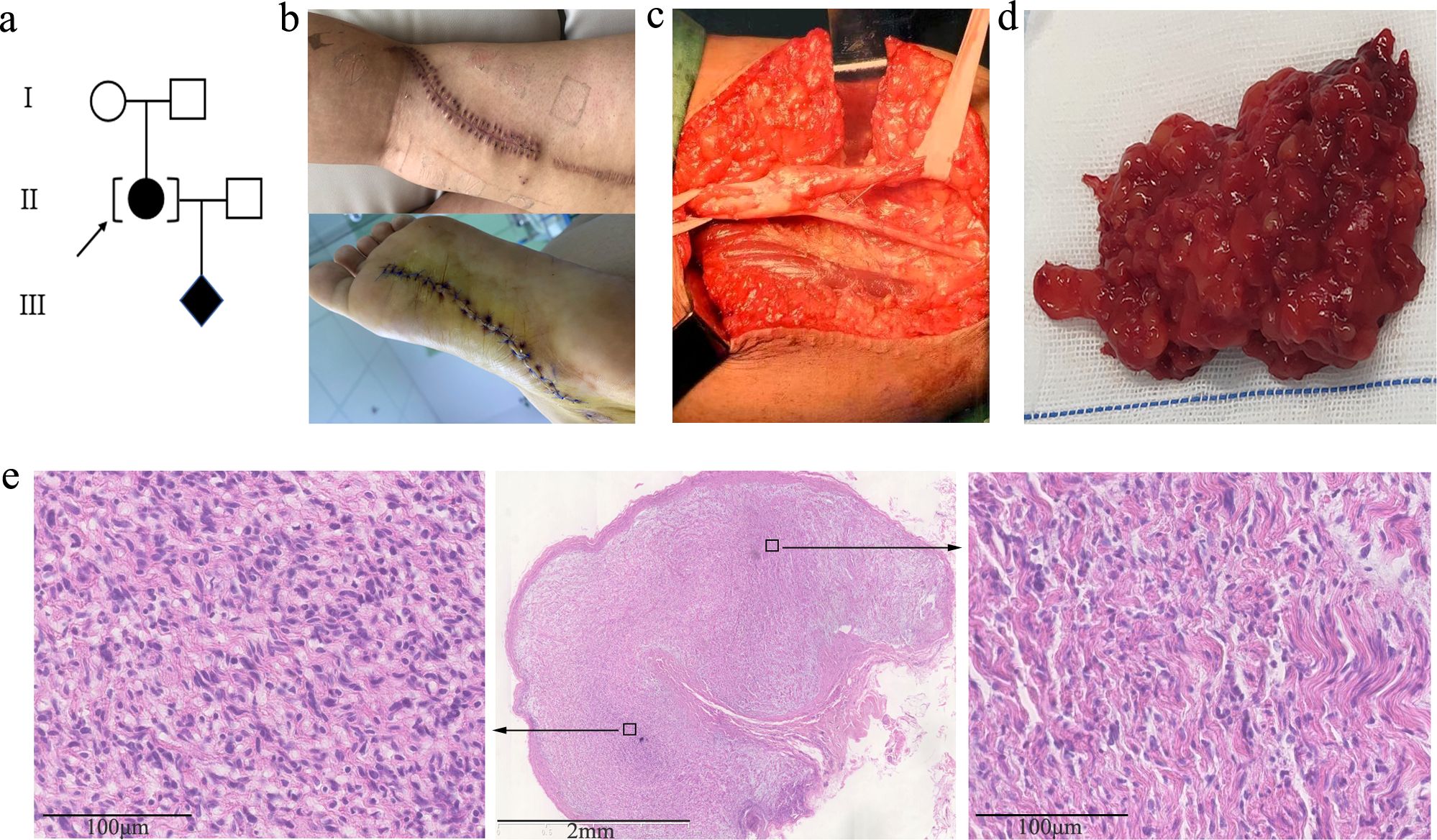
Figure 1. (A) Family pedigree of the case. (B) Photos of recent surgical incision sutures on the left leg and sole. (C) Tumor resection from the left sciatic nerve during surgery. (D) Representative gross plexiform neurofibromas tissue excised from the left leg of the proband. (E) Low (2.5×)-magnification image (middle) of histopathology by HE staining. The perineurium can be seen around the plexiform nodules. High (20×)-magnification images of local high (left) and low (right) cell density areas are framed with black squares and displayed by the black arrows. The tumor cells are spindle-shaped, with wavy nuclei and loose stroma, which is accompanied by obvious mucinous degeneration.
After obtaining written informed consent, the peripheral blood sample of the proband was collected for whole exome sequencing (WES) to seek for potential genetic defects. A small heterozygous 9p21.3 deletion involving CDKN2A, CDKN2A-DT, and exon 1 of ANRIL was suspected from the decreased read number in the WES data (Figure 2A). The deletion was predicted to be 28 kb based on the spanning of captured exons (human assembly GRCh37/hg19 chr9:21,967,137_21,995,160). For confirmation, five pairs of primers against the CDKN2A-coding region were designed for quantitative PCR (qPCR) analysis, showing that all the amplified fragments were decreased to half levels of the internal control (Figure 2B). According to the American College of Medical Genetics and Genomics (ACMG) guideline, the deletion was ranked as “pathogenic” for completely covering the established haploinsufficiency gene CDKN2A, which is related to the susceptibility to melanoma and neural system tumor syndrome (OMIM 155755), melanoma-pancreatic cancer syndrome (OMIM 606719), and melanoma, cutaneous malignant, 2 (OMIM 155601). No variation was found in the neurofibromatosis type 1 and 2 (NF1 and NF2) genes in the WES data, and MLPA (SALSA MLPA Probemix P081, P082, and P044) was also performed to exclude exon deletions or duplications in NF1 and NF2 (data not shown). We have also carefully inspected other putative variants of known genes that are correlated with neurofibroma and melanoma in the raw data of WES. All the variants with allele frequency below 0.01 are listed in Supplementary Table S1. None of them is ranked as “pathogenic” or “likely pathogenic”. Collectively, the heterozygous 9p21.3 microdeletion involving CDKN2A, CDKN2A-DT, and exon 1 of ANRIL should be responsible for the multiple PNFs in the proband. The patient was suggested to avoid smoking and alcohol, take sun protection, receive human papillomavirus (HPV) vaccination, and monitor tumors with regular follow-up.
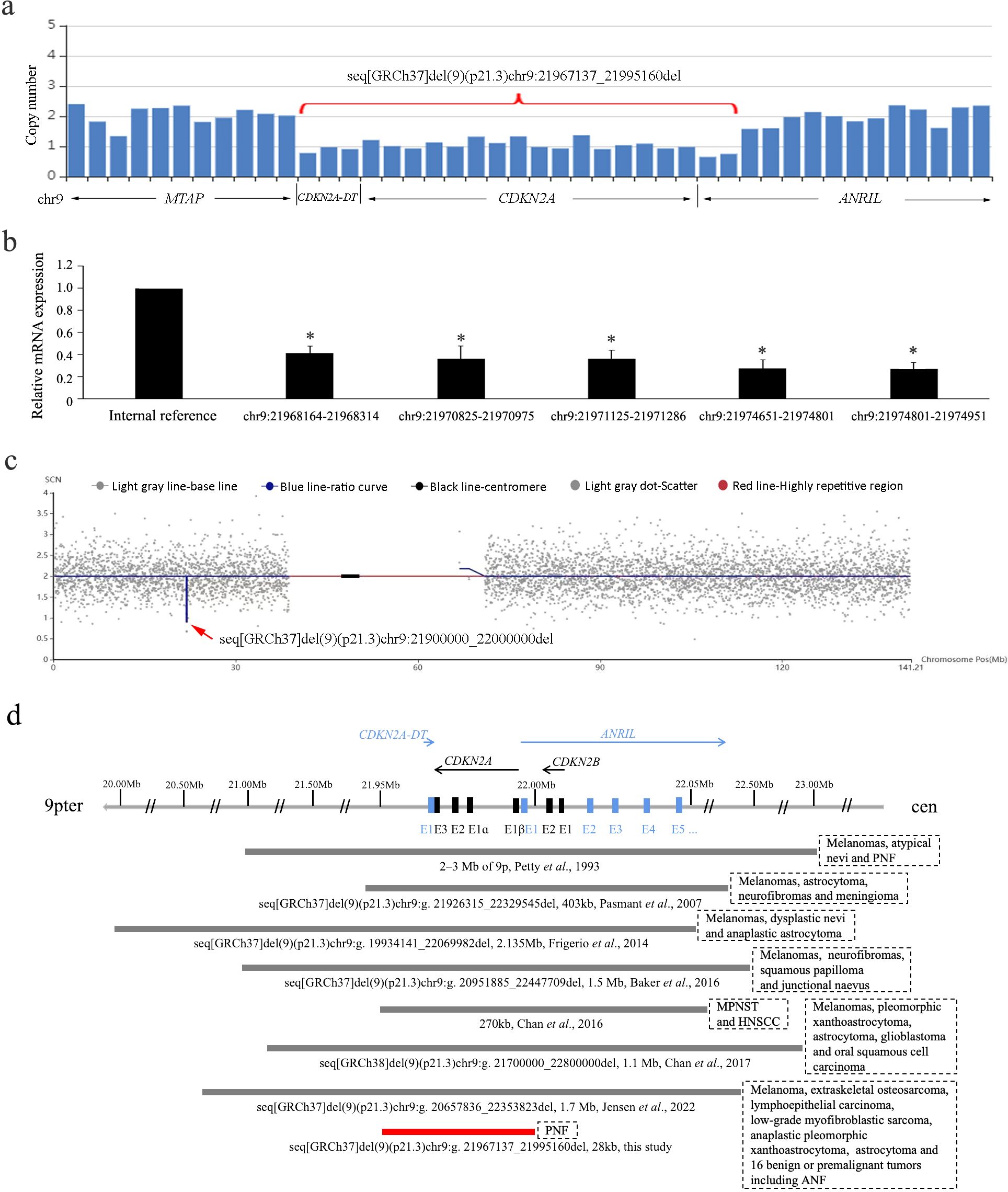
Figure 2. (A) Copy number loss of CDKN2A, CDKN2A-DT, and exon 1 of ANRIL by captured CNV analysis of the WES data. (B) Haploinsufficiency of CDKN2A gene by quantitative PCR using five pairs of primers. Data are collected in triplicate. *P < 0.05 in the ANOVA compared with internal reference. (C) Microdeletion at chromosome 9p21.3 spanning 21,900,000 to 22,000,000 by CNV-seq. The blue line indicates the ratio curve of the copy number in the patient, and the deletion is highlighted by a red arrow. (D) Schematic diagram of the reported germline 9p21.3 microdeletions involving the CDKN2A/CDKN2B/ANRIL locus. The bar lengths represent the deletion sizes. Our case is the smallest one not involving CDKN2B (indicated in red). PNF, plexiform neurofibromas; ANF, atypical neurofibromas; MPNST, malignant peripheral nerve sheath tumor; HNSCC, head and neck squamous cell carcinoma.
The couple requested for prenatal diagnosis for this torturous disease, and amniocentesis was performed at her 18 gestational weeks. Karyotype analysis of amniocytes displayed normal results, whereas copy number variation-sequencing (CNV-seq) showed a 0.1-Mb heterozygous deletion on chromosome 9p21.3 (GRCh37/hg19 chr9:21,900,000_22,000,000) (Figure 2C), covering the last exon of MTAP, CDKN2A, and CDKN2A-DT and exon 1 of ANRIL. The WES data of MTAP were rechecked in the gravida, showing normal read numbers of exon 8 and the 3'UTR region, which was confirmed by qPCR (data not shown). Considering the different resolution of WES and CNV-seq technologies, the deletion in the fetus should be maternally inherited. For posttest genetic counseling, the patient was informed of an affected fetus with the early-onset cancer predisposition syndrome. After in-depth consideration about the increased risk of premalignant and malignant tumors, the couple terminated the pregnancy at 20 gestational weeks with informed choice. For 50% chance of inheriting the small deletion, preimplantation genetic testing (PGT) was suggested for her next pregnancy using the remaining DNA of amniocytes for haplotype construction. By the time of a recent follow-up, the woman had been pregnant after PGT. Amniocentesis was also performed for prenatal diagnosis, showing normal results of karyotyping and CNV-seq.
A total of eight cases with 9p21.3 microdeletions have been reported in literature (including the present case) (Figure 2D). They are from different ethnic origins, among which Caucasian was the most frequent (3/8). The deletion sizes ranged from 28 kb to more than 2 Mb. All the deletions encompassed the entire CDKN2A/CDKN2B/ANRIL locus and the flanking genes except for our case. Phenotypically, melanoma was characteristic that occurred in 75% (6/8) of the cases. The two cases without melanoma were from Singapore and China. Other cutaneous implications included atypical/dysplastic nevi or junctional naevus (3/8), and squamous papilloma or carcinoma (3/8). Neural system tumors of various types occurred in all the cases, including neurofibromas (4/8), astrocytoma (4/8), peripheral nerve sheath tumor (2/8), and meningiomas (1/8). Moreover, other tumors were also reported in one patient including extraskeletal osteosarcoma, low-grade myofibroblastic sarcoma, and lymphoepithelial carcinoma. Comparatively, our case with the smallest deletion had the mildest symptom of PNF only and, together with the other Eastern Asian origin, had no melanoma.
Germline 9p21.3 deletions involving the contiguous CDKN2A/CDKN2B/ANRIL region exhibit cancer predisposition syndrome with characteristics overlapping NF1, familial atypical multiple mole melanoma syndrome (FAMMM), and Li–Fraumeni syndrome (LFS) (7, 8). The proband was previously thought to have NF1 for her early onset and rapid increase of neurofibromas. Yet, no cafe-au-lait spots, axillary/inguinal freckling, or Lisch nodule were observed after examination. No small sequence variation or exon deletion/duplication was found in NF1 and NF2 by WES and MLPA. By going through the WES data prudently, we found decreased reads of the contiguous CDKN2A, CDKN2A-DT, and exon 1 of ANRIL, which support a small heterozygous 9p21.3 deletion spanning 28 kb. The core protein-coding CDKN2A gene was validated for heterozygous deletion by qPCR using five pairs of primers, which substantiated the microdeletion as “pathogenic” for covering this established haploinsufficiency gene. It is usually challenging to recognize small CNVs by WES through short-read next generation. For patients with multiple neurofibromas, there is a necessity to look over the captured CNV on 9p21.3.
Our case is the smallest germline 9p21.3 deletion involving CDKN2A but not CDKN2B. It is well known that CDKN2A is the most frequently mutated high-risk gene in familial melanoma. Carriers have a risk of melanoma that is greater than a 65-fold increase compared with the normal population, and a lifetime penetrance for melanoma of 60% to 90% (11). Owning to the common embryologic precursor of the neuroectoderm, some germline CDKN2A mutations are prone to develop both melanomas and neural system tumors (12, 13). It is interesting that our patient was found with neither melanoma nor any presymptom of dysplastic nevus. No other cutaneous tumors were observed, either. By reviewing literature, only one Singaporean case with germline 9p21.3 deletion involving CDKN2A, CDKN2B, and partial MTAP and ANRIL genes had no melanoma but squamous cell carcinoma in the head and neck (8). The additive role of CDKN2B and other contiguous genes might contribute to the development of melanoma, since p15 expression encoded by CDKN2B has been shown to differentiate nevus from melanoma (14). Meanwhile, the ethnic background of Eastern Asian origin, environmental factors, and the strong willingness to take sun protection could not be ignored. More Asian cases are needed for further correlation, and the possibility of late-onset melanoma in these cases cannot be ruled out.
By literature reviewing, all the previously reported cases with germline 9p21.3 deletion encompassed the entire CDKN2A/CDKN2B/ANRIL locus. The tumor spectrum was broad, including but not limited to malignant melanomas and neural system tumors (astrocytoma, neurofibromas, schwannomas, and meningiomas). An Iraqi case with 1.7-Mb deletion involving FOCAD, IFN gene cluster, MTAP, CDKN2A, CDNK2A-DT, CDKN2B, and ANRIL had multiple malignancies including melanoma, extraskeletal osteosarcoma, lymphoepithelial carcinoma, low-grade myofibroblastic sarcoma, anaplastic pleomorphic xanthoastrocytoma, astrocytoma, and 16 benign or premalignant tumors (10), while the Italian case with a larger 2.135-Mb deletion presented with fewer tumors including melanomas, dysplastic nevi, and anaplastic astrocytoma (6). The tumor proneness could not be simply related to the size of the deletions, especially for genes with limited knowledge of the associated cancer risks. The interaction with other genomic loci may also play a role. Comparatively, our patient carried the smallest 28-kb deletion involving CDKN2A, CDNK2A-DT, and the first exon of ANRIL. CDNK2A-DT is a small antisense non-coding RNA gene located at the 3'UTR region of CDKN2A, with an unknown role in tumorigenesis. ANRIL is a large antisense non-coding RNA gene (126-kb region, consisting of 19–21 exons) with the first exon located in the promoter region of CDKN2A, regulating cancer progression through epigenetic silencing of other genes in this cluster (15). It is not sure whether the deletion of its first exon could completely disrupt the transcription of ANRIL and abolish its role in tumorigenesis. Our proband had only PNF with benign nature at present, strengthening the contributive effect of at least CDKN2B and possibly ANRIL in tumorigenesis.
It is a clinical challenge for posttest genetic counseling of cancer predisposition syndromes that related to reproductive choice. The ACMG guideline for prenatal WES recommended reporting of some late-onset genes including cancer genes as secondary findings. PNF is a kind of histologically benign peripheral nerve sheath tumor. However, it causes functional damage, deformities, and pain and increases the risk of transformation into atypical neurofibromas (ANF) and further into aggressive malignant peripheral nerve sheath tumor (MPNST) (16, 17). Our patient had suffered a lot including 11 times of surgical resections, the requirement for prenatal diagnosis by the couple was allowed, and the informed choice of termination of the pregnancy of an affected fetus was also respected. Since the gravida was adopted with unavailability of pedigree tracing and deletion was much smaller than 1 Mb, haplotype construction was achieved using the remaining amniocyte DNA and a pregnancy of an unaffected fetus was obtained after PGT and subsequent prenatal diagnosis.
In conclusion, we described the tumorous characteristics of multiple PNFs without melanoma in a pregnant woman carrying the smallest germline 9p21.3 microdeletion involving CDNK2A but not CDNK2B. Our data expand the genotype–phenotype correlation of 9p21.3 microdeletion, highlight 9p21.3 deletion as a potential differential diagnosis for neurofibroma, and emphasize the importance of CNV analysis on the WES data wherein small deletions might be easily overlooked. We also share our experience in prenatal diagnosis, genetic counseling, and reproductive choice for the affected individuals.
The datasets presented in this article are not readily available due to privacy-related concerns. Requests to access the datasets should be directed to the corresponding authors.
The studies involving humans were approved by Ethics Committee of Shengjing Hospital of China Medical University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
YZ: Writing – original draft, Funding acquisition. XL: Writing – original draft, Supervision. HG: Writing – original draft, Methodology. CQ: Writing – review & editing, Conceptualization. XlL: Writing – review & editing, Funding acquisition.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the grants from the National Natural Science Foundation (32300488), Natural Science Foundation of Liaoning Province (2022-MS-208), Science and Technology Foundation of Liaoning Province (2023JH2/20200102), and Talent project of Reproductive Hospital of China Medical University.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2025.1437093/full#supplementary-material
1. Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell. (1995) 83:993–1000. doi: 10.1016/0092-8674(95)90214-7
2. Sharpless NE, DePinho RA. The INK4A/ARF locus and its two gene products. Curr Opin Genet Dev. (1999) 9:22–30. doi: 10.1016/S0959-437X(99)80004-5
3. Pasmant E, Laurendeau I, Héron D, Vidaud M, Vidaud D, Bièche I. Characterization of a germ-line deletion, including the entire INK4/ARF locus, in a melanoma-neural system tumor family: identification of ANRIL, an antisense noncoding RNA whose expression coclusters with ARF. Cancer Res. (2007) 67:3963–9. doi: 10.1158/0008-5472.CAN-06-2004
4. Kong Y, Hsieh CH, Alonso LC. ANRIL: A lncRNA at the CDKN2A/B Locus With Roles in Cancer and Metabolic Disease. Front Endocrinol (Lausanne). (2018) 9:405. doi: 10.3389/fendo.2018.00405
5. Petty EM, Gibson LH, Fountain JW, Bolognia JL, Yang-Feng TL, Housman DE, et al. Molecular definition of a chromosome 9p21 germ-line deletion in a woman with multiple melanomas and a plexiform neurofibroma: implications for 9p tumor-suppressor gene(s). Am J Hum Genet. (1993) 53:96–104.
6. Frigerio S, Disciglio V, Manoukian S, Peissel B, Della Torre G, Maurichi A, et al. A large de novo 9p21.3 deletion in a girl affected by astrocytoma and multiple melanoma. BMC Med Genet. (2014) 15:59. doi: 10.1186/1471-2350-15-59
7. Baker MJ, Goldstein AM, Gordon PL, Harbaugh KS, Mackley HB, Glantz MJ, et al. An interstitial deletion within 9p21.3 and extending beyond CDKN2A predisposes to melanoma, neural system tumours and possible haematological Malignancies. J Med Genet. (2016) 53:721–7. doi: 10.1136/jmedgenet-2015-103446
8. Chan SH, Lim WK, Michalski ST, Lim JQ, Ishak NDB, Met-Domestici M, et al. Germline hemizygous deletion of CDKN2A-CDKN2B locus in a patient presenting with Li-Fraumeni syndrome. NPJ Genom Med. (2016) 1:16015. doi: 10.1038/npjgenmed.2016.15
9. Chan AK, Han SJ, Choy W, Beleford D, Aghi MK, Berger MS, et al. Familial melanoma-astrocytoma syndrome: synchronous diffuse astrocytoma and pleomorphic xanthoastrocytoma in a patient with germline CDKN2A/B deletion and a significant family history. Clin Neuropathol. (2017) 36:213–21. doi: 10.5414/NP301022
10. Jensen MR, Stoltze U, Hansen TVO, Bak M, Sehested A, Rechnitzer C, et al. 9p21.3 Microdeletion involving CDKN2A/2B in a young patient with multiple primary cancers and review of the literature. Cold Spring Harb Mol Case Stud. (2022) 8(4):a006164. doi: 10.1101/mcs.a006164
11. Bishop DT, Demenais F, Goldstein AM, Bergman W, Bishop JN, Bressac-de-Paillerets B, et al. Geographical variation in the penetrance of CDKN2A mutations for melanoma. J Natl Cancer Inst. (2002) 94:894–903. doi: 10.1093/jnci/94.12.894
12. Sargen MR, Merrill SL, Chu EY, Nathanson KL. CDKN2A mutations with p14 loss predisposing to multiple nerve sheath tumours, melanoma, dysplastic naevi and internal Malignancies: a case series and review of the literature. Br J Dermatol. (2016) 175:785–9. doi: 10.1111/bjd.2016.175.issue-4
13. Prowse AH, Schultz DC, Guo S, Vanderveer L, Dangel J, Bove B, et al. Identification of a splice acceptor site mutation in p16INK4A/p14ARF within a breast cancer, melanoma, neurofibroma prone kindred. J Med Genet. (2003) 40:e102. doi: 10.1136/jmg.40.8.e102
14. Taylor LA, O’Day C, Dentchev T, Hood K, Chu EY, Ridky TW, et al. p15 expression differentiates nevus from melanoma. Am J Pathol. (2016) 186:3094–9. doi: 10.1016/j.ajpath.2016.08.009
15. Hjazi A, Ghaffar E, Asghar W, Alauldeen Khalaf H, Ikram Ullah M, Mireya-Romero-Parra R, et al. CDKN2B-AS1 as a novel therapeutic target in cancer: Mechanism and clinical perspective. Biochem Pharmacol. (2023) 213:115627. doi: 10.1016/j.bcp.2023.115627
16. Carrió M, Gel B, Terribas E, Zucchiatti AC, Moliné T, Rosas I, et al. Analysis of intratumor heterogeneity in Neurofibromatosis type 1 plexiform neurofibromas and neurofibromas with atypical features: Correlating histological and genomic findings. Hum Mutat. (2018) 39:1112–25. doi: 10.1002/humu.2018.39.issue-8
Keywords: 9p21.3 deletion, plexiform neurofibromas, genetic counseling, case report, CDKN2A
Citation: Zhang Y, Li X, Gao H, Qiu C and Liu X (2025) Case report: The smallest 9p21.3 microdeletion involving CDKN2A but not CDKN2B causes multiple plexiform neurofibromas. Front. Oncol. 15:1437093. doi: 10.3389/fonc.2025.1437093
Received: 23 May 2024; Accepted: 21 January 2025;
Published: 19 February 2025.
Edited by:
Elisa Frullanti, University of Siena, ItalyReviewed by:
Lukas Rasulić, University of Belgrade, SerbiaCopyright © 2025 Zhang, Li, Gao, Qiu and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoliang Liu, bGl1eGxAc2otaG9zcGl0YWwub3Jn; Chuang Qiu, cWl1Y0Bzai1ob3NwaXRhbC5vcmc=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.