 Xiaoman Zheng1
Xiaoman Zheng1 Wenshuang Ding
Wenshuang Ding Weijie Zhong
Weijie Zhong- 1Department of Geriatrics, Hematology & Oncology Ward, Guangzhou First People’s Hospital, Guangdong Medical University, Guangzhou, Guangdong, China
- 2Department of Pathology, Guangzhou First People’s Hospital, Guangdong Medical University, Guangzhou, Guangdong, China
Hodgkin lymphoma variant of Richter syndrome (HL-type RS) is a very rare disease, in which chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL) is transformed into novel Hodgkin lymphoma. The most important prognostic factor of HL-type RS is the clonal relationship between HL-type RS and the preexisting CLL/SLL. Detailed confirmation of clonally unrelated HL-type RS cases have not been reported. To the best of our knowledge, this is the first case of HL-type RS confirmed as clone independent by a detailed comparison of immunoglobulin gene rearrangement clones and gene mutations. A 76-year-old man, diagnosed with SLL 1 year before transformation was treated with Zanubrutinib 3 months before transformation. When diagnosed with HL-type RS, he presented with symptoms of hemophagocytic syndrome. Positive therapeutic effects were achieved using a modified rituximab-doxorubicin, bleomycin, vinblastine, dacarbazine regimen in combination with Zanubrutinib. We also discuss a thorough review of the relevant literature we performed to help us better understand this rare disease.
1 Introduction
Richter syndrome (RS) is defined as the development of an aggressive lymphoma in patients with chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL). The majority of cases transform into diffuse large B-cell lymphoma (DLBCL), with the remainder transforming into Hodgkin lymphoma (HL) or other aggressive lymphomas, known as the DLBCL variant (DLBCL-type RS) and the HL variant (HL-type RS), respectively (1, 2). The survival rates of the two types of RS differ markedly, with a median overall survival (OS) of 5.9 months for DLBCL-type RS and 30.8 months for HL-type RS. The prognosis of HL-type RS is significantly better (3), but the median OS of HL-type RS was still significantly lower than that of primary classical HL (4). HL-type RS is rare, and the most important prognostic factor of HL-type RS is the clonal relationship between HL-type RS and the preexisting CLL/SLL. The clonally related HL-type RS has a significantly worse prognosis, while the clonally unrelated HL-type RS has a relatively better prognosis (5).
Currently, few cases of HL-type RS have been reported, no case of confirmed clonally unrelated HL-type RS has been reported, and there are also a lack of literature reviews on HL-type RS. Here, we present a recent case of clonally unrelated HL-type RS, which was confirmed by PCR and a large panel of next-generation sequencing (NGS). The patient experienced a good outcome. A detailed review of relevant literature was performed and is discussed.
2 Case presentation
A 76-year-old man was admitted to our hospital department on May 19, 2022, due to recurring abdominal pain for 5 days, with no fever, night sweats, or weight loss. He had a past medical history of coronary heart disease, hypertension, and was a hepatitis B virus carrier. An enlarged lymph node with an approximate size of 4 × 2.5 cm was palpated in the right inguinal region. Laboratory results were presented in Table 1. An enhanced computed tomography (CT) scan of the whole abdomen revealed multiple enlarged and fused lymph nodes in the abdominal cavity, retroperitoneum, pelvic cavity, right inguinal region, and right diaphragmatic angle area. The largest was located in the hepatogastric gap and measured approximately 5.3 × 3.0cm. Complete excision biopsy of the right inguinal lymph node was performed on May 31, 2022. The pathological result was showed in Table 1 (Figure 1). Whole-body positron emission tomography (PET)/CT result was also showed in Table 1. Bone marrow cytology and biopsy showed no bone marrow invasion. The bone marrow karyotype analysis was 46 XY, inv (9) (p11q13)c (20). The NGS (222 genes) results of lymph node paraffin tissue are shown in Table 2. The 222 genes panel included UNC13D gene, but no other familial hemophagocytic lymphohistiocytosis related genes. The clonal analysis results of immunoglobulin (Ig) gene rearrangement are shown in Table 3. Combined with the results of PET/CT, blood routine, lymph node pathology, and bone marrow examination, the diagnosis was SLL (Lugano stage IIIA). The patient did not meet the indications of initiating treatment and was instructed to be followed up in the clinic.
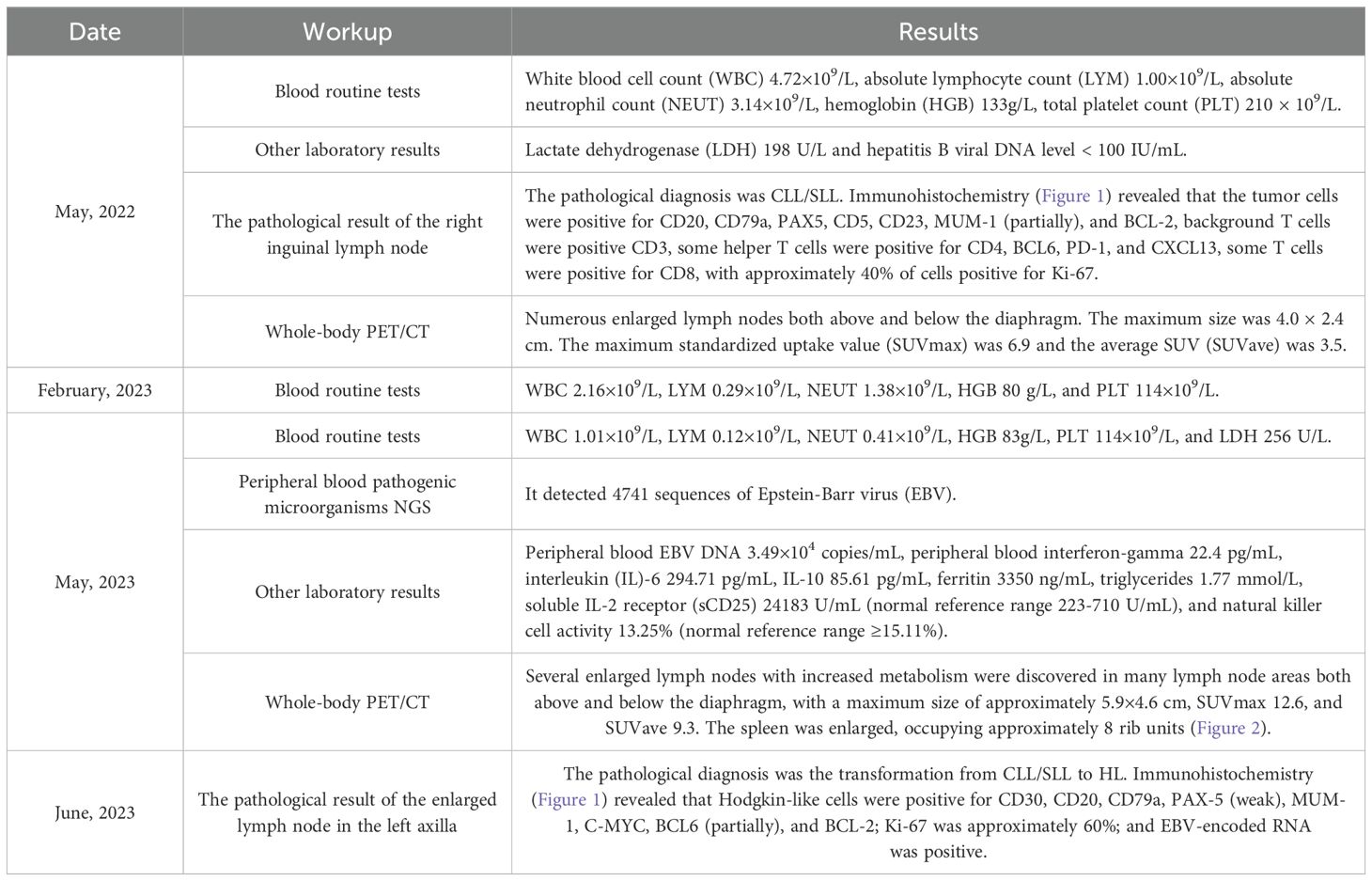
Table 1. Results of laboratory workup.
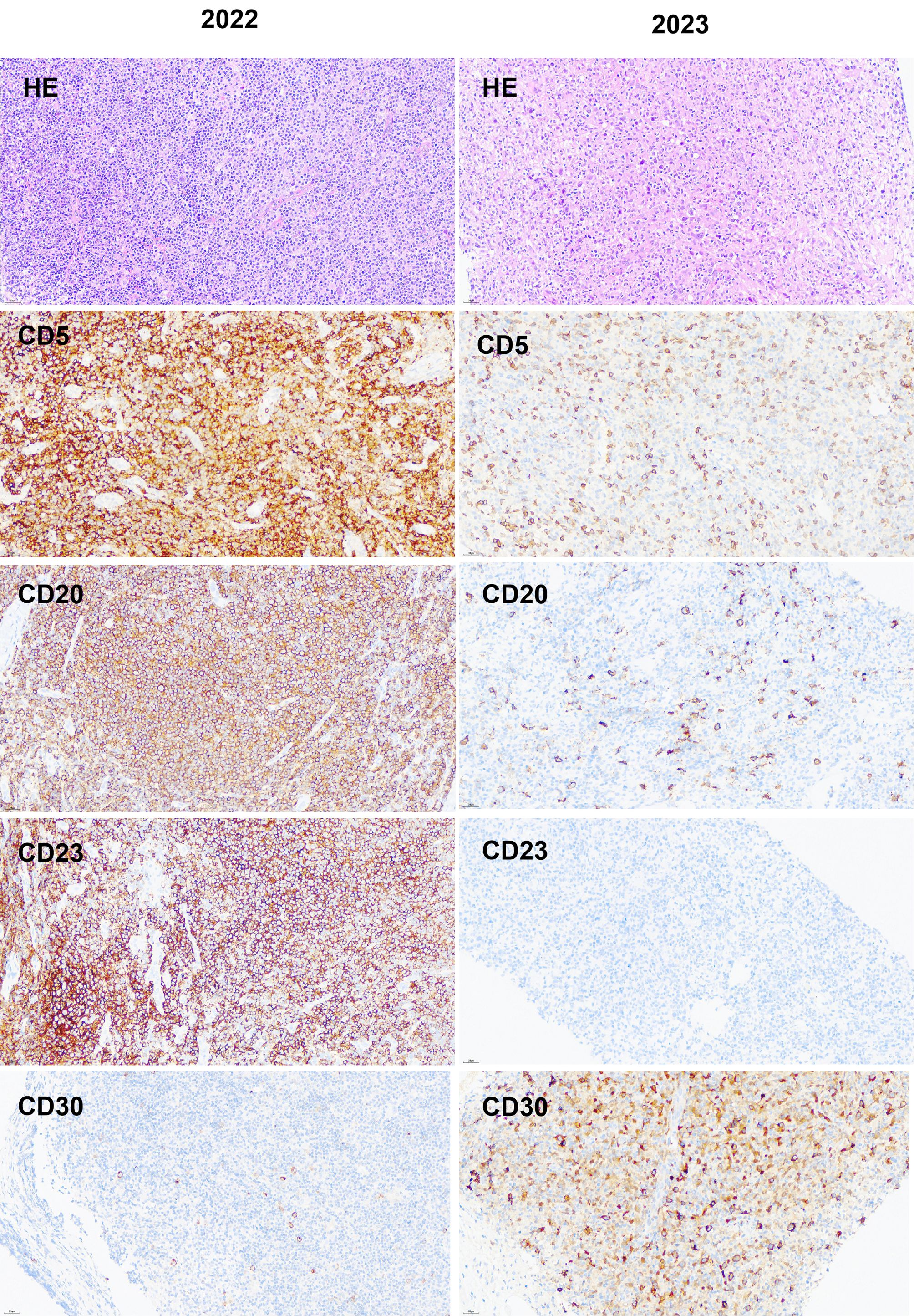
Figure 1. Comparison of immunohistochemical images from two lymph node paraffin tissues.
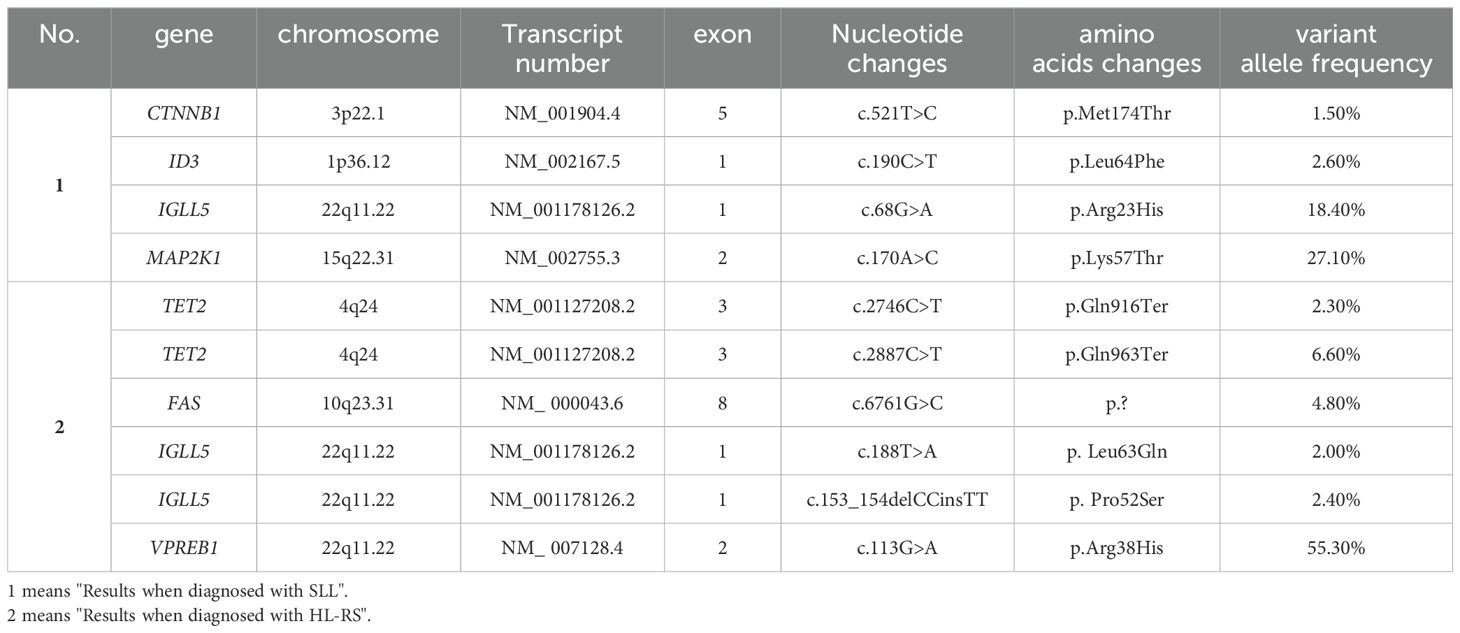
Table 2. Comparison of two NGS (222 genes) results from lymph node paraffin tissue.

Table 3. Comparison of two immunoglobulin gene rearrangement results from lymph node paraffin tissue.
On February 20, 2023, the patient was readmitted to our hospital due to recurrent fever for 3 days, and multiple blood routine tests showed progressive decrease in whole blood cells (Table 1). Repeated CT plain scans of the chest and whole abdomen suggested multiple lymph nodes were enlarged compared to the previous ones. Re-examination of bone marrow cytology and immunophenotyping showed no significant abnormal population. We considered that the patient’s SLL had progressed and he met the indication for initiating treatment. Oral administration of Zanubrutinib capsules 160mg bid was initiated on March 3, 2023. Within the first three months of taking Zanubrutinib, the superficial enlarged lymph nodes had shrunk and the blood routine tests had basically returned to normal.
However, on May 25, 2023, the patient was admitted to our hospital again due to repeated high fever for 1 week, with persistent high fever and a maximum body temperature of 39-40°C, requiring the use of antipyretic drugs to reduce the fever. Laboratory results were presented in Table 1. No hemophagocytic cells were found in bone marrow cell morphology and immunophenotyping examinations. Whole-body PET/CT result was showed in Table 1 (Figure 2). According to the 2004 diagnostic criteria for hemophagocytic lymphohistiocytosis (HLH), the patient met 6 of 8 criteria, and the diagnosis of HLH was clear. Dexamethasone, ruxolitinib, and human immunoglobulin were immediately administered due to the patient’s critical condition.
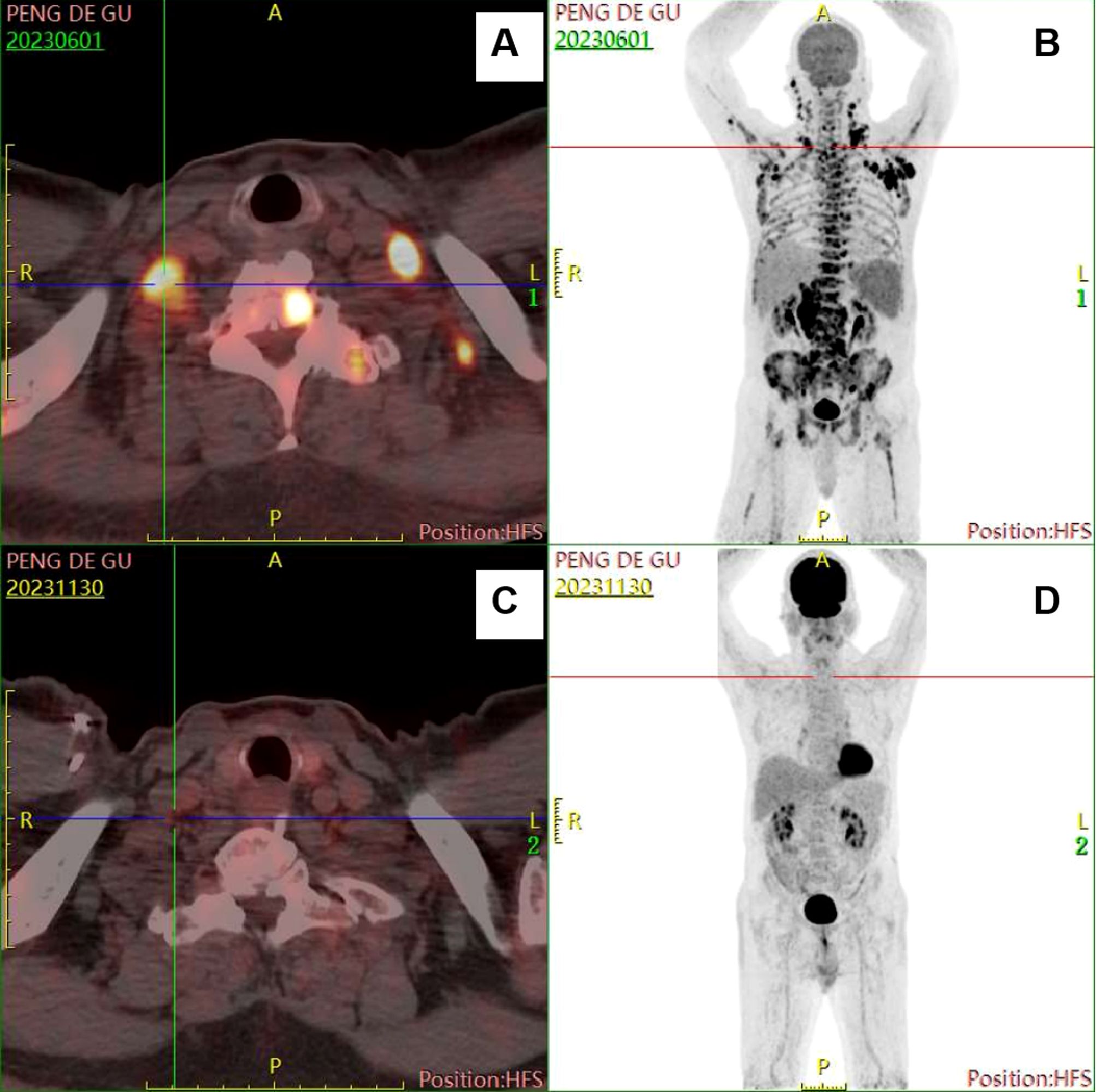
Figure 2. Comparison of images from two PET/CT examinations. (A, B) images when diagnosed with HL-type RS. (C, D) images at the end of chemotherapy treatment.
On June 5, 2023, a biopsy was performed on the enlarged lymph node in the left axilla. The pathological result was showed in Table 1 (Figure 1). The results of NGS (222 genes) of lymph node puncture tissue are shown in Table 2. The results of Ig gene rearrangement clone analysis are shown in Table 3. Based on the significant increase in LDH, symptoms, PET-CT and new pathological results, the diagnosis was that SLL had transformed into HL (stage IIIB, IPS 5 points), namely RS, accompanied by EBV infection.
According to the results presented in Tables 2, 3, the Ig gene rearrangements and mutated genes detected in the two tests were completely unrelated, indicating that the transformed HL was clonally unrelated to the primary SLL. Due to the second pathological results indicating CD20 positive and the patient’s advanced age of 76 years, an 8-course modified rituximab-doxorubicin, bleomycin, vinblastine, dacarbazine (R-ABVD) chemotherapy regimen (rituximab 600mg intravenous infusion d0, doxorubicin liposomes 40 mg intravenous infusion d1, bleomycin 15,000 units intravenous infusion d1, vincristine 3 mg intravenous infusion d1, and dacarbazine injection 500 mg intravenous infusion d1 every 21 days). After one course of chemotherapy, HLH-related symptoms were significantly relieved, and the patient no longer had a fever. The HLH therapeutic efficacy reached complete remission (CR) after 3 courses of chemotherapy. The changes in the peripheral blood cytokine levels are shown in Figure 3. The therapeutic effect was evaluated to be CR by whole-body enhanced CT scan after 4 courses of chemotherapy. After 8 courses of chemotherapy, whole-body PET/CT indicated that a complete metabolic remission was achieved (Figure 2). The patient is currently maintaining a state of sustained CR. Maintenance therapy of Zanubrutinib 160mg bid was continued during and after chemotherapy.
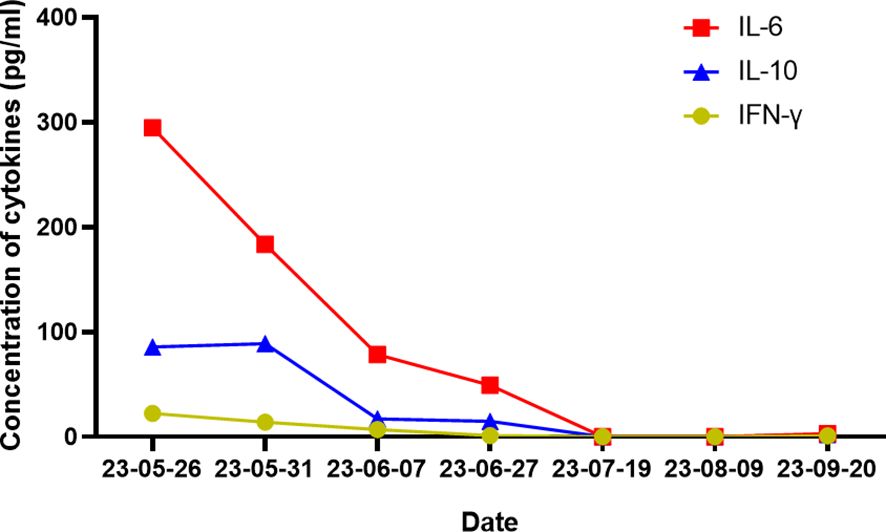
Figure 3. Images of changes in multiple cytokines.
3 Discussion and literature review
RS, also known as Richter’s Transformation, is a term defined as a specific form of CLL/SLL transforming into aggressive lymphoma, with an incidence rate of approximately 2-10% yearly (6, 7). More than 90% of RS cases transform to DLBCL, with the remainder transforming to HL and other rare forms of lymphomas (8). The incidence of HL-type RS in CLL/SLL population was 0.4-0.7%, accounting for less than 5% of RS cases (9), and the annual incidence rate is 0.05% (8).
HL-type RS is rare in clinical conditions. Up to now, only approximately 100 cases of HL-type RS have been reported in the international literature (8, 9). while no more than 5 cases have been reported in China (10). Among these 5 cases, none were tested for clonal relation before and after transformation, or for clonal relation only by IgH rearrangements through PCR. So, these 5 cases have obvious limitations. Moreover, there are very few literature reviews or reviews on HL-type RS. This elderly patient is the first reported case of clone independent HL-type RS, confirmed by detailed PCR and a big panel of NGS (222 genes). We promptly diagnosed HLH and HL-type RS and administered a modified R-ABVD regimen combined with Zanubrutinib, which achieved a very good outcome. As of the time of publication, the patient has been followed up for more than 15 months starting from the diagnosis of RS and is currently in a continuous CR state.
HL-type RS is more common in the elderly, mainly in males (male-to-female ratio approximately 3:1 to 6:1) (11), with a median age of 64-72 years at the time of HL-type RS diagnosis (8, 9, 11–14) and a median time from CLL/SLL to HL-type RS diagnosis of 3.2-7.5 years (9, 11–14). The common clinical symptoms of HL-type RS include lymph node enlargement, B symptoms, fever, splenomegaly, hepatomegaly, fatigue, and night sweats (13). Nearly half (47%) of HL-type RS patients experience elevated LDH levels (13), 82.9-87% of patients are clinically staged Ann Arbor stage III-IV, and 65% of patients have an International Prognostic Score > 4 (11, 15). Patients with HL-type RS often have EBV infection, with a reported EBV positivity rate of 67% (11), and EBV positivity rate in pathological sections of up to 71% (16). This case was also an elderly male, who was 76 years old when he was diagnosed with HL-type RS. The interval from SLL to HL-type RS progression was only about 1 year, which was significantly shorter than the average level reported in the literature. The patient’s symptoms at the time of transformation were fever, pancytopenia, enlarged lymph nodes, B symptoms, splenomegaly, and markedly elevated LDH level, combined with EBV infection. The diagnosis was HLH. Chaker et al. also reported an EBV positive case of HL-type RS presenting as HLH (17). Therefore, it is important to identify and diagnose HLH promptly, while also being vigilant for the occurrence of RS.
The clonal correlation before and after transformation is crucial for the treatment and prognosis of RS. So far, the clonal correlation between DLBCL-type RS and CLL/SLL has been fully elucidated. A previous study found through NGS that mutations in TP53, NOTCH1, MYC, and CDKN2A genes were closely associated with 90% of DLBCL-type RS cases (1). Moreover, a recent study confirmed through single-cell sequencing that the MYC BACH2/JUNB/RGS2, and IFR9/PRDX1 pathways, amino acid metabolism, proteasome pathway, and immune escape are all involved in the occurrence and development of DLBCL-type RS (18). However, the clonal correlation between HL-type RS and CLL/SLL has rarely been reported, and only a few studies have analyzed the clonal correlation between HL-type RS and CLL/SLL by detecting the IgH rearrangement or IgHV mutation rate by PCR. So, the clonal correlation between HL-type RS and CLL/SLL, and the biological characteristics of HL-type RS are still unknown (1, 14). Dujardin et al. detected IgH rearrangements by PCR and found that 7 of 12 HL-type RS patients had clonal correlation with pre-transformation CLL/SLL, who were identical to classical RS and had a poor prognosis. The other 5 HL-type RS patients had no clonal correlation with pre-transformation CLL/SLL, and these patients were usually EBV positive and had a better prognosis (5). Mauro et al. also used PCR and fluorescence in situ hybridization techniques and found that 18 of 28 HL-type RS patients had no IgHV mutations, 13 of 25 had no chromosomal abnormalities, 6 of 25 had 13q-, 6/25 had +12, 1/25 had 17p-, and 1of 14 had TP53 mutations (14). Xiao et al. used two methods, laser capture microdissection and PCR, to demonstrate that the expression of ZAP-70 in CLL cells from pathological sections correlated with the clonal correlation of HL-type RS, whereas EBV status or morphological patterns were not associated with the clonal correlation (16). HL-type RS transformed from ZAP-70-positive CLL was not related to CLL clones, whereas HL-type RS transformed from ZAP-70-negative CLL was related to CLL clones, and rarely could clonally-independent HL-type RS cells be derived from ZAP-70-negative CLL (16, 19). For the first time in our case of HL-type RS, detailed comparison of Ig gene rearrangement clonal and gene mutation analyses were performed in pathological tissues before and after transformation, using both PCR and NGS in a large panel. Before transformation, IgH and IgK rearrangement clones were positive, IgL rearrangement clone was negative, and CTNNB1, ID3, IGLL5, and MAP2K1 gene mutations were detected. After transformation, IgH rearrangement clone was positivity, IgK and IgL rearrangement clones were negative, and TET2, FAS, IGLL5, and VPREB1 gene mutations were detected (Tables 1, 2). It can be clearly seen from the results that the HL-type RS in this case was not related to the SLL clone before transformation, and that this patient harbored an EBV infection and had a good outcome after chemotherapy, consistent with a report in the literature (5). Detailed Ig gene rearrangement clonal analysis and gene mutations data before and after the transformation of this patient will enrich the data on the biological characteristics of HL-type RS.
Therapeutic advances in RS in recent years have mainly focused on DLBCL-type RS. Ongoing clinical research continues to explore new therapies or drugs including chimeric antigen receptor-T therapy, bispecific antibodies, Bruton tyrosine kinase inhibitors, Programmed cell death protein 1/Programmed death-ligand 1 inhibitors, Venetoclax, and antibody coupled drugs, with some breakthroughs realized (20, 21). However, relatively few new drugs can be used for HL-type RS. Given the extremely low prevalence of HL-type RS, clinical trials of new drugs are impossible (1). Because of similarities with primary HL, HL-type RS was usually treated with ABVD or modified ABVD chemotherapy regimens as the first-line regimens, with treatment response rates of 40-60% and median OS of approximately 4 years for HL-type RS (8, 9, 13, 15). A recent study, which is one of the largest single-center real-world population-based cohort studies to date (11), found that 32 patients with HL-type RS had a median OS of 31 months, with 2-year progression-free survival (PFS)and OS of 47% and 57%, respectively. The median OS was significantly higher than the previously reported median OS of 8-9.4 months for DLBCL-type RS (22, 23), but lower than the median OS for primary HL. The 5-year OS of advanced primary HL has reached 87% (4). However, patients who were able to tolerate chemotherapy with standard ABVD or modified ABVD regimens showed an improved 2-year PFS and OS of 70% and 74%, respectively, which was similar to the 2-year OS of elderly primary HL patients reported by the same researchers earlier (24). However, the median age of the cohort population was as high as 71 years, and more than one-third of the patients could not tolerate the ABVD regimen. As a result, the survival of the entire population was significantly worse than that of primary HL. Another multicenter study from the United States, with the largest number of HL-type RS patients to date, reported a total of 94 patients enrolled in the study, with a median OS of up to 65 months and a 2-year OS of 72%. The patients who were able to tolerate the standard ABVD-based therapy had a median OS of up to 13.2 years, far exceeding the median OS of the entire HL-type RS population. This may be due to the higher proportion of people aged <65 years who could tolerate the ABVD regimen in this study (25). The collective evidence confirms that the ABVD regimen is the standard first-line regimen for well-tolerated HL-type RS patients. The main possible adverse event of this regimen is severe pulmonary toxicity caused by exposure to bleomycin. If mid-term PET/CT reveals remission and no significant changes, bleomycin can be removed and the AVD regimen used after two cycles of the ABVD regimen (26). Concerning new drugs, both PD1 inhibitors and Brentuximab vedotin (an anti-CD30 monoclonal antibody coupled to monomethyl auristatin E as an ADC) have proven effective in primary refractory relapsed (R/R) HL, but there is insufficient data regarding their use in R/R HL-type RS (27, 28). Brentuximab vedotin in combination with AVD (A+AVD) has been used in the first-line treatment of advanced primary HL with superior results to the ABVD regimen (29). A recent case report applied A+AVD for the first-line treatment of a case of HL-type RS with good results (30). Another case of R/R HL-type RS resistant to Bendamustine, which progressed and subsequently received sequential treatment with Brentuximab vedotin and Pembrolizumab, with encouraging results (31). Based on these findings, PD1 inhibitors and Brentuximab vedotin might become important choices for the first- or second-line treatment of Hodgkin HL-type RS in the future. Given the older onset age of HL-type RS, very few patients could undergo hematopoietic stem cell transplantation (HSCT), and there is still a lack of evidence to support the role of HSCT. In the largest cohort of HL-type RS cases reported to date, only 7 of 94 patients received HSCT after the first CR, but their 2-year OS was not significantly better than that of HL-type RS patients who did not receive HSCT (25).
In this case, although the patient was diagnosed with RS at the age of 76-years-of-age, we chose a modified R-ABVD regimen based on the pathological results, which prolonged the chemotherapy intervals of the ABVD regimen as well as reducing the doses of various drugs and the total number of cycles, in combination with Zanubrutinib to control SLL. The treatment achieved very satisfactory results.
Up to now, after more than 15 months of follow-up, the patient is still in CR, similar to the aforementioned results from the literature. In the future, if the disease progresses, PD1 inhibitors and Brentuximab vedotin will be the second-line treatment choice.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Ethics statement
The studies involving humans were approved by the Ethics Committee of Guangzhou First People’s Hospital, Guangdong Medical University. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. The requirement of ethical approval was waived by the Ethics Committee of Guangzhou First People’s Hospital, Guangdong Medical University for the studies involving animals because We have got an ethical approval. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
XZ: Data curation, Formal analysis, Investigation, Methodology, Project administration, Writing – original draft. WD: Data curation, Formal analysis, Investigation, Resources, Validation, Writing – review & editing. ZZ: Investigation, Methodology, Project administration, Resources, Writing – review & editing. JL: Data curation, Investigation, Methodology, Project administration, Resources, Writing – review & editing. WZ: Conceptualization, Funding acquisition, Methodology, Project administration, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Guangzhou Planned Project of Science and Technology, China (2023A03J0963).
Acknowledgments
We thank International Science Editing (http://www.internationalscienceediting.com) for editing this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Rossi D, Spina V, Gaidano G. Biology and treatment of Richter syndrome. Blood. (2018) 131:2761–72. doi: 10.1182/blood-2018-01-791376
2. Parry E, Leshchiner I, Guièze R, Johnson C, Tausch E, Parikh S, et al. Evolutionary history of transformation from chronic lymphocytic leukemia to Richter syndrome. Nat Med. (2023) 29:158–69. doi: 10.1038/s41591-022-02113-6
3. Abrisqueta P, Delgado J, Alcoceba M, Oliveira A, Loscertales J, Hernández-Rivas J, et al. Clinical outcome and prognostic factors of patients with Richter syndrome: real-world study of the Spanish Chronic Lymphocytic Leukemia Study Group (GELLC). Br J haematology. (2020) 190:854–63. doi: 10.1111/bjh.v190.6
4. Siegel R, Miller K, Fuchs H, Jemal A. Cancer statistics, 2021. CA: Cancer J Clin. (2021) 71:7–33. doi: 10.3322/caac.21654
5. Dujardin F, Lefrancq T, Bléchet C, Boni-Boka M, Sénecal D, Desmoulins I, et al. Hodgkin's disease variant of Richter's syndrome. Two cases and literature review. Annales pathologie. (2008) 28:311–6. doi: 10.1016/j.annpat.2007.11.009
6. Swerdlow S, Campo E, Pileri S, Harris N, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. (2016) 127:2375–90. doi: 10.1182/blood-2016-01-643569
7. Parikh S, Kay N, Shanafelt T. How we treat Richter syndrome. Blood. (2014) 123:1647–57. doi: 10.1182/blood-2013-11-516229
8. Parikh S, Habermann T, Chaffee K, Call T, Ding W, Leis J, et al. Hodgkin transformation of chronic lymphocytic leukemia: Incidence, outcomes, and comparison to de novo Hodgkin lymphoma. Am J Hematol. (2015) 90:334–8. doi: 10.1002/ajh.23939
9. Bockorny B, Codreanu I, Dasanu C. Hodgkin lymphoma as Richter transformation in chronic lymphocytic leukaemia: a retrospective analysis of world literature. Br J haematology. (2012) 156:50–66. doi: 10.1111/j.1365-2141.2011.08907.x
10. Sha Y, Jiang R, Miao Y, Qiu T, Qin S, Qiu J, et al. Clonality relatedness and molecular characteristics of Richter transformation. Zhonghua xueyexue zazhi. (2022) 43:841–7. doi: 10.3760/cma.j.issn.0253-2727.2022.10.007
11. Zhu K, Jamroz A, Huang S, Villa D, Freeman C, Scott D, et al. Outcomes of Hodgkin variant Richter transformation in chronic lymphocytic leukaemia and small lymphocytic lymphoma in British Columbia. Br J haematology. (2022) 198:684–92. doi: 10.1111/bjh.v198.4
12. Drozd-Sokołowska J, Zaucha J, Żółtak T, Jamroziak K, Grzybowska-Izydorczyk O, Witkowska M, et al. Hodgkin lymphoma transformation of chronic lymphocytic leukemia-A real life data from the Polish Lymphoma Research Group. Hematological Oncol. (2019) 37:383–91. doi: 10.1002/hon.2624
13. Tsimberidou A, O'Brien S, Kantarjian H, Koller C, Hagemeister F, Fayad L, et al. Hodgkin transformation of chronic lymphocytic leukemia: the M. D. Anderson Cancer Center experience. Cancer. (2006) 107:1294–302. doi: 10.1002/cncr.v107:6
14. Mauro F, Galieni P, Tedeschi A, Laurenti L, Del Poeta G, Reda G, et al. Factors predicting survival in chronic lymphocytic leukemia patients developing Richter syndrome transformation into Hodgkin lymphoma. Am J Hematol. (2017) 92:529–35. doi: 10.1002/ajh.24714
15. Tadmor T, Shvidel L, Goldschmidt N, Ruchlemer R, Fineman R, Bairey O, et al. Hodgkin's variant of Richter transformation in chronic lymphocytic leukemia; a retrospective study from the Israeli CLL study group. Anticancer Res. (2014) 34:785–90.
16. Xiao W, Chen W, Sorbara L, Davies-Hill T, Pittaluga S, Raffeld M, et al. Hodgkin lymphoma variant of Richter transformation: morphology, Epstein-Barr virus status, clonality, and survival analysis-with comparison to Hodgkin-like lesion. Hum Pathol. (2016) 55:108–16. doi: 10.1016/j.humpath.2016.04.019
17. Chaker L, Segeren C, Bot F, Maartense E. Haemophagocytic syndrome and Hodgkin's disease variant of Richter's syndrome after fludarabine for CLL. Eur J haematology. (2010) 85:91–2. doi: 10.1111/j.1600-0609.2010.01444.x
18. Li H, Yuan L, Wang P, Sheng Y, Fu Z, Peng H. Clonal architecture and single-cell transcriptome landscape in Richter's syndrome. Br J haematology. (2023) 202:1055–60. doi: 10.1111/bjh.v202.5
19. Tzankov A, Fong D. Hodgkin's disease variant of Richter's syndrome clonally related to chronic lymphocytic leukemia arises in ZAP-70 negative mutated CLL. Med Hypotheses. (2006) 66:577–9. doi: 10.1016/j.mehy.2005.09.007
20. Briski R, Taylor J. Treatment of Richter transformation of chronic lymphocytic leukemia in the modern era. Cancers. (2023) 15:1857. doi: 10.3390/cancers15061857
21. Bajwa A, Voorhees T, Kittai A. Cellular therapy advances in chronic lymphocytic leukemia and Richter's syndrome. Curr problems Cancer. (2022) 46:100827. doi: 10.1016/j.currproblcancer.2021.100827
22. Wang Y, Tschautscher M, Rabe K, Call T, Leis J, Kenderian S, et al. Clinical characteristics and outcomes of Richter transformation: experience of 204 patients from a single center. Haematologica. (2020) 105:765–73. doi: 10.3324/haematol.2019.224121
23. Al-Sawaf O, Robrecht S, Bahlo J, Fink A, Cramer P, V Tresckow J, et al. Richter transformation in chronic lymphocytic leukemia (CLL)-a pooled analysis of German CLL Study Group (GCLLSG) front line treatment trials. Leukemia. (2021) 35:169–76. doi: 10.1038/s41375-020-0797-x
24. Cheng P, Villa D, Gerrie A, Freeman C, Slack G, Gascoyne R, et al. The outcome of older adults with classic Hodgkin lymphoma in British Columbia. Blood Adv. (2022) 6:5924–32. doi: 10.1182/bloodadvances.2022008258
25. Stephens D, Boucher K, Kander E, Parikh S, Parry E, Shadman M, et al. Hodgkin lymphoma arising in patients with chronic lymphocytic leukemia: outcomes from a large multi-center collaboration. Haematologica. (2021) 106:2845–52. doi: 10.3324/haematol.2020.256388
26. Martin W, Ristow K, Habermann T, Colgan J, Witzig T, Ansell S. Bleomycin pulmonary toxicity has a negative impact on the outcome of patients with Hodgkin's lymphoma. J Clin Oncol. (2005) 23:7614–20. doi: 10.1200/JCO.2005.02.7243
27. Zhang Y, Xing Z, Mi L, Li Z, Zhu J, Wei T, et al. Novel agents for relapsed and refractory classical Hodgkin lymphoma: A review. Front Oncol. (2022) 12:929012. doi: 10.3389/fonc.2022.929012
28. Gualberto A. Brentuximab Vedotin (SGN-35), an antibody-drug conjugate for the treatment of CD30-positive Malignancies. Expert Opin investigational Drugs. (2012) 21:205–16. doi: 10.1517/13543784.2011.641532
29. Ansell S, Radford J, Connors J, Długosz-Danecka M, Kim W, Gallamini A, et al. Overall survival with brentuximab vedotin in stage III or IV Hodgkin's lymphoma. New Engl J Med. (2022) 387:310–20. doi: 10.1056/NEJMoa2206125
30. Miki K, Ogasawara R, Sugimura S, Sugita J, Nozu R, Kojima K, et al. A case of Hodgkin lymphoma-type Richter syndrome presenting as small-intestinal perforation. Int J Hematol. (2023) 118:766–71. doi: 10.1007/s12185-023-03655-2
Keywords: Hodgkin lymphoma variant of Richter syndrome, chronic lymphocytic leukemia, small lymphocytic lymphoma, hemophagocytic syndrome, clonal relationship
Citation: Zheng X, Ding W, Zhu Z, Li J and Zhong W (2024) Clonally unrelated HL-type RS manifested as hemophagocytic syndrome: a case report and literature review. Front. Oncol. 14:1472560. doi: 10.3389/fonc.2024.1472560
Received: 29 July 2024; Accepted: 15 November 2024;
Published: 05 December 2024.
Edited by:
Jeffrey J. Pu, Harvard Medical School, United StatesReviewed by:
Maurizio Aricò, Dept. of Pediatrics, ItalyArtem Oganesyan, Hematology Center After R. Yeolyan, Armenia
Copyright © 2024 Zheng, Ding, Zhu, Li and Zhong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weijie Zhong, ZXl3ZWlqaWV6aG9uZ0BzY3V0LmVkdS5jbg==