 Shehryar R. Sheikh1,2
Shehryar R. Sheikh1,2 Laura J. Klesse3†
Laura J. Klesse3† Ross Mangum4†
Ross Mangum4† Ashley Bui3†
Ashley Bui3† Benjamin I. Siegel5,6†Mohamed S. Abdelbaki7†
Benjamin I. Siegel5,6†Mohamed S. Abdelbaki7† Neha J. Patel8*† for the National Pediatric Cancer Foundation (NPCF) Central Nervous System Task Force
Neha J. Patel8*† for the National Pediatric Cancer Foundation (NPCF) Central Nervous System Task Force- 1Department of Neurosurgery, Cleveland Clinic, Cleveland, OH, United States
- 2Department of Molecular Medicine, Case Western Reserve University, Cleveland, OH, United States
- 3Department of Pediatrics, Division of Hematology/Oncology, UT Southwestern Medical Center, Dallas, TX, United States
- 4Center for Cancer and Blood Disorders, Phoenix Children’s Hospital, Phoenix, AZ, United States
- 5Brain Tumor Institute, Children’s National Hospital, Washington, DC, United States
- 6Division of Neurology, Children’s National Hospital, Washington, DC, United States
- 7Division of Hematology and Oncology, Department of Pediatrics, School of Medicine, Washington University, St. Louis, MO, United States
- 8Department of Pediatric Hematology-Oncology and Blood and Marrow Transplant, Cleveland Clinic, Cleveland, OH, United States
Pediatric low-grade gliomas (pLGGs) are the most common brain tumors in children. Many patients with unresectable tumors experience recurrence or long-term sequelae from standard chemotherapeutics. This mini-review explores the emerging role of MEK inhibitors in the management of pLGGs, highlighting their potential to transform current treatment paradigms. We review the molecular basis for therapeutic MEK inhibition in the context of pLGG, provide an evidence base for the use of the major MEK inhibitors currently available in the market for pLGG, and review the challenges in the use of MEKi inhibitors in this population.
1 Introduction
Pediatric low-grade gliomas (pLGGs) are the most common brain tumors in children, constituting approximately 30% of all central nervous system (CNS) tumors in this population (1, 2). The majority of children with pLGG survive well into adulthood (3, 4); this realization has led to a gradual shift in our conceptualization of pLGG into a chronic disease paradigm wherein emphasis is placed on reducing long-term morbidity to optimize functional outcomes (5). Since the early 1990s, chemotherapy (particularly the combination of carboplatin and vincristine) has been the mainstay of treatment in United States (6). The 5-year progression-free survival with chemotherapy in patients with sporadic pLGG is around 40% and around 65-85% in patients with neurofibromatosis type 1 (NF-1) associated pLGG (7, 8). Though specific chemotherapy protocols vary across the globe, no regimen has shown consistently superior outcomes compared to these historical benchmarks.
Throughout the past decade, multiple landmark studies have provided novel insights into the molecular landscape of pLGGs. The understanding that this disease harbors fewer somatic driver alterations relative to other cancers has made molecularly targeted therapies a particularly exciting avenue. Specifically, the role of mitogen-activated protein kinase (MEK), a serine-threonine kinase in the classic RAS-MAPK signaling cascade, has become a keen area of focus for therapeutic inhibition. In this mini-review, we provide a primer on the role of MEK inhibition in pLGG. The text is organized into the following sections: 1) we recapitulate the key molecular biology mechanisms that underlie pLGG pathogenesis and thereby lay molecular rationale for MEK inhibition as a treatment paradigm, 2) we review the major MEK inhibitors available in the market and present the results of key clinical trials, and 3) we discuss key considerations in the contemporary clinical use of MEK inhibition in pLGG.
2 Role of MAPK/ERK in pLGG pathogenesis
Over the last decade, multiple large genomic studies have identified the key somatic driver events for pLGG (9–13). The most important discovery from these studies has been that the disease is driven primarily by alterations in the MAPK/ERK (mitogen-activated protein kinase/extra-cellular signal-regulated kinase) pathway (8). Canonically, MAPK/ERK is a critical signaling pathway regulating multiple cellular processes including growth, proliferation, differentiation, and survival. In physiological conditions, the pathway is initiated by an extracellular signal (e.g. growth factors, cytokines, hormones) binding to a receptor tyrosine kinase (RTK) which causes dimerization and autophosphorylation. The activated RTK then recruits and activates RAS, a GTPase, which in turn activates RAF, a protein kinase (including the notable isoform BRAF). RAF in turn phosphorylates and activates MEK, another kinase enzyme. MEK activates ERK which translocates into the nucleus where it phosphorylates transcription factors thereby altering gene expression. Relatedly, neurofibromin (a protein encoded by the NF1 gene) accelerates deactivation of RAS thus acting as a negative regulator of the pathway (Figure 1).
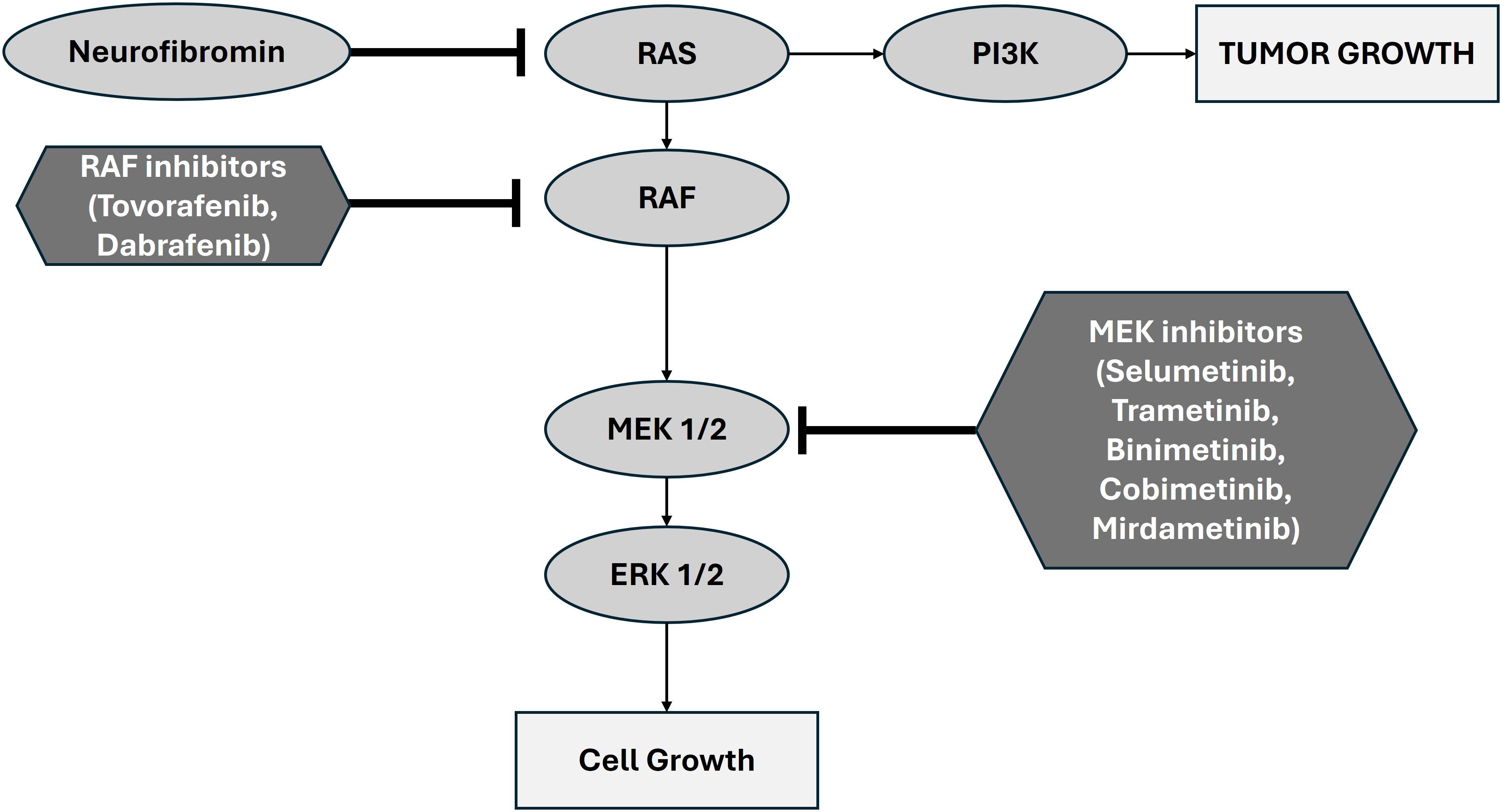
Figure 1. Schematic representation of the canonical MAPK/ERK pathway showing the role of MEK1/2 and the basis of therapeutic inhibition. Adapted from Zhao et al., 2014 (19).
Structural variations in the BRAF gene are the most common somatic driver event for sporadic (non-NF1-related) pLGG, found in roughly 70% of cases (13, 14). These fusion events create abnormal gene products such as KIAA1549-BRAF which lead to the constitutive activation of the MAPK pathway. A smaller, but still significant, subset of pLGGs harbor single nucleotide polymorphisms in BRAF (most frequently BRAFV600E) which result in upregulated protein activity (15). One in five children with NF1 develop pLGG in the central nervous system, with approximately one third occurring in the optic pathway (16). Over 90% of pLGGs in NF1 patients harbor bi-allelic inactivation of NF1 without the BRAF mutations that are common in sporadic cases (17). A subset of NF1-assocaited pLGGs carry a higher risk of progression and treatment-resistance. This clinical heterogeneity is thought to be driven by acquisition of additional molecular alterations (18). For example, alterations in ATRX, CDK2A, and TP53, genes typically associated with high grade gliomas, are found infrequently in NF1-associated pLGGs and may portend more aggressive behavior.
2.1 Rationale for MEK inhibition
Given the central role of the MAPK pathway in pLGG pathogenesis, targeting MEK, a key kinase in this pathway, presents a rational therapeutic strategy. MEK inhibitors can potentially interrupt the aberrant signaling cascade driving pLGG pathogenesis, thereby halting tumor progression and inducing tumor regression in some cases. MEK inhibition is promising because of the potential for target specificity in comparison with classic chemotherapy (carboplatin and vincristine) raising the possibility of more limited toxicities and side effects. Furthermore, there is a key distinction between MEK1/2 and other protein targets within the MAPK/ERK pathway; MEK1/2 are kinases that activate ERK1 and ERK 2 but do not have any other known physiological substrates (19). In contrast, ERK1/2 catalyzes phosphorylation of countless cytoplasmic and nuclear targets for a diverse array of cellular functions. MEK1/2 thus serves as a relatively selective (and thus strategic) targetable bottleneck in tumorigenesis.
Though we will focus here on the role of MEK inhibition, it is worthwhile to note that in 2024 the FDA approved a highly selective type II RAF kinase inhibitor (Tovorafenib) pursuant to the results of a multicenter, open-label, single-arm trial in children with relapsed or refractory pLGG harboring an activating BRAF alteration (FIREFLY-1, NCT04775485) (20). The approval of Tovorafenib serves as an encouraging proof of concept supporting a focus on highly selective targeting of the RAS/RAF/MAPK pathway in pLGG. Relatedly, LOGGIC/FIREFELY-2 (NCT05566795) is currently enrolling patients and will serve as a Phase 3 trial investigating the efficacy of Tovorafenib monotherapy compared with chemotherapy in children with newly diagnosed LGG harboring an activating RAF mutation (21).
3 Major MEK inhibitors that have been clinically adopted
Multiple trials have experimented with the use of MEK inhibitors in different cancers that harbor mutation in the MEK pathway. Here, we review the available MEK inhibitors and the published clinical data on their role in pLGG (Table 1):
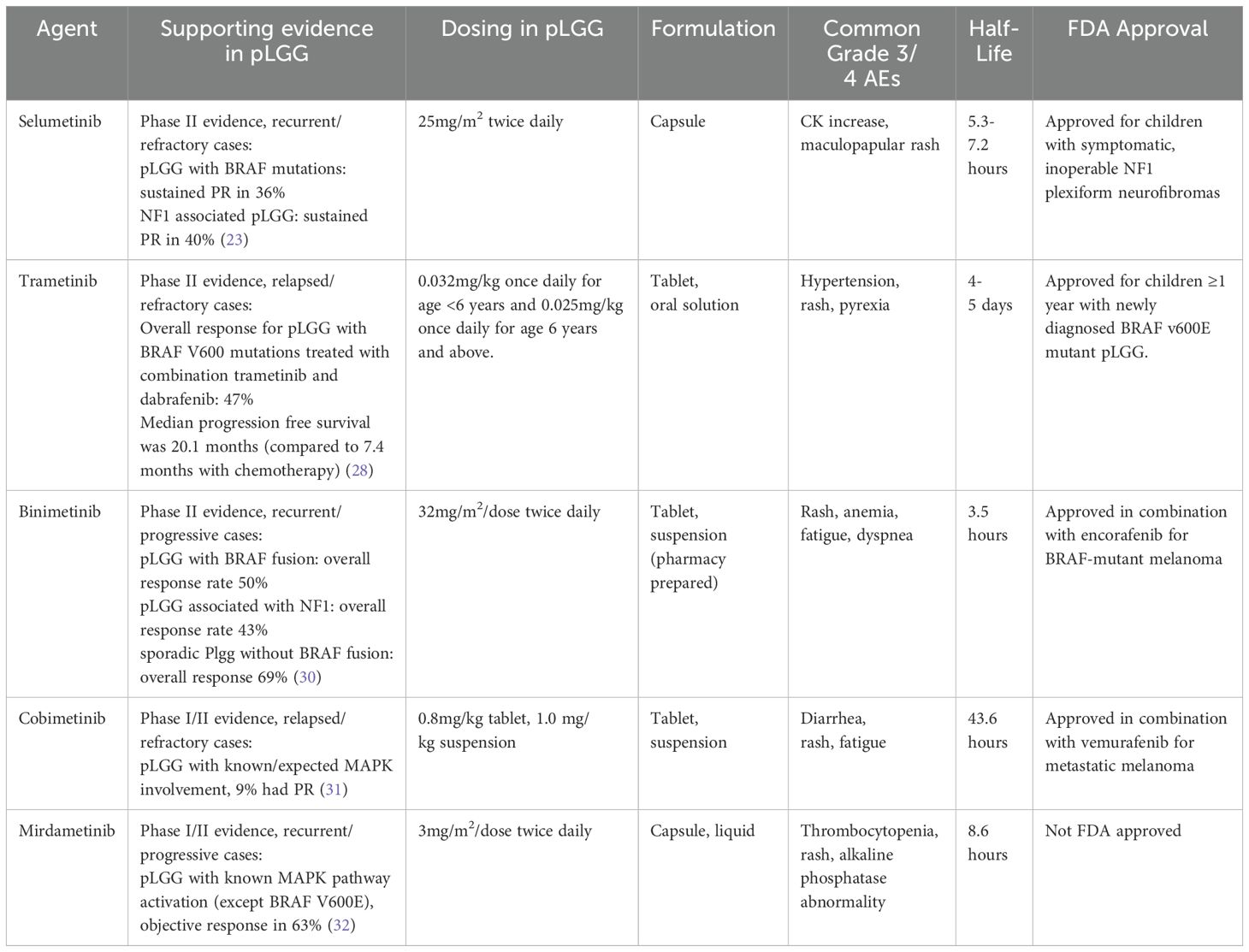
Table 1. Summary of major MEK inhibitors which have been trial in for pLGG. Adapted in part from de Blank et al. (34).
3.1 Selumetinib
Selumetinib is an orally available non-ATP-competitive small molecular inhibitor of MEK1/2. In 2017, a phase I trial of selumetinib in pediatric patients with recurrent or refractory pLGG was published by the Pediatric Brain Tumor Consortium (PBTC). The trial established a recommended phase 2 dose (RP2D) and reported a 2-year progression-free survival (PFS) at RP2D of 69% (SE 9.8%) (22). In a subsequent phase II trial in children with BRAF-aberrant or NF1-associated recurrent, refractory, or progressive LGG, imaging response rates were in the 30-40% range (23). Of the patients with BRAF aberrations (n=25), 36% achieved sustained partial response (PR) (median follow-up 36.4 months). Similarly, patients with NF1-associated tumors (n=25) achieved 40% sustained PR (medial follow-up 48.6 months). The study also reported on visual outcomes in 10 patients with optic pathway gliomas (all NF1-associated); 8 reported stable vision and 2 reported improvement. Elevated creatinine phosphokinase and maculopapular rash were the most frequent grade 3 or higher adverse events, both occurring in about 10% of patients. Notably, 36% of patients required a dose reduction due to toxicity, either grade 2 or grade 3. Although common, elevation of creatine phosphokinase was not symptomatic in most treated patients and the requirement for dose reduction based solely on this laboratory finding is an ongoing discussion in the treating community. The results of this phase II trial generated great excitement given that the tumor response with selumetinib therapy appeared comparable to that with chemotherapy but with significant advantages including oral administration without the need for central lines, no immunosuppression, less hair loss, and reduction in number of clinic visits (24). Results in a separate stratum of the same phase II trial that involved recurrent optic pathway or hypothalamic LGG in patients without NF1 (n=25) showed that 24% had PR, 56% had stable disease (SD), and 20% had disease progression (25). Visual acuity improved in 26% and was stable in the rest.
These results led to the current phase III, non-inferiority trials through the Children’s Oncology Group (COG) comparing selumetinib with standard-of-care chemotherapy (carboplatin and vincristine) for newly diagnosed pLGG. ACNS1821 (NCT03871257) is being conducted in children with NF1-associated pLGG, while ACNS1833 (NCT04166409) is being conducted in children with non-NF1 and non-BRAFV600E mutant pLGG.
Though not in the context of pLGG, we note that compelling long term safety data for the use of selumetinib comes from the SPRINT trial (NCT01362803) conducted in the context of inoperable symptomatic plexiform neurofibromas (PN) in children with NF1. In the phase I study (n=24), investigators had reported only 1 grade 4 AE (asymptomatic elevated CPK). In the phase II study (n=50), three grade 4 AEs had been reported (CPK increase, hyperuricemia, and skin ulceration). In a five-year follow-up report after publication of phase II results, investigators reported that there had been no new or concerning safety signals in patients from the phase I or phase II studies (26).
3.2 Trametinib + Dabrafenib
Trametinib is a reversible, orally bioavailable, allosteric inhibitor of MEK1/2. In 2022, the FDA approved the combination of trametinib and dabrafenib (a BRAF inhibitor) in a tumor-agnostic fashion for the treatment of patients ≥ 6 years of age with unresectable/metastatic BRAF V600E-mutant solid tumors that have progressed after prior treatment and have no satisfactory alternative. As part of the supporting data for this approval, Bouffet et al. reported initial results from NCT02124772, a phase I/II study in children with relapsed/refractory BRAF V600-mutant pLGG treated with either trametinib monotherapy or in combination with dabrafenib (27). The PR rate was 15% in the monotherapy group (n=13) and 25% in the combination therapy group (n=36). Median PFS with combination therapy was 36.9 months, compared to 16.4 months with monotherapy. In the monotherapy group, common adverse events were paronychia (54%), diarrhea (46%) and dry skin (46%). Adverse events led to discontinuation in 54% of patients. In the combination group, the most common adverse events were pyrexia (50%) and dry skin (42%). Adverse events led to discontinuation of combination therapy in 22% of patients. A follow-up phase II trial, NCT02684058, studied the use of combination trametinib + dabrafenib (T+D) as first-line treatment for BRAF V600-mutant pLGG compared with chemotherapy (C+V) (28). Seventy-three children were treated with MAPK inhibition (T+D) and 37 with chemotherapy (C+V). The trial reported a higher overall response rate (ORR) in the group treated with MAPK inhibition (47% vs 11%, p< 0.001) and higher median PFS (20.1 months versus 7.4 months, p<0.001) when compared to the chemotherapy group. The most frequent adverse events in the T+D group were pyrexia (68%), headache (47%), and vomiting (34%), but there were fewer grade ≥3 adverse events in the T+D group and fewer discontinuations (4% versus 18%) as opposed to the carboplatin and vincristine group. Subsequent to the emergence of these data, the FDA approved D+T for children ≥1 year of age with newly diagnosed BRAF V600E mutant pLGG in March 2023. Given these data, the combination of trametinib and dabrafenib is now considered the standard of care for this select group of patients.
The utility of trametinib as a single agent in pLGGs without BRAFV600E alterations has not been well studied and currently only retrospective cohort data are available (29–31). Manoharan et al. reported on a retrospective cohort of patients treated with trametinib for progressive pLGGs and demonstrated a small percentage of PR and minor responses (MR) with most patients having SD on therapy (31).
Additional studies focused on trametinib monotherapy are currently underway, including PLGG-MEKTRIC, a large prospective trial comparing daily trametinib monotherapy with weekly vinblastine in children with non-NF1 associated pLGG and without BRAFV600E mutation in France (NCT05180825), and TRAM-01, a phase 2 open-label basket trial in Canada (NCT03363217), is testing trametinib as a single agent in pediatric patients with progressing/refractory glioma or plexiform neurofibroma with MAPK.ERK pathway activation.
3.3 Binimetinib
Binimetinib is an oral selective MEK 1/2 inhibitor currently FDA approved in the US for use in combination with BRAF inhibitor encorafenib in adult patients with unresectable BRAFV600 altered melanoma and non-small cell lung cancer. A multi-institutional phase II trial was conducted to evaluate the use of binimetinib in children with previously treated radiographically progressive pLGG (32). Radiographic response rates at 1 year were: 50% in pLGG with BRAF mutation (total n=28, 12 PR, 2 MR), 43% in NF1-associated pLGG (total n=21, 5 PR, 4 MR), and 69% in sporadic pLGG without BRAF mutation (total n = 29, 10 PR, 10 MR). The most common toxicities, as with other MEK inhibitors, were predominantly dermatologic and therapy had to be discontinued in 22% of patients, while 49% required a dose reduction due to toxicity.
3.4 Cobimetinib
Cobimetinib is an orally active, highly selective small molecule inhibitor of MEK1. iMATRIX-cobi (NCT02639546) was a multicenter phase I/II study investigating cobimetinib in pediatric patients with relapsed or refractory solid tumors with known/expected MAPK pathway involvement (33). Thirty-two patients with LGG were enrolled. Three patients (9%) had PR and 18 (56%) had SD (median follow-up 15 months). Eleven percent of patients (n=6) experienced adverse events requiring discontinuation from drug. It should be noted that 5 out of 6 of these patients experienced an ocular toxicity: one patient had grade 2 retinal detachment, 2 had chorioretinopathy (1 grade 2, 1 grade 4), one had grade 1 pigment epithelial detachment, and one had grade 1 serous retinal detachment. The higher incidence of ocular toxicities with this agent is notable given the interest in using MEKi for optic pathway gliomas wherein the tolerance for such adverse events is particularly low.
3.5 Mirdametinib
Mirdametinib is an oral small molecule allosteric inhibitor of MEK1/2. Though not yet available in the market, a New Drug Application (NDA) for the agent has recently been accepted by the FDA. SJ901 (NCT04923126) is a multi-arm phase I/II trial of mirdametinib in recurrent/progressive MEKi-naïve pLGG (excluding BRAFV600); results from phase I have recently been published (34). In 23 enrolled patients with median follow-up of 14.6 months, only one dose limiting toxicity (grade 3 thrombocytopenia) was seen. Twelve (63%) of the nineteen patients with measurable tumors achieved and objective response (OR) (of these, 1 major, 6 partial, and 5 minor). RP2D has been established (3mg/m2/dose BID) and phase II is ongoing.
4 Key considerations in clinical use
4.1 Patient selection
Determining appropriate selection of patients is perhaps the most challenging aspect of translating our burgeoning understanding of the molecular basis of pLGG into clinical practice. Not all pLGG – even with very similar genetic profiles - are likely to benefit equally from MEK inhibition and evidence-based decision-making paradigms are scant. Sigaud et al. have recently published results for a MAPKi sensitivity score (MSS) (35). Using pLGG transcriptomic data from large databases, they were able to show that computational models could predict the response of cell lines to drugs inhibiting different aspects of the MAPK pathway including MEKi. A key finding of their work was that accurate predictions relied not only on genetic information from the tumor cells but also the tumor immune microenvironment. Higher susceptibility scores correlated with higher immune cell infiltration (i.e. high expression in microglia compartment in single-cell RNA sequencing). This latter finding underscores the enormous complexity involved in predicting how a given patient will respond to MAPK inhibition based on molecular markers alone. Though not yet prospectively validated and thus not currently generalizable to routine clinical practice, the Sigaud method allows us to be optimistic about the development of computational tools that could be used in concert with the genetic profiling which is increasingly routine in the care of pLGG patients to make more personalized data-driven treatment recommendations.
4.2 Treatment duration and post-discontinuation re-growth
There is no consensus on the appropriate duration of treatment with MEK inhibitors for pLGG, though 2 years has become a common yardstick in clinical practice as this was the duration of most pLGG focused trials(though without a clear scientific rationale) (36). Use of MEK inhibitors is not limited to the pediatric population, however, and their use has been longstanding in adult tumors, particularly in melanoma. An increasingly common paradigm is to cease therapy after reaching prolonged CR. This notion is based on small cohort studies wherein most patients who achieved prolonged CR were monitored long term (8-36 months) without recurrence (37). It is challenging, however, to translate this principle into the care of pLGG patients treated with MEKi as CR is rare in our patients. Differences in tumor biology and behavior require adjustments to treatment strategy. Some pLGGs will remain stable for many years in a state of clinical senescence. Prolonged exposure to MEKi and the potential toxicities may not be warranted for this length of time and is likely not sustainable for most patients. Furthermore, the phenomenon of clinical senescence greatly complicates our ability to confidently ascertain whether tumor stability is related to therapeutic MEK inhibition as opposed to the natural history of the tumor.
A related concern in pLGG, not seen in most adult indications, is the rapid regrowth of disease soon after discontinuation. In the phase II selumetanib trial discussed earlier, 56% of patients with sporadic pLGG had disease progression with the majority occurring after therapy discontinuation. More strikingly still, in the NF1-associated pLGG stratum (n=25), only one patient progressed while on therapy while seven progressed after therapy discontinuation (23, 24). These studies have raised concern about the need for prolonged therapy in a subset of patients with recurrent, progressive disease and the possible merits of a slow wean of the drug. Such a ‘slow wean’ approach for MEKi has recently been endorsed in a consensus statement by Canadian experts (38).
4.3 Retreatment
Retreatment of patients with the same or alternative MEK inhibitors after progression with discontinuation of drug has become more accepted in clinical practice. There is some initial evidence supporting this type of re-retreatment. In the adult melanoma literature for instance, retreatment with combination MEK and BRAF inhibition is reasonable and is associated with at least temporary response (39). Specifically, in pLGG, preliminary evidence suggests that retreatment with selumetinib in patients who progress after initial therapy may be effective in restoring response or stability (40, 41). A key area of ongoing investigation is whether the addition of anti-resistance agents to MAPKi regimens may be a useful adjunct in retreatment paradigms. A current phase I/II trial through the PBTC (PBTC-055, NCT04201457) is testing addition of hydroxychloroquine to the existing combination of trametinib and dabrafenib (T + D + HCQ) in patients previously treated with a RAF or MEK inhibitor. The addition of HCQ is based on preclinical evidence showing that the agent can inhibit treatment induced autophagy which is a known pathway to treatment resistance in pediatric gliomas (42). Safety data for 18 evaluable subjects enrolled in the phase I component were comparable to prior MEK inhibitor trials; there were 2 dose-limiting toxicities, both grade 3 rash, and the highest dose level of HCQ was declared the RP2D (43).
4.4 Intermittent dosing
There has been substantial interest in the idea of using intermittent dosing (or ‘drug holidays’) as a way of mitigating treatment related toxicities while potentially decreasing resistance to MAPK inhibition. Again, population specific evidence for pLGG is lacking but reports from adult studies may be at least partially informative. A randomized phase II trial in adult melanoma patients investigated continuous versus intermittent dosing of combination trametinib and dabrafenib in metastatic or unresectable BRAFV600E- mutant melanoma (44). There were no significant differences in terms of toxicity or OS however patients in the continuous dosing arm had improved PFS. In another trial, continuous dosing of combination cobimetinib and vemurafenib (a Type 1 BRAF inhibitor) was compared with intermittent dosing in patients with advanced BRAF-mutant melanoma (45). Again, patients in the intermittent dosing arm did not have improved PFS or substantially reduced toxicities. Though these adult melanoma studies are imperfect proxies for the pLGG context given the significant difference in the disease pathophysiology and natural history, they do provide evidence for the notion that an intermittent dosing approach should be viewed with caution at this time. Clinical trials testing the specific question of whether intermittent dosing may be beneficial for pediatric glioma patients are underway; NCT03326388 (INSPECT) is a phase I/II study in children with NF1-associated tumors (inoperable plexiform neurofibromas and recurrent optic pathway gliomas) being treated with intermittent dosing of selumetinib. Notably, the recently published ReNeu trial found that children with NF-1 associated inoperable symptomatic plexiform neurofibromas treated with intermittent dosing of Mirdametinib (3 weeks on/1 week off) met the primary efficacy endpoint, with ORR of 52% (46).
4.5 Potential impact on fertility
Given the long term survival of most pLGG patients, there has been a sustained interest in establishing the impact of targeted therapies (including MEKi) on fertility, but compelling data in human subjects remain scant. Concerns about the potential toxicity to fertility come from animal studies. In female rats, cobimetinib exposure increases apoptosis and necrosis of cells in the corpus luteum and vaginal epithelium while also causing testicular degeneration in male dogs (47). Trametinib has also been associated with reduced corpus leuteum cells in female rats, but no observed effect in male reproductive tissues (47). To our knowledge, there are currently no data in humans to better inform the long-term risks to fertility associated with MEKi use (48).
5 Discussion
While chemotherapeutics have been the mainstay for the treatment of pLGG since the early 1990s, our understanding of the molecular drivers of the disease has deepened markedly in the last decade leading to the widespread appreciation of the role of targeted therapies. Unlike other cancers, pLGG is driven by relatively few somatic driver mutations primarily impacting the MAPK/ERK pathway. MEK inhibition has now been evaluated in multiple phase I/II trials in both sporadic and NF1 associated pLGG and early results suggest that for appropriately selected patients outcomes are likely to be as good as (if not superior to) those with conventional chemotherapies. The emergence of the MAPK pathway alterations and the activity of targeted agents has made molecular characterization of the tumor a required aspect of diagnosis and management. Several questions remain, however, in the continued use of this targeted therapy. Identification of the patients most likely to benefit, the durability of therapy, the length of optimal therapy, and the process for therapy discontinuation remain clinical challenges. Much work is necessary to resolve these challenges before the treatment paradigm can shift to one wherein molecular targeted therapies are the standard treatment. Combination of MEKi with cytotoxic chemotherapy or combination of MEKi with other targeted therapies such as PD1 inhibition may be exciting future avenues that are yet to be explored.
Author contributions
SS: Conceptualization, Investigation, Visualization, Writing – original draft, Writing – review & editing. LK: Writing – review & editing. RM: Writing – review & editing. AB: Writing – review & editing. BS: Writing – review & editing. MA: Writing – review & editing. NP: Conceptualization, Investigation, Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ward E, DeSantis C, Robbins A, Kohler B, Jemal A. Childhood and adolescent cancer statistics, 2014. CA: Cancer J Clin. (2014) 64:83–103. doi: 10.3322/caac.21219
2. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro-oncology. (2021) 23:1231–51. doi: 10.1093/neuonc/noab106
3. Bandopadhayay P, Bergthold G, London WB, Goumnerova LC, Morales La Madrid A, Marcus KJ, et al. Long-term outcome of 4,040 children diagnosed with pediatric low-grade gliomas: an analysis of the Surveillance Epidemiology and End Results (SEER) database. Pediatr Blood cancer. (2014) 61:1173–9. doi: 10.1002/pbc.24958
4. Krishnatry R, Zhukova N, Guerreiro Stucklin AS, Pole JD, Mistry M, Fried I, et al. Clinical and treatment factors determining long-term outcomes for adult survivors of childhood low-grade glioma: a population-based study. Cancer. (2016) 122:1261–9. doi: 10.1002/cncr.v122.8
5. Fangusaro J, Jones DT, Packer RJ, Gutmann DH, Milde T, Witt O, et al. Pediatric low-grade glioma: state-of-the-art and ongoing challenges. Neuro-oncology. (2024) 26:25–37. doi: 10.1093/neuonc/noad195
6. Packer RJ, Lange B, Ater J, Nicholson HS, Allen J, Walker R, et al. Carboplatin and vincristine for recurrent and newly diagnosed low-grade gliomas of childhood. J Clin Oncol. (1993) 11:850–6. doi: 10.1200/JCO.1993.11.5.850
7. Manoharan N, Liu KX, Mueller S, Haas-Kogan DA, Bandopadhayay P. Pediatric low-grade glioma: Targeted therapeutics and clinical trials in the molecular era. Neoplasia. (2023) 36:100857. doi: 10.1016/j.neo.2022.100857
8. Packer RJ, Pfister S, Bouffet E, Avery R, Bandopadhayay P, Bornhorst M, et al. Pediatric low-grade gliomas: implications of the biologic era. Neuro Oncol. (2017) 19:750–61. doi: 10.1093/neuonc/now209
9. Jones DT, Hutter B, Jäger N, Korshunov A, Kool M, Warnatz H-J, et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet. (2013) 45:927–32. doi: 10.1038/ng.2682
10. Zhang J, Wu G, Miller C, Tatevossian R, Dalton J, Tang B, et al. St. Jude Children’s Research Hospital–Washington University Pediatric Cancer Genome Project. Wholegenome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet. (2013) 45:602–12. doi: 10.1038/ng.2611
11. Bandopadhayay P, Ramkissoon LA, Jain P, Bergthold G, Wala J, Zeid R, et al. MYB-QKI rearrangements in angiocentric glioma drive tumorigenicity through a tripartite mechanism. Nat Genet. (2016) 48:273–82. doi: 10.1038/ng.3500
12. Ryall S, Zapotocky M, Fukuoka K, Nobre L, Stucklin AG, Bennett J, et al. Integrated molecular and clinical analysis of 1,000 pediatric low-grade gliomas. Cancer Cell. (2020) 37:569–83.e5. doi: 10.1016/j.ccell.2020.03.011
13. Jones D, Kocialkowski S, Liu L, Pearson D, Ichimura K, Collins V. Oncogenic RAF1 rearrangement and a novel BRAF mutation as alternatives to KIAA1549: BRAF fusion in activating the MAPK pathway in pilocytic astrocytoma. Oncogene. (2009) 28:2119–23. doi: 10.1038/onc.2009.73
14. Pfister S, Janzarik WG, Remke M, Ernst A, Werft W, Becker N, et al. BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J Clin Invest. (2008) 118:1739–49. doi: 10.1172/JCI33656
15. Lassaletta A, Zapotocky M, Mistry M, Ramaswamy V, Honnorat M, Krishnatry R, et al. Therapeutic and prognostic implications of BRAF V600E in pediatric low-grade gliomas. J Clin Oncol. (2017) 35:2934–41. doi: 10.1200/JCO.2016.71.8726
16. Kotch C, de Blank P, Gutmann DH, Fisher MJ. Low-grade glioma in children with neurofibromatosis type 1: surveillance, treatment indications, management, and future directions. Child's Nervous System. (2024) 40:3241–50. doi: 10.1007/s00381-024-06430-8
17. Fisher MJ, Jones DT, Li Y, Guo X, Sonawane PS, Waanders AJ, et al. Integrated molecular and clinical analysis of low-grade gliomas in children with neurofibromatosis type 1 (NF1). Acta neuropathologica. (2021) 141:605–17. doi: 10.1007/s00401-021-02276-5
18. D'Angelo F, Ceccarelli M, Tala, Garofano L, Zhang J, Frattini V, et al. The molecular landscape of glioma in patients with Neurofibromatosis 1. Nat Med. (2019) 25:176–87. doi: 10.1038/s41591-018-0263-8
19. Zhao Y, Adjei AA. The clinical development of MEK inhibitors. Nat Rev Clin Oncol. (2014) 11:385–400. doi: 10.1038/nrclinonc.2014.83
20. Kilburn LB, Khuong-Quang D-A, Hansford JR, Landi D, van der Lugt J, Leary SES, et al. The type II RAF inhibitor tovorafenib in relapsed/refractory pediatric low-grade glioma: the phase 2 FIREFLY-1 trial. Nat Med. (2024) 30:207–17. doi: 10.1038/s41591-023-02668-y
21. van Tilburg CM, Kilburn LB, Crotty E, Smith AA, Perreault S, Franson AF, et al. LOGGIC/FIREFLY-2: A phase 3, randomized trial of tovorafenib vs. chemotherapy in pediatric and young adult patients with newly diagnosed low-grade glioma harboring an activating RAF alteration. Am Soc Clin Oncol. (2023). doi: 10.1200/JCO.2023.41.16_suppl.TPS10067
22. Banerjee A, Jakacki RI, Onar-Thomas A, Wu S, Nicolaides T, Young Poussaint T, et al. A phase I trial of the MEK inhibitor selumetinib (AZD6244) in pediatric patients with recurrent or refractory low-grade glioma: a Pediatric Brain Tumor Consortium (PBTC) study. Neuro-oncology. (2017) 19:1135–44. doi: 10.1093/neuonc/now282
23. Fangusaro J, Onar-Thomas A, Poussaint TY, Wu S, Ligon AH, Lindeman N, et al. Selumetinib in paediatric patients with BRAF-aberrant or neurofibromatosis type 1-associated recurrent, refractory, or progressive low-grade glioma: a multicentre, phase 2 trial. Lancet Oncol. (2019) 20:1011–22. doi: 10.1016/S1470-2045(19)30277-3
24. Bouffet E. Selumetinib in paediatric low-grade glioma: a new era? Lancet Oncol. (2019) 20:900–1. doi: 10.1016/S1470-2045(19)30304-3
25. Fangusaro J, Onar-Thomas A, Poussaint TY, Wu S, Ligon AH, Lindeman N, et al. A phase II trial of selumetinib in children with recurrent optic pathway and hypothalamic low-grade glioma without NF1: a Pediatric Brain Tumor Consortium study. Neuro Oncol. (2021) 23:1777–88. doi: 10.1093/neuonc/noab047
26. Gross AM, Dombi E, Wolters PL, Baldwin A, Dufek A, Herrera K, et al. Long-term safety and efficacy of selumetinib in children with neurofibromatosis type 1 on a phase 1/2 trial for inoperable plexiform neurofibromas. Neuro-Oncology. (2023) 25:1883–94. doi: 10.1093/neuonc/noad086
27. Bouffet E, Geoerger B, Moertel C, Whitlock JA, Aerts I, Hargrave D, et al. Efficacy and safety of trametinib monotherapy or in combination with dabrafenib in pediatric BRAF V600–mutant low-grade glioma. J Clin Oncol. (2023) 41:664–74. doi: 10.1200/JCO.22.01000
28. Bouffet E, Hansford J, Garré ML, Hara J, Plant-Fox A, Aerts I, et al. Primary analysis of a phase II trial of dabrafenib plus trametinib (dab+ tram) in BRAF V600–mutant pediatric low-grade glioma (pLGG). Am Soc Clin Oncol. (2022). doi: 10.1056/NEJMoa2303815
29. Selt F, van Tilburg CM, Bison B, Sievers P, Harting I, Ecker J, et al. Response to trametinib treatment in progressive pediatric low-grade glioma patients. J Neurooncol. (2020) 149:499–510. doi: 10.1007/s11060-020-03640-3
30. Kondyli M, Larouche V, Saint-Martin C, Ellezam B, Pouliot L, Sinnett D, et al. Trametinib for progressive pediatric low-grade gliomas. J Neurooncol. (2018) 140:435–44. doi: 10.1007/s11060-018-2971-9
31. Manoharan N, Choi J, Chordas C, Zimmerman MA, Scully J, Clymer J, et al. Trametinib for the treatment of recurrent/progressive pediatric low-grade glioma. J Neurooncol. (2020) 149:253–62. doi: 10.1007/s11060-020-03592-8
32. Robison N, Pauly J, Malvar J, Gardner S, Allen J, Margol A, et al. LTBK-04. Late Breaking Abstract: MEK162 (binimetinib) in children with progressive or recurrent low-grade glioma: a multi-institutional phase II and target validation study. Neuro Oncol. (2022) 24:i191–2. doi: 10.1093/neuonc/noac079.716
33. Trippett T, Toledano H, Campbell Hewson Q, Verschuur A, Langevin AM, Aerts I, et al. Cobimetinib in pediatric and young adult patients with relapsed or refractory solid tumors (iMATRIX-cobi): A multicenter, phase I/II study. Target Oncol. (2022) 17:283–93. doi: 10.1007/s11523-022-00888-9
34. Robinson GW, Vinitsky A, Bag AK, Chiang J, Li Q, Lin T, et al. LGG-53. Results from the phase 1 and phase 1 expansion cohorts of SJ901: A phase 1/2 trial of single-agent mirdametinib (PD-0325901) in children, adolescents, and young adults with low-grade glioma. Neuro-Oncology. (2024) 26:129–30. doi: 10.1093/neuonc/noae064.443
35. Sigaud R, Albert TK, Hess C, Hielscher T, Winkler N, Kocher D, et al. MAPK inhibitor sensitivity scores predict sensitivity driven by the immune infiltration in pediatric low-grade gliomas. Nat Commun. (2023) 14:4533. doi: 10.1038/s41467-023-40235-8
36. de Blank PMK, Gross AM, Akshintala S, Blakeley JO, Bollag G, Cannon A, et al. MEK inhibitors for neurofibromatosis type 1 manifestations: Clinical evidence and consensus. Neuro-Oncology. (2022) 24:1845–56. doi: 10.1093/neuonc/noac165
37. Warburton L, Meniawy TM, Calapre L, Pereira M, McEvoy A, Ziman M, et al. Stopping targeted therapy for complete responders in advanced BRAF mutant melanoma. Sci Rep. (2020) 10:18878. doi: 10.1038/s41598-020-75837-5
38. Erker C, Vanan MI, Larouche V, Nobre L, Cacciotti C, Vairy S, et al. Canadian consensus for treatment of BRAF V600E mutated pediatric and AYA gliomas. Curr Oncol. (2024) 31:4022–9. doi: 10.3390/curroncol31070299
39. Amann V, Hoffmann D, Mangana J, Dummer R, Goldinger S. Successful retreatment with combined BRAF/MEK inhibition in metastatic BRAFV 600-mutated melanoma. J Eur Acad Dermatol Venereology. (2017) 31:1638–40. doi: 10.1111/jdv.2017.31.issue-10
40. Fangusaro JR, Onar-Thomas A, Poussaint TY, Wu S, Ligon AH, Lindeman NI, et al. LTBK-01. Updates on the phase II and re-treatment study of AZD6244 (selumetinib) for children with recurrent or refractory pediatric low grade glioma: a pediatric brain tumor consortium (PBTC) study. Neuro-Oncology. (2018) 20:i214–i. doi: 10.1093/neuonc/noy109
41. Fangusaro J, Jones DT, Packer RJ, Gutmann DH, Milde T, Witt O, et al. Pediatric low-grade glioma: State-of-the-art and ongoing challenges. Neuro-Oncology. (2023) 26:25–37. doi: 10.1093/neuonc/noad195
42. Mulcahy Levy JM, Zahedi S, Griesinger AM, Morin A, Davies KD, Aisner DL, et al. Autophagy inhibition overcomes multiple mechanisms of resistance to BRAF inhibition in brain tumors. eLife. (2017) 6:e19671. doi: 10.7554/eLife.19671
43. Hoffman LM, Levy JM, Kilburn L, Billups C, Stokes V, McCourt E, et al. EPCT-01. Pediatric Brain Tumor Consortium (PBTC)-055: A phase I study of trametinib and hydroxychloroquine (HCQ) for BRAF-fusion or Neurofibromatosis type-1 (NF1)-associated pediatric gliomas. Neuro-Oncology. (2022) 24:i35–i. doi: 10.1093/neuonc/noac079.129
44. Algazi AP, Othus M, Daud AI, Lo RS, Mehnert JM, Truong T-G, et al. Continuous versus intermittent BRAF and MEK inhibition in patients with BRAF-mutated melanoma: a randomized phase 2 trial. Nat Med. (2020) 26:1564–8. doi: 10.1038/s41591-020-1060-8
45. Gonzalez-Cao M, Mayo de Las Casas C, Oramas J, Berciano-Guerrero MA, de la Cruz L, Cerezuela P, et al. Intermittent BRAF inhibition in advanced BRAF mutated melanoma results of a phase II randomized trial. Nat Commun. (2021) 12:7008. doi: 10.1038/s41467-021-26572-6
46. Moertel CL, Hirbe AC, Shuhaiber HH, Viskochil D, Sidhu A, Bielamowicz KJ, et al. ReNeu: A pivotal phase 2b trial of mirdametinib in children and adults with neurofibromatosis type 1 (NF1)-associated symptomatic inoperable plexiform neurofibroma (PN). Am Soc Clin Oncol. (2024). doi: 10.1200/JCO.2024.42.16_suppl.3016
47. Walter JR, Xu S, Paller AS, Choi JN, Woodruff TK. Oncofertility considerations in adolescents and young adults given a diagnosis of melanoma: Fertility risk of Food and Drug Administration-approved systemic therapies. J Am Acad Dermatol. (2016) 75:528–34. doi: 10.1016/j.jaad.2016.04.031
48. Helgadottir H, Matikas A, Fernebro J, Frödin J-E, Ekman S, Rodriguez-Wallberg KA. Fertility and reproductive concerns related to the new generation of cancer drugs and the clinical implication for young individuals undergoing treatments for solid tumors. Eur J Cancer. (2024) 202. doi: 10.1016/j.ejca.2024.114010
Keywords: pediatric low grade glioma, PLGG, MEK, MAPK, targeted therapy, MEKi
Citation: Sheikh SR, Klesse LJ, Mangum R, Bui A, Siegel BI, Abdelbaki MS and Patel NJ (2024) The role of MEK inhibition in pediatric low-grade gliomas. Front. Oncol. 14:1503894. doi: 10.3389/fonc.2024.1503894
Received: 29 September 2024; Accepted: 04 December 2024;
Published: 20 December 2024.
Edited by:
Giuseppe Maria Milano, Bambino Gesù Children’s Hospital (IRCCS), ItalyReviewed by:
Angela Mastronuzzi, Bambino Gesù Children’s Hospital (IRCCS), ItalyFrancesco Bruno, University Hospital of the City of Health and Science of Turin, Italy
Helen Toledano, Schneider Children’s Medical Center, Israel
Copyright © 2024 Sheikh, Klesse, Mangum, Bui, Siegel, Abdelbaki and Patel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Neha J. Patel, cGF0ZWxuMjJAY2NmLm9yZw==
†These authors have contributed equally to this work and share senior authorship